

Summary
Background
CNS relapse of acute lymphocytic leukaemia is difficult to treat. Durable remissions of relapsed or refractory B-cell acute lymphocytic leukaemia have been observed following treatment with CD19-directed chimeric antigen receptor (CAR) T cells; however, most trials have excluded patients with active CNS disease. We aimed to assess the safety and activity of CAR T-cell therapy in patients with a history of CNS relapsed or refractory B-cell acute lymphocytic leukaemia.
Methods
In this post-hoc analysis, we included 195 patients (aged 1–29 years; 110 [56%] male and 85 [44%] female) with relapsed or refractory CD19-positive acute lymphocytic leukaemia or lymphocytic lymphoma from five clinical trials (Pedi CART19, 13BT022, ENSIGN, ELIANA, and 16CT022) done at the Children’s Hospital of Philadelphia (Philadelphia, PA, USA), in which participants received CD19-directed CAR T-cell therapy between April 17, 2012, and April 16, 2019. The trials required control of CNS disease at enrolment and infusion and excluded treatment in the setting of acute neurological toxic effects (>grade 1 in severity) or parenchymal lesions deemed to increase the risk of neurotoxicity. 154 patients from Pedi CART19, ELIANA, ENSIGN, and 16CT022 received tisagenlecleucel and 41 patients from the 13BT022 trial received the humanised CD19-directed CAR, huCART19. We categorised patients into two strata on the basis of CNS status at relapse or within the 12 months preceding CAR T-cell infusion—either CNS-positive or CNS-negative disease. Patients with CNS-positive disease were further divided on the basis of morphological bone marrow involvement—either combined bone marrow and CNS involvement, or isolated CNS involvement. Endpoints were the proportion of patients with complete response at 28 days after infusion, Kaplan-Meier analysis of relapse-free survival and overall survival, and the incidence of cytokine release syndrome and neurotoxicity.
Findings
Of all 195 patients, 66 (34%) were categorised as having CNS-positive disease and 129 (66%) as having CNS-negative disease, and 43 (22%) were categorised as having isolated CNS involvement. The median length of follow-up was 39 months (IQR 25–49) in the CNS-positive stratum and 36 months (18–49) in the CNS-negative stratum. The proportion of patients in the CNS-positive stratum with a complete response at 28 days after infusion was similar to that in the CNS-negative stratum (64 [97%] of 66 vs 121 [94%] of 129; p=0 ·74), with no significant difference in relapse-free survival (60% [95% CI 49–74] vs 60% [51–71]; p=0·50) or overall survival (83% [75–93] vs 71% [64–79]; p=0·39) at 2 years between the two groups. Overall survival at 2 years was significantly higher in patients with isolated CNS involvement compared with those with bone marrow involvement (91% [82–100] vs 71% [64–78]; p=0·046). The incidence and severity of neurotoxicity (any grade, 53 [41%] vs 38 [58%]; grade 1, 24 [19%] vs 20 [30%]; grade 2, 14 [11%] vs 10 [15%]; grade 3, 12 [9%] vs 6 [9%], and grade 4, 3 [2%] vs 2 [3%]; p=0·20) and cytokine release syndrome (any grade, 110 [85%] vs 53 [80%]; grade 1, 12 [9%] vs 2 [3%]; grade 2, 61 [47%] vs 38 [58%]; grade 3, 18 [14%] vs 7 [11%] and grade 4, 19 [15%] vs 6 [9%]; p=0·26) did not differ between the CNS-negative and the CNS-positive disease strata.
Interpretation
Tisagenlecleucel and huCART19 are active at clearing CNS disease and maintaining durable remissions in children and young adults with CNS relapsed or refractory B-cell acute lymphocytic leukaemia or lymphocytic lymphoma, without increasing the risk of severe neurotoxicity; although care should be taken in the timing of therapy and disease control to mitigate this risk. These preliminary findings support the use of these CAR T-cell therapies for patients with CNS relapsed or refractory B-cell acute lymphocytic leukaemia.
Funding
Children’s Hospital of Philadelphia Frontier Program.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy has transformed treatment for children and young adults with relapsed or refractory B-cell acute lymphocytic leukaemia, with 1-year relapse-free survival rates approaching 60%.1-4 Evidence for the safety and efficacy of CD19-directed CAR T-cell therapy for CNS relapse, however, remains scarce. CNS involvement complicates around 20% of relapses, and a history of CNS disease is associated with decreased overall survival after relapse.5,6 The standard treatment for CNS relapse includes intensive chemotherapy and cranial radiation, which has risks of adverse long-term learning and growth effects, and toxic effects on the endocrine system. The standard treatment for patients with high-risk CNS relapse is haematopoietic stem-cell transplantation (HSCT), which is also associated with clinically significant morbidity and mortality.7-9 Alternative approaches are needed, particularly for patients with CNS relapse after cranial radiation or HSCT.
Given the systemic and potentially devastating neurological toxic effects10-12 observed with CD19-directed CAR T-cell therapy for bone marrow disease, determining the safety and efficacy of this treatment for patients with CNS disease is essential. Case series, although limited by small numbers of participants, have shown tolerability of CAR T-cell therapy in patients with a history of CNS disease, without disproportionate neurological complications.13-16 Preliminary data from patients with acute lymphocytic leukaemia have also suggested activity2,14,16,17 and durability3,18 in CNS leukaemia control, with biological plausibility supported by the detection of CAR by PCR in cerebrospinal fluid (CSF).14,16,19 Robust evidence of efficacy, particularly in patients who have isolated CNS relapse, however, is scarce.
To examine the safety and activity of CD19-directed CAR T-cell therapy for CNS relapsed B-cell acute lymphocytic leukaemia, we did a post-hoc analysis of children and young adults given tisagenlecleucel (also known as CTL019) or the similar humanised CD19-directed CAR, huCART19,20 with relapsed or refractory acute lymphocytic leukaemia with and without CNS involvement.
Methods
Study design and participants
Eligible patients were children and young adults aged 1–29 years with relapsed or refractory CD19-positive acute lymphocytic leukaemia or lymphocytic lymphoma from five completed clinical trials (Pedi CART19 [NCT01626495],2 13BT022 [NCT02374333],20 16CT022 [NCT02906371],21 ENSIGN [NCT02228096],22 and ELIANA [NCT02435849]3) done at the Children’s Hospital of Philadelphia (Philadelphia, PA, USA), in which they received CD19-directed CAR T-cell therapy between April 17, 2012, and April 16, 2019 (n=195). These non-randomised phase 1/2 trials enrolled patients with CD19-positive acute lymphocytic leukaemia or lymphocytic lymphoma (Pedi CART19, 13CT022, and 16CT022 only) that was refractory, in second or greater relapse, or relapsed after HSCT. Most patients (n=154) received tisagenlecleucel, an anti-CD19 CAR containing a 4-1BB costimulatory signal.23 Patients from the 13BT022 trial (n=41) received huCART19, in which the anti-CD19 single-chain variable fragment has been humanised.20 CNS status before infusion was classified as CNS1 (CSF with fewer than 5 white blood cells per μL and no blasts), CNS2 (CSF with fewer than 5 white blood cells per μL and positive for leukaemic blasts), and CNS3 (CSF with ≥5 white blood cells per μL and positive for blasts or parenchymal or cranial nerve involvement). All trials permitted the inclusion of patients with CNS disease that had cleared, and excluded patients with bulky intracranial disease that did not improve. However, PEDI CART19, ENSIGN, and ELIANA trials excluded patients with active CNS3 disease, whereas 13BT022 and 16CT022 trials permitted inclusion of patients with active CNS3 disease that was controlled on therapy, as did Pedi CART19 following a protocol amendment (see appendix pp 2–3 for detailed inclusion and exclusion criteria for infusion in each trial). This post-hoc pooled clinical trial analysis was approved by the institutional review board of the Children’s Hospital of Philadelphia. Patients or their guardians provided written informed consent for treatment in each respective clinical trial.
Procedures
Patient demographics, baseline characteristics, and adverse event reports for neurotoxicity and cytokine release syndrome were obtained from the clinical trial databases. Patient clinical history, including previous radiation treatment, disease status at referral, and neurological history, was manually extracted from medical records, including clinical trial referral and enrolment records. Medications administered following infusion and details of other post-infusion toxicity management were electronically extracted from electronic medical records.
All patients were required to have CSF evaluations before enrolment in the trials. Patients underwent staging lumbar puncture and bone marrow aspirate and biopsy with multiparameter flow cytometry assessment of minimal residual disease within 5 days before infusion, with the exception of 23 patients who underwent staging disease evaluation at enrolment and received bridging chemotherapy before infusion. Pre-infusion disease burden was defined on the basis of the highest blast percentage of the three measurements, with a bone marrow disease burden of M1 defined as less than 5% lymphoblasts, M2 as 5–25% lymphoblasts, and M3 as greater than 25% lymphoblasts.
We categorised patients into two strata on the basis of CNS status at relapse or within the 12 months preceding CAR T-cell infusion—either CNS-positive disease or CNS-negative disease. Patients with CNS-positive disease were further divided on the basis of morphological bone marrow involvement—either combined bone marrow and CNS involvement (M1 bone marrow) or isolated CNS involvement. In secondary analyses, the isolated CNS stratum was compared with bone marrow involvement (ie, all other included patients with bone marrow or combined bone marrow and CNS involvement). Patients were included in the CNS-positive stratum24,25 if they had CNS3 or persistent CNS2 disease (leukaemic blasts observed in at least two consecutive lumbar punctures) at the time of their most recent relapse or within 12 months before infusion (n=66). Patients who had CNS3 disease at initial diagnosis that cleared during induction therapy and did not have CNS recurrence were included in the CNS-negative stratum (n=1). Patients with a single CNS2 CSF finding at infusion, who otherwise did not meet criteria for inclusion in the CNS-positive stratum, remained in the CNS-negative stratum. Patients were considered as having isolated CNS involvement if they met the aforementioned criteria for the CNS-positive stratum and they had a bone marrow status of M1 at their most recent relapse.
After seizures were observed, antiepileptic drugs were given as prophylaxis after infusion to patients with a history of seizure, stroke, methotrexate neurotoxicity, neurological deficits, focal CNS lesions, or at the discretion of the treating physician, beginning after 30 patients had received treatment in the earliest trial (Pedi CART19).
Cytokine release syndrome was graded according to the Penn scale.26 Other adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), versions 3.0 and 4.03. Neurotoxicity included any of the following neurological adverse event terms on the CTCAE and was categorised as encephalopathy (encephalopathy, depressed level of consciousness, lethargy, somnolence, memory impairment, confusion, delirium, agitation, irritability, hallucinations, altered mood, or a change in mental status), speech impairment (aphasia, dysphasia, dysarthria, or word-finding difficulty), movement disorder (ataxia, involuntary movements, muscle weakness, or tremor), vision changes (blurred vision or diplopia), cranial nerve disorder (any symptom involving a cranial nerve), and seizure. Specific neurological comorbidities of clinical interest (a previous history of stroke, seizure, methotrexate neurotoxicity, or neurological deficit) were analysed individually and as a composite endpoint. Adverse events were defined as serious when they were life-threatening or resulted in hospitalisation or prolongation of hospitalisation, congenital anomaly or disability (or required intervention to prevent), or death, or when considered an important medical event by the clinical investigator. Encephalopathy was considered an important medical event and reported as a serious adverse event in these trials.
Outcomes
Endpoints were the proportion of patients with a complete response at 28 days after infusion, relapse-free survival and overall survival during the follow-up period, and the incidence of cytokine release syndrome and neurotoxicity in the first 8 weeks after infusion. A complete response was defined as a morphological bone marrow disease burden of M1 with trilineage haematopoiesis and no evidence of extramedullary disease, including CNS disease. Patients who died without evidence of disease before their first post-infusion disease assessment were considered not evaluable. Relapse-free survival was defined as the time from the first disease assessment after infusion until morphological relapse or death in patients with a complete response following infusion. Patients were censored at the time of receiving HSCT or another alternate cancer-directed therapy or at the last known contact, whichever occurred first. No deaths had occurred before relapse in these patients. Overall survival was defined as the time from infusion to death in all infused patients, with patients censored at the last known contact. Time to onset of serious neurological adverse events was defined as the time from infusion to the start of the event in days.
Statistical analysis
Patients were followed up for analysis of relapse and survival outcomes to the data cutoff of Jan 1, 2021. All infused patients included in this study were evaluated for safety. Patient characteristics were compared by CNS stratum by use of Fisher’s exact tests for categorical variables and Wilcoxon rank-sum test for continuous variables. Post-infusion toxicity management, complete response rate, and the incidence of cytokine release syndrome and neurotoxicity events were summarised as frequencies and proportions by CNS stratum, which were compared by use of the Fisher’s exact test. Time to onset of serious adverse neurotoxicity events and the duration of these events were summarised as medians (IQRs) by CNS stratum, which were compared by use of the Wilcoxon rank-sum test. Kaplan-Meier curves of relapse-free survival and overall survival were plotted by CNS stratum and were compared with log-rank tests. 95% CIs were calculated on the basis of the log-log survival, with the variance estimated using Greenwood formula Because few patients were followed up for more than 48 months, follow-up for relapse-free survival and overall survival was censored at 48 months. To explore risk factors for neurotoxicity, logistic regression models were fit for the incidence of three dichotomised outcomes: any neurotoxicity, any grade 3–4 neurotoxicity, and any seizure. Cox proportional hazards regression models were used to explore predictors of relapse-free survival and overall survival, with the proportional hazard assumptions assessed with log-log plots. Univariate regression models were constructed to identify factors associated with the outcomes of interest. Factors associated with outcomes at p<0·1, and factors of clinical interest (regardless of statistical association), were then included in the multivariate regression model. A multivariate model was not constructed for seizure because of the small number of events. Statistical analyses were done using SAS software, version 9.4. A two-sided p value of less than 0·05 was considered to indicate a significant difference.
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
Of 222 patients enrolled in all five studies, 195 paediatric and young adult patients with relapsed or refractory acute lymphocytic leukaemia or lymphocytic lymphoma infused with CD19-directed CAR T cells between April 17, 2012, and April 16, 2019 were included in this study (figure 1). The remaining 27 patients were not infused. 66 (34%) patients with CNS involvement at the time of their last relapse or within 12 months before infusion were included in the CNS-positive stratum; 43 (65%) of these patients were included in the isolated CNS involvement stratum. Compared with patients in the CNS-negative stratum, significantly more patients in the CNS-positive stratum had a previous history of receiving CNS radiation (39 [59%] of 66 vs 14 [11%] of 129; p<0·0001), were in a second or greater relapse at referral (50 [76%] vs 59 [46%]; p<0·0001), had detectable CNS disease at infusion (nine [14%] vs nine [7%]; p=0·021), and had a bone marrow disease burden of less than 0·01% at infusion (44 [67%] vs 30 [23%]; p<0·0001; table 1). 42 (64%) patients in the CNS-positive stratum received seizure prophylaxis following infusion compared with only 22 (17%) patients in the CNS-negative stratum (p<0·0001). Aside from these differences, distributions of patient demographic and clinical characteristics, including neurological comorbidities, were similar between these two strata (table 1). These patterns were similar for patients with isolated CNS involvement compared with all other included patients. 14 (7%) of all 195 patients had CNS2 disease at infusion, and four (2%) patients had CNS3 disease at infusion (table 1).
Figure 1: Flow diagram for participants in trials included in the analysis.
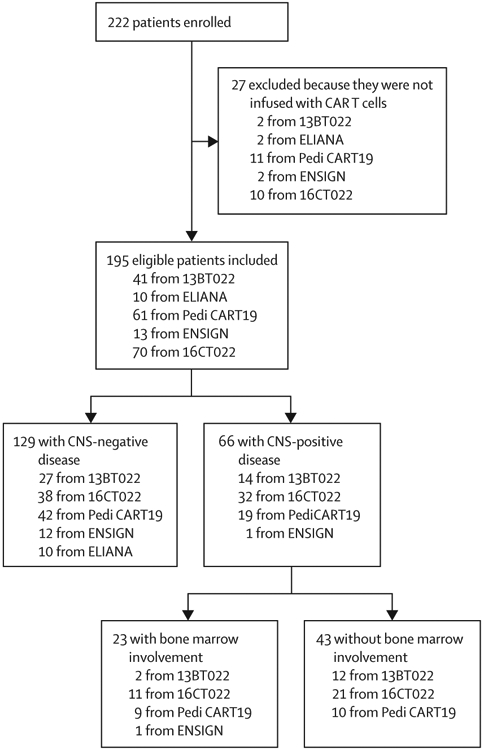
Patients were pooled from five clinical trials (Pedi CART19 [NCT01626495], 13BT022 [NCT02374333], ENSIGN [NCT02228096], ELIANA [NCT02435849], and 16CT022 [NCT02906371]). Patients were stratified to the CNS-positive stratum if they had a CNS status of CNS3 or persistent CNS2 at the time of their most recent relapse or within 12 months before CART-cell infusion.
CAR=chimeric antigen receptor.
Table 1:
Demographics and baseline clinical characteristics by stratum
| All patients (n=195) |
CNS stratification | CNS categorisation by bone marrow involvement | |||||
|---|---|---|---|---|---|---|---|
| CNS-negative stratum (n=129) |
CNS-positive stratum (n=66) |
p value | Patients with bone marrow and combined bone marrow CNS involvement (n=152) |
Patients with isolated CNS involvement* (n=43) |
p value | ||
| Age at infusion, years | 11·3 (7·7–16·5) | 12·4 (7·6–17·0) | 10·2 (7·9–15·0) | 0·63 | 12·3 (7·8–17·0) | 10·2 (8·0–13·7) | 0·35 |
| Age group, years | |||||||
| <3 | 8 (4%) | 4 (3%) | 4 (6%) | 0·76 | 4 (3%) | 4 (9%) | 0·29 |
| 3–<10 | 72 (37%) | 47 (36%) | 25 (38%) | ·· | 57 (38%) | 15 (35%) | |
| 10–<18 | 82(42%) | 56 (43%) | 26 (39%) | ·· | 64 (42%) | 18 (42%) | ·· |
| ≥18 | 33 (17%) | 22 (17%) | 11 (17%) | ·· | 27 (18%) | 6 (14%) | ·· |
| Sex | ·· | ·· | ·· | 0·94 | ·· | ·· | 0·80 |
| Male | 110 (56%) | 73 (57%) | 37 (56%) | ·· | 85 (56%) | 25 (58%) | ·· |
| Female | 85 (44%) | 56 (43%) | 29 (44%) | ·· | 67 (44%) | 18 (42%) | ·· |
| Previous haematopoietic stem-cell transplantation | 94 (48%) | 59 (46%) | 35 (53%) | 0·33 | 73 (48%) | 21 (49%) | 0·93 |
| Previous radiation treatment | |||||||
| Cranial | 53 (27%)† | 14 (11%) | 39 (59%) | <0·0001 | 23 (15%) | 30 (70%) | <0·0001 |
| Total body irradiation | 83 (43%) | 52 (40%) | 31 (47%) | 0·37 | 66 (43%) | 17 (40%) | 0·65 |
| Disease status at referral | ·· | ·· | ·· | <0·0001 | ·· | 0·0002 | |
| Primary refractory | 27 (14%) | 26 (20%) | 1 (2%) | ·· | 27 (18%) | 0 | ·· |
| First relapse | 59 (30%) | 44 (34%) | 15 (23%) | ·· | 50 (33%) | 9 (21%) | ·· |
| Second or greater relapse | 109 (56%) | 59 (46%) | 50 (76%) | ·· | 75 (49%) | 34 (79%) | ·· |
| Neurological comorbidity | |||||||
| Stroke | 4 (2%) | 3 (2%) | 1 (2%) | >0·99 | 3 (2%) | 1 (2%) | >0·99 |
| Seizure | 26 (13%) | 14 (11%) | 12 (18%) | 0·15 | 18 (12%) | 8 (19%) | 0·25 |
| Methotrexate toxicity | 15 (8%) | 11 (9%) | 4 (6%) | 0·54 | 12 (8%) | 3 (7%) | >0·99 |
| Neurological deficit | 13 (7%) | 7 (5%) | 6 (9%) | 0·37 | 8 (5%) | 5 (12%) | 0·17 |
| Bone marrow disease burden before infusion | ·· | ·· | ·· | <0·0001 | ·· | ·· | <0·0001 |
| <0·01% | 74 (38%) | 30 (23%) | 44 (67%) | ·· | 39 (26%) | 35 (81%) | ·· |
| 0·01–4·99% (M1) | 30 (15%) | 22 (17%) | 8 (12%) | ·· | 24 (16%) | 6 (14%) | ·· |
| 5–25% (M2) | 19 (10%) | 17 (13%)‡ | 2 (3%) | ·· | 19 (13%) | 0 | ·· |
| >25% (M3) | 72 (37%) | 60 (47%)‡ | 12 (18%)‡ | ·· | 70 (46%) | 2 (5%)§ | ·· |
| CNS status before infusion | ·· | ·· | ·· | 0·021 | ·· | ·· | 0·054 |
| CNS1 | 177 (91%) | 120 (93%)‡ | 57 (86%)‡ | ·· | 139 (91%) | 38 (88%) | ·· |
| CNS2 | 14 (7%) | 9 (7%)‡ | 5 (8%) | ·· | 12 (8%) | 2 (5%) | ·· |
| CNS3¶ |
4 (2%) | 0 | 4 (6%) | ·· | 1 (1%) | 3 (7%) | ·· |
| Seizure prophylaxis | 64 (33%) | 22 (17%) | 42 (64%) | <0·0001 | 33 (22%) | 31 (72%) | <0·0001 |
Data are median (IQR) or n (%), unless otherwise specified. CAR=chimeric antigen receptor. CSF=cerebrospinal fluid.
Defined at relapse or refractory evaluation.
Three patients received cranial radiation for the current relapse before enrolment, seven patients received focal radiation (orbital [n=4], spinal or cauda equina [n=2], and cerebellar [n=1]) for the current relapse, and the remaining 43 patients received previous cranial radiation either as prophylaxis or for treatment of a previous relapse.
Includes patients in whom pre-infusion bone marrow evaluation and lumbar puncture were done at enrolment (3–6 weeks before CAR T-cell infusion) and who received bridging chemotherapy in the interim period (see appendix [p 1] for the numbers of patients in each group).
Two patients in the isolated CNS stratum developed marrow disease during the bridging period.
These four patients with CNS3 pre-infusion showed residual enhancement by MRI (optic nerve [n=1]; cranial nerve V, cranial nerve VII, and cerebellar lesion [n=1]; or cauda equina nerve roots [n=1]) or leukaemic blasts in the CSF (6 white blood cells per μL [n=1]).
No difference in the proportion of patients with a complete response at 28 days after infusion was observed between the CNS-positive and the CNS-negative disease strata (64 [97%] of 66 vs 121 [94%] of 129; p=0·74; table 2). Compared with bone marrow involvement, a similar proportion of patients in the isolated CNS involvement stratum had a complete response (42 [98%] of 43; table 2). Of the 18 (9%) patients with CNS2 or CNS3 disease at infusion, evaluation at 28 days showed that 15 (83%) patients had CNS1 disease; of the remaining three patients, one was not evaluable (they died due to toxicity before day 28), and two had CNS2 disease (both of whom had CNS1 disease by 3 months without further therapy).
Table 2:
Disease outcomes after anti-CD19 CAR T-cell infusion by stratum
| All patients (n=195) | CNS stratification | CNS categorisation by bone marrow involvement | |||||
|---|---|---|---|---|---|---|---|
| CNS-negative stratum (n=129) |
CNS-positive stratum (n=66) |
p value | Patients with bone marrow and combined bone marrow CNS involvement (n=152) |
Patients with isolated CNS involvement (n=43) |
p value | ||
| Disease response at day 28 | |||||||
| Complete response | 185 (95%) | 121 (94%) | 64 (97%) | 0·74 | 143 (98%) | 42 (98%) | 0·35 |
| No response | 7 (4%) | 6 (5%) | 1 (2%) | ·· | 7 (5%) | 0 | ·· |
| Not evaluable* | 3 (2%) | 2 (2%) | 1 (2%) | ·· | 2 (1%) | 1 (2%) | ·· |
| Patients with relapse | 72/185 (39%) | 45/121 (37%) | 27/64 (42%) | 0·51 | 56/143 (39%) | 16/42 (38%) | 0·90 |
| CNS status at relapse | ·· | ·· | ·· | 0·0066 | ·· | ·· | 0·0062 |
| CNS1 | 45/72(63%) | 33/45 (73%) | 12/27 (44%) | ·· | 38/56 (68%) | 7/16 (44%) | ·· |
| CNS2 | 4/72 (6%) | 1/45 (2%) | 3/27 (12%) | ·· | 1/56 (2%) | 3/16 (19%) | ·· |
| CNS3 | 7/72 (10%) | 1/45 (2%) | 6/27 (22%) | ·· | 3/56 (5%) | 4/16 (25%) | ·· |
| Unknown | 16/72 (22%) | 10/45 (22%) | 6/27 (22%) | ·· | 14/56 (25%) | 2/16 (12%) | ·· |
| Follow-up duration, months | 37 (21–49) | 36 (18–49) | 39 (25–49) | 0·73 | 36 (18–49) | 40 (29–52) | 0·38 |
Data are n (%), n/N (%),or median (IQR), unless otherwise specified. CSF=cerebrospinal fluid.
Three patients died before evaluation at day 28 from sequelae of cytokine release syndrome, coagulopathy, and infection without clear evidence of progressive disease.
The median length of follow-up was 39 months (IQR 25–49) in the CNS-positive stratum and 36 months (18–49) in the CNS-negative disease stratum. The overall proportion of patients who had CNS relapse after CAR T-cell infusion was 4% (seven of 169 patients; table 2); an additional four (6%) patients had CNS2 status at the time of bone marrow relapse. There was no difference in the proportion of patients who relapsed after CAR T-cell infusion in the CNS-positive stratum compared with the CNS-negative stratum (27 [42%] of 66 vs 45 [37%] of 129; p=0·51), although patients in the CNS-positive stratum were significantly more likely to have CNS3 disease at relapse (p=0·0066; table 2). No difference in relapse-free survival between the CNS-positive stratum (60% [95% CI 49–74] at 2 years) and the CNS-negative stratum (60% [51–71] at 2 years) was observed (p=0·50; figure 2A). In a separate analysis of patients with isolated CNS involvement, relapse-free survival at 2 years was 66% (52–82) compared with 58% (50–68) for patients with bone marrow involvement (p=0·15; figure 2B); 16 (38%) of 43 patients with isolated CNS involvement relapsed after CAR T-cell infusion compared with 56 (39%) of 152 with bone marrow involvement (table 2). Evaluation of risk factors for relapse using univariate analysis of relapse-free survival showed that a bone marrow disease burden of M3 at infusion (hazard ratio [HR] 4·515 [95% CI 2·606–7·824]; p<0·0001), presence of CNS blasts at infusion (HR 2·060 [1·083–3·920]; p=0·028), and female sex (HR 1·781 [1·114–2·848]; p=0·016) were significantly associated with a higher risk, whereas a history of brain radiation was associated with lower risk (HR 0·548 [0·317–0·948]; p=0·031; appendix p 4). In the multivariate analysis, only a bone marrow disease burden of M3 at infusion was associated with a significantly higher risk of relapse (HR 5·354 [95% CI 2·778–10·321]; p<0·0001), and being aged 18 years or older was associated with a significantly lower risk of relapse (0·456 [0·211–0·987]; p=0·046; appendix p 4). CNS2 or CNS3 disease at infusion was not associated with a significantly higher risk of relapse; however, due to the small sample size, there was limited power to detect a statistical difference. The log-log plots suggested no violation of the proportional hazards assumption. Of the 14 patients who were CNS2 at infusion, four (29%) had continuous remission, one (7%) received HSCT during remission, eight (57%) had morphological bone marrow relapse (CNS status at relapse was CNS1 in five patients, CNS3 in one patient, and unknown in two patients), and one (7%) patient was not evaluable (they died due to toxicity before day 28). Of four patients who had CNS3 disease at infusion, two (50%) were in continuous remission, and two (50%) had morphological bone marrow relapse (one patient had CNS1 and the other patient had CNS3 disease at relapse).
Figure 2: Relapse-free survival and overall survival by CNS status.

(A) Relapse-free survival by strata (CNS-positive stratum vs the CNS-negative stratum) in patients who had a complete response. Data were censored for allogeneic haematopoietic transplantation (one patient in the CNS-positive stratum and 19 in the CNS-negative stratum) or other alternative therapy (three in the CNS-positive stratum and 18 in the CNS-negative stratum) given during remission and at 48 months of follow-up. (B) Relapse-free survival in patients with isolated CNS involvement compared with those with bone marrow or combined bone marrow and CNS involvement; 20 patients in the bone marrow or combined bone marrow and CNS involvement stratum received HSCT, and two patients in the isolated CNS stratum and 19 in the bone marrow or combined bone marrow and CNS involvement stratum received alternative therapy), with follow-up censored at 48 months. Overall survival in the CNS-positive stratum versus the CNS-negative stratum (C) and in patients with isolated CNS involvement compared with those with bone marrow or combined bone marrow and CNS involvement (D). In A–D, tick marks indicate the time of censoring and dashed lines represent 95% CIs.
Overall survival did not differ between the CNS-positive stratum and the CNS-negative stratum (83% [95% CI 75–93] vs 71% [64–79] at 2 years; p=0·39; figure 2C); however, overall survival in patients who had isolated CNS involvement was significantly higher than in those who did not (91% [82–100] vs 71% [64–78] at 2 years; p=0·046; figure 2D). 21 (32%) of 66 patients in the CNS-positive stratum, 48 (37%) of 129 patients in the CNS-negative stratum, and ten (23%) of 43 patients in the isolated CNS stratum died. Following univariate analysis of risk factors for overall survival, bone marrow disease burden of M3 at infusion (HR 6·54 [95% CI 3·28–13·04], p<0·0001), a history of brain radiation (0·55 [0·30–1·01], p=0·053), age (10 to <18 years 0·42 [0·24–0·74], p=0·0027; and 18 years or older 0·48 [0·23–1·00], p=0·049), female sex (1·73 [1·06–2·81], p=0·027), treatment received (huCART19 vs tisagenlecleucel 0·47 [0·23–0·99], p=0·048), and having CNS2 or CNS3 disease at infusion (2·10 [1·07–4·11], p=0·032) were selected on the basis of p values for inclusion in the multivariate model (appendix p 4). The CNS-positive stratum was also included in the multivariate model on the basis of clinical interest. In the final model, higher marrow disease burden was significantly associated with an increased risk of death, and being aged 10 years or older was associated with a lower risk of death (appendix p 4). Patients with M3 disease had a higher risk of death than those with no detectable bone marrow disease (<0·01% lymphoblasts) at the time of infusion (HR 6·97 [95% CI 3·09–15·74]; p<0·0001), with no violation of the proportional hazards assumption.
The overall incidence of neurotoxicity did not differ between the CNS-positive stratum and the CNS-negative stratum (p=0·20), and there was no difference in the number of neurological serious adverse events reported between these two strata (p=0·75; table 3), nor in treatment with dexamethasone (p>0·99; appendix p 5). Compared with the CNS-negative stratum, more patients in the CNS-positive stratum reported vision changes (three [2%] of 129 vs seven [11%] of 66; p=0·023), speech impairment (three [2%] vs six [9%]; p=0·023), and movement disorder (five [4%] vs four [6%]; p=0·042). There was no difference in the incidence of encephalopathy, cranial nerve disorder, or seizure between the two strata (table 3).
Table 3:
Cytokine release syndrome and neurotoxic adverse events by CNS stratum
| CNS-negative stratum (n=129) |
CNS-positive stratum (n=66) |
p value | |
|---|---|---|---|
| Cytokine release syndrome | ·· | ·· | 0·26 |
| 0 | 19 (15%) | 13 (20%) | ·· |
| 1 | 12 (9%) | 2 (3%) | ·· |
| 2 | 61 (47%) | 38 (58%) | ·· |
| 3 | 18 (14%) | 7 (11%) | ·· |
| 4 | 19 (15%) | 6 (9%) | ·· |
| Any neurotoxicity | ·· | ·· | 0·20 |
| 0 | 76 (59%) | 28 (41%) | ·· |
| 1 | 24 (19%) | 20 (30%) | ·· |
| 2 | 14 (11%) | 10 (15%) | ·· |
| 3 | 12 (9%) | 6 (9%) | ·· |
| 4 | 3 (2%) | 2 (3%) | ·· |
| Encephalopathy | ·· | ·· | 0·36 |
| 0 | 82 (64%) | 35 (53%) | ·· |
| 1 | 20 (16%) | 16 (24%) | ·· |
| 2 | 13 (10%) | 7 (11%) | ·· |
| 3 | 13 (10%) | 6 (10%) | ·· |
| 4 | 1 (1%) | 2 (3%) | ·· |
| Seizure | ·· | ·· | 0·71 |
| 0 | 119 (92%) | 61 (92%) | ·· |
| 1 | 1 (1%) | 0 | ·· |
| 2 | 5 (4%) | 4 (6%) | ·· |
| 3 | 1 (1%) | 1 (2%) | ·· |
| 4 | 3 (2%) | 0 | ·· |
| Vision changes | ·· | ·· | 0·023 |
| 0 | 126 (98%) | 59 (89%) | ·· |
| 1 | 2 (2%) | 6 (9%) | ·· |
| 2 | 1 (1%) | 1 (2%) | ·· |
| 3 | 0 | 0 | ·· |
| 4 | 0 | 0 | ·· |
| Speech impairment | ·· | ·· | 0·023 |
| 0 | 126 (98%) | 60 (91%) | ·· |
| 1 | 3 (2%) | 2 (3%) | ·· |
| 2 | 0 | 1 (2%) | ·· |
| 3 | 0 | 3 (5%) | ·· |
| 4 | 0 | 0 | ·· |
| Movement disorder | ·· | ·· | 0·42 |
| 0 | 124 (96%) | 6 (9%) | ·· |
| 1 | 5 (4%) | 1 (2%) | ·· |
| 2 | 0 | 3 (5%) | ·· |
| 3 | 0 | 0 | ·· |
| 4 | 0 | 0 | ·· |
| Cranial nerve disorder | ·· | ·· | 0·26 |
| 0 | 127 (98%) | 65 (98%) | ·· |
| 1 | 2 (2%) | 0 | ·· |
| 2 | 0 | 0 | ·· |
| 3 | 0 | 1 (2%) | ·· |
| 4 | 0 | 0 | ·· |
| Neurological serious adverse event | ·· | ·· | 0·75 |
| 0 | 102 (79%) | 48 (73%) | ·· |
| 1 | 1 (1%) | 1 (2%) | ·· |
| 2 | 12 (9%) | 9 (14%) | ·· |
| 3 | 11 (9%) | 6 (9%) | ·· |
| 4 | 3 (2%) | 2 (3%) | ·· |
Data are n (%). No grade 5 adverse events were reported.
Neurological serious adverse events reported within 8 weeks of infusion included encephalopathy (n=39), seizure (n=15), movement disorder (n=1), and speech impairment (n=1). There was no difference in the time to development of encephalopathy in the CNS-negative stratum compared with the CNS-positive stratum (median 6 days [IQR 5–9] vs 5 days [5–6]; p=0·58) or duration of encephalopathy (6 days [4–11] vs 4 days [3–6]; p=0·19; appendix p 5). No neurological serious adverse events occurred more than 8 weeks after infusion.
When evaluating risk factors for the occurrence of any grade neurotoxicity in the univariate and multivariate analyses, only a history of CNS-relapsed or refractory disease and a bone marrow disease burden of M3 at infusion (univariate odds ratio [OR] 2·581 [95% CI 1·324–5·032]; p=0·030; multivariate OR 5·720 [2·415–13·546]; p=0·0033) were significantly associated with an increased risk (appendix pp 5–6). Patients in the CNS-positive stratum were more likely to develop any symptoms of neurotoxicity than those in the CNS-negative stratum (OR 3·42 [1·44–8·12]; p=0·0053), and patients with a bone marrow disease burden of M2 (5·46 [1·66–17·97]; p=0·063) or M3 (5·72 [2·42–13·55]; p=0·0033) at infusion were more likely to develop neurotoxicity than those with no detectable bone marrow disease burden at infusion (appendix p 6).
Examination of the risk factors for developing grade 3 or 4 neurotoxicity by univariate analysis showed that only neurological comorbidity (OR 2·708 [95% CI 1·080–6·791]; p=0·034) and a bone marrow disease burden of M3 at infusion (7·315 [2·040–26·228]; p=0·0014) were associated with an increased risk, both of which were maintained as significant risk factors in the multivariate analysis (1·572 [0·729–3·391]; p=0·25; 5·720 [2·415–13·546]; p=0·0033; appendix p 5). Patients with a bone marrow disease burden of M3 at infusion had a significantly higher risk of developing severe neurotoxicity (ie, grade 3 or 4) compared with those with 25% or less bone marrow blasts at infusion (OR 11·79 [95% CI 3·60–38·54]; p<0·0001), as did patients with a history of a neurological comorbidity compared with those without (4·15 [1·43–12·05]; p=0·0088; appendix p 6). Risk of severe neurotoxicity did not differ between CNS-positive and CNS-negative strata (2·38 [0·76–7·49]; p=0·14).
There was no difference in the incidence or severity of seizure between the CNS-negative stratum and the CNS-positive stratum (p=0·71), with ten (8%) of 129 patients in the CNS-negative stratum and five (8%) of 66 patients in the CNS-positive stratum reporting a seizure (table 3). Nine (14%) of 64 patients (five in the CNS-positive stratum, four with a history of seizure, and four with previous methotrexate toxicity) who received prophylactic antiepileptics had a seizure compared with six (5%) of 131 patients who did not. Time to the seizure event did not differ in patients who had CNS-positive disease (median 6 days [IQR 5–11]) compared with patients who had CNS-negative disease (9 days [8–12]; p=0·54). Four (3%) patients in the CNS-negative stratum had multiple seizure events, including two patients with status epilepticus, and three (2%) patients required intubation in the context of their seizure event. In the CNS-positive stratum, three (5%) patients had multiple seizure events, one (2%) had an episode of status epilepticus, and two (3%) required intubation. The use of rescue benzodiazepine (p>0·99) and treatment-dose antiepileptic medications (p=0·77) was similar across the two strata (appendix p 5).
In the univariate analyses exploring risk factors for seizure, a history of methotrexate toxicity was associated with a significantly increased risk (OR 5·59 [95% CI 1·53–20·43]; p=0·0093), as was history of neurological deficit (4·25 [1·03–17·53]; p=0·045) and neurological comorbidity (6·68 [2·23–20·07]; p=0·0007). Although OR estimates were large for high bone marrow disease burden at infusion, a previous history of seizure or stroke, or having isolated CNS involvement did not significantly change the risk of seizure (appendix p 7).
There was no difference in the incidence or severity of cytokine release syndrome between the CNS-negative stratum and the CNS-positive stratum (p=0·26; table 3), nor was there difference in the administration of medications used for the management of cytokine release syndrome between these strata, including tocilizumab and intravenous steroids (appendix p 5). An association between severe cytokine release syndrome and neurotoxicity was observed, with 18 (72%) of 25 patients with grade 3 cytokine release syndrome, and 22 (88%) of 25 patients with grade 4 cytokine release syndrome also having neurotoxicity. Similar concordance was observed between severe cytokine release syndrome and severe neurotoxicity, with 15 (60%) of 25 patients with grade 4 cytokine release syndrome also having severe neurotoxicity (appendix p 7).
Discussion
To our knowledge this is the first study of children and young adults with relapsed or refractory acute lymphocytic leukaemia or lymphocytic lymphoma, given CD19-directed CAR T-cell products tisagenlecleucel or huCART19, to show equivalent activity and safety of these treatments in patients with and without CNS involvement at relapse. This study extends the findings of several case-series suggesting CAR T-cell activity in leukaemia and lymphoma with CNS involvement, despite these studies being limited by small patient numbers.13-16 It should be noted that, although most patients included in our analysis were in CNS remission at the time of infusion due to bridging intrathecal chemotherapy, they had not received consolidative therapy for CNS relapse before receiving CAR T cells. Although second remissions can often occur for first isolated CNS involvement,6 and even subsequent remissions are common,6 CNS involvement in the multiply-relapsed setting, particularly after radiation and HSCT, is challenging to treat and generally leads to a poor outcome.27 Therefore, the observed durable remissions in a population enriched for second or greater relapses (50 [76%] of 66), relapse after cranial irradiation (39 [59%]), and relapse after HSCT (35 [53%]) represents a major advance in the treatment of patients with CNS relapse acute lymphocytic leukaemia.
Additional analyses in this cohort also revealed several risk factors for relapse after CAR T-cell therapy. The most impactful prognostic factor for relapse was pre-infusion bone marrow disease burden, with a burden of M3 associated with a higher risk of relapse than a burden of less than 0·01%. Patients received bridging chemotherapy and lymphodepleting chemotherapy before CAR T-cell infusion; therefore, pre-infusion disease burden might indicate chemotherapy response. This association could reflect the underlying refractory biology of the leukaemia and suggests that chemorefractory leukaemias are more likely to relapse after CAR T-cell therapy; however, this notion requires further investigation. Other groups have published similar findings. Park and colleagues28 reported a higher risk of relapse in patients with more than 5% bone marrow blasts following treatment with 19-28z CAR T cells compared with those with less than 5% bone marrow blasts. Conversely, in our study, patients with negative minimal residual disease at the pre-infusion assessment showed improved relapse-free survival and overall survival outcomes, and those with isolated CNS involvement showed improved overall survival compared with all other patients, which is probably associated with the low bone marrow disease burden in this patient group. Notably, although older age (ie, 10 years and older) is associated with a higher risk of relapse after front-line therapy for acute lymphocytic leukaemia and worse outcomes in relapse than a younger age (ie, 1–10 years), we found that older age at infusion was not associated with worse outcomes after CAR T-cell therapy compared with younger ages, and that patients aged 10–29 years had a lower risk of relapse in the multivariate analysis. Larger studies are needed to establish whether the observed age effect is dependent on other factors that our exploratory subgroup analyses were not powered to detect, or if older age is a true independent protective factor.
Another key finding of this study is equivalent risk of severe neurotoxicity in patients with and without a history of CNS involvement. In early CAR T-cell trials, the emergence of neurotoxicity as a prominent adverse event raised safety concerns for patients with CNS disease and neurological comorbidities. Therefore, both efficacy and toxicity concerns led most CAR T-cell trials to exclude patients with active CNS disease. In this study, patients in the CNS-positive stratum were more likely to have neurotoxicity than those in the CNS-negative stratum; however, grade 1 and 2 neurological events were the largest contributor to this difference, and there was no increased risk of grade 3 or 4 neurotoxicity. However, after adjusting for other risk factors, including pre-infusion bone marrow disease burden, the point estimate of risk for grade 3 or 4 neurotoxicity increased, but not significantly. Notably, the presence of CNS2 or CNS3 disease at infusion did not significantly increase the risk of neurotoxicity. The results of our study, together with others,29,30 suggest a strong association between high bone marrow disease burden and the risk of cytokine release syndrome and neurotoxicity; therefore, the identified association between CNS disease and neurotoxicity might be mitigated by the low burden of bone marrow disease observed in this stratum.
Previous studies have shown an increased risk of neurotoxicity in patients with neurological comorbidities.29,30 Our study did not show a significantly increased risk of neurotoxicity in patients with a previous history of seizure, stroke, methotrexate neurotoxicity, or neurological deficit, probably, in part, due to the small sample size for each individual event. However, when combined into a composite endpoint, neurological comorbidity was significantly associated with an increased risk of high-grade neurotoxicity. Similarly, previous methotrexate neurotoxicity, neurological deficit, and composite neurological comorbidity were significantly associated with an increased risk of seizure. Even though OR estimates suggested an increased risk of both neurotoxicity and seizure with a previous history of stroke or seizure, the low incidence of these events precluded definitive conclusions. Additionally, the prophylactic use of antiepileptics in patients with a history of seizure, stroke, methotrexate neurotoxicity, neurological deficit, or focal CNS lesions could have affected the incidence of seizure.
The strengths of this study include its large sample size, robust toxicity and outcome data, and long follow-up period. This study was a post-hoc analysis of pooled clinical trial data; therefore, it is limited by the retrospective nature and variability in the population. The variation in characteristics of the population strengthens the ability to do risk factor analyses and to generalise the results to the whole population of patients with acute lymphocytic leukaemia or lymphocytic lymphoma, but also limits the size of subgroup analyses. Neurotoxicity risk factors and outcomes are rare, and the subgroups are small, leading to unstable estimates and wide CIs in some analyses; these estimates should be interpreted with caution and not be considered as confirmatory. It should also be noted that the five included trials required control of CNS disease, and excluded treatment in the setting of acute neurological toxicities greater than grade 1 or parenchymal lesions deemed to increase the risk of neurotoxicity. Few patients (14 [7%] of 195) had CNS2 disease and even fewer (four [2%]) had CNS3 disease at infusion. Despite these safety measures, there was an increased risk of any grade neurotoxicity, reinforcing that care should be taken in the timing of infusion and disease control to reduce the risk of severe neurotoxicity. Additionally, only patients receiving tisagenlecleucel or huCART19 (CAR T cells constructed with a 4-1BB costimulatory domain),2,23 were included in this analysis, limiting the generalisability to other CAR T-cell constructs, most notably those with CD28 costimulatory domains, which have previously been associated with rare cases of life-threatening or fatal cerebral oedema.11
In conclusion, this study shows that tisagenlecleucel or huCART19 CAR T-cell therapy is effective at clearing CNS disease and maintaining durable remissions in acute lymphocytic leukaemia with CNS involvement. The overall incidence of CNS relapse was low. Our data suggest that patients with CNS disease that is adequately controlled before infusion could be safely given CD19 CAR T cells, with no increased risk of severe neurotoxicity.
Data sharing
Individual deidentified participant data that underlie the results reported in this Article will only be shared if the following conditions are met: the investigators provide a methodologically sound proposal with approved aims; publication of the requested data does not compromise an ongoing trial or study; there is a strong scientific rationale for the data to be used for the requested purpose; and investigators, who have invested time and effort into developing these trials, have a period of exclusivity in which to pursue their own aims with the data before key trial data are made available to others. Proposals should be directed to the corresponding author, and data requestors will need to sign a data access agreement to gain access.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed on July 13, 2021, using the search terms "chimeric antigen receptor T cell", "leukemia", "lymphoma", "central nervous system", "neurotoxicity", "CAR T", "CAR-T", "B-ALL", "B-LLy", "ALL", "LLy", and "CNS". We searched for articles on CAR T-cell therapy for the treatment of CNS disease in patients with relapsed and refractory B-cell leukaemia and lymphoma, published from database inception to April 30, 2021, with no language restrictions. Only two prospective studies, ChiCTR-OPN-17013507 and NCT03064269, were identified, each of which had enrolled a total of three patients and had used different CAR T-cell constructs. Four case-series were identified, each presenting five or fewer patients with CNS manifestations of leukaemia or lymphoma, who were typically given tisagenlecleucel. All identified studies suggested CAR T-cell activity in leukaemia and lymphoma with CNS involvement, but they did not have sufficient sample size to examine the effects of other factors on outcomes or the granularity necessary to understand the associated neurotoxicity risk. We therefore concluded that there were no robust data to council the families of patients with CNS disease about the risk of neurotoxicity, risk of relapse, or the probability of overall survival associated with this treatment modality, nor to guide clinicians in the choice of CAR T-cell therapy as a treatment in the setting of CNS relapse.
Added value of this study
To our knowledge, this post-hoc analysis of participants from five clinical trials represents the largest analysis of children and young adults (aged 1–29 years) with acute lymphocytic leukaemia given anti-CD19 CAR T-cell therapies for relapsed or refractory CNS disease, including a small group of patients with evidence of active CNS disease at the point of infusion. In addition, to our knowledge, this study is the first to show equivalent activity and safety of this therapy in patients with CNS-relapsed or refractory disease and patients without a recent history of CNS involvement.
Implications of all the available evidence
Tisagenlecleucel is approved and indicated in the USA, Europe, Canada, Australia, and Japan for the treatment of multiply relapsed or refractory B-cell acute lymphocytic leukaemia in paediatric and young adult patients. Understanding the use of this therapy in patients with proximate CNS involvement is a priority. The activity and safety data from this secondary analysis extend these findings and support the use of the CD19-directed CAR T-cell therapies tisagenlecleucel and huCART19 in children and young patients with relapsed or refractory leukaemia with CNS disease.
Acknowledgments
This study was supported by the Children’s Hospital of Philadelphia Frontier Program. The clinical trials included in this post-hoc pooled analysis were supported by clinical trial awards funded by Novartis Pharmaceuticals and a research alliance between the University of Pennsylvania, Novartis Pharmaceuticals, and the Children’s Hospital of Philadelphia Frontier Program. ABL, SAG, and SLM are supported by grants from the National Institutes of Health grant (5K12CA076931–22 to ABL, and 5P01CA214278–04 to SAG and SLM). The authors thank the Cancer Immunotherapy Program clinical research team and Mark Ramos at the Children’s Hospital of Philadelphia (who provided automatically abstracted data from Epic [Epic Systems Corporation, Verona, WI, USA]). Initial data from this study were presented as an oral presentation at the American Society of Clinical Oncology Annual Meeting in 2020.
Footnotes
Declaration of interests
CC has served as a consultant for Novartis Pharmaceuticals. CHJ is an inventor of intellectual property, licensed by the University of Pennsylvania to Novartis; has received patent royalties; is a scientific co-founder of Tmunity Therapeutics and DeCART Therapeutics, for which he has founder’s stock but no income; and is an advisor for AC Immune, Bluesphere Bio, Cellares, Celldex, Cabaletta, Carisma, Kiadis, Viracta, and Ziopharm. SAG has received research or clinical trial support, or both, from Novartis, Servier, and Kite, and has participated in consulting, in study steering committees, or in scientific or clinical advisory boards for Novartis, Cellectis, Adaptimmune, Eureka, TCR2, Juno, GlaxoSmithKline, Vertex, Humanigen, CBMG, Janssen/JnJ, Jazz Pharmaceuticals, Allogene, Cabaletta, and Roche. SRR has received research funding from and has served as a consultant for Pfizer, and a family member owns stock in Optinose. SLM has served as a consultant for Novartis Pharmaceuticals, Kite Pharma, and Wugen, and receives clinical trial funding from Novartis Pharmaceuticals. All other authors declare no competing interests.
Contributor Information
Allison Barz Leahy, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Penn Center for Cancer Care Innovation, University of Pennsylvania, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Haley Newman, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Yimei Li, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Biostatistics, Epidemiology, and Informatics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Hongyan Liu, Department of Biomedical and Health Informatics, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Regina Myers, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Amanda DiNofia, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Joseph G Dolan, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Colleen Callahan, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Diane Baniewicz, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Kaitlin Devine, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Lisa Wray, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Richard Aplenc, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Carl H June, Center for Cellular Immunotherapies, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA; The Parker Institute for Cancer Immunotherapy, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Stephan A Grupp, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Susan R Rheingold, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Shannon L Maude, Division of Oncology and Cancer Immunotherapy Program, Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA; Center for Cellular Immunotherapies, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
References
- 1.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385: 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med 2014; 371: 1507–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. New Engl J Med 2018; 378: 439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19CAR T cells of defined formulation and dose in children and young adults. Blood 2017; 129: 3322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun W, Malvar J, Sposto R, et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: a therapeutic advances in childhood leukemia & lymphoma study. Leukemia 2018; 32: 2316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen K, Devidas M, Cheng S-C, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia 2008; 22: 2142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Follin C, Erfurth EM. Long-term effect of cranial radiotherapy on pituitary-hypothalamus area in childhood acute lymphoblastic leukemia survivors. Cun Treat Option Oncol 2016; 17: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldsby RE, Liu Q, Nathan PC, et al. Late-occurring neurologic sequelae in adult survivors of childhood acute lymphoblastic leukemia: a report from the childhood cancer survivor study. J Clin Oncol 2009; 28: 324–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eapen M, Zhang M-J, Devidas M, et al. Outcomes after HLA-matched sibling transplantation or chemotherapy in children with acute lymphoblastic leukemia in a second remission after an isolated central nervous system relapse: a collaborative study of the Children’s Oncology Group and the Center for International Blood and Marrow Transplant Research. Leukemia 2008; 22: 281–86. [DOI] [PubMed] [Google Scholar]
- 10.Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity associated with CD19-targeted CAR-T cell therapies. CNS Drugs 2018; 32: 1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert MJ. Severe neurotoxicity in the phase 2 trial of JCAR015 in adult B-ALL (ROCKET Study): analysis of patient, protocol and product attributes. Society for Immunotherapy of Cancer annual meeting; National Harbor, MD, USA; Nov 8–12, 2017 (oral presentation). [Google Scholar]
- 12.Turtle CJ, Hanafi L-A, Berger C, et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016; 126: 2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frigault MJ, Dietrich J, Martinez-Lage M, et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood 2019; 134: 860–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He X, Xiao X, Li Q, et al. Anti-CD19 CAR-T as a feasible and safe treatment against central nervous system leukemia after intrathecal chemotherapy in adults with relapsed or refractory B-ALL. Leukemia 2019; 33: 2102–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubinstein JD, Nelson AS, Krupski C, et al. Chimeric antigen receptor T-cell therapy in patients with neurologic comorbidities. Pediatr Blood Cancer 2020; 67: e28199. [DOI] [PubMed] [Google Scholar]
- 16.Rheingold SR, Chen L, Maude SL, et al. Efficient trafficking of chimeric antigen receptor (CAR)-modified T cells to CSF and induction of durable CNS remissions in children with CNS/combined relapsed/refractory ALL. Blood 2015; 126: 3769. [Google Scholar]
- 17.Maude SL, Teachey DT, Rheingold SR, et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. Proc Am Soc Clin Oncol 2018; 34: 3011 (abstr). [Google Scholar]
- 18.Rubinstein JD, Krupski C, Nelson AS, O’Brien MM, Davies SM, Phillips CL. Chimeric antigen receptor T-cell therapy in patients with multiply relapsed or refractory extramedullary leukemia. Biol Blood Marrow Tr 2020. 26: e280–85. [DOI] [PubMed] [Google Scholar]
- 19.Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 2017; 130: 2317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers RM, Li Y, Leahy AB, et al. Humanized CD19-targeted chimeric antigen receptor (CAR) T cells in CAR-naive and CAR-exposed children and young adults with relapsed or refractory acute lymphoblastic leukemia. J Clin Oncol 2021; published online June 22. 10.1200/JCO.20.03458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadauke S, Myers RM, Li Y, et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol 2021; 39: 920–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maude SL, Pulsipher MA, Boyer MW, et al. Efficacy and safety of CTL019 in the first US phase II multicenter trial in pediatric relapsed/refractory acute lymphoblastic leukemia: results of an interim analysis. Blood 2016; 128 (suppl 1): 2801 (abstr). [Google Scholar]
- 23.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen EC, Devidas M, Chen S, et al. Dexamethasone and High-dose methotrexate improve outcome for children and young adults with high-risk B-acute lymphoblastic leukemia: a report from Children’s Oncology Group study AALL0232. J Clin Oncol 2016; 34: 2380–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salzer WL, Burke MJ, Devidas M, et al. Toxicity associated with intensive postinduction therapy incorporating clofarabine in the very high-risk stratum of patients with newly diagnosed high-risk B-lymphoblastic leukemia: a report from the Children’s Oncology Group study AALL1131. Cancer 2018; 124: 1150–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol 2018; 11: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamdi A, Mawad R, Bassett R, et al. Central nervous system relapse in adults with acute lymphoblastic leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Tr 2014; 20: 1767–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med 2018; 378: 449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gofshteyn JS, Shaw PA, Teachey DT, et al. Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann Neurol 2018; 84: 537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gust J, Hay KA, Hanafi L-A, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 2017; 7: 1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.