

Abstract
The muscle-intrinsic clock machinery is required for the maintenance of muscle growth, remodeling and function. Our previous studies demonstrated that the essential transcription activator of the molecular clock feed-back loop, Brain and Muscle Arnt-Like 1(Bmal1), plays a critical role in myogenic progenitor behavior to promote and regenerative myogenesis. Using genetic approaches targeting Bmal1 in the DMDmdx (mdx) dystrophic mouse model, here we report that the loss of Bmall function significantly accelerated dystrophic disease progression. In contrast to the mild dystrophic changes in mdx mice, the genetic loss-of-function of Bmal1 aggravated muscle damage in this dystrophic disease background, as indicated by persistently elevated creatine kinase levels, increased injury area and reduced muscle grip strength. Mechanistic studies revealed that markedly impaired myogenic progenitor proliferation and myogenic response underlie the defective new myofiber formation in the chronic dystrophic milieu. Taken together, our study identified the function of pro-myogenic clock gene Bmal1 in protecting against dystrophic damage, suggesting the potential for augmenting Bmal1 function to ameliorate dystrophic or degenerative muscle diseases.
Keywords: circadian clock, muscular dystrophy, muscle injury, regeneration, regenerative myogenesis
Graphical Abstract
Introduction
Repeated contraction-induced degeneration-regeneration cycles, a hallmark of dystrophic diseases such as Duchene muscular dystrophy (DMD), lead to progressive loss of muscle mass and function [1, 2]. Effective means to augment regenerative repair to promote muscle growth and improve muscle mass could ameliorate dystrophic pathophysiology and potentially benefit patients with DMD [3–5]. Therefore, better understanding of molecular events that promote regenerative capacity could offer novel options to preserves functional muscle mass and ameliorate muscular dystrophy.
The circadian clock that drives 24-hour rhythms in physiology comprise of a transcriptional-translational feedback loop that ultimately coordinates physiological processes with external timing cues [6]. In addition to the central clock residing in the suprachiasmatic nuclei entrained directly to light input, nearly all tissue and cell types in our body possess cell-autonomous clock rhythms that are normally driven by central clock. Skeletal muscle possesses intrinsic clock activity, which can be shifted by timing of exercise besides being driven by the central clock output. The clock transcription negative feedback loop underlies the ~24 hour rhythms in behavior and physiology. Brain and Muscle Arnt-Like 1 (Bmal1), a muscle-enriched clock transcription activator, is essential for driving molecular clock transcription together with its heterodimer partner, Circadian Locomotor Output Cycles Kaput (CLOCK). Transcription activation of the repressors of the clock loop, including Periods (Per) and Cryptochromes (Cry), leads to subsequent inhibition of transcription to form the feedback loop. Various posttranscriptional and posttranslational mechanisms further contribute to establish the circadian oscillation cycles.
Skeletal muscle possesses cell-autonomous molecular clocks that are critical for temporal control of muscle function by entraining locomotor activity [7–9]. Recent studies indicated that the muscle-intrinsic clock is required for maintenance of muscle mass, growth and metabolic regulation [10, 11]. This temporal element is involved in muscle mass regulation through sarcomeric structural organization and myogenic progenitor behaviors [12–15]. We demonstrated previously that the essential clock activator Bmal1 promotes regenerative myogenesis and prevents sarcopenia [14, 16–18]. More recently, our study revealed that Rev-erbα, a ligand-dependent nuclear receptor and transcriptional repressor of Bmal1 in the core clock loop[19, 20], suppresses myogenic progenitor proliferation and differentiation [21]. In addition, Cry2 was found to be critical for circadian regulation of myogenic differentiation [22]. The circadian clock circuit may exert temporal control of myogenic progression, and thus represent a potential novel pathway for muscle-wasting disease intervention.
Utilizing the mdx mice as a DMD disease model, we generated double-null mutant of Bmal1 with the DMD disease background to directly test whether Bmal1 protects against dystrophic muscle injury. Our study revealed that the loss of Bmal1 significantly exacerbates dystrophic pathology in the mdx mice, demonstrating that Bmal1 maintains muscle regenerative capacity in muscular dystrophy.
Materials and Methods
Animals
Animals were maintained in the Beckman Research Institute of City of Hope vivarium under a constant 12:12 light dark cycle, with lights on at 7:00 AM (ZT0). All experiments were approved by the IACUC committee of the Beckman Research Institute of City of Hope. Bmal1-null (Arntltm1Bra, Stock No: 009100) and mdx mutant mice (Dmdmdx, Stock No: 001801) were originally purchased from the Jackson Laboratory. Mice were crossed for three generations to obtain double homozygote mutants.
RNA extraction and quantitative reverse-transcriptase PCR analysis
Trizol (Invitrogen) and RNeasy miniprep kits (Qiagen) were used to isolate total RNA from snap-frozen muscle tissues and cells, respectively. cDNA was generated using q-Script cDNA Supermix kit (Quanta Biosciences) and quantitative PCR was performed using a Roche 480 Light Cycler with Perfecta SYBR Green Supermix (Quanta Biosciences). Relative expression levels of target genes were determined using the comparative Ct method and normalized to 36B4 as internal control. Primer sequence are listed in Suppl. Table S1.
Hematoxylin and eosin histology
Muscles were collected and fixed with 10% neutral-buffered formalin for 72 hours, washed in 70% alcohol prior to embedding in paraffin. 10μm paraffin-embedded muscle cross-sections were processed for deparaffinization using xylene, rehydrated in ethanol, and stained with hematoxylin and eosin. Muscle cross section area (CSA) was calculated by outlining the myofiber boundary to measure area size. Total of four representative 10X fields from each muscle were counted and plotted for muscle diameter size distribution.
Immunoblot analysis
20-40 μg of total protein were resolved on SDS-PAGE gel, transferred to nitrocellulose membrane, and used for immunoblotting. Immunoblots were developed by chemiluminescence kit (Pierce Biotechnology). Source and dilution information of primary antibodies are included as Suppl. Table S2. Appropriate specific secondary antibodies were used at a dilution of 1:3000.
Skeletal muscle histology and immunofluorescence staining
Muscles were snap frozen in liquid-nitrogen cooled isopentane and embedded in OCT, as described previously [23]. 10μm thick cryosections at the middle region of TA or gastrocnemius muscles, or diaphragm were fixed using 4% paraformaldehyde for 10 mins and permeabilized by 0.2% Triton X-100 in PBS. Endogenous IgG was blocked using non-specific mouse IgG (M.O.M Immunodetection kit, VECTOR Laboratories) for 30 mins, and further blocking using blocking reagent from M.O.M kit was carried out prior to primary antibody incubation at 4°C overnight. The primary antibodies used for immunofluorescence staining and the dilutions are listed (Suppl. Table S2). Images were captured using Echo Microscope at indicated magnifications.
Evans Blue Dye staining
Evans blue dye (EBD) was diluted in PBS to a final concentration of 10 mg/ml, sterilized and given via intraperitoneal injection at a dose of 50 μl of diluted EBD per 10 g of body weight. Frozen TA, gastrocnemius, quadricep and diaphragm muscle samples were collected 24 hours after injection. Frozen sections were further stained using laminin antibody for immunofluorescence imagining. Number of EBD-positive fibers were counted using 4X field images to stitch the entire cross section, and divided by the total number of myofibers for analysis of the proportion of necrotic myofibers.
Masson’s Trichrome staining
Masson’s Trichrome staining was performed using the Masson’s Trichrome Stain Kit according to manufacturer’s instructions (Polyscience Inc.). Briefly, frozen sections were dehydrated in ethanol, and then sequentially processed in Bouin’s fixative at 60°C for 1 hour, Weigert’s Iron hematoxylin for 10 minutes, Biebrich Scarlet-Acid Fuchsin for 5 minutes, Phosphotungstic/phosphomolybdic acid for 10 minutes, followed by washing in acetic acid.
Serum creatine kinase assay
Retro-orbital blood was collected in mice at 8 to 30 weeks of age as indicated in the specific groups and serum was frozen at −80°C until analysis. Total serum creatine kinase (CK) activity were analyzed using commercial Creatine Kinase Activity Assay Kit (Sigma) according to the manufacturer’s instructions.
Grip strength test
A digital grip strength meter with an axial force transducer (Harvard Apparatus) was used to measure forelimb grip strength with a metal bar attachment. Briefly, the mouse was lifted by the tail and allowed to grasp the bar with front paws kept at height of the bar. Tail was gently pulled back the mouse at a constant slow speed until the grasp is broken. Pulling for the grip strength were repeated five times by one minute apart for each mouse. The peak strength value was recorded and normalized by body weight.
Statistical analysis
Data was expressed as Mean ± SEM. The differences between groups were determined by two-sided, unpaired Student’s t test, or two-way ANOVA between multiple groups. Minimum of three biological replicates were used to perform statistical analysis. P values less than 0.05 were considered statistically significant.
Results
Dynamic regulation of Bmal1 during mdx disease progression and loss of clock regulation in Bmal1/Dmd mutants
Dystrophic disease pathology in mdx mice displays a progressive phenotype with a severe degenerative phase starting at 7-8 weeks of age, followed by regenerative repair and a repetitive injury-induced chronic fibrotic response in older animals [2, 24]. We first examined the regulation of the Bmal1 in mdx mice at 8, 20 and over 26 weeks of age. Bmal1 protein was persistently induced in mdx mice as compared to normal wild-type controls (WT) at the same age. This Bmal1 induction could be due to the myogenic injury stimuli in the dystrophic muscle as we reported in muscle regeneration previously[18], although its levels decline with age in both groups (Fig. 1A). To determine the role of Bmal1 in the pathogenesis of muscle dystrophy, we generated mice with global genetic targeting of Bmal1 (BmKO) in the dystrophic disease background by crossing with the DMD mdx mutant mice1. Bmal1 protein expression was absent in distinct muscle types in the BmKO/mdx mice, including quadricep (Quad), gastrocnemius (GN) and Tibialis Anterior (TA) that we examined (Fig. 1B). The loss of Bmal1 and its effects on core clock gene expression was determined by RT-qPCR analysis in BmKO/mdx, using gastrocnemius as a representative lower limb muscle (Fig. 1C). Interestingly, Bmal1 mRNA level was significantly down-regulated in mdx mice as compared to that of age-matched wild-type controls at 20 weeks of age. As expected, a direct Bmal1 target gene Nrld1 (Rev-erbα), was nearly absent in the BmKO/mdx mice. Expression of additional core clock genes in these mice further demonstrated the loss of the clock circuit regulation, as indicated by marked reductions of Period 2 (Per2) and D-element Binding Protein (Dbp). We also examined clock gene expression in diaphragm, the muscle exhibiting most severe dystrophic damage in mdx mice (Fig. 1F). A similar pattern of attenuated core clock gene expression, including Rev-erbα and Dbp, was observed in BmKO/mdx as compared to that of the age-matched mdx mutants.
Figure 1.
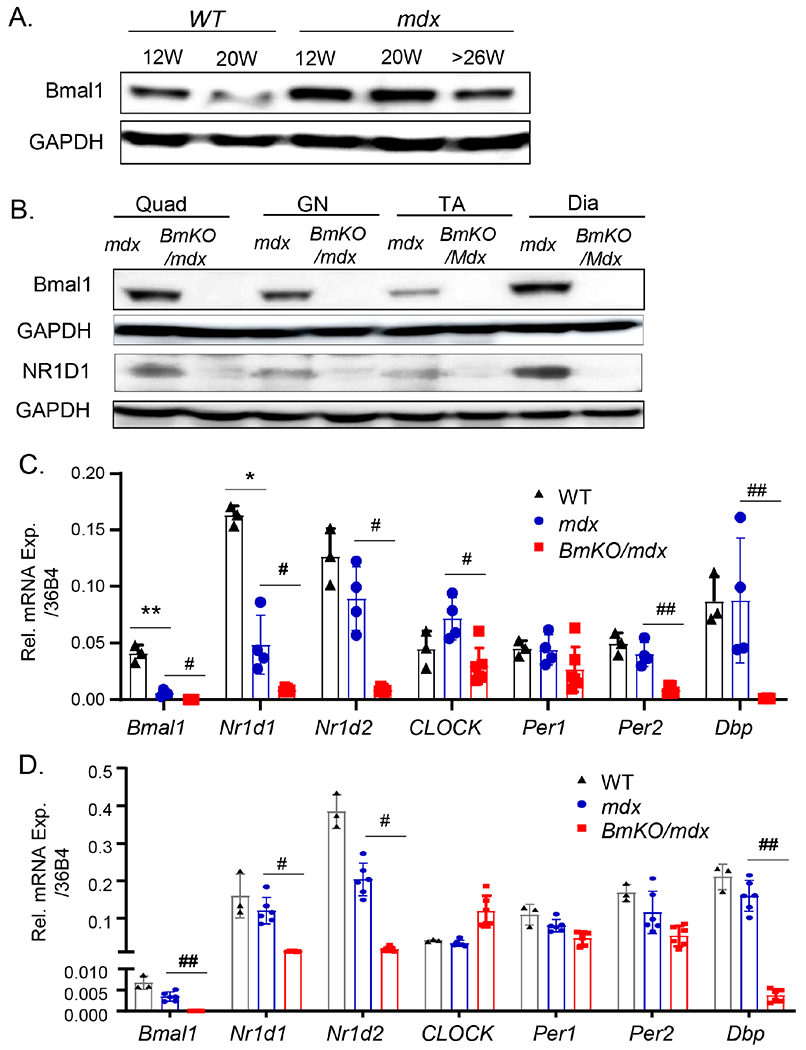
Dynamic regulation of Bmal1 during mdx disease progression. (A) Immunoblot analysis of Bmal1 protein in gastrocnemius muscle of WT and mdx mutants at different ages as indicated. Pooled samples of 4-6 mice for each group were used. (B) Western validation of loss of Bmal1, together with Bmal1 target gene NR1D1, in quadriceps (Quad), gastrocnemius (GN), TA, and diaphragm (Dia) muscles of mdx and BmKO/mdx DKO mice. Pooled samples of four mice each group were used. (C, D) RT-qPCR analysis of clock gene expression in representative muscle groups, TA (C) and diaphragm (D), of 12-week-old male wild-type (n=3), mdx mutant (n=4) and BmKO/mdx mice (n=6). Relative mRNA expression was expressed as mean ±SEM with normalization to 36B4. *, ** P≤0.05 or 0.01 vs. WT; #, ##: P≤0.05 or 0.01 BmKO/mdx vs. mdx mice.
Loss of Bmal1 in dystrophic mdx mutant reduces muscle mass, function and exacerbates muscle damage
At 20 weeks of age, a significant reduction of total body weight of ~8.5 grams in the BmKO/mdx double mutants was observed as compared to that of the mdx at a comparable age (Fig. 2A). The lower body weight was largely attributed to loss of muscle mass, as indicated by a ~9.5% lower lean mass indicated by NMR analysis (Fig. 2B). In younger adult mice at 12 weeks of age, analysis of dissected individual muscle groups revealed significantly reduced weights of diaphragm and gastrocnemius in BmKO/mdx than that of mdx controls, whereas soleus displayed a tendency toward lower weight but not the TA muscle (Fig. 2C). To test whether reduced muscle mass in BmKO/mdx mice attenuates muscle function, we preformed grip strength test in 20-24 week-old mice and found that strength performance was significantly compromised to nearly 50% of that of the mdx controls (Fig. 2D). Based on the pro-myogenic functions of Bmal1 and its role in muscle structural maintenance, we postulated that the reduced muscle mass and functional deficits in BmKO/mdx mice could be due to exacerbated muscle damage. We thus examined the level of plasma creatine kinase activity as a global assessment of the extent of muscle injury due to loss of membrane integrity-related leaking of muscle enzymes. As shown in Fig. 2E as compared to age-matched mdx mice, a striking nearly 2-3 fold increase in creatine kinase activities was observed across different ages of BmKO/mdx mice examined, indicating markedly elevated muscle damage than the baseline dystrophic injury in mdx muscle. Based on this finding, we performed immunofluorescence staining using anti-mouse IgG to evaluate sarcolemma integrity and necrotic areas indictive of muscle damage. As shown in Fig. 2F, in transverse stitched whole cross sections of TA, there was a substantial increase in IgG infiltration area with necrotic myofibers that comprised approximately 30% of the TA in 20-week-old BmKO/mdx mice as compared to mdx. A surprising finding was the significant drop-off of survival of the BmKO/mdx mutant cohort as compared to mdx mice over the course of 40 weeks (Fig. 2G). This reduction of survival began in mice as young as 3 weeks old, which accelerated between 5-10 weeks of age, possibly due to the muscle injury and compromised function. Approximately 40% of BmKO/mdx mice died prematurely at younger than 8 weeks of age, as compared to that of largely no premature death in mdx mice. Together, these results indicated a severe dystrophic consequence of muscle wasting with impaired function and aggravated muscle damage due to loss of Bmal1 in the mdx dystrophin-null background.
Figure 2.
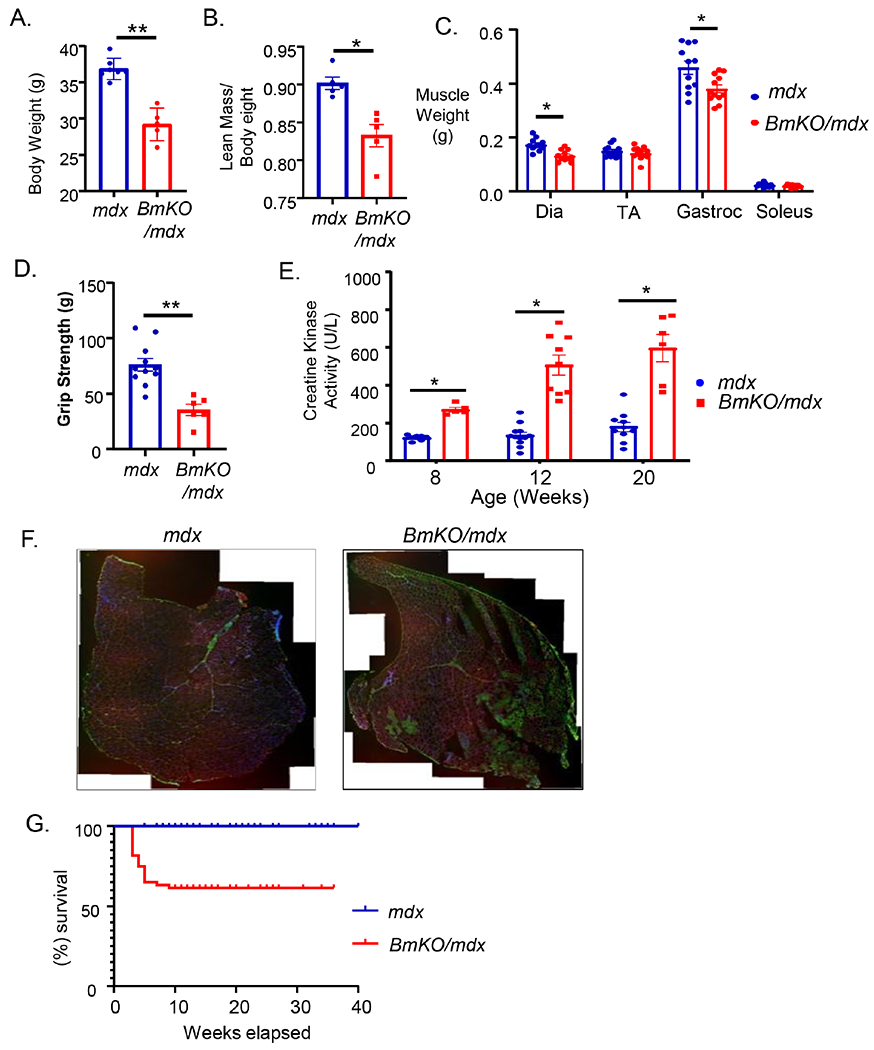
Loss of Bmal1 in mdx mice impairs muscle mass and attenuates muscle strength. (A-C) reduced total body weight (A), lean muscle mass (B) as measured by NMR analysis in 20-week-old mdx mutant and BmKO/mdx mice (n=5/group). (C) Wet tissue weight of dissected individual muscle groups in 20-week-old mdx and BmKO/mdx mice (n=11/group). (D) Muscle function as measured by forearm grip strength in 20-week-old mdx mutant (n=11) and BmKO/mdx mice (n=6). (E) Bmal1-deficiency in mdx mice progressively increases creatine kinase activity in mice of 8, 12 and 20 weeks of age (n=5-10/group). (F) Representative images of IgG immunofluorescence staining in complete stitched TA muscle crossed sections of mdx and BmKO/mdx mice. (Blue: nuclear DAPI, Green: IgG staining of necrotic myofibers, Red: laminin). (G) Kaplan-Meyer survival curve analyses of lifespan in BmKO/mdx mice (n=77) as compared with mdx mice (n=60).
Severe muscle injury in Bmal1/mdx double mutant diaphragm
Due to the continuous respiratory work-load, diaphragm is the most severely affected muscle in dystrophic disease and the pathology is progressive with age[1, 24]. We thus first examined the extent of muscle damage in the diaphragm in the BmKO/mdx mice at 8, 12 and 20 weeks of age. As compared to age-matched mdx controls, H&E histology revealed marked reductions of diaphragm thickness along with less regenerative areas in BmKO/mdx mice of 8 and 12 weeks of age, but not at 20 weeks (Fig. 3A). Quantitative measurement of diaphragm thickness confirmed ~30-50% reductions of thickness in 8 and 12-weeks-old BmKO/mdx mice (Fig. 3B), consistent with the lower diaphragm weight observed. The comparable diaphragm thickness at 20 weeks of age could be due to compensatory hypertrophy with disease progression that was not evident in younger mice. Consistent with the reduced diaphragm thickness and weight, central nucleated myofibers in 8 and 12 weeks-old BmKO/mdx diaphragms, indicative of neo-myofiber formation due to injury, were markedly attenuated as compared to mdx mutants, while the abundance of new myofibers did not differ at 20 weeks of age (Fig. 3C). We further analyzed myofiber size distribution by measuring cross-section areas in diaphragm at distinct ages. Interestingly, although only a tendency toward smaller myofiber distribution was observed at 8-weeks-old BmKO/mdx mice (Fig. 3D), a significant left shift of fiber size distribution was evident in Bmal1-deficient mdx diaphragm at older ages of 12 and 20 weeks (Fig. 3E & 3F). Given that the small fibers analyzed on the size distribution represent newly regenerated myofibers, this result suggests potentially heightened regenerative response due to aggravated muscle injury in BmKO/mdx mice. Thus, Bmal1 deficiency in a DMD disease background led to pronounced dystrophic changes in diaphragm.
Figure 3.
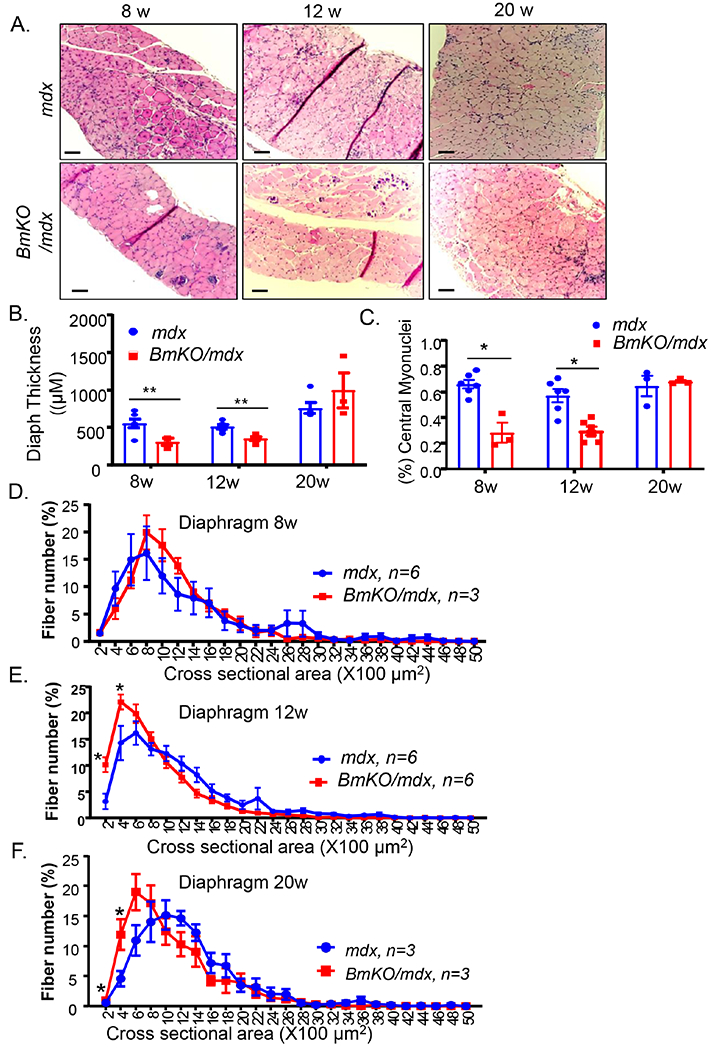
Reduced diaphragm thickness and central nuclei-containing myofibers in BmKO/mdx mice. (A) Representative images of H/E staining of diaphragm from 8, 12 and 20 weeks-old mdx and BmKO/mdx mice at 4X magnification. Scale bar: 200 μm. (B, C) Diaphragm thickness (B) and the percentage of central nuclei-containing myofibers (C) as measured from H/E histology cross sections in 8, 12 and 20-weeks-old mdx and BmKO/mdx mice (n=4-6/group). (D-F) Distribution of diaphragm cross-section area as percentage of total myofibers in 8 (D), 12 (E) and 20-weeks-old (F) mdx and BmKO/mdx mice (n=4-6/group).
To determine the extent of muscle damage in BmKO/mdx diaphragm, we used Evan’s blue dye (EBD) and IgG immunostaining that specifically identified the necrotic injury area. As indicated in Fig. 4A, areas of overlapping Evans blue dye and IgG staining were markedly expanded in BmKO/mdx, as compared to the limited injury seen in mdx controls at 12 weeks of age. Furthermore, we examined fibrosis as the chronic consequence of muscle damage in these mice. Masson’s Trichrome staining revealed markedly elevated collagen deposition in 20-week-old BmKO/mdx diaphragm as compared to mdx (Fig. 4B), possibly secondary to chronic muscle damage. We next tested whether a compromised myogenic response in BmKO/mdx accounts for the increased muscle damage. Analysis of total eMyHC protein level in diaphragm muscle extract revealed a tendency of reduction of new myofiber formation as compared to controls (Fig. 4C & 4D). Interestingly, the expression levels of key myogenic factors, Myod1, Myogenin and Myf5 in BmKO/mdx mice, were similar to the mdx mutants (Fig. 4E). However, the key cell cycle inhibitor p21 was markedly elevated, suggesting potentially impaired cell proliferation response in the Bmal1/mdx double mutant (Fig. 4F).
Figure 4.
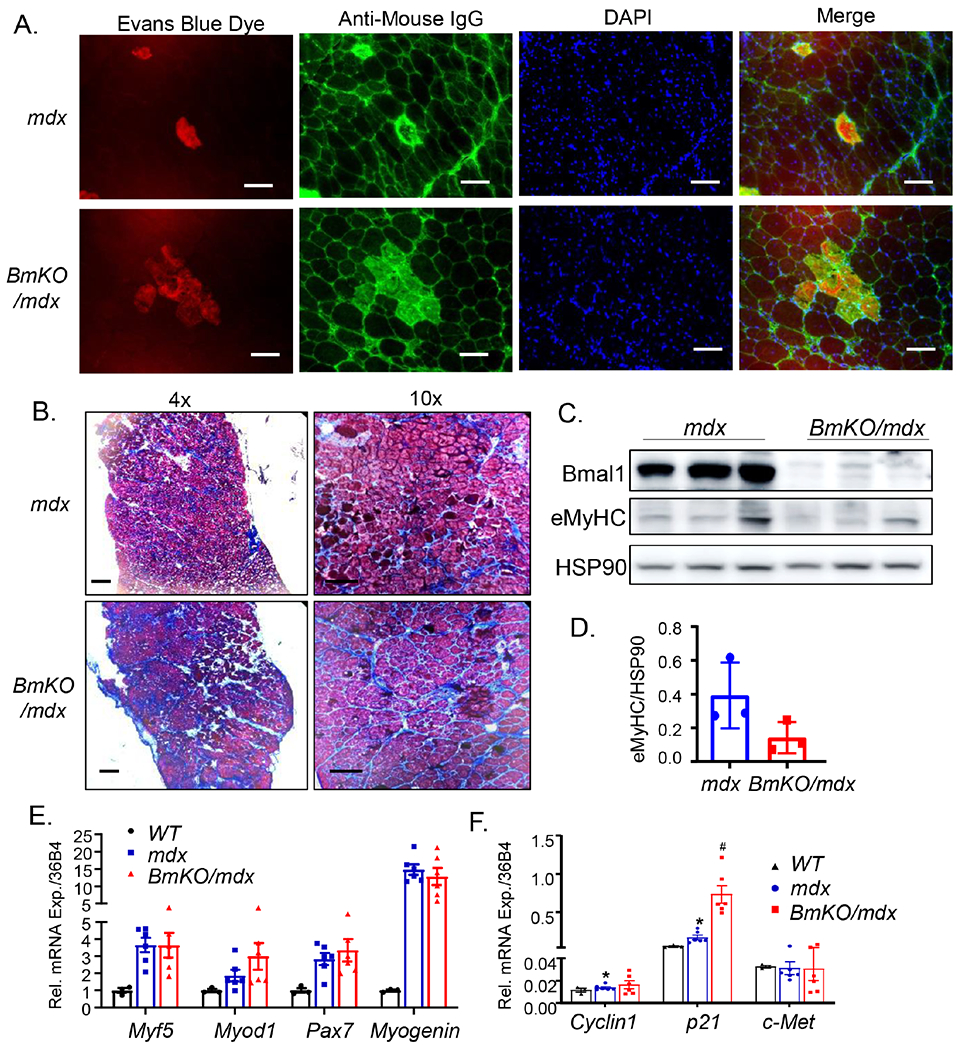
Bmal1 deficiency exacerbates muscle damage in mdx mice diaphragm. (A) Representative images of Evans blue dye staining (Red) and IgG infiltration (Green) to identify damage areas in 12-weeks-old mdx and BmKO/mdx mice. (B) Representative images of Mason’s trichrome staining for identification of fibrosis in 20-week-old mdx and BmKO/mdx mice at 4X and 10X magnification. Scale bar: 200 μm. (C, D) Immunoblot analysis of embryonic myosin heavy chain (eMyHC) expression (C) and quantification (D) in 20-weeks-old mdx and BmKO/mdx mice with HSP90 as loading control. Pooled sample of four mice per lane were used for each group. (E, F) RT-qPCR analysis of genes involved in myogenic response (E), and satellite cell proliferation (F) in 20-weeks-old WT (n=3), mdx (n=6) and BmKO/mdx mice (n=6) diaphragm, with normalization to 36B4 level. *, ** P≤0.05 or 0.01 vs. WT, #, ##: P≤0.05 or 0.01 BmKO/mdx vs. mdx mice.
Loss of Bmal1 exacerbates TA muscle damage in mdx mice
In comparison to the pronounced dystrophic damage in diaphragm, limb muscles in mdx mice are relatively spared and present mild disease pathology3. We next examined muscle injury in TA as the representative limb muscle group in BmKO/mdx vs. mdx controls at different ages. The TA muscle in both groups of mice displayed substantially variable muscle fiber size, as expected of chronic injury-induced regenerative repair in the dystrophic muscle. Bmal1 deficiency in the BmKO/mdx mice led to markedly pronounced inflammatory infiltration as compared to the mild presentation in mdx controls at 8 and 12 weeks of age, as indicated by the histological analysis (Fig. 5A). There were abundant myofibers with central nuclei in the TA both cohorts of mice, indicative of extensive new myofiber formation due to injury. Interestingly, the loss of Bmal1 did not significantly affect central nuclei-containing myofibers at 8, 12 or 20 weeks-old in the dystrophic genetic background (Fig. 5B). Analysis of myofiber cross section area revealed that TA muscle fibers in 8 (Fig. 5C) or 12-weeks-old (Fig. 5D) BmKO/mdx mice exhibited marked shifts toward smaller myofiber distribution, accompanied by correspondingly lower percentages of larger fibers. At 20 weeks of age, however, only a tendency for a leftward shift was detected in the double mutants, although was not statistically different with mdx mice (Fig. 5E). This reduced muscle fiber size in BmKO/mdx is in line with the observation of diminished muscle mass and previous findings in the Bmal1-null mice1.
Figure 5.
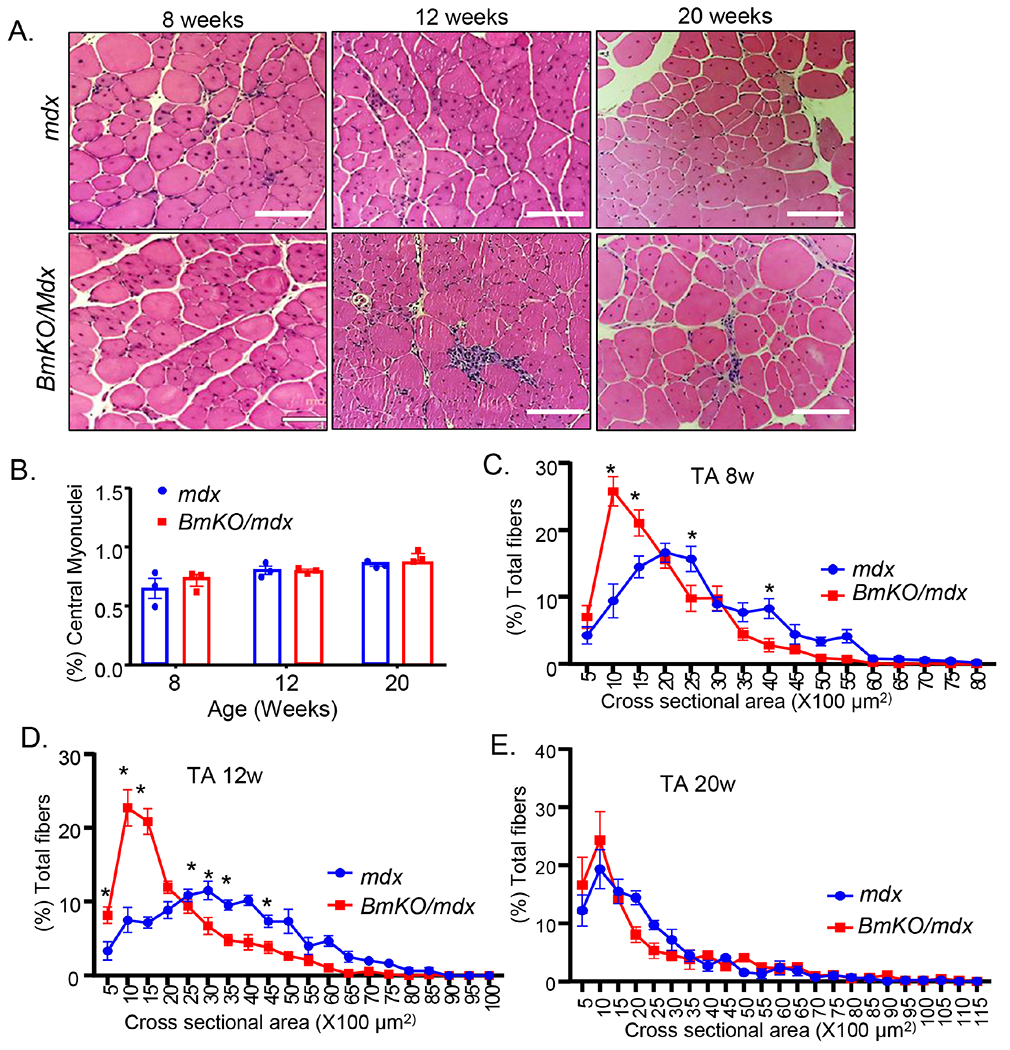
Loss of Bmal1 exacerbates muscle damage in TA of dystrophic mice. (A) Representative H/E images of TA cross section of 8, 12 and 20-weeks-old mdx and BmKO/mdx mice (10X). Scale bar: 100 μm. (B) Quantification of the percent of central nuclei-containing myofibers in TA cross section in mdx and BmKO/mdx mice at indicated ages (n=3-5/group). (C-E) TA muscle cross-section area distribution as percentage of total myofibers in 8 (C), 12 (D) and 20 weeks-old (E) mdx and BmKO/mdx mice (n=5-6/group). *, ** P≤0.05 or 0.01 BmKO/mdx vs. mdx control.
Bmal1 deficiency in mdx mice impairs regenerative repair by attenuating neomyofiber formation and satellite cell proliferation
Based on findings of severe muscle damage and wasting in mdx mice lacking Bmal1, we tested whether impaired regenerative myogenesis underlies these changes. We thus performed immunostaining using embryonic myosin heavy chain (eMyHC) to identify newly regenerated myofibers in TA muscle of 12-weeks-old mice. In comparison with mdx controls, eMyHC-positive staining fibers were markedly attenuated in Bmal1/mdx double mutants (Fig. 6A), with quantitative analysis demonstrating a nearly 2/3 reduction of eMyHC-positive fibers (Fig. 6B). In addition, analysis of myogenic factors involved in regenerative myogenesis in BmKO/mdx vs. mdx controls revealed significantly attenuated expressions of myogenin and the marker of nascent myofiber, eMyHC (Fig. 6C), indicating a defective regenerative repair response. Furthermore, we used Pax7 and Ki67 double staining to identify the number of total and proliferative satellite cells, respectively (Fig. 6D). There was a clear reduction of number of Pax7-positive cells in mdx mice lacking Bmal1, and the quantification revealed that both the abundance of total Pax7+ and Pax7+/Ki67+ double positive proliferative satellite cells were markedly lower in BmKO/mdx double mutant (Fig. 6E). Together, these findings highlight a severe loss of new myofiber regeneration in these mice due to impaired satellite cell proliferation. Consistent with these results, gene expression survey found marked down-regulation of two important factors involved in satellite cell proliferation, cyclin D1 and c-Met, together with significantly elevated expression of a key cell cycle inhibitor, p21 (Fig. 6F). These changes in cell cycle regulators, potential direct or indirect targets of Bmal1, may underlies the defective satellite cell proliferation in mice with Bmal1 deficiency in a dystrophic disease background.
Figure 6.
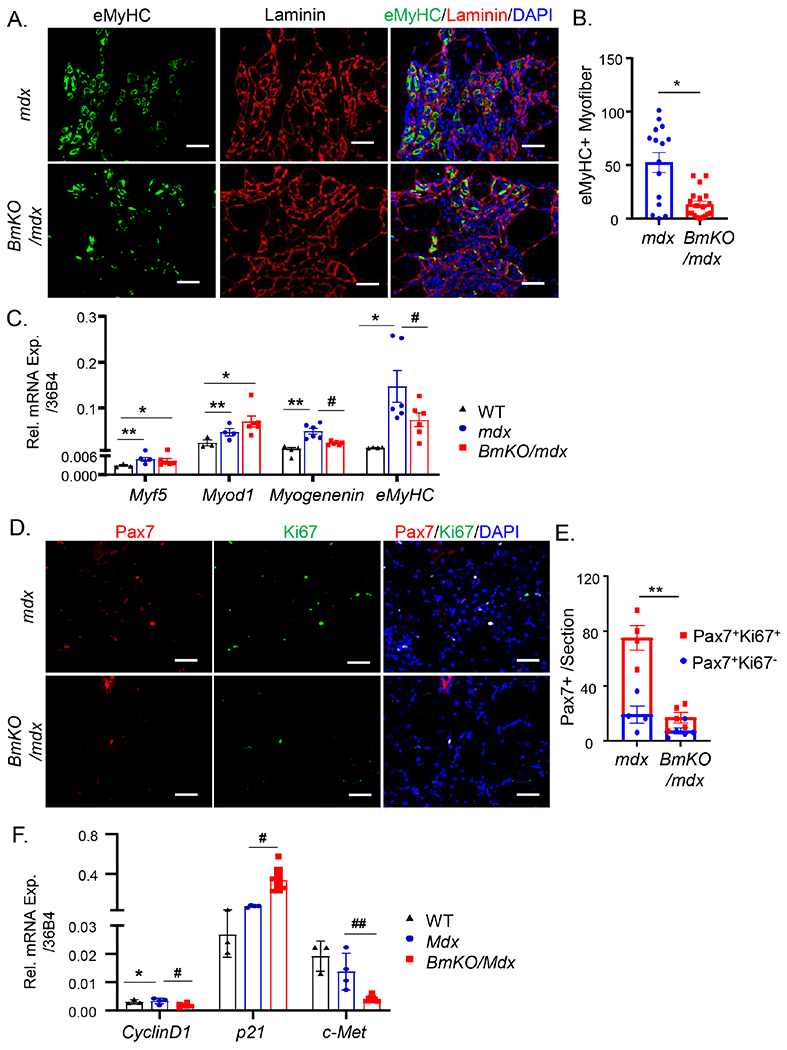
Bmal1 deficiency in mdx mice impairs regenerative repair by attenuating neo-myofiber formation and satellite cell proliferation. (A, B) Representative images of embryonic myosin heavy chain (eMyHC, green), laminin (red) and DAPI (blue) in TA from mdx and BmKO/mdx mice (A), and the quantification of fibers with eMyHC expression as a percentage of total fibers from mdx and BmKO/mdx TA cross sections (B). Scale bar: 50 μm. (C) RT-qPCR analysis of myogenic factor expression in TA of WT (n=3), mdx mice (n=7) and BmKO/mdx mice (n=4). (D, E) Representative images of Pax7 (Red), Ki67 (Green) and DAPI in TA sections from mdx (n=4) and BmKO/mdx mice (n=5, D), and quantification of Pax7+ myofibers with or without Ki67 co-staining in TA cross sections (E). Scale bar: 50 μm. (F) RT-qPCR analysis of key factors involved in satellite cells proliferative response in TA muscle of WT (n=3), mdx mice (n=7) and BmKO/mdx mice (n=4). *, ** P≤0.05 or 0.01 vs. WT, #, ##: P≤0.05 or 0.01 BmKO/mdx vs. mdx mice.
Discussion
The current mainstay of dystrophic disease treatment strategies still relies on anti-inflammatory therapy to preserve muscle function despite the underlying cause of repetitive muscle damage and resultant loss of muscle regenerative capacity [25]. Better understanding of molecular pathways contributing to dystrophic disease development is imperative for uncovering novel targets for interventions. Through genetic ablation of Bmal1 in mdx dystrophic disease model, our current study revealed an important function of this key clock regulator in promoting regenerative myogenesis to protect against dystrophic muscle loss. Maintaining Bmal1 function could be a new target to promote muscle regenerative capacity in muscle wasting disease conditions.
The muscle-intrinsic circadian clock plays an important role in maintaining muscle mass and its contractile functions [7, 10]. How muscle clock function can be applied to muscle disease conditions has not been directly tested. Consistent with our prior findings that Bmal1 promotes myogenic progenitor properties [14] and regenerative myogenesis [26], the current study indicate that loss of Bmal1 significantly exacerbates the regenerative response in the chronic injury milieu of the mdx mice. Likely due to the defective proliferative response of the myogenic muscle stem cell population, neo-myofiber formation was significantly attenuated in Bmal1/mdx-double null mutant mice and this compromised regenerative capability may underlie the exacerbated disease phenotype. In addition to Bmal1, clock repressor Rev-erbα play an antagonistic role in suppressing the proliferative and differentiative capacities of Bmal1 [21]. Cry2, another major negative component of the core clock loop, was recently reported to positively regulate myogenesis [22]. Thus, the clock circuit may coordinately modulate muscle stem cell properties, although the specific mechanisms involved could be entirely distinct due to regulation of clock-controlled output genes. As a direct target gene of Bmal1, Rev-erbα expression was suppressed in Bmal1/mdx mice. Thus, it is possible that certain effect of loss of Bmal1 on satellite cells could be counteracted by the loss of Rev-erbα negative control of muscle stem cell proliferation. Nonetheless, the aggravated muscle damage in BmKO/mdx mice suggests a dominant effect of Bmal1 regulation within this system. It remains to be seen whether the modulation of additional clock regulators may also apply to dystrophic disease conditions. In addition, the effect of protective effects of Bmal1 in dystrophic disease also implicates that further studies are warranted to test whether augmenting clock regulatory effects on muscle regenerative capacity can protect against broader muscle wasting conditions.
The loss of Bmal1 leads to significant muscle wasting with impaired muscle function in the chronic degenerative-regenerative injury milieu of the mdx mice. In agreement of the severe muscle damage in Bmal1−/−/mdx mice, fibrotic changes of the muscle, particularly in the diaphragm was aggravated. Interstitial fibrosis of the muscle in the mdx occurs as a result of the injury-induced inflammation. As muscle injury persists in the dystrophic condition, exaggerated macrophage activation and inflammatory stimuli results in fibrotic response that replaces effective muscle mass and impairs contractile capacity of the muscle [27]. The heightened fibrosis observed in Bmal1−/−/mdx mutant mice is likely a chronic consequence of muscle damage-induced inflammatory milieu. Circadian clock components are known to modulate both the innate and adaptive immune response [28, 29]. Particularly, Bmal1 regulation of macrophage or T cell functions, both integral to muscle regenerative repair, may affect dystrophic pathology and disease progression [30–33]. Our analysis of macrophage marker gene expression F4/80 and various cytokines in TA or diaphragm did not reveal significance differences between mdx mice with or without Bmal1 (data not shown). Nonetheless, cell-type specific Bmal1 loss-of-function studies may dissect the precise mechanisms involved in muscle stem cell or immune cell regulation in dystrophic disease.
The cell-autonomous muscle clock is required to maintain muscle mass and its contractile functions [7, 10]. The muscle wasting and the functional deficits displayed by the Bmal1/mdx-double null mutants is in line with previous studies. However, the significantly reduced survival, occurring early in the lifespan of young adult mice, was very striking. The mechanisms leading to the early death in these mice remains unknown. We found marked dystrophic muscle wasting and functional deficits in distinct groups of muscles. During the course of maintaining the Bmal1/mdx-double null cohort, there were a high frequency of these mice that required euthanasia due to severe muscle wasting, suggesting that impaired muscle function and potential resultant ambulatory deficits may have contributed, at least in part, the mortality rate. As the most severe muscle damage in the DMD model occurred in the diaphragm due to mechanical stress, compromised respiratory function caused by diaphragm pathology may also contribute to early mortality. Interestingly, recent studies reveal that circadian disruption due to misalignments of endogenous clock with environmental lightings, such as shift work, is associated with the development of sarcopenia and clock re-setting by exercise may prevent muscle wasting [34]. These findings, together with our current genetic model, lends further support that maintaining proper circadian clock functions could be potentially important for muscle mass preservation in muscle wasting conditions beyond dystrophic disease. Besides the known functions of muscle clock network on myogenic progenitor behavior, immune function or sarcomeric structure or metabolic regulations [35, 36], it remains to be seen whether clock may regulate additional aspects of muscle mass or function to mitigate muscle diseases. As our study suggests, preserving or boosting Bmal1 function could be a potential therapeutic target for dystrophic muscle disease. Additional mechanisms involved in clock regulation of muscle growth and muscle mass maintenance could be worth exploring.
The findings from our current study of loss of Bmal1 function in a dystrophic disease model suggest that Bmal1 or the Bmal1-driven clock activity could be fine-tuned to facilitate optimal muscle stem cell function that could be applicable for muscle disease therapy. Testing promoting Bmal1 function to prevent or treat muscle diseases may ultimately lead to the discovery of a novel drug target for dystrophic or degenerative muscle wasting conditions.
Supplementary Material
Highlights.
Genetic loss of Bmal1 function in the dystrophic disease background of mdx mice led to progressive aggravated muscle damage, muscle wasting and impaired muscle function.
Bmal1 deficiency in mdx mice led to severe muscle injury in diaphragm with defective regenerative response.
Bmal1 function is required for promoting satellite cell proliferation and regenerative myogenesis to protect against dystrophic muscle damage.
Acknowledgement
We thank the Shared Resources Core Facility of City of Hope for their expert technical assistance in histology. This project was supported by grant from Muscular Dystrophy Association 381294 to KM.
Non-standard Abbreviations:
- Bmal1
Brain and Muscle Arnt-like Protein 1
- CLOCK
Circadian Locomotor Output Cycles Kaput
- DMD
Duchene Muscular Dystrophy
- TA
Tibialis Anterior
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT AUTHORSHIP STATEMENT
HG: data curation and investigation, formal analysis, manuscript writing and editing; XX, YL and SC: data curation and investigation; KM: formal analysis, project administration, manuscript writing and editing, and funding acquisition.
Declaration of Interest: None
Reference
- 1.DiMario JX, Uzman A, and Strohman RC, Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev Biol, 1991. 148(1): p. 314–21. [DOI] [PubMed] [Google Scholar]
- 2.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, and Sweeney HL, Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A, 1993. 90(8): p. 3710–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, and Khurana TS, Functional improvement of dystrophic muscle by myostatin blockade. Nature, 2002. 420(6914): p. 418–21. [DOI] [PubMed] [Google Scholar]
- 4.Gehrig SM, Ryall JG, Schertzer JD, and Lynch GS, Insulin-like growth factor-I analogue protects muscles of dystrophic mdx mice from contraction-mediated damage. Exp Physiol, 2008. 93(11): p. 1190–8. [DOI] [PubMed] [Google Scholar]
- 5.Barton ER, Morris L, Musaro A, Rosenthal N, and Sweeney HL, Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol, 2002. 157(1): p. 137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi JS, Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet, 2017. 18(3): p. 164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi Y, Cho J, No MH, Heo JW, Cho EJ, Chang E, Park DH, Kang JH, and Kwak HB, Re-Setting the Circadian Clock Using Exercise against Sarcopenia. Int J Mol Sci, 2020. 21(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eastman CI, Hoese EK, Youngstedt SD, and Liu L, Phase-shifting human circadian rhythms with exercise during the night shift. Physiol Behav, 1995. 58(6): p. 1287–91. [DOI] [PubMed] [Google Scholar]
- 9.Wolff G and Esser KA, Scheduled exercise phase shifts the circadian clock in skeletal muscle. Med Sci Sports Exerc, 2012. 44(9): p. 1663–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee S and Ma K, Circadian clock regulation of skeletal muscle growth and repair. F1000Res, 2016. 5: p. 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harfmann BD, Schroder EA, and Esser KA, Circadian rhythms, the molecular clock, and skeletal muscle. J Biol Rhythms, 2015. 30(2): p. 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews JL, Zhang X, McCarthy JJ, McDearmon EL, Hornberger TA, Russell B, Campbell KS, Arbogast S, Reid MB, Walker JR, Hogenesch JB, Takahashi JS, and Esser KA, CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc Natl Acad Sci U S A, 2010. 107(44): p. 19090–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo B, Chatterjee S, Li L, Kim JM, Lee J, Yechoor VK, Minze LJ, Hsueh W, and Ma K, The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J, 2012. 26(8): p. 3453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatterjee S, Nam D, Guo B, Kim JM, Winnier GE, Lee J, Berdeaux R, Yechoor VK, and Ma K, Brain and muscle Arnt-like 1 is a key regulator of myogenesis. J Cell Sci, 2013. 126(Pt 10): p. 2213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nam D, Chatterjee S, Yin H, Liu R, Lee J, Yechoor VK, and Ma K, Novel Function of Rev-erbalpha in Promoting Brown Adipogenesis. Sci Rep, 2015. 5: p. 11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, and Antoch MP, Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev, 2006. 20(14): p. 1868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDearmon EL, Patel KN, Ko CH, Walisser JA, Schook AC, Chong JL, Wilsbacher LD, Song EJ, Hong HK, Bradfield CA, and Takahashi JS, Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science, 2006. 314(5803): p. 1304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatterjee S, Yin H, Nam D, Li Y, and Ma K, Brain and muscle Arnt-like 1 promotes skeletal muscle regeneration through satellite cell expansion. Exp Cell Res, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Yin L, Wu N, and Lazar MA, Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl Recept Signal, 2010. 8: p. e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, and Lazar MA, Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science, 2007. 318(5857): p. 1786–9. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee S, Yin H, Li W, Lee J, Yechoor VK, and Ma K, The Nuclear Receptor and Clock Repressor Rev-erbalpha Suppresses Myogenesis. Sci Rep, 2019. 9(1): p. 4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowe M, Lage J, Paatela E, Munson D, Hostager R, Yuan C, Katoku-Kikyo N, Ruiz-Estevez M, Asakura Y, Staats J, Qahar M, Lohman M, Asakura A, and Kikyo N, Cry2 Is Critical for Circadian Regulation of Myogenic Differentiation by Bclafl-Mediated mRNA Stabilization of Cyclin D1 and Tmem176b. Cell Rep, 2018. 22(8): p. 2118–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams KL, Castanon-Cervantes O, Evans JA, and Davidson AJ, Environmental circadian disruption elevates the IL-6 response to lipopolysaccharide in blood. J Biol Rhythms, 2013. 28(4): p. 272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charge SB and Rudnicki MA, Cellular and molecular regulation of muscle regeneration. Physiol Rev, 2004. 84(1): p. 209–38. [DOI] [PubMed] [Google Scholar]
- 25.Loboda A and Dulak J, Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol Rep, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatterjee S, Yin H, Nam D, Li Y, and Ma K, Brain and muscle Arnt-like 1 promotes skeletal muscle regeneration through satellite cell expansion. Exp Cell Res, 2015. 331(1): p. 200–10. [DOI] [PubMed] [Google Scholar]
- 27.Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, and Munoz-Canoves P, Aberrant repair andfibrosis development in skeletal muscle. Skelet Muscle, 2011. 1(1): p. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter SJ, Durrington HJ, Gibbs JE, Blaikley J, Loudon AS, Ray DW, and Sabroe I, A matter of time: study of circadian clocks and their role in inflammation. J Leukoc Biol, 2016. 99(4): p. 549–60. [DOI] [PubMed] [Google Scholar]
- 29.Scheiermann C, Gibbs J, Ince L, and Loudon A, Clocking in to immunity. Nat Rev Immunol, 2018. 18(7): p. 423–437. [DOI] [PubMed] [Google Scholar]
- 30.Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, Farrow SN, Else KJ, Singh D, Ray DW, and Loudon AS, The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc Natl Acad Sci U S A, 2012. 109(2): p. 582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitchen GB, Cunningham PS, Poolman TM, Iqbal M, Maidstone R, Baxter M, Bagnall J, Begley N, Saer B, Hussell T, Matthews LC, Dockrell DH, Durrington HJ, Gibbs JE, Blaikley JF, Loudon AS, and Ray DW, The clock gene Bmal1 inhibits macrophage motility, phagocytosis, and impairs defense against pneumonia. Proc Natl Acad Sci U S A, 2020. 117(3): p. 1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spengler ML, Kuropatwinski KK, Comas M, Gasparian AV, Fedtsova N, Gleiberman AS, Gitlin II, Artemicheva NM, Deluca KA, Gudkov AV, and Antoch MP, Core circadian protein CLOCK is a positive regulator of NF-kappaB-mediated transcription. Proc Natl Acad Sci U S A, 2012. 109(37): p. E2457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Yang Z, Niu Z, Peng J, Li Q, Xiong W, Langnas AN, Ma MY, and Zhao Y, MOP3, a component of the molecular clock, regulates the development of B cells. Immunology, 2006. 119(4): p. 451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi YI, Park DK, Chung JW, Kim KO, Kwon KA, and Kim YJ, Circadian rhythm disruption is associated with an increased risk of sarcopenia: a nationwide population-based study in Korea. Sci Rep, 2019. 9(1): p. 12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin H, Li W, Chatterjee S, Xiong X, Saha P, Yechoor V, and Ma K, Metabolic-sensing of the skeletal muscle clock coordinates fuel oxidation. FASEB J, 2020. 34(5): p. 6613–6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wefers J, van Moorsel D, Hansen J, Connell NJ, Havekes B, Hoeks J, van Marken Lichtenbelt WD, Duez H, Phielix E, Kalsbeek A, Boekschoten MV, Hooiveld GJ, Hesselink MKC, Kersten S, Staels B, Scheer F, and Schrauwen P, Circadian misalignment induces fatty acid metabolism gene profiles and compromises insulin sensitivity in human skeletal muscle. Proc Natl Acad Sci U S A, 2018. 115(30): p. 7789–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.