

Abstract
Interfering with the self-assembly of virus nucleocapsids is a promising approach for the development of novel antiviral agents. Applied to hepatitis B virus (HBV), this approach has led to several classes of capsid assembly modulators (CAMs) that target the virus by either accelerating nucleocapsid assembly or misdirecting it into non-capsid-like particles, thereby inhibiting the HBV replication cycle. Here, we have assessed the structures of early nucleocapsid assembly intermediates, bound with and without CAMs, using molecular dynamics simulations. We find that distinct conformations of the intermediates are induced depending on whether the bound CAM accelerates or misdirects assembly. Specifically, the assembly intermediates with bound misdirecting CAMs appear flattened relative to those with bound accelerators. Finally, the potency of CAMs within the same class was studied. We find that increased contacts with the capsid protein and favorable binding energies inferred from free-energy perturbation calculations are indicative of increased potency.
Graphical Abstract
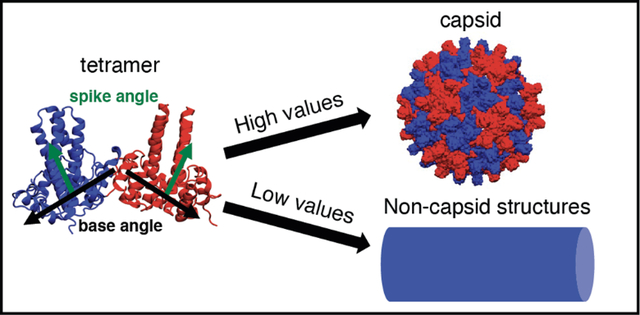
Introduction
Hepatitis B virus (HBV) is the leading cause of liver disease, including cirrhosis, hepatocellular carcinoma and liver failure.1 The chronic infection affects roughly 250 million people worldwide and results in approximately 800,000 deaths each year.1 A promising orthogonal approach for eliminating the infection is to target the HBV nucleocapsid.2–10 Capsid assembly modulators (CAMs) are small molecules that affect capsid assembly by interacting with the capsid proteins.2–7,9,10 Different assembly effects, such as acceleration or misdirection, have been achieved by different CAMs.2–10 It was also discovered that CAMs can both inhibit virus replication and interfere with cccDNA synthesis, suggesting that they could help eliminate the virus from hepatocytes more efficiently, thereby leading to an effective cure.2,3,11–13 However, a better understanding of HBV capsid assembly and the interference mechanisms of novel CAMs is needed for improved rational drug design efforts.
HBV capsid protein primarily exists as a homodimer in solution11 and its N-terminal domain (Cp149) is sufficient for forming regular capsids, while the C-terminal region contains an arginine rich domain implicated in packaging pregenomic RNA (pgRNA).14,15 The Cp149 dimer consists of two domains: the dimerization interface and the assembly interface (Figure 1A), with the latter interface forming inter-dimer contacts during capsid assembly.16,17 Previous studies proposed that the Cp149 dimers trigger capsid assembly by adopting an energetically unfavorable “assembly-active” conformation, which in turn leads to assembly nucleation (Figure 1C).18–21 It was also concluded that the assembly is nucleated by the formation of a hexamer, a triangular trimer of dimers, which is the rate-limiting step, and is followed by successive addition of dimers or other small intermediates, e.g., tetramers or hexamers, until the complete nucleocapsid is formed.18–20
Figure 1:
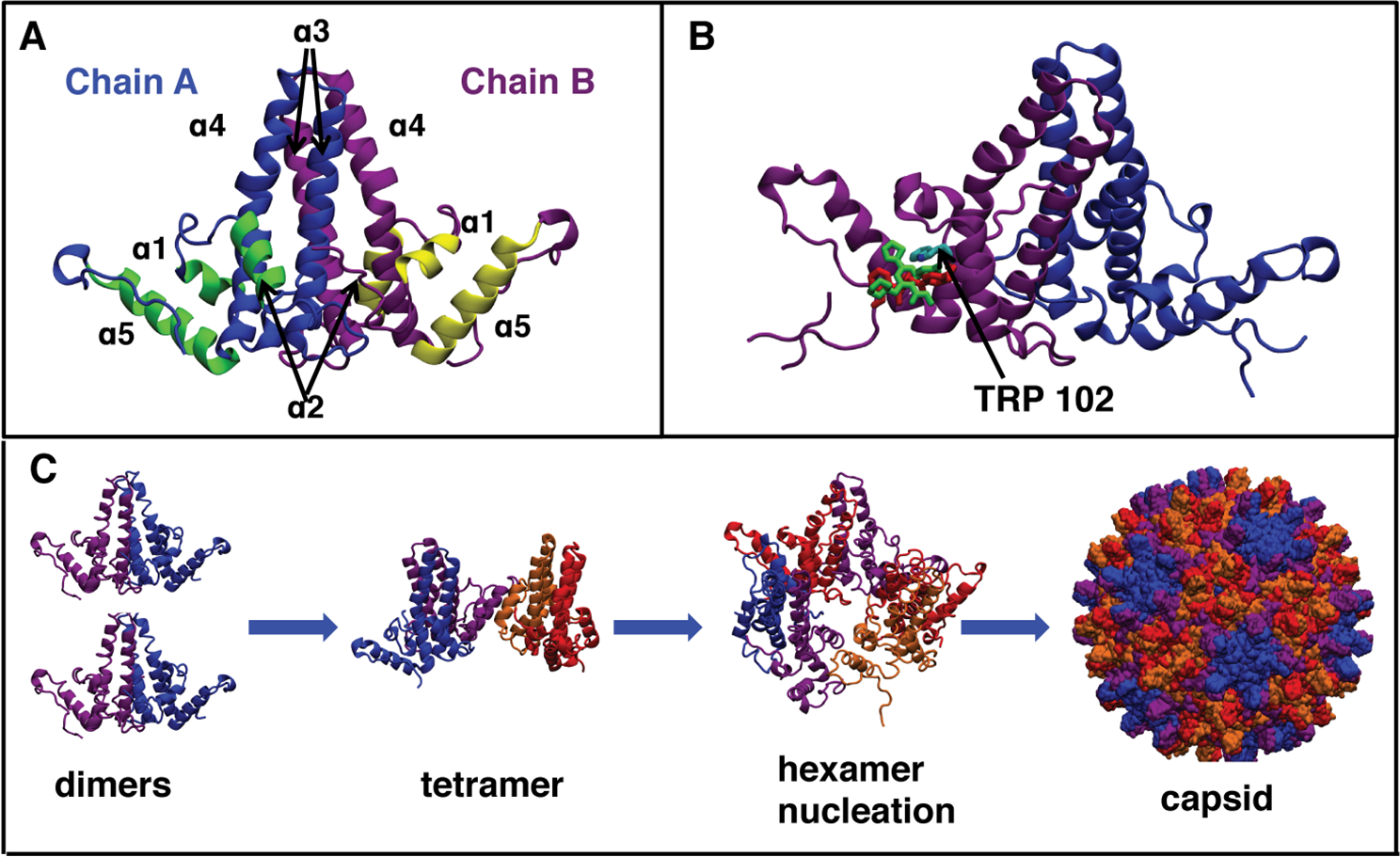
HBV capsid structure and assembly. (A) Structure of Cp149 dimer taken from the capsid structure (PDB code 3J2V). The names of helices α1–5 are indicated; yellow and green helices form the assembly interface, while the dimerization interface helices, α3-α4, are colored blue and purple respectively. (B) Overlap of GLS4 and AT130 binding sites after alignment of the bound protein structures (PDB codes 5E0I and 4G93, respectively). Both compounds occupy a similar space in the HAP pocket and interact with Trp102. (C) HBV capsid assembly process based on experimental data and mathematical modeling.18,30–32
Several factors, such as ions,18–20 mutations22–24 and CAMs2–7 alter kinetics and/or thermodynamics of HBV capsid assembly, potentially preventing the formation of normal capsids and, in some cases, localizing the capsid in the cytoplasm.25 It has been suggested that the kinetic effects are caused by increased concentration of the “assembly-active” dimer conformation,19,20 while thermodynamic effects are caused by more favorable inter-dimer contacts.3,26,27 CAMs that alter HBV capsid assembly are divided into several structural and mechanistic classes (Figure S1). Heteroaryldihydropyrimidines (HAPs) misdirect capsid assembly into non-capsid structures,2,3 while phenylpropenamides (PPAs)4,5 and sulfamoyl benzamides (SBAs)6–8 induce formation of capsids lacking the viral pgRNA. Although both PPAs and SBAs cause formation of empty capsids, it has been shown that some PPAs, e.g., AT130, also increase the assembly rate of Cp149.5 In contrast, no changes in assembly rates were observed for Cp149 assembly with and without SBAs, suggesting that SBAs and PPAs alter the capsid assembly differently.6,7,28 More recently a novel CAM class, based on a glyoxamidopyrrolo backbone (GLP-26), has been shown to robustly inhibit HBV replication and modify the assembly of the HBV nucleocapsid.25,29 Addition of GLP-26 to Cp149 prior to assembly resulted in the formation of small, spherical, misshapen particles, distinct from structures observed for either ATs or HAPs.25
Recent work utilizing ligands derived from HAP, PPA, and SBA classes have shown potent inhibition of extracellular HBV DNA, reduction in cccDNA, low cytotoxicity, with several having progressed to pre-clinical through phase II clinical trials.33,34 These new ligands include GLS4, a HAP compound, as well as several PPA and SBA compounds. Research continues to discover novel chemotypes such as phthalazinones35 and pyrazoles.34 Moreover, GLP-26, based on a glyoxamidopyrrole backbone, displays robust low-nanomolar activity in vitro and demonstrated reduction of HBV DNA and other HBV markers in a humanized HBV mouse model,25,29 and one of its related derivatives (ALG-184) is in Phase 1b clinical development.34
Crystal and cryogenic electron microscopy (cryo-EM) structures show that all CAMs bind in the same HAP pocket at the inter-dimer interface (Figure 1B).7,26,27,36 Slightly altered dimer–dimer orientations and several hydrophobic contacts between the CAM and protein residues in the pocket were observed, explaining the more energetically favorable dimer-dimer contacts upon CAM-binding observed experimentally3,26,27. Nevertheless, further studies are needed to elucidate the enhanced assembly kinetics and the misdirection of assembly caused by HAP compounds. While the known structures were obtained from CAMs binding to either preformed capsids or to a hexamer of the assembly-incompetent Y132A mutant,23 it is possible that CAMs induce distinct structural changes in early assembly intermediates. Although these intermediates have been detected by mass spectroscopy,30–32 a paucity of information is available about their structure, their dynamics, and how these properties are altered upon CAM binding.
Both capsids and transient assembly intermediates can be studied by means of molecular dynamics (MD) simulations. It was previously shown that HAPs decrease the structural fluctuations of the Cp149 hexamer37 and flatten free tetramers and the hexameric units in the assembled capsid.38,39 Additionally, prior MD simulations of Cp149 capsids showed that they are highly flexible, and that both CAMs and mutations can alter capsid dynamics.24,40 Recently, MD simulations of stress-induced capsid disassembly were used to identify weak points as well as conserved residues whose interactions are readily disrupted.41 Here, using MD simulations, we have assessed the structure and dynamics of Cp149 tetramers and hexamers in the apo state as well as in the presence of CAMs from the four structural classes of CAMs with distinct effects on the assembly (HAPs, PPAs, SBAs and GLPs). Noticeable structural changes in these intermediates were observed for several classes of compounds, explaining their unique effects on assembly. In addition, we have performed hydrogen bonding analysis and free energy perturbation (FEP) calculations in order to investigate the influence of structural changes on the binding affinity and its correlation to ligand potency.
Results
Conformational changes in early assembly intermediates observed by MD
We performed MD simulations of a truncated wild type (WT) Cp149 tetramer and hexamer, as well as tetramers with either Y132A or V124W mutations (summarized in Table 1). These mutations where chosen because they alter the assembly mechanism in distinct, yet known, ways with the former inhibiting and the latter accelerating assembly.22,23 Thus, we are able to distinguish conformational preferences of the tetramer in the case of capsid assembly inhibition or acceleration. Although HBV nucleocapsids contain four quasi-equivalent tetramers and two quasi-equivalent hexamers, we have determined that the ABCD tetramer and the CDCDCD hexamer are the best starting points for tetramer and hexamer simulations (see Supporting Information for an explanation of the nomenclature).
Table 1:
List of simulated systems, BAY stands for BAY41-4109, “tetra” stands for tetramer and “hexa” stands for hexamer. The middle column indicates which PDB structure was used as a starting state, with the exception of GLP compounds, which started from a docked structure.29 The last column lists the total simulation time for each system; in aggregate, the combined simulation time is ∼11 μs. All simulations were performed at pH 7 and with 0.15 M NaCl.
| System | Structure | number of simulations × time (ns) |
|---|---|---|
| Apo | ||
| apo tetra | 3J2V | 700+1000+2×150 |
| apo hexa sym | 3J2V | 2×150 |
| apo hexa asym | 3J2V | 3×150 |
| tetra V124W | 3J2V | 2×150 |
| tetra Y132A | 3J2V | 2×150 |
| apo tetra | 5E0I | 2×150 |
| apo hexa | 5E0I | 2×150 |
| tetra Y132A | 5E0I | 2×150 |
| HAP | ||
| tetra with HAP1, HAP4 HAP7, HAP12, GLS4 or BAY | 5E0I | 2×150 |
| hexa with 3 GLS4 | 5E0I | 2×150 |
| hexa with 1 GLS4 | 5E0I | 2×150 |
| AT | ||
| tetra with AT130 | 4G93 | 2×150 |
| tetra with AT61 | 4G93 | 3×150 |
| hexa with 3 AT130 | 4G93 | 2×150 |
| hexa with 1 AT130 | 4G93 | 2×150 |
| SBA | ||
| tetra with SBA_R01, DRV23 | 5T2P | 2×150 |
| tetra with DRV01 | 5T2P | 3×150 |
| GLP | ||
| tetra with GLP-26, Comp2 | 5T2P | 2×150 |
| tetra with GLP-17A, GLP-17C | 5T2P | 3×150 |
| hexa with 1 GLP-26 | 5T2P | 3×150 |
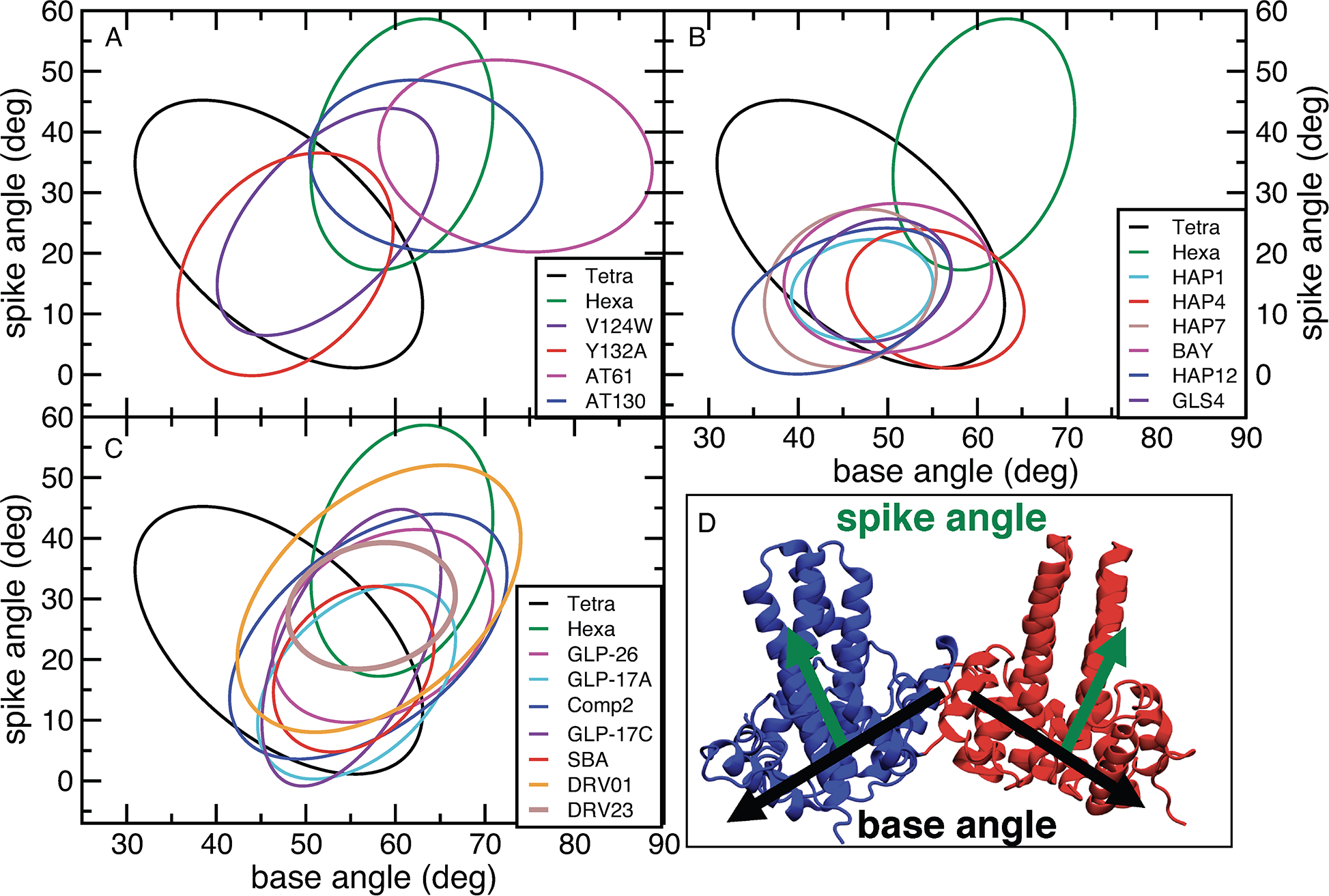
Significant differences in inter-dimer orientation were observed for the hexamer in comparison to the tetramer. These structural changes are well-described by changes in spike and base angles of the tetrameric unit (Figure 2D). The spike angle was calculated between the dimerization interfaces (α3 and α4 helices) of each dimer and describes the “bending” of the tetrameric unit. The base angle was calculated from the positions of the interface-forming α5 helices in each dimer and describes the “opening” and “closure” of the tetrameric unit. To illustrate the observed structural differences, the distributions of spike and base angles for each system were projected on a two-dimensional scatter plot, and standard deviations ellipses (SDEs; Methods) were used to illustrate the spread of the distributions in our simulations.42 The SDEs for the studied systems were quantitatively compared by calculating their fractional overlap area (FOA) and reported in Table 2. The values of FOA can range from 0% (no overlap) to 100% (perfect overlap). Values over 50% indicate notable structural similarity, while values close to 0% indicate significant structural differences.
Figure 2:
Comparison of standard deviation ellipses (SDEs) for spike and base angle distributions for selected simulations. SDEs are centered on the average values of a distribution. Their width and height are based on the standard deviation of the corresponding variables, while their rotation is based on the correlation between two variables. The ellipses shown here are scaled to envelop 90% of the sampled distributions. The locations of the starting structures in the graphs are shown in Figure S9. With the exception of Hexa SDE, all other SDEs are for tetramer structures. (A) SDEs of base and spike angles for the simulated apo structures and for PPA compounds, which accelerate assembly. (B) SDEs of base and spike angles for selected bound misdirecting CAMs. (C) SDEs of base and spike angles for SBAs, which do not alter the empty capsid assembly, and GLPs, which cause formation of misformed capsids. In (B) and (C) the results for the apo tetramer and hexamer are added for comparison. (D) Definitions of spike and base angles. See Methods and Supporting Information for details.
Table 2:
Fractional area overlaps for SDEs in %. Each row shows the fractional overlap area of the total SDE for that system with the systems specified in each column. To estimate the differences between the independent simulations for each system, we calculated the overlap area between the SDE of each independent simulation and the SDE from the combined simulations as a fraction of the latter SDE area. Averages of these fractional areas from all independent simulations are shown as diagonal values in bold. The following abbreviations are used: Tetra (T), Hexa (H) Comp2 (C2), DRV01 (01), DRV23 (23), BAY41-4109 (BAY).
| System | Apo | HAP | AT | SBA | GLP | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T | H | Y132A T | V124W T | 1 T | 4 T | 7 T | 12 T | BAY T | GLS4 T | GLS4 H | 130 T | 61 T | 130 H | SBA T | 01 T | 23 T | 17A T | 17C T | C2 T | 26 T | 26 H | |
|
| ||||||||||||||||||||||
| Apo | ||||||||||||||||||||||
|
| ||||||||||||||||||||||
| T | 72 | 14 | 52 | 45 | 21 | 33 | 33 | 3 | 45 | 27 | 15 | 07 | 0 | 0 | 33 | 47 | 16 | 40 | 41 | 51 | 32 | 24 |
| H | 20 | 70 | 20 | 45 | 0 | 7 | 2 | 2 | 11 | 3 | 4 | 66 | 40 | 27 | 23 | 86 | 41 | 27 | 38 | 62 | 54 | 43 |
| Y132A T | 82 | 21 | 65 | 72 | 34 | 28 | 63 | 61 | 61 | 42 | 03 | 12 | 0 | 1 | 38 | 64 | 27 | 43 | 50 | 68 | 43 | 18 |
| V124W T | 72 | 48 | 71 | 60 | 31 | 27 | 43 | 39 | 54 | 39 | 5 | 38 | 14 | 2 | 42 | 90 | 44 | 47 | 63 | 87 | 63 | 39 |
|
| ||||||||||||||||||||||
| HAP | ||||||||||||||||||||||
|
| ||||||||||||||||||||||
| 100 T | 96 | 0 | 100 | 91 | 87 | 56 | 100 | 100 | 100 | 91 | 0 | 0 | 0 | 0 | 51 | 68 | 3 | 60 | 60 | 89 | 38 | 7 |
| 4 T | 90 | 12 | 48 | 47 | 33 | 77 | 41 | 46 | 71 | 46 | 48 | 0 | 0 | 0 | 71 | 62 | 13 | 84 | 76 | 78 | 53 | 62 |
| 7 T | 80 | 3 | 98 | 66 | 53 | 37 | 69 | 88 | 80 | 61 | 0 | 0 | 0 | 0 | 37 | 53 | 9 | 44 | 47 | 67 | 31 | 6 |
| 12 T | 67 | 4 | 86 | 56 | 49 | 38 | 81 | 76 | 69 | 57 | 3 | 0 | 0 | 0 | 36 | 45 | 7 | 43 | 44 | 59 | 29 | 10 |
| BAY T | 96 | 16 | 81 | 72 | 46 | 55 | 69 | 65 | 78 | 57 | 18 | 3 | 0 | 0 | 61 | 70 | 20 | 68 | 70 | 88 | 51 | 32 |
| GLS4 T | 100 | 8 | 98 | 91 | 72 | 63 | 91 | 94 | 100 | 91 | 5 | 0 | 0 | 0 | 62 | 80 | 15 | 70 | 72 | 98 | 53 | 18 |
| GLS4 H | 65 | 13 | 8 | 13 | 0 | 80 | 0 | 6 | 38 | 6 | 83 | 1 | 0 | 0 | 50 | 41 | 7 | 59 | 45 | 56 | 44 | 100 |
|
| ||||||||||||||||||||||
| AT | ||||||||||||||||||||||
|
| ||||||||||||||||||||||
| 130 T | 11 | 76 | 13 | 42 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 75 | 63 | 31 | 15 | 90 | 37 | 19 | 32 | 69 | 56 | 42 |
| 61 T | 0 | 34 | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 46 | 58 | 15 | 1 | 45 | 11 | 3 | 8 | 31 | 23 | 18 |
| 130 H | 0 | 100 | 3 | 70 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 97 | 64 | 90 | 12 | 100 | 55 | 20 | 49 | 76 | 67 | 76 |
|
| ||||||||||||||||||||||
| SBA | ||||||||||||||||||||||
|
| ||||||||||||||||||||||
| SBA T | 85 | 4 | 61 | 69 | 28 | 67 | 38 | 41 | 74 | 43 | 28 | 22 | 3 | 6 | 87 | 89 | 42 | 100 | 95 | 100 | 82 | 63 |
| 01 T | 45 | 56 | 39 | 55 | 14 | 22 | 21 | 19 | 32 | 21 | 9 | 51 | 35 | 8 | 34 | 53 | 31 | 37 | 44 | 75 | 59 | 38 |
| 23 T | 51 | 87 | 53 | 88 | 2 | 15 | 12 | 9 | 30 | 13 | 5 | 68 | 27 | 31 | 52 | 100 | 86 | 58 | 84 | 100 | 99 | 70 |
|
| ||||||||||||||||||||||
| GLP | ||||||||||||||||||||||
|
| ||||||||||||||||||||||
| 17A T | 81 | 37 | 55 | 61 | 26 | 63 | 37 | 38 | 65 | 39 | 27 | 23 | 5 | 8 | 79 | 78 | 38 | 66 | 90 | 93 | 72 | 61 |
| 17C T | 72 | 46 | 56 | 71 | 23 | 50 | 34 | 35 | 58 | 34 | 18 | 33 | 11 | 16 | 65 | 81 | 48 | 78 | 53 | 92 | 75 | 55 |
| C2 T | 57 | 46 | 48 | 61 | 21 | 32 | 30 | 29 | 46 | 29 | 14 | 45 | 28 | 16 | 43 | 87 | 36 | 51 | 58 | 59 | 69 | 48 |
| 26 T | 51 | 59 | 44 | 65 | 13 | 32 | 21 | 21 | 39 | 23 | 16 | 53 | 30 | 20 | 52 | 98 | 51 | 57 | 68 | 100 | 82 | 62 |
| 26 H | 39 | 47 | 18 | 40 | 2 | 37 | 4 | 7 | 24 | 8 | 36 | 40 | 23 | 23 | 40 | 64 | 36 | 48 | 50 | 71 | 62 | 42 |
A wide range of base angles (31–63°) was observed for the tetramer (Figure 2A and Table 3). In comparison, the hexamer simulations displayed a narrower range (49–72°), centered around 60°, as expected for a planar, symmetric hexamer. The closed hexameric structure does not have any free dimer interfaces, which likely contributes to smaller ranges of sampled base angles. The hexamer also adopted larger spike angles than the tetramer (17–59° compared to 1–45°, respectively). The tetramer FOA with the hexamer is only 14%, indicative of significant structural differences in terms of spike and base angles for the two systems. The FOA with the hexamer increases significantly to 48% for the assembly-enhancing V124W mutant tetramer (Figure 2A), which could decrease the energetic barrier against nucleus formation.22 In contrast, the assembly-incompetent Y132A mutant tetramer23 showed similar FOA with the hexamer state as the WT tetramer (21%). If Y132A inhibited assembly by inducing greater structural differences between hexamer and tetramer structures, we would expect a decreased FOA in comparison to the WT state. Because the FOA does not change, our results suggest that the assembly inhibition by this mutation may not be caused by an altered inter-dimer orientation. To ensure that our results are not dependent on the initial structures, we also simulated several systems using different starting states (Table 1) and arrived at the same conclusions (see Repeated Simulations in Supporting Information). Furthermore, extended simulations of the apo tetramer led to similar base and spike angles, as well as RMSD in comparison to the shorter 150-ns runs (Figures S11, S12, S16).
Table 3:
The range of sampled base and spike angles in deg for all simulated systems. The values are based on the calculated SDEs. The PDB used for each run or set of runs is indicated in parentheses.
| System (PDB) | Base | Spike (deg) | System (PDB) | Base | Spike (deg) |
|---|---|---|---|---|---|
|
| |||||
| Apo (3J2V and 5E0I) | Apo (3J2V and 5E0I) | ||||
|
| |||||
| WT Tetra (3J2V) | 31–63 | 1–45 | WT Hexa Asym (3J2V) | 49–72 | 6–59 |
| WT Hexa Sym (3J2V) | 51–71 | 17–59 | Y132A Tetra (3J2V) | 40–74 | −4–65 |
| V124 Tetra (3J2V) | 40–65 | 6–44 | WT Tetra (5E0I) | 36–62 | −7–50 |
| WT Hexa (5E0I) | 47–74 | 4–60 | Y132A Tetra (5E0I) | 35–59 | 1–36 |
|
| |||||
| HAP (5E0I) | HAP (5E0I) | ||||
|
| |||||
| Tetra HAP1 | 39–55 | 6–22 | Tetra HAP4 | 45–65 | 1–24 |
| Tetra HAP7 | 36–55 | 1–27 | Tetra HAP12 | 33–57 | 0–24 |
| Tetra BAY41-4109 | 38–62 | 4–28 | Tetra GLS4 | 41–57 | 5–26 |
| Hexa 1 GLS4 | 55–66 | −2–23 | Hexa 3 GLS4 | 57–63 | −1–17 |
|
| |||||
| AT (4G93) | AT (4G93) | ||||
|
| |||||
| Tetra AT130 | 50–76 | 20–49 | Tetra AT61 | 58–89 | 20–52 |
| Hexa 1 AT130 | 56–66 | 26–49 | Hexa 3 AT130 | 52–69 | 22–47 |
|
| |||||
| SBA and GLP (5T2P) | SBA and GLP (5T2P) | ||||
|
| |||||
| Tetra SBA_R01 | 46–64 | 5–32 | DRV01 | 42–74 | 8–52 |
| Tetra DRV23 | 48–67 | 18–39 | Tetra GLP-17A | 45–65 | 1–33 |
| Tetra GLP-17C | 45–64 | −1–41 | Tetra Comp2 | 41–72 | 4–44 |
| Tetra GLP-26 | 46–71 | 10–42 | Hexa 1 GLP-26 | 53–68 | −8–44 |
Bound CAMs alter the structure of the early assembly intermediates
In addition to simulations of the apo state, we have also simulated Cp149 tetramers with the following bound HAP compounds: BAY41–4109, HAP1, HAP4, HAP7, HAP12, and GLS4 (Figure S1A). GLS4 and HAP12 are the most potent HAP compounds, while HAP4 and HAP7 are some of the least potent ones (Table 4).3,28 Furthermore, simulations of the PPAs AT130 and AT61, which accelerate capsid assembly,4,5 and of three SBA compounds, SBA_R01, DRV01 and DRV23, which do not alter assembly of Cp149,6,7 were performed (Figures S1B and C). Finally, four GLP compounds: GLP-17A, GLP17-C, GLP-26, and Comp2, which induce formation of spherical, albeit misshapen capsids were simulated (Figure S1D).25,29
Table 4:
Hydrophobic contacts (HC) and hydrogen bonds (HB) between studied CAMs and capsid protein. Standard deviations based on two separate runs are also shown. The EC50s are also shown for compounds if they were compared to others in the same class in the referenced study.
| Molecule | HC (Count) | HB (Count) | EC50 μM |
|---|---|---|---|
|
| |||
| HAP | |||
|
| |||
| HAP1 | 156.1±1.7 | 1.11±0.02 | 0.133 |
| HAP4 | 177.9±0.2 | 1.07±0.08 | 1.93 |
| HAP7 | 186.3±3.0 | 1.12±0.02 | ≫103 |
| HAP12 | 179.0±7.0 | 1.02±0.01 | 0.0123 |
| BAY41-4109 | 186.0±1.9 | 1.05±0.03 | 0.1236 |
| GLS4 | 174.3±3.5 | 1.0±0.02 | 0.00128 |
|
| |||
| AT | |||
|
| |||
| AT130 | 167.5±2.9 | 1.51±0.03 | 2.55 |
| AT61 | 171.11±20.8 | 0.95±0.59 | 18.55 |
|
| |||
| SBA | |||
|
| |||
| SBA_R01 | 155.8±12.8 | 1.65±0.24 | NA |
| DRV01 | 155.0±1.1 | 1.09±0.34 | 2.96 |
| DRV23 | 176.0±0.6 | 2.43±0.03 | 0.86 |
|
| |||
| GLP | |||
|
| |||
| GLP-17A | 181.2±2.9 | 3.3±0.05 | 0.00429 |
| GLP-17C | 169±8.9 | 1.82±0.17 | 0.5829 |
| Comp2 | 166.9±4.3 | 1.74±0.14 | ≫10 |
| GLP-26 | 181.8±0.1 | 3.3±0.004 | 0.00329 |
With the exception of HAP4 and BAY41–4109, the remaining HAP compounds exhibit very similar base and spike angle distributions with FOAs of 49–100% (Figure 2B). The distributions of the base (≤24°) and spike (≤26°) angles are significantly narrower than those observed in the apo simulation (Table 3). Moreover, the averaged base and spike angles are significantly smaller than in the case of the apo hexamer, while the spike angles are also smaller than those of the apo tetramer (Figure 2B and Table 3). The BAY41-4109- and HAP4-bound tetramer displayed spike angles similar to other HAPs, while the base angles were somewhat larger (Figure 2B and Table 3). In addition, BAY41–4109 had a moderate FOA with other active HAPs (46–67%), while HAP4 had ≤50%FOA overlap with all other HAPs except BAY41–4109. HAP4 is one of the less potent HAPs (Table 4) and the observed difference in base angles could contribute to the lower potency. Conversely, no significant differences in structural distributions were observed between the less potent HAP7 and the most active HAPs, HAP12 and GLS4 (FOAs > 60%).
In contrast, the structures observed in AT130 and AT-61 bound simulations are remarkably different from all HAP-bound runs (all FOAs 2–0%), with larger base (50–89°) and spike (20–52°) angles (Figure 2A). The AT130 conformations are more “hexamer-like”, based on their overlap with the ones from apo hexamer simulations (FOA 76%), explaining the assembly accelerating effects of AT130. AT-61 has a smaller overlap with the hexamer (FOA 34%), which could contribute to its lower potency.
Simulations of SBAs showed some variance, in particular for DRV01 in comparison to the SBA_R01 and DRV23 (Figure 2C). The latter two compounds have a significant overlap with GLP compounds (FOAs 58–100%) and a narrow distribution of base (≤ 20°) and spike (≤ 27°) angles. In contrast, DRV01 has wider base (32°) and angle (44°) distribution ranges and a little overlap with the other SBAs (FOA ≤ 34%). In fact it only has high overlap with Comp2 (FOA 75%), and a moderate overlap with apo hexamer and GLP-26 (FOAs 56% and 59%, respectively). DRV01 is significantly less active than DRV23 (Table 4), which could explain the observed differences. There are also moderate differences between SBA_R01 and DRV23. Notably, SBA_R01 has a high similarity to apo tetramer, and the outlier HAPs (HAP4 and BAY41–4109) (FOAs of 85%, 67% and 74%, respectively), whereas DRV23 has a higher overlap with apo hexamer and AT130 (FOAs in ranges of 87% and 68%, respectively).
Finally, the simulations of the four GLPs (Figure 2C) resulted in similar SDEs (Tables 2 and 3, FOAs 51–100%). The tetramers of these compounds look distinct from all non-outlier HAPs (FOA ≤ 39%). All GLPs have at least some overlap with both apo tetramer and apo hexamer, although the ranges vary (FOAs 37%−81%). There are also significant overlaps between many GLP and SBA compounds (FOAs 37%−98%). Our results suggest that GLPs and SBAs form similar tetramer structures, despite differences in the final assembly.
We also investigated whether similar structural changes could be observed in hexamers with CAMs bound. As in the tetramer case, the hexamers with three distinct CAMs bound, namely AT130, GLS4 and GLP-26, all look very different (Figure 3). In contrast to tetramers and apo-hexamers, these hexamers displayed a narrow range of base angles (52–67°). However, the range of observed spike angles closely resembles that for the tetramers with the same bound compound (20–50° and 0–22° for AT130 and GLS4, respectively (Table 3. A wider range of spike angles was sampled for the GLP-26 hexamer (Figure 3). The general trend for reduced conformational sampling in CAM-bound hexamers as mirrored in the reduced ranges of observed base and spike angles and RMSD (Figures S11–S13), suggests stabilization of the hexameric unit by bound CAMs, in agreement with experimental data and previous simulations.3,5,37 Although a hexamer has three dimer interfaces, binding of only one CAM is sufficient to induce the observed conformational changes (Figure S10). This agrees with the experimental observation that HAP compounds can prevent formation of most capsids at concentrations that are lower than those of Cp149.28 Changes in tetramer and hexamer structures due to distinct base and spike angles are illustrated in Figure 4. HAP-induced structures with small base and spike angle are very flat, and increases in either base or spike angles exacerbate the curvature. In contrast, structures with large base and spike angles, such as those seen for the apo hexamer and the AT130-bound intermediates are highly curved.
Figure 3:
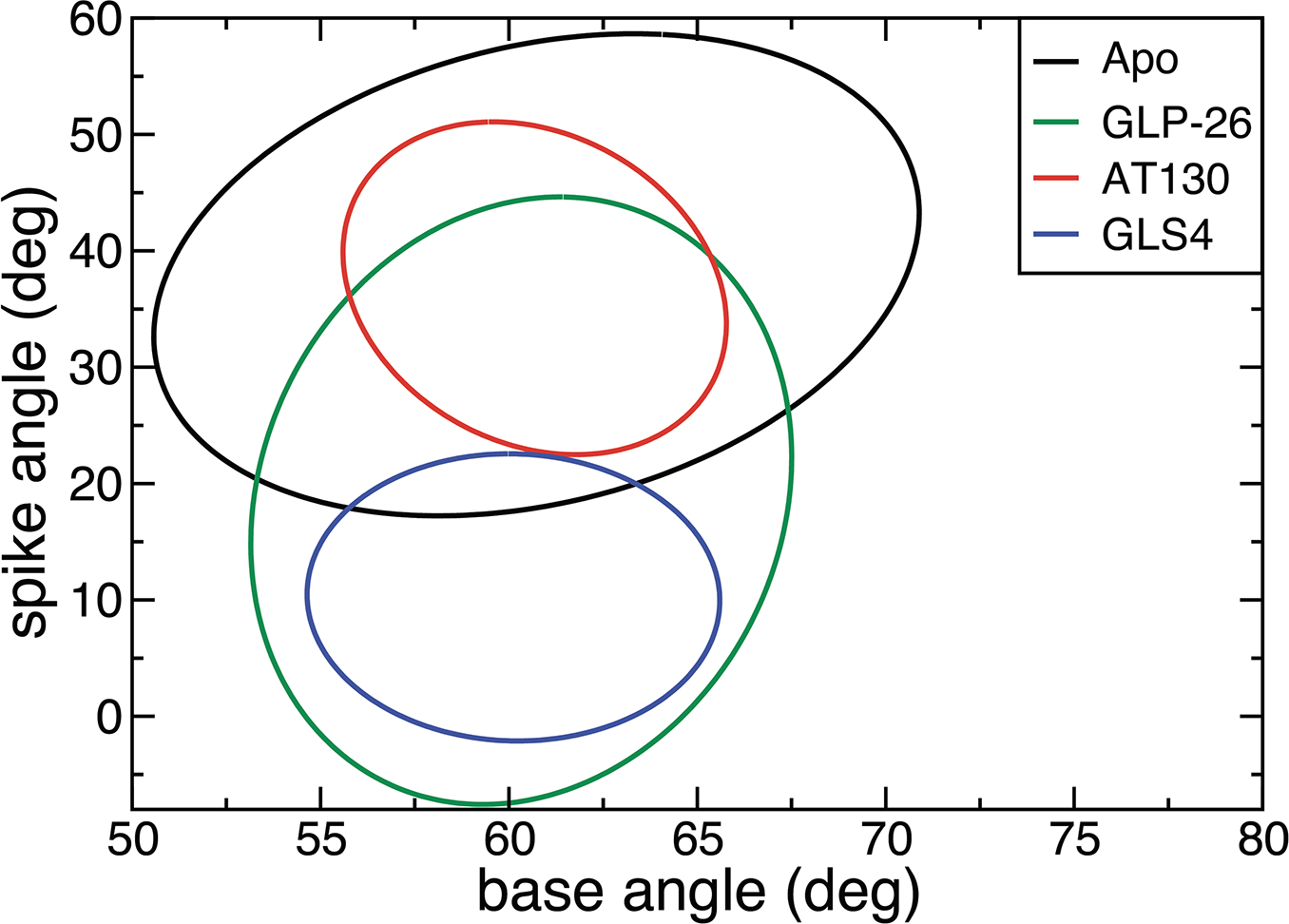
Standard deviation ellipses for hexamers with one bound GLS4, GLP-26, or AT130. Apo hexamer is added for comparison.
Figure 4:
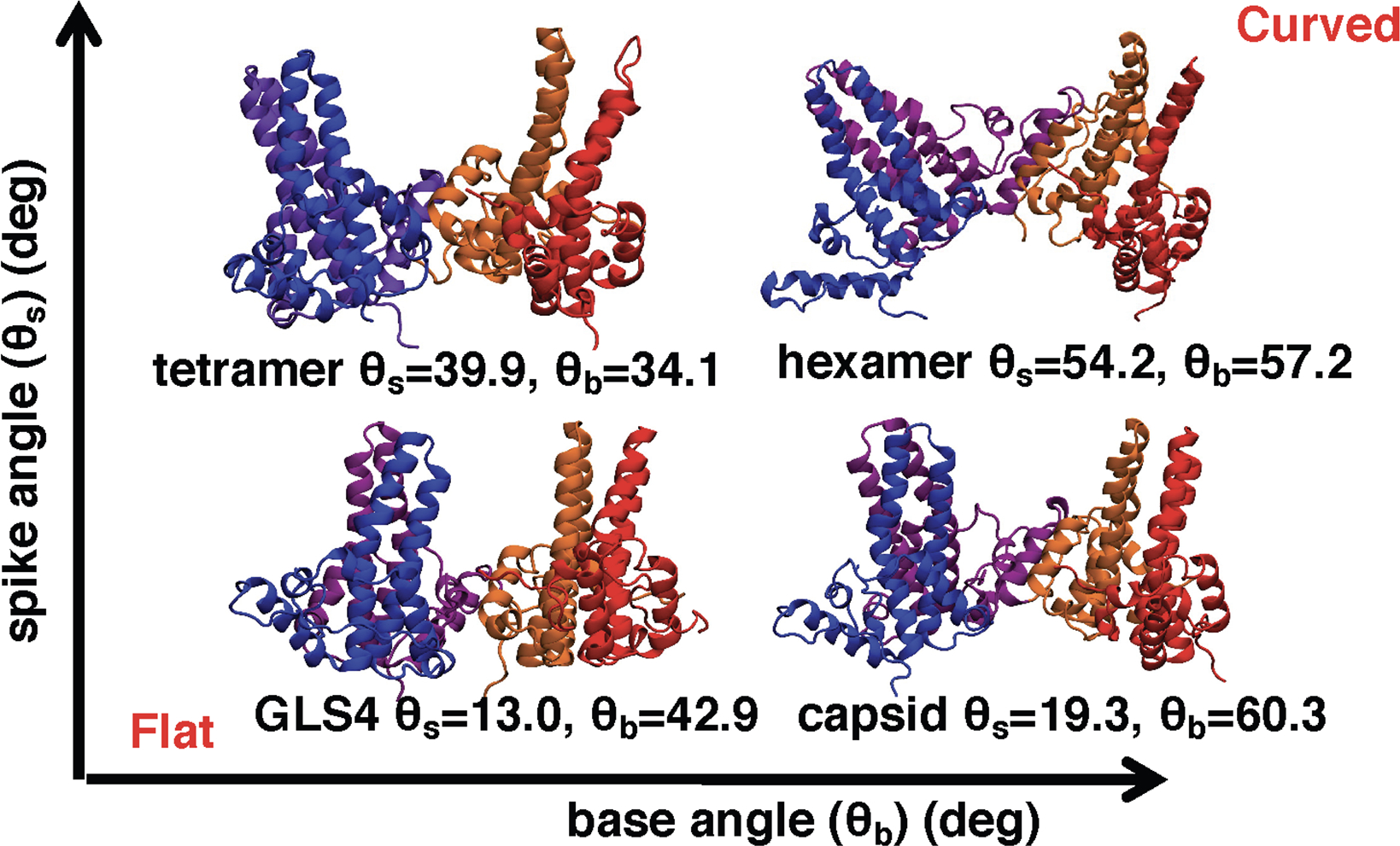
Selected snapshots of tetramers from our simulations illustrate the effect of spike and base angles on the curvature of the assembly intermediates. See Figure 2D for base and spike definitions. θb and θs correspond to the values of base and spike angles for each structure, respectively. Increases in either spike or base angle induce more curvature in the tetramer structures, with the most curved structures having large base and spike angles.
Activities within the same CAM class are dependent on their binding affinity to the capsid protein
Although standard MD simulations can elucidate the mechanistic differences between different classes of CAMs, the cause of the different potencies within the same class is less clear. We hypothesized that different potencies could be attributed to different binding affinities for the core protein tetramer. To test this hypothesis, we first analyzed the hydrogen bonds (HBs) and hydrophobic contacts (HCs) between the CAMs and the protein (Table 4). For GLPs a significant decrease in both HCs and HBs was observed for the less active Comp2 and GLP17-C, in comparison to the more potent GLP17-A and GLP-26. In addition, among the SBA compounds the less active DRV01 has fewer HBs than the more active DRV23. However, no significant differences in HBs or HCs were observed for the two PPAs AT130 and AT61, although the fluctuations of these interactions were significantly higher for the less active AT61. For HAPs, significant differences in HBs were not observed, while for HCs there was only a significant drop for HAP1, which did not correlate with potency.
A more detailed analysis of HBs revealed that the HAPs and PPAs primary formed an HB with the Trp102 side chain. It was maintained in over 90% of all HAP simulation frames, and in 48% and 34% of all AT130 and AT61 frames, respectively (Table S3). In comparison, HBs with up to three different residues were observed for GLPs and SBAs, namely the Trp102 side chain, Thr128 side chain and Leu140 backbone (Figure 5). In agreement with their lower potency, Comp2 and DRV01 display a significant decrease in HBs with Trp102 and Thr128. HB with Thr128 is completely eliminated for Comp2 due to addition of a methyl group (Figure S1D and Table S3). Hydrogen bonding with Leu140 is absent or low in SBA_R01, DRV01, and GLP-17C.
Figure 5:
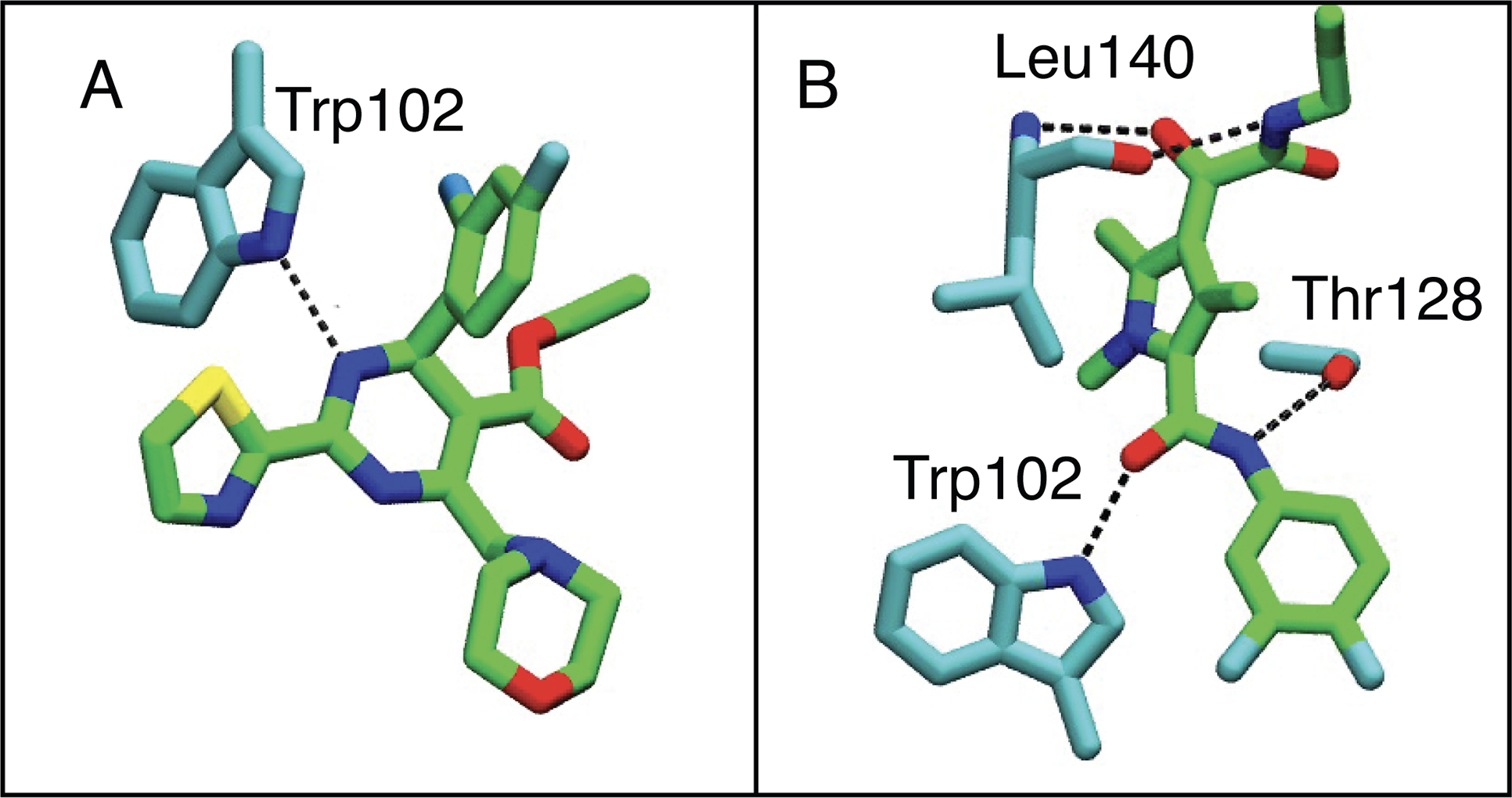
Comparison of hydrogen bonding with HBV capsid protein between GLP/SBA derivatives, and HAP compounds. Hydrogens are omitted for clarity. The CAM is colored in green and protein residues are colored in cyan. (A) Hydrogen bonding with GLS4. (B) Hydrogen bonding with GLP-26.
Since we could not identify systematic differences between the HAP compounds based on their HB or HC counts (Table 4), or hydrogen bonding with Trp102, we hypothesized that the potency differences resulted from other factors underlying the binding affinity. Therefore, we tested if differences in binding could be identified by FEP calculations (see Figure 6 for setup). The calculated free-energy differences are compared to the ones derived from experimental EC50s (Figure 6).3,28 We assume that Boltzmann-weighted binding free energies are proportional to the experimental EC50s and, thus, use the equation:
| (1) |
As shown in Figure 6, there is very good agreement (within 0.6 kcal/mol) between calculated and experimentally determined binding free energies, indicating that these quantities represent the largest factor responsible for the differences in the potency of HAP compounds.
Figure 6:
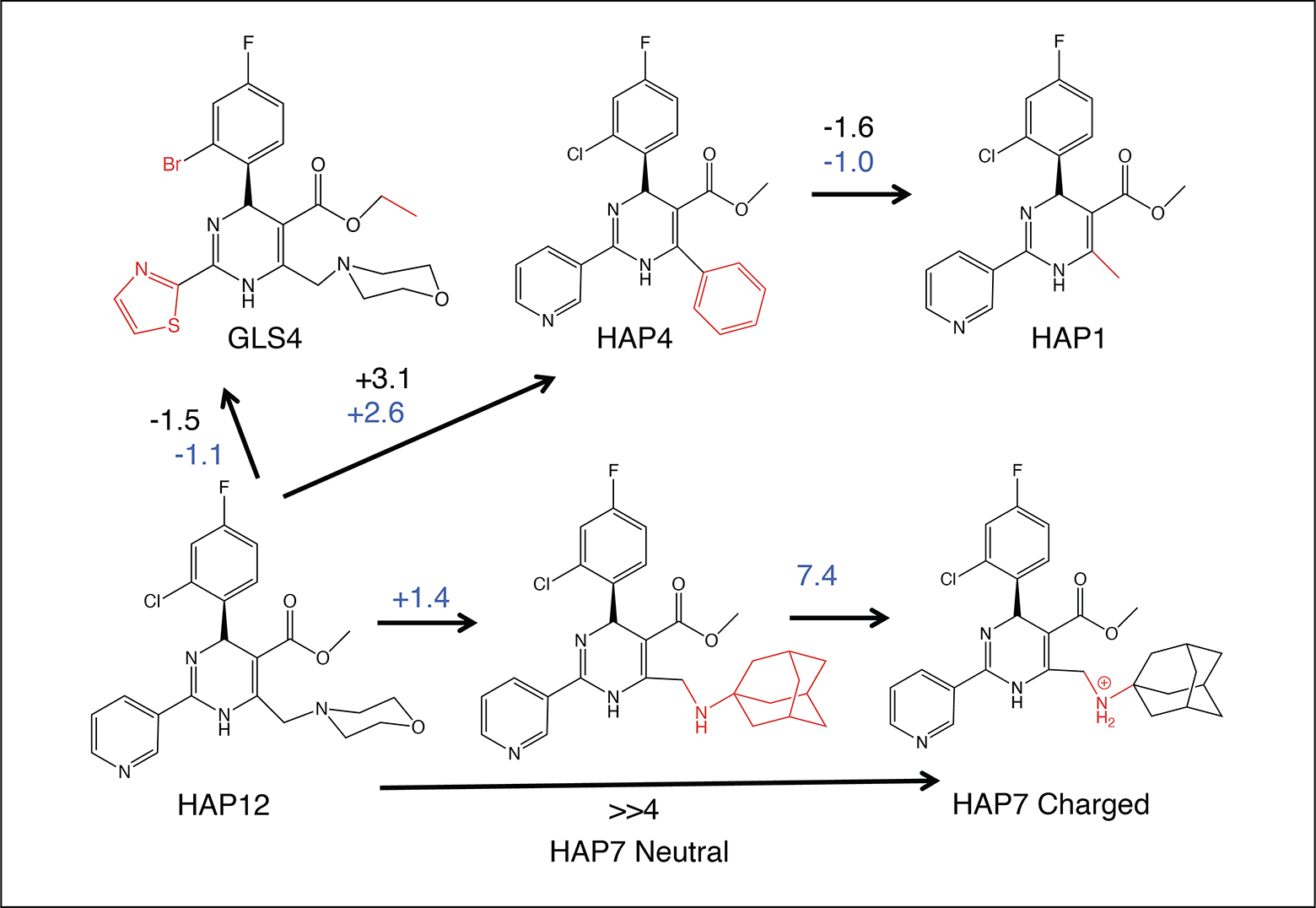
Setup and results of our FEP calculations. The structural differences from HAP12 are shown in red. All differences in binding free energies are shown in kcal/mol. Black values were calculated from experimental EC50s from Table 4 using Eq. 1, while blue values were obtained from FEP calculations.
It should be noted that HAP7, HAP12 and GLS4 have a tertiary amine group that could be protonated at neutral pH. Given that the binding of charged compounds in the inter-dimer hydrophobic pocket is expected to be unfavorable,3 we initially assumed that the amine of R2 group (Figure S1) of HAP7, HAP12 and GLS4 is not ionized. However, our FEP calculations comparing binding of neutral HAP7 and HAP12 did not agree with the experimental data (Figure 6 and Table 5). Therefore, we considered the possibility that different protonation states could contribute to the different potencies of these two compounds. Our pKa calculations using DFT (see Supporting Information) show that pKas for HAP12 and HAP7 on the R2 group nitrogen (Figure S1) are 4.5 and 9.5 at 310 K, respectively. Consequently, almost no HAP12 and almost all of HAP7 would be protonated at pH 7.0. We proceeded with an FEP calculation between neutral and protonated HAP7 and finding that the binding free energy is 7.4 kcal/mol less favorable for the protonated state. The total binding free energy difference between neutral HAP12 and protonated HAP7 was 8.8 kcal/mol, in agreement with experimental estimates of >>4 kcal/mol.3 Our calculations show that relative FEP calculations are a reliable tool for predicting the potency of CAMs with similar structures and also suggest that protonated amine groups may be detrimental to CAM potency.
Table 5:
Comparison of experimental binding free energies, relative to HAP12 (EC50 = 0.012 μM),3,28 to the ones calculated with FEP. The EC50s used for calculating experimental binding free energies according to Eq. 1 are also added. Compound structures are displayed in Figure S1, while Figure 6 shows the transformations used in FEP calculations.
| Name | EC50 (μM) | ΔΔGbind,HAP12 Exp (kcal/mol) | ΔΔGbind,HAP12 FEP (kcal/mol) |
|---|---|---|---|
| HAP1 | 0.13 | 1.5 | 1.6 |
| HAP4 | 1.9 | 3.1 | 2.6 |
| HAP7 (Neutral) | NA | NA | 1.4 |
| HAP7 (Charged) | ≫10 | ≫4 | 8.8 |
| GLS4 | 0.001 | −1.5 | −1.1 |
Discussion
Nucleocapsid assembly is governed by a weak association between core protein sub-units and is highly sensitive to the assembly conditions.19,43,44 Mass spectroscopy experiments show high concentrations of both tetramers and hexamers during capsid assembly,30–32 suggesting that they are both important assembly intermediates. It has been proposed that for HBV, the assembly is nucleated by an intermediate formed by three dimers, a triangular hexamer shown in Figure 1C.18–20 Although it is not clear if hexamer closure is sufficient for nucleation, or if an additional structural change after closure is required.
Our simulations revealed that tetramers and hexamers of the capsid protein sample different inter-dimer orientations and that the differences are well-described by base and spike angles between the dimers (Figures 2A and 2D). The tetramer exhibits great structural flexibility in terms of both base and spike angles and RMSD (Figures S11 and S12), suggesting that only some of the tetramer conformations are able to incorporate another dimer to form an open hexamer, followed by hexamer closure. In view of our results, we hypothesize that the “assembly active” conformation is a tetramer that adopts a more “hexamer-like” conformation, in agreement with the theory of allosteric assembly.19,20,44 According to this theory the “assembly active” conformation is rare under regular assembly conditions and is more frequent under conditions that enhance the assembly kinetics. We show that the frequency of “hexamer-like” conformations is increased for the V124W mutant in comparison to WT Cp149, rationalizing the acceleration of the capsid assembly kinetics by this mutant.
CAMs can either accelerate capsid assembly or misdirect it into non-capsid structures.2–5 HAP compounds lead to the formation of tubes and sheets instead of regular capsids,2,3 while AT130 and SBAs cause the formation of regular capsids lacking viral pgRNA.4,5 Finally, recently discovered GLPs induce spherical misshaped particles.25 Experiments have shown that all compounds except those belonging to the SBA class lead to more energetically favorable inter-dimer association and increase the rate of nucleation and assembly, as measured from the depletion of dimers in solution.3,25,28,45 While crystal structures show stabilization of the inter-dimer interfaces by CAMs through additional hydrophobic contacts,7,27,36 they do not account for the observed kinetic effects, nor how HAPs misdirect capsid assembly, or how GLPs change the shape of the capsids.
The results presented here demonstrate that HAPs and PPAs introduce distinct changes in the structures of early Cp assembly intermediates (Figures 2B and 2C), as measured by changes in base and spike angles, which rationalizes their distinct effects on the assembly. Intermediates with small base and spike angles, as observed for HAP-bound tetramers, appeared flatter than the apo tetramer, explaining why they ultimately form non-spherical assemblies (Figure 4).3 In contrast, intermediates with bound PPAs have large base and spike angles, and their shape is more curved than the apo tetramer (Figure 4), which results in the formation of spherical capsids.5 Similar trends in curvature and spike angles were observed for tetramers and hexamers with the same CAM. Flattening was also observed in the previous simulations of HBV capsids with bound HAP1 and tetramers with other HAP compounds.38,39 Finally, for GLP- and SBA-bound structures, the base and spike angles were larger than those for the apo tetramer, yet smaller than those for the AT130-bound simulations (Figure 2). Although SBAs and GLPs have slightly different effects on assembly, with the former resulting in formation of normal capsids and the latter causing formation of misshaped spherical capsids,25,28 we could not establish a difference between the two chemotypes by looking at the base and spike angle distributions. It is possible that more subtle changes are responsible for their distinct effects on the assembly.
Our results also provide some insight into the assembly nucleation. If nucleation was caused by structural transitions after hexamer closure, we would expect GLS4-, AT130-, and GLP-26-bound hexamers to be structurally similar, as all of these compounds are shown to enhance nucleation.3,25,28,45 However, significant structural changes were observed for hexamers with different CAMs, as measured by differences in the spike angles (Figure 3), hence suggesting that hexamer closure is a more likely nucleation event. In line with our results, a recent study showed that an open hexamer is one of the most abundant assembly intermediates.46 The results from our simulations could be confirmed by determining the collision cross-section (Ω) of intermediates in the presence of misdirecting and accelerating CAMs, as was previously done by Uetretcht et al. for intermediates during unmodified assembly.30 Based on our data, we predict distinct changes in Ω for assembly intermediates in the presence of accelerating and misdirecting CAMs.
We have also looked at differences in Cp149 interactions for CAMs with different potencies within the same class. Increases in both hydrogen bonding and hydrophobic contacts with Cp were observed for the more potent SBAs and GLPs in comparison to the less potent ones (Table 4). In particular, hydrogen bonding with Trp102 and Thr128 appears to be important for potency based on our data (Table S3). For HAPs and PPAs, few differences in hydrogen bonding and hydrophobic contacts were observed, which did not correlate with differences in potencies. However, relative binding free energies, calculated using FEP, were highly correlated with experimental EC50 values (Figure 6) for HAP compounds. We conclude that FEP calculations are more reliable than regular MD and subsequent analysis for predicting CAM potency. Still, in some cases hydrogen bond and hydrophobic contact analysis of MD simulations can be sufficient. Based on our results, we propose that standard MD simulations and structural analysis may be used for predicting the mode of action of novel CAMs, while FEP could be used for predicting changes in CAM potency in response to smaller structural modifications.
Conclusions
Our work demonstrates the utility of studying early capsid assembly intermediates, in particular their inter-subunit motions, in order to better understand the assembly of nucleocapsids and how to interfere with it. We show that tetrameric and hexameric nucleocapsid assembly intermediates of HBV adopt distinct tertiary structures, which limits the rate of capsid assembly nucleation. We propose that assembly nucleation is initiated by capsid-protein tetramers adopting a “hexamer-like” conformation, characterized by larger base and spike angles. Certain mutations such as V124W, as well as binding of assembly-accelerating CAMs, increase the frequency of such tetramer conformations. In contrast, structure-misdirecting CAMs induce the formation of flat assembly intermediates with low spike angles, which explains their effects on assembly. Furthermore, the rich and nuanced behavior of HBV CAM-mediated viral inhibition is demonstrated. Whereas the traditional HAPs and PPAs operate in agreement with a conformational shift mechanism as captured in base and spike angles of the complexes, SBAs and GLPs appear to display a more subtle mechanism that is not fully captured by the spike and base angles alone. This observation is in line with the variations in capsid formation of GLPs and SBAs. Moreover, within a given class, we have performed FEP calculations and showed the correlation between binding affinity and HBV inhibition potency. Taken together, the use of MD to characterize the effects of CAMs upon the structure and dynamics of early assembly intermediates, followed by detailed FEP calculations, provides a valuable tool in the rational design of HBV antiviral agents.
Methods
Structure Preparation
The following structures were used in our simulations: 3J2V,47 4G93,27 5E0I,36 and 5T2P.7 3J2V is the latest structure of the WT HBV capsid, 4G93 is the structure of AT130 bound to preformed HBV capsids, in which several native cysteines were mutated into alanines, while the last two structures are those of the hexameric Y132A mutant with bound drugs. 5E0I was crystallized with bound NVR-010–001-E2, which differs from GLS4 by a missing methyl group (Figure S1), whereas 5T2P was crystallized with bound SBA_R01. 5T2P was also used for simulations of the GLP compounds using a docked structure.29 Structure preparation and MD protocols are described in the Supporting Information. Table 1 indicates which structure was used for each simulated system. In total, 28 simulation systems were constructed for ~11 μs of MD simulations (Table 1).
Molecular Dynamics
NAMD2.1248 was used for all simulations except for that of the apo tetramer starting from the 5E0I structure, for which AMBER1649 was used. The CHARMM36 force field was employed for all systems.50 Additional simulation details are provided in Supporting Information. For the apo systems, the energy of all atoms was minimized at once, while for the systems with bound compounds a two-step minimization was used. In the first energy minimization step only water and ions were unrestrained, followed by an unrestrained energy minimization for all the atoms. Previously, we found that a two-step minimization can increase the compound stability in the binding pocket.51 After minimization, a two-step equilibration was performed for all systems. During the first step, water and ions were equilibrated for 0.5 ns while restraining the protein and the CAM. During the second step, 1-ns-long equilibration step, the restraints were removed from the CAM and protein side chains. Harmonic force constants of 2 kcal·mol−1·Å−2 were used for restraints in all cases. See Table 1 for the length and number of production runs for each system.
Analysis of simulations
The first 10 ns of each production run were discarded prior to any analysis, after which the trajectory frames, stored at a frequency of 0.5 ns, were analyzed. The following definitions were used for base and spike angles: the base angle was calculated based on the positions of the α5 helices, while the spike angle was calculated based on the positions of the α3 and α4 helices (see Supporting Information). For the hexamer simulations, three base and spike values were obtained from each trajectory frame and added to the analysis. Another group recently used a similar analysis independently.39 Because the top parts of helices α3 and α4 are very flexible, only the bottom parts of these helices were used for spike angle calculations, as described in the Supporting Information. The geometric center of the backbone atoms was used for all base and spike calculations. For each system, the data from 2×150 ns simulations was combined and projected on a two-dimensional scatter plot. Standard deviation ellipses (SDEs) were drawn for each system to enable easier comparison of the sampled structures. In a two-dimensional plot SDEs are centered at the average values of the two variables, while the relative height and width are determined by the standard deviations of these variables.42 The rotation of the SDE is calculated from correlation coefficient between two variables, also called covariance, and the total ellipse size is scaled to encompass a specific percentage of the provided distribution,42 which corresponds to the confidence level of the ellipse. We chose to plot ellipses corresponding to 90% confidence level.42 Comparison of SDEs between the two simulations of each system are shown in Figures S15–S18. Additionally, distributions of base and spike angles are shown as scatter plots in Figures S19–S23. Because there is no mathematical formula to calculate overlap area between two ellipses we used numerical integration to calculate all overlap area for FOA calculations. For hydrophobic contacts all contacts between carbons and/or halogens within 6.0 Å were counted. For hydrogen bond analysis we only included polar atoms (O, N, S, Cl, Br, F), and the cutoffs for the donor-acceptor distance and donor-hydrogen-acceptor angle were 3.5 Å and 145.0°, respectively.
Free energy perturbation (FEP) calculations
The relative binding free energies of a series of four substrates, namely HAP1, HAP4, HAP7 and GLS4, to Cp149 was determined with respect to HAP12 using the free energy perturbation (FEP) method.52,53 Towards this end, point mutation of the substrates was carried out in bulk water (unbound state) and at the binding site (bound state). Considering the nature of the point mutations, the reaction path was stratified into 50 stages of equal widths. Each alchemical transformation was run for 15 ns in the unbound state, and for 15 ns in the bound state, except for the transformation of HAP12 into HAP7, for which sampling was increased to 40 ns in bulk water, and 80 ns in the protein (see Table S4). The dual-topology paradigm was utilized, whereby a common scaffold is sought, and the chemical moieties characteristic of the initial and the final states of the transformation coexist, yet do not interact.54 See Supporting Information for additional details.
Experimental
Activity of Comp2 was measured as previously described.25,29 No inhibition of HBV DNA expression was observed up to 10 μM. Therefore it is concluded that this compound is inactive.
Supplementary Material
Acknowledgement
This work was supported by the National Institutes of Health grants R01-AI-148740 (to JCG), R01-AI-132833 (to RFS), and in part by 5P30-AI-50409 (to RFS). Computational resources were provided through the Extreme Science and Engineering Discovery Environment (XSEDE; TG-MCB130173), which is supported by the National Science Foundation (NSF; ACI-1548562). This work also used the Hive cluster, which is supported by the NSF (1828187) and is managed by PACE at the Georgia Institute of Technology.
Abbreviations
- HBV
hepatitis Beta virus
- CAM
capsid assembly modulator
- Cp
capsid protein
- WT
wild type
- HAP
heteroaryldihydropyramidine
- PPA
phenylpropenamide
- SBA
sulfamoyl benzamidine
- GLP
glyoxamidopyrrole
- EM
electron microscopy
- MD
molecular dynamics
- FEP
free-energy perturbation
- SDE
standard deviation ellipses
- FOA
fractional area overlap
- RMSD
root mean square deviation
- EC
effective concentration
- HB
hydrogen bonds
- HC
hydrophobic contacts
Footnotes
Conflict of Interest Disclosure
Drs. Schinazi, Amblard and Bassit along with Emory University are entitled to equity and royalties related to products licensed to Aligos Therapeutics, Inc. being further evaluated in the research described in this paper. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
Supporting Information Available
Additional computational details on structure preparation, molecular dynamics protocol, free-energy perturbation calculations and computational pKa calculations. Exact definitions of base and spike angles. Discussion of quasi-equivalent structures in the HBV capsid. Data for additional simulations not shown here for repeated Apo WT and Y132A systems. Table with averages of base and spike angles for all studied systems. RMSD graphs for the simulated systems. Comparison of the data between the different simulations of each system. Scatter plots of base and spike angles for each system. Molecular formula strings of all mentioned compounds.
References
- (1).Schweitzer A; Horn J; Mikolajczyk RT; Krause G; Ott JJ Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [DOI] [PubMed] [Google Scholar]
- (2).Deres K; Schröder CH; Paessens A; Goldmann S; Hacker HJ; Weber O; Krämer T; Niewöhner U; Pleiss U; Stoltefuss J; Graef E; Koletzki D; Masantschek RNA; Reimann A; Jaeger R; Gross R; Beckermann B; Schlemmer K-H; Haebich D; Rübsamen-Waigmann H Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893–896. [DOI] [PubMed] [Google Scholar]
- (3).Bourne C; Lee S; Venkataiah B; Lee A; Korba B; Finn MG; Zlotnick A Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J. Virol. 2008, 82, 10262–10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Delaney WE; Edwards R; Colledge D; Shaw T; Furman P; Painter G; Locarnini S Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 2002, 46, 3057–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Feld JJ; Colledge D; Sozzi V; Edwards R; Littlejohn M; Locarnini SA The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 2007, 76, 168–177. [DOI] [PubMed] [Google Scholar]
- (6).Campagna MR; Liu F; Mao R; Mills C; Cai D; Guo F; Zhao X; Ye H; Cuconati A; Guo H; Chang J; Xu X; Block TM; Guo J-T Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J. Virol. 2013, 90, 3994–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhou Z; Hu T; Zhou X; Wildum S; Garcia-Alcalde F; Xu Z; Wu D; Mao Y; Tian X; Zhou Y; Shen F; Zhang Z; Tang G; Najera I; Yang G; Shen HC; Young JA; Qin N Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci. Rep. 2017, 7, 42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sari O; Boucle S; Cox BD; Ozturk T; Russell OO; Bassit L; Amblard F; Schinazi RF Synthesis of sulfamoylbenzamide derivatives as HBV capsid assembly effector. Eur. J. Med. Chem. 2017, 138, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cole AG Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr. Opin. Pharmacol. 2016, 30, 131–137. [DOI] [PubMed] [Google Scholar]
- (10).Yang L; Liu F; Tong X; Hoffmann D; Zuo J; Lu M Treatment of chronic hepatitis B virus infection using small molecule modulators of nucleocapsid assembly: Recent advances and perspectives. ACS Infect. Dis. 2019, 5, 713–724. [DOI] [PubMed] [Google Scholar]
- (11).Seeger C; Mason WS Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Allweiss L; Dandri M The role of cccDNA in HBV maintenance. Viruses 2017, 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schinazi RF; Ehteshami M; Bassit L; Asselah T Towards HBV curative therapies. Liver Int. 2018, 38 Suppl 1, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Birnbaum F; Nassal M Hepatitis B virus nucleocapsid assembly: Primary structure requirements in the core protein. J. Virol. 1990, 64, 3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nassal M The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Conway JF; Cheng N; Zlotnick A; Wingfield PT; Stahl SJ; Steven AC Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997, 386, 91–94. [DOI] [PubMed] [Google Scholar]
- (17).Wynne SA; Crowther RA; Leslie AGW The crystal structure of the human hepatitis B virus capsid. Mol. Cell 1999, 3, 771–780. [DOI] [PubMed] [Google Scholar]
- (18).Zlotnick A; Johnson JM; Wingfield PW; Stahl SJ; Endres D A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 1999, 38, 14644–14652. [DOI] [PubMed] [Google Scholar]
- (19).Ceres P; Zlotnick A Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry 2002, 41, 11525–11531. [DOI] [PubMed] [Google Scholar]
- (20).Stray SJ; Ceres P; Zlotnick A Zinc ions trigger conformational change and oligomerization of hepatitis B virus capsid protein. Biochemistry 2004, 43, 9989–9998. [DOI] [PubMed] [Google Scholar]
- (21).Packianathan C; Katen SP; Dann CE; Zlotnick A Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J. Virol. 2010, 84, 1607–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bourne CR; Katen SP; Fulz MR; Packianathan C; Zlotnick A A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry 2009, 48, 1736–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tan Z; Maguire ML; Loeb DD; Zlotnick A Genetically altering the thermodynamics and kinetics of hepatitis B virus capsid assembly has profound effects on virus replication in cell culture. J. Virol. 2013, 87, 3208–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ruan L; Hadden JA; Zlotnick A Assembly properties of hepatitis B virus core protein mutants correlate with their resistance to assembly–directed antivirals. J. Virol. 2018, 92, e01082–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Amblard F; Boucle S; Bassit L; Cox B; Sari O; Tao S; Chen Z; Ozturk T; Verma K; Russell O; Rat V; de Rocquigny H; Fiquet O; Boussand M; Di Santo J; Strick-Marchand H; Schinazi RF Novel hepatitis B virus capsid assembly modulator induces potent antiviral responses in vitro and in humanized mice. Antimicrob. Agents Chemother. 2020, 64, e01701–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bourne CR; Finn MG; Zlotnick A Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J. Virol. 2006, 80, 11055–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Katen SP; Tan Z; Chirapu SR; Finn M; Zlotnick A Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 2013, 21, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ren Q; Liu X; Luo Z; Li J; Wang C; Goldmann S; Zhang J; Zhang Y Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (GLS4). Bioorg. Med. Chem. 2017, 25, 1042–1056. [DOI] [PubMed] [Google Scholar]
- (29).Amblard F; Boucle S; Bassit L; Chen Z; Sari O; Cox B; Verma K; Ozturk T; Ollinger-Russell O; Schinazi RF Discovery and structure activity relationship of glyoxamide derivatives as anti-hepatitis B virus agents. Bioorg. Med. Chem. 2021, 31, 115952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Uetrecht C; Barbu IM; Shoemaker GK; van Duijn E; Heck AJ Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat. Chem. 2011, 3, 126–132. [DOI] [PubMed] [Google Scholar]
- (31).Lutomski CA; Lyktey NA; Pierson EE; Zhao Z; Zlotnick A; Jarrold MF Multiple pathways in capsid assembly. J. Am. Chem. Soc. 2018, 140, 5784–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Shepherd DA; Holmes K; Rowlands DJ; Stonehouse NJ; Ashcroft AE Using ion mobility spectrometry-mass spectrometry to decipher the conformational and assembly characteristics of the hepatitis B capsid protein. Biophys. J. 2013, 105, 1258–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nijampatnam B; Liotta DC Recent advances in the development of HBV capsid assembly modulators. Curr. Opin. Chem. Biol. 2019, 50, 73–79. [DOI] [PubMed] [Google Scholar]
- (34).Bassit L; Ono SK; Schinazi RF Moving fast toward hepatitis B virus elimination. Adv. Exp. Med. Biol. 2021, 1322, 115–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen W; Liu F; Zhao Q; Ma X; Lu D; Li H; Zeng Y; Tong X; Zeng L; Liu J; Yang L; Zuo J; Hu Y Discovery of phthalazinone derivatives as novel hepatitis B virus capsid inhibitors. J. Med. Chem. 2020, 63, 8134–8145. [DOI] [PubMed] [Google Scholar]
- (36).Klumpp K; Lam AM; Lukacs C; Vogel R; Ren S; Espiritu C; Baydo R; Atkins K; Abendroth J; Liao G; Efimov A; Hartman G; Flores OA High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 15196–15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rath SL; Liu H; Okazaki S; Shinoda W Identification of factors promoting HBV capsid self-assembly by assembly-promoting antivirals. J. Chem. Inf. Model. 2018, 58, 328–337. [DOI] [PubMed] [Google Scholar]
- (38).Perilla JR; Hadden JA; Goh BC; Mayne CG; Schulten K All-atom molecular dynamics of virus capsids as drug targets. J. Phys. Chem. Lett. 2016, 7, 1836–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Liu H; Okazaki S; Shinoda W Heteroaryldihydropyrimidines alter capsid assembly by adjusting the binding affinity and pattern of the hepatitis B virus core protein. J. Chem. Inf. Model. 2019, 59, 5104–5110. [DOI] [PubMed] [Google Scholar]
- (40).Hadden JA; Perilla JR; Schlicksup CJ; Venkatakrishnan B; Zlotnick A; Schulten K All-atom molecular dynamics of the HBV capsid reveals insights into biological function and cryo-EM resolution limits. eLife 2018, 7, e32478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ghaemi Z; Gruebele M; Tajkhorshid E Molecular mechanism of capsid disassembly in hepatitis B virus. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2102530118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wang B; Shi W; Miao Z Confidence analysis of standard deviational ellipse and its extension into higher dimensional Euclidean space. PLoS One 2015, 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Perlmutter JD; Hagan MF Mechanisms of virus assembly. Ann. Rev. Phys. Chem. 2015, 66, 217–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lazaro GR; Hagan MF Allosteric control of icosahedral capsid assembly. J. Phys. Chem. B 2016, 120, 6306–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Katen SP; Chirapu SR; Finn MG; Zlotnick A Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem. Biol. 2010, 5, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Oliver RC; Potrzebowski W; Najibi SM; Pedersen MN; Arleth L; Mahmoudi N; Andr I Assembly of capsids from hepatitis B virus core protein progresses through highly populated intermediates in the presence and absence of RNA. ACS Nano 2020, 14, 10226–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yu X; Jin L; Jih J; Shih C; Hong Zhou Z 3.5 Å cryo-EM structure of hepatitis B virus core assembled from full-length core protein. PLoS One 2013, 8, e69729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable molecular dynamics with NAMD. J. Comp. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Case DA; Betz RM; Cerutti DS; Cheatham TE III; Darden TA; Duke RE; Giese TJ; Gohlke H; Goetz AW; Homeyer N; Izadi S; Janowski P; Kaus J; Kovalenko A; Lee TS; LeGrand S; Li P; Lin C; Luchko T; Luo R; Madej B; Mermelstein D; Merz KM; Monard G; Nguyen H; Nguyen HT; Omelyan I; Onufriev A; Roe DR; Roitberg A; Sagui C; Simmerling CL; Botello-Smith WM; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Xiao L; Kollman PA AMBER 2016; University of California, San Francisco, 2016. [Google Scholar]
- (50).Best RB; Zhu X; Shim J; Lopes PE; Mittal J; Feig M; MacKerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pavlova A; Gumbart JC Parametrization of macrolide antibiotics using the force field toolkit. J. Comput. Chem. 2015, 36, 2052–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Landau LD Statistical physics; The Clarendon Press: Oxford, 1938. [Google Scholar]
- (53).Pohorille A; Jarzynski C; Chipot C Good practices in free-energy calculations. J. Phys. Chem. B 2010, 114, 10235–10253. [DOI] [PubMed] [Google Scholar]
- (54).Zwanzig RW High-temperature equation of state by a perturbation method. I. Nonpolar gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.