

Abstract
The cytoskeleton is a complex of detergent-insoluble components of the cytoplasm playing critical roles in cell motility, shape generation, and mechanical properties of a cell. Fibrillar polymers—actin filaments, microtubules, and intermediate filaments—are major constituents of the cytoskeleton, which constantly change their organization during cellular activities. The actin cytoskeleton is especially polymorphic, as actin filaments can form multiple higher-order assemblies performing different functions. Structural information about cytoskeleton organization is critical for understanding its functions and mechanisms underlying various forms of cellular activity. Because of the nanometer-scale thickness of cytoskeletal fibers, electron microscopy (EM) is a key tool to determine the structure of the cytoskeleton.
This article describes application of rotary shadowing (or platinum replica) EM (PREM) for visualization of the cytoskeleton. The procedure is applicable to thin cultured cells growing on glass coverslips and consists of detergent extraction (or mechanical “unroofing”) of cells to expose their cytoskeleton, chemical fixation to provide stability, ethanol dehydration and critical point drying to preserve three-dimensionality, rotary shadowing with platinum to create contrast, and carbon coating to stabilize replicas. This technique provides easily interpretable three-dimensional images, in which individual cytoskeletal fibers are clearly resolved and individual proteins can be identified by immunogold labeling. More importantly, PREM is easily compatible with live cell imaging, so that one can correlate the dynamics of a cell or its components, e.g., expressed fluorescent proteins, with high-resolution structural organization of the cytoskeleton in the same cell.
Keywords: Electron microscopy, Cytoskeleton, Critical point drying, Rotary shadowing, Actin, Microtubules, Immunogold, Correlative microscopy
1. Introduction
Electron microscopy (EM) has been instrumental in discovering the cytoskeleton in the first place and also in investigating its structural organization in different cells and conditions. The initial progress in the cytoskeletal studies closely paralleled the development of EM techniques. Thus, the introduction of heavy metal fixation led to the discovery of actin filaments in non-muscle cells [1], while the discovery of microtubules [2] was made possible after the introduction of aldehyde fixation [3].
A great value of EM is its ability to obtain structural information at a high-resolution level, which for biological samples is limited by a sample preparation procedure rather than by the power of a transmission electron microscope (TEM). Vacuum in the TEM column and electron beam irradiation impose strict restrictions on how samples should be prepared, which in turn greatly affect the quality of images and the rate of success. A large number of different EM protocols have been developed over the years to improve the quality of samples and the amount of collected information and to avoid artifacts. Each technique has its pluses and minuses, making it more suitable for some applications than for others.
The thin sectioning technique was initially a dominant way to visualize the cytoskeleton [4, 5]. It involves the embedding of chemically fixed specimens into a resin followed by thin sectioning to allow for beam penetration. Contrast is generated by positive staining of the sections with heavy metal salts, and the limited ability of stains to bind bioorganic material reduces the resolution of this technique. Thin sections provide a 2D view of the sample at a single plane, and a series of sections is required to retrieve the 3D information. Such reconstruction works well with relatively large and simple objects but is not efficient in revealing the details of complex and delicately organized cytoskeletal structures. Among cytoskeleton components, actin filaments are notoriously difficult to preserve and visualize by thin section EM. Although actin filament bundles can be reasonably well visualized in plastic-embedded samples after appropriate fixation, delicate actin filament networks, such as those in lamellipodia, in the bulk cytoplasm, or at the surface of membrane organelles, remain largely uninterpretable even in best examples [6–9].
Different versions of whole mount EM have been used to investigate the structural organization of the cytoskeleton in its entirety. Thus, the structural arrangement of actin filaments in lamellipodia was first visualized by the negative staining EM of cultured cells [10]. In this technique, partially permeabilized cells growing on EM grids are immersed into a heavy metal stain solution, which is blotted off shortly after, and the samples are dried in open air. The dried stain generates a dark amorphous background on which the structures appear as translucent shapes. Negative staining EM provides high resolution and allows one to see the thin regions of a cell all the way through. In combination with electron tomography, this approach can successfully reveal structural organization of branched actin filament networks [11–13]—the cytoskeleton structures that are particularly challenging for high-resolution structural studies. Weaknesses of the negative staining EM include partial sample flattening during air drying, relatively low contrast, and low stability of the samples. In cryo EM technique, the samples are quickly frozen (to prevent ice crystal formation) and viewed while still embedded in amorphous ice, either as whole mounts or after cryosectioning, so that they remain hydrated and the proteins retain their natural conformation [14–16]. To view frozen samples, a TEM should be equipped with a chilled sample holder, and electron beam power and the observation time should be minimized to keep the specimen frozen. No contrasting procedures are used in this technique except for the specimen’s own contrast. Because of low intrinsic contrast and sensitivity of samples to irradiation, cryo EM has been most useful for analyses of samples suitable for averaging, such as purified macromolecules [16, 17]. Application of cryo EM to intact cells is challenging due to limited opportunity of applying averaging approaches, although subtomogram averaging has been successfully used for imaging of a few suitable subcellular structures [14]. Cryo EM, especially in combination with electron tomography, can image only small regions of the cell due to lengthy image acquisition and sample damage during imaging, so that a larger context of the imaged region is often unavailable. Another limitation of cryoEM is its incompatibility with immunochemistry [18]. Because of higher intrinsic contrast of lipids, membrane organelles are more permissive for cryo EM analyses in intact cells [19–24], as compared to purely proteinaceous structures, such as the cytoskeleton. Among cytoskeletal components, microtubules are more detectable by cryo EM [25–27] than actin filaments, while actin filament bundles are easier to visualize and analyze [28, 29] than branched actin networks [30, 31].
In metal replica EM, heavy metals are evaporated onto a 3D sample at an angle, which reveals its surface topography [32, 33]. The quality of the samples is greatly enhanced if rotary, and not unilateral, coating is used, as it helps to avoid deep featureless shadows. As metal coating is not cohesive, it is subsequently stabilized by a layer of carbon, which keeps metal grains together and is fairly transparent for the electron beam. The coated sample, or just a metal-carbon replica, is subsequently removed from its original support and placed onto EM grids. The resolution of replica EM is quite high, but it depends on the metal grain size, the thickness of the coating, and the angle of shadowing. Platinum is the most popular metal, as it provides a good compromise between the grain size and an ease of evaporation. The replica technique was initially introduced to study freeze-fractured samples [34], but it is applicable for a large range of samples, such as single molecules [35–37], cells [33, 38–48], and tissues [49–51]. This approach can reveal the 3D structure in great detail, but it is limited by the depth of shadowing penetration.
Given that PREM (platinum replica electron microscopy) reveals only the sample surface, analyses of the cytoskeleton can be accomplished only after the cytoskeleton is exposed to metal shadowing. This is typically achieved by detergent extraction, which reproducibly exposes the entire cytoskeleton of the cell. For best results, the extraction conditions need to be carefully adjusted to properly preserve the cytoskeleton architecture. Nonionic detergent Triton X-100 is most commonly used to dissolve cell membranes, whereas preservation of the cytoskeleton relies on the composition of the buffer and is improved by various stabilizing supplements, such as phalloidin for actin filaments, taxol for microtubules, and nonspecific stabilizers polyethelete glycol or glutaraldehyde [33, 46, 52–54]. Since detergent extraction dissolves all cellular membranes, analyses of the membrane-associated cytoskeleton are typically accomplished using mechanical rupture of the cell (unroofing), which can expose cell interior without dissolving membranes. In two main unroofing approaches employed for cytoskeleton studies, the mechanical impact is delivered either through an ultrasonic burst [44, 47, 55–57] or by applying an adhesive material to the cell surface and subsequently peeling it off [48, 57–61]. Mechanical rupture methods are not highly reproducible, but successful samples excellently reveal interactions between the cytoskeleton and membrane organelles at high resolution.
In most cells, the cytoskeleton is heavily dominated by actin filaments, which largely conceal other cytoskeletal components, such as microtubules, intermediate filaments, myosin II bipolar filaments, etc. Such cytoskeleton components can be uncovered by treating the chemically or mechanically exposed cytoskeletons with the actin-severing protein gelsolin, which dissolves actin filaments without perturbing non-actin structures [53, 62–65]. For our studies of the cytoskeleton in cultured cells, we chose the PREM approach, in which either detergent extraction or unroofing is used to expose the cytoskeleton; chemical fixation helps to preserve the sample structure; ethanol dehydration followed by critical point drying (CPD) preserves the cell’s 3D organization; and rotary shadowing with platinum creates contrast. Over the years, we have found a good combination of individual steps to develop a reliable and relatively simple protocol that consistently produces highly informative images with excellent yield that can be combined with immunochemistry [33, 38, 39, 42, 48, 53, 54, 62, 66, 67]. However, this approach is not universal but is limited to relatively thin samples attached to glass surfaces. Also, because of extensive fixation and dehydration, it can achieve the molecular level of resolution only for very large molecules, such as myosin II [62], plectin [63], or spectrin [68], but is optimal for analyses of the fine cytoskeletal architecture with a single filament resolution at the scale of a whole cell.
As EM, in general, cannot work with live samples, investigators can only guess the kind of activity the cell was involved in at the moment of fixation and what it would do next. A partial solution for this problem is provided by correlative light and EM (CLEM), in which the dynamics of a living cell is followed by time-lapse optical imaging, and the same sample is subsequently analyzed by EM. Our PREM protocol made it possible to perform CLEM routinely, as it allowed us to obtain high-quality structural information for a cell of interest with high probability [38, 46, 48, 53, 54, 66]. Other labs also successfully used CLEM in combination with PREM using similar or slightly modified protocols [43, 47, 69, 70]. Several other EM techniques have also been used in a correlative approach [15], including resin-embedded samples [21, 71–75], cryoEM [28, 29, 31], and negatively contrasted cells [12, 76].
In this article, I present detailed protocols for PREM of the cytoskeleton and its extensions, such as immunogold labeling, correlative PREM, and actin depletion by gelsolin. Given that the key steps ensuring high quality of PREM samples are the same for the cytoskeletal preparations prepared by detergent extraction and by unroofing, only detergent-extracted cells are used here as an example. However, mechanically ruptured cells prepared by various unroofing protocols are also compatible with these procedures [48, 56, 57].
2. Materials
2.1. Cell Culture and Extraction
Small (6–12 mm) coverslips that can be made by cutting regular coverslips with a diamond pencil. Trapezoidal (square with one oblique side) coverslips lacking mirror symmetry are helpful to easily determine the cell-containing side. Commercially available 12 mm round coverslips are also acceptable (see Note 1).
Phosphate-buffered saline (PBS) with Ca2+ and Mg2+(-commercially available).
PEM buffer: 100 mM PIPES (free acid), pH 6.9 (adjust with KOH), 1 mM MgCl2, and 1 mM EGTA. Make working buffer from 2× stock solution, which can be stored up to 1 month at 4 °C (see Note 2).
Extraction solution: 1% Triton X-100 in PEM buffer supplemented (optionally) with 1–4% polyethylene glycol (PEG) (MW 20,000–40,000), 2–10 μM phalloidin, and/or 2–10 μM taxol (paclitaxel) (see Note 3). Use a stirrer and allow 15–20 min to dissolve PEG. Extraction solutions can be stored for up to 3 days at 4 °C, but phalloidin and taxol should be added before use. Stock solutions (1000×) of phalloidin and taxol are made in dimethylsulfoxide (DMSO) and stored at −20 °C in aliquots.
2.2. Fixation
Glutaraldehyde solution: 2% glutaraldehyde (EM grade, Sigma) in 0.1 M sodium cacodylate, pH 7.3. The working solution can be stored at 4 °C for up to 1 week, 2× stock solution of sodium cacodylate is stable at 4 °C. Caution: Glutaraldehyde is toxic and volatile, so a fume hood should be used when working; sodium cacodylate is toxic.
Tannic acid solution: 0.1% tannic acid (Fisher) in distilled water (see Note 4). Use within a day.
Aqueous uranyl acetate solution: 0.2% uranyl acetate in distilled water. Use a stirrer to dissolve. Store at room temperature. Caution: Uranyl acetate is toxic.
2.3. Dehydration and Critical Point Drying (CPD)
Graded ethanol solutions: 10%, 20%, 40%, 60%, 80%, and 100% ethanol in distilled water.
Alcohol uranyl acetate solution: 0.2% uranyl acetate in 100% ethanol. Use a stirrer to dissolve. Use within a day. Caution: Uranyl acetate is toxic.
Dehydrated ethanol: Wash molecular sieves (4 Å, 8–12 mesh) (Fisher) with several changes of water to remove dust. Dry in the air, bake at 160 °C overnight, cool down, and add to a bottle of 100% ethanol (~50 g per 500 mL). Seal with Parafilm and store at room temperature. Do not shake, as the beads are fragile and easily generate dust.
CPD sample holder and scaffolds: A holder with a lid and two scaffolds are homemade with a stainless-steel wire mesh. The holder should fit into the chamber of the CPD apparatus. A scaffold is required to maintain the holder above the stirring bar during stirring. It should fit a beaker in which dehydration will be processed, e.g., a 50 mL glass beaker.
CPD device: We use Samdri PVT-3D (Tousimis) CPD with manual operation, but other devices are also appropriate.
Carbon dioxide: Use liquid dehydrated CO2 (bone dry grade) in a tank with a siphon (deep tube) and a water and oil-absorbing filter (Tousimis). Siphonized tanks make it possible to take the liquid phase of CO2 from the bottom of the tank.
2.4. Platinum and Carbon Coating
Vacuum evaporator: We use Auto 306 coater (Boc Edwards) and Carbon Coating equipped with a water-cooled diffusion pumping system, carbon and metal evaporation sources, a rotary stage, and a thickness monitor.
Metals for evaporation (Ted Pella): Tungsten wire (0.76 mm), platinum wire (0.2 mm), and carbon rods (3 mm).
2.5. Preparation of Replicas
Hydrofluoric acid (HF): 5–10% HF in distilled water is prepared from the concentrated acid (49%). Do not use glassware to handle acid-containing solutions. Caution: It is extremely volatile and toxic; it causes severe skin burns. Use a fume hood and wear gloves.
Platinum loop: The optimal loop diameters are 3–5 mm.
EM grids: Formvar-coated EM grids with low mesh size (e.g., 50) to provide a large viewing area. Other options are acceptable.
2.6. Immunogold PREM
Immunogold buffer: 20 mM Tris–HCl, pH 8.0, 0.5 M NaCl, and 0.05% Tween 20. For dilution of antibodies, the buffer is supplemented with 1% bovine serum albumin (BSA); for washing, 0.1% BSA is added. Stock solutions (5×) are stable at 4 °C for several months. Sodium azide can be added to the stock solution to prevent microbial contamination. Caution: Sodium azide is toxic.
Quenching solution: 2 mg/mL sodium borohydrate (NaBH4) in PBS. Use immediately.
Blocking solution: 1 mg/mL glycine (or lysine) in PBS. Stable at 4 °C.
2.7. Correlative PREM
Marked coverslips: Homemade coverslips with reference marks are prepared by evaporating gold through a finder grid placed in the middle of a 22 × 22 square or 25 mm round coverslip. A variety of finder grids are available commercially. Baking (160 °C overnight) of gold-coated coverslips is necessary for the firm adhesion of gold to glass (see Note 5). For light microscopy, choose cells on the clear footprint of the finder grid.
Light microscopy: The light microscopic system should be equipped with an environmental chamber to maintain normal cell behavior; it should allow for fast exchange of culture medium to extraction solution to quickly stop cellular activity during imaging. A simple option is to use open dishes on a heated stage with the cells growing in a bicarbonate-free medium. We use UNO Stage Top Incubator from Okolab, which has an easily removable chamber lid.
2.8. Actin Depletion
3. Methods
In a basic form, PREM can be used to study the cytoskeleton architecture in a cell population. In an advanced form, it can be combined with immunogold staining to detect specific proteins in the cytoskeleton (Fig. 1), with light microscopy to correlate the cytoskeleton organization with cell behavior or with the distribution and dynamics of fluorescent probes (Figs. 1 and 2) and with actin depletion to fully reveal the organization of microtubules, intermediate filaments, and myosin II filaments (Fig. 3). The basic PREM procedure consisting of detergent extraction, fixation, dehydration, CPD, metal shadowing, and preparation of replicas is described first, and it is followed by the description of the advanced applications.
Fig. 1.
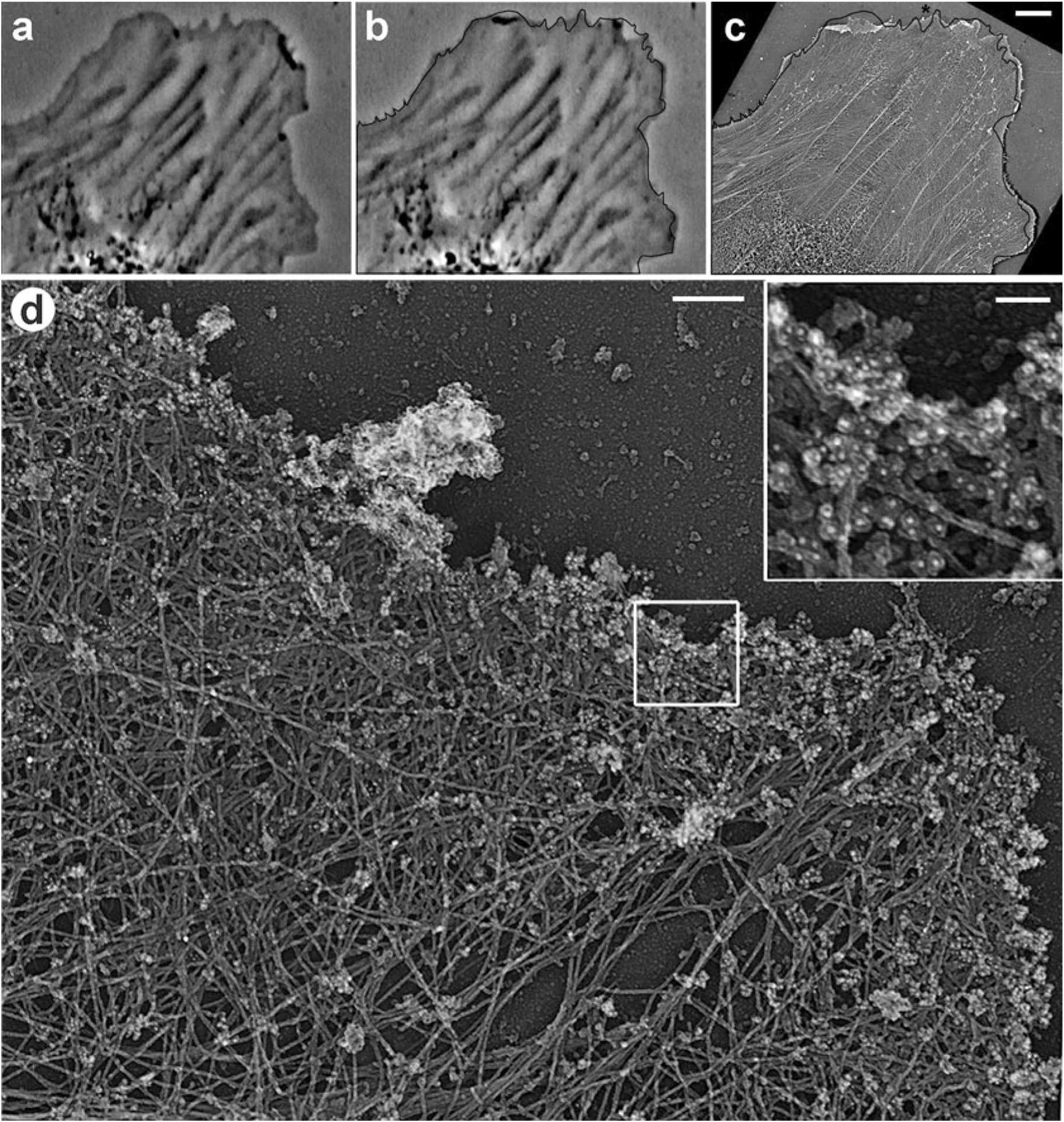
Correlative phase-contrast and PREM of cultured Rat-2 fibroblast combined with immunogold staining of ADF/cofilin. (a, b) Frames from time-lapse sequence showing the last live cell image (b) and an image 12 s earlier (a). Black line in (b) shows the cell edge outline from (a). (c) Low magnification PREM image of the cell overlaid with the cell outline, as in (b). (d) PREM of the protruding edge (asterisk in c) comprising a lamellipodium filled with dense actin network. Before the PREM processing, the sample was immunogold labeled with cofilin antibody; inset in (d) shows gold particles as white dots. Scale bars: 5μm (a–c), 200 nm (d), 50 nm (inset in d)
Fig. 2.
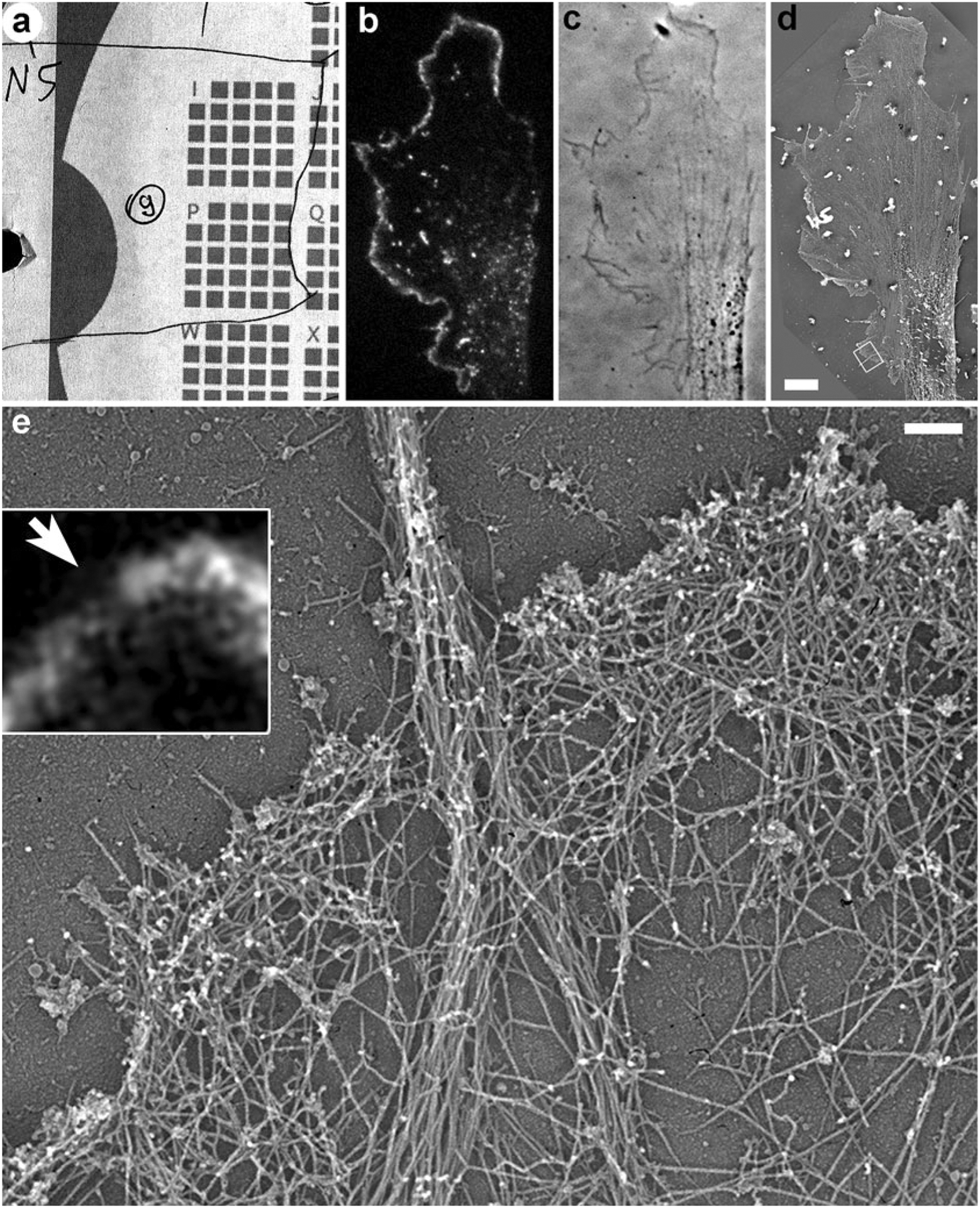
Correlative fluorescence microscopy and PREM of cultured B16F1 mouse melanoma cell expressing EGFP-capping protein. (a) Map showing position of the cell (number 9 in a circle) relative to the reference marks on the coverslip. (b) Fluorescence image of the cell showing localization of EGFP-capping protein to the edge of lamellipodia and puncta in lamella. (c) Phase contrast image of the same cell. (d) Low magnification PREM image of the same cell. Box indicates a region enlarged in (e). (e) High magnification PREM of the boxed region from (d) showing actin filament bundle in a filopodium in the center and dense branched network of actin filaments in lamellipodia. Inset shows the same region by fluorescence microscopy. Bright fluorescence corresponds to lamellipodia, while the dim region (arrow) corresponds to the filopodium. Scale bars: 5μm (b–d), 200 nm (e)
Fig. 3.
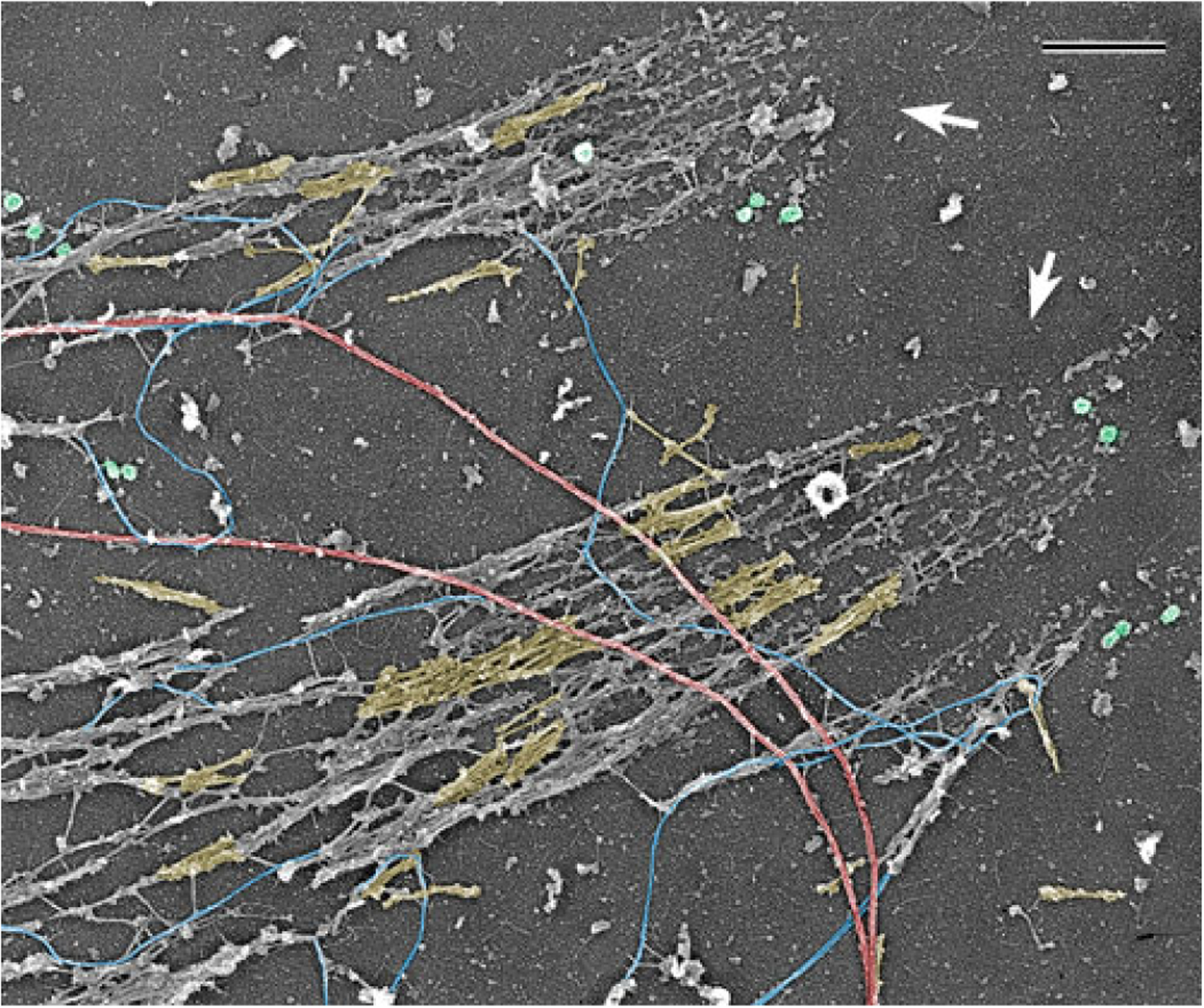
PREM of the cytoskeleton of a cultured PtK1 potoroo epithelial cell after depletion of actin filaments by gelsolin treatment. Image shows a peripheral cell region containing two large focal adhesions (arrows). Abundant myosin II filaments, a subset of which is highlighted in yellow, accumulate along the actin-depleted stress fibers extending away from focal adhesions, while some individual myosin II filaments can be seen elsewhere in the cytoplasm. Exposed microtubules (red), intermediate filaments (blue), and clathrin-coated vesicles (green) are also visible. Scale bar: 500 nm
A major source of artifacts in PREM is the failure to perform a genuine CPD, which may occur if wet samples are transiently exposed to air, or water is not fully exchanged to ethanol or ethanol to liquid CO2, or if the dried samples absorb ambient humidity. In order to get a high-quality preparation, it is critical not to allow a liquid-gas interface to touch the samples at any point during the procedure. Practically, it means keeping the cells away from air while they are wet and away from water while they are dry. Changes of solutions need to be done quickly, with a layer of liquid always being retained above the cells. After drying, the cells should be kept at low humidity until they are coated with carbon.
3.1. Cell Culture and Extraction
Detergent extraction is used to expose the internal cytoskeletal structures, while the carrier buffer is designed to maximally preserve them until fixation. Additional preservation may be achieved using specific and nonspecific stabilizers.
Put small glass coverslips into a culture dish and plate the cells. Cell culture conditions are specific for each system and are not discussed here. If several coverslips are placed into the same dish, make sure that they do not overlap and that there are no bubbles underneath.
When the cells are ready, gently remove the culture medium from the dish either (a) using a pipette, or (b) by pouring it out, or (c) by taking a coverslip out with forceps and placing it into the next solution; quickly rinse with the pre-warmed to 37 °C PBS (see Note 6).
Immediately, but gently, add extraction solution equilibrated to room temperature to the dish with coverslips (or place the coverslip into another dish with the extraction solution if the coverslip was taken out); gently stir the dish with the extraction solution to ensure that the detergent instantly reaches all the cells (see Note 7). Incubate for 3–5 min at room temperature.
Rinse cells with PEM buffer two to three times, for a few seconds each time, at room temperature (see Note 8).
3.2. Fixation
Chemical fixation provides cytoskeletal structures with physical resistance against damage by subsequent procedures, especially dehydration and CPD. It is a three-step procedure using different fixatives: glutaraldehyde, tannic acid, and uranyl acetate.
After the last PEM rinse, add glutaraldehyde solution and incubate for at least 20 min at room temperature. If necessary, the samples can be stored at this stage at 4 °C for up to 3 weeks. Take care to prevent evaporation during storage.
Transfer coverslips to another container with tannic acid solution, or change solutions in the same dish (see Note 9). Make sure that the cells remain covered with liquid during transfer. No washing is necessary before this step. Incubate for 20 min at room temperature.
Take the coverslips out of the tannic acid solution one by one, rinse by dipping sequentially into two water-filled beakers, and place in a new plate with distilled water. Do not keep the coverslips out of the solution longer than necessary. Incubate for 5 min (see Note 10).
Take the coverslips out of the water one by one, rinse twice again by dipping into water, and place in a new plate with aqueous uranyl acetate solution. Avoid drying during transfer. Incubate for 20 min at room temperature.
Wash off uranyl acetate solution with distilled water by transferring the samples or exchanging the solutions.
3.3. Dehydration and CPD
Drying of the samples is necessary to expose the surfaces for metal coating in a vacuum. However, plain drying in the open air generates major structural distortions. When the liquid-gas interface passes through the samples, the forces of the surface tension that are enormous at the cellular scale flatten the samples. During CPD, the temperature and pressure of a liquid are raised above its critical point, at which the phase boundary and surface tension do not exist. In this state, the liquid can well be considered as compressed gas. When the pressure is released, the samples remain dry with the 3D organization intact because they never experienced the surface tension (see Note 11). Carbon dioxide has reasonably low values of critical point pressure and temperature that can be tolerated by biological samples. However, a direct transfer of the samples from water to CO2 is not possible, and ethanol, which is freely miscible with both water and CO2, is used as an intermediate. For dehydration and CPD, the coverslips are stacked in the sample holder with pieces of lens tissue as spacers and processed simultaneously.
Cut lens tissue with loosely arranged fibers into pieces fitting the size of the CPD sample holder or a little larger (see Note 12).
Place the CPD sample holder into a wide container with distilled water. Put a piece of lens tissue at the bottom of the holder, place a coverslip with the cell side up on the lens tissue, and cover with another piece of lens tissue. Continue loading the coverslips into the holder, alternating them with pieces of lens tissue. All loading should be done under water. Make sure that the coverslips are minimally exposed to air during loading. Do not overload the holder, as it will interfere with the exchange of solutions (see Note 13). Keep track of sample identity based on its position in the holder.
Put a stirrer bar into a 50 mL glass beaker, place a scaffold over it, and add 10% ethanol in the amount sufficient to cover the CPD holder. Quickly transfer the CPD holder with samples from the water-filled container into the beaker. Place the beaker on a magnetic stirrer, and stir for at least 5 min.
Prepare another beaker with a stirrer bar and a scaffold and add 20% ethanol. Transfer the CPD holder from the first beaker into the second, and stir for at least 5 min.
Continue dehydration by transferring the CPD holder sequentially through the remaining graded ethanols, at least 5 min in each: 40%, 60%, 80%, and 100% (twice). Alternate the two beakers with their scaffolds and stirrer bars (see Note 14).
Place the CPD holder into a beaker with 0.2% uranyl acetate in ethanol to fully cover it. Incubate for 20 min. Stirring is not necessary.
Transfer the CPD holder through beakers with 100% ethanol (twice) and dehydrated 100% ethanol dried over a molecular sieve (twice), as in steps 3–5. Stir for at least 5 min in each.
Fill the chamber of the CPD apparatus with dehydrated ethanol, just sufficient to cover the CPD holder. Place the holder in the ethanol and close the lid. Operate the CPD according to the manufacturer’s instructions or following the procedure below (see Note 15).
Open the CO2 tank. Cool down the CPD chamber to 10–15 °C to keep the CO2 in a liquid state. Maintain this temperature until the heating step (see Note 16).
Open the inlet valve on the CPD to allow the CO2 to fill the chamber. If the device is equipped with a magnetic stirrer, turn it on and keep it running till the heating step. Wait for 5 min. This is a mixing step, during which the ethanol in the chamber, the sample holder, and the samples are equilibrated with the liquid CO2 from the tank.
With the inlet valve still open, open the outlet valve slightly until you clearly see that the liquid exchange is happening in the chamber, but do not allow the level of the liquid to go below the top of the sample holder. Wait for 30 s. This is a washing step, when the ethanol–CO2 mixture is released from the chamber and replaced with pure CO2 from the tank. If the CPD is not equipped with a magnetic stirrer, shake the CPD manually during the washing to mix the chamber contents better (see Note 17).
Close the outlet valve. Wait for 5 min.
Repeat steps 11 and 12 nine more times (total of ten washes). After the last wash, wait until the chamber is completely filled with CO2; that may take less than 5 min.
Close both the inlet and outlet valves and turn on the heat. As the chamber is isolated, heating will raise both the temperature and the pressure. Wait until both parameters exceed the critical values for CO2 (critical pressure = 1072 psi or 73 atm, critical temperature = 31 °C) and reach values of ~1250 psi (85 atm) and ~40 °C. If one value is reached sooner than the other, maintain the former at a steady-state level by turning the heater or the outlet valve on and off until both the values are reached.
Open the outlet valve slightly to slowly release the pressure until it reaches the atmospheric pressure. It should take about 10 min (see Note 18).
Open the CPD chamber, remove the sample holder and immediately put it into a desiccator.
3.4. Platinum and Carbon Coating
Platinum shadowing generates the contrast of the samples. The angle and the thickness of the coating are critical parameters influencing the quality of the image. Lower angles provide higher contrast but do not penetrate deep into the sample. Thinner coats provide higher resolution, but lower contrast. For cellular studies, we shadow platinum at a ~45°angle with rotation to achieve a ~2 nm thickness of the coat, which is controlled by the thickness monitor. Carbon is applied from the top of the samples with a thickness of 3.5–5 nm. The basic steps of coating are listed below. Use the equipment manual for detailed operation.
Open the coating chamber of the vacuum evaporator. Load the evaporation materials, platinum and carbon. Adjust the angles of coating.
Mount the samples onto the rotary stage of the vacuum evaporator using a double-sided tape. This will prevent the dislodging of the samples during rotation (see Note 19). To prevent damage of the samples by ambient humidity, perform mounting as quickly as possible (see Note 20).
Pump down the coating chamber till ~5 × 10−6 mbar.
Turn on the stage rotation, shadow with platinum (2 nm) and then with carbon (3.5–5 nm) (see Note 21).
Vent the coating chamber. Remove the samples together with the mounting tape and place in a Petri dish. The samples can be safely stored at ambient conditions at this stage.
3.5. Preparation of Replicas
The release of the replicas from the coverslips is achieved by floating of Replicas the coverslips onto the surface of hydrofluoric acid solution, which dissolves glass. After that, the replicas are washed and mounted on EM grids.
While the coverslips are still attached to the sticky tape, scratch the coated surface of each coverslip with a needle or a razor blade to make regions fitting the size of an EM grid.
Fill the wells of a 12-well plate with ~10% HF almost to the top, which makes replica handling easier.
Detach a coverslip from the sticky tape and place it on the surface of the HF solution, so that the coverslip remains floating. Wait until the glass sinks, leaving the platinum replica floating. The replica will fall apart along the scratches made in step 1 (see Note 22).
Fill the wells of another 12-well plate with distilled water, and add a trace amount of a detergent to decrease the surface tension of water. Stock solution of the detergent is prepared by dissolving ~1 drop of detergent in ~20 mL of water. To make a working solution, dip a platinum loop into the stock solution, take it out (this will create a film on the loop), and dip the loop into a water-filled well. The final concentration of detergent is ~10−6% (see Note 23).
Using a platinum loop, transfer the replica pieces onto the surface of the detergent-containing water. Wait for 1 min or more.
Fill the wells of another 12-well plate with distilled water without detergent. Transfer the replica pieces onto the surface of the water. Wait for 1 min or more.
Pick up the replica pieces onto Formvar-coated EM grids with the lower side of the replica attached to the Formvar film. The technique of replica mounting is similar to the way thin sections are picked up onto grids. Fasten a grid in a pair of forceps, partially submerge the grid into water at a ~45° angle, bring the grid close to a piece of replica and allow them to make contact; then, gently pull the grid out of the water making sure that the replica piece remains attached to, and spreads over, the grid (see Note 24). Dry grids.
Examine the samples in TEM. Present the images in inverse contrast (as negatives) because it gives a more natural view of the structure, as if illuminated with scattered light.
3.6. Immunogold PREM
Structural information has much greater value if the identity of the structures is known. Immunostaining is a conventional way to identify cellular components. For EM purposes, the antibodies are labeled with electron-dense markers. A popular marker, colloidal gold, has a higher electron density than platinum and thus is appropriate for PREM. For successful immunogold PREM, a primary antibody should work after glutaraldehyde fixation, which optimally preserves the structure (see Note 25).
After glutaraldehyde fixation (Subheading 3.2, step 1), wash the samples with three changes of PBS. Incubate for at least 5 min in the last change.
Quench samples with NaBH4 in PBS for 10 min at room temperature. Shake off the bubbles occasionally. Rinse with PBS as in step 1.
Incubate with blocking solution for 20 min at room temperature.
Apply primary antibody at concentration giving bright staining by light microscopy. Incubate for 30–45 min at room temperature. Wash with PBS as in step 1.
Rinse once in immunogold buffer with 0.1% BSA. Apply gold-conjugated secondary antibody diluted ~1:10 in immunogold buffer with 1% BSA. Incubate overnight at room temperature in a sealed container in moist conditions (see Note 26).
Rinse in immunogold buffer with 0.1% BSA as in step 1, and perform the remaining steps starting from Subheading 3.2, step 1 (see Note 27).
3.7. Correlative Light Microscopy and PREM (CLEM)
CLEM combines the advantages of both microscopic techniques, namely, the high spatial resolution of EM and the high temporal resolution of optical live imaging. In this procedure, the cell dynamics is recorded by light microscopy, and then the same cell is analyzed by EM. The correlative analysis is extremely important from at least two points of view: to control for potential artifacts and to establish functional connections between the cytoskeletal organization and the cell’s motile behavior or the dynamics of cytoskeletal components [79, 80]. Modifications of the basic procedure as required for correlative PREM are described below.
- Cell Culture and Light Microscopy.
- Grow cells in dishes with marked coverslips mounted over a hole in the bottom of the dish. To make a dish, drill a 18 mm hole in the bottom of a 35 mm dish; polish the edges to remove any burrs; apply a thin layer of vacuum grease just outside the edges of the hole; place a marked coverslip symmetrically over the hole with the coated side facing up; and press firmly to spread the grease until it forms a clear circle and all the air bubbles are gone (see Note 28).
- While imaging a region of interest, mark its position on a map with a pattern of reference marks.
- Because of the large difference in the resolution of light microscopy and PREM, perform light microscopy at the highest possible resolution.
- PREM is able to reveal even minor photodamage, not recognizable at the light microscopic level; therefore, keep the illumination of the samples to a minimum.
Extraction: Change the culture medium to extraction solution as soon as possible after the acquisition of the last live image, and take another image after extraction. It will serve as a reference to correlate the light and PREM images.
Fixation: Perform all the fixation and washing steps with the marked coverslips still attached to the dishes, by exchanging solutions.
- Dehydration: Before loading the coverslips into the CPD sample holder, excise the central marked area of the coverslip as following:
- Wipe away the immersion oil from the bottom of the dish using dry cotton swabs first, followed by ethanol-soaked swabs.
- Detach the marked coverslip from the bottom of the dish, and immediately place it into a water-filled 100 mm Petri dish.
- Lightly press down the coverslip to allow the vacuum grease on the underside to secure the coverslip in the dish. Make sure that grease does not contaminate the region of interest.
-
Use a diamond pencil as a cutter and a razor blade as a ruler to cut off the greasy margins of the coverslip. Do not press the pencil hard, but instead make several light cuts along the same line, guided by the razor blade, until the margin detaches. Move it out of the way and cut off another margin. This procedure should be done with the coverslip completely submerged in the water (see Note 29). When done, transfer the excised central region to another water-filled container. Use a new dish for the next coverslip, as the remaining glass crumbs may cause shattering of the coverslip.All subsequent processing, including coating, is performed as in the basic procedure.
- Preparation of replicas: After the samples are coated, the region of interest needs to be specifically recovered for PREM analysis as follows:
- Immobilize a coated coverslip in the middle of a 100 mm Petri dish, with two pieces of double-sided tape positioned under opposite corners of the coverslip, so that the region of interest is not obstructed and the coverslip is not attached too strongly.
- Under a dissection microscope, localize the cells of interest using locator marks. Dried and shadowed cells have sufficient contrast for their shape to be seen even at low magnification. If necessary, use a regular light microscope with a low power lens.
- Using a razor blade (or a needle), make cuts in the platinum–carbon layer around the region of interest (see Note 30). To facilitate the separation of this region from the rest of the replica after HF treatment, make additional cuts connecting the region of interest with the edges of the coverslip.
- Make a drawing on the map to depict the exact shape of the outlined region of interest, which will help to identify it from among the other replica pieces during replica preparation.
- Perform the washing and mounting on grids, as described in the basic protocol, handling only the replica piece with the region of interest. While mounting on a grid, use a dissection microscope to make sure that the cell of interest does not go to a grid bar; or use single-hole grids.
3.8. Actin Depletion
Depletion of abundant actin filaments from the cytoskeletons by gelsolin treatment exposes other normally obscured cytoskeletal structures, thus permitting detailed analyses of their organization in cells (see Note 31)
After detergent extraction (Subheading 3.1, step 4), rinse coverslips once with gelsolin buffer supplemented with 10 μM taxol to preserve microtubules (see Note 32).
Make small drops of gelsolin solution on a piece of Parafilm placed on the bottom of a Petri dish, one drop per coverslip at sufficient distances from one another (see Note 33).
Place a coverslip onto the gelsolin drop with cell side up and press the coverslip down to allow the gelsolin solution to completely cover the coverslip surface. Close the dish and incubate 1 h at room temperature in moist conditions (see Note 34).
Rinse three times with PEM buffer and proceed to sample fixation (Subheading 3.2, step 1).
4. Notes
Small coverslips allow for better exchange of solutions during dehydration and CPD and thus for better quality of samples at the end.
Stock solutions with a concentration of more than 2× relative to the working concentration would change pH significantly after dilution to the working concentration. Free acid PIPES is not soluble in water and forms a milky suspension but becomes soluble upon neutralization. KOH granules can be used for neutralization initially, until the solution almost clears. However, remember to allow enough time for the granules to dissolve before adding more. Finish the pH adjustment with 1 N KOH. KOH is preferable over NaOH because K+-containing buffers more faithfully imitate the cytoplasm composition.
PEG is a nonspecific stabilizer of the cytoskeleton; phalloidin and taxol are specific stabilizers of actin filaments and microtubules, respectively.
Low molecular weight tannic acid (MW ~322), also called digallic acid, which was originally manufactured by Mallinckrodt Inc. under the product number 1764, has been shown to yield best results in preserving cells structures, including fibrillar components of the cytoplasm [81] and the actin cytoskeleton, in particular [82], as compared with high molecular weight tannins (≥1400). We are still using the Mallinckrodt brand of low molecular weight tannic acid that was previously offered by Fisher but currently discontinued. Digallic acid is now offered by other companies, e.g., Santa Cruz Biotechnology, and appears to be a valid substitute, although we did not test it.
Commercially available etched coverslips are not suitable for PREM, as the etched marks are not visible in TEM.
Rinsing with PBS is optional, but if omitted, the extraction solution at the next step should be added in sufficient quantity to overcome the potentially harmful effects from the remaining medium and serum.
High viscosity of the extraction solution makes it poorly miscible with PBS or medium, in which cells were submerged in the preceding step. Without stirring, slow diffusion of the detergent would lead to gradual extraction and result in severe cell distortion and cytoskeleton disruption. On the other hand, vigorous stirring would disrupt the cytoskeleton mechanically. Thus, a fine compromise should be found. The choice of the extraction solution depends on a cell type and a goal. For a new experimental system, try different options in preliminary experiments. A basic extraction solution (Triton X-100 in PEM) produces better clarity of the cytoskeleton but makes it easier to damage the cells during extraction. If using this protocol, handle the samples extremely gently, and use phalloidin and taxol to better preserve the actin filaments and microtubules, respectively. The addition of PEG to the extraction solution allows for better preservation of the structure, but it also retains many cytoskeleton-associated components, which may partially obscure the filament arrangement. Such an effect is increased with increasing PEG concentration and molecular weight, but PEGs in the range of 20,000–40,000 act similarly. We typically use 2% PEG (35,000). For extremely fragile and poorly attached cells, low concentrations of glutaraldehyde can be added to the extraction solution as stabilizing supplements. In this case, the detergent and fixative compete with each other, and the results depend on their ratio. The extraction solution containing 0.5% Triton X-100 and 0.25% glutaraldehyde in PEM buffer worked well in our experiments.
For PEG-containing extraction solutions, use a longer washing time, at least 1 min in each change. If the stabilizing drugs are used during extraction, add them also to the rinsing buffer in a fourfold to fivefold lower concentration.
It is convenient to use a multiwell plate with numbered wells (24-well for 6–8 mm coverslips or 12-well for 9–12 mm coverslips) to transfer the samples. This makes it possible to combine samples from different experiments for PREM processing while keeping parallel samples in the original container as a backup.
Uranyl acetate and tannic acid react with each other and form a precipitate. Extensive washing is important to avoid the formation of debris on the samples.
Two alternative procedures have been used to dry samples before metal coating: freeze-drying [41, 49, 57, 83–87] and air-drying from an organic solvent hexamethyldisilazane (HMDS) [47, 88–90]. Freeze-drying, when successful, results in excellent sample quality but is limited by a low success rate. In some published images of freeze-dried samples, neighboring filaments appear fused suggesting inadequate drying. This problem might arise from insufficient rate of ice sublimation or partial sample melting. Air-drying from HMDS is becoming popular due to its simplicity. As compared with water, drying from HMDS causes less sample distortion because HMDS has low, but not zero, surface tension. Although CPD and HMDS drying are believed to be equally good, direct comparisons of these two procedures were conducted only using microscopy techniques that were insufficient to resolve individual cytoskeletal filaments [91–93]. We directly compared sample quality by PREM of cytoskeletal preparations prepared using either CPD or air-drying from HMDS. We found that the samples were comparable at low and medium magnifications, but at high magnifications required to resolve individual actin filaments, the quality of HDMS-dried samples was clearly inferior. Specifically, in CPD samples, actin filaments had uniform diameters and were clearly distinguishable from one another even in tight bundles. In contrast, in the HDMS-dried samples, actin filaments often fused with each other and with an underlying surface. Similar artifacts can also be seen in published PREM images obtained after HMDS drying.
Pieces of lens tissue slightly larger, than the holder’s bottom area, will make minor wrinkles which promote looser packing of the coverslips in the holder and facilitate the liquid exchange.
The acceptable number of samples for a load depends on the sizes of both the holder and the coverslips. For an 18 × 12 mm holder and ~7 × 7 mm coverslips, the maximum load is 12. For larger coverslips, the load should be decreased. Larger holders may accept more samples, especially if the coverslips are staggered.
It is not necessary to dry the beakers before the next incubation, as the ethanol concentration may not be exact, except for 100% ethanol, when it is better to dry the beakers and scaffolds. Incubation for 5 min is minimal. For larger coverslips or greater loads, increase the incubation time.
The process of CPD is most commonly used for scanning EM and production of microelectronics. Consequently, the protocols suggested by the manufacturers or incorporated into automated procedures of CPDs are designed for those applications. PREM, however, is more demanding in terms of sample quality. We adjusted the CPD processing to fully remove all traces of ethanol from the samples before bringing the CO2 to the critical point; this helps to eliminate minor artifacts that appear as a fusion of closely positioned filaments in the cytoskeleton. The CPD operation described here is applicable to manual CPDs, such as Samdri PVD-3D (Tousimis), which we use in the lab, or to semi-automatic CPDs switched to a manual mode of operation, e.g., Samdri-795 (Tousimis).
Lower temperatures are acceptable, but the diffusion of ethanol from the samples will be slower, so longer washing time is needed. Warming up the chamber till the ambient temperature is allowed if the outlet valve of the CPD is closed, and the CO2 remains pressurized and in liquid form. However, it is important to cool down the chamber back to 15 °C before opening the outlet valve for purging out the ethanol–CO2 mixture.
Letting the liquid level go below the samples will irreversibly damage them. On the other hand, a too low rate of liquid exchange is also a mistake. Adjust the outlet valve to get a steady-state liquid level, about halfway from the top of the holder to the top of the chamber. This will also make the liquid mixing more efficient. Although the shaking step sounds a bit amusing, it does make difference by helping to remove the ethanol from the samples.
Fast release of pressure may cause condensation of CO2 back to liquid state and ruin the dried samples.
Conventional Scotch double-sided tape becomes too sticky in a vacuum, preventing the safe detachment of the samples after coating. To avoid this problem, sandwich the double-sided tape between the glued parts of two Post-It notes, with the sticky sides exposed.
Humidity in the room should be below 35%; the 35–50% humidity level may be acceptable, but much caution and the speedy mounting of the samples is required; humidity >50% is not acceptable. Try to run a powerful dehumidifier in the latter case.
If the evaporator is not equipped with a thickness monitor, the thickness of the coating may be adjusted in the preliminary experiments based on the amount of coating material loaded (for platinum) or used (carbon) for evaporation.
To safely float a coverslip, grab it with the forceps from the top for parallel edges, lift the coverslip, and carefully place it onto the liquid surface, keeping it in a horizontal position. Practice first by placing a coverslip onto a clean solid surface. Alternatively, a coverslip can be placed first on a platinum loop bent at 90° angle and then loaded horizontally onto the liquid surface.If the replica does not fall apart along the scratches, use the platinum loop to reach the replica from below, lightly touch it, and pull or shake it to detach it from other pieces. Extreme care should be used not to ruin the replicas with these manipulations.
Water has much greater surface tension than HF, which may cause severe replica breakage, if detergent is not added. Test the detergent concentration before applying it to the samples. An overdose of detergent causes shrinkage and even drowning of the replicas. Stock solutions should be changed at least every 2 weeks. Old detergents leave contamination on the samples, looking like semi-transparent films between filaments. House-hold non-colored detergent, such as Ivory, works fine. Triton X-100 can also be used, but it should be prepared fresh every time.
Sometimes, replicas appear to be repelled by the grid, making it difficult to establish the initial contact between a replica piece and a grid. Try to gently guide a piece of replica to the wall of the well to restrict its motility, and then pick it up. However, there is a danger of smashing the replica against the wall with this approach. Treating grids with glow discharge prior to capturing replicas also helps.
The efficiency of staining may be improved if the cells are fixed with a lower (e.g., 0.2%) glutaraldehyde concentration before staining. For some antibodies that do not work after glutaraldehyde fixation, it may be possible to stain unfixed samples by incubating them with primary antibodies diluted in PEM with phalloidin and taxol for 10–15 min, then fixing with glutaraldehyde, quenching, washing and staining with a secondary antibody.
Gold size of 10–20 nm is optimal for this technique, as smaller particles are poorly visible and larger particles are too disruptive for an image.
Second glutaraldehyde fixation helps retaining gold-conjugated secondary antibody during subsequent procedures.
The coverslips can be mounted either inside or outside the dish, but inside mounting is more convenient at later stages, when the centerpiece of the coverslip needs to be cut out. For mounting, use a minimal amount of grease, just sufficient to seal the dish; excessive grease causes complications at later stages. Commercially available glass-bottom dishes can also be used, but it may be difficult to remove the coverslip from the bottom for PREM processing.
Cutting under water is more difficult than in the air; therefore, use a sharp diamond pencil and avoid glass crumbs.
To reduce the effect of shaky hands, hold a razor blade with one hand with the sharp blade corner pointing down; stabilize the blade by putting the index finger of the other hand onto the blunt blade corner pointing up; rest the forearms on the table and the other fingers of both hands on the microscope stage and/or dish edges; keep the blade above the sample and find its unfocused image in the microscope; slowly bring down the sharp corner of the blade until it almost comes to focus; bring the blade corner to a region where a cut is to be made; and under microscope control, bring it down to the sample and make a scratch.
It appears that a similar goal of depleting actin filaments could be achieved using actin-depolymerizing drugs, such as latrunculins or cytochalasins. However, these drugs need to be applied to living cells, which severely affects cell physiology and causes multiple structural rearrangements of the cytoskeleton. In contrast, gelsolin is applied to cytoskeletal preparations, not to living cells, and non-actin structures retain their endogenous organization in the course of treatment [46, 64, 65].
Taxol should be added to all solutions for preservation of microtubules, because otherwise all microtubules will be destroyed by the Ca2+-containing gelsolin buffer. If using calcium-independent N-terminal fragment of gelsolin [94], rather than the full length gelsolin, it is possible to use PEM buffer instead of gelsolin buffer as vehicle. Yet, addition of taxol greatly helps in proper preservation of microtubules in either case.
Hydrophobic surface of Parafilm prevents gelsolin solution from spreading. The volume of drops depends on the size of coverslips and should be sufficient to cover the coverslip surface.
One option to generate moist conditions is to attach a water-soaked filter paper to the lid to the Petri dish, shake off extra water, and close the lid. Another option is to put wet filter paper around the dish periphery.
Acknowledgments
The author acknowledges the current support from NIH grant R01 GM 095977.
References
- 1.Wohlfarth-Bottermann KE (1962) Weitrei-chende fibrillare Protoplasmadifferenzierungen und ihre Bedeutung fur die Protoplasmastromung. I. Elektronenmikroskopischer Nachweis und Feinstruktur. Protoplasma 54:514–539 [Google Scholar]
- 2.Ledbetter MC, Porter KR (1963) A “microtubule” in plant cell fine structure. J Cell Biol 19 (1):239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabatini DD, Bensch K, Barrnett RJ (1963) Cytochemistry and electron microscopy. The preservation of cellular ultrastructure and enzymatic activity by aldehyde fixation. J Cell Biol 17:19–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abercrombie M, Heaysman JE, Pegrum SM (1971) The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Exp Cell Res 67(2):359–367 [DOI] [PubMed] [Google Scholar]
- 5.Wohlfarth-Bottermann KE (1964) Differentiations of the ground cytoplasm and their significance for the generation of the motive force of ameboid movement. In: Allen RD, Kamiya N (eds) Primitive motile systems in cell biology. Academic, pp 79–109 [Google Scholar]
- 6.Kukulski W, Schorb M, Kaksonen M, Briggs JA (2012) Plasma membrane reshaping during endocytosis is revealed by time-resolved electron tomography. Cell 150(3):508–520. 10.1016/j.cell.2012.05.046 [DOI] [PubMed] [Google Scholar]
- 7.Luduena MA, Wessells NK (1973) Cell locomotion, nerve elongation, and microfilaments. Dev Biol 30(2):427–440 [DOI] [PubMed] [Google Scholar]
- 8.Tilney LG, Derosier DJ, Mulroy MJ (1980) The organization of actin filaments in the stereocilia of cochlear hair cells. J Cell Biol 86 (1):244–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu M, Huang B, Graham M, Raimondi A, Heuser JE, Zhuang X, De Camilli P (2010) Coupling between clathrin-dependent endocytic budding and F-BAR-dependent tubulation in a cell-free system. Nat Cell Biol 12 (9):902–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Small JV, Isenberg G, Celis JE (1978) Polarity of actin at the leading edge of cultured cells. Nature 272(5654):638–639 [DOI] [PubMed] [Google Scholar]
- 11.Damiano-Guercio J, Kurzawa L, Mueller J, Dimchev G, Schaks M, Nemethova M, Pokrant T, Bruhmann S, Linkner J, Blanchoin L, Sixt M, Rottner K, Faix J (2020) Loss of Ena/VASP interferes with lamellipodium architecture, motility and integrin-dependent adhesion. elife 9:e55351. 10.7554/eLife.55351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mueller J, Szep G, Nemethova M, de Vries I, Lieber AD, Winkler C, Kruse K, Small JV, Schmeiser C, Keren K, Hauschild R, Sixt M (2017) Load adaptation of lamellipodial actin networks. Cell 171(1):188–200.e116. 10.1016/j.cell.2017.07.051 [DOI] [PubMed] [Google Scholar]
- 13.Sturner T, Tatarnikova A, Mueller J, Schaffran B, Cuntz H, Zhang Y, Nemethova M, Bogdan S, Small V, Tavosanis G (2019) Transient localization of the Arp2/3 complex initiates neuronal dendrite branching in vivo. Development 146(7):dev171397. 10.1242/dev.171397 [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty S, Jasnin M, Baumeister W (2020) Three-dimensional organization of the cytoskeleton: a cryo-electron tomography perspective. Protein Sci 29(6):1302–1320. 10.1002/pro.3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopek BG, Paez-Segala MG, Shtengel G, Sochacki KA, Sun MG, Wang Y, Xu CS, van Engelenburg SB, Taraska JW, Looger LL, Hess HF (2017) Diverse protocols for correlative super-resolution fluorescence imaging and electron microscopy of chemically fixed samples. Nat Protoc 12(5):916–946. 10.1038/nprot.2017.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murata K, Wolf M (2018) Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim Biophys Acta 1862(2):324–334. 10.1016/j.bbagen.2017.07.020 [DOI] [PubMed] [Google Scholar]
- 17.Stark H, Chari A (2016) Sample preparation of biological macromolecular assemblies for the determination of high-resolution structures by cryo-electron microscopy. Microscopy 65 (1):23–34. 10.1093/jmicro/dfv367 [DOI] [PubMed] [Google Scholar]
- 18.Lucic V, Rigort A, Baumeister W (2013) Cryoelectron tomography: the challenge of doing structural biology in situ. J Cell Biol 202 (3):407–419. 10.1083/jcb.201304193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel BD, Schaffer M, Albert S, Asano S, Plitzko JM, Baumeister W (2015) In situ structural analysis of Golgi intracisternal protein arrays. Proc Natl Acad Sci USA 112 (36):11264–11269. 10.1073/pnas.1515337112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer TD, Dash PK, Liu J, Waxham MN (2018) Morphology of mitochondria in spatially restricted axons revealed by cryo-electron tomography. PLoS Biol 16(9):e2006169. 10.1371/journal.pbio.2006169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman DP, Shtengel G, Xu CS, Campbell KR, Freeman M, Wang L, Milkie DE, Pasolli HA, Iyer N, Bogovic JA, Stabley DR, Shirinifard A, Pang S, Peale D, Schaefer K, Pomp W, Chang CL, Lippincott-Schwartz J, Kirchhausen T, Solecki DJ, Betzig E, Hess HF (2020) Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science 367(6475):eaaz5357. 10.1126/science.aaz5357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurley JH, Nogales E (2016) Next-generation electron microscopy in autophagy research. Curr Opin Struct Biol 41:211–216. 10.1016/j.sbi.2016.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorgens DM, Inman JL, Wojcik M, Robertson C, Palsdottir H, Tsai WT, Huang H, Bruni-Cardoso A, Lopez CS, Bissell MJ, Xu K, Auer M (2017) Deep nuclear invaginations are linked to cytoskeletal filaments—integrated bioimaging of epithelial cells in 3D culture. J Cell Sci 130(1):177–189. 10.1242/jcs.190967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Cuellar LK, Forster F, Hyman AA, Plitzko JM, Baumeister W (2016) Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 351 (6276):969–972. 10.1126/science.aad8857 [DOI] [PubMed] [Google Scholar]
- 25.Chakraborty S, Mahamid J, Baumeister W (2020) Cryoelectron tomography reveals nanoscale organization of the cytoskeleton and its relation to microtubule curvature inside cells. Structure 28(9):991–1003.e1004. 10.1016/j.str.2020.05.013 [DOI] [PubMed] [Google Scholar]
- 26.Grange M, Vasishtan D, Grunewald K (2016) Cellular electron cryo tomography and in situ sub-volume averaging reveal the context of microtubule-based processes. J Struct Biol 197(2):181–190. 10.1016/j.jsb.2016.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul DM, Mantell J, Borucu U, Coombs J, Surridge KJ, Squire JM, Verkade P, Dodding MP (2020) In situ cryo-electron tomography reveals filamentous actin within the microtubule lumen. J Cell Biol 219(9): e201911154. 10.1083/jcb.201911154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson KL, Page C, Swift MF, Suraneni P, Janssen ME, Pollard TD, Li R, Volkmann N, Hanein D (2017) Nano-scale actin-network characterization of fibroblast cells lacking functional Arp2/3 complex. J Struct Biol 197 (3):312–321. 10.1016/j.jsb.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marston DJ, Anderson KL, Swift MF, Rougie M, Page C, Hahn KM, Volkmann N, Hanein D (2019) High Rac1 activity is functionally translated into cytosolic structures with unique nanoscale cytoskeletal architecture. Proc Natl Acad Sci USA 116(4):1267–1272. 10.1073/pnas.1808830116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jasnin M, Crevenna AH (2016) Quantitative analysis of filament branch orientation in Listeria actin comet tails. Biophys J 110 (4):817–826. 10.1016/j.bpj.2015.07.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jasnin M, Beck F, Ecke M, Fukuda Y, Martinez-Sanchez A, Baumeister W, Gerisch G (2019) The architecture of traveling actin waves revealed by cryo-electron tomography. Structure 27(8):1211–1223.e1215. 10.1016/j.str.2019.05.009 [DOI] [PubMed] [Google Scholar]
- 32.Heuser J (1981) Preparing biological samples for stereomicroscopy by the quick-freeze, deep-etch, rotary-replication technique. Methods Cell Biol 22:97–122 [DOI] [PubMed] [Google Scholar]
- 33.Svitkina TM (2017) Platinum replica electron microscopy: imaging the cytoskeleton globally and locally. Int J Biochem Cell Biol 86:37–41. 10.1016/j.biocel.2017.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steere RL (1957) Electron microscopy of structural detail in frozen biological specimens. J Cell Biol 3(1):45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friend JE, Sayyad WA, Arasada R, McCormick CD, Heuser JE, Pollard TD (2018) Fission yeast Myo2: molecular organization and diffusion in the cytoplasm. Cytoskeleton (Hoboken) 75(4):164–173. 10.1002/cm.21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heuser J (1989) Protocol for 3-D visualization of molecules on mica via the quick-freeze, deep-etch technique. J Electron Microsc Tech 13(3):244–263 [DOI] [PubMed] [Google Scholar]
- 37.Loesser KE, Franzini-Armstrong C (1990) A simple method for freeze-drying of macromolecules and macromolecular complexes. J Struct Biol 103(1):48–56 [DOI] [PubMed] [Google Scholar]
- 38.Chikina AS, Svitkina TM, Alexandrova AY (2019) Time-resolved ultrastructure of the cortical actin cytoskeleton in dynamic membrane blebs. J Cell Biol 218(2):445–454. 10.1083/jcb.201806075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Efimova N, Svitkina TM (2018) Branched actin networks push against each other at adherens junctions to maintain cell-cell adhesion. J Cell Biol 217(5):1827–1845. 10.1083/jcb.201708103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henson JH, Ditzler CE, Germain A, Irwin PM, Vogt ET, Yang S, Wu X, Shuster CB (2017) The ultrastructural organization of actin and myosin II filaments in the contractile ring: new support for an old model of cytokinesis. Mol Biol Cell 28(5):613–623. 10.1091/mbc.E16-06-0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heuser JE, Kirschner MW (1980) Filament organization revealed in platinum replicas of freeze-dried cytoskeletons. J Cell Biol 86 (1):212–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ong K, Svitkina T, Bi E (2016) Visualization of in vivo septin ultrastructures by platinum replica electron microscopy. Methods Cell Biol 136:73–97. 10.1016/bs.mcb.2016.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott BL, Sochacki KA, Low-Nam ST, Bailey EM, Luu Q, Hor A, Dickey AM, Smith S, Kerkvliet JG, Taraska JW, Hoppe AD (2018) Membrane bending occurs at all stages of clathrin-coat assembly and defines endocytic dynamics. Nat Commun 9(1):419. 10.1038/s41467-018-02818-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sochacki KA, Shtengel G, van Engelenburg SB, Hess HF, Taraska JW (2014) Correlative super-resolution fluorescence and metal-replica transmission electron microscopy. Nat Methods 11(3):305–308. 10.1038/nmeth.2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Svitkina TM, Shevelev AA, Bershadsky AD, Gelfand VI (1984) Cytoskeleton of mouse embryo fibroblasts. Electron microscopy of platinum replicas. Eur J Cell Biol 34(1):64–74 [PubMed] [Google Scholar]
- 46.Svitkina TM, Verkhovsky AB, Borisy GG (1995) Improved procedures for electron microscopic visualization of the cytoskeleton of cultured cells. J Struct Biol 115(3):290–303 [DOI] [PubMed] [Google Scholar]
- 47.Vassilopoulos S, Gibaud S, Jimenez A, Caillol G, Leterrier C (2019) Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. Nat Commun 10(1):5803. 10.1038/s41467-019-13835-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang C, Svitkina TM (2019) Ultrastructure and dynamics of the actin-myosin II cytoskeleton during mitochondrial fission. Nat Cell Biol 21(5):603–613. 10.1038/s41556-019-0313-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirokawa N (1989) Quick-freeze, deep-etch electron microscopy. J Electron Microsc (Tokyo) 38 Suppl:S123–S128 [PubMed] [Google Scholar]
- 50.Meyer HW, Richter W (2001) Freeze-fracture studies on lipids and membranes. Micron 32 (6):615–644 [DOI] [PubMed] [Google Scholar]
- 51.Suleiman H, Zhang L, Roth R, Heuser JE, Miner JH, Shaw AS, Dani A (2013) Nanoscale protein architecture of the kidney glomerular basement membrane. elife 2:e01149. 10.7554/eLife.01149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Small JV (1981) Organization of actin in the leading edge of cultured cells: influence of osmium tetroxide and dehydration on the ultrastructure of actin meshworks. J Cell Biol 91(3 Pt 1):695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Svitkina TM, Borisy GG (1998) Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol 298:570–592. 10.1016/S0076-6879(98)98045-4 [DOI] [PubMed] [Google Scholar]
- 54.Svitkina T (2007) Electron microscopic analysis of the leading edge in migrating cells. Methods Cell Biol 79:295–319. 10.1016/S0091-679X(06)79012-4 [DOI] [PubMed] [Google Scholar]
- 55.Collins A, Warrington A, Taylor KA, Svitkina T (2011) Structural organization of the actin cytoskeleton at sites of clathrin-mediated endocytosis. Curr Biol 21(14):1167–1175. 10.1016/j.cub.2011.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heuser J (2000) The production of ‘cell cortices’ for light and electron microscopy. Traffic 1 (7):545–552 [DOI] [PubMed] [Google Scholar]
- 57.Morone N, Usukura E, Narita A, Usukura J (2020) Improved unroofing protocols for cryo-electron microscopy, atomic force microscopy and freeze-etching electron microscopy and the associated mechanisms. Microscopy (Oxf) 69(6):350–359. 10.1093/jmicro/dfaa028 [DOI] [PubMed] [Google Scholar]
- 58.Brands R, Feltkamp CA (1988) Wet cleaving of cells: a method to introduce macromolecules into the cytoplasm. Application for immunolo-calization of cytosol-exposed antigens. Exp Cell Res 176(2):309–318 [DOI] [PubMed] [Google Scholar]
- 59.Massaad MJ, Oyoshi MK, Kane J, Koduru S, Alcaide P, Nakamura F, Ramesh N, Luscinskas FW, Hartwig J, Geha RS (2014) Binding of WIP to actin is essential for T Cell actin cytoskeleton integrity and tissue homing. Mol Cell Biol 34(23):4343–4354. 10.1128/MCB.00533-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peitsch CF, Beckmann S, Zuber B (2016) iMEM: isolation of plasma membrane for cryoelectron microscopy. Structure 24 (12):2198–2206. 10.1016/j.str.2016.09.016 [DOI] [PubMed] [Google Scholar]
- 61.Sato F, Asakawa H, Fukuma T, Terada S (2016) Semi-in situ atomic force microscopy imaging of intracellular neurofilaments under physiological conditions through the ‘sandwich’ method. Microscopy 65 (4):316–324. 10.1093/jmicro/dfw006 [DOI] [PubMed] [Google Scholar]
- 62.Shutova MS, Spessott WA, Giraudo CG, Svitkina T (2014) Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Curr Biol 24(17):1958–1968. 10.1016/j.cub.2014.07.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Svitkina TM, Verkhovsky AB, Borisy GG (1996) Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol 135(4):991–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verkhovsky AB, Borisy GG (1993) Non-sarcomeric mode of myosin II organization in the fibroblast lamellum. J Cell Biol 123 (3):637–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verkhovsky AB, Svitkina TM, Borisy GG (1995) Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J Cell Biol 131(4):989–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Svitkina TM, Borisy GG (2006) Correlative light and electron microscopy studies of cytoskeletal dynamics. In: Celis J (ed) Cell biology: a laboratory handbook, vol 3. vol 28, 3rd edn. Elsevier, pp 277–285 [Google Scholar]
- 67.Efimova N, Yang C, Chia JX, Li N, Lengner CJ, Neufeld KL, Svitkina TM (2020) Branched actin networks are assembled on microtubules by adenomatous polyposis coli for targeted membrane protrusion. J Cell Biol 219(9): e202003091. 10.1083/jcb.202003091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Efimova N, Korobova F, Stankewich MC, Moberly AH, Stolz DB, Wang J, Kashina A, Ma M, Svitkina T (2017) BetaIII spectrin is necessary for formation of the constricted neck of dendritic spines and regulation of synaptic activity in neurons. J Neurosci 37 (27):6442–6459. 10.1523/JNEUROSCI.3520-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ripoll L, Heiligenstein X, Hurbain I, Domingues L, Figon F, Petersen KJ, Dennis MK, Houdusse A, Marks MS, Raposo G, Delevoye C (2018) Myosin VI and branched actin filaments mediate membrane constriction and fission of melanosomal tubule carriers. J Cell Biol 217(8):2709–2726. 10.1083/jcb.201709055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sochacki KA, Dickey AM, Strub MP, Taraska JW (2017) Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nat Cell Biol 19(4):352–361. 10.1038/ncb3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Avinoam O, Schorb M, Beese CJ, Briggs JA, Kaksonen M (2015) Endocytic sites mature by continuous bending and remodeling of the clathrin coat. Science 348(6241):1369–1372. 10.1126/science.aaa9555 [DOI] [PubMed] [Google Scholar]
- 72.Harterink M, Vocking K, Pan X, Soriano Jerez EM, Slenders L, Freal A, Tas RP, van de Wetering WJ, Timmer K, Motshagen J, van Beuningen SFB, Kapitein LC, Geerts WJC, Post JA, Hoogenraad CC (2019) TRIM46 organizes microtubule fasciculation in the axon initial segment. J Neurosci 39(25):4864–4873. 10.1523/JNEUROSCI.3105-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kukulski W, Picco A, Specht T, Briggs JA, Kaksonen M (2016) Clathrin modulates vesicle scission, but not invagination shape, in yeast endocytosis. elife 5:e16036. 10.7554/eLife.16036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A, Schwab Y, Gross CT (2018) Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 9 (1):1228. 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wesolowska N, Avilov I, Machado P, Geiss C, Kondo H, Mori M, Lenart P (2020) Actin assembly ruptures the nuclear envelope by prying the lamina away from nuclear pores and nuclear membranes in starfish oocytes. elife 9:e49774. 10.7554/eLife.49774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nemethova M, Auinger S, Small JV (2008) Building the actin cytoskeleton: filopodia contribute to the construction of contractile bundles in the lamella. J Cell Biol 180 (6):1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shutova M, Yang C, Vasiliev JM, Svitkina T (2012) Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLoS One 7(7):e40814. 10.1371/journal.pone.0040814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Verkhovsky AB, Surgucheva IG, Svitkina TM, Tint IS, Gelfand VI (1987) Organization of stress fibers in cultured fibroblasts after extraction of actin with bovine brain gelsolin-like protein. Exp Cell Res 173(1):244–255 [DOI] [PubMed] [Google Scholar]
- 79.Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG (2003) Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol 160(3):409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang C, Czech L, Gerboth S, Kojima S, Scita G, Svitkina T (2007) Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol 5(11):e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simionescu N, Simionescu M (1976) Galloyl-glucoses of low molecular weight as mordant in electron microscopy. I. Procedure, and evidence for mordanting effect. J Cell Biol 70 (3):608–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maupin P, Pollard TD (1983) Improved preservation and staining of HeLa cell actin filaments, clathrin-coated membranes, and other cytoplasmic structures by tannic acid-glutaraldehyde-saponin fixation. J Cell Biol 96 (1):51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bishai EA, Sidhu GS, Li W, Dhillon J, Bohil AB, Cheney RE, Hartwig JH, Southwick FS (2013) Myosin-X facilitates Shigella-induced membrane protrusions and cell-to-cell spread. Cell Microbiol 15(3):353–367. 10.1111/cmi.12051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bridgman PC, Dailey ME (1989) The organization of myosin and actin in rapid frozen nerve growth cones. J Cell Biol 108(1):95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hartwig JH, Shevlin P (1986) The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J Cell Biol 103 (3):1007–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weng L, Enomoto A, Miyoshi H, Takahashi K, Asai N, Morone N, Jiang P, An J, Kato T, Kuroda K, Watanabe T, Asai M, Ishida-Takagishi M, Murakumo Y, Nakashima H, Kaibuchi K, Takahashi M (2014) Regulation of cargo-selective endocytosis by dynamin 2 GTPase-activating protein girdin. EMBO J 33(18):2098–2112. 10.15252/embj.201488289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE (2012) Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148 (5):973–987. 10.1016/j.cell.2011.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Elkhatib N, Bresteau E, Baschieri F, Rioja AL, van Niel G, Vassilopoulos S, Montagnac G (2017) Tubular clathrin/AP-2 lattices pinch collagen fibers to support 3D cell migration. Science 356(6343):aal4713. 10.1126/science.aal4713 [DOI] [PubMed] [Google Scholar]
- 89.Ferrari R, Martin G, Tagit O, Guichard A, Cambi A, Voituriez R, Vassilopoulos S, Chavrier P (2019) MT1-MMP directs force-producing proteolytic contacts that drive tumor cell invasion. Nat Commun 10 (1):4886. 10.1038/s41467-019-12930-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Franck A, Laine J, Moulay G, Lemerle E, Trichet M, Gentil C, Benkhelifa-Ziyyat S, Lacene E, Bui MT, Brochier G, Guicheney P, Romero N, Bitoun M, Vassilopoulos S (2019) Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Mol Biol Cell 30(5):579–590. 10.1091/mbc.E18-11-0718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Braet F, De Zanger R, Wisse E (1997) Drying cells for SEM, AFM and TEM by hexamethyldisilazane: a study on hepatic endothelial cells. J Microsc 186(Pt 1):84–87. 10.1046/j.1365-2818.1997.1940755.x [DOI] [PubMed] [Google Scholar]
- 92.Bray DF, Bagu J, Koegler P (1993) Comparison of hexamethyldisilazane (HMDS), Peldri II, and critical-point drying methods for scanning electron microscopy of biological specimens. Microsc Res Tech 26(6):489–495. 10.1002/jemt.1070260603 [DOI] [PubMed] [Google Scholar]
- 93.Sokolova AI, Pavlova ER, Khramova YV, Klinov DV, Shaitan KV, Bagrov DV (2019) Imaging human keratinocytes grown on electrospun mats by scanning electron microscopy. Microsc Res Tech 82(5):544–549. 10.1002/jemt.23198 [DOI] [PubMed] [Google Scholar]
- 94.Kwiatkowski DJ, Janmey PA, Yin HL (1989) Identification of critical functional and regulatory domains in gelsolin. J Cell Biol 108 (5):1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]