

Abstract
Isoniazid (INH) remains the most safe and cost-effective drug for the treatment and prophylaxis of tuberculosis. The use of INH has increased over the past years, largely as a result of the coepidemic of human immunodeficiency virus infection. It is frequently given chronically to critically ill patients who are coprescribed multiple medications. The ability of INH to elevate the concentrations in plasma and/or toxicity of coadministered drugs, including those of narrow therapeutic range (e.g., phenytoin), has been documented in humans, but the mechanisms involved are not well understood. Using human liver microsomes (HLMs), we tested the inhibitory effect of INH on the activity of common drug-metabolizing human cytochrome P450 (CYP450) isoforms using isoform-specific substrate probe reactions. Incubation experiments were performed at a single concentration of each substrate probe at its Km value with a range of INH concentrations. CYP2C19 and CYP3A were inhibited potently by INH in a concentration-dependent manner. At 50 μM INH (∼6.86 μg/ml), the activities of these isoforms decreased by ∼40%. INH did not show significant inhibition (<10% at 50 μM) of other isoforms (CYP2C9, CYP1A2, and CYP2D6). To accurately estimate the inhibition constants (Ki values) for each isoform, four concentrations of INH were incubated across a range of five concentrations of specific substrate probes. The mean Ki values (± standard deviation) for the inhibition of CYP2C19 by INH in HLMs and recombinant human CYP2C19 were 25.4 ± 6.2 and 13 ± 2.4 μM, respectively. INH showed potent noncompetitive inhibition of CYP3A (Ki = 51.8 ± 2.5 to 75.9 ± 7.8 μM, depending on the substrate used). INH was a weak noncompetitive inhibitor of CYP2E1 (Ki = 110 ± 33 μM) and a competitive inhibitor of CYP2D6 (Ki = 126 ± 23 μM), but the mean Ki values for the inhibition of CYP2C9 and CYP1A2 were above 500 μM. Inhibition of one or both CYP2C19 and CYP3A isoforms is the likely mechanism by which INH slows the elimination of coadministered drugs, including phenytoin, carbamazepine, diazepam, triazolam, and primidone. Slow acetylators of INH may be at greater risk for adverse drug interactions, as the degree of inhibition was concentration dependent. These data provide a rational basis for understanding drug interaction with INH and predict that other drugs metabolized by these two enzymes may also interact.
Isoniazid (INH), introduced in the 1950s, continues to be one of the most safe and cost-effective agents used to treat tuberculosis (9). Because of the increasing incidence of tuberculosis that has resulted from the coepidemic of human immunodeficiency virus infection (32), the use of INH in the prevention (9, 17) and treatment (9) of tuberculosis is on the rise. INH is often given chronically to critically ill patients who are on multiple medications to treat tuberculosis and AIDS-related mycobacterial disease (18, 45), raising the potential for adverse drug-drug interactions.
In humans, INH has been found to decrease the clearance of several drugs, including phenytoin (see, e.g., references 3, 24, and 30), carbamazepine (54, 58, 59), diazepam (36), triazolam (37), vincristine (7), primidone (48), and acetaminophen (10, 35), sufficiently to require a reduction of the dose or discontinuation of INH. Upon consideration of the primary metabolic pathways of the drugs affected and the enzymes catalyzing them, many of these drug-drug interactions appear to be attributable to pharmacokinetic changes that can be understood in terms of alterations of multiple hepatic drug metabolic pathways catalyzed by the cytochrome P450 (CYP450) system (2). In fact, the ability of INH to inhibit the hepatic CYP450 system in vitro in rat liver microsomes has been described (25, 34). In animals (6), it has been shown that INH effectively inhibits para-hydroxylation of phenytoin in vivo. Despite the strong link between INH drug interactions and inhibition of the CYP450 system and despite its widespread use for more than 4 decades, our knowledge is incomplete with respect to which specific CYP450 isoforms are inhibited, making prediction of drug interactions with INH difficult. The only isoform that has been studied in detail in human and animal models is CYP2E1, for which INH has been reported to have a biphasic effect (induction and inhibition) (8). This property of INH may explain the increased risk of hepatotoxicity of coadministered compounds (e.g., ethanol and acetaminophen) (10, 33, 35), but it is unlikely to explain the clinically documented interactions with other drugs, as most of them appear to be catalyzed by CYP isoforms other than CYP2E1 (e.g., phenytoin by CYP2C9 and CYP2C19 and carbamazepine by CYP3A and CYP2C8 [2, 28]).
In order to enable physicians to improve predictions about which drugs might interact with INH, the present study was undertaken to evaluate the inhibitory potency of INH on six common human drug-metabolizing CYP450 isoforms in vitro.
MATERIALS AND METHODS
Chemicals.
Isoniazid, phenytoin, phenacetin, acetaminophen, chlorpropamide, midazolam, dextromethorphan, chlorzoxazone, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, NADP, and the disodium salt of EDTA were purchased from Sigma Chemical Co. (St. Louis, Mo.). S-Mephenytoin, 4-hydroxy-S-mephenytoin, 6-hydroxychlorzoxazone, 4-hydroxymidazolam, and 4-methylhydroxytolbutamide were purchased from Ultrafine Chemicals (Manchester, United Kingdom). Levallorphan was obtained from U.S. Pharmacopeia Convention (Rockville, Md.). Flurbiprofen and 4′-hydroxyflurbiprofen were provided by Timothy Tracy, University of West Virginia School of Pharmacy. Dextrorphan and 3-methoxymorphinan were purchased from Hoffman-La Roche, Inc. (Nutley, N.J.). N-(4-Hydroxyphenyl)butamide was kindly provided by John Strong (Division of Clinical Pharmacology, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Rockville, Md.). Omeprazole was a generous gift from Tommy Andersson (Clinical Pharmacology, Astra Haessle AB, Moelndal, Sweden). Other reagents were of high-pressure liquid chromatography (HPLC) grade.
HLMs and recombinant human CYP450s.
The human liver microsomes used were prepared from human liver tissue that was medically unsuitable for liver transplantation and frozen at −80°C within 3 h of cross-clamp time. The characteristics of liver donors, procedure for preparation of microsomal fractions, and their CYP450 contents have been described in detail elsewhere (19). The microsomal pellets were resuspended in a reaction buffer (0.1 M Na+ and K+ phosphate, 1.0 mM EDTA, 5.0 mM MgCl2, pH 7.4) to a protein concentration of 10 mg/ml (stock) and were kept at −80°C until used. Protein concentrations were determined as described by Pollard et al. (41). Detailed protocols for the measurement of each CYP isoform activity using isoform-specific substrate reaction probes and their apparent kinetic parameters (Km and Vmax values) have been described in our earlier work (11, 23, 47).
Baculovirus-insect cell-expressed human CYP2C19 and CYP2C9 (with reductase) were purchased from Gentest Corporation (Woburn, Mass.) and stored at −80°C. Protein concentrations and CYP450 contents were as supplied by the manufacturer.
Inhibition of CYP450 by INH.
The inhibitory effects of INH on the activities of CYP1A2, -2C19, -2C8, -2D6, -2E1, and -3A were tested in HLMs using probes selective for each isoform. The reaction probes used were as follows: phenacetin O-deethylation for CYP1A2 (49); tolbutamide 4-methylhydroxylation (43) and flurbiprofen 4′-hydroxylation (53) for CYP2C9; S-mephenytoin 4′-hydroxylation (60) and omeprazole 4′-hydroxylation (20) for CYP2C19; dextromethorphan O-demethylation for CYP2D6 (5); midazolam 4-hydroxylation (51), omeprazole sulfone formation (20), and dextromethorphan N-demethylation (16) for CYP3A; and chlorzoxazone 6-hydroxylation for CYP2E1 (40). Using incubation conditions specific to each isoform that were linear for time, substrate, and protein concentrations as detailed in our previous publications (11, 23, 47), isoform-specific substrate probes were incubated in duplicate at 37°C with HLMs and an NADPH-generating system in the absence (control) or presence of various concentrations (0 to 250 μM) of INH. Unless otherwise specified, a 5-min preincubation was carried out before the reaction was initiated by adding HLMs. For each inhibition study, preliminary experiments were carried out by incubating a single isoform-specific substrate concentration around its Km with a range of INH concentrations (0 to 250 μM) or a single INH concentration (50 μM). The 50 μM (∼6.86-μg/ml) INH concentration used in these experiments was included to represent clinically relevant INH concentrations in plasma during therapy (14). Simulation of the preliminary data thus obtained was then used to generate appropriate substrate and INH concentrations for the determination of exact inhibition constants (Ki values) for each isoform. Dixon plots for the inhibition by INH of each substrate probe were performed with two HLMs.
After termination of the incubation reactions with appropriate reagents, samples were centrifuged and injected into an HPLC system either directly or after reconstitution with mobile phase. The concentrations of the metabolites and internal standards were measured by HPLC with UV or fluorescent detection specific for each assay. Rates of production of each metabolite from the substrate probes were quantified by using the ratio of the area under the curve for the metabolite to the area under the curve for each internal standard using an appropriate standard curve. The rates of metabolite formation from substrate probes in the presence of INH were compared with those for controls in which the inhibitor was replaced with vehicle.
HPLC.
Instruments used for HPLC were controlled by a Waters (Milford, Mass.) Millennium 2010 Chromatography Manager and included a Waters model 510 or 600 HPLC pump, Waters 710B or 717 autosampler, Waters 490 or 484 UV detector, and Spectrovision FD-300 dual monochromator fluorescence detector (Groton Technology Inc., Concord, Mass.). Full chromatographic conditions for each assay have been described elsewhere (11, 23, 47).
Enzyme assays.
We measured the activities of CYP1A2, -2C19, -2C9, -2D6, -2E1, and -3A, using specific marker reaction probes. Incubation conditions and HPLC methods for measurement of each activity of each CYP are validated and have been routinely used in our laboratory, and details have been published in a number of our earlier papers (11, 23, 47). The INH final concentrations used in the microsomal incubation ranged from 5 to 250 μM. Concentrations of substrates were as follows: dextromethorphan, 10 to 60 μM; S-mephenytoin, 10 to 40 μM; omeprazole, 25 to 150 μM; tolbutamide, 15 to 75 μM; flurbiprofen, 12 to 25 μM; chlorzoxazone, 5 to 25 μM; phenacetin, 25 to 50 μM; and midazolam, 10 to 60 μM.
CYP2C19-catalyzed S-mephenytoin 4-hydroxylation and omeprazole 5-hydroxylation were measured as described earlier (11, 23). The CYP2D6-catalyzed dextromethorphan O-demethylation to dextrorphan was assayed as described by Broly et al. (5). The measurement of CYP3A activity was done using dextromethorphan N-demethylation (16), midazolam 4-hydroxylation (20), and omeprazole sulfone formation (20). The CYP1A2-catalyzed O-deethylation of phenacetin to acetaminophen was measured by a method described by Tassaneeyakul et al. (49) with modifications (23). The effect of INH on the rate of CYP2E1-catalyzed chlorzoxazone 6-hydroxylation was evaluated by the method described by Peter et al. (40). CYP2C9-mediated 4-methylhydroxylation of tolbutamide was determined in both HLMs and recombinant human CYP2C9 and CYP2C19 as described by Relling et al. (43) with slight modifications (23). The incubation method for tolbutamide 4-methylhydroxylation was the same as those for CYP2D6 and CYP3A (dextromethorphan assay) described above except that the incubation time was 1 h in the case of tolbutamide.
Tolbutamide has been widely used to probe the activity of CYP2C9 in vivo and in vitro (31), but recent reports implicate CYP2C19 in its metabolism (1, 26). Because of the known interaction of INH with phenytoin (3, 24, 30), a substrate of CYP2C9 and CYP2C19 (1, 28, 31), we characterized in more detail the inhibitory effect of INH on CYP2C19 and CYP2C9. We used three approaches. First, we tested whether tolbutamide is metabolized by recombinant human CYP2C19 and CYP2C9 isoforms and whether INH has similar inhibitory effects in HLMs and in recombinant enzymes. The second protocol involved use of flurbiprofen 4′-hydroxylation as an additional probe (53) to define the degree of inhibition of CYP2C9 in HLMs by INH. The assay of this reaction was performed as described by Tracy et al. (53). Lastly, to test for any mechanism-based inhibition of CYP2C9 by INH, HLMs were preincubated with an NADPH-generating system without or with 100 and 250 μM INH for 0, 5, 15, and 30 min. After initiation of the reaction by addition of tolbutamide (50 μM), the incubation mixture was further incubated for 1 h.
Data analysis.
The reaction velocity of each substrate probe in the presence of INH was expressed as the percentage of the control velocity with no INH present. Approximate initial inhibition constants (Ki values) were calculated from experiments that were conducted using a single substrate and multiple INH concentrations with the use of the equation, assuming competitive inhibition
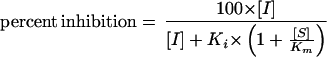 |
where I is INH concentration, Ki is the inhibition constant, S is the substrate concentration, and Km is the substrate concentration at half of the maximum velocity (Vmax) of the reaction. Estimates for kinetic parameters from this analysis were used to generate computer-simulated optimal concentrations of substrate and INH for the determination of Dixon plots. The inhibition data from Dixon plots were fitted to appropriate nonlinear regression models of enzyme inhibition, and accurate Ki values were calculated (WinNonlin version 1.5; Scientific Consulting, Inc., Apex, N.C.). The Ki values obtained from visual inspection of Dixon or secondary Lineweaver-Burk plots served as initial estimates for this determination. An appropriate model and mechanism of inhibition were decided graphically and from parameters of the model (e.g., dispersion of residuals and standard errors of the parameter estimates).
RESULTS
We tested the inhibitory effect of INH on the activities of common drug-metabolizing human CYP450 isoforms, as assessed by isoform-specific substrate probe reactions. First, preliminary experiments that involved incubation of a single concentration of each substrate reaction probe around its respective Km value with multiple INH concentrations (0 to 250 μM) in HLMs were conducted. The data summarized in Fig. 1 demonstrate that INH at a single (50 μM) (Fig. 1A) or multiple (0 to 250 μM) (Fig. 1B) concentrations consistently inhibited the activities of CYP2C19 and CYP3A relative to those of the other isoforms tested. The data in Fig. 1 were then used to simulate appropriate ranges of substrate and inhibitor concentrations to construct Dixon plots for the inhibition of each isoform by INH in HLMs or recombinant CYP450s for precise estimation of inhibition constants (Ki values).
FIG. 1.

Inhibition of CYP450 isoforms by INH at 50 μM (A) and 0 to 250 μM (B) in HLMs. The activity of each isoform was measured by isoform-specific substrate reaction probes at approximately their respective Km values: 60 μM for phenacetin O-deethylation (CYP1A2); 25 μM for dextromethorphan (CYP2D6); 25 μM for midazolam 4-hydroxylation (CYP3A); 50 μM for omeprazole (CYP2C19); 50 μM for tolbutamide (CYP2C9); and 25 μM for chlorzoxazone (CYP2E1). Data represent averages of duplicates.
CYP2C19 activity.
As shown in Fig. 1A, INH (50 μM) inhibited the activity of CYP2C19-catalyzed omeprazole 5-hydroxylation by about 40%. The degree of inhibition was dependent on the INH concentration used (34% at 25 μM, 42% at 50 μM, and 70% at 250 μM INH) (Fig. 1B). This inhibition of CYP2C19 by INH was similar in HLMs and recombinant human CYP2C19 for both substrates (omeprazole and S-mephenytoin) tested (Fig. 2). Representative Dixon plots for the inhibition of CYP2C19 in HLMs and recombinant CYP2C19 are demonstrated in Fig. 3A and B, respectively. Clearly, INH is a potent competitive inhibitor, with mean Ki values of 25 and 13 μM, respectively (Table 1).
FIG. 2.

Inhibitory effect of INH (0 to 250 μM) on CYP2C19-catalyzed omeprazole 5-hydroxylation in HLMs and recombinant human CYP2C19. Data represent averages of duplicate determinations.
FIG. 3.

Dixon plots for the inhibition of CYP2C19-catalyzed omeprazole 5-hydroxylation by INH (0 to 250 μM) in HLMs (A) and S-mephenytoin 4-hydroxylation by INH (0 to 100 μM) in recombinant human CYP2C19 (B). Each point represents the average of duplicate determinations.
TABLE 1.
Inhibitory potency of INH for CYP450 isoformsa
| Substrate reaction probe | CYP450 isoform | Ki, μM (SD) | Mechanism of inhibition |
|---|---|---|---|
| S-Mephenytoin 4-hydroxylation | CYP2C19 | 13 (2.4) | Competitive |
| Omeprazole 5-hydroxylation | CYP2C19 | 25.4 (6.20) | Competitive |
| Omeprazole sulfone formation | CYP3A | 51.8 (2.5) | Noncompetitive |
| Midazolam 4-hydroxylation | CYP3A | 75.9 (7.8) | Noncompetitive |
| Chlorzoxazone 6-hydroxylation | CYP2E1 | 110 (33) | Noncompetitive |
| Dextromethorphan O-demethylation | CYP2D6 | 126 (23) | Competitive |
| Phenacetin O-deethylation | CYP1A2 | >1,000 | |
| Tolbutamide 4-methylhydroxylationb | CYP2C9 | 102 (17) | Noncompetitive |
| Flurbiprofen 4′-hydroxylation | CYP2C9 | >500 | Noncompetitive |
| Dextromethorphan N-demethylation | CYP3A | 474 (103) | Competitive |
To calculate the inhibition constants (Ki values) for each isoform, data obtained from three HLMs to construct Dixon plots were fitted to an appropriate nonlinear regression enzyme inhibition model. The mechanism of inhibition was decided graphically and from the enzyme inhibition models (see Materials and Methods).
In recombinant CYP2C9.
CYP3A activity.
The inhibition of CYP3A by INH was first assessed using a single concentration of midazolam, at approximately the Km, as a substrate probe in HLMs. As shown in Fig. 1, INH potently inhibited the activity of CYP3A in a concentration-dependent manner. At therapeutically relevant concentrations of INH (e.g., 50 μM [14, 52, 57]), over 40% of CYP3A activity was inhibited. Since the degree of inhibition of CYP3A may be substrate dependent, we tested the ability of INH to inhibit CYP3A in HLMs using three different substrate probe reactions: midazolam 4-hydroxylation, omeprazole sulfone formation, and dextromethorphan N-demethylation. Representative Dixon plots for the inhibition of CYP3A-catalyzed reactions are shown in Fig. 4, and the corresponding Ki values derived from these data are listed in Table 1. INH showed comparable noncompetitive inhibition of omeprazole sulfone formation (Ki, ∼52 μM) and midazolam 4-hydroxylation (Ki, ∼76 μM), but it was six to nine times weaker as a competitive inhibitor of dextromethorphan N-demethylation.
FIG. 4.

Dixon plots for the inhibition of CYP3A-catalyzed omeprazole sulfone formation (A), midazolam 4-hydroxylation (B), and dextromethorphan N-demethylation (C) by INH in HLMs. The INH concentrations used were 0 to 100 μM (A and B) and 0 to 250 μM (C). Each point represents the average of duplicate determinations.
CYP2C9 activity.
As demonstrated in Fig. 1, inhibition of tolbutamide 4-methylhydroxylation by INH in HLMs was minimal. Even at supratherapeutic concentrations of INH, the inhibition did not exceed 15%. As revealed in Fig. 5A, INH had little effect on flurbiprofen 4-hydroxylation (another probe of CYP2C9) or tolbutamide 4-methylhydroxylation in HLMs. The Dixon plot for the inhibition of tolbutamide 4-methylhydroxylation by INH (Fig. 5B) was constructed using recombinant human CYP2C9. The Ki value derived from this Dixon plot (>100 μM) was four and eight times higher than that obtained for the inhibition of CYP2C19 in HLMs and recombinant human CYP2C19, respectively (Table 1). To explore the potential for mechanism-based inhibition, we preincubated INH with microsomes from human livers in the presence of an NADPH-generating system before adding tolbutamide as a substrate probe. There was no additional effect of INH on the catalytic activity of CYP2C9 due to preincubation (Fig. 5C).
FIG. 5.

Inhibitory effect of INH on CYP2C9-catalyzed 4-methylhydroxylation and flurbiprofen 4-hydroxylation. (A) Inhibition of CYP2C9 (with tolbutamide [TB] [20 and 40 μM] and flurbiprofen [FB] [13 and 25 μM] as substrates) by INH (0 to 250 μM) in HLMs; (B) Dixon plots for the inhibition by INH of CYP2C9 (with tolbutamide) in recombinant human CYP2C9; (C) inhibitory effect of INH (0 to 250 μM) on CYP2C9 (with tolbutamide [50 μM]) with preincubation (PI) or without preincubation in HLMs (see Materials and Methods for details). Data are averages for duplicate incubations.
CYP2E1 activity.
INH is a moderate inhibitor of CYP2E1-catalyzed chlorzoxazone 6-hydroxylation in HLMs (Fig. 1 and 6). At therapeutically relevant INH concentrations, the degree of inhibition was <20%. Since this finding contradicted our initial expectations, we performed a positive control inhibition experiment using the same HLMs with diethyldithiocarbamate, a known CYP2E1 inhibitor. As revealed in Fig. 6A, diethyldithiocarbamate was indeed a strong inhibitor of CYP2E1-catalyzed chlorzoxazone 6-hydroxylation relative to inhibition by INH. The Dixon plot for the inhibition of CYP2E1 by INH (Fig. 6B) showed that INH is a competitive inhibitor in HLMs, with a Ki value of 110 μM.
FIG. 6.

Inhibition of CYP2E1-mediated chlorzoxazone 6-hydroxylation by INH. (A) Percent inhibition of CYP2E1 activity (with chlorzoxazone [25 μM]) by INH and diethyldithiocarbamate (DEDTC) as a positive control in HLMs; (B) Representative Dixon plots for the inhibition of CYP2E1 activity by INH (25 to 250 μM) in HLMs. Data represent averages of duplicates.
CYP2D6 and CYP1A2 activities.
INH showed weak inhibition of CYP2D6 (Fig. 1), with a Ki value of >100 μM (Table 1). The representative Dixon plot for the inhibition of CYP2D6 by INH (Fig. 7A) and analysis of the parameters of the enzyme inhibition model suggest that the type of inhibition is competitive. The ability of INH to inhibit CYP1A2-mediated phenacetin O-deethylation was negligible when phenacetin at a single concentration (50 μM) was incubated with INH at multiple concentrations. At the highest INH concentration tested (250 μM), the formation of acetaminophen was inhibited by only ∼16%. Since we did not find any significant inhibition of CYP1A2 at two concentrations of phenacetin (Fig. 7B), we did not attempt to conduct further analysis. The Ki value given in Table 1 is an approximate estimate from these data using the equation in the “Data analysis” section above.
FIG. 7.

Inhibition of CYP2D6-catalyzed dextromethorphan O-demethylation and CYP1A2-catalyzed O-deethylation by INH in HLMs. (A) Dixon plots for the inhibition of CYP2D6 (INH, 0 to 250 μM); (B) percent inhibition of CYP1A2 (with phenacetin [25 and 50 μM] as a substrate) (INH, 0 to 250 μM). Each point represents the average of duplicate determinations.
DISCUSSION
The ability of INH to cause adverse drug interactions with a number of drugs whose clearance is dependent on the CYP450 system (Table 2) (2, 46, 57) has been well documented, but the specific isoforms inhibited by INH are not fully understood. In this study we provide the first comprehensive assessment of inhibition of six drug-metabolizing CYP450 isoforms by INH and provide the scientific basis with which to explain documented drug interactions with INH and predict new ones. This is particularly timely because a number of newly developed drugs for the treatment of HIV and opportunistic infections as well as other diseases (e.g., depression and drug-resistant tuberculosis) are likely to be coprescribed with INH.
TABLE 2.
Drug interactions potentially involving inhibition of CYP450s by INH
| Drug affected | CYP450(s) involved | Type of study and subjects | Pharmacokinetic changes observeda | Side effects observed | INH dose (mg/day) | Reference(s) |
|---|---|---|---|---|---|---|
| Phenytoin | CYP2C9, CYP2C19 | Several studies and case reports | Increased levels in serum, more in SA | Ataxia, hyperreflexia, nystagmus, tremor | 300–600 | 3, 24, 30 |
| Carbamazepeine | CYP3A, CYP2C8 | Case report | Increased levels in plasma | Drowsiness, ataxia, nystagmus, headache, vomiting | 200–300 | 54, 58, 59 |
| Diazepam | CYP3A, CYP2C19 | Randomized, crossover design | No change in Vd, increased t1/2 (33%), decreased CL (26%), more effect in SA | Sedation, respiratory depression | 180 | 36 |
| Triazolam | CYP3A, CYP2C19 | Placebo controlled | Increased t1/2 (32%) | None reported | 180 | 37 |
| Primidone | Unknown | Case study, female | Increased t1/2 (60%), increased Css (83%), decreased plasma PB and PEMA levels | None reported | 300 | 48 |
| Ethosuximide | CYP3A | Case report | Increased level in serum | Increased psychotic behavior | 300 | 55 |
| Vitamin D | Vitamin D hydroxylase | Parallel | Decreased 1,25(OH)2D, increased PTH, decreased Ca2+ and PO4 | None reported | 300 | 4 |
| Antipyrine | CYP1A2, CYP3A, CYP2C19 | Parallel | Decreased CL (18%) | None reported | 300 | 4 |
| Chlorzoxazoneb | CYP2E1 | Placebo controlled | Increased or decreased CL, more effect in SA than in FA | None reported | 300 | 27 |
| Acetaminophenb | CYP2E1, CYP1A2 | Placebo controlled | Increased or decreased CL, more effect in SA than in FA | Increased hepatotoxicity? | 300 | 61 |
| Isoflurane and enflurane | CYP2E1 | Placebo controlled | Increased defluorination, more effect in SA than in FA | Decreased effectiveness | 300 | 46 |
| Theophylline | CYP1A2, CYP3A, CYP2E1 | Placebo controlled, case report | No change or decrease in CL (16 to 23%) | Seizure, nausea and vomiting, palpitation | 300–600 | 46 |
| Vincristine | CYP3A | Case report | NA | Tingling of fingers, weakness in limbs | 300 | 7 |
| Warfarin | CYP2C9 | Single case report | NA | Increased PT and bleeding | 600 | 46 |
PB, phenobarbital; PEMA, phenylethylmalonamide; Vd, volume of distribution; t1/2, terminal elimination half-life; CL, clearance; SA, slow acetylators; FA, fast acetylators; NA, not available; PTH, prothrombin time; 1,25(OH)2D, 1,25-hydroxy-vitamin D.
Changes in CL depend on the time after INH administration.
Our data demonstrate that INH is a strong inhibitor of CYP2C19, which is genetically a polymorphic enzyme that is involved in the metabolism of several clinically important drugs, including phenytoin, omeprazole, antidepressants (e.g., citalopram), proguanil, and nelfinavir (15, 29). If our inhibition data can predict in vivo situations, we expect that INH will alter the pharmacokinetics of drugs that are substrates of CYP2C19. The clinical relevance of INH-induced inhibition of CYP2C19 can be best illustrated by examining the INH-phenytoin clinical interaction documented in the literature. Phenytoin toxicity associated with elevated concentrations in plasma is the most clearly and frequently documented interaction described so far for patients taking INH concomitantly (Table 2). In animal experiments, it has been shown that INH inhibits parahydroxylation of phenytoin (6, 8, 25). Although CYP2C9 has been suggested to be a major enzyme responsible for the oxidation of phenytoin (31), evidence that also implicates CYP2C19 as an important determinant of phenytoin pharmacokinetics, particularly at higher concentrations is evolving (1, 28). We have shown recently that the antiplatelet drug ticlopidine, a preferential inhibitor of CYP2C19 (with little effect on CYP2C9) in vivo and in vitro (13, 22, 50), significantly impairs the elimination of phenytoin in humans (12). Other drugs that are potent CYP2C19 (but not CYP2C9) inhibitors, including omeprazole, felbamate, and topiramate, have been reported to increase the concentrations in plasma and/or toxicity of phenytoin (28). Furthermore, we have no evidence from the literature that INH inhibits the elimination of CYP2C9 substrates other than phenytoin. One case of increased warfarin (at 10 mg/day) toxicity (increased prothrombin time, hematuria, and bleeding gums) caused by INH was described in 1977 (46). This adverse drug interaction occurred after the patient inadvertently took 10 extra doses of INH (600 mg/day) but did not occur at a regular therapeutic dose of INH (300 mg/day). The fact that there is only one case report of high-dose INH-warfarin drug interaction compared to the frequent reports of phenytoin interaction despite the wider use of INH and warfarin (a CYP2C9 substrate) (31) suggests that therapeutic doses of INH have little effect on CYP2C9 substrate drugs. These and our in vitro data lead us to believe that inhibition of CYP2C19-mediated metabolism of phenytoin by INH provides a clear explanation for the increased risk of clinical phenytoin toxicity during INH coadministration. This means that CYP2C19 is an important enzyme in phenytoin metabolism, particularly when the activity of the alternative pathway (CYP2C9) is low. Although our data have particular relevance to elimination of phenytoin owing to its saturable pharmacokinetic properties (28), we would also expect INH to alter the therapeutic and toxic actions of other CYP2C19 substrate drugs, including omeprazole, diazepam, citalopram, nelfinavir, and proguanil (15, 29).
The inhibition of CYP3A activity by INH occurred at concentrations close to the therapeutic range and may partly explain some of the drug interactions reported in the literature with INH, which include interactions with carbamazepine, ethosuximide, and vincristine (Table 2). In view of the major role of CYP3A in drug metabolism in the liver and intestine and because of the likelihood of coadministration of INH with substrates of CYP3A (e.g., protease inhibitors), our finding may have considerable clinical importance. While the inhibition was relatively potent when omeprazole sulfone formation or midazolam hydroxylation was used as probe reactions, it is important to note that inhibition of dextromethorphan N-demethylation was weak and competitive in nature. This reemphasizes the fact that inhibition of CYP3A-mediated metabolism is substrate dependent, or it could indicate that dextromethorphan N-demethylation is a nonspecific probe of CYP3A (56).
The weak inhibition by INH of CYP2E1-mediated chlorzoxazone 6-hydroxylation that we observed in this study does not concur with in vivo studies. Studies in vivo have shown unequivocally that INH has a dual effect on the activity of CYP2E1, i.e., inhibition and induction (8, 27, 38). It decreases the clearances of substrates of CYP2E1 (chlorzoxazone, acetaminophen, ethanol, and halogenated anesthetic agents [Table 2]) through competitive binding to the active site of the enzyme when an adequate concentration of INH in plasma is available. It functions as an inducer of CYP2E1, probably through ligand stabilization, once the drug is stopped and the concentration in plasma reaches undetectable levels (8). Therefore, the pharmacokinetic consequence of interaction with CYP2E1 could be a decrease, no change, or an increase of clearance of a substrate probe, depending on the dose of INH and the time between the last ingestion of INH and the ingestion of the substrate probe (8). There are a number of possible explanations for the lack of a strong effect of INH on the activity of CYP2E1 in vitro.
First, it is possible that INH is a mechanism-based inhibitor of CYP2E1. In fact, CYP2E1 is implicated in INH-induced hepatotoxicity (44), suggesting that INH or a metabolite may be a substrate of CYP2E1 and that this metabolite, by being a substrate of this isoform, may be a competitive inhibitor of CYP2E1. Preincubations of INH with HLMs and an NADPH-generating system before the addition of the substrate are unlikely to reveal a mechanism-based interaction, because the primary metabolism of INH in humans does not involve CYP450s (57). Alternatively, the effect of known synthetic metabolites of INH on the activity of CYP2E1 may provide information on the role of specific metabolites, but we have little knowledge on the therapeutic concentrations of most INH metabolites. Second, INH may be quickly depleted compared to chlorzoxazone in our in vitro incubation, leading to incomplete inhibition. We did not measure the time course of INH disappearance in the HLM incubation mixtures. Further study is required to clarify all of these possibilities, as this may provide the bases not only for inhibition of CYP2E1 but also for the mechanisms by which INH produces hepatotoxicity or increases the risk of hepatotoxicity of other coadministered drugs.
We have shown that INH is an inhibitor of two important CYP450 isoforms in vitro, and these data provide the basis with which to identify potential drug interactions during INH therapy. INH-phenytoin interactions documented in the literature appear to be mediated through inhibition of CYP2C19. Inhibition of CYP2C19 and/or CYP3A is the likely mechanism by which INH slows the elimination of several coadministered drugs (Table 2), including carbamazepine (CYP3A), diazepam (CYP3A and CYP2C19), triazolam (CYP3A), and vincristine (CYP3A). INH also inhibits the metabolism of drugs such as primidone and ethosuximide (Table 2), but because of the lack of knowledge on the isoforms catalyzing these drugs, it is difficult to assign a mechanism for these interactions.
Besides the effect of INH on xenobiotics, there is a possibility that chronic INH administration may significantly inhibit endobiotic metabolism by CYP2C19, CYP3A, or other enzymes and modify their biochemical actions. Thus, INH has been reported to inhibit the metabolism of vitamin D and cortisol in humans (4) and leukotriene oxidation in animals (39).
In humans, INH undergoes N acetylation by N-acetyltransferase, and genetic polymorphic expression of this enzyme is the main cause for high interindividual variability in INH pharmacokinetics (52, 57). Two distinct subgroups (slow and fast acetylators) that differ in their ability to metabolize INH have been identified among humans. Unlike for other polymorphisms (e.g., CYP2D6 and CYP2C19), the proportion of slow acetylators is very high (up to 80%) in certain populations (42). Our data indicate that INH inhibits the activity of CYP2C19 and CYP3A in a concentration-dependent manner and that slow acetylators would therefore experience greater changes in the elimination of other drugs. Certainly, ethnic background, acetylation status, and dose should be taken in to account when monitoring INH-drug interactions.
Second, INH-drug interaction is likely to be greater with drugs whose metabolic pathways involve CYP2C19 and CYP3A or both. These include HIV type 1 protease inhibitors (e.g., nelfinavir), proton pump inhibitors (e.g., omeprazole), benzodiazepines (e.g., diazepam), and proguanil with drugs of a low therapeutic range (e.g., phenytoin and carbamazepine) or nonlinear pharmacokinetics (e.g., phenytoin). Lastly, these interactions are likely to be significant when INH is used as a single agent for the prophylaxis of tuberculosis or when it is administered in the absence of rifampin or other enzyme inducers. This fact is best illustrated by the small INH-phenytoin interaction in patients who were also on rifampin (21). In this setting, a dramatic reduction of serum phenytoin concentration was observed, as the induction effect of rifampin far outweighs the inhibitory effect of INH. It appears particularly important to monitor INH-drug interactions when the inducer is discontinued while the patient continues to take INH with a drug whose metabolism is susceptible to induction of CYP2C19 or CYP3A. The lack of a significant effect of INH on the activity of CYP2E1 in vitro does not concur with in vivo data, suggesting involvement of INH metabolites, and has implications for its own metabolism and hepatotoxicity.
ACKNOWLEDGMENT
This work was supported in part by a Center for Education and Therapeutics grant from the Agency for Health Care Policy and Research, Washington, D.C.
REFERENCES
- 1.Bajpai M, Roskos L K, Shen D D, Levy R H. Roles of cytochrome P4502C9 and cytochrome P4502C19 in the stereoselective metabolism of phenytoin to its major metabolite. Drug Metab Dispos. 1996;24:1401–1403. [PubMed] [Google Scholar]
- 2.Bertz R J, Granneman G R. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet. 1997;32:210–258. doi: 10.2165/00003088-199732030-00004. [DOI] [PubMed] [Google Scholar]
- 3.Brennan R W, Dehejia H, Kutt H, Verebely K, McDowell F. Diphenylhydantoin intoxication attendant to slow inactivation of isoniazid. Neurology. 1970;20:687–693. doi: 10.1212/wnl.20.7.687. [DOI] [PubMed] [Google Scholar]
- 4.Brodie M J, Boobis A R, Hillyard C J, Abeyasekera G, MacIntyre I, Park B K. Effect of isoniazid on vitamin D metabolism and hepatic monooxygenase activity. Clin Pharmacol Ther. 1981;30:363–367. doi: 10.1038/clpt.1981.173. [DOI] [PubMed] [Google Scholar]
- 5.Broly F, Libersa C, Lhermitte M, Bechtel P, Dupuis B. Effect of quinidine on the dextromethorphan O-demethylase activity of microsomal fractions from human liver. Br J Clin Pharmacol. 1989;28:29–36. doi: 10.1111/j.1365-2125.1989.tb03502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buttar H S, Wong L T, Moffatt J H. Effect of isoniazid on the metabolism of 14C-diphenylhydantoin in rats. Arch Int Pharmacodyn Ther. 1978;235:9–18. [PubMed] [Google Scholar]
- 7.Chan J D. Pharmacokinetic drug interactions of vinca alkaloids: summary of case reports. Pharmacotherapy. 1998;18:1304–1307. [PubMed] [Google Scholar]
- 8.Chien J Y, Thummel K E, Slattery J T. Pharmacokinetic consequences of induction of CYP2E1 by ligand stabilization. Drug Metab Dispos. 1997;25:1165–1175. [PubMed] [Google Scholar]
- 9.Cohn D L. Treatment and prevention of tuberculosis in HIV-infected persons. Infect Dis Clin N Am. 1994;8:399–412. [PubMed] [Google Scholar]
- 10.Crippin J S. Acetaminophen hepatotoxicity: potentiation by isoniazid. Am J Gastroenterol. 1993;88:590–592. [PubMed] [Google Scholar]
- 11.Desta Z, Kerbusch T, Soukhova N, Richard E, Ko J W, Flockhart D A. Identification and characterization of human cytochrome P450 isoforms interacting with pimozide. J Pharmacol Exp Ther. 1998;285:428–437. [PubMed] [Google Scholar]
- 12.Donahue S, Flockhart D A, Abernethy D R. Ticlopidine inhibits phenytoin clearance. Clin Pharmacol Ther. 1999;66:563–568. doi: 10.1053/cp.1999.v66.103277001. [DOI] [PubMed] [Google Scholar]
- 13.Donahue S R, Flockhart D A, Abernethy D R, Ko J-W. Ticlopidine inhibition of phenytoin metabolism mediated by potent inhibition of CYP2C19. Clin Pharmacol Ther. 1997;62:572–577. doi: 10.1016/S0009-9236(97)90054-0. [DOI] [PubMed] [Google Scholar]
- 14.Evans D A P, Manley K A, McKusick V A. Genetic control of isoniazid metabolism in man. Br Med J. 1960;2:485–491. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flockhart D A. Drug interactions and the cytochrome P450 system. The role of cytochrome P450 2C19. Clin Pharmacokinet. 1995;29(Suppl. 1):45S–52S. doi: 10.2165/00003088-199500291-00008. [DOI] [PubMed] [Google Scholar]
- 16.Gorski J C, Jones D R, Wrighton S A, Hall S D. Characterization of dextromethorphan N-demethylation by human liver microsomes. Contribution of the cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol. 1994;48:173–182. doi: 10.1016/0006-2952(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 17.Gourevitch M N, Hartel D, Selwyn P A, Schoenbaum E E, Klein R. Effectiveness of isoniazid chemoprophylaxis for HIV-infected drug users at high risk for active tuberculosis. AIDS. 1999;13:2069–2074. doi: 10.1097/00002030-199910220-00009. [DOI] [PubMed] [Google Scholar]
- 18.Grange J M, Winstanley P A, Davies P D. Clinically significant drug interactions with antituberculosis agents. Drug Saf. 1994;11:242–251. doi: 10.2165/00002018-199411040-00003. [DOI] [PubMed] [Google Scholar]
- 19.Harris J W, Rahman A, Kim B R, Guengerich F P, Collins J M. Metabolism of taxol by human hepatic microsomes and liver slices: participation of cytochrome P450 3A4 and an unknown P450 enzyme. Cancer Res. 1994;54:4026–4035. [PubMed] [Google Scholar]
- 20.Karam W G, Goldstein J A, Lasker J M, Ghanayem B I. Human CYP2C19 is a major omeprazole 5-hydroxylase, as demonstrated with recombinant cytochrome P450 enzymes. Drug Metab Dispos. 1996;24:1081–1087. [PubMed] [Google Scholar]
- 21.Kay L, Kampmann J P, Svendsen T L, Vergman B, Hansen J E, Skovsted L, Kristensen M. Influence of rifampicin and isoniazid on the kinetics of phenytoin. Br J Clin Pharmacol. 1985;20:323–326. doi: 10.1111/j.1365-2125.1985.tb05071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko J-W, Desta Z, Soukhova N, Tracy T, Flockhart D A. In vitro inhibition of the cytochrome P450 (CYP450) system by the antiplatelet drug ticlopidine: potent effect on CYP2C19 and CYP2D6. Br J Clin Pharmacol. 2000;49:343–351. doi: 10.1046/j.1365-2125.2000.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ko J-W, Soukhova N, Thacker D, Chen P, Flockhart D A. Evaluation of omeprazole and lansoprazole as inhibitors of cytochrome P450 isoforms. Drug Metab Dispos. 1997;25:853–862. [PubMed] [Google Scholar]
- 24.Kutt H, Brennan R, Dehejia H, Verebely K. Diphenylhydantoin intoxication. A complication of isoniazid therapy. Am Rev Respir Dis. 1970;101:377–384. doi: 10.1164/arrd.1970.101.3.377. [DOI] [PubMed] [Google Scholar]
- 25.Kutt H, Verebely K. Metabolism of diphenylhydantoin by rat liver microsomes. I. Characteristics of the reaction. Biochem Pharmacol. 1970;19:675–686. doi: 10.1016/0006-2952(70)90230-3. [DOI] [PubMed] [Google Scholar]
- 26.Lasker J M, Wester M R, Aramsombatdee E, Raucy J L. Characterization of CYP2C19 and CYP2C9 from human liver: respective roles in microsomal tolbutamide, S-mephenytoin, and omeprazole hydroxylations. Arch Biochem Biophys. 1998;353:16–28. doi: 10.1006/abbi.1998.0615. [DOI] [PubMed] [Google Scholar]
- 27.Leclercq I, Desager J P, Horsmans Y. Inhibition of chlorzoxazone metabolism, a clinical probe for CYP2E1, by a single ingestion of watercress. Clin Pharmacol Ther. 1998;64:144–149. doi: 10.1016/S0009-9236(98)90147-3. [DOI] [PubMed] [Google Scholar]
- 28.Levy R H. Cytochrome P450 isoenzymes and antiepileptic drug interactions. Epilepsia. 1995;36(Suppl. 5):S8–S13. doi: 10.1111/j.1528-1157.1995.tb06007.x. [DOI] [PubMed] [Google Scholar]
- 29.Lillibridge J H, Lee C A, Pithavala Y K, Sandoval T M, Wu E Y, Zhang K E, et al. The role of CYP2c19 in the formation of nelfinavir hydroxy-butylamide (M8): in vitro/in vivo correlation. ISSX Proc. 1998;13:55. [Google Scholar]
- 30.Miller R R, Porter J, Greenblatt D J. Clinical importance of the interaction of phenytoin and isoniazid: a report from the Boston Collaborative Drug Surveillance Program. Chest. 1979;75:356–358. doi: 10.1378/chest.75.3.356. [DOI] [PubMed] [Google Scholar]
- 31.Miners J O, Birkett D J. Cytochrome P4502C9: an enzyme of major importance in drug metabolism. Br J Clin Pharmacol. 1998;45:525–538. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore M, McCray E, Onorato I M. Cross-matching TB and AIDS registries: TB patients with HIV co-infection, United States, 1993–1994. Public Health Rep. 1999;114:269–277. doi: 10.1093/phr/114.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morimoto M, Hagbjork A L, Nanji A A, Ingelman-Sundberg M, Lindros K O, Fu P C, et al. Role of cytochrome P4502E1 in alcoholic liver disease pathogenesis. Alcohol. 1993;10:459–464. doi: 10.1016/0741-8329(93)90065-v. [DOI] [PubMed] [Google Scholar]
- 34.Muakkassah S F, Bidlack W R, Yang W C. Mechanism of the inhibitory action of isoniazid on microsomal drug metabolism. Biochem Pharmacol. 1981;30:1651–1658. doi: 10.1016/0006-2952(81)90393-2. [DOI] [PubMed] [Google Scholar]
- 35.Nolan C M, Sandblom R E, Thummel K E, Slattery J T, Nelson S D. Hepatotoxicity associated with acetaminophen usage in patients receiving multiple drug therapy for tuberculosis. Chest. 1994;105:408–411. doi: 10.1378/chest.105.2.408. [DOI] [PubMed] [Google Scholar]
- 36.Ochs H R, Greenblatt D J, Roberts G-M, Dengler H J. Diazepam interaction with antituberculosis drugs. Clin Pharmacol Ther. 1981;29:671–678. doi: 10.1038/clpt.1981.94. [DOI] [PubMed] [Google Scholar]
- 37.Ochs H R, Greenblatt D J, Knuchel M. Differential effect of isoniazid on triazolam and oxazepam conjugation. Br J Clin Pharmacol. 1983;16:743–746. doi: 10.1111/j.1365-2125.1983.tb02256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Shea D, Kim R B, Wilkinson G R. Modulation of CYP2E1 activity by isoniazid in rapid and slow N-acetylators. Br J Clin Pharmacol. 1997;43:99–103. doi: 10.1111/j.1365-2125.1997.tb00039.x. [DOI] [PubMed] [Google Scholar]
- 39.Parthe S, Hagmann W. Inhibition of leukotriene omega-oxidation by isonicotinic acid hydrazide (isoniazid) Eur J Biochem. 1990;187:119–124. doi: 10.1111/j.1432-1033.1990.tb15284.x. [DOI] [PubMed] [Google Scholar]
- 40.Peter R, Bocker R, Beaune P H, Iwaski M, Guengerich F P, Xang C S. Hydroxylation of chlorzoxazone as a specific probe of cytochrome P450 IIE1. Chem Res Toxicol. 1990;3:566–573. doi: 10.1021/tx00018a012. [DOI] [PubMed] [Google Scholar]
- 41.Pollard H B, Menard R, Brabdt H A, Pazolzs C J, Creutz C E, Ramu A. Application of Bradford's assay to adrenal gland subcellular fractions. Anal Biochem. 1978;86:761–763. doi: 10.1016/0003-2697(78)90805-9. [DOI] [PubMed] [Google Scholar]
- 42.Relling M V. Polymorphic drug metabolism. Clin Pharm. 1989;8:852–863. [PubMed] [Google Scholar]
- 43.Relling M V, Aoyama T, Gonzalez F J, Meyer U A. Tolbutamide and mephenytoin hydroxylation by human cytochrome P450s in the CYP2C subfamily. J Pharmacol Exp Ther. 1990;252:442–447. [PubMed] [Google Scholar]
- 44.Sarich T C, Adams S P, Petricca G, Wright J M. Inhibition of isoniazid-induced hepatotoxicity in rabbits by pretreatment with an amidase inhibitor. J Pharmacol Exp Ther. 1999;289:695–702. [PubMed] [Google Scholar]
- 45.Schluger N W. Issues in the treatment of active tuberculosis in human immunodeficiency virus-infected patients. Clin Infect Dis. 1999;28:130–135. doi: 10.1086/515088. [DOI] [PubMed] [Google Scholar]
- 46.Self T H, Chrisman C R, Baciewicz A M, Bronze M S. Isoniazid drug and food interactions. Am J Med Sci. 1999;317:304–311. doi: 10.1097/00000441-199905000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Shin J G, Soukhova N, Flockhart D A. Effect of antipsychotic drugs on human liver cytochrome P-450 (CYP) isoforms in vitro: preferential inhibition of CYP2D6. Drug Metab Dispos. 1999;27:1078–1084. [PubMed] [Google Scholar]
- 48.Sutton G, Kupferberg H J. Isoniazid as an inhibitor of primidone metabolism. Neurology. 1975;25:1179–1181. doi: 10.1212/wnl.25.12.1179. [DOI] [PubMed] [Google Scholar]
- 49.Tassaneeyakul W, Birkett D J, Veronese M E, McManus M E, Tukey R H, Quattrochi L C, et al. Specificity of substrate and inhibitor probes for human cytochrome P450 1A1 and 1A2. J Pharmacol Exp Ther. 1993;265:401–407. [PubMed] [Google Scholar]
- 50.Tateishi T, Kumai T, Watanabe M, Nakura H, Tanaka M, Kobayashi S. Ticlopidine decreases the in vitro activity of CYP2C19 as measured by omeprazole metabolism. Br J Clin Pharmacol. 1999;47:454–457. doi: 10.1046/j.1365-2125.1999.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thummel K E, Shen D D, Podoll T D, Kunze K L, Trager W F, Hartwell P S, Raisys V A, et al. Use of midazolam as a human cytochrome P450 3A probe. I. In vitro-in vivo correlations in liver transplant patients. J Pharmacol Exp Ther. 1994;271:549–556. [PubMed] [Google Scholar]
- 52.Tiitinen H. Isoniazid and ethionamide serum levels and inactivation in Finnish subjects. Scand J Resp Dis. 1969;50:110–124. [PubMed] [Google Scholar]
- 53.Tracy T S, Marra C, Wrighton S A, Gonzalez F J, Korzekwa K R. Studies of flurbiprofen 4′ hydroxylation: additional evidence suggesting the sole involvement of cytochrome P450 2C9. Biochem Pharmacol. 1996;52:1305–1309. doi: 10.1016/0006-2952(96)00501-1. [DOI] [PubMed] [Google Scholar]
- 54.Valsalan V C, Cooper G L. Carbamazepine intoxication caused by interaction with isoniazid. Br Med J. 1982;285:261–262. doi: 10.1136/bmj.285.6337.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Wieringen A, Vrijlandt C M. Ethosuximide intoxication caused by interaction with isoniazid. Neurology. 1983;33:1227–1228. doi: 10.1212/wnl.33.9.1227. [DOI] [PubMed] [Google Scholar]
- 56.von Moltke L L, Greenblatt D J, Grassi J M, Granda B W, Venkatakrishnan K, Schmider J, et al. Multiple human cytochromes contribute to biotransformation of dextromethorphan in-vitro: role of CYP2C9, CYP2C19, CYP2D6, and CYP3A. J Pharm Pharmacol. 1998;50:997–1004. doi: 10.1111/j.2042-7158.1998.tb06914.x. [DOI] [PubMed] [Google Scholar]
- 57.Weber W W, Hein D W. Clinical pharmacokinetics of isoniazid. Clin Pharmacokinet. 1979;4:401–422. doi: 10.2165/00003088-197904060-00001. [DOI] [PubMed] [Google Scholar]
- 58.Wright J M, Stokes E F, Sweeney V P. Isoniazid-induced carbamazepine toxicity and vice versa: a double drug interaction. N Engl J Med. 1982;307:1325–1327. doi: 10.1056/NEJM198211183072107. [DOI] [PubMed] [Google Scholar]
- 59.Wright J M. Carbamazepine-isoniazid interaction. Pediatrics. 1983;71:139. [PubMed] [Google Scholar]
- 60.Wrighton S A, Stevens J C, Becker G W, VandenBranden M. Isolation and characterization of human liver cytochrome P450 2C19: correlation between 2C19 and S-mephenytoin 4′-hydroxylation. Arch Biochem Biophys. 1993;306:240–245. doi: 10.1006/abbi.1993.1506. [DOI] [PubMed] [Google Scholar]
- 61.Zand R, Nelson S D, Slattery J T, Thummel K E, Kalhorn T F, Adams S P, Wright J M. Inhibition and induction of cytochrome P4502E1-catalyzed oxidation by isoniazid in humans. Clin Pharmacol Ther. 1993;54:142–149. doi: 10.1038/clpt.1993.125. [DOI] [PubMed] [Google Scholar]