

Abstract
CRK and CRKL encode cytoplasmic adaptors that contribute to the etiology of congenital heart disease. Neural crest cells (NCCs) are required for cardiac outflow tract (OFT) septation and aortic arch formation. The roles of Crk/Crkl in NCCs during mouse cardiovascular development remain unknown. To test this, we inactivated Crk and/or Crkl in NCCs. We found that the loss of Crk, rather than Crkl, in NCCs resulted in double outlet right ventricle, while loss of both Crk/Crkl in NCCs resulted in severe defects with earlier lethality due to failed OFT septation and severe dilation of the pharyngeal arch arteries (PAAs). We found that these defects are due to altered cell morphology resulting in reduced localization of NCCs to the OFT and failed integrity of the PAAs, along with reduced expression of Integrin signaling genes. Further, molecular studies identified reduced differentiation of vascular smooth muscle cells that may in part be due to altered Notch signaling. Additionally, there is increased cellular stress that leads to modest increase in apoptosis. Overall, this explains the mechanism for the Crk/Crkl phenotype.
Introduction
CRK and CRKL (CRK-like) together comprise the CRK adaptor gene family, and they encode intracellular cytoplasmic adaptor molecules, which act in many signaling pathways (1). CRK/CRKL have src homolog region 2 (SH2) and SH3 domains that are needed for protein–protein interactions that transduce extracellular signals (2,3). Both CRK and CRKL are ubiquitously expressed in all cells and tissues (4,5). These proteins are involved in many biological processes including cell migration, proliferation, survival and differentiation (1). Overexpression of CRK/CRKL serve as cancer oncogenes (6); however, our main interest is in their function in cardiovascular development, particularly, the molecular pathogenesis of congenital heart disease (CHD) caused by inactivation of CRK and CRKL.
Both CRK and CRKL are human congenital disease genes. CRK is in the chromosome 17p13.3 region and haploinsufficiency of genes in this region causes Miller–Dieker syndrome (MDS). Some affected MDS individuals have CHD including double outlet right ventricle (DORV), tetralogy of Fallot (TOF), D-transposition of the great arteries, atrial septal defect and ventricular septal defects (VSDs) (7–10). CRKL is in the chromosome 22q11.2 region and haploinsufficiency of genes in this region causes 22q11.2 deletion syndrome (22q11.2DS). Approximately 65% of affected 22q11.2DS individuals have CHD, and among them, most have cardiac outflow tract (OFT) defects such as persistent truncus arteriosus and TOF as well as aortic arch anomalies (11,12). Since most affected individuals have cardiac OFT defects, it is important to explain their embryonic origin.
The cardiac OFT, including the aortic arch, forms within the embryonic pharyngeal apparatus. This temporary structure is composed of individual arches on either side that form dynamically, each containing a pharyngeal arch artery (PAA). The PAAs join the cardiac OFT at the aortic sac in which PAAs 3–6 become remodeled to form the aortic arch with arterial branches (13). The cardiac OFT undergoes septation by fusion of the dorsal aortic sac wall protrusion (ASP) and distal cardiac OFT cushions to form the aorta and pulmonary trunk (14,15).
Studies of mouse models showed that both Crk−/− or Crkl−/− global null mutant embryos exhibited late-gestation embryonic lethality with cardiac OFT defects and vascular abnormalities (16–19). Dilation in the aortic arch was observed in Crk−/− null mouse embryos (16). The embryonic mesoderm is required for normal cardiac OFT and vascular formation in the pharyngeal apparatus (20). Inactivation of both genes in mouse mesodermal cells results in early-gestation embryonic lethality with severe cardiac OFT and vascular angiogenesis defects (19). Taken together, these observations suggest that Crk/Crkl play unique and shared roles in mouse cardiovascular development. Neural crest cell (NCC) deficiency causes some of the observed defects in Crk/Crkl null mutant mice (21–23). However, the NCC-specific functions of Crk and Crkl for mouse cardiovascular development are unknown thus far.
NCCs delaminate from the neural tube and migrate to various regions of the embryo, and they can differentiate into many cell types, including vascular smooth muscle cells (24–26). In this study, we used the Wnt1-Cre line (27) to inactivate Crk and/or Crkl in the NCC lineage in the mouse. We found that cardiovascular development is sensitive to the dosage of Crk and Crkl in NCCs. Inactivation of Crk, but not Crkl, in NCCs resulted in cardiovascular defects, but inactivation of both Crk/Crkl caused lethality during early mid-gestation with failed cardiac OFT septation and dilated PAAs. We examined the NCC lineage as they migrated to the distal pharyngeal arches. We found that the distal pharyngeal arches appeared normal, but there was severely reduced NCC number in the dorsal wall of aortic sac and cardiac OFT. We noticed that the cellular shape became rounded with less protrusions, along with reduction of expression of Integrin signaling genes in the OFT and PAAs. We further identified reduced differentiation of vascular smooth muscle cells that may occur due to altered Notch signaling. We also identified modest increase in apoptosis downstream of increased expression of cellular stress markers. These cellular and molecular changes result in the lack of OFT septation and dilation of PAAs observed in these embryos.
Results
Inactivation of both Crk and Crkl in NCCs results in failed cardiac OFT septation and dilation of PAAs in mouse embryos
To investigate the function of Crk and Crkl in NCCs, we generated Crk and Crkl single and double conditional knockout (cko) embryos using the Wnt1-Cre driver and floxed alleles (16,27). Wnt1 is an early expressed NCC marker gene and the Wnt1-Cre line is commonly used to mediate gene inactivation in NCCs (27,28). Approximately 60% of Crk cko embryos had DORV with an obligate VSD (N = 15; Fig. 1A). All Wnt1-Cre/+; Crkf/+ littermates had normal hearts (N = 5; Fig. 1A). We obtained only 6.8% Crk cko mice compared to the expected 25% at postnatal day P20 (total N = 103; Table 1). This is consistent with the frequency of DORV in Crk cko embryos. All Crk−/− embryos had a DORV (19) as we confirmed (N = 7; 100% penetrance; Supplementary Material, Fig. S1). Thus, Crk cko embryos had the same heart defect but with reduced penetrance as compared to global null embryos.
Figure 1.
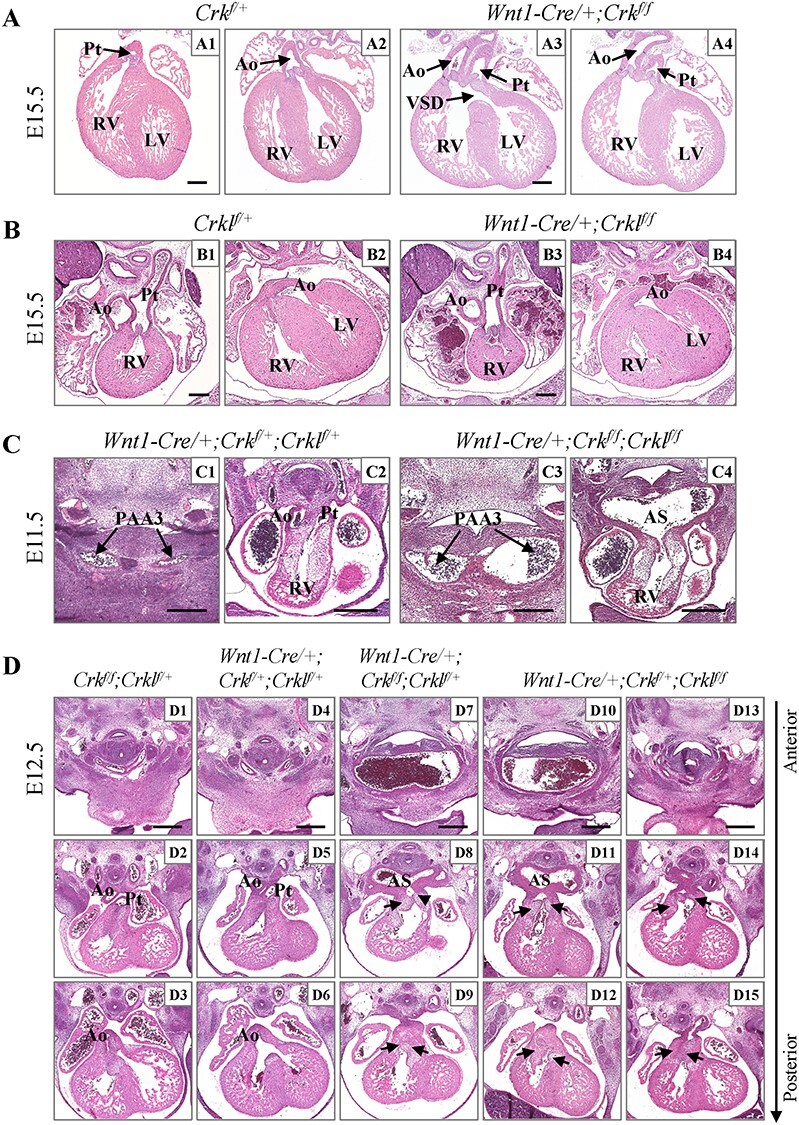
Dosage-dependent requirement of Crk and Crkl in NCCs for cardiac OFT and aortic arch development. (A) Cardiac OFT alignment defects in H&E sections from Crk cko embryos. DORV with VSD was observed in Crk cko embryos (A3; N = 15). (B) Normal heart in Crkl cko embryos (N = 7). (C) All Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous embryos had normal anatomy (C1–C2; N = 6). OFT septation defect with dilated PAA3 (C3) and dilated aortic sac (C4) were found in Crk/Crkl cko embryos (N = 9, 100% penetrance). (D) Cardiac OFT septation and vascular defects in Wnt1-Cre/+; Crkf/f; Crklf/+ (N = 6) and Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant embryos (N = 9). Dilated common carotid arteries were shown in (D7, D10). Defective OFT septation was shown (D8, D9, D11, D12, D14, D15). Black arrows in (D) indicate shared valves for the aorta and pulmonary trunk. VSD, ventricular septal defect; PAA3, third pharyngeal arch artery; AS, aortic sac; Ao, aorta; Pt, pulmonary trunk; RV, right ventricle; LV, left ventricle. Scale bar: 250 μm.
Table 1.
Analysis of the Mendelian ratio of Wnt1-Cre/+; Crkf/f pups at postnatal day 20
| Wnt1-Cre/+; Crkf/f | Wnt1-Cre/+; Crkf/+ | Crkf/f | Crkf/+ | Total number | |
|---|---|---|---|---|---|
| Number Percentage |
7 6.8% |
31 30.1% |
42 40.8% |
23 22.3% |
103 |
A Crkl flox allele was generated before analyzing Crkl cko mutants (Supplementary Material, Fig. S2). The Crkl cko embryos had normal hearts at E15.5 (N = 7; Fig. 1B) and we obtained a normal Mendelian ratio of Crkl cko mice at P20 (N = 143; Table 2). This contrasts with the phenotype in Crkl−/− embryos, which die by E16.5 and half of them had either DORV or an overriding aorta (17,18). The data indicate that inactivation of Crkl in NCCs alone is not sufficient to cause a cardiac phenotype. We found that the relative expression of Crk is 2-fold higher than Crkl in cardiac NCCs in Wnt1-Cre/+; GFPf/+ embryos at E10.5 (Supplementary Material, Fig. S2B and C), which may partially explain why Crk cko mutants had a cardiovascular phenotype, while Crkl cko mutants did not.
Table 2.
Analysis of the Mendelian ratio of Wnt1-Cre/+; Crklf/f pups at postnatal day 20
| Wnt1-Cre/+; Crklf/f | Wnt1-Cre/+; Crklf/+ | Crklf/f | Crklf/+ | Total number | |
|---|---|---|---|---|---|
| Number Percentage |
36 25.2% |
42 29.3% |
31 21.7% |
34 23.8% |
143 |
To test for shared functions of Crk and Crkl in NCCs, we examined the cardiovascular phenotypes of Wnt1-Cre/+; Crkf/f; Crklf/f (Crk/Crkl cko), Wnt1-Cre/+; Crkf/f; Crklf/+, Wnt1-Cre/+; Crkf/+; Crklf/f and Wnt1-Cre/+; Crkf/+; Crklf/+ embryos. Both Crk and Crkl are ubiquitously expressed in all cells at all developmental stages (Supplementary Material, Fig. S3A). We confirmed that the genes were inactivated in Crk/Crkl cko embryos at E10.5 (Supplementary Material, Fig. S3).
We found that Crk/Crkl cko embryos died between E11.5 and E12.5 (Table 3, Supplementary Material, Table S1), which is earlier than in global Crk or Crkl null mutant embryos. Histological analysis showed that Crk/Crkl cko embryos exhibited dilated third and fourth PAAs, dilated aortic sac as well as OFT septation defects (N = 9, 100% penetrance) at E11.5, compared to the Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous littermates (N = 6) which exhibited normal structures, were viable and survived normally (Fig. 1C; Table 3). The Wnt1-Cre/+; Crkf/+; Crklf/+ and Crk/Crkl cko embryos could be obtained from the same litter; therefore, we used Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous embryos as the control in most of the experiments.
Table 3.
Summary of cardiovascular phenotypes in Crk and/or Crkl null and NCC cko embryos
| Genotypes | Embryonic lethality | Dilated common carotid artery | Cardiac OFT defect |
|---|---|---|---|
| Crkl−/− | E16.5 (17,18) |
No | DORV, TOF, VSD (50% penetrance) (17,18) |
| Crk−/− | E13.5–E15.5 (16) | Yes (16) | DORV, VSD (19) (N = 7, 100% penetrance at E15) |
| Wnt1-Cre/+; Crklf/f | No | No (N = 7) | No (N = 7) |
| Wnt1-Cre/+; Crkf/f | E16.5–P20 | No (N = 5) | DORV, VSD (N = 15, 60% penetrance at E15.5) |
| Wnt1-Cre/+; Crkf/+; Crklf/+ | No | No (N = 7, E12.5) | No (N = 7, E12.5) |
| Wnt1-Cre/+; Crkf/f; Crklf/+ | E12.5–E14 | Yes (N = 6, 100% penetrance at E12.5) |
OFT septation defect (N = 6, 100% penetrance at E12.5) |
| Wnt1-Cre/+; Crkf/+; Crklf/f | E12.5–E14 | Yes (N = 9, 78% penetrance at E12.5) |
OFT septation defect (N = 9, 100% penetrance at E12.5) |
| Wnt1-Cre/+; Crkf/f; Crklf/f | E11.5–E12.5 | Yes (N = 9, 100% penetrance at E11.5) |
OFT septation defect (N = 9, 100% penetrance at E11.5) |
Most of the Wnt1-Cre/+; Crkf/f; Crklf/+ and Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant embryos died by E14.5 (Table 3, Supplementary Material, Tables S1–3). Those embryos also exhibited dilated PAAs and an OFT septation defect at E11.5 but were less severe as compared to the Crk/Crkl cko littermates (Fig. 1C; Supplementary Material, Fig. S4C). The third PAA forms the common carotid arteries (CCAs) and the fourth PAA forms a critical part of the aortic arch and the subclavian arteries (13). The distal cardiac OFT is physically connected to the third and fourth PAAs via the aortic sac. The presence of both dilated PAAs/aortic sac and OFT septation defects in Wnt1-Cre/+; Crkf/f; Crklf/+, Wnt1-Cre/+; Crkf/+; Crklf/f three allele and Crk/Crkl cko embryos made us wonder if the dilated vessels and OFT septation defects occurred equally. At E12.5, Wnt1-Cre/+; Crkf/f; Crklf/+ and Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant embryos exhibited severely dilated CCAs and an OFT septation defect (Fig. 1D; Table 3). The aorta and pulmonary trunk failed to become separated, and they shared valves that are derived from cardiac OFT cushions (Fig. 1D). All six Wnt1-Cre/+; Crkf/f; Crklf/+ embryos examined had dilated CCAs and an OFT septation defect, while nine Wnt1-Cre/+; Crkf/+; Crklf/f embryos examined had an OFT septation defect but only seven of them had dilated CCAs (78% penetrance; Fig. 1D; Table 3). We found that Wnt1-Cre/+; Crkf/f; Crklf/+ embryos exhibited more severely dilated CCAs and an OFT septation defect than Wnt1-Cre/+; Crkf/+; Crklf/f littermates (Fig. 1D). The data suggest that Crk is more important than Crkl in NCCs and that the OFT septation defect and dilated CCAs do not always co-occur. The Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous (N = 7) and control (Wnt1-Cre negative, N = 11) embryos had normal structures (Fig. 1D; Table 3).
Besides the cardiac OFT and aortic arch, we found that Crk and Crkl are also required for peripheral vascular development in the head, neural tube and craniofacial regions leading to blood hemorrhages when inactivated (Supplementary Material, Fig. S5). As for other aspects of cardiovascular development, peripheral vascular development is more sensitive to Crk than Crkl in NCCs (Supplementary Material, Fig. S5). Craniofacial hemorrhages have been noted previously in global Crk null embryos (16).
Crk and Crkl are required for allocating NCCs to form the dorsal ASP and cardiac OFT cushions
Next, we tested whether NCCs are present in the aortic sac and cardiac OFT in mutant embryos (Fig. 2). The dorsal ASP (also known as the dorsal wall protrusion of aortic sac) (23) forms between E9.5 and E10.5 (Fig. 2A). Following this, the aortopulmonary septum forms by E11.5 (Fig. 2A). We used the Rosa26-GFP flox allele as a reporter to label NCCs (29). In control embryos, few NCCs were observed in the dorsal aortic sac wall and distal cardiac cushions at E9.5 (Fig. 2A). By E10.5 and later, many more NCCs were observed in those regions and the ASP is present (Fig. 2A). At E9.5, there was no obvious difference in these structures between Crk/Crkl cko and control embryos (both N = 4, Supplementary Material, Fig. S4A). At E10.5, Crk/Crkl cko embryos (N = 6) exhibited a much thinner dorsal aortic sac wall, absent or shorter dorsal ASP and hypoplastic OFT cushions, versus the control littermates (N = 6, Fig. 2B). At this stage, less NCCs were observed in the dorsal aortic sac wall, dorsal ASP and OFT cushions in Crk/Crkl cko (N = 15) versus control (N = 9) embryos (Fig. 2B). The Crk/Crkl cko (N = 7) and Wnt1-Cre/+; Crkf/f; Crklf/+/Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant embryos (N = 8) exhibited a thinner dorsal aortic sac wall, shorter dorsal ASP and hypoplastic OFT cushions, compared to either the Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous or Wnt1-Cre negative embryos at this stage (Supplementary Material, Fig. S4B). Defects were usually more severe in Crk/Crkl cko than Wnt1-Cre/+; Crkf/f; Crklf/+/Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant littermates. As compared to PAA defects, there was no obvious difference between Wnt1-Cre/+; Crkf/f; Crklf/+ and Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant embryos and between two types of controls (Wnt1-Cre/+; Crkf/+; Crklf/+ or Wnt1-Cre negative) littermates.
Figure 2.
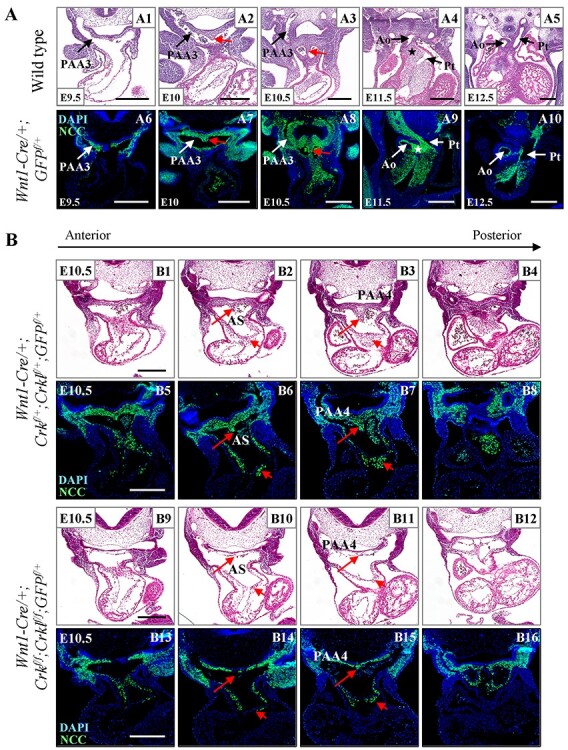
Crk and Crkl are required for allocating NCCs to form the dorsal ASP and cardiac OFT cushions. (A) Transverse H&E and immunofluorescence (stained with anti-GFP antibody) sections from wild-type embryos at E9.5–E12.5. Red arrows in (A) indicate dorsal ASP. Stars in (A) indicate position of the aortopulmonary septum. (B) Defective formation of dorsal ASP and hypoplastic OFT cushions in Crk/Crkl cko embryos. Transverse sections with same methods as for 2A, from control and Crk/Crkl cko embryos, respectively. Red arrows with long tails in (B) indicate dorsal ASP and red arrows with short tails in (B) indicate one of OFT cushions. We observed a thinner dorsal aortic sac wall, much shorter dorsal ASP with less NCCs and fewer NCCs in OFT cushions in Crk/Crkl cko embryos (B14, B15). At least six embryos for each genotype were analyzed. VSD, ventricular septal defect; PAA3, third pharyngeal arch artery; PAA4, fourth pharyngeal arch artery; AS, aortic sac; Ao, aorta; Pt, pulmonary trunk. Scale bar: 250 μm.
Abnormal migration of NCC lineages in Wnt1-Cre/+; Crkf/f; Crklf/+, Wnt1-Cre/+; Crkf/+; Crklf/f and Crk/Crkl cko embryos
We then examined NCC distribution in mutant versus control embryos (Fig. 3). NCCs were present normally in the pharyngeal arches in Crk/Crkl cko and control embryos at E8.5 and E9 (Supplementary Material, Fig. S6). Whole mount immunofluorescence staining followed by clearing of embryos was performed at E9.5 and E10.5 to better visualize the NCC populations. NCC migratory streams caudal to the otic vesicle are termed post-otic NCC streams. There were slightly fewer NCCs in the post-otic streams at E9.5 and E10.5 in Crk/Crkl cko versus control embryos (N = 4 for each genotype; Fig. 3A; Supplementary Material, Fig. S6). There was no obvious difference in the NCC populations in the distal pharyngeal arches in Crk/Crkl cko versus control embryos at E9.5 and E10.5 (Fig. 3A; sSupplementary Material, Fig. S6). We then more carefully analyzed the NCC number in pharyngeal arches 3–6 plus the cardiac OFT after FACS purification. There was a small reduction (~17% reduction calculated by mean of two groups) of the total number of NCCs in these regions in Crk/Crkl cko versus Wnt1-Cre/+; GFPf/+ embryos at E10.5 (Supplementary Material, Fig. S7). Thus, the small reduction of NCC number in the distal pharyngeal arches alone might not explain the severe phenotype in Crk/Crkl cko embryos.
Figure 3.
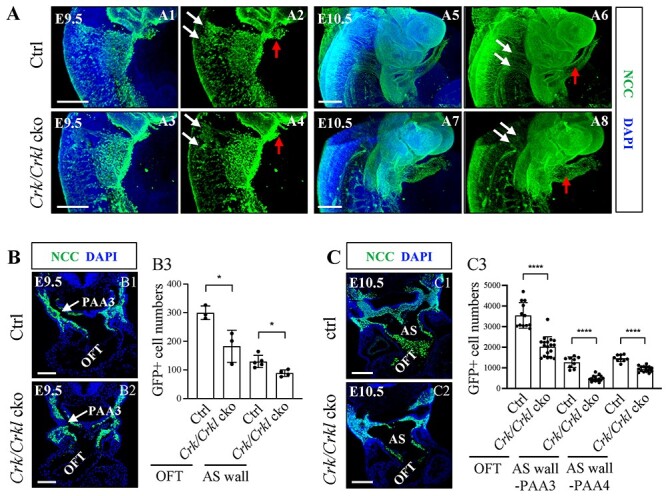
Greatly reduced NCC localization in the aortic sac wall and OFT in Crk/Crkl cko embryos. (A) Sagittal view and the three-dimensional (3D) reconstruction of whole mount GFP (green) embryos. Slightly fewer migratory NCCs in streams in Crk/Crkl cko versus Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous control embryos. White and red arrows in (A) indicate migrating NCC streams and cardiac OFT, respectively. Defective migrating NCC streams were shown in (A4, A8). N = 4 for each genotype. (B, C) Less NCCs in cardiac OFT and dorsal aortic sac wall regions in Crk/Crkl cko versus control embryos at E9.5 (B) and E10.5 (C) as shown by immunofluorescence (B1–2, C1–2). Quantification of NCC numbers in cardiac OFT and dorsal aortic sac wall regions in Crk/Crkl cko versus control embryos at E9.5 (B3) and E10.5 (C3). Each dot in graph represented one embryo (B3, C3). The graphs (B3, C3) were plotted with mean and standard deviation. Two-tailed Student’s t-test was used for the statistical analysis. *: 0.01 < P < 0.05. ****: P < 0.0001. The genotype of the control embryos in (A–C) is Wnt1-Cre/+; Crkf/+; Crklf/+. PAA3, third pharyngeal arch artery; AS, aortic sac. Scale bar in (A1–4, C1–2): 200 μm. Scale bar in (A5–8): 500 μm. Scale bar in (B1, B2): 100 μm.
We next focused on the aortic sac and cardiac OFT regions. The distal cardiac OFT appeared dilated in Crk/Crkl cko versus control embryos (Fig. 3A). We next quantified NCC numbers in the aortic sac and cardiac OFT on tissue sections. At E9.5, we observed few NCCs in the dorsal aortic sac wall and cardiac OFT in Crk/Crkl cko versus control embryos (Fig. 3B). At E10.5, we observed far fewer NCCs in the dorsal aortic sac wall (plus dorsal ASP) and cardiac OFT in Crk/Crkl cko versus control embryos (Fig. 3C; Supplementary Material, Fig. S8). These observations suggest that NCCs are present in the distal pharyngeal arches but significantly fewer NCCs entered the dorsal wall of aortic sac, dorsal ASP and cardiac OFT region in Crk/Crkl cko embryos. For the Wnt1-Cre/+; Crkf/f; Crklf/+ and Wnt1-Cre/+; Crkf/+; Crklf/f three allele mutant embryos, significantly reduced number of NCCs was also seen in the dorsal aortic sac wall and cardiac OFT regions at E10.5, compared to controls (Supplementary Material, Fig. S8).
Normal cell proliferation and increased apoptosis in Crk/Crkl cko embryos
We tested whether the reduced number of NCCs observed in the cardiac OFT and dorsal aortic sac wall in Crk/Crkl cko embryos was due to defective NCC proliferation (Fig. 4). Using 5-ethynyl-2′-deoxyuridine (EdU) labeling and anti-phospho-histone H3 (pH3) antibody staining methods, there was no significant difference between Crk/Crkl cko and control embryos in the cardiac OFT and dorsal aortic sac wall regions at E10.5 (Fig. 4A–C; Supplementary Material, Fig. S9A and B).
Figure 4.
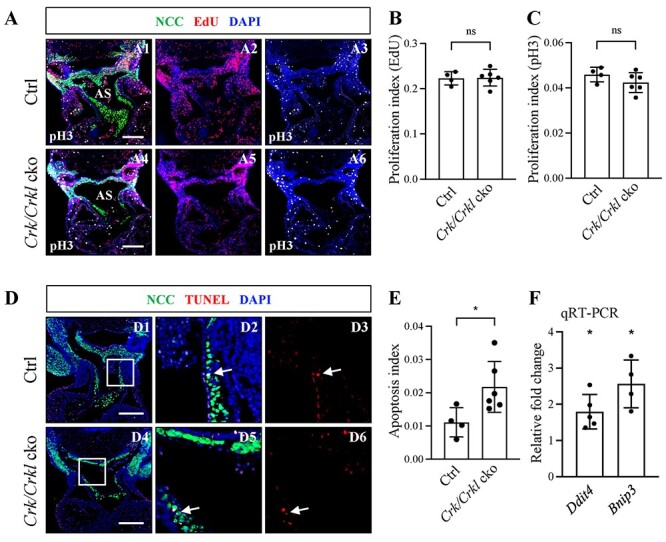
Analysis of proliferation and apoptosis NCCs in Crk/Crkl cko versus control embryos at E10.5. (A) Transverse immunofluorescence sections stained with anti-GFP, anti-pH3 antibodies and for EdU from Crk/Crkl cko versus Wnt1-Cre/+; Crkf/+; Crklf/+ double heterozygous control embryos (A1–6). (B, C) Quantification of the ratio of EdU (B) and pH3 (C) positive NCCs with respect to the total NCC cell number in the cardiac OFT of Crk/Crkl cko versus control embryos. (D) Immunofluorescence TUNEL assay of apoptosis (D1–D6) from Crk/Crkl cko versus control embryos, with zoomed in images (D2, D3, D5, D6) indicated in white box in (D1) and (D4), respectively. White arrows indicate the TUNEL positive NCCs. (E) Quantification of TUNEL positive NCCs with respect to the total NCC cell number in the OFT. Each dot in graph represented one embryo (B, C, E). The genotype of the control embryos in (A–E) is Wnt1-Cre/+; Crkf/+; Crklf/+. (F) qRT-PCR shows that Ddit4 and Bnip3 were significantly increased in FACS purified NCCs from Crk/Crkl cko versus Wnt1-Cre/+; GFPf/+ embryos. Each dot for qRT-PCR represents one biological replicate (pooled 4–6 embryos). The graphs (B, C, E, F) were plotted with mean and standard deviation. Two-tailed Student’s t-test was used for the statistical analysis. *: 0.01 < P < 0.05. ns, no significance; PAA3 and PAA4, third and fourth pharyngeal arch arteries; AS, aortic sac. Scale bar in (A and D): 200 μm.
Next, we checked whether apoptosis of NCCs was increased by TUNEL staining. At E10.5, in mutant and control embryos, very few cells underwent apoptosis (Fig. 4D). Nonetheless, we saw a small but significant increase in apoptosis of NCCs in the cardiac OFT in Crk/Crkl cko versus control littermates (Fig. 4E). We did not see any significant difference in the dorsal aortic sac wall region (Supplementary Material, Fig. S9C). Both DDIT4 (DNA damage inducible transcript 4) and BNIP3 (Bcl-2 family protein BCL2/adenovirus E1B interacting protein 3) have been reported to induce cell apoptosis under cellular stress, including hypoxia and DNA damage (30,31). We examined Ddit4 and Bnip3 expression by qRT-PCR using FACS purified NCCs from the distal pharyngeal apparatus plus heart, and the data showed that both genes were significantly increased in cardiac NCCs in Crk/Crkl cko versus controls (Fig. 4F). Overall, our data indicate that the reduced number of NCCs cannot be explained exclusively by increased apoptosis.
Besides NCCs, the second heart field (SHF) mesoderm is critically important in forming the cardiac OFT and aortic arch. SHF progenitors are adjacent to migrating NCCs in the OFT, and it is critical for elongating the OFT and forming the myocardium surrounding the septated aorta and pulmonary trunk (32–34). To examine whether inactivation of Crk/Crkl in NCCs will affect the myocardium of the OFT, we performed immunofluorescence staining of alpha-smooth muscle actin (αSMA), which marks muscle cells (Supplementary Material, Fig. S10). Our data showed that there were no obvious differences of myocardium wall thickness and fluorescent intensity of αSMA staining in the OFT in Crk/Crkl cko versus control embryos at E10.5, indicating that the SHF development in Crk/Crkl cko embryos was grossly normal.
Defective NCC differentiation into vascular smooth muscle cells in Crk/Crkl cko embryos
To determine whether PAA defects observed in Crk/Crkl cko embryos were caused by defective differentiation of NCCs into vascular smooth muscle cells, we examined αSMA expression at E11.5 (Fig. 5). We found that αSMA expression in PAAs 3–6 and the distal cardiac OFT were decreased in Crk/Crkl cko versus control embryos (both N = 4; Fig. 5A). There was some expression of αSMA in non-NCCs in the fourth PAA in Crk/Crkl cko embryos (Fig. 5A). Further, αSMA was unevenly expressed in PAAs 3–6 and distal cardiac OFT in Crk/Crkl cko embryos (Fig. 5A). In addition, Acta2 (αSMA), Tagln (SM22) and Actc1 (Actin, alpha cardiac muscle 1) were significantly decreased in expression in NCCs isolated from the distal pharyngeal apparatus plus cardiac OFT in Crk/Crkl cko versus controls at E10.5 (Fig. 5B). Taken together, Crk and Crkl are required for smooth muscle differentiation in the cardiac OFT and PAAs.
Figure 5.
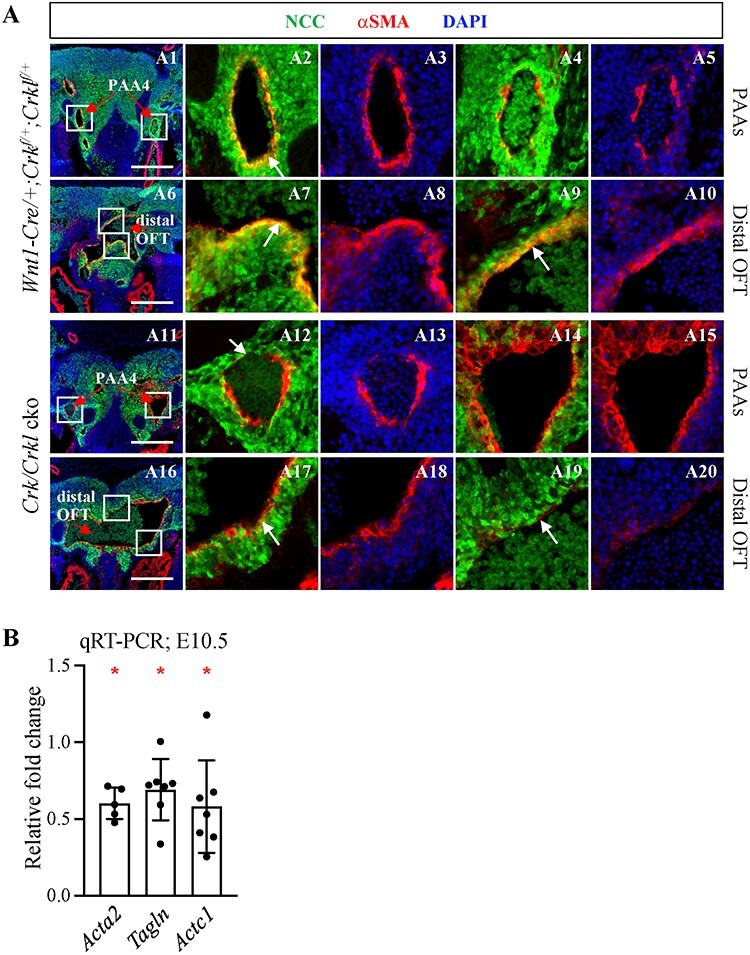
Reduced smooth muscle differentiation of NCCs in Crk/Crkl cko versus control embryos. (A) Frontal immunofluorescence sections stained with anti-GFP, anti-αSMA antibodies showing reduced vascular smooth muscle differentiation of NCCs in Crk/Crkl cko versus control (Wnt1-Cre/+; Crkf/+; Crklf/+) embryos at E11.5. (A2–5), (A7–10), (A12–15) and (A17–20) are zoomed in images of white boxes indicated in (A1), (A6), (A11) and (A16), respectively. Red arrows in (A1, A11) and (A6, A16) indicate PAA4 and distal cardiac OFT, respectively. White arrows in (A2, A12) and (A7, A9, A17, A19) indicate NCC-derived smooth muscle cell layer around PAA4 and distal cardiac OFT, respectively. Reduced smooth muscle cells among NCCs could be seen in Crk/Crkl cko embryos (A12, A17, A19). Scale bar in (A): 200 μm. (B) qRT-PCR shows the relative fold change of Acta2, Tagln and Actc1 in FACS purified NCCs from Crk/Crkl cko versus Wnt1-Cre/+; GFPf/+ embryos. The graph was plotted with mean and standard deviation. Each dot in graphs represented one biological replicate (pooled 4–6 embryos). Two-tailed Student’s t-test was used for the statistical analysis. *: 0.01 < P < 0.05.
Cell morphology changes in Crk/Crkl cko embryos
Reduced differentiation alone may not explain the presence of severely dilated PAAs. Further, mildly increased apoptosis cannot explain the severe reduced numbers of NCCs in the OFT. In the literature, there is strong in vitro evidence for changes in cell shape (shrunken and rounded shape) that disrupts cell–cell interactions and can cause reduced migration when CRK and/or CRKL is inactivated (35,36). To test for cell shape changes, we performed a detailed histological analysis, and we found that the cells in the dorsal aortic sac wall and cardiac OFT in Crk/Crkl cko versus control embryos had more rounded shape with less cytosol at E10.5 (both N = 4; Fig. 6A). We further stained filamentous actin with phalloidin, and there were less and shorter cellular protrusions in NCCs in the cardiac OFT region in Crk/Crkl cko versus control embryos at this stage (both N = 4; Fig. 6B). Integrin signaling is well known for its role in regulating focal adhesion formation with cell shape changes and cell migration (37,38). We examined expression of Integrin signaling genes Flna (ligand; filamin/ECM), Fn1 (ligand; fibronectin); Itga5 (subunit of integrin), Itgb1 (subunit of integrin) and Ilk (key mediator of Integrin signaling) and found that all were significantly reduced except for Fn1, in cardiac NCCs isolated from the distal pharyngeal apparatus plus cardiac OFT of Crk/Crkl cko embryos, compared to controls at E10.5 (Fig. 6C).
Figure 6.
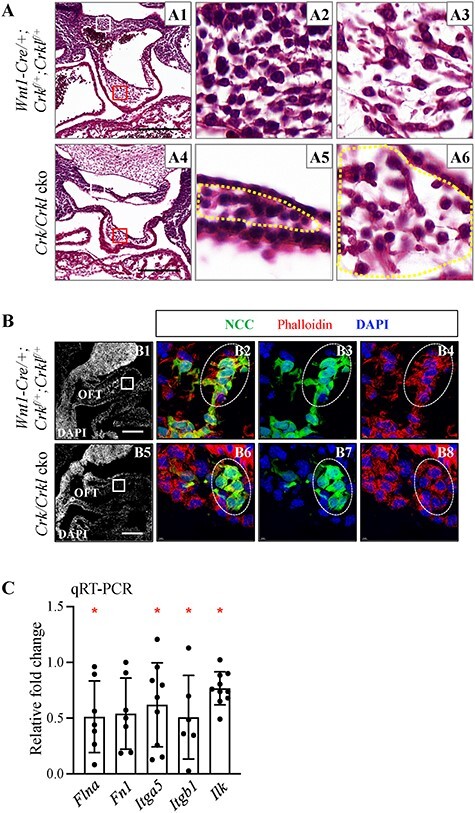
Altered NCC cell morphology in Crk/Crkl cko versus control embryos at E10.5. (A) Transverse H&E sections from Wnt1-Cre/+; Crkf/+; Crklf/+ (control) (A2, A3) and Crk/Crkl cko (A5, A6) embryos showing more rounded shape in four-allele cko embryos. (A2, A5; A3, A6) are zoomed in images of white or red boxes from (A1) and (A4), respectively. Based on the analysis of NCC distribution using immunofluorescence staining on sections at E10.5, all cells in A2 as well as A5 (cells in region marked by yellow dashes) and most cells in A3 as well as A6 (cells in region marked by yellow dashes) are NCCs. (B) Sagittal immunofluorescence sections stained with anti-GFP antibody and Phalloidin from Crk/Crkl cko and control embryos. (B2–4) and (B6–8) are zoomed in images of white boxes indicated in (B1) and (B5), respectively. Four embryos for each genotype were analyzed in (A, B). PAA4, fourth pharyngeal arch artery. Scale bar: 200 μm. (C) qRT-PCR shows the relative fold change of expression of several Integrin signaling genes in FACS purified NCCs from Crk/Crkl cko versus Wnt1-Cre/+; GFPf/+ embryos. The graph was plotted with mean and standard deviation. Each dot in graphs represents one biological replicate (pooled 4–6 embryos). Two-tailed Student’s t-test was used for the statistical analysis. *: 0.01 < P < 0.05.
Reduced Notch signaling in both smooth muscle cells and endothelial cells in Crk/Crkl cko embryos
Notch signaling mediates NCC differentiation to vascular smooth muscle needed for cardiovascular development (39–41). In vitro studies showed that αSMA acts as the direct downstream target of Notch signaling (42,43). Since we observed reduced αSMA expression, we investigated whether Notch signaling is altered in Crk/Crkl cko embryos. For this, we examined the cleaved NOTCH1 intracellular domain (NICD; marker for activated Notch signaling). At E9.5, no obvious change of NICD expression was observed in the distal cardiac OFT and PAAs between Crk/Crkl cko and control embryos (data not shown). At E10.5, decreased NICD expression was observed in the endothelial cells (OFT only; inner single cell layer around the vessel lumen) and surrounding smooth muscle cells in the distal cardiac OFT as well as third and fourth PAAs in Crk/Crkl cko versus control embryos (both N = 4; Fig. 7A). Alternation of NICD expression in the endothelium in Crk/Crkl cko embryos may be secondary since they are not derived from the NCC lineage. By qRT-PCR, we found that Notch signaling pathway genes Jag1 (ligand) and Notch1 (receptor), as well as downstream effectors, Hes1 and Hey1, were significantly reduced but not two other downstream effectors, Hes5 and Hey2, in cardiac NCCs from the distal pharyngeal apparatus plus cardiac OFT in Crk/Crkl cko embryos versus controls at E10.5 (Fig. 7B). Hes5 and Hey2 were downregulated in the Crk/Crkl cko versus control samples in most biological replicates. However, the relative changes of Hes5 and Hey2 in control versus Crk/Crkl cko samples did not show statistically significant differences. This is in contrast to Hes1 and Hey1, which were significantly reduced. We suggest that lack of a statistically significant reduction of expression of Hes5 and Hey2 might be due to large variations among replicates, which is caused by low expression of Hes5 and Hey2 in NCCs. The qRT-PCR data showed that the relative expression of Hes5 and Hey2 as detected by a higher delta Ct value were much lower compared to the expression of Hes1 and Hey1 in the control samples (data not shown). Overall, our data suggest that Notch signaling is reduced in NCCs in Crk/Crkl cko embryos.
Figure 7.
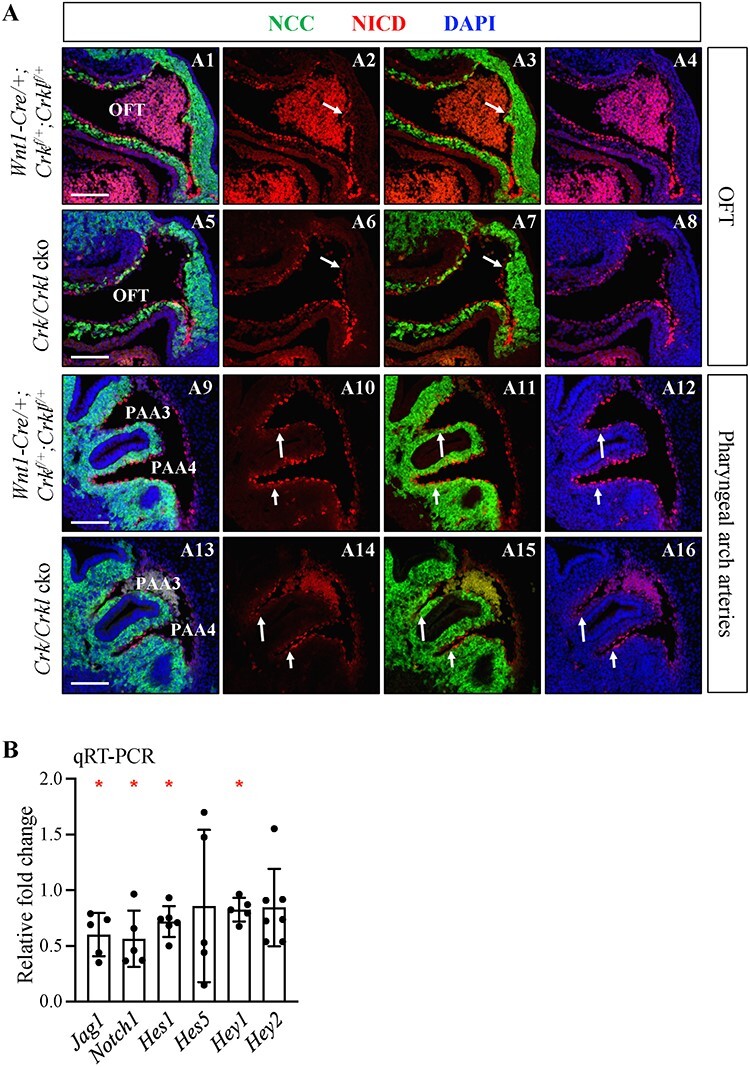
Reduced Notch signaling in both smooth muscle cells and endothelial cells in Crk/Crkl cko versus control embryos at E10.5. (A) Sagittal immunofluorescence on sections stained with anti-GFP, anti-NICD antibodies of cardiac OFT and PAAs from Crk/Crkl cko and Wnt1-Cre/+; Crkf/+; Crklf/+ (control) embryos. White arrows (A2, A3, A6, A7) indicate NICD staining in distal cardiac OFT endothelial cells. We observed reduced NICD staining in distal cardiac OFT endothelial cells and NCC derived smooth muscle cells in Crk/Crkl cko embryo (A5–8). White arrows in (A10–12, A14–16) indicate NICD staining in smooth muscle cells from PAA3 and PAA4. Note reduced NICD staining in smooth muscle cells from PAA3 and PAA4 regions in Crk/Crkl cko embryo (A13–16). Blood cells with autofluorescence signals were seen in A1–4 and A13–16. Four embryos for each genotype were analyzed. Scale bar in (A): 100 μm. (B) qRT-PCR shows that several Notch signaling genes were increased in expression in FACS purified NCCs from Crk/Crkl cko versus Wnt1-Cre/+; GFPf/+ embryos. The graph was plotted with mean and standard deviation. Each dot in graph (B) represented one biological replicate (pooled 4–6 embryos). Two-tailed Student’s t-test was used for the statistical analysis. *: 0.01 < P < 0.05.
Discussion
In this report, we show that Crk/Crkl have shared, essential dosage sensitive functions in NCCs needed to form the cardiac OFT junctional complex and the aortic arch. When both genes are inactivated, NCCs fail to migrate to the OFT, further the PAAs become severely dilated. Cellular and molecular studies suggest that changes in cell shape, perhaps due to decreased Integrin signaling causes reduced localization of NCCs in the OFT and dilated PAAs. Further, decreased vascular smooth muscle differentiation may be caused by impaired Notch signaling. We also identified increase in cellular stress and modest increase in apoptosis in NCCs. When taken together, these changes may explain the basis of the observed phenotypes.
NCCs are more sensitive to loss of Crk than Crkl for cardiovascular development
We found that NCC inactivation of Crk alone disrupts cardiac OFT development, but mice with NCC inactivation of Crkl are normal. The cause of DORV observed in the Crk cko embryos may be due to reduced numbers of NCCs that contributed to the cardiac OFT. We suggest this because the same cardiac defect was also observed in NCC-deficient Splotch2H mutants (23). Although both Crk and Crkl are ubiquitously expressed, one reason for the above phenotypic difference observed could be because Crk is expressed 2-fold higher than Crkl in NCCs. Further, Crk encodes two alternative splicing variants, CRK I and CRK II, while Crkl only encodes one protein, and therefore, these two forms may have additional functions (1). Supporting slightly different functions, global inactivation of either Crk or Crkl results in somewhat different cardiac phenotypes (16,17). To further identify different functions, gene expression profiling was performed in primary mouse embryonic fibroblasts in which either Crk, or Crkl, or both genes were inactivated with some differences in gene ontology terms noted (19).
We suggest that CRK may contribute to CHD in individuals affected with MDS. MDS is an extremely rare disease with limited cases reported. Deletion sizes vary with most including deletion of CRK. The majority of affected individuals have lissencephaly, which is a type of severe brain defect, and they rarely survive beyond 2 years of age. We found that in the mouse, Crk is expressed higher than Crkl. This would suggest that individuals with deletion or mutation of CRK would be more strongly associated with CHD than for CRKL. However, this has not been the case for MDS versus 22q11.2DS. We suggest that this may be due to the rarity and severity of MDS. Cardiac defects have been reported in some cases, but perhaps secondary to brain malformations (7–10,44). Therefore, we do not know yet for sure whether CRK is the major gene contributing to CHD in MDS individuals.
For 22q11.2DS, haploinsufficiency of TBX1 seems to be the main driver of CHD, while CRKL contributes to the overall phenotype (45–47). Further, 20–30% of affected individuals with smaller nested deletions around CRKL that do not include TBX1 have cardiac malformations (18). This supports the idea that CRKL is a disease gene on its own. Although, in the mouse, Crkl is expressed at a lower level than Crk, 22q11.2DS occurs much more commonly and is better described than MDS. Therefore, it is possible that differences in CHD frequency in both diseases could be due to ascertainment biases. Further, we found differences in Crk versus Crkl expression in mouse models but this does not mean that the relative expression of these genes are the same in humans. Future studies will need to be done to identify individuals with isolated mutations in CRK and CRKL in association with sporadic CHD. This would help to provide direct evidence that CRK and CRKL are causative genes for heart malformations in humans.
Cellular mechanism for Crk/Crkl function in cardiovascular development
Based upon our phenotypic and cell behavioral analyses, we generated a model for Crk/Crkl function in cardiovascular development (Fig. 8A). In Fig. 8A, we show the region in the embryo where the PAAs join the aortic sac and cardiac OFT. Within the aortic sac, a dorsal ASP forms and migrates to fuse with the distal cushions of the cardiac OFT to form the separated aorta and pulmonary trunk (14,15). These are defective when Crk/Crkl are inactivated, due to the presence of fewer NCCs entering these structures (Fig. 8A). It is important to know the cellular mechanisms as to why there are fewer NCCs in the OFT when Crk/Crkl are inactivated. Our data support the idea that cellular shape is altered, and this affects migration of NCCs into the OFT. Since NCCs are present in the pharyngeal arches but are reduced in the OFT, we suggest that these changes are due to defective response to localized signaling medicated by CRK/CRKL. Many in vitro studies have showed that inactivation of either Crk or Crkl or both genes causes decreased or totally blocked cell migration due to their crucial roles in regulating the cellular cytoskeletal network and cell shape (6). We also found modest increase in apoptosis of NCCs in the cardiac OFT of Crk/Crkl cko embryos together with significant upregulation of cellular stress-induced apoptosis genes, Ddit4 and Bnip3. The observed increased apoptosis in NCCs is likely the primary defect since loss of Crk/Crkl in cultured fibroblasts also resulted in an increase of cell death (48). The impaired cell migration reduced differentiation to vascular smooth muscle in NCCs along with modest increase in apoptosis that could, when taken together, contribute to the overall phenotype.
Figure 8.
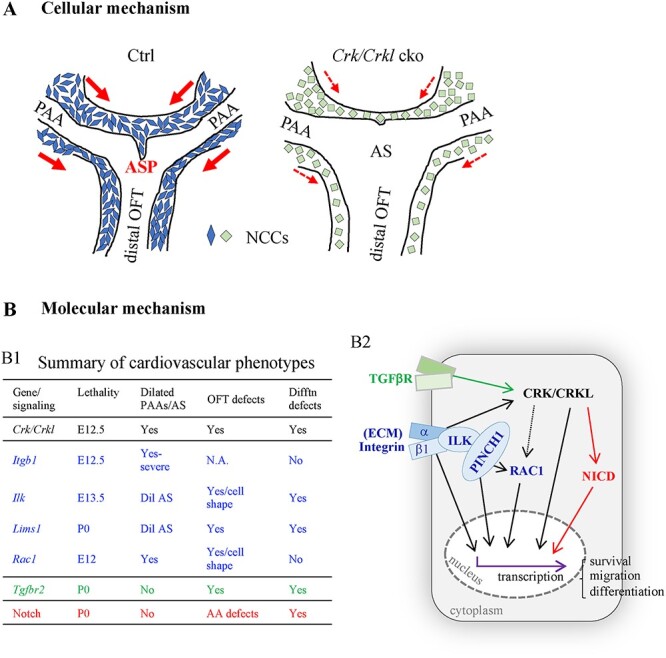
Schematic models of Crk/Crkl function in the development of cardiac OFT and aortic arch. (A) Model showing rounded cell shape, altered gene expression (green color) and reduced NCC contribution to the dorsal aortic sac wall, dorsal ASP and cardiac OFT in Crk/Crkl cko versus control (Wnt1-Cre/+; Crkf/+; Crklf/+) embryos at E10.5. We observed a shorter or missing ASP and dilated aortic sac in Crk/Crkl cko embryos. Red arrows indicate direction of migration of NCCs from the pharyngeal arches to either the dorsal aortic sac wall or cardiac OFT, with thinner lines in Crk/Crkl cko embryos representing less migration. AS: aortic sac. (B) Model showing molecular mechanism of CRK/CRKL function for cardiac OFT and PAA formation. (B1) Summary of the cardiovascular phenotypes for the mutants with NCC inactivation of Crk/Crkl as well as genes in Integrin [Itgb1, Ilk, Lims1 (PINCH1), Rac1; blue color], TGFβ (Tgfbr2; green color) and Notch signaling pathways (red color) (39,49–53,67,89). Dilated PAAs and/or aortic sac were observed in Crk/Crkl as well as Integrin signaling gene mutants with Itgb1 mutants showed the most severe phenotype. OFT septation defects were observed in Crk/Crkl, Ilk, Lims1, Rac1 and Tgfbr2 mutants. Altered cell morphology/shape was observed in the OFT cushions in Ilk and Rac1 mutants. Impaired vascular smooth muscle differentiation was observed in Crk/Crkl, Ilk, Lims1 and Tgfbr2 mutants as well as Notch mutants. Dil AS, dilated aortic sac; Difftn, differentiation; P0, postnatal day 0 (at birth); AA defects, aortic arch defects. (B2) Model of signaling pathways mediated by CRK/CRKL in cardiac NCCs (gray box) during the cardiovascular development. We suggest that CRK/CRKL mediate Integrin, TGFB and Notch signaling required for proper cardiac NCC behaviors. Integrin signaling regulates many cellular activities by modulating the cellular cytoskeletal network downstream of the extracellular matrix (ECM) (37,38). CRK/CRKL and Integrin signaling regulate the cytoskeleton network and gene expression via either small GTPase RAC1 or other molecules (60–62), which might affect cell migration, survival as well as differentiation (indicated as black arrows). The black dashed arrow indicates that the connection is not proven in vivo in our study. Notch and TGFβ signaling are required for vascular smooth muscle cell differentiation (39,67). Our data and the literature (64–66,90–92) support that CRK/CRKL indirectly regulates Notch signaling (indicated in red arrows) and may act downstream of TGFβ signaling pathway (indicated in green arrow) as well as the Integrin signaling pathway (indicated in black arrows) for smooth muscle differentiation. TGFβR, TGFβ receptor.
Molecular mechanism for cardiac OFT and PAA formation mediated by Crk/Crkl
The severely dilated PAAs and aortic sac in Crk/Crkl double conditional mutants are the main factors causing their death by early mid-gestation. We searched the literature for mice with similar phenotypes to identify the genetic pathway that these genes function. Interestingly, mutant mouse embryos with loss of several genes in the Integrin signaling pathway including Itgb1 (integrin β1), Ilk (integrin linked kinase), Lims1 (LIM and senescent cell antigen-like domains 1; also name as ‘Pinch1’) and Rac1 (Rac family small GTPase 1) in NCCs displayed defective cardiac OFT septation, aortic sac and/or PAA dilation with various severity, the similar as in Crk/Crkl cko embryos (Fig. 8B1) (49–53). In this report, we found several Integrin signaling pathway genes decreased in expression including Flna, Itga5, Itgb1 and Ilk in Crk/Crkl cko embryos. Therefore, it is possible that the defects in common between integrin and Crk/Crkl genes could be because they act in the same genetic pathway.
Integrin signaling regulates multiple cellular behaviors that are relevant to Crk/Crkl function. The main cellular function is to organize the cytoskeletal network and to activate downstream intracellular signaling (Fig. 8B2) (37,38). Integrins are heterodimeric cell surface receptors containing α and β subunits. In mammals, half of the α, β subunit combinations contain the β1 subunit (54). ILK is an intracellular serine/threonine protein kinase, and it physically interacts with integrin β1 (55). ILK also physically interacts with the cellular adaptor protein PINCH1 (56), thereby mediating Integrin/ILK dependent signaling (57–59) (Fig. 8B2). The small Rho GTPase protein, RAC1, can be activated through Integrin signaling that affects transcription and cell behavior (60,61) (Fig. 8B2). In addition to transcriptional changes, translocation of CRK/CRKL to focal adhesions mediates integrin-induced cell migration via activating RAC1 (62). Thus, literature and mouse studies on these genes suggest the important roles of integrin β1—CRK/CRKL—RAC1 signaling for PAA/aortic sac development. It is also possible that CRK/CRKL instead act through other proteins, as noted by the dashed line in Fig. 8B2. Also, CRK/CRKL seems to act in parallel or more broadly than ILK/PINCH1 because CRK/CRKL mutant embryos have more severe defects.
We found that loss of Crk/Crkl in NCCs resulted in impaired smooth muscle differentiation (Fig. 8B1). There are several possible molecular mechanisms that can explain this, and three include Integrin, Notch and Tgfβ signaling (Fig. 8B2). Conditional inactivation of Ilk and Lims1 resulted in impaired differentiation (Fig. 8B1). Notch signaling is also required in NCCs for normal vascular smooth muscle differentiation (39,40,63). We found impaired Notch signaling in Crk/Crkl cko mutants. These pathways also intersect, with evidence for Integrin and Notch interactions. Studies in Drosophila also showed that Integrin signaling modulates Notch activity during epithelial cell differentiation via regulating intracellular trafficking and/or processing of Notch (64). Chick studies showed that Integrin signaling regulates Notch activity via controlling downstream Wnt signaling during the genesis of somites (65). In addition to Integrin and Notch signaling, both in vitro and in vivo studies showed that TGFβ signaling plays important role in inducing NCC differentiation into smooth muscle cells (66,67). Increased CRKL phosphorylation was observed in a NCC stem cell line during smooth muscle differentiation in response to TGFβ signaling (67). Further, reduced Smad2/3 phosphorylation, which mediates TGFβ signaling, was observed in the cardiac OFT cushions of embryos with NCC mediated inactivation of Integrin signaling pathway genes, Lims1 and Ilk (49,53). This suggests that there is a connection between Integrin and TGFβ signaling. We found that when Crk/Crkl is inactivated, proper signaling cannot take place leading to reduced differentiation (Fig. 8B2). Another possible pathway is mitogen-activated protein kinases (MAPK) that promote differentiation and other cell behaviors (68). ERK1/2 (extracellular signal regulated kinases 1/2) and JNK (c-JUN N-terminal kinases) signaling are two canonical intracellular MAPK pathways in mammals mediated by CRK/CRKL (69–73). However, we did not see obvious alternation of the phosphorylation of ERK1/2 and c-JUN in Crk/Crkl cko embryos, supporting the other pathways above in mediating signaling, although more work in the future needs to be done to rule out this and other possible signaling pathways.
One question is whether decreased smooth muscle differentiation in NCCs can result in dilation of the PAAs and aortic sac. Upon examination of the literature, it seems that defective vascular smooth muscle differentiation can occur without vascular dilation in mouse models (39–41,67,74–77). Another possibility is reduced cell number can cause dilation. However, even when smooth muscle cell number was reduced in half by diphtheria toxin treatment, vessel dilation did not occur (78). This suggests other mechanisms besides failed differentiation or reduced smooth muscle cell number must be responsible for the vascular dilation phenotype in this model. One possibility is that there is a reduction of focal adhesions between cells. It is known that Integrin signaling is important to form and maintain focal adhesions (38). Although we did not specifically examine focal adhesions, we found that in Crk/Crkl cko embryos, cells had more rounded shapes, and this is consistent with such defects. Another possibility is that mutant cells have a defective response to change in the local ECM environment. Mouse studies indicate that loss of integrin β1 in vascular smooth muscle cells affect ECM assembly which, in part, causes less resistance to the high shear stress in PAAs and the distal cardiac OFT leading to dilation (51,79–81). In humans, mutations in ECM proteins in the TGFβ pathway cause aneurysm (dilated blood vessels) in adults (Fig. 8B2) (82–86). CRKL might act as a downstream molecule of TGFβ signaling and mediate a possible crosstalk between TGFβ and Integrin signaling for differentiation and to support blood vessel integrity (Fig. 8B2).
In conclusion, our work highlights the dosage requirements of Crk and Crkl in NCCs for normal development of the cardiac OFT and aortic arch. We also provide a cellular and molecular model to explain the mechanisms leading to failed OFT septation and dilation of the aortic sac with PAAs. Since both CRK and CRKL are human CHD-related genes for Miller–Dieker and 22q11.2 deletion syndrome, respectively, our work provides a molecular basis for better understanding the pathogenesis of the heart defects in those deletion syndromes.
Materials and Methods
Mouse strains
The following mouse alleles used in this study have been previously described: Wnt1-Cre/+ (27), Crkf/f (flox = f) (16), Crklf/f (18), Rosa26-GFPf/f (29) and Crklneo/+ (18). Crklf/f and Crk null alleles were generated in our lab. To generate the Crk null allele, we crossed Crkf/f mice with Zp3-Cre mice (Jackson laboratory, stock No: 003651) and screened resulting animals by PCR. Homozygous Crk+/− mice were obtained in F2 generation (Supplementary Material, Fig. S1), backcrossed with the wild type (Swiss Webster background) for two generations and then maintained in a mixed background (C57BL/6, 129SvEv and Swiss Webster). Mouse crosses and PCR strategies for generating Crklf/f and Crk+/− mouse alleles are available upon request.
Crk−/− embryos were generated by intercrossing Crk+/− mice. Wnt1-Cre/+; Crkf/f embryos (or mice) were generated by crossing male Wnt1-Cre/+; Crkf/+ mice with female Crkf/f mice. Wnt1-Cre/+; Crklf/f embryos (or mice) were generated by crossing male Wnt1-Cre/+; Crklf/+ mice with female Crklf/f mice. Most Crk/Crkl cko embryos were generated by crossing male Wnt1-Cre/+; Crkf/+; Crklf/+mice with female Crkf/f; Crklf/f or Crkf/f; Crklf/f; GFPf/f mice. Few Wnt1-Cre/+; Crkf/f; Crklf/+ and Wnt1-Cre/+; Crkf/+; Crklf/f embryos were generated by crossing male Wnt1-Cre/+; Crkf/+ or Wnt1-Cre/+; Crklf/+ mice with female Crkf/f; Crklf/f mice. The Crkf/f; Crklf/f mice were intercrossed for 2 years and maintained in a mixed background (C57BL/6, 129SvEv and Swiss Webster).
All mouse studies were carried out according to NIH protocols approved by the Institutional Animal Care of Use Committee at Albert Einstein College of Medicine (Albert Einstein College of Medicine, Bronx, NY; https://einsteinmed.org/administration/animal-care-use-committee/). The protocol number is #00001034. Carbon dioxide (CO2) inhalation was used for euthanasia as directed by the NIH and Albert Einstein College of Medicine.
Primers used for RT-qPCR
Details of each primer is listed in Supplementary Material, Table S4.
Mouse embryo heart histology
Mouse embryos were collected at various embryonic stages by vaginal plug checking; E0.5 indicates plug date. For E10.5 and younger embryos, their stages were defined by counting numbers of somite pairs: E8.5 (7–10 somite pairs), E9.0 (14–18 somite pairs), E9.5 (20–26 somite pairs) and E10.5 (30–35 somite pairs). For E11.5 and older embryos, their stages were defined by checking females for vaginal plugs in the morning.
Mouse embryos were collected in phosphate-buffered saline (PBS), then fixed by 4% paraformaldehyde (PFA) in PBS for 1 h (for E9.5 and E10.5 embryos) or 2 h (for E11.5–E12.5 embryos) at 4°C. After fixation, the embryos were dehydrated through graded series of ethanol (70, 95 and 100% ethanol × 2), then cleared in xylene and embedded in paraffin. The thickness of all paraffin sections was 10 μm. All hematoxylin and eosin (H&E) stained histological sections were performed in the Einstein Histology and Comparative Pathology Core Facility (Albert Einstein College of Medicine, Bronx, NY; https://einsteinmed.org/research/shared-facilities/cores/15/histology-and-comparative-pathology/) using standard protocols. All mutant and control embryos analyzed were obtained from at least three independent mice crosses and male mice (father). Images were taken by using scanners from Einstein Analytical Imaging Facility (Albert Einstein College of Medicine, Bronx, NY; https://einsteinmed.org/research/shared-facilities/analytical-imaging-facility/) or by light microscopy.
Immunofluorescence staining on paraffin sections
Embryos at different stages were isolated and imbedded in paraffin the same way as above. The paraffin sections were deparaffinized in xylene, then rehydrated in graded series of ethanol (100% × 2, 95%, 70%, distilled water) and followed by boiled in citric acid-based antigen retrieval unmasking solution (Vector laboratories, H-3300) for 15 min. Then sections were washed by 0.1% PBST (0.1% Triton X-100 in PBS) and blocked in 5% donkey serum (Sigma, D9663; diluted in 0.1% PBST) for 1 h at room temperature (RT). After blocking, sections were incubated in primary antibodies for overnight at 4°C, washed by 0.1% PBST, incubated in secondary antibodies for 1 h at RT, washed by 0.1% PBST again and mounted by mounting medium (Vector laboratories, H-1500). Primary antibodies used included: goat anti-GFP (1:200; Abcam, ab6673), rabbit anti-phospho-Histone H3 (Ser10) (1: 200; Millipore, 06-570), mouse anti-Crk (1: 200; BD, 610036), rabbit anti-alpha smooth muscle actin (αSMA) antibody (1: 200; Abcam, ab5694) and rabbit anti-cleaved Notch1 (1: 200, cell signaling technology, 4147). Secondary antibodies used included: donkey anti-goat IgG Alexa Fluor 488 (1: 200, Invitrogen, A11055), donkey anti-rabbit IgG Alexa Fluor 647 (1: 200, Invitrogen, A31573), donkey anti-mouse IgG Alexa Fluor 568 (1: 200, Invitrogen, A10037), donkey anti-rabbit IgG Alexa Fluor 555 (1: 200, Invitrogen, A31572) and donkey anti-rabbit IgG biotin (1:200, Invitrogen, 31821).
For immunofluorescence staining of anti-cleaved Notch1 antibody, tyramide signal amplification (TSA) was performed. Sections were treated with 3% hydrogen peroxide (H2O2) for 10 min at RT to quench the endogenous peroxidase activity after the antigen retrieval step. After secondary antibody (donkey anti-rabbit IgG biotin) incubation, sections were washed by 0.1% PBST and incubated with Streptavidin-HRP Conjugate (1:200, PerkinElmer, NEL750001EA) for 1 h at room temperature. Then sections were washed by 0.1% PBST and incubated with Cyanin 3 for 5 min diluted in TSA Plus Fluorescein detection kit (1:500, PerkinElmer, NEL741001KT). Images were taken from an apotome light microscope.
Immunofluorescence staining on whole mount embryos
The protocol of whole mount immunofluorescence staining on embryos was described (87). Briefly, embryos with desired stages were isolated and fixed in 4% PFA in PBS overnight at 4°C. After washing with PBS, embryos were incubated in 0.1% PBST overnight, blocked in 10% donkey serum overnight and incubated in primary antibody for 3–4 days. Then embryos were washed with 0.1% PBST (1 h × 5). Then embryos were incubated with secondary antibody together with DAPI (Thermo Scientific, 62248) for 3–4 days. Embryos were then dehydrated through graded series of methanol and cleared in BABB solution (benzyl benzoate: benzyl alcohol = 2: 1 in volume). All steps were done at 4°C.
For phalloidin immunofluorescence staining, embryos were stained with phalloidin (0.05 mg/ml, Sigma, P1951) for 2 h after secondary antibody incubation. Then after washing with 0.1% PBST, embryos were cryoprotected in 30% sucrose in PBS for overnight at 4°C and embedded in OCT. Frozen sections were washed in PBS to remove OCT and then mounted by mounting media (Vector laboratories, H-1500).
Images of embryos and sections were taken from either Nikon CSU-W1 Spinning Disk confocal microscope or Leica SP8 confocal microscope from Einstein Analytical Imaging Facility (Albert Einstein College of Medicine, Bronx, NY; https://einsteinmed.org/research/shared-facilities/analytical-imaging-facility/). Images were processed by ImarisView 9.6.0 software.
TUNEL and cell proliferation assays on paraffin sections
Apoptotic cell death was examined by TdT-mediated dUTP-X nick end labeling (TUNEL) method. TUNEL assays on sections were performed after secondary antibodies staining by using In Situ Cell Death Detection Kit TMR red (Millipore, 12156792910). Sections were incubated for 1 h at RT to develop fluorescent signals.
Cell proliferation was assessed by two methods: one was immunofluorescence of using anti-phospho-Histone H3 (Ser10) antibody, which is a mitosis marker, and the other was immunofluorescence staining of 5-Ethynyl-2′-deoxyuridine (EdU), which is used to detect cells undergoing DNA synthesis. EdU was injected in pregnant females (intraperitoneal injection; 5 μg EdU/gram of mice weight) 2 h before dissection. Detection of EdU signal on sections was performed after secondary antibody staining using the Click-iT™ EdU Cell Proliferation Kit (Thermo Fisher Scientific, C10338). Sections were incubated for 30 min at RT to develop fluorescent signals.
RNAscope in situ hybridization on paraffin sections
RNAscope in situ hybridization was performed on paraffin sections as previously described (88). Embryos at E10.5 were fixed in 4% PFA in PBS for overnight at 4°C and imbedded in paraffin. Crkl probe was custom designed from Advanced Cell Diagnostics. The eGFP probe (400281) probe was also from Advanced Cell Diagnostics. RNAscope Multiplex Fluorescent Reagent kit v2 (Advanced Cell Diagnostics, 323100) and TSA Plus Fluorescein detection kit were used for the detection of the fluorescent signal.
Cell number quantification on tissue sections
Cell numbers were counted for all NCCs, phospho-histone H3 (pH 3) positive NCCs, EdU positive NCCs and TUNEL positive NCCs in OFT and dorsal aortic sac wall regions on serial transverse sections of embryos at E9.5 and E10.5. When counting cells, sections from all embryos at same stage were matched by position within the embryo. For embryos at E10.5, for the OFT region, we counted all GFP+ (positive: +), pH 3 + GFP+, EdU + GFP+ and TUNEL+GFP+ cells in each section within OFT of embryos with various genotypes as desired. For embryos at E9.5, for the OFT region, we counted all GFP+ in each section within OFT of embryos with various genotypes as desired. For embryos at E10.5 for the dorsal aortic sac wall region, we counted all GFP+, pH 3 + GFP+, EdU + GFP+ and TUNEL+GFP+ cells in five serial sections (total 50 μm thickness) along both third and fourth PAAs of embryos with various genotypes as desired. And there were no overlapping sections counted between third and fourth PAAs. For the analysis of apoptosis, we also counted 16 serial sections (total 160 μm thickness) between third and fourth PAAs of embryos with various genotypes as desired. For embryos at E9.5, for the dorsal aortic sac wall region, we counted all GFP+ cells in four serial sections (total 40 μm thickness) along third PAAs positions of embryos with various genotypes as desired. ImageJ software was used for counting cell numbers. At least three embryos per genotype from at least three independent litters were used for each experiment.
Cell sorting and qRT-PCR
The tissue including pharyngeal arches 3–6 and whole heart was micro-dissected from E10.5 (30–35 somite) embryos and then incubated in 0.25% Trypsin-EDTA (Gibco, 25-200-056) together with DNase I (50 units/ml) for 8 min to dissociate tissue to cell suspension. NCCs were collected by flow cytometer from the Einstein Flow Cytometry Core Facility (Albert Einstein College of Medicine, Bronx, NY; https://einsteinmed.org/research/facilities/facs/page.aspx) and directly stored in −80°C freezer for short-term storage. Total RNA was isolated using TRIzol reagent (Invitrogen, 15596018) and cDNA was synthesized using the Cells-to-cDNA II Kit (Invitrogen, AM1722). Approximately 100 000 NCCs (from 4 to 6 embryos) were pooled and used for per RNA sample. For analysis of qRT-PCR results, Rplp0 was used as the internal control and the control/cko samples pair used for calculating relative fold change had similar somite numbers.
Statistical analysis
The graphs were plotted with mean and standard deviation using Prism 9 software. Two tailed unpaired t-tests were used for statistical analysis.
Supplementary Material
Acknowledgements
We thank Tom Curran from Children’s Research Institute (Children’s Mercy Kansas City) for the gift of Crk flox mouse strain. We also thank Jianbo Wang from University of Alabama and our colleague Christopher De Bono for advice in analyzing histological sections and interpreting phenotypes.
Conflict of Interest statement. The authors declare no competing or financial interests.
Contributor Information
Lijie Shi, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Silvia E Racedo, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Alexander Diacou, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Taeju Park, Children's Mercy Research Institute, Children's Mercy Kansas City, Kansas City, MO 64108, USA; Department of Pediatrics, University of Missouri-Kansas City School of Medicine, Kansas City, MO 64108, USA.
Bin Zhou, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Bernice E Morrow, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10461, USA.
Funding
Our work was funded by National Institutes of Health (P01 HD070454 to B.E.M., B.Z.; R01 HL132577 to B.E.M., B.Z.); American Heart Association Predoctoral Fellowship (19PRE34380071 to L.S.).
References
- 1. Feller, S.M. (2001) Crk family adaptors-signalling complex formation and biological roles. Oncogene, 20, 6348–6371. [DOI] [PubMed] [Google Scholar]
- 2. Sadowski, I., Stone, J.C. and Pawson, T. (1986) A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol. Cell. Biol., 6, 4396–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mayer, B.J., Hamaguchi, M. and Hanafusa, H. (1988) A novel viral oncogene with structural similarity to phospholipase C. Nature, 332, 272–275. [DOI] [PubMed] [Google Scholar]
- 4. Matsuda, M., Tanaka, S., Nagata, S., Kojima, A., Kurata, T. and Shibuya, M. (1992) Two species of human CRK cDNA encode proteins with distinct biological activities. Mol. Cell. Biol., 12, 3482–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. ten Hoeve, J., Morris, C., Heisterkamp, N. and Groffen, J. (1993) Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene, 8, 2469–2474. [PubMed] [Google Scholar]
- 6. Park, T. (2021) Crk and CrkL as therapeutic targets for cancer treatment. Cell, 10:739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greenberg, F., Courtney, K.B., Wessels, R.A., Huhta, J., Carpenter, R.J., Rich, D.C. and Ledbetter, D.H. (1988) Prenatal diagnosis of deletion 17p13 associated with DiGeorge anomaly. Am. J. Med. Genet., 31, 1–4. [DOI] [PubMed] [Google Scholar]
- 8. Saltzman, D.H., Krauss, C.M., Goldman, J.M. and Benacerraf, B.R. (1991) Prenatal diagnosis of lissencephaly. Prenat. Diagn., 11, 139–143. [DOI] [PubMed] [Google Scholar]
- 9. Thomas, M.A., Duncan, A.M., Bardin, C. and Kaloustian, V.M. (2004) Lissencephaly with der(17)t(17; 20)(p13.3; p12.2)mat. Am. J. Med. Genet. A, 124A, 292–295. [DOI] [PubMed] [Google Scholar]
- 10. Chen, C.P., Liu, Y.P., Lin, S.P., Chen, M., Tsai, F.J., Chen, Y.T., Chen, L.F., Hwang, J.K. and Wang, W. (2010) Ventriculomegaly, intrauterine growth restriction, and congenital heart defects as salient prenatal sonographic findings of Miller-Dieker lissencephaly syndrome associated with monosomy 17p (17p13.2--> pter) in a fetus. Taiwan. J. Obstet. Gynecol., 49, 81–86. [DOI] [PubMed] [Google Scholar]
- 11. Burn, J. and Goodship, J. (1996) Developmental genetics of the heart. Curr. Opin. Genet. Dev., 6, 322–325. [DOI] [PubMed] [Google Scholar]
- 12. Peyvandi, S., Lupo, P.J., Garbarini, J., Woyciechowski, S., Edman, S., Emanuel, B.S., Mitchell, L.E. and Goldmuntz, E. (2013) 22q11.2 deletions in patients with conotruncal defects: data from 1,610 consecutive cases. Pediatr. Cardiol., 34, 1687–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hiruma, T., Nakajima, Y. and Nakamura, H. (2002) Development of pharyngeal arch arteries in early mouse embryo. J. Anat., 201, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Webb, S., Qayyum, S.R., Anderson, R.H., Lamers, W.H. and Richardson, M.K. (2003) Septation and separation within the outflow tract of the developing heart. J. Anat., 202, 327–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson, R.H., Chaudhry, B., Mohun, T.J., Bamforth, S.D., Hoyland, D., Phillips, H.M., Webb, S., Moorman, A.F., Brown, N.A. and Henderson, D.J. (2012) Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc. Res., 95, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park, T.J., Boyd, K. and Curran, T. (2006) Cardiovascular and craniofacial defects in Crk-null mice. Mol. Cell. Biol., 26, 6272–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guris, D.L., Fantes, J., Tara, D., Druker, B.J. and Imamoto, A. (2001) Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet., 27, 293–298. [DOI] [PubMed] [Google Scholar]
- 18. Racedo, S.E., McDonald-McGinn, D.M., Chung, J.H., Goldmuntz, E., Zackai, E., Emanuel, B.S., Zhou, B., Funke, B. and Morrow, B.E. (2015) Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am. J. Hum. Genet., 96, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imamoto, A., Ki, S., Li, L., Iwamoto, K., Maruthamuthu, V., Devany, J., Lu, O., Kanazawa, T., Zhang, S., Yamada, T. et al. (2020) Essential role of the Crk family-dosage in DiGeorge-like anomaly and metabolic homeostasis. Life Sci. Alliance, 3:e201900635; 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saga, Y., Kitajima, S. and Miyagawa-Tomita, S. (2000) Mesp1 expression is the earliest sign of cardiovascular development. Trends Cardiovasc. Med., 10, 345–352. [DOI] [PubMed] [Google Scholar]
- 21. Conway, S.J., Henderson, D.J. and Copp, A.J. (1997) Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development, 124, 505–514. [DOI] [PubMed] [Google Scholar]
- 22. Epstein, J.A., Li, J., Lang, D., Chen, F., Brown, C.B., Jin, F., Lu, M.M., Thomas, M., Liu, E., Wessels, A. et al. (2000) Migration of cardiac neural crest cells in Splotch embryos. Development, 127, 1869–1878. [DOI] [PubMed] [Google Scholar]
- 23. Bradshaw, L., Chaudhry, B., Hildreth, V., Webb, S. and Henderson, D.J. (2009) Dual role for neural crest cells during outflow tract septation in the neural crest-deficient mutant Splotch(2H). J. Anat., 214, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirby, M.L., Gale, T.F. and Stewart, D.E. (1983) Neural crest cells contribute to normal aorticopulmonary septation. Science, 220, 1059–1061. [DOI] [PubMed] [Google Scholar]
- 25. Bronner-Fraser, M. and Fraser, S.E. (1988) Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature, 335, 161–164. [DOI] [PubMed] [Google Scholar]
- 26. Dupin, E., Creuzet, S. and Le Douarin, N.M. (2006) The contribution of the neural crest to the vertebrate body. Adv. Exp. Med. Biol., 589, 96–119. [DOI] [PubMed] [Google Scholar]
- 27. Jiang, X., Rowitch, D.H., Soriano, P., McMahon, A.P. and Sucov, H.M. (2000) Fate of the mammalian cardiac neural crest. Development, 127, 1607–1616. [DOI] [PubMed] [Google Scholar]
- 28. Echelard, Y., Vassileva, G. and McMahon, A.P. (1994) Cis-acting regulatory sequences governing Wnt-1 expression in the developing mouse CNS. Development, 120, 2213–2224. [DOI] [PubMed] [Google Scholar]
- 29. Mao, X., Fujiwara, Y., Chapdelaine, A., Yang, H. and Orkin, S.H. (2001) Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood, 97, 324–326. [DOI] [PubMed] [Google Scholar]
- 30. Vande Velde, C., Cizeau, J., Dubik, D., Alimonti, J., Brown, T., Israels, S., Hakem, R. and Greenberg, A.H. (2000) BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol., 20, 5454–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brugarolas, J., Lei, K., Hurley, R.L., Manning, B.D., Reiling, J.H., Hafen, E., Witters, L.A., Ellisen, L.W. and Kaelin, W.G., Jr. (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev., 18, 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelly, R.G., Brown, N.A. and Buckingham, M.E. (2001) The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell, 1, 435–440. [DOI] [PubMed] [Google Scholar]
- 33. Mjaatvedt, C.H., Nakaoka, T., Moreno-Rodriguez, R., Norris, R.A., Kern, M.J., Eisenberg, C.A., Turner, D. and Markwald, R.R. (2001) The outflow tract of the heart is recruited from a novel heart-forming field. Dev. Biol., 238, 97–109. [DOI] [PubMed] [Google Scholar]
- 34. Waldo, K.L., Kumiski, D.H., Wallis, K.T., Stadt, H.A., Hutson, M.R., Platt, D.H. and Kirby, M.L. (2001) Conotruncal myocardium arises from a secondary heart field. Development, 128, 3179–3188. [DOI] [PubMed] [Google Scholar]
- 35. Antoku, S., Saksela, K., Rivera, G.M. and Mayer, B.J. (2008) A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J. Cell Sci., 121, 3071–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park, T.J. and Curran, T. (2014) Essential roles of Crk and CrkL in fibroblast structure and motility. Oncogene, 33, 5121–5132. [DOI] [PubMed] [Google Scholar]
- 37. Hynes, R.O. (1992) Integrins: versatility, modulation, and signaling in cell adhesion. Cell, 69, 11–25. [DOI] [PubMed] [Google Scholar]
- 38. Giancotti, F.G. and Ruoslahti, E. (1999) Integrin signaling. Science, 285, 1028–1032. [DOI] [PubMed] [Google Scholar]
- 39. High, F.A., Zhang, M., Proweller, A., Tu, L., Parmacek, M.S., Pear, W.S. and Epstein, J.A. (2007) An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J. Clin. Invest., 117, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Manderfield, L.J., High, F.A., Engleka, K.A., Liu, F., Li, L., Rentschler, S. and Epstein, J.A. (2012) Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation, 125, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mead, T.J. and Yutzey, K.E. (2012) Notch pathway regulation of neural crest cell development in vivo. Dev. Dyn., 241, 376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noseda, M., Fu, Y., Niessen, K., Wong, F., Chang, L., McLean, G. and Karsan, A. (2006) Smooth muscle alpha-actin is a direct target of Notch/CSL. Circ. Res., 98, 1468–1470. [DOI] [PubMed] [Google Scholar]
- 43. Doi, H., Iso, T., Sato, H., Yamazaki, M., Matsui, H., Tanaka, T., Manabe, I., Arai, M., Nagai, R. and Kurabayashi, M. (2006) Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. J. Biol. Chem., 281, 28555–28564. [DOI] [PubMed] [Google Scholar]
- 44. Nagamani, S.C., Zhang, F., Shchelochkov, O.A., Bi, W., Ou, Z., Scaglia, F., Probst, F.J., Shinawi, M., Eng, C., Hunter, J.V. et al. (2009) Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J. Med. Genet., 46, 825–833. [DOI] [PubMed] [Google Scholar]
- 45. Yagi, H., Furutani, Y., Hamada, H., Sasaki, T., Asakawa, S., Minoshima, S., Ichida, F., Joo, K., Kimura, M., Imamura, S. et al. (2003) Role of TBX1 in human del22q11.2 syndrome. Lancet, 362, 1366–1373. [DOI] [PubMed] [Google Scholar]
- 46. Gong, W., Gottlieb, S., Collins, J., Blescia, A., Dietz, H., Goldmuntz, E., McDonald-McGinn, D.M., Zackai, E.H., Emanuel, B.S., Driscoll, D.A. et al. (2001) Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J. Med. Genet., 38, E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McDonald-McGinn, D.M., Sullivan, K.E., Marino, B., Philip, N., Swillen, A., Vorstman, J.A., Zackai, E.H., Emanuel, B.S., Vermeesch, J.R., Morrow, B.E. et al. (2015) 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers., 1, 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Park, T., Koptyra, M. and Curran, T. (2016) Fibroblast growth requires CT10 regulator of kinase (Crk) and Crk-like (CrkL). J. Biol. Chem., 291, 26273–26290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang, X., Sun, Y., Schneider, J., Ding, J.H., Cheng, H., Ye, M., Bhattacharya, S., Rearden, A., Evans, S. and Chen, J. (2007) Pinch1 is required for normal development of cranial and cardiac neural crest-derived structures. Circ. Res., 100, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thomas, P.S., Kim, J., Nunez, S., Glogauer, M. and Kaartinen, V. (2010) Neural crest cell-specific deletion of Rac1 results in defective cell-matrix interactions and severe craniofacial and cardiovascular malformations. Dev. Biol., 340, 613–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Turlo, K.A., Noel, O.D., Vora, R., LaRussa, M., Fassler, R., Hall-Glenn, F. and Iruela-Arispe, M.L. (2012) An essential requirement for beta1 integrin in the assembly of extracellular matrix proteins within the vascular wall. Dev. Biol., 365, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arnold, T.D., Zang, K. and Vallejo-Illarramendi, A. (2013) Deletion of integrin-linked kinase from neural crest cells in mice results in aortic aneurysms and embryonic lethality. Dis. Model. Mech., 6, 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dai, X., Jiang, W., Zhang, Q., Xu, L., Geng, P., Zhuang, S., Petrich, B.G., Jiang, C., Peng, L., Bhattacharya, S. et al. (2013) Requirement for integrin-linked kinase in neural crest migration and differentiation and outflow tract morphogenesis. BMC Biol., 11, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Humphries, J.D., Byron, A. and Humphries, M.J. (2006) Integrin ligands at a glance. J. Cell Sci., 119, 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hannigan, G.E., Leung-Hagesteijn, C., Fitz-Gibbon, L., Coppolino, M.G., Radeva, G., Filmus, J., Bell, J.C. and Dedhar, S. (1996) Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature, 379, 91–96. [DOI] [PubMed] [Google Scholar]
- 56. Tu, Y., Li, F., Goicoechea, S. and Wu, C. (1999) The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol. Cell. Biol., 19, 2425–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hobert, O., Moerman, D.G., Clark, K.A., Beckerle, M.C. and Ruvkun, G. (1999) A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans. J. Cell Biol., 144, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clark, K.A., McGrail, M. and Beckerle, M.C. (2003) Analysis of PINCH function in Drosophila demonstrates its requirement in integrin-dependent cellular processes. Development, 130, 2611–2621. [DOI] [PubMed] [Google Scholar]
- 59. Liang, X., Zhou, Q., Li, X., Sun, Y., Lu, M., Dalton, N., Ross, J., Jr. and Chen, J. (2005) PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Mol. Cell. Biol., 25, 3056–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berrier, A.L., Martinez, R., Bokoch, G.M. and LaFlamme, S.E. (2002) The integrin beta tail is required and sufficient to regulate adhesion signaling to Rac1. J. Cell Sci., 115, 4285–4291. [DOI] [PubMed] [Google Scholar]
- 61. Price, L.S., Leng, J., Schwartz, M.A. and Bokoch, G.M. (1998) Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell, 9, 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li, L., Guris, D.L., Okura, M. and Imamoto, A. (2003) Translocation of CrkL to focal adhesions mediates integrin-induced migration downstream of Src family kinases. Mol. Cell. Biol., 23, 2883–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. High, F.A., Lu, M.M., Pear, W.S., Loomes, K.M., Kaestner, K.H. and Epstein, J.A. (2008) Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. U. S. A., 105, 1955–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gomez-Lamarca, M.J., Cobreros-Reguera, L., Ibanez-Jimenez, B., Palacios, I.M. and Martin-Bermudo, M.D. (2014) Integrins regulate epithelial cell differentiation by modulating Notch activity. J. Cell Sci., 127, 4667–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rallis, C., Pinchin, S.M. and Ish-Horowicz, D. (2010) Cell-autonomous integrin control of Wnt and Notch signalling during somitogenesis. Development, 137, 3591–3601. [DOI] [PubMed] [Google Scholar]
- 66. Chen, S. and Lechleider, R.J. (2004) Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ. Res., 94, 1195–1202. [DOI] [PubMed] [Google Scholar]
- 67. Wurdak, H., Ittner, L.M., Lang, K.S., Leveen, P., Suter, U., Fischer, J.A., Karlsson, S., Born, W. and Sommer, L. (2005) Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev., 19, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim, E.K. and Choi, E.J. (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta, 1802, 396–405. [DOI] [PubMed] [Google Scholar]
- 69. Brewer, J.R., Mazot, P. and Soriano, P. (2016) Genetic insights into the mechanisms of Fgf signaling. Genes Dev., 30, 751–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lemmon, M.A. and Schlessinger, J. (2010) Cell signaling by receptor tyrosine kinases. Cell, 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Larsson, H., Klint, P., Landgren, E. and Claesson-Welsh, L. (1999) Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain-containing adaptor protein Crk. J. Biol. Chem., 274, 25726–25734. [DOI] [PubMed] [Google Scholar]
- 72. Tanaka, S., Ouchi, T. and Hanafusa, H. (1997) Downstream of Crk adaptor signaling pathway: activation of Jun kinase by v-Crk through the guanine nucleotide exchange protein C3G. Proc. Natl. Acad. Sci. U. S. A., 94, 2356–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Girardin, S.E. and Yaniv, M. (2001) A direct interaction between JNK1 and CrkII is critical for Rac1-induced JNK activation. EMBO J., 20, 3437–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vallejo-Illarramendi, A., Zang, K. and Reichardt, L.F. (2009) Focal adhesion kinase is required for neural crest cell morphogenesis during mouse cardiovascular development. J. Clin. Invest., 119, 2218–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kaartinen, V., Dudas, M., Nagy, A., Sridurongrit, S., Lu, M.M. and Epstein, J.A. (2004) Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development, 131, 3481–3490. [DOI] [PubMed] [Google Scholar]
- 76. Wang, X. and Astrof, S. (2016) Neural crest cell-autonomous roles of fibronectin in cardiovascular development. Development, 143, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang, J., Xiao, Y., Hsu, C.W., Martinez-Traverso, I.M., Zhang, M., Bai, Y., Ishii, M., Maxson, R.E., Olson, E.N., Dickinson, M.E. et al. (2016) Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development, 143, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Clarke, M.C., Figg, N., Maguire, J.J., Davenport, A.P., Goddard, M., Littlewood, T.D. and Bennett, M.R. (2006) Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med., 12, 1075–1080. [DOI] [PubMed] [Google Scholar]
- 79. Shojima, M., Oshima, M., Takagi, K., Torii, R., Nagata, K., Shirouzu, I., Morita, A. and Kirino, T. (2005) Role of the bloodstream impacting force and the local pressure elevation in the rupture of cerebral aneurysms. Stroke, 36, 1933–1938. [DOI] [PubMed] [Google Scholar]
- 80. Huo, Y., Guo, X. and Kassab, G.S. (2008) The flow field along the entire length of mouse aorta and primary branches. Ann. Biomed. Eng., 36, 685–699. [DOI] [PubMed] [Google Scholar]
- 81. Berry, C.L., Greenwald, S.E. and Rivett, J.F. (1975) Static mechanical properties of the developing and mature rat aorta. Cardiovasc. Res., 9, 669–678. [DOI] [PubMed] [Google Scholar]
- 82. Dietz, H.C., Cutting, G.R., Pyeritz, R.E., Maslen, C.L., Sakai, L.Y., Corson, G.M., Puffenberger, E.G., Hamosh, A., Nanthakumar, E.J., Curristin, S.M. et al. (1991) Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature, 352, 337–339. [DOI] [PubMed] [Google Scholar]
- 83. Pope, F.M., Martin, G.R., Lichtenstein, J.R., Penttinen, R., Gerson, B., Rowe, D.W. and McKusick, V.A. (1975) Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc. Natl. Acad. Sci. U. S. A., 72, 1314–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Loeys, B.L., Schwarze, U., Holm, T., Callewaert, B.L., Thomas, G.H., Pannu, H., De Backer, J.F., Oswald, G.L., Symoens, S., Manouvrier, S. et al. (2006) Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med., 355, 788–798. [DOI] [PubMed] [Google Scholar]
- 85. van de Laar, I.M., Oldenburg, R.A., Pals, G., Roos-Hesselink, J.W., de Graaf, B.M., Verhagen, J.M., Hoedemaekers, Y.M., Willemsen, R., Severijnen, L.A., Venselaar, H. et al. (2011) Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet., 43, 121–126. [DOI] [PubMed] [Google Scholar]
- 86. van de Laar, I.M., van der Linde, D., Oei, E.H., Bos, P.K., Bessems, J.H., Bierma-Zeinstra, S.M., van Meer, B.L., Pals, G., Oldenburg, R.A., Bekkers, J.A. et al. (2012) Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet., 49, 47–57. [DOI] [PubMed] [Google Scholar]
- 87. Ramirez, A. and Astrof, S. (2020) Visualization and analysis of pharyngeal arch arteries using whole-mount immunohistochemistry and 3D reconstruction. J. Vis. Exp., 157, e60797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang, F., Flanagan, J., Su, N., Wang, L.C., Bui, S., Nielson, A., Wu, X., Vo, H.T., Ma, X.J. and Luo, Y. (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn., 14, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fuchs, S., Herzog, D., Sumara, G., Buchmann-Moller, S., Civenni, G., Wu, X., Chrostek-Grashoff, A., Suter, U., Ricci, R., Relvas, J.B. et al. (2009) Stage-specific control of neural crest stem cell proliferation by the small rho GTPases Cdc42 and Rac1. Cell Stem Cell, 4, 236–247. [DOI] [PubMed] [Google Scholar]
- 90. De Joussineau, C., Soule, J., Martin, M., Anguille, C., Montcourrier, P. and Alexandre, D. (2003) Delta-promoted filopodia mediate long-range lateral inhibition in Drosophila. Nature, 426, 555–559. [DOI] [PubMed] [Google Scholar]
- 91. Estrach, S., Ambler, C.A., Lo Celso, C., Hozumi, K. and Watt, F.M. (2006) Jagged 1 is a beta-catenin target gene required for ectopic hair follicle formation in adult epidermis. Development, 133, 4427–4438. [DOI] [PubMed] [Google Scholar]
- 92. Cohen, M., Georgiou, M., Stevenson, N.L., Miodownik, M. and Baum, B. (2010) Dynamic filopodia transmit intermittent Delta-Notch signaling to drive pattern refinement during lateral inhibition. Dev. Cell, 19, 78–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.