

Abstract
Exploring the new therapeutic indications of known drugs for treating COVID-19, popularly known as drug repurposing, is emerging as a pragmatic approach especially owing to the mounting pressure to control the pandemic. Targeting multiple targets with a single drug by employing drug repurposing known as the polypharmacology approach may be an optimised strategy for the development of effective therapeutics. In this study, virtual screening has been carried out on seven popular SARS-CoV-2 targets (3CLpro, PLpro, RdRp (NSP12), NSP13, NSP14, NSP15, and NSP16). A total of 4015 approved drugs were screened against these targets. Four drugs namely venetoclax, tirilazad, acetyldigitoxin, and ledipasvir have been selected based on the docking score, ability to interact with four or more targets and having a reasonably good number of interactions with key residues in the targets. The MD simulations and MM-PBSA studies showed reasonable stability of protein-drug complexes and sustainability of key interactions between the drugs with their respective targets throughout the course of MD simulations. The identified four drug molecules were also compared with the known drugs namely elbasvir and nafamostat. While the study has provided a detailed account of the chosen protein-drug complexes, it has explored the nature of seven important targets of SARS-CoV-2 by evaluating the protein-drug complexation process in great detail.
Graphical abstract
Drug repurposing strategy against SARS-CoV2 drug targets. Computational analysis was performed to identify repurposable approved drug candidates against SARS-CoV2 using approaches such as virtual screening, molecular dynamics simulation and MM-PBSA calculations. Four drugs namely venetoclax, tirilazad, acetyldigitoxin, and ledipasvir have been selected as potential candidates.

Supplementary Information
The online version contains supplementary material available at 10.1007/s12039-022-02046-0.
Keywords: Polypharmacology, Antivirals, Drug repurposing, Virtual screening, MM-PBSA
Introduction
The severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2) resulted in a pandemic1–3 and continues to cause turmoil in our everyday lives.4 Tackling the challenges caused by viral infections often involves the development of therapeutics which not only control the pristine form of the virus but also its variants.5 The SARS-CoV-2 pandemic triggered a collapse of the healthcare system and has instigated the search for therapeutics. Though there are few therapies available for the treatment of SARS-CoV-2 infection, the effective cure is still a matter of question.6–9 The major factor of this enhanced transmission may be traced to the emergence of new mutant strains.10,11 Around 30 SARS-CoV-2 proteins have been reported so far12 and the manifestation of mutations in terms of infectivity, transmission, replication, and transcription is an intriguing fact.13–15 The genome sequence of SARS-CoV-2 is 96.2% identical to the bat CoV RaTG13 sequence, although it shares 79.5% identity to SAR-CoV.16
SARS-CoV-2 encodes four structural proteins, and sixteen non-structural proteins (NSPs). The four structural proteins make up a mature SARS-CoV-2 viz, envelope (E), membrane (M), nucleocapsid (N) and spike (S).17 The Spike (S) homotrimeric glycoprotein located on the virion surface allows entry of the virus into the human cells and employs the cellular transmembrane serine protease 2 (TMPRSS2) for S protein priming which helps in internalization of the virus.18,19
Figure 1 displays the viral targets selected in the current study based on their role in viral replication and RNA synthesis. The 3 cysteine-like protease (3CLpro)20 and papain-like protease (PLpro)21 are involved in the cleaving of polyprotein to release independent functional proteins, where the former, due to its high sequence similarity among the coronavirus, may be studied for developing a general-purpose antiviral drug. Further RNA-dependent RNA polymerase (RdRp (NSP12)),22 Helicase (NSP13),23 Exoribonuclease (NSP14)24 and Endoribonuclease (NSP15)25 play a pivotal role in SARS-CoV-2 replication and RNA synthesis, while 2'-O-methyltransferase (NSP16) stabilizes the RNA, and protect the viral RNA from host immune response.26 Thus, the current study has a robust representation of proteins involved in RNA synthesis, cleaving of polyprotein to release functional proteins, and replication. Spike proteins, which are essentially responsible for the viral entry was not included as they have been extensively studied, besides the current study focuses on selecting the proteins involved in viral transcription and translation. Thus, the chosen proteins, as depicted in Figure 1 are capable of providing common features for developing multi-targeting drugs against COVID-19.
Figure 1.


Cartoon representation of the selected protein targets (A) 3CLpro PDB ID:6M03 (B) PLpro PDB ID: 6W9C (C) NSP12: Modelled (D) NSP13: Modelled, (E) NSP14: Modelled, (F) NSP15 PDB ID: 6W01, (G) NSP16 PDB ID: 6W4H, along with labelled domains of each protein. The selected binding site residues are encircled and highlighted which depict the amino acid residues of the binding site residues.
The 3CLpro protein consists of three domains (domain I, domain II, domain III) where domain I and domain II have a five-stranded antiparallel β-barrel structure with a chymotrypsin-like folding scaffold, whereas the domain III has a five α-helices cluster.27,28 PLpro protein has two domains which are ubiquitin-like (UBL) domain in N-terminal and it is necessary for pathway antagonism and ubiquitin-specific protease (USP) domain in C-terminal which contains the protease active sites.29,30 The structure of NSP12 protein contains RdRp domain, RdRp associate nucleotidyltransferase (NiRAN) domain and an interface domain connecting the two domains and these domains, consist of the active sites and thus, play a key functional role of NSP12 protein.31 There are five domains for NSP13 which consist of zinc-binding domain (ZBD) which is involved in coordinating three structural zinc ions, a helical stalk domain, a beta-barrel 1B domain and two RecA like helicase subdomains which are 1A and 2A. This RecA like” helicase subdomains contain residues that are involved in binding of nucleotides and also hydrolysis.32 The NSP14 protein structure contains two domains which are exoribonuclease (ExoN), in N-terminal and N7‐methyltransferase (N7‐MTase) domain in C-terminal. The replication proofreading activity of Exon domain makes it a potential target for developing inhibitors against SARS-CoV-2.33 NSP15 consists of three domains which are N-terminal domain (ND), middle domain (MD), and C-terminal catalytic NendoU domain (NendoU). The EndoU domains are involved in biological functions which are associated with RNA processing and also in innate immune response.34 Finally, the NSP16 consist of a methyltransferase (MTase) domain and the alpha-D helix in MTase domain is important for SAM-binding (S-adenosyl-L-methionine-dependent methyltransferase) and RNA cap-binding for methyltransferase activity.35
Vaccine and drug development for the prevention and treatment of COVID-19 has become an area of interest for the scientific community worldwide. Several vaccines have been developed globally, but fail to eradicate the virus completely due to the emergence of new viral strains. Thus, employing drug repurposing36 with a polypharmacology37 approach may decipher the process to tackle the therapeutic emergency. As a response to the COVID-19 pandemic, several studies have utilized various molecular modeling approaches for repurposing pre-existing drugs.38–41 There are a few repurposed drugs available in the market for COVID-19 treatment, however, several limitations have been observed with the existing drugs.9,42–44 To examine the relevance of the selected seven targets, a docking study has been performed on the 15 clinically repurposed drugs that were extensively employed for the COVID-19 treatment (Figure 2 and Figure S1, SI). Figure 2 provides an Autodock Vina analysis of the 105 complexes (all complexes of 15 clinically used drugs with 7 targets) and 83 of these combinations have shown fair binding affinity.
Figure 2.

The distribution of docking scores of known popular drugs with seven SARS-CoV-2 drug targets. The drugs are shown in X-axis and the proteins are shown on Y-axis. The yellow-blue color spectrum shows increasing binding affinity. The drug molecules which have a docking score of less than −6 kcal/mol are represented with white space in corresponding proteins.
This supports the selection of drug targets having a good ability to identify the promiscuous candidates in the docking study. Extensive docking analysis was carried out on the known drug candidates, as provided in the drug bank and drug central using Autodock Vina45 to see the interactions and binding affinity of 4015 approved drugs against well-known seven NSPs of SARS-CoV-2, namely 3CLpro, PLpro, RdRp, NSP13, NSP14, NSP15, and NSP16. Analysis of virtual screening results followed by 100 ns MD simulation and MM-PBSA calculations indicate that venetoclax, tirilazad, acetyldigitoxin and ledipasvir showed reasonable stability and retain consistent interaction with their respective multiple targets. The schematic workflow adopted in this study has been displayed in Figure 3.
Figure 3.

Schematic representation of workflow adopted in this study to identify repurposable drug candidates against multiple SARS-CoV-2 targets.
Selection of target proteins and their structural details
A total of seven SARS-CoV-2 protein targets have been selected in this study based on their role in COVID-19 pathophysiology, especially viral RNA replication. The therapeutic targets were selected based on (i) the significance of particular proteins in viral replication and transcription, (ii) the presence of a binding pocket that presents hotspots for the drug development, and (iii) availability of viral protein with a good resolution from RCSB Protein Data Bank (PDB) database (https://www.rcsb.org).46 After analyzing the information on bound inhibitor, missing residues and resolution of the structure, four crystal structures of SARS-CoV-2 were taken from PDB. Table S1 (SI) provides the PDB ID, active site residues of the chosen targets namely: (i) 3CLpro (ii) PLpro (iii) NSP15, (iv) NSP16 and three homology modeled proteins namely: (v) NSP12 (vi) NSP13 (Helicase), (vii) NSP14 were obtained from SWISSMODEL server47 (https://swissmodel.expasy.org) due to unavailability of resolved crystal structures during this study.
The 3CLpro is a key enzyme of the virus, as it cleaves viral replicase polyprotein at 11 sites and generates NSP 4 to 16 that are essential for viral replication. The CLpro active site contains the catalytic dyad Cis145 and His41. 3CLpro of SARS-CoV-2 shares 96% similarity with that of SARS-CoV.48 The PLpro (multi-domain protein) cleaves viral replicase polyprotein at three sites to release NSP 1 to NSP3 which antagonize the host innate immunity. The PLpro sequence identity between SARS-CoV-2 and SARS-CoVis 83%.49
The RdRp enzyme (NSP12) is a core catalytic subunit essential for viral RNA synthesis replication. RdRp proteins have been targeted for the development of potential antiviral molecules against different viruses including hepatitis C virus (HCV),50 ZIKA virus (ZIKV),51 and coronaviruses (CoVs).52 The nucleoside triphosphate (NTP) entry channel for RdRp of SARS-CoV-2 is formed by a stretch of amino acid Ser759 to Asp761 and other amino acid residues such as Lys545, Arg553 and Arg555. RdRp of SARS-CoV-2 is highly similar to other coronaviruses such as SARS-CoV with which it has 96% sequence identity.53
NSP13 belongs to the 1B helicase superfamily with NTPase and it is involved in duplex RNA/DNA unwinding and 5′-RNA capping activities.22 The site-directed mutagenesis studies on SARS-CoV NSP13 have revealed that six residues, Lys288, Ser289, Asp374, Glu375, Gln404, and Arg567 are essential for NTPase activity.54 Due to their essential role in viral replication and conservation across all coronavirus species, NSP13 has emerged as a potential target for antiviral drug development.22,54
It has been observed that NSP14 is key for viral mRNA stability as it evades the host immune response.24,55 The residues Ile332, Phe367, Val389, Asp331, Gly333, Trp292, Phe401, Tyr420, Phe426, Thr428, and Phe506 are important for N7-MTase activity. In the present study, the SAM binding domain of NSP14 has been considered as a binding pocket for drug binding.55 Processes such as capping of viral mRNA are catalyzed by NSP16, which shares above 90% similarity among the novel and older SARS-coronavirus.
Uridylate-specific endoribonuclease (NSP15) cleaves 3′ of uridylates via a ribonuclease A (RNase-A) and it restrains exposure to the host immune system.25 It has very high sequence similarity with NSP15 protein of SARS and MERS and the catalytic C-terminal domain (active site) of the enzyme-containing the catalytic triad His235, His250 and Lys290.34,56 Encouragingly due to the SAM binding site's conserved nature, inhibitors targeting this pocket may be developed as pan-antiviral inhibitors.57 The binding pocket of the selected seven SARS-CoV-2 proteins active sites are shown in Figure 1 and Table S1, SI.
Computational
Homology modelling of NSP12, NSP13 and NSP14 protein structures
The modelled structure of NSP12 (YP_009724389.1), NSP13 (YP_009724389.1) and NSP14 (BAE93399.1) protein was downloaded from SWISS-Model repository (https://swissmodel.expasy.org/repository/).58 The selected modelled structures were validated using Ramachandran (RC) plot59 and ERRAT60 validation tools available in the SAVES server (https://saves.mbi.ucla.edu/).
Preparation of protein structures
All the seven selected SARS-CoV-2 proteins were prepared in Autodock Tools (ADT version 1.5.7rc1) by removing solvent molecules and non-interacting ions, adding polar hydrogens and partial charge assignment. The protein structures were saved in Autodock PDBQT. Based on the information of active site residues and their adjacent residues, the grid box size was generated. Table S2 (SI) provides the grid box parameters for all the seven SARS-CoV-2 protein targets.
Preparation of drug structures
A total of 4015 known drug candidates retrieved from DrugBank61 and DrugCentral62 were considered for the present study. Redundant molecules and ion-containing molecules were removed manually during the SMILES notations inspection. Further, the geometry optimization was performed using the raccoon.py script of MGL tools and converted to 3D structures. The selected non-redundant molecules were further converted to PDBQT format.
Virtual screening
The virtual screening was performed using AutodockVina1.1.2. The grid box was set up around the active site of the protein (Table S2, SI). Total nine conformers were generated and the best conformer was identified based on docking score and the binding pose. The top-ranked 20 candidates for each target were identified from virtual screening based on the high docking score. The top 25 drugs were prioritized according to the calculated physicochemical properties (Table S3, SI) and pharmacokinetic properties (Table S4, SI). This analysis leads to the identification of 25 drug candidates, which bind to at least two or more of the selected seven targets, by imposing a threshold value of -6.0 kcal/mol.
Further, the drugs having good docking scores and interactions with four or more targets were selected and subjected to post-docking analysis. Discovery studio visualizer63 was used to analyze the protein-drug complex interactions. The schematic representation of the workflow adopted in this study has been shown in Figure 3.
Molecular dynamics simulations and MM-PBSA calculations
As described in the foregoing sections, 18 protein-drug complexes have been identified and subjected to MD simulations. MD simulations were performed with Gromacs 5.0.4 package64 using CHARMM force field65 and SPC water model.66 The topological parameters of the drug molecules were generated using the CGenFF server.67 MD simulations were performed under periodic boundary conditions (PBCs) with a cubical box maintaining a distance of 1.0 nm and 1.5 nm and the boundary edges between the protein-drug complexes depending on the size of the proteins. The protein-drug complexes were solvated with SPC water molecules and the system was neutralized by adding the counterions into the solvated box depending on the charge of the system. Then, the systems were subjected to 1000 step steepest descent minimization followed by 2000 conjugate-gradient minimization for initial energy minimization with the frozen drug to avoid a further structural clash in the solvated system. Then, the whole system was subjected to 5000 steepest descent minimizations followed by 6000 conjugate-gradient minimizations with a maximum step size of 0.01 fs for final energy minimization. After subjecting the systems to position restraint equilibrations, heat under canonical ensemble from 0 to 303 K for 500 ps was generated using a modified Berendsen thermostat.68 Later, the systems were equilibrated for 1 ns under isothermal-isobaric conditions (with a constant pressure of 1.0 bar). Finally, the production run of 100 ns was performed without any restraints, followed by an integration time step of 2 fs. The coordinates were saved every 2 ps under constant conditions of 300 K temperature and 1 atm pressure.69 The LINCS algorithm70 was used to restrain the bond lengths and the long-range electrostatics were calculated using the particle mesh Ewald (PME),71 while the SETTLE algorithm72 was employed to constrain the geometry of water molecules. The trajectory of each system was analyzed using Origin Pro73 and VMD software.74 Molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) method using g_mmpbsa package,75 was employed to calculate the binding free energies of protein-drug complexes using the following equation.
| 1 |
Where, Ecomplex, Eprotein and Edrug are the total MMPBSA energy of protein-drug complex, total solution free energies of the isolated protein and drug molecules, respectively. The total free energy of each individual can be expressed using equation 2.
| 2 |
Where, z is either protein, drug or protein-drug complex. <EMM> is the ensemble average value of the molecular mechanic’s potential energies in a vacuum. T and S represent absolute temperature and entropy, respectively and together TS contributes to the entropic contribution to the total free energy in a vacuum. <EMM> is the sum of both bonded and nonbonded interactions of the molecules and <Esolvation> is the average free energy of solvation, consisting of polar (Epolar) and nonpolar (Enonpolar) components.
The Poisson-Boltzmann equation is solved using a linear model to estimate the polar solvation energy (Epolar)whereas the nonpolar part of the solvation energy (Enonpolar) is estimated using solvent accessible surface area (SASA) method and can be expressed using equation 3.
| 3 |
Where, γ is a parameter representing surface tension of the solvent, A is the calculated SASA of the molecule and b is a fitting parameter. The last 20 ns trajectory (i.e., 80 ns to 100 ns) was used for the MMPBSA calculation. MmPbSaStat.py program was used to calculate the binding energies and MmPbSaDecomp.py was used to extract the residue-specific contributions towards binding.75 In this current investigation, we retrieved 400 frames from 80–100 ns trajectories from all the eighteen complexes, to calculate the binding free energy. To analyse the docking score distribution of all the four drugs with their respective targets, different conformers were generated from the 100 ns trajectories by taking a pose at interval of 10 ns. The drug molecules were removed and protein was prepared without changing their conformation. The putative 180 (18 * 10) points generated along the trajectory of MD simulations were subjected to docking.
Results and Discussion
In the current study, a virtual screening analysis of the known drug molecules obtained from Drug bank61 and Drug central62 was carried out against the seven SARS-CoV2 viral targets (3CLpro, PLpro, NSP12, NSP13, NSP14, NSP15 & NSP16). The top 20 compounds which amount to the list of top 0.4% of the molecules have been selected for further studies. It was observed that none of the known drug molecules which are popularly used in the treatment of COVID-19 were listed out in the top 0.4% (Figure 2) though most of them show a fair binding with multiple targets. Around 25 of the known drugs appear to have significant binding affinity for at least 2 or more targets as shown in Table 1. Among these, 4 drugs namely venetoclax, acetyldigitoxin, tirilazad and ledipasvir were considered for further studies. The two drugs ergotamine and dihydroergotamine were omitted from the study as they appear to be less promising candidates for drug repurposing. Thus, the 18 selected complexes were subjected to MD simulations and MM-PBSA calculations. In addition, an analysis of the docking poses, RMSF, RMSD, H-bond have been carried out along the 100 ns MD trajectory of these 18 complexes, which provided interesting insights into the nature of binding.
Table 1.
List of 25 drug molecules and their therapeutic indications.
| Sl. No. | Drugs | Known indication | Targets |
|---|---|---|---|
| 1. | Dihydroergotamine | Migraine | 3CLpro, PLpro, NSP13, NSP14, NSP15, NSP16 |
| 2. | Ergotamine | Migraine | 3CLpro, PLpro, Nsp12, NSP13, NSP14, NSP15, NSP16 |
| 3. | Venetoclax | Anticancer | 3CLpro, PLpro, NSP12, NSP13, NSP14, NSP16 |
| 4. | Acetyldigitoxin | Cardiovascular | 3CLpro, NSP13, NSP14, NSP16 |
| 5. | Dihydroergocristine | Migraine | NSP12, NSP13, NSP15, NSP16 |
| 6. | Ledipasvir | Antiviral | PLpro, NSP12, NSP14, NSP16 |
| 7. | Tirilazad | Cardiovascular | 3CLpro, PLpro, NSP12, NSP16 |
| 8. | Antrafenine | Antiinflammatory | PLpro, NSP14, NSP15 |
| 9. | Conivaptan | Hyponatremia | NSP13, NSP14, NSP15 |
| 10. | Entrectinib | Anticancer | 3CLpro, PLpro, NSP16 |
| 11. | Paritaprevir | Antiviral | 3CLpro, NSP13, NSP16 |
| 12. | Ubrogepant | Migraine | 3CLpro, PLpro, NSP15 |
| 13. | Bisdequalinium | Antibacterial | NSP12, NSP13 |
| 14. | Digitoxin | Cardiovascular | NSP13, NSP14 |
| 15. | Eltrombopag | Anemia | NSP14, NSP16 |
| 16. | Glycyrrhizic acid | Antidiabetic | NSP13, NSP15 |
| 17. | Quinupristin | Antibacterial | NSP12, NSP16 |
| 18. | Teicoplaninaglycone | Antibacterial | 3CLpro, NSP16 |
| 19. | Temoporfin | Anticancer | 3CLpro, PLpro |
| 20. | Zinostatin | Anticancer | NSP14, NSP15 |
| 21. | Guamecycline | Anticancer | 3CLpro, NSP12 |
| 22. | Anazolene | Antiallergic | NSP15, NSP14 |
| 23. | Deslanoside | Cardiovascular | NSP12, NSP13 |
| 24. | Dexamethasone | Anticancer | NSP15, 3CLpro |
| 25. | Lumacaftor | Cystic fibrosis | NSP15, PLpro |
The last column lists the SARS-CoV-2 targets which show noticeable binding affinity with the drug as indicated by the docking score.
Homology modeling
From the models available at the SWISS-Model repository, model template PDB ID: 6M71 was chosen for NSP12 which showed 99% sequence identity with ERRAT value of 92%. Ramachandran plot of the modelled structure reveals that there is nearly 91% favored region, 7.7% allowed region and 0.7% outlier region. For NSP13, model template PDB ID: 5RLC was selected which showed 96% sequence identity and also ERRAT value of 96%. From the RC plot, it can be seen that the selected model showed 89.6% favored region, 9.8% allowed region and 0.2% outlier region. Similarly, for NSP14, model template PDB ID: 5NFY was selected which had 90% sequence identity with ERRAT value of 90% and the RC plot validates the selected model having 86.0% favored region, 11.6% allowed region and 1.7% outlier region. The template models were selected based on their sequence identity and query coverage which was followed by calculating the RC plot with ERRAT value that validated the model structure for further study. The RC plot and ERRAT values are shown in Table S5, SI.
Virtual screening analysis
Docking studies were performed for over 4015 approved drugs on the binding pocket of selected SARS-CoV-2 target proteins. The distribution of binding energies of 4015 approved drugs for the seven viral targets is shown in Figure 4. In this study, the top 20 drug molecules (Table 1) from each of the seven targets were identified based on the highest docking score obtained from AutodockVina. Venetoclax was found to have significant interaction with 6 targets while tirilazad, acetyldigitoxin and ledipasvir interacted significantly with 4 targets. Thus, the identified 18 complexes were subjected to MD simulations.
Figure 4.

The distribution of docking scores of different drug molecules with seven selected SARS-CoV-2 drug targets. The drugs are shown in X-axis and the proteins are shown in Y-axis. The yellow-green color spectrum shows increasing binding affinity. The drug molecules which has less than − 6 kcal/mol or no docking score are represented with white space in corresponding proteins. The four selected drugs are highlighted in boldface.
Molecular dynamics simulations
Molecular dynamics (MD) simulations for the top 18 protein-drug complexes were carried out to study the conformational stability and sustainability of the protein-ligand complex. MD simulations can effectively validate the results obtained from molecular docking approaches, by examining the variation of the binding interactions in the complexes at various points along the trajectory. Similarly, the computed RMSD values will indicate the stability and rigidity of the complex (Figure 5). Figure 5A shows the RMSD for the seven targets in presence of venetoclax. NSP14 showed an increase in RMSD after 80 ns suggesting conformational flexibility. It was observed that NSP14 showed larger conformational flexibility when it is bound to venetoclax while in the case of other drugs such as acetyldigitoxin, and ledipasvir, the protein RMSD shows a convergence suggesting conformational stability. Significant changes in conformational nature were observed for PLpro bound to tirilazad, as for the remaining cases, the structural changes in proteins were observed to be less significant. In Figure S2 (SI), the apo protein backbone RMSD has been illustrated. It can be observed that the protein structure has been largely intact even after binding of the drug molecules, considered in the study. Figure 5B illustrate the variations in the RMSD values corresponding to the drugs when they are bound to different protein targets. Here the convergence reveal that the drugs are in the binding pocket of the target protein during the simulation time period. However, substantial fluctuations in the RMSD are an indication pointing to the possibility that the drug obtained an alternative conformation in the binding site. Depending upon the relative free energy differences, different conformations are sampled either less frequently or often. Even though the docking protocol gives relative energies of different docking poses, the discussion is usually based on the most stable binding pose. So, computing the free energies using configurations obtained by molecular dynamics simulations is necessary to obtain an accurate estimate for the binding free energies. It is worth noting that tirilazad in PLpro target displayed fluctuations in RMSD suggesting its conformational flexibility. However, ledipasivir exhibited less significant conformational flexibility in its targets. Since the binding site microenvironments are specific for each target, different conformations may be required for optimal binding with different targets. RMSFs are the measures of conformational flexibility of various residues and larger values for RMSFs suggest larger conformational changes while near-zero values suggest their rigid nature. RMSFs for various target proteins bound to different drugs are shown in Figure S2, SI. The protein bound to tirilazad showed the highest RMSF value suggesting that this induced a larger conformational change in the target protein. In NSP14, ledipasivir and venetoclax induce larger structural changes in protein in comparison to drug acetyldigitoxin. In the remaining targets, different drugs were observed to exhibit similar effects of the RMSFs (Figure S3, SI). The compactness in the overall protein can be compared from the radius of gyration which supports the RMSD results (Figure 5).
Figure 5.


RMSD plot generated from 100 ns trajectory (A) Protein backbone plots are shown in the left panel (B) Ligand RMSD plots of four selected drugs with its respective proteins are shown in the right panel.
In order to validate our findings, we also performed MD simulation for the reported drug molecules elbasvir and nafamostat (interacting with all the targets of venetoclax). Comparison with the known reference molecules, elbasvir and nafamostat, reveals a high similarity to the shortlisted drug molecules in the study.
Hydrogen bond (H-bond) interaction is one of the major players in governing drug stability at the protein's active site. All the 18 protein-ligand complexes reveal the existence of multiple hydrogen bonds, with the number of such bonds fluctuating between two to five (Figure S4, SI). Analysing the frames along the molecular dynamics trajectory indicate that the number of hydrogen bonds were fluctuating between three to four, among the six complexes of the venetoclax. Similarly, the number of hydrogen bonds in the complexes involving tirilazad, acetyldigitoxin and ledipasvir ranges from two to three, three to four and two to five, respectively.
Analysis of non-covalent interactions of the protein-drug complex along the MD trajectory
The interaction pattern of the initial docked complex before MD simulation and after MD simulation of venetoclax, tirilazad, acetyldigitoxin and ledipasvir in complex with the respective protein targets were analysed. The interactions at 100 ns are depicted in Table S6 (SI). In the initial venetoclax-3CLpro complex, there was one H-bond, one π-σ bond, one C-H….O bond, two π-alkyl, one alkyl interactions as shown in Figure 6A and Table S6, SI. However, H-bond with residue Thr24, one π-σ bond with Thr25, one π-alkyl with Met49, one alkyl with Met165 were retained after 100ns MD simulations. In addition, one new H-bond with Thr26, two π-alkyl bonds with His41 and one alkyl with Cys145 were exhibited in this complex. Though, C-H bond with Ser46 was replaced by Asn142 during MD simulation.
Figure 6.

2D representation depicting docking interactions of venetoclax with SARS-CoV-2 protein targets (A) 3CLpro (−9.1 kcal/mol) (B) PLpro (−8.2 kcal/mol) (C) NSP12 (−10.2 kcal/mol) (D) NSP13 (−10.8 kcal/mol) (E) NSP14 (−12.1 kcal/mol) and (F) NSP16 (−11.8 kcal/mol).
Venetoclax-PLpro complex displayed five H-bonds, one C-H….O bond, one π-π T-shaped bond, one π-alkyl and three alkyl interactions were found in the initial docked complex as shown in Figure 6B. On the other hand, none of the interactions was retained except one C-H….O interaction with Asp164 during MD simulation. However, some new interactions were established after MD simulations such as two H-bonds with Arg166, one alkyl interaction with Pro298, and one C-H interaction with Lys157 in venetoclax-PLpro complex (Table S6, SI). Venetoclax was not able to retain H-bonds, CH….N bond (His810), C-H….O (Lys798, Asp618) bond, alkyl, π-π T-shaped, π-anion and halogen interactions with NSP12 after MD simulations. However, venetoclax retained only two π-alkyl bonds with His810 and Lys798 as depicted in Figure 6C and Table S6 (SI). Venetoclax complex maintained only two π-alkyl interactions with Tyr180 and Pro408 residues of NSP13 during MD simulation and for venetoclax-NSP14 complex, none of the interactions was reserved which were existed in the initial docked complex. Furthermore, there were some new interactions were observed with NSP14 such as one H-bond with Gln262, one π-π stacked interaction with Lys288 and one π-alkyl interaction with Val290 as shown in Figure 6D and Table S6 (SI).
After MD simulation of venetoclax-NSP16 complex, only one H-bond with Asp6873 was retained whereas some new interactions formed such as π-π T-shaped bonds with residues Leu6898, Pro6932 and Val6937, and one π-anion interaction with Ser6872 residue of NSP16 (Table S6, SI).
From the initial docked complex of tirilazad-3CLpro, it can be observed that there is one H-bond, one cation-π, two π-alkyl and seven alkyl interactions as shown in Figure 7A and Table S7 (SI). However, after 100 ns of simulation, only one H bond with residue Arg188 was retained. Interaction of tirilazad with PLpro showed one H-bond, two C-H…O bond, one π-σ bond, two π-anion, one π-alkyl, and three alkyl interactions were observed as shown in Table S7, SI and Figure 7B. Furthermore, one π-anion with Asp164, one π-alkyl with Tyr264, two alkyl interactions with Leu162 and Pro248 were intact after 100 ns MD simulations (Table S7, SI). In addition to one C-H…O bond with Asp164, one new C-H….O bond with Pro248 was observed after MD simulation. However, in tirilazad-PLpro complex, tirilazad missed H-bond with Trp800, π-sulfur interaction with Cys813 and one π-alkyl bond with Phe812 of NSP12 after MD simulations (Figure 7C and Table S7, SI). Whereas, tirilazad retained two alkyl bonds out of six with Lys593 and Val588, one π-alkyl bond with Phe594 and C-H….O bond with Thr591 as depicted in Table S7, SI. A new alkyl bond was also formed with Leu758 of NSP12. In the case of initial tirilazad-NSP16 complex, there were two H-bonds, two C-H bond, three alkyl, one π-alkyl and one π-anion interactions present (Table S7, SI and Figure 7D). However, one H-bond with Lys6968, one C-H….O bond with Try6930, one alkyl interaction with Met6840, one π–alkyl interaction with Tyr6930 and one π–anion interaction with Asp6879 were retained after MD simulations. Additionally, some new interactions were observed such as one H-bond with Ser7000, two C-H bonds with Ser6999 and Asp6897, and two π–alkyl interactions with Met6929 and Leu6898.
Figure 7.

2D representation depicting docking interactions of tirilazad with SARS-CoV-2 protein targets (A) 3CLpro (−8.7 kcal/mol) (B) PLpro (−7.9 kcal/mol) (C) NSP12 (−9.7 kcal/mol) and (D) NSP16 (−11.4 kcal/mol).
In the acetyldigitoxin-3CLpro complex, initially, there were seven H-bonds and five alkyl interactions as shown in Figure 8A and Table S8, SI. However, only four H-bonds with residue Asn142, Gly143, His163 and Glu166, and four alkyl interactions with Met49, Cys145, Leu27, Met165 were retained after 100ns MD simulations. In addition, one new H-bond with Thr26 was found in this complex. The acetyldigitoxin with NSP13 docked complex exhibited five H-bonds, two C-H bond and five alkyl interactions as shown in Figure 8B and Table S8, SI. On the other hand, none of the H-bond was retained but three new H-bonds with Thr380, Asp383 and Ser539 were found in post-MD interactions. One C-H….O bond with Ser535 and two alkyl bonds with Ala312 and Cys309 were reserved and two new alkyl interactions with Ala407 and Arg339 were also observed in post-MD complex of Acetyldigitoxin-Nsp13 as mentioned in Table S8, SI). In theacetyldigitoxin-NSP14 complex, (Figure 8C and Table S8, SI) one new H-bond with Leu144 and two new alkyl interactions with His378 and His401 were observed other than existing interactions initial complex. Only one alkyl interaction with Phe504 was retained whereas one new alkyl interactions with His345 of NSP14 was established after MD simulations (Table S8, SI). In acetyldigitoxin-NSP16 complex, initially, there were four H-bonds, one C-H….O bond, four alkyl, and two π-alkyl interactions were present (Table S8, SI and Figure 8D). Furthermore, one new H-bond with Thr6970, one C-H….O bond with Gly6946, and two π–alkyl interactions with Pro6932 and Leu6898 were established other than existed interactions in acetyldigitoxin-NSP16 complex after MD simulations (Table S8, SI).
Figure 8.

2D representation depicting docking interactions of acetyldigitoxin with SARS-CoV-2 protein targets (A) 3CLpro (− 9.6 kcal/mol) (B) NSP13 (− 10.5 kcal/mol) (C) NSP14 (− 12.0 kcal/mol) and (D) NSP16 (− 11.3 kcal/mol). The docking score obtained from Autodock Vina for each protein are given in squared bracket.
Initially, there were two H-bonds, one C-H….O bond, one π-alkyl, two alkyl, one π-anion and one halogen interaction in the ledipasvir-PLpro complex, as shown in Table S9 (SI) and Figure 9A. On the other hand, after MD simulation, some new interactions were observed (one H-bond with residue Glu203, two alkyl interactions with Tyr264 and Leu162, and two C-H bonds with Thr301 and Asp164). However, one π-alkyl interaction with Tyr264, one π-anion and one halogen interaction with Glu167 was retained during MD simulation of the ledipasvir-PLpro complex.
Figure 9.

2D representation depicting docking interactions of ledipasvir with SARS-CoV-2 protein targets (A) PLpro (− 8.2 kcal/mol) (B) NSP12 (− 10.2 kcal/mol) (C) NSP14 (− 11.8 kcal/mol) and (D) NSP16 (− 11.4 kcal/mol). The docking score obtained from Autodock Vina for each protein are given in squared bracket.
Ledipasvir-NSP12 complex exhibited three C-H….O bond, one π-σ bond, one π-anion, one π-donor H-bond, and four alkyl interactions (Table S9, SI and Figure 9B). However, one π-σ interaction with Ala688, one π-anion with Asp761, and two alkyl interactions with Lys500 and Ala685 were retained after 100 ns MD simulations (Table S9, SI). Additionally, two new C-H bonds with Arg569 and Tyr689 were observed in the ledipasvir-NSP12 complex after MD simulations. But ledipasvir-NSP12 complex missed π-donor H-bond after molecular dynamics.
In the ledipasvir-NSP14, initially, four H-bonds, one C-H….O bond, two alkyl, eight π-alkyl, one π-π T-shaped bond and one halogen interactions were present (Table S9 (SI) and Figure 9C). However, five π-alkyl interactions with Pro429, Val290, Pro335 and Trp292, one C-H bond with Try6930, one π-π T-shaped bond with Phe367 were retained after MD simulations. One new alkyl interaction with Trp385 and two new C-H bonds with Val389 and Phe426 were observed in the post-MD ledipasvir-NSP14 complex.
In the ledipasvir-NSP16 complex, initially, there are seven H-bonds, one C-H….O bond, two π-σ bonds, two π-alkyl, one π-donor H-bond and one halogen interactions were present (Table S9 (SI) and Figure 9D). However, only three H-bond with Asp6873, Asp6912, and Phe6947, one π-σ bond with Leu6898, and one halogen interaction with Tyr6930 were engaged after MD simulations. But three H-bonds, one π-σ bond, π-alkyl and π-anion bonds were not retained in the post-MD complex. Additionally, few new interactions were examined such as four H-bond with Lys6844, Asn6841, Lys6898 and Glu6945, one π-σ bond with Met6929, and one C-H….O bonds with Glu7001 during the MD simulation of the ledipasvir-NSP16 complex. To compare with the known drugs, a study has been undertaken with elbasvir and nafamostat, which reveal that interaction with the active site residues are virtually the same as that of the 4 other selected candidates in the aforementioned section (the corresponding 2D interaction diagram of elbasvir and nafamostat has been illustrated in Figures 10, 11).
Figure 10.

2D representation depicting docking interactions obtained from Autodock Vina of elbasvir with SARS-CoV-2 protein targets (A) 3CLpro (− 8.1 kcal/mol) (B) PLpro (− 8.3 kcal/mol) (C) NSP12 (− 9.5 kcal/mol) (D) NSP13 (− 9.6 kcal/mol) (E) NSP14 (− 11.2 kcal/mol) and (F) NSP16 (− 9.3 kcal/mol).
Figure 11.

2D representation depicting docking interactions obtained from Autodock Vina of nafamostat with SARS-CoV-2 protein targets (A) 3CLpro (− 8.3 kcal/mol) (B) PLpro (− 7.7 kcal/mol) (C) NSP12 (− 8.1 kcal/mol) (D) NSP13 (− 7.6 kcal/mol) (E) NSP14 (− 10.1 kcal/mol) and (F) NSP16 (− 9.2 kcal/mol).
To examine the nature of interactions evolving during molecular dynamics simulations, we ventured on collecting frames at the interval of 10 ns, for each of the 18 complexes. The 180 frames, thus retried were subjected to docking with two objectives in mind; a) the stability of the complex and b) the variation of binding energy during the MD simulation. Figure 12 illustrates the variations of the binding energies of all the complexes, while the detailed analysis of the exact count of the non-covalent interactions76,77 at each of the frames was documented in the supporting information (Table S6-S9, SI). The electrostatic potential map of all the drug molecules along with the interacting amino acids are depicted individually in the six figures given in the supporting information (Figures S5-S10, SI).
Figure 12.

The docking score distributions of different conformers generated from regular time intervals of MD trajectories of (A) venetoclax, (B) tirilazad, (C) acetyldigitoxin and (D) ledipasvir complexes respectively. About 10 poses were extracted from the 100 ns trajectories by taking a pose at interval of 10 ns. The drug molecules were removed and protein was prepared without changing their conformation. The respective drug molecules were docked in the active site of 18 * 10 conformer.
From the docking study carried out at an interval of 10 ns from 100 ns trajectories, it was observed that among the six protein targets interacting with venetoclax, NSP16, and NSP14 have shown substantial variations in the docking scores along the trajectory, which may be traced to the fluctuating number of hydrogen bonds and other non-covalent interactions (Figure 12). The current exercise was carried out to see whether the protein-ligand complexes are intact or not during the simulations. Clearly, it was observed that in most cases the protein-ligand complexes are intact, albeit mostly losing some of the binding interactions as a function of time. While most of the protein-drug complexes retain the bulk of the binding affinity, even after the 100 ns simulation, the venetoclax complex with NSP16 drifting of the docking score may be traced to the loss of as many as 3 hydrogen bonds during the time evolution of the trajectory. The variations in the docking scores can be explained by comparing with the loss (mostly) or gain of the number of hydrogen bonds during the various frames of the MD trajectory (see Table S6-S9, SI).
MM-PBSA binding free energy calculation
The binding affinity of the drug obtained from Autodock Vina is based on the single protein-drug configuration corresponding to the least energy binding pose of the drug within the binding site of a target protein. However, the drug can have multiple effective conformations within the binding site and the reliable binding free energy estimate should be obtained as the sum over these. The binding free energies computed using MM-PBSA78 the multi-targeting potential of this target approach corresponds to average binding free energies obtained over various protein-ligand configurations from MD trajectories. Even though the calculation of binding free energies from MM-PBSA approach is computationally very demanding, this approach has been successful in ranking protein-ligand complexes.78–80 However, it is worth noting that the MM-PBSA approaches were shown to yield binding free energies which are quantitatively much larger when compared to docking energies.79,80 MM-PBSA approach has also been used to identify various lead compounds for COVID-19. However, the current study differs in the aspect that we aim at identifying drugs that are having multi-targeting potential.
Below we discuss the binding free energies computed for the four selected drugs with their high-affinity targets. A recent study on remdesivir interaction with RNA dependent RNA polymerase using MM-PBSA approach reported binding free energy of −78.5 kcal/mol.81,82 Since remdesivir is one of the recommended drugs for the treatment of COVID-19, we use this value as a reference to comment on the relative binding free energies for the drugs studied in this work. The binding free energies computed using MM-PBSA approach for venetoclax further confirms the multi-targeting potential of this target. Table 2 shows the computed binding free energies for this drug with 6 different targets and binding free energies are in the range -56 kcal/mol to -103 kcal/mol. If we analyze the individual contributions to total binding free energies it can be clearly established that the protein-drug interactions are majorly driven by hydrophobic interactions. In general, the sum of electrostatic and polar solvation energy is positive and so electrostatic interactions are acting against the complex formation. The non-polar solvation free energies are negative suggesting that they contribute to the stabilization of protein-drug complex formation. Based on the reference value, it is observed that the drug, venetoclax has superior binding affinity for the 4 targets such as 3CLpro, NSP14, NSP12, NSP13 (as the binding free energies were lower than the reference value, −78.5 kcal/mol) while it has moderate affinities for the remaining two targets. Similarly, the tirilazad showed significantly larger binding affinities for the two targets PLpro and NSP16 when compared to the remaining targets, NSP12 and 3CLpro. Ledipasvir showed larger binding affinities for the targets, NSP12, NSP14 and NSP16 when compared to the target, PLpro. However, the drug acetyldigitoxin showed only moderate binding affinities for the four targets 3CLpro, NSP13, NSP14 and NSP16. Even though this drug showed superior binding affinities for all targets based on the scoring function as implemented in Autodock Vina, with the rescoring using MM-PBSA binding free energies, we only observe moderate binding. The binding free energies of these molecules are essentially in the same range, compared to that of the known drug molecules elbasvir and nafamostat. The validation study by taking known drug molecules as reference ones increase the confidence levels in the application of these models. Thus, the first level screening using Autodock Vina and rescoring using more reliable scoring functions based on MM-PBSA binding free energies can be a potential protocol to remove false positives and to improve the reliability of the results lead compounds. The study suggests that the multilevel scoring approach using two or more scoring functions (with varying accuracy) sequentially can be effectively used to identify lead compounds for therapeutics. Overall, the current study using Autodock Vina and MM-PBSA based scoring functions suggest that the drugs, venetoclax, tirilazad and ledipasvir have multi-targeting potential and can be used to combat the current COVID-19 challenge which is posing a major threat to world healthcare systems due to its ability to mutate at a faster rate. Table 2 displayed the average MM-PBSA energy (kcal/mol) terms for the binding of four multi-targeting candidates.
Table 2.
MM-PBSA analyses of the four selected drug candidates against the multiple targets of SARS-CoV-2.
| Drug name | Protein | vdW. energy | Elec. energy | Polar solvation energy | SASA energy | Binding energy |
|---|---|---|---|---|---|---|
|
Venetoclax |
3CLpro | − 187.08 | − 24.72 | 129.59 | − 21.10 | − 103.32 |
| PLpro | − 133.73 | − 28.05 | 120.60 | − 15.06 | − 56.24 | |
| NSP12 | − 131.44 | − 25.94 | 81.83 | − 13.03 | − 88.58 | |
| NSP13 | − 278.11 | − 52.01 | 275.11 | − 33.15 | − 88.17 | |
| NSP14 | − 245.48 | − 46.32 | 174.52 | − 24.35 | − 141.63 | |
| NSP16 | − 188.05 | − 39.82 | 181.72 | − 21.29 | − 67.447 | |
|
Tirilazad |
3CLpro | − 121.73 | − 27.08 | 110.30 | − 13.06 | − 52.59 |
| PLpro | − 151.48 | − 6.45 | 72.31 | − 18.39 | − 104.01 | |
| NSP12 | − 102.36 | − 9.23 | 56.26 | − 10.32 | − 30.68 | |
| NSP16 | − 135.24 | − 18.43 | 77.64 | − 15.40 | − 91.44 | |
|
Acetyldigitoxin |
3CLpro | − 94.62 | − 32.69 | 99.41 | − 12.83 | − 40.73 |
| NSP13 | − 100.81 | − 39.69 | 117.42 | − 14.40 | − 37.28 | |
| NSP14 | − 59.15 | − 14.75 | 59.30 | − 8.70 | − 23.30 | |
| NSP16 | − 38.70 | − 12.68 | 64.74 | − 10.45 | − 55.57 | |
|
Ledipasvir |
PLpro | − 114.89 | − 28.08 | 87.50 | − 14.59 | − 70.03 |
| NSP12 | − 241.98 | − 79.58 | 207.79 | − 29.28 | − 116.06 | |
| NSP14 | − 273.26 | − 48.60 | 223.06 | − 33.70 | − 132.50 | |
| NSP16 | − 259.25 | − 100.71 | 266.84 | − 29.25 | − 122.38 | |
|
Elbasvir |
3CLpro | − 167.08 | − 21.72 | 119.59 | − 19.20 | − 99.32 |
| PLpro | − 158.14 | − 27.30 | 118.92 | − 17.14 | − 83.65 | |
| NSP12 | − 194.16 | − 40.30 | 178.91 | − 25.30 | − 80.85 | |
| NSP13 | − 194.25 | − 54.67 | 185.75 | − 22.94 | − 86.11 | |
| NSP14 | − 294.48 | − 96.95 | 306.26 | − 35.04 | − 120.21 | |
| NSP16 | − 198.05 | − 49.83 | 191.72 | − 29.30 | − 77.54 | |
|
Nafamostat |
3CLpro | − 38.44 | − 5.63 | 20.90 | − 5.90 | − 28.45 |
| PLpro | − 0.002 | 21.87 | 13.50 | − 0.19 | 35.18 | |
| NSP12 | − 123.44 | − 23.64 | 71.32 | − 11.43 | − 78.48 | |
| NSP13 | − 123.10 | 314.19 | 394.97 | − 19.55 | 566.50 | |
| NSP14 | − 92.99 | − 82.88 | 112.56 | − 12.90 | − 76.22 | |
| NSP16 | − 168.05 | − 19.82 | 161.72 | − 11.29 | − 47.447 |
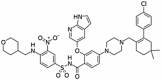
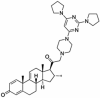
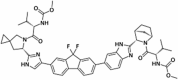
Each of the 18 complexes was subjected to 100ns MD simulation. The energy terms (in kcal/mol) were calculated from the data obtained from the last 20 ns trajectory.
vdW.: Van der Waals; Elec.: Electrostatic; SASA: solvent accessible surface area.
Venetoclax interaction with the SARS-CoV-2 targets
After considering a number of factors discussed in the foregoing sections, one may come to the conclusion that venetoclax has been a highly potential drug candidate from polypharmocology point of view, and essentially viral infections it can be a very promising option. A differential analysis has been carried out in order to validate and examine the robustness of the study carried out. As a first step, we have calculated the distance between the interacting catalytic residues with the venetoclax. The average distance was in between 0.2 nm–3 nm. In case of Plpro, the distance has been fluctuated between 2 nm–4 nm. This analysis clearly shows that the venetcolax has been bound well within the binding pocket (Figure S11, SI). Moreover, we have performed the PCA – FEL analysis for the venetoclax MD complexes. The first 10 eigenvectors projections of proteins were extracted and analyzed for their cosine content and FEL was defined based on PC1 and PC2 having a cosine content less than or equal to 0.2. The FEL of 6 venetoclax complexes was mapped from two PCs based on cosine content analysis in order to get minimum energy and energetically favored conformations. The energetically favored conformed were retrieved from the MD trajectories and interactions were analyzed and depicted in Figure S12, SI.
Conclusions
In the present study, various molecular modeling strategies were applied to identify promising multi-targeting repurposable potential drug candidates against SARS-CoV-2 infection. An effective polypharmacology approach has been employed in this study, by considering seven of the essential proteins involved in SARS-CoV-2 infection, 3CLpro, PLpro, RdRp (NSP12), NSP13, NSP14, NSP15, and NSP16, to develop efficient therapeutics capable of confronting the resistant strains. Some of the polypharmacological hits reported in this study have never been considered for COVID19 treatment. After identifying the known drugs which displayed high promiscuity in binding with several of the druggable targets, we tried to filter the top 20 hits from each target. Four drugs namely, venetoclax, tirilazad, acetyldigitoxin and ledipasvir were selected based on high docking score, visual examination of interactions, good binding pose and drugs showing interaction with four or more targets. The extensive MD simulation and MM-PBSA analysis showed the stability of protein-drug complexes and the sustainability of the key interactions with the respective targets. The van der Waals and electrostatic energies were found to have the prime driving forces to the overall negative binding free energy and stabilities. Our proposed repurposing candidates are exhibited multi-targeting potential which may assist the experimental study with higher chances to identify new antiviral drugs against SARS-CoV-2. The current study reveals that targets of key importance in COVID-19, display substantial binding with several of the known drugs, which indicates that polypharmacology approaches may be optimal choices. While vaccine development is indispensable, there is tremendous pressure to develop efficient anti-viral molecules, especially as one witnesses the emergence of a number of immune escape variants.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
GNS thanks J C Bose National Fellowship of DST New Delhi. HS thanks for an Institutional postdoctoral Fellowship. EJ thanks DST Inspire Fellowship. KK thanks UGC for Junior Research Fellowship. We acknowledge CSIR-4PI for providing supercomputing facility.
Supplementary Information (SI)
Tables S1–S9 and Figures S1–S12 are available at www.ias.ac.in/chemsci.
References
- 1.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. Access on: 16 December 2021
- 2.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pedersen SF, Ho YC. SARS-CoV-2: a storm is raging. J. Clin. Investig. 2020;130:2202. doi: 10.1172/JCI137647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reid AH, Taubenberger JK. The origin of the 1918 pandemic influenza virus: a continuing enigma. J. Gen. Virol. 2003;84:2285. doi: 10.1099/vir.0.19302-0. [DOI] [PubMed] [Google Scholar]
- 5.Lotfi M, Hamblin MR, Rezaei N. COVID-19: transmission, prevention, and potential therapeutic opportunities. Clin. Chim. Acta. 2020;508:254. doi: 10.1016/j.cca.2020.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menni C, Klaser K, May A, Polidori L, Capdevila J, Louca P, Sudre CH, Nguyen LH, Drew DA, Merino J, Hu C. Vaccine side-effects and SARS-CoV-2 infection after vaccination in users of the COVID Symptom Study app in the UK: a prospective observational study. Lancet Infect. Dis. 2021;21:939. doi: 10.1016/S1473-3099(21)00224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho P, Zheng JQ, Wu CC, Hou YC, Liu WC, Lu CL, Zheng CA, Lu KC, Chao YC. Perspective Adjunctive Therapies for COVID-19: beyond antiviral therapy. Int. J. Med. Sci. 2021;18:314. doi: 10.7150/ijms.51935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borba M G S, Val F F A, Sampaio V S, Alexandre M A A, Melo G C, Brito M, Mourao M P G, Sousa J D B, Silva D, Guerra M V F, Hajjar L A, Pinto R C, Balierio A A A, Pacheco A G F, Santos J D O, Naveca F G, Xavier M S, Siqueria A M, Schwarzbold A, Croda J, Nogueria M L, Romero G A S, Croda J, Nogueria M L, Romero G A S, Bassat Q, Fontes C J, Albuquerque B C, Ribeiro C T, Monteiro W M, Lacerda M V G and CloroCovid-19 Team 2020 Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection: a randomized clinical trial JAMA Netw. Open.3 e208857 [DOI] [PMC free article] [PubMed]
- 9.Chorin E, Dai M, Shulman E, Wadhwani L, Bar-Cohen R, Barbhaiya C, Aizer A, Holmes D, Bernstein S, Spinelli M, Park DS. The QT interval in patients with COVID-19 treated with hydroxychloroquine and azithromycin. Nat. Med. 2020;26:808. doi: 10.1038/s41591-020-0888-2. [DOI] [PubMed] [Google Scholar]
- 10.Singh J, Samal J, Kumar V, Sharma J, Agrawal U, Ehtesham NZ, Sundar D, Rahman SA, Hira S, Hasnain SE. Structure-function analyses of new SARS-CoV-2 variants B. 1.1. 7, B. 1.351 and B. 1.1. 28.1: clinical, diagnostic, therapeutic and public health implications. Viruses. 2021;13:439. doi: 10.3390/v13030439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang R, Chen J, Gao K, Hozumi Y, Yin C, Wei GW. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants Commun. Biol. 2021;15:1. doi: 10.1038/s42003-021-01754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arya R, Kumari S, Pandey B, Mistry H, Bihani SC, Das A, Prashar V, Gupta GD, Panicker L, Kumar M. Structural insights into SARS-CoV-2 proteins. J. Mol. Biol. 2021;433:166725. doi: 10.1016/j.jmb.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hossain MU, Bhattacharjee A, Emon MT, Chowdhury ZM, Ahammad I, Mosaib MG, Moniruzzaman M, Rahman MH, Islam MN, Ahmed I, Amin MR. Novel mutations in NSP-1 and PLPro of SARS-CoV-2 NIB-1 genome mount for effective therapeutics. J. Genet. Eng. Biotechnol. 2021;19:1. doi: 10.1186/s43141-021-00152-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banoun H. Evolution of SARS-CoV-2: review of mutations, role of the host immune system. Nephron. 2021;28:1. doi: 10.1159/000515417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fehr AR, Perlman S, Maier HJ, Bickerton E, Britton P. An overview of their replication and pathogenesis; section 2 genomic organization. Methods Mol Biol. 2015;1282:1. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Liu Z, Chen Z, Huang X, Xu M, He T, Zhang Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020;92:667. doi: 10.1002/jmv.25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F. Cell entry mechanisms of SARS-CoV-2. Pro. Natl. Acad. Sci. U.S.A. 2020;117:11727. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 21.Osipiuk J, Azizi SA, Dvorkin S, Endres M, Jedrzejczak R, Jones KA, Kang S, Kathayat RS, Kim Y, Lisnyak VG, Maki SL. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:1. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng Q, Peng R, Yuan B, Wang M, Zhao J, Fu L, Qi J, Shi Y. Structural basis of SARS-CoV-2 polymerase inhibition by Favipiravir. Innov. J. 2021;28:100080. doi: 10.1016/j.xinn.2021.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Malone B, Llewellyn E, Grasso M, Shelton PM, Olinares PD, Maruthi K, Eng ET, Vatandaslar H, Chait BT, Kapoor TM. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020;182:1560. doi: 10.1016/j.cell.2020.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saramago M, Bárria C, Costa VG, Souza CS, Viegas SC, Domingues S, Lousa D, Soares CM, Arraiano CM, Matos RG. New targets for drug design: importance of nsp14/nsp10 complex formation for the 3′-5′exoribonucleolytic activity on SARS-CoV-2. FEBS J. 2021;288:5130. doi: 10.1111/febs.15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pillon MC, Frazier MN, Dillard LB, Williams JG, Kocaman S, Krahn JM, Perera L, Hayne CK, Gordon J, Stewart ZD, Sobhany M. Cryo-EM structures of the SARS-CoV-2 endoribonuclease Nsp15 reveal insight into nuclease specificity and dynamics. Nat. Commun. 2021;12:1. doi: 10.1038/s41467-020-20608-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosas-Lemus M, Minasov G, Shuvalova L, Inniss NL, Kiryukhina O, Brunzelle J, Satchell KJ. High-resolution structures of the SARS-CoV-2 2′-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020;13:651. doi: 10.1126/scisignal.abe1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tahir Ul Qamar M, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm Anal. 2020;10:313. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira JC, Rabeh WM. Biochemical and biophysical characterization of the main protease, 3-chymotrypsin-like protease (3CLpro) from the novel coronavirus SARS-CoV 2 Sci. Rep. 2020;10:22200. doi: 10.1038/s41598-020-79357-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu Z, Huang B, Tang J, Liu S, Liu M, Ye Y, Liu Z, Xiong Y, Zhu W, Cao D, Li J, Niu X, Zhou H, Zhao YJ, Zhang G, Huang H. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Hu T, Hua T, Zhang B, Yang X, Li J, Yang H, Liu Z, Xu W, Guddat LW, Wang Q, Lou Z, Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newman JA, Douangamath A, Yadzani S, Yosaatmadja Y, Aimon A, Brandão-Neto J, Dunnett L, Gorrie-Stone T, Skyner R, Fearon D, Schapira M, von Delft F, Gileadi O. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021;12:4848. doi: 10.1038/s41467-021-25166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tahir M. Coronavirus genomic nsp14-ExoN, structure, role, mechanism, and potential application as a drug target. J. Med. Virol. 2021;93:4258. doi: 10.1002/jmv.27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim Y, Jedrzejczak R, Maltseva NI, Wilamowski M, Endres M, Godzik A, Michalska K, Joachimiak A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020;29:1596. doi: 10.1002/pro.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Su C, Ke M, Jin X, Xu L, Zhang Z, Wu A, Sun Y, Yang Z, Tien P, Ahola T, Liang Y, Liu X, Guo D. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2'-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 2011;7:e1002294. doi: 10.1371/journal.ppat.1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohari MH, Sastry GN. FDA approved drugs complexed to their targets: evaluating pose prediction accuracy of docking protocols. Sci. Signal. 2012;18:4263. doi: 10.1007/s00894-012-1416-1. [DOI] [PubMed] [Google Scholar]
- 37.Janardhan S, John L, Prasanthi M, Poroikov VV, Sastry GN. A QSAR and molecular modelling study towards new lead finding: polypharmacological approach to Mycobacterium tuberculosis SAR QSAR. Environ Res. 2017;28:815. doi: 10.1080/1062936X.2017.1398782. [DOI] [PubMed] [Google Scholar]
- 38.Kandeel M, Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. doi: 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinzi L, Tinivella A, Caporuscio F, Rastelli G. Drug repurposing and polypharmacology to fight SARS-CoV-2 through inhibition of the main protease. Front Pharmacol. 2021;12:84. doi: 10.3389/fphar.2021.636989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Singh B, Kumari P, Kumar PV, Agnihotri G, Khan S, Kant Beuria T, Syed GH, Dixit A. Identification of multipotent drugs for COVID-19 therapeutics with the evaluation of their SARS-CoV2 inhibitory activity. Comput. Struct. Biotechnol. J. 2021;19:1998. doi: 10.1016/j.csbj.2021.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar N, Sarma H, Sastry GN. Repurposing of approved drug molecules for viral infectious diseases: a molecular modelling approach. J. Biomol. Struct. Dyn. 2021;22:1. doi: 10.1080/07391102.2021.1905558. [DOI] [PubMed] [Google Scholar]
- 42.Ippolito M, Cortegiani A. Length of remdesivir treatment in patients with severe COVID-19. Breathe. 2021;17:200276. doi: 10.1183/20734735.0276-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta AK, Parker BM, Priyadarshi V, Parker J. Cardiac adverse events with remdesivir in COVID-19 infection. Cureus. 2020;12:e11132. doi: 10.7759/cureus.11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghazy RM, Almaghraby A, Shaaban R, Kamal A, Beshir H, Moursi A, Ramadan A, Taha SH. A systematic review and meta-analysis on chloroquine and hydroxychloroquine as monotherapy or combined with azithromycin in COVID-19 treatment. Sci. Rep. 2020;10:1. doi: 10.1038/s41598-020-77748-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;30:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berman H, Henrick K, Nakamura H. Announcing the worldwide protein data bank. Nat. Struct. Mol. Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 47.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA, Rempfer C, Bordoli L, Lepore R. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:296. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. Microbiology. 2000;81:853. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 49.Rut W, Lv Z, Zmudzinski M, Patchett S, Nayak D, Snipas SJ, El Oualid F, Huang TT, Bekes M, Drag M, Olsen SK. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020;42:4596. doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shirota Y, Luo H, Qin W, Kaneko S, Yamashita T, Kobayashi K, Murakami S. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 2002;13:11149. doi: 10.1074/jbc.M111392200. [DOI] [PubMed] [Google Scholar]
- 51.Godoy AS, Lima GM, Oliveira KI, Torres NU, Maluf FV, Guido RV, Oliva G. Crystal structure of Zika virus NS5 RNA-dependent RNA polymerase. Nat. Commun. 2017;27:1. doi: 10.1038/ncomms14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Subissi L, Posthuma CC, Collet A, Zevenhoven-Dobbe JC, Gorbalenya AE, Decroly E, Snijder EJ, Canard B, Imbert I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. U.S.A. 2014;37:E3900. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jia Z, Yan L, Ren Z, Wu L, Wang J, Guo J, Zheng L, Ming Z, Zhang L, Lou Z, Rao Z. Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47:6538. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krishnan DA, Sangeetha G, Vajravijayan S, Nandhagopal N, Gunasekaran K. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Inform. Med. Unlocked. 2020;20:100392. doi: 10.1016/j.imu.2020.100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dostalik P, Krafcikova P, Silhan J, Kozic J, Chalupska D, Chalupsky K, Boura E. Structural analysis of the OC43 coronavirus 2′-O-RNA methyltransferase. J. Virol. 2021;19:00463. doi: 10.1128/JVI.00463-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nencka R, Silhan J, Klima M, Otava T, Krafcikova P, Boura E. Coronaviral RNA-methyltransferases: function, structure and inhibition. Nucleic Acids Res. . 2022;50:635. doi: 10.1093/nar/gkab1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:296. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 60.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wishart DS. DrugBank and its relevance to pharmacogenomics. Pharmacogenomics. 2008;9:1155. doi: 10.2217/14622416.9.8.1155. [DOI] [PubMed] [Google Scholar]
- 62.Ursu O, Holmes J, Knockel J, Bologa CG, Yang JJ, Mathias SL, Nelson SJ, Oprea TI. DrugCentral: online drug compendium. Nucleic Acids Res. 2016;26:993. doi: 10.1093/nar/gkw993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Discovery Studio Visualizer Software 2012, Version 4.0. http://www.accelrys.com accessed on 12 October 2021
- 64.Abraham M J, Murtola T, Schulz R, Páll S, Smith J C, Hess B and Lindahl E 2015 GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers SoftwareX.19
- 65.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. 2001;43:9954. doi: 10.1021/jp003020w. [DOI] [Google Scholar]
- 67.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012;52:3155. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berendsen H J, Postma J P and van Gunsteren W F Hermans J 1981 Intermolecular Forces B Pullman (Ed.) p. 331–342
- 69.Berendsen H J 1999 Computational Molecular Dynamics: Challenges, Methods, Ideas P Deuflhard, J Hermans, B Leimkuhler, A E Mark, S Reich, A D Skeel (Eds.) (Berlin, Heidelberg: Springer) p. 3–36
- 70.Hess B, Bekker H, Berendsen HJ, Fraaije JG. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 71.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 72.Miyamoto S, Kollman PA. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952. doi: 10.1002/jcc.540130805. [DOI] [Google Scholar]
- 73.Origin (Pro) 8.5, OriginLab Corporation, Northampton, MA, USA.
- 74.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 75.Kumari R and Kumar R 2014 Open-Source Drug Discovery Consortium, Lynn A. g_mmpbsa—a GROMACS tool for high-throughput MM-PBSA calculations J. Chem. Inf. Model.54 1951 [DOI] [PubMed]
- 76.Mahadevi AS, Sastry GN. Cooperativity in noncovalent interactions. Chem. Rev. 2016;116:2775. doi: 10.1021/cr500344e. [DOI] [PubMed] [Google Scholar]
- 77.Kumar N, Saha S, Sastry GN. Towards developing a criterion to characterize non-covalent bonds: a quantum mechanical study. Phys. Chem. Chem. Phys. 2021;23:8478. doi: 10.1039/D0CP05689H. [DOI] [PubMed] [Google Scholar]
- 78.Kuhn B, Gerber P, Schulz-Gasch T, Stahl M. Validation and use of the MM-PBSA approach for drug discovery. J. Med. Chem. 2005;48:4040. doi: 10.1021/jm049081q. [DOI] [PubMed] [Google Scholar]
- 79.Huang K, Luo S, Cong Y, Zhong S, Zhang JZ, Duan L. An accurate free energy estimator: based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity. Nanoscale. 2020;12:10737. doi: 10.1039/C9NR10638C. [DOI] [PubMed] [Google Scholar]
- 80.Chen F, Liu H, Sun H, Pan P, Li Y, Li D, Hou T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016;18:22129. doi: 10.1039/C6CP03670H. [DOI] [PubMed] [Google Scholar]
- 81.El Hassab MA, Shoun AA, Al-Rashood ST, Al-Warhi T, Eldehna WM. Identification of a new potential SARS-COV-2 RNA-dependent RNA polymerase inhibitor via combining fragment-based drug design, docking, molecular dynamics, and MM-PBSA calculations. Front Chem. 2020;30:915. doi: 10.3389/fchem.2020.584894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Srivastava HK, Sastry GN. Molecular dynamics investigation on a series of HIV protease inhibitors: assessing the performance of MM-PBSA and MM-GBSA approaches. J. Chem. Inf. Model. 2012;52:3088. doi: 10.1021/ci300385h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.