

Abstract
(1) Background: Epigenome-wide association studies (EWAS) in peripheral blood have repeatedly found associations between tobacco smoking and aberrant DNA methylation (DNAm), but little is known about DNAm signatures of smoking in the human brain, which may contribute to the pathophysiology of addictive behavior observed in chronic smokers. (2) Methods: We investigated the similarity of DNAm signatures in matched blood and postmortem brain samples (n = 10). In addition, we performed EWASs in five brain regions belonging to the neurocircuitry of addiction: anterior cingulate cortex (ACC), Brodmann Area 9, caudate nucleus, putamen, and ventral striatum (n = 38–72). (3) Results: cg15925993 within the LOC339975 gene was epigenome-wide significant in the ACC. Of 16 identified differentially methylated regions, two (PRSS50 and LINC00612/A2M-AS1) overlapped between multiple brain regions. Functional enrichment was detected for biological processes related to neuronal development, inflammatory signaling and immune cell migration. Additionally, our results indicate the association of the well-known AHRR CpG site cg05575921 with smoking in the brain. (4) Conclusion: The present study provides further evidence of the strong relationship between aberrant DNAm and smoking.
Keywords: DNA methylation, tobacco smoking, neurocircuitry, EWAS, addiction
1. Introduction
Tobacco smoking has a strong impact on human health and has been identified as a risk factor for a variety of diseases [1,2]. While the mechanisms are still unclear, it has been assumed that epigenetic factors, especially DNA methylation (DNAm) changes, play a role in the pathophysiology of smoking-associated diseases [3,4]. To date, most epigenetic association studies (EWASs) in the context of smoking have been conducted in peripheral blood, largely given the convenient, less-invasive sampling procedure in comparison to other tissues. The largest EWAS meta-analysis of smoking in blood (n = 15,907) found 18,760 cytosine-phosphate-guanine (CpG) sites associated with smoking at epigenome-wide significance [5]. Annotation of these CpG sites to genes pointed towards differential methylation for almost one-third of all genes in the human genome, indicating an extensive effect of smoking on DNAm levels.
Findings from blood have been partially replicated in other tissues such as the lungs [6,7] and adipose tissue [8]. As the brain is involved in the development and maintenance of tobacco use disorder (TUD), DNAm changes in the brain are of interest but knowledge on smoking-associated DNAm signatures remains sparse. One EWAS has examined DNAm signatures of smoking in the nucleus accumbens (NAc, n = 221) using postmortem tissue, finding seven CpG sites to be associated with smoking at epigenome-wide significance [9]. None of these were found to be significant in an EWAS of smoking in blood, which suggests heterogeneity of associations between tissues [9]. Deciphering these tissue-specific and tissue-shared methylation patterns could provide insights into a potential proxy function of blood for predicting differential methylation in the brain.
In substance use disorders (SUDs), a whole neurocircuitry of addiction consisting of multiple cortical, striatal, and limbic brain regions exhibits functional changes [10]. Cortical regions such as the prefrontal and the anterior cingulate cortex (ACC) are involved in executive control and are especially important in the preoccupation/anticipation stage of the addiction cycle [10]. The striatum is subdivided into a ventral (VS) and a dorsal part with the latter comprising the caudate nucleus (CN) and the putamen (PUT). The VS is thought to be related to reward processing [11], whereas CN and PUT are involved in sensorimotor processing and the habitual behavior observed in later stages of addiction [12].
To investigate differential methylation in the context of smoking, methylation changes within brain regions which are part of the neurocircuitry of addiction need to be assessed. In the present study, we investigated the tissue-specificity of smoking-associated methylation signatures by performing EWASs of smoking and evaluating the similarity of smoking-associated methylation patterns between blood and multiple brain regions (ACC, Brodmann Area 9 (BA9), CN, PUT and the VS). Based on the results, we performed downstream analyses including the assessment of differentially methylated regions (DMRs), gene ontology (GO) enrichment analysis, and GWAS enrichment analysis.
2. Materials and Methods
2.1. Samples
Postmortem human brain tissue was obtained from the New South Wales Tissue Resource Center (NSWTRC, University of Sydney, Sydney, Australia), as part of a previous study on alcohol use disorder (AUD) [13]. Information on smoking was available for a total of 304 postmortem brain samples originating from 80 European American tissue donors. For each donor, tissue from at least one of the five brain regions (ACC, BA9, CN, PUT, and VS) was available. BA9 brain samples from an additional 12 subjects were obtained from the University of Texas Health Science Center at Houston (UTHealth, Prof. Consuelo Walss-Bass). Here, sample donors were of Asian (n = 1), Black (n = 1), Hispanic (n = 2), and White (n = 8) ethnicities. For 10 of the 12 subjects, matched blood samples were available. Total sample sizes of different brain regions ranged from 38 to 72 (Supplementary Table S1). Inclusion criteria were age > 18 and no history of severe psychiatric, neurodevelopmental, or substance use disorder (except AUD and TUD). These criteria as well as smoking status were assessed in next-of-kin interviews. Subjects included in the present study had a smoking status of either current smoking or never smoking prior to death. A descriptive summary of phenotypic information on tissue donors is shown in Table 1.
Table 1.
Demographic data of tissue donors.
| NSWTRC | UTHealth | |||||
|---|---|---|---|---|---|---|
| Characteristic | Smokers | Non-Smokers | p | Smokers | Non-Smokers | p |
| n | 50 | 30 | 7 | 5 | ||
| Age (years) | 56.14 (9.00) | 57.83 (10.50) | 0.47 | 54.86 (9.48) | 50.20 (10.80) | 0.46 |
| Sex (M/F) | 40/10 | 18/12 | 0.09 | 6/1 | 5/0 | 1.00 |
| PMI (hours) | 32.17 (15.22) | 34.20 (18.09) | 0.61 | 31.00 (10.96) | 33.62 (9.69) | 0.67 |
| AUD (% cases) | 72.00 | 20.00 | 1.89 × 10−5 | 71.43 | 40.00 | 0.62 |
For quantitative measures, standard deviation (±SD) is provided in brackets. NSWTRC, New South Wales Tissue Research Center; UTHealth, University of Texas Health Science Center at Houston; PMI, postmortem interval; AUD, alcohol use disorder, p: p-value of a t-Test/Chi-squared test assessing the significance of differences between smokers and non-smokers.
2.2. DNA Extraction, DNAm Analysis, and Quality Control
Extraction of DNA, sample randomization, epigenome-wide DNAm analysis, and quality control of methylation data was performed as described in Zillich et al. [13]. In brief, epigenome-wide DNAm was determined using the Infinium Human Methylation EPIC BeadChip (Illumina, San Diego, CA, USA) and quality control was performed with a customized and updated version of the CPACOR pipeline [14]. Samples from the NSWTRC were processed separately for each brain region in either a single or in two batches (ACC and PUT: one batch, BA9, CN and VS: two batches). The UTHealth samples were processed in a single batch after randomization according to sex, AUD status, and tissue. Methylation data were quantile-normalized.
2.3. Statistical Analyses
All statistical analyses were performed using R version 3.6.1 [15]. A schematic workflow depicting the different analysis levels within the present study is shown in Figure 1.
Figure 1.
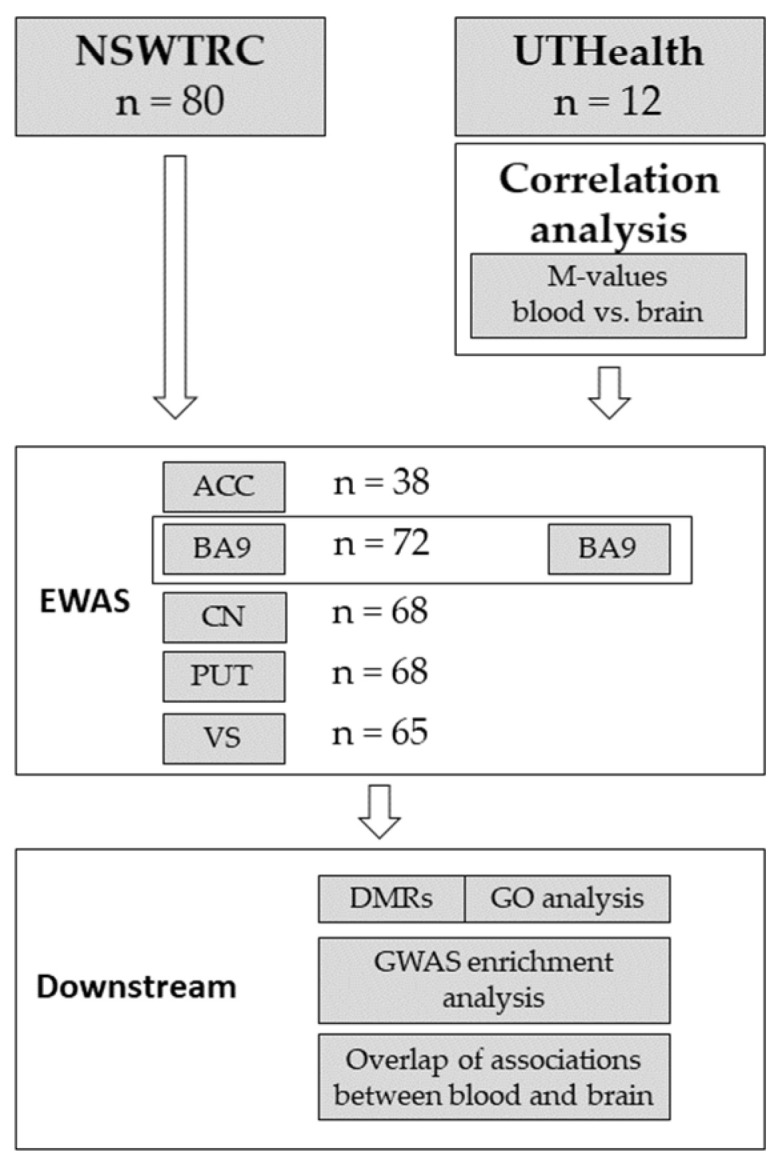
Analysis flowchart.
2.3.1. Between-Tissue Correlation
M-values of methylation were generated by logit-transformation of β-values as described by Du, Zhang [16]. For each of the 632,086 CpG sites remaining after QC, average M-values were calculated separately in blood and brain samples. Correlation of mean M-values between blood and brain (n = 10) was determined using the Pearson correlation method.
2.3.2. EWAS in Five Brain Regions
Separate EWASs were performed to identify smoking associated CpG sites in each of the five brain regions (ACC, BA9, CN, PUT, VS). The EWASs were restricted to samples originating from donors with European American ancestry to reduce confounding by ancestry. A linear regression model with M-values specified as the dependent variable and smoking status as a predictor was used. As covariates, sex, age, postmortem interval (PMI), AUD status, batch, and the first 10 principal components of control probes (PCcp) were included. Neuronal cell fractions were estimated using the Houseman approach [17] with the dorsolateral prefrontal cortex reference dataset [18], and included as a covariate. For BA9, tissue from both resources (NSWTRC and UTHealth) was combined in the EWAS, whereas for all other brain regions, tissue samples from NSWTRC were used exclusively. In the EWAS of smoking in BA9, the first four genotype principal components (PCgeno) were used as additional covariates to correct for genotype differences between sample donors. Genotype data of NSWTRC samples were generated using the Illumina Infinium OMNI5 array (Illumina, San Diego, CA, USA). The Illumina Infinium Global Screening Array (Illumina, San Diego, CA, USA) was used for genotyping of UTHealth samples. Genotype QC was performed as described by Turner, Armstrong [19]. Sample sizes used in the EWAS were n = 38 (ACC), n = 65 (VS), n = 68 (CN and PUT), and n = 72 (BA9). Prior to EWAS, a variance inflation factor (VIF) analysis was performed to investigate multicollinearity in the linear model. If a VIF larger than 10 was detected, one of the correlated covariates was removed in a way so that the final models for each brain region were most comparable to each other (Supplementary Information 1). p-values obtained in the association analyses were FDR-adjusted for multiple testing using the Benjamini-Hochberg procedure [20].
2.3.3. Differentially Methylated Regions
Differentially methylated regions were identified using the comb-p software (v. 0.50.2) [21]. The following DMR definition was used: seed p-values < 0.01 for a minimum number of two CpG sites within a 500 bp genomic window. Correction for multiple testing was performed using the Sĭdaák method as implemented in comb-p.
2.3.4. Gene Ontology (GO) Enrichment Analysis
Functional enrichment of genes harboring smoking-associated CpG sites within GO terms was investigated using missMethyl [22] (v. 1.20.4). Probes were annotated to genes according to the Illumina EPIC manifest file (http://webdata.illumina.com.s3-website-us-east-1.amazonaws.com/downloads/productfiles/methylationEPIC/infinium-methylationepic-v-1-0-b4-manifest-file-csv.zip) as downloaded on 10 August 2018. Analysis was restricted to CpG sites with association p-values < 0.001.
2.3.5. GWAS Enrichment Analysis
To investigate a potential overlap between polygenic and polyepigenetic signal, we performed GWAS enrichment analysis. Gene sets were derived from EWAS results for each of the five brain regions. For this, CpG sites exhibiting a nominally significant (p < 0.05) association with smoking were annotated to genes. Enrichment of these gene sets within GWAS signals of different smoking phenotypes and SUDs was investigated using Multi-marker Analysis of GenoMic Annotation (MAGMA, v. 1.08b) [23]. Summary statistics for smoking phenotypes were obtained from Liu [24]: (1) smoking initiation (ever/never, median n/SNP = 632,802), (2) age of initiation (median n/SNP = 262,990), (3) smoking cessation (median n/SNP = 312,821), and (4) cigarettes per day (median n/SNP = 263,954). For SUDs, GWASs on AUD (n = 313,959) [25], opioid use disorder (OUD, dependent vs. unexposed, n = 37,003) [26], and cannabis use disorder (CUD, n = 374,287) [27] were tested. Summary statistics for problematic alcohol use (PAU, n = 435,563) [25] were also included. Significance level was adjusted to Bonferroni correction for multiple testing of the 8 GWASs (n = 8, p < 6.25 × 10−3).
3. Results
While sample sizes ranged between 38 and 72 per brain region, tissue of 92 donors was included in the present study. Sample characteristics are displayed in Table 1 and a detailed breakdown of batches and brain regions is available in Supplementary Table S1, along with the causes of death in Supplementary Table S2.
3.1. Correlation of Methylation Levels
Hierarchical clustering based on M-values of all CpG sites (n = 632,086) revealed a clear distinction based on tissue of origin (Figure 2A). At the same time, the between-tissue correlation between averaged methylation levels of blood and brain was high, with a Pearson correlation of r = 0.91, p < 2.2 × 10−16 (Figure 2B).
Figure 2.

(A) Hierarchical clustering of matched blood and Brodmann Area 9 (BA9) brain samples (n = 10) based on methylation M-values (row normalized) of the n = 632,086 CpG sites remaining after quality control. Next to the tissue type, smoking status, alcohol use disorder status and ethnicity are displayed as additional phenotypic variables. (B) Pearson correlation of mean M-values in the between-subject correlation analysis using matched blood and BA9 brain samples.
3.2. Epigenome-Wide Association Studies
To expand the analysis of methylation signatures of smoking to the neurocircuitry of addiction, we performed EWASs of smoking in five brain regions. In the ACC, one CpG site (cg152925993) was differentially methylated at epigenome-wide significance between smokers and non-smokers (b = 0.53, se = 0.07, p = 7.33 × 10−8, FDR = 0.048, n = 38). This CpG site was annotated to the long non-coding RNA (lncRNA) LOC339975. No epigenome-wide significant CpG sites were identified in the other brain regions. The strongest association in BA9 was observed for cg22909901 (b = 0.34, se = 0.06, p = 9.99 × 10−7, FDR = 0.652, n = 72) which was annotated to MCF2L2/B3GNT5. In the ventral striatum, cg09139806 in RNF220 was most significant (b = −0.534, se = 0.10, p = 1.15 × 10−6, FDR = 0.364, n = 65). In the dorsal striatum, the strongest associations with smoking were observed for cg07022048 in KRT7 for the caudate nucleus (b = −0.22, se = 0.04, p = 2.66 × 10−7, FDR = 0.173, n = 68) and for cg18712580 in STK38L (b = −0.64, se = 0.12, p = 1.87 × 10−6, FDR = 1, n = 68) in the putamen. A summary table including EWAS sample sizes, genomic inflation factors and resulting FDR-thresholds is provided in Supplementary Table S3. Manhattan plots for ACC, BA9, and VS are provided in Figure 3 and for CN and PUT in Supplementary Figure S1A,B. Supplementary Table S4a–e summarize the top 10 associations from each brain region.
Figure 3.

Manhattan plots for the EWAS of smoking in (A) anterior cingulate cortex (ACC), (B) Brodmann Area 9 (BA9), and (C) the ventral striatum (VS). Top 10 smoking-associated CpG sites for each region and CpG sites contributing to differentially methylated regions (DMRs) are highlighted in green. Annotation to gene names was performed where possible. Names of genes harboring a DMR are highlighted in green. The horizontal line depicts epigenome-wide significance (5% FDR, for further information, see Supplementary Table S3).
3.3. Differentially Methylated Regions
We identified a total of 16 DMRs in the five brain regions that were associated with tobacco smoking. Two DMRs were observed in multiple brain regions. One was a region consisting of 5 probes in PRSS50 in ACC and VS, and another contained the LINC00612/A2M-AS1 genes in both dorsal striatal regions (CN and PUT). DMRs for each brain region are summarized in Table 2 and are highlighted using green gene names in the Manhattan plots.
Table 2.
Differentially methylated regions (DMRs) identified in the five brain regions.
| Chr | CpG | Gene | Dir 1 | N Probes | Z P | Z Sidak p | Start | End | |
|---|---|---|---|---|---|---|---|---|---|
| ACC | 3 | cg02834909 | PRSS50 | + | 5 | 1.72 × 10−8 | 7.48 × 10−5 | 46,759,438 | 46,759,589 |
| BA9 | 3 | cg22909901 |
MCF2L2; B3GNT5 |
+ | 2 | 3.71 × 10−8 | 3.19 × 10−4 | 182,981,632 | 182,981,708 |
| 6 | cg23161317 | ZNF192P1 | − | 6 | 3.17 × 10−9 | 1.20 × 10−5 | 28,129,313 | 28,129,486 | |
| 6 | cg23681866 | HCG4B | − | 5 | 1.48 × 10−9 | 6.66 × 10−6 | 29,895,116 | 29,895,261 | |
| CN | 1 | cg10703826 | TBX15 | − | 7 | 5.96 × 10−10 | 2.03 × 10−6 | 119,532,044 | 119,532,234 |
| 12 | cg26114124 |
LINC00612; A2M-AS1 |
− | 8 | 3.02 × 10−12 | 5.60 × 10−9 | 9,217,510 | 9,217,860 | |
| 16 | cg06751612 |
PIEZO1; LOC100289580 |
+ | 2 | 1.96 × 10−7 | 4.38 × 10−3 | 88,798,826 | 88,798,855 | |
| 5 | cg11916729 | SCGB3A1 | + | 6 | 2.81 × 10−7 | 1.82 × 10−3 | 180,018,465 | 180,018,565 | |
| PUT | 12 | cg02883147 |
LINC00612; A2M-AS1 |
− | 5 | 9.86 × 10−9 | 3.58 × 10−5 | 9,217,669 | 9,217,860 |
| 19 | cg24716275 | ZNF264 | − | 6 | 7.80 × 10−10 | 4.17 × 10−6 | 57,702,371 | 57,702,501 | |
| VS | 3 | cg00817731 | PRSS50 | + | 5 | 1.71 × 10−7 | 7.52 × 10−4 | 46,759,438 | 46,759,589 |
| 4 | cg08064687 | INPP4B | − | 2 | 7.43 × 10−9 | 8.65 × 10−5 | 143,326,485 | 143,326,542 | |
| 6 | cg14654363 | - | − | 8 | 1.00 × 10−9 | 4.28 × 10−6 | 28,601,365 | 28,601,520 | |
| 6 | cg27596495 | HCRTR2 | + | 4 | 1.37 × 10−8 | 6.00 × 10−5 | 55,039,232 | 55,039,383 | |
| 7 | cg06036947 | ADAP1 | − | 2 | 5.34 × 10−8 | 4.60 × 10−4 | 949,758 | 949,835 | |
| 7 | cg07972322 |
C7orf49; TMEM140 |
− | 5 | 4.84 × 10−10 | 1.23 × 10−6 | 134,832,770 | 134,833,032 |
DMRs associated with tobacco smoking in the anterior cingulate cortex (ACC), Brodmann Area 9 (BA9), caudate nucleus (CN), putamen (PUT) and ventral striatum (VS). Genes exhibiting DMRs within multiple brain regions are highlighted in bold type. Dir 1: DMR characterized by hypermethylation (+) or hypomethylation (−) of probes.
3.4. Gene-Ontology Analysis
In the ACC, smoking-associated CpG sites were enriched for GO terms related to neurodevelopment, cell growth, and morphogenesis. Enriched GO terms in BA9 were related to dendritic spine development and chromatin modification. In the VS, neuron-specific pathways were enriched, as well as processes associated with the regulation of vessel development. In both dorsal striatal regions, genes harboring differentially methylated CpG sites were enriched for GO terms related to immune pathways. After correction for multiple testing, no GO terms remained statistically significant. Result tables for the top 10 associated GO terms are shown in Supplementary Table S5a–e.
3.5. GWAS Enrichment Analysis
Gene sets consisting of smoking-associated CpG sites were overrepresented in GWAS summary statistics of smoking traits, such as smoking initiation, age of initiation, and cigarettes per day. Furthermore, significant enrichment for other SUDs, such as cannabis-, alcohol-, and opioid use disorder was observed. Statistical significance of enrichment for all tested gene-sets is displayed in Figure 4.
Figure 4.

Summary of the results from GWAS enrichment analysis. For each brain region and each GWAS, the statistical significance of enrichment is shown. Dashed line represents nominal significance (p < 5 × 10−2) while the solid line depicts the significance threshold after Bonferroni correction for multiple testing (n = 8, p < 6.25 × 10−3). AUD, alcohol use disorder; PAU, problematic alcohol use; CUD, cannabis use disorder; OUD, opioid use disorder.
3.6. Consistency of Smoking-Associated CpG Sites in EWAS Results of Blood and Brain
We examined differential methylation of CpG sites in the brain, which have previously been suggested to predict smoking status in peripheral blood. When we investigated the nine available CpG sites from the prediction model by Maas et al. [28], we found no consistent differential methylation across the five brain regions. For two of them, nominally significant associations were detected. The AHRR CpG site cg05575921, even used as a single-marker smoking status predictor, was significantly associated with smoking in the ACC (p = 0.038) and PUT (p = 0.033). cg21566642, an intergenic CpG site, was associated with smoking in BA9 (p = 0.018). Full results of the consistency analysis are listed in Table 3.
Table 3.
Associations of the predictor CpG sites in blood and brain.
| CpG Site | Gene | Blood * (n = 15,907) |
ACC (n = 38) |
BA9 (n = 72) |
CN (n = 68) |
PUT (n = 68) |
VS (n = 65) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | pval | β | pval | β | pval | β | pval | β | pval | β | pval | ||
| cg05575921 | AHRR | −0.180 | 4.55 × 10−26 | −0.310 | 0.038 | −0.158 | 0.163 | −0.247 | 0.059 | −0.220 | 0.033 | −0.030 | 0.827 |
| cg03636183 | F2RL3 | −0.095 | 1.12 × 10−20 | −0.117 | 0.118 | −0.010 | 0.834 | −0.056 | 0.198 | −0.016 | 0.619 | −0.080 | 0.116 |
| cg09935388 | GFI1 | −0.083 | 3.14 × 10−17 | −0.162 | 0.345 | −0.183 | 0.132 | 0.163 | 0.206 | −0.060 | 0.595 | 0.142 | 0.369 |
| cg12876356 | GFI1 | −0.057 | 1.49 × 10−15 | 0.022 | 0.838 | −0.041 | 0.516 | 0.028 | 0.640 | 0.025 | 0.705 | 0.028 | 0.730 |
| cg12803068 | MYO1G | 0.063 | 9.06 × 10−23 | 0.075 | 0.556 | −0.045 | 0.598 | 0.101 | 0.163 | 0.082 | 0.292 | 0.014 | 0.810 |
| cg13039251 | PDZD2 | 0.030 | 1.36 × 10−15 | 0.060 | 0.673 | −0.051 | 0.519 | 0.109 | 0.217 | 0.118 | 0.186 | −0.014 | 0.894 |
| cg01940273 | - | −0.082 | 2.03 × 10−34 | −0.015 | 0.895 | −0.035 | 0.569 | −0.008 | 0.903 | 0.007 | 0.885 | −0.013 | 0.851 |
| cg15693572 | - | 0.053 | 3.85 × 10−11 | −0.120 | 0.297 | 0.068 | 0.478 | 0.113 | 0.227 | −0.009 | 0.912 | −0.030 | 0.800 |
| cg21566642 | - | −0.126 | 4.22 × 10−25 | −0.205 | 0.202 | −0.176 | 0.018 | 0.039 | 0.707 | −0.147 | 0.119 | 0.048 | 0.628 |
For the nine CpG sites derived from Maas, Vidaki [28], effect sizes and p-values of the association analyses with smoking in blood (* EWAS from Joehanes, Just [5]) and the five brain regions are summarized. Nominally significant results are highlighted in bold type. Β, EWAS effect size; pval, EWAS p-value; ACC, anterior cingulate cortex; BA9, Brodmann Area 9; CN, caudate nucleus; PUT, putamen; VS, ventral striatum.
4. Discussion
In the present study, we investigated the consistency of smoking-associated methylation signatures in blood and brain, examined differential methylation in the brain associated with smoking status, and performed several EWAS downstream analyses in five brain regions related to the neurocircuitry of addiction. The strong overall correlation of methylation levels in matched blood and brain samples is in line with previous findings of high cross-tissue correlation coefficients [29,30].
For the EWAS of smoking in the ACC, one epigenome-wide significant CpG site was observed within LOC339975. We identified a total of 16 differentially methylated regions associated with smoking status. A DMR in PRSS50 was shared between the ACC and the VS. A functional role of PRSS50 in the brain has not been systematically evaluated so far. However, in cancer cells, knockdown of PRSS50 resulted in impaired cell proliferation and increased levels of apoptosis [31]. Also, promoter hypermethylation of PRSS50 was detected in an EWAS of age-related macular degeneration (AMD) in blood and retinal tissue [32]. In the present study, also hypermethylation was observed for the PRSS50 DMR in the ACC and the VS. The second smoking-associated DMR shared between two regions of the brain (CN and PUT) was annotated to LINC00612/A2M-AS1. Both lncRNAs, LINC00612 and A2M-AS1, are involved in inflammatory processes. An anti-inflammatory function disrupted by smoking has been discovered for LINC00612 in the lungs [33], while A2M-AS1 has been linked to interleukin 1 receptor signaling in cardiomyocytes [34]. Further studies need to investigate if these lncRNAs are also involved in inflammatory signaling in the brain. Functional enrichment of inflammatory and immune-related processes was also supported by the results of the GO enrichment analysis: in PUT, genes harboring smoking-associated CpG sites were enriched in immune cell migration pathways. In the ACC, BA9, and the VS, GO enrichment analysis revealed pathway enrichment related to neuronal development and morphogenesis. Given the direct influence of nicotine on immune cell function [35] and neuronal development [36], results from the present study may point towards an additional epigenetic effect of smoking on these cellular processes.
GWAS enrichment analysis revealed significant overrepresentation of EWAS-derived gene-sets within GWAS signals of several smoking phenotypes and SUDs. This points towards a fraction of genes detected by GWAS and EWAS contributing to the development and maintenance of tobacco smoking and SUDs. Further research needs to uncover how genetic and epigenetic mechanisms collectively contribute to the disease course in SUDs.
The well-known AHRR CpG site cg05575921 was associated with smoking in the ACC and PUT and was consistently hypomethylated in all investigated brain regions. As hypomethylation and significant association with smoking has previously also been detected for cg05575921 in blood [5], the lungs [6] and in adipose tissue [8], it may be a common locus representing smoking status across tissues.
In contrast to the strong general correlation of methylation levels between tissues, associations with smoking were heterogenous between tissues. Blood samples might thus be limited in the extent to which they can function as a proxy for smoking-associated differential methylation in the brain. Concurrently, a potential specificity of associations implies that specific methylation signatures of smoking in the brain could contribute to the pathophysiology of TUD. This is also supported by the overlap of polygenic and poly-epigenetic signals identified for smoking phenotypes. Nevertheless, smoking could also have a non-specific effect on the entire organism and further studies are needed to investigate the functional relevance of smoking-associated differential methylation within the neurocircuitry of addiction and its contribution to the development and maintenance of TUD.
Several limitations need to be addressed. First, a large fraction of tissue donors was diagnosed with AUD prior to death. Despite adjustment for AUD in the linear model, we cannot rule out residual confounding by alcohol consumption and future studies should investigate smoking independent of other SUDs. However, a recent Mendelian randomization study has shown that a smoking-specific risk on other disorders remains after correcting for the genetic risk for alcohol consumption [37]. Second, smoking status was assessed based on next-of-kin reports which are known to be less precise than direct measurement of the nicotine metabolite cotinine [38]. Smoking status and TUD status often overlap, but TUD was not specifically assessed in the present study. A recent EWAS of smoking status and TUD found both specific and shared methylation signals between smoking status and lifetime TUD [39], which underlines the importance of investigating both traits simultaneously. Third, due to multiple testing, even with our largest sample size of N = 72, the EWAS of smoking in the brain is still insufficiently powered. At the same time, postmortem brain tissue is scarce which makes it challenging to obtain enough samples.
The present study identified smoking-associated DNAm changes in the neurocircuitry of addiction related to immunological and neurodevelopmental processes. However, certain DNAm signatures might represent a predisposition to tobacco smoking rather than a consequence of it. Follow-up studies should thus investigate tissues of donors deceased at different stages of TUD, which may enable the identification of changes in DNAm levels during the disease course and differentiate between predispositions and consequences of smoking. The functional consequences of smoking-associated changes in DNAm levels should also be addressed in a more comprehensive design, for example using a multi-omics approach integrating methylation and transcriptomic data. Ultimately, deeper insight into methylation changes within the neurocircuitry of addiction could lay the foundation for better understanding of the pathomechanisms of tobacco use disorder.
Acknowledgments
We thank Elisabeth Röbel and Claudia Schäfer-Arnold for technical assistance. Tissues were received from the New South Wales Brain Tissue Resource Centre at the University of Sydney which is supported by the University of Sydney. Research reported in this publication was supported by the National Institute of Alcohol Abuse and Alcoholism of the National Institutes of Health under Award Number R28AA012725 and R01DA044859. The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health. We are grateful for the invaluable donations and participation from families, as well as for the generous collaboration of the medical examiners at the Harris County Institute of Forensic Sciences.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jpm12040566/s1. Information 1: Linear models used for the EWAS of smoking in different brain regions; Figure S1: EWAS of smoking in CN and PUT; Table S1: Number of samples for each brain region and blood investigated in the present study; Table S2: Summary table displaying cause id death of sample donors; Table S3: EWAS sample sizes, number of assessed CpG sites, genomic inflation factores, and 5% FDR thresholds; Table S4a–e: Top 10 smoking-associated CpG sites in the five brain regions; Table S5a–e: Top 10 GO terms of the five brain regions.
Author Contributions
Conceptualization, C.W.-B., R.S. and M.R.; data curation, E.P., F.S., G.R.F., J.C.F. and L.S.; formal analysis, L.Z. and E.P.; funding acquisition, G.R.F., A.C.H., S.H.W., C.W.-B., R.S. and M.R.; investigation, L.Z., E.P., G.R.F., J.C.F., M.M.F., S.H.W. and R.S.; methodology, L.Z., E.P., F.S. and J.F.; project administration, M.M.N., S.H.W., C.W.-B. and R.S.; resources, M.M.N.; supervision, J.F., S.H.W. and M.R.; writing—original draft, L.Z. and E.P.; writing—review and editing, L.Z., E.P., F.S., J.F., G.R.F., J.C.F., M.M.F., L.S., A.C.H., M.M.N., S.H.W., C.W.-B., R.S. and M.R. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept “A systems-medicine approach towards distinct and shared resilience and pathological mechanisms of substance use disorders” (01ZX01909 to Rainer Spanagel, Marcella Rietschel, Stephanie H Witt, Anita C Hansson), “Towards Targeted Oxytocin Treatment in Alcohol Addiction (Target-OXY)” (031L0190A to Jerome C Foo). ERA-NET program: Psi-Alc (FKZ: 01EW1908), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 402170461—TRR 265 to Rainer Spanagel, Anita C Hansson and Marcella Rietschel [40], R01DA044859 to Consuelo Walss-Bass, and MH121580 to Gabriel R. Fries.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki; and was approved by the Ethics Committee II of the Medical Faculty Mannheim (2009-238N-MA) and the University of Texas Health Science Center at Houston (HSC-MS-15-0247).
Informed Consent Statement
Biomaterial was used from anonymous donors of the New South Wales Tissue Resource Centre at the University of Sydney, Sydney Australia.
Data Availability Statement
Raw data and summary statistics for all analyses are available on request.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Reitsma M.B., Kendrick P.J., Ababneh E., Abbafati C., Abbasi-Kangevari M., Abdoli A., Abedi A., Abhilash E.S., Abila D.B., Aboyans V., et al. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990–2019: A systematic analysis from the Global Burden of Disease Study 2019. Lancet. 2021;397:2337–2360. doi: 10.1016/S0140-6736(21)01169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization . WHO Report on the Global Tobacco Epidemic, 2019: Offer Help to Quit Tobacco Use. World Health Organization; Geneva, Switzerland: 2019. [Google Scholar]
- 3.Zong D., Liu X., Li J., Ouyang R., Chen P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenet. Chromatin. 2019;12:65. doi: 10.1186/s13072-019-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breitling L.P. Current Genetics and Epigenetics of Smoking/Tobacco-Related Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2013;33:1468–1472. doi: 10.1161/ATVBAHA.112.300157. [DOI] [PubMed] [Google Scholar]
- 5.Joehanes R., Just A.C., Marioni R.E., Pilling L.C., Reynolds L.M., Mandaviya P.R., Guan W., Xu T., Elks C.E., Aslibekyan S., et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stueve T.R., Li W.-Q., Shi J., Marconett C.N., Zhang T., Yang C., Mullen D., Yan C., Wheeler W., Hua X., et al. Epigenome-wide analysis of DNA methylation in lung tissue shows concordance with blood studies and identifies tobacco smoke-inducible enhancers. Hum. Mol. Genet. 2017;26:3014–3027. doi: 10.1093/hmg/ddx188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundar I.K., Yin Q., Baier B.S., Yan L., Mazur W., Li D., Susiarjo M., Rahman I. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin. Epigenet. 2017;9:38. doi: 10.1186/s13148-017-0335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai P.-C., Glastonbury C., Eliot M.N., Bollepalli S., Yet I., Castillo-Fernandez J., Carnero-Montoro E., Hardiman T., Martin T., Vickers A., et al. Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health. Clin. Epigenetics. 2018;10:126. doi: 10.1186/s13148-018-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markunas C.A., Semick S.A., Quach B.C., Tao R., Deep-Soboslay A., Carnes M.U., Bierut L.J., Hyde T.M., Kleinman J.E., Johnson E.O., et al. Genome-wide DNA methylation differences in nucleus accumbens of smokers vs. nonsmokers. Neuropsychopharmacology. 2021;46:554–560. doi: 10.1038/s41386-020-0782-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koob G., Volkow N. Neurocircuitry of Addiction. Am. Psychiatr. Assoc. Publ. Textb. Subst. Use Disord. Treat. 2021;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volkow N.D., Morales M. The Brain on Drugs: From Reward to Addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 12.Balleine B.W., Delgado M.R., Hikosaka O. The Role of the Dorsal Striatum in Reward and Decision-Making. J. Neurosci. 2007;27:8161–8165. doi: 10.1523/JNEUROSCI.1554-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zillich L., Frank J., Streit F., Friske M.M., Foo J.C., Sirignano L., Heilmann-Heimbach S., Dukal H., Degenhardt F., Hoffmann P., et al. Epigenome-wide association study of alcohol use disorder in five brain regions. Neuropsychopharmacology. 2021;47:832–839. doi: 10.1038/s41386-021-01228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehne B., Drong A.W., Loh M., Zhang W., Scott W.R., Tan S.-T., Afzal U., Scott J., Jarvelin M.-R., Elliott P., et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015;16:37. doi: 10.1186/s13059-015-0600-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2019. [Google Scholar]
- 16.Du P., Zhang X., Huang C.-C., Jafari N., Kibbe A.W., Hou L., Lin S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houseman E.A., Accomando W.P., Koestler D.C., Christensen B.C., Marsit C.J., Nelson H.H., Wiencke J.K., Kelsey K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaffe A.E., Kaminsky Z.A. FlowSorted.DLPFC.450k: Illumina Human Methylation Data on Sorted Frontal Cortex Cell Populations, R Package Version 1.24.0. Bioconductor; Seattle, WA, USA: 2020. [Google Scholar]
- 19.Turner S., Armstrong L.L., Bradford Y., Carlson C.S., Crawford D.C., Crenshaw A.T., De Andrade M., Doheny K.F., Haines J.L., Hayes G., et al. Quality Control Procedures for Genome-Wide Association Studies. Curr. Protoc. Hum. Genet. 2011;68:1.19.1–1.19.18. doi: 10.1002/0471142905.hg0119s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. [(accessed on 21 February 2022)];J. R. Stat. Soc. Ser. B Methodol. 1995 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. Available online: http://www.jstor.org/stable/2346101. [DOI] [Google Scholar]
- 21.Pedersen B.S., Schwartz D.A., Yang I.V., Kechris K.J. Comb-p: Software for combining, analyzing, grouping and correcting spatially correlated p-values. Bioinformatics. 2012;28:2986–2988. doi: 10.1093/bioinformatics/bts545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phipson B., Maksimovic J., Oshlack A. missMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32:286–288. doi: 10.1093/bioinformatics/btv560. [DOI] [PubMed] [Google Scholar]
- 23.de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu M., Jiang Y., Wedow R., Li Y., Brazel D.M., Chen F., Datta G., Davila-Velderrain J., McGuire D., Tian C., et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019;51:237–244. doi: 10.1038/s41588-018-0307-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H., Sealock J.M., Sanchez-Roige S., Clarke T.-K., Levey D.F., Cheng Z., Li B., Polimanti R., Kember R.L., Smith R.V., et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci. 2020;23:809–818. doi: 10.1038/s41593-020-0643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polimanti R., Psychiatric Genomics Consortium Substance Use Disorders Workgroup. Walters R.K., Johnson E.C., McClintick J.N., Adkins A.E., Adkins D.E., Bacanu S.-A., Bierut L.J., Bigdeli T.B., et al. Leveraging genome-wide data to investigate differences between opioid use vs. opioid dependence in 41,176 individuals from the Psychiatric Genomics Consortium. Mol. Psychiatry. 2020;25:1673–1687. doi: 10.1038/s41380-020-0677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson E.C., Demontis D., Thorgeirsson E.T., Walters R.K., Polimanti R., Hatoum A.S., Sanchez-Roige S., Paul E.S., Wendt F.R., Clarke T.-K., et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry. 2020;7:1032–1045. doi: 10.1016/S2215-0366(20)30339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maas S.C.E., BIOS Consortium. Vidaki A., Wilson R., Teumer A., Liu F., Van Meurs J.B.J., Uitterlinden A.G., Boomsma D.I., De Geus E.J.C., et al. Validated inference of smoking habits from blood with a finite DNA methylation marker set. Eur. J. Epidemiol. 2019;34:1055–1074. doi: 10.1007/s10654-019-00555-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braun P.R., Han S., Hing B., Nagahama Y., Gaul L.N., Heinzman J.T., Grossbach A.J., Close L., Dlouhy B.J., Howard M., et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl. Psychiatry. 2019;9:47. doi: 10.1038/s41398-019-0376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horvath S., Zhang Y., Langfelder P., Kahn R.S., Boks M.P., Van Eijk K., Van den Berg L.H., Ophoff R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou L., Bao Y.-L., Zhang Y., Wu Y., Yu C.-L., Huang Y.-X., Sun Y., Zheng L.-H., Li Y.-X. Knockdown of TSP50 inhibits cell proliferation and induces apoptosis in P19 cells. IUBMB Life. 2010;62:825–832. doi: 10.1002/iub.390. [DOI] [PubMed] [Google Scholar]
- 32.Oliver V.F., Jaffe A.E., Song J., Wang G., Zhang P., Branham K.E., Swaroop A., Eberhart C.G., Zack D.J., Qian J., et al. Differential DNA methylation identified in the blood and retina of AMD patients. Epigenetics. 2015;10:698–707. doi: 10.1080/15592294.2015.1060388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo J., Li L., Hu D., Zhang X. LINC00612/miR-31-5p/Notch1 Axis Regulates Apoptosis, Inflammation, and Oxidative Stress in Human Pulmonary Microvascular Endothelial Cells Induced by Cigarette Smoke Extract. Int. J. Chronic Obstr. Pulm. Dis. 2020;15:2049–2060. doi: 10.2147/COPD.S255696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song X.-L., Zhang F.-F., Wang W.-J., Li X.-N., Dang Y., Li Y.-X., Yang Q., Shi M.-J., Qi X.-Y. LncRNA A2M-AS1 lessens the injury of cardiomyocytes caused by hypoxia and reoxygenation via regulating IL1R2. Genes Genom. 2020;42:1431–1441. doi: 10.1007/s13258-020-01007-6. [DOI] [PubMed] [Google Scholar]
- 35.Qiu F., Liang C.-L., Liu H., Zeng Y.-Q., Hou S., Huang S., Lai X., Dai Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget. 2017;8:268–284. doi: 10.18632/oncotarget.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dwyer J.B., McQuown S.C., Leslie F.M. The dynamic effects of nicotine on the developing brain. Pharmacol. Ther. 2009;122:125–139. doi: 10.1016/j.pharmthera.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosoff D.B., Smith G.D., Mehta N., Clarke T.-K., Lohoff F.W. Evaluating the relationship between alcohol consumption, tobacco use, and cardiovascular disease: A multivariable Mendelian randomization study. PLOS Med. 2020;17:e1003410. doi: 10.1371/journal.pmed.1003410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis S.J., Cherry N.M., Niven R.M., Barber P.V., Wilde K., Povey A.C. Cotinine levels and self-reported smoking status in patients attending a bronchoscopy clinic. Biomarkers. 2003;8:218–228. doi: 10.1080/1354750031000120125. [DOI] [PubMed] [Google Scholar]
- 39.Nagamatsu S.T., Pietrzak R.H., Xu K., Krystal J.H., Gelernter J., Montalvo-Ortiz J.L. Dissecting the Epigenomic Differences between Smoking and Nicotine Dependence in a Veteran Cohort. medRxiv. 2021 doi: 10.1111/adb.13259. [DOI] [PubMed] [Google Scholar]
- 40.Heinz A., Kiefer F., Smolka M.N., Endrass T., Beste C., Beck A., Liu S., Genauck A., Romund L., Banaschewski T., et al. Addiction Research Consortium: Losing and regaining control over drug intake (ReCoDe)—From trajectories to mechanisms and interventions. Addict. Biol. 2020;25:e12866. doi: 10.1111/adb.12866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data and summary statistics for all analyses are available on request.