

Abstract
α-Alkynyldiazomethanes, generated in situ from the corresponding sulfonyl hydrazones in the presence of a base, can serve as effective metalloradicophiles in Co(II)-based metalloradical catalysis (MRC) for asymmetric cyclopropanation of alkenes. With D2-symmetric chiral amidoporphyrin 2,6-DiMeO-QingPhyrin as the optimal supporting ligand, the Co(II)-based metalloradical system can efficiently activate different α-alkynyldiazomethanes at room temperature for highly asymmetric cyclopropanation of a broad range of alkenes. This catalytic radical process provides a general synthetic tool for stereoselective construction of alkynyl cyclopropanes in high yields with high both diastereoselectivity and enantioselectivity. Combined computational and experimental studies offer several lines of evidence in support of the underlying stepwise radical mechanism for the Co(II)-catalyzed olefin cyclopropanation involving a unique α-metalloradical intermediate that is associated with two resonance forms of α-Co(III)-propargyl radical and γ-Co(III)-allenyl radical. The resulting enantioenriched alkynyl cyclopropanes, as showcased with several stereospecific transformations, may serve as valuable chiral building blocks for stereoselective organic synthesis.
Graphical Abstract
INTRODUCTION
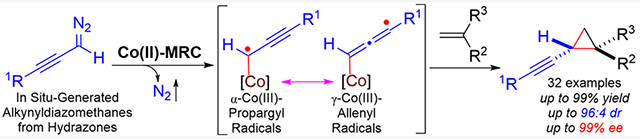
Radical reactions have been increasingly exploited as complementary synthetic methods to ionic reactions in modern organic synthesis as they enjoy intrinsic attributes such as high reactivity and tolerance of functional groups.1 Among significant challenges facing this endeavor is the control of reactivity as well as selectivities in the reactions of free organic radicals, especially enantioselectivity.2 Rising to these challenges, metalloradical catalysis (MRC) has emerged as a conceptually new approach to the development of stereoselective radical reactions through catalytic generation of metal-supported organic radicals as key catalytic intermediates.3–5 As stable 15e-metalloradicals with well-defined low-spin d7-configuration, Co(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)] have been recognized as a family of open-shell catalysts that are effective for asymmetric cyclopropanation of alkenes with diazo compounds.6 During the Co(II)-catalyzed cyclopropanation, the initially generated α-metalloalkyl radical intermediates from metalloradical activation of diazo compounds, which are centrally situated inside the pocketlike environment of the chiral porphyrin ligands,7 can be precisely governed to perform a sequence of homolytic reactions such as radical addition and radical substitution with olefin substrates, leading to productive formation of cyclopropanes with effective control of diastereoselectivity and enantioselectivity. Except for a few recent examples of using α-aryldiazomethanes,8 Co(II)-based radical cyclopropanation has so far been mostly involved with the use of acceptor- and acceptor/acceptor-substituted diazo compounds as metalloradicophiles.6 In all the previous cases, the key α-Co(III)-supported C-centered radical intermediates are stabilized by C(sp2)-based carbonyl or aryl substituents through potential H-bonding interactions with the amide units of the catalyst, facilitating the reactivity and stereoselectivity of the catalytic radical process. It was unclear if Co(II)-based metalloradical system could also be applicable to other types of diazo compounds with substituents beyond C(sp2)-based carbonyl and aryl groups. Specifically, we were intrigued to learn whether α-alkynyldiazomethanes, a common type of diazo compounds containing C(sp)-based alkynyl substituents, could be employed as potential metalloradicophiles by Co(II)-based metalloradical system for radical olefin cyclopropanation (Scheme 1). As α-alkynyldiazomethanes 1′ are typically generated in situ from corresponding hydrazones 1 under basic conditions,9 it would be crucially important to match the rates between the diazomethane generation and the ensuing metalloradical activation in order to preclude their thermal decomposition to form pyrazole and azine side products. Apart from the concern with the effectiveness for initially generated α-Co(III)-propargyl radicals I to the olefin substrates to control the enantioselectivity for the first C–C bond formation? What also under question is the control of chemoselectivity as well as the diastereoselectivity during the final step of radical substitution of γ-Co(III)-alkyl radicals II for the second C—C bond formation. Considering the presence of the C≡C triple bond, could γ-Co(III)-alkyl radicals II undergo the desired 3-exo-tet radical cyclization chemoselectively over the potentially competitive 4-exo-dig and 5-endo-dig cyclization, leading to diastereoselective construction of cyclopropanes? We hoped to address these and related issues through judicious development of Co(II)-based metalloradical catalyst by fine-tuning the environments of D2-symmetric chiral amidoporphyrin ligand. If implemented successfully, then it would lead to the development of a new catalytic radical process for asymmetric olefin cyclopropanation with in situ-generated α-alkynyldiazomethanes to construct chiral alkynyl cyclopropanes. In addition to serving as useful intermediates for stereoselective organic synthesis, chiral alkynyl cyclopropanes occur as common substructures in many important natural products and drug molecules (see Figure S1).11
Scheme 1.
Proposed Pathway for Radical Cyclopropanation of Alkenes with α-Alkynyldiazomethanes via Co(II)-MRC
Metal-catalyzed asymmetric cyclopropanation of alkenes with α-alkynyldiazomethanes offers a potentially general approach for stereoselective construction of valuable chiral alkynyl cyclopropanes.12–14 While donor/acceptor-substituted diazo compounds alkynyldiazoacetates were successfully metalloradical activation of α-alkynyldiazomethanes 1’, the additional issue of regioselectivity could arise from two potential reactivity modes of the resulting α-metalloalkyl radical intermediates that are associated with the two resonance forms of α-Co(III)-propargyl radicals I and γ-Co(III)-allenyl radicals I’.10 Furthermore, in the absence of the aforementioned H-bonding interactions, what elements could be utilized during the subsequent radical addition of the employed by Davies and co-workers as carbene precursors for Rh2-catalyzed asymmetric cyclopropanation,15 donor-substituted diazo compounds α-alkynyldiazomethanes, in contrast, have been scarcely explored.16 A single example of cyclopropanation of trans-stilbene with the use of α-alkynyldiazomethane could be found in a recent report by Bi and co-workers on Ag-based catalytic system involving in situ generation of diazo compounds from N-nosylhydrazones.16c This underdevelopment is mainly attributed to the inherent propensity of α-alkynyldiazomethanes toward the formation of the stable aromatic pyrazoles.17 To circumvent the problem, Echavarren and co-workers recently reported an innovative two-step approach for synthesis of alkynyl cyclopropanes from alkenes based on Rh2-catalyzed decarbenation of 7-alkynyl cycloheptatrienes as alkynylcarbene precursors.18 To the best of our knowledge, there has been no previous report on catalytic system for asymmetric olefin cyclopropanation with α-alkynyldiazomethanes for stereoselective synthesis of chiral alkynyl cyclopropanes. As a new application of Co(II)-based metalloradical catalysis (MRC), we herein report the development of the first asymmetric catalytic system for olefin cyclopropanation that can effectively utilize α-alkynyldiazomethanes through in situ generation from the corresponding sulfonyl hydrazones in the presence of base. Through identification of an optimal D2-symmetric chiral amidoporphyrin (D2-Por*) as the supporting ligand, the Co(II)-based catalytic system is capable of efficiently activating different alkynyldiazomethanes at room temperature to cyclopropanate a wide range of alkenes with varied electronic and steric properties, delivering alkynyl cyclopropanes in high yields with excellent control of both diastereoselectivity and enantioselectivity. We show the importance of catalyst development through fine-tuning of the ligand environment in achieving high reactivity as well as stereoselectivities. We present combined computational and experimental studies that shed light on the underlying stepwise radical mechanism of the Co(II)-catalyzed cyclopropanation. To demonstrate the synthetic applications of the new catalytic system, we showcase several examples of stereoselective transformations of the resulting enantioenriched alkynyl cyclopropanes.
RESULTS AND DISCUSSION
Catalyst Development.
At the outset of the project, α-(phenylethynyl)diazomethane (1a′), which was in situ generated from corresponding trisylhydrazone 1a in the presence of KH, was designated as the representative α-alkynyldiazomethane for asymmetric radical cyclopropanation of styrene (2a) using Co(II)-based metalloradical catalysts [Co(Por)] (Scheme 2; see Table S1). It was found that simple achiral metalloradical catalyst [Co(P1)] (P1 = tetraphenylporphyrin) could activate the diazo compound to cyclopropanate styrene, giving desired alkynyl cyclopropane 3a in low yield (36%) with inferior control of diastereoselectivity (14% de). With the employment of the Co(II) complex of D2h-symmetric achiral amidoporphyrin [Co(P2)] (P2 = 3,5-DitBu-IbuPhyrin) as the catalyst,19 the catalytic cyclopropanation reaction was significantly enhanced, forming cyclopropane 3a in a higher yield (84%) with improved diastereoselectivity (56% de) in favor of the trans-isomer. To evaluate the feasibility of asymmetric induction during the catalytic process, Co(II) complexes of D2-symmetric chiral amidoporphyrins were employed as the catalysts. When first-generation chiral metalloradical catalyst [Co(P3)] (P3 = 3,5-DitBu-ChenPhyrin)6a was used, the catalytic reaction produced cyclopropane 3a in an almost quantitative yield (98%) with the same level of diastereoselectivity (58% de) while exhibiting significant enantioselectivity (53% ee). To improve the stereoselectivities of the catalytic process, we then applied metalloradical catalyst [Co(P4)] (P4 = 3,5-DitBu-ZhuPhyrin),20 which is devised to achieve conformational rigidity through unique intramolecular H-bonding interactions in (S)-(−)-2-tetrahydrofurancarboxamide units. As expected, the catalytic reaction by [Co(P4)] brought about improved enantioselectivity (59% ee) but reduced diastereoselectivity (52% de) and significantly decreased yield (34%) as a result of the more sterically encumbered ligand environment. These results prompted us to explore second-generation chiral metalloradical catalysts bearing cyclopropanecarboxyamides with two contiguous stereogenic centers in the hope of further enhancing the asymmetric induction of the catalytic process. Gratifyingly, when [Co(P5)] (P5 = 3,5-DitBu-QingPhyrin) was used as the catalyst for the reaction,6g cyclopropane 3a was produced in high yield (80%) with excellent enantioselectivity (94% ee) albeit with low diastereoselectivity (20% de). Inspired by this positive outcome, we then employed the analogous catalyst [Co(P6)] (P6 = 2,6-DiMeO-QingPhyrin) bearing the same chiral amide units but with different achiral meso-aryl groups to further enhance the rigidity of the chiral environment. To our delight, the catalytic reaction by [Co(P6)] indeed significantly improved both diastereoselectivity (74% de) and yield (90%) while maintaining the excellent enantioselectivity (96% ee). The remarkable difference in performance among the metalloradical catalysts indicates that subtle alteration in ligand environment can give rise to significant improvement in catalytic reactivity as well as stereoselectivities, manifesting the effectiveness of catalyst development in controlling the radical process.
Scheme 2. Ligand Effect on Co(II)-Based Catalytic System for Olefin Cyclopropanation with Alkynyldiazomethanea.
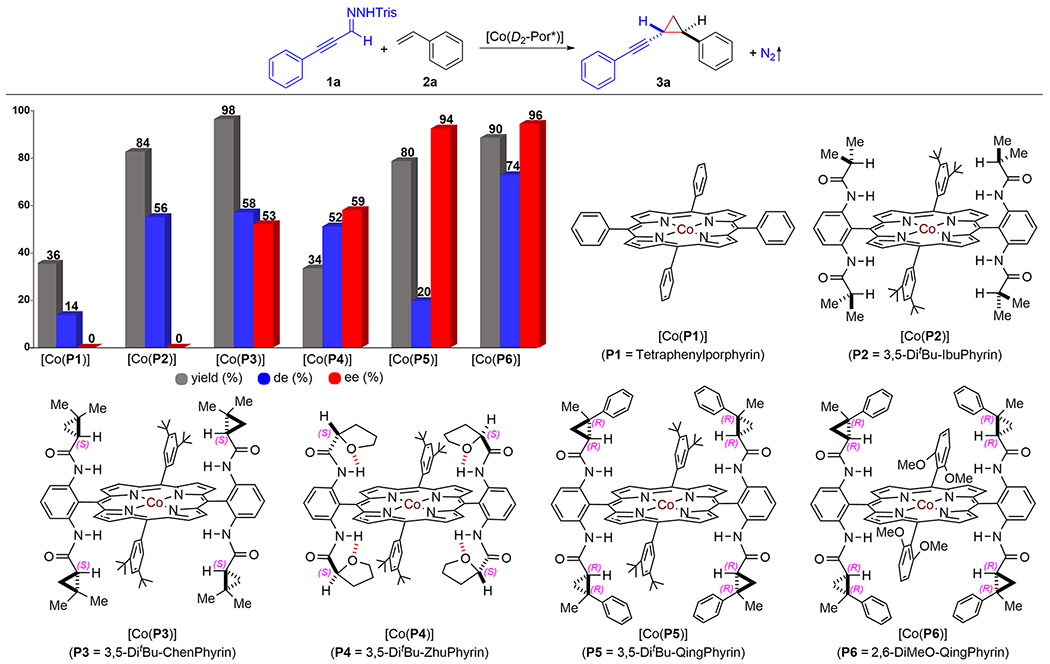
a Carried out with 1a (0.10 mmol) and 2a (0.20 mmol) in the presence of KH (0.40 mmol) by [Co(Por)] (2 mol %) in ethyl acetate (0.6 mL) at 22 °C for 24 h; Tris = 2,4,6-triisopropylbenzenesulfonyl; isolated yields; diastereomeric excess (de) determined by 1H NMR; enantiomeric excess (ee) of trans-isomer determined by chiral HPLC.
Substrate Scope.
Under the optimized conditions, the scope and versatility of [Co(P6)]-catalyzed asymmetric cyclopropanation were evaluated using different α-alkynyldiazomethanes through in situ generation from the corresponding sulfonyl hydrazones 1 under the basic condition with various olefins (Table 1). Similar to α-alkynyldiazomethane 1a’, α-(arylethynyl)diazomethanes bearing substituents with varied electronic and steric properties on the phenyl group could be effectively employed as metalloradicophiles for the Co(II)-based catalytic system as shown for asymmetric cyclopropanation of styrene (2a) as the representative olefin substrate. For example, α-(arylethynyl)diazomethanes bearing alkyl substituents at different aryl positions such as p-Me and o-iPr could be efficiently activated by [Co(P6)] at room temperature to cyclopropanate styrene, producing desired alkynyl cyclopropanes 3b and 3c in high yields with good diastereoselectivities and excellent enantioselectivities (entries 1 and 2). Furthermore, [Co(P6)] could activate α-(arylethynyl)diazomethanes containing an electron-donating–OMe group at all the three different positions of the aryl group for the cyclopropanation reaction, allowing stereoselective construction of corresponding alkynyl cyclopropanes 3d—3f in high yields with high diastereoselectivities and excellent enantioselectivities (entries 3—5). It is interesting to note that the observed diastereoselectivity increases when the position of MeO-substituent in cyclopropanes 3d—3f moves from para- to the meta- to the ortho-position. Likewise, α-(arylethynyl)diazomethanes bearing electron-withdrawing groups could also be suitable metalloradicophiles as exemplified by asymmetric synthesis of alkynyl cyclopropanes 3g and 3h in high yields with excellent control of stereoselectivities (entries 6 and 7). Moreover, naphthalenecontaining α-alkynyldiazomethanes could also be effectively activated by [Co(P6)] under the same conditions for asymmetric cyclopropanation reaction, leading to high-yielding formation of corresponding alkynyl cyclopropane 3i with both high diastereoselectivity and enantioselectivity (entry 8). In addition to α-(arylethynyl)diazomethanes, the Co(II)-based metalloradical system was shown to be applicable to α-alkynyldiazomethanes containing certain nonaryl substituents as demonstrated by catalytic asymmetric cyclopropanation of styrene with alkyl substituted diazo derivative, generating desired cyclopropane 3j with high stereoselectivities albeit in moderate yield (entry 9). Moreover, a triisopropylsilyl (TIPS)-substituted diazo derivative can also serve as suitable substrate, furnishing the corresponding cyclopropane 3k in good yield with high diastereoselectivity and enantioselectivity (entry 10).
Table 1.
Scope of Catalytic Asymmetric Cyclopropanation of Alkenes with α-Alkynyldiazomethanes by [Co(P6)]a
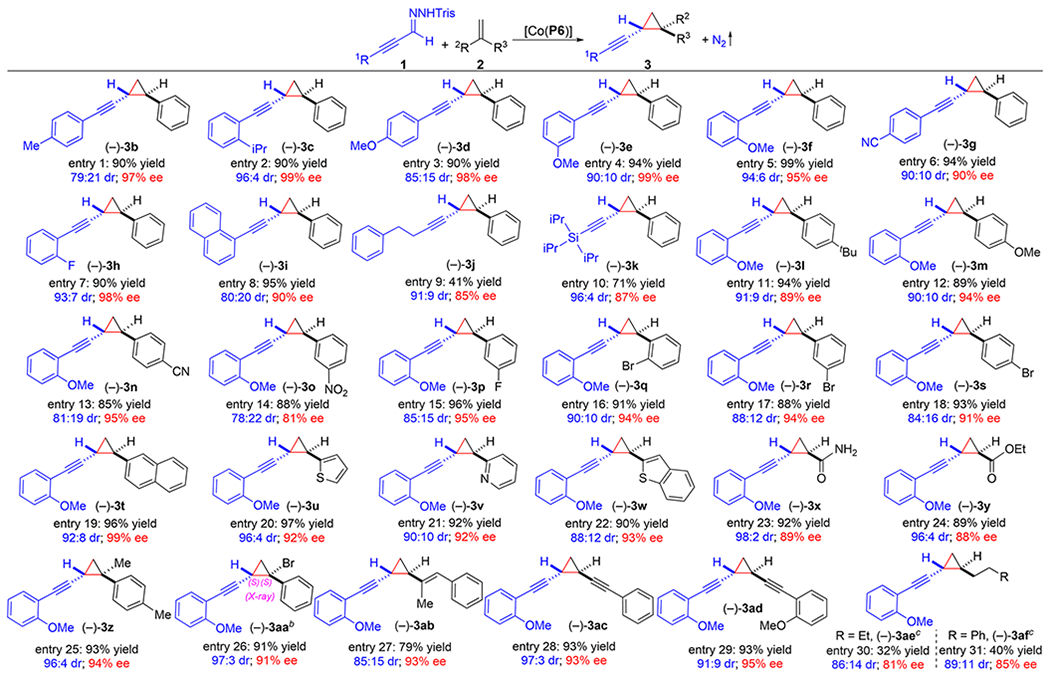
|
Carried out with 1 (0.10 mmol) and 2 (0.20 mmol) in the presence of KH (0.40 mmol) by [Co(P6)] (2 mol %) in ethyl acetate (0.6 mL) at 22°C for 24 h; Tris = 2,4,6-triisopropylbenzenesulfonyl; isolated yields; diastereomeric ratio (dr) determined by 1H NMR analysis of reaction mixture; enantiomeric excess (ee) of trans-isomer determined by chiral HPLC.
Absolute configuration determined by X-ray crystallography.
Carried out with 2 (1.00 mmol) at 80 °C.
In addition to being capable of using different α-alkynyldiazomethanes, the Co(II)-based system for asymmetric cyclopropanation was found to be suitable to a wide range of alkenes under the optimized conditions (Table 1). Like styrene, its derivatives bearing various substituents such as –tert-Bu, –OMe, –CN, and –NO2 groups at different positions could be reliably cyclopropanated by [Co(P6)] with α-alkynyldiazomethane 1a’, generating corresponding alkynyl cyclopropanes 3l–3o in similarly high yields with effective control of diastereoselectivities and enantioselectivities (entries 11–14). Additionally, halogenated aromatic olefins could also serve as suitable substrates in [Co(P6)]-catalyzed asymmetric cyclopropanation as exemplified by highly stereoselective generation of alkynyl cyclopropanes 3p–3s with halogen atoms at various positions (entries 15–18). Furthermore, extended aromatic olefins like 2-vinylnaphthalene could be also effectively applied to the catalytic system, affording alkynyl cyclopropane 3t in excellent yield with high diastereoselectivity and outstanding enantioselectivity (entry 19). Moreover, the Co(II)-based cyclopropanation was shown to be compatible with heteroaryl alkenes such as those containing thiophene, pyridine, and benzothiophene, affording corresponding heteroaryl cyclopropanes 3u–3w in high yields with high stereoselectivities (entries 20–22). To further highlight the unique feature of Co(II)-based metalloradical catalysis, even electron-deficient olefins such as acrylamide and ethyl acrylate, which are challenging substrates for catalytic cyclopropanation systems involving electrophilic metallocarbene intermediates, could be effectively cyclopropanated by [Co(P6)] to form desired products 3x and 3y in high yields with high stereoselectivities (entries 23 and 24). In addition to monosubstituted olefins, 1,1-disubstituted olefins like α-methylstyrene and α-bromostyrene could serve as suitable substrates as well, allowing for highly stereoselective construction of trisubstituted cyclopropanes 3z and 3aa with excellent control of the newly generated quaternary carbon stereogenic centers (entries 25 and 26). The absolute configuration of the major enantiomer of alkynyl cyclopropane 3aa was established as (S,S) by X-ray crystallography. When conjugated dienes and enynes were used as the substrates, [Co(P6)] could regio- and chemoselectively cyclopropanate the terminal double bonds, leading to exclusive formation of cyclopropanes 3ab–3ad in high yields with high stereoselectivities without affecting the internal alkene and alkyne units (entries 27–29). It is interesting to note the C2-symmetric structure of bisalkynyl cyclopropane 3ad. Notably, [Co(P6)]-based catalytic system proved to be even effective for asymmetric cyclopropanation of unactivated alkyl-substituted olefins, affording alkynylcyclopropanes 3ae and 3af in moderate yields with high stereoselectivities (entries 30 and 31).
Mechanistic Studies.
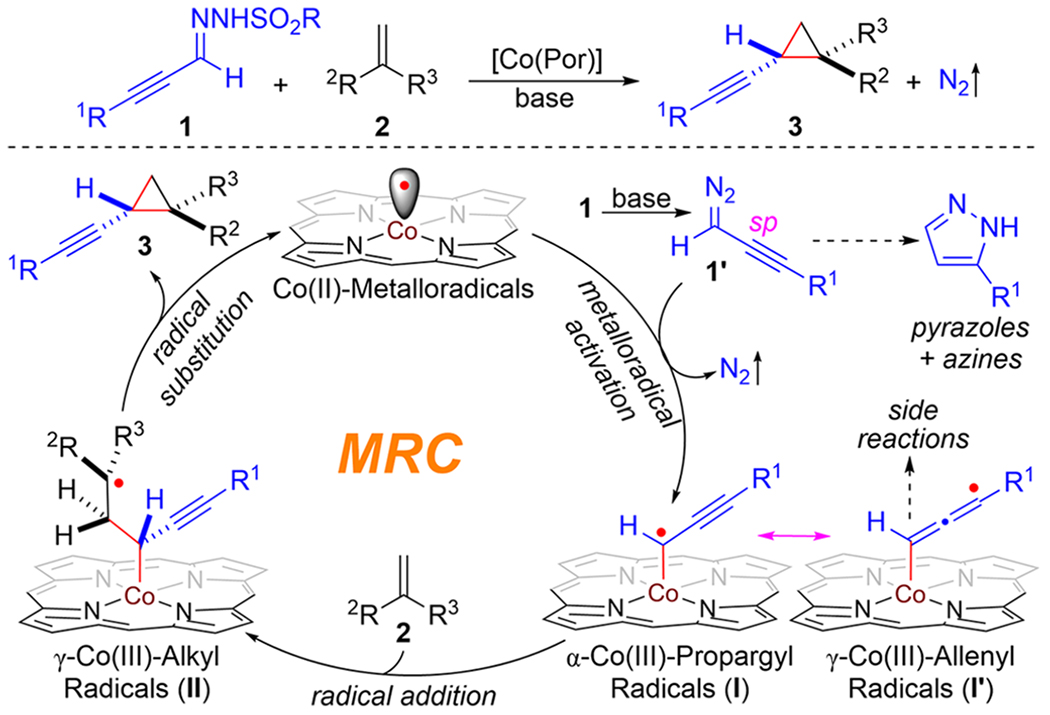
Combined computational and experimental studies were carried out to shed light on the underlying stepwise radical mechanism of Co(II)-based catalytic system for asymmetric cyclopropanation (Scheme 3). First, density functional theory (DFT) calculations were conducted to examine the details of the catalytic pathway and associated energetics for asymmetric cyclopropanation of styrene (2a) with (phenylethynyl)diazomethane (1a’) by metalloradical catalyst [Co(P6)] (A) (Scheme 3A; see the Supporting Information for details). The DFT calculations reveal the initial formation of intermediate B resulting from the binding of diazomethane 1a’ by catalyst [Co(P6)] through a network of noncovalent attractive interactions, including H-bonding and π-stacking interactions, as illustrated in the DFT-optimized structure (Scheme 3A). The binding event, which is slightly exergonic by 1.2 kcal/mol, positions the α-carbon atom of 1a’ in close proximity to the Co(II)-metalloradical center of [Co(P6)] (C⋯Co: ~2.792 Å) for the subsequent activation. Bound diazomethane 1a’ is then further activated by catalyst [Co(P6)] to generate α-Co(III)-propargyl radical intermediate C with the release of dinitrogen as byproduct. The metalloradical activation, which is exergonic by 18.2 kcal/mol, is found to be the rate-determining step associated with a relatively high but accessible activation barrier (ΔG‡TS1 = 16.3 kcal/mol). As displayed by the spin plot of intermediate C (Scheme 3A), the spin density mainly distributes on α- and γ-carbon atoms in similar amounts (α-C: 0.59; γ-C: 0.41), which can be represented as two resonance forms of α-Co(III)-propargyl radical C and γ-Co(III)-allenyl radical C’. To rationalize the observed regioselectivity of the catalytic reaction, we calculated the energetics associated with subsequent radical addition to alkene 2a by both α-Co(III)-propargyl radical form C and γ-Co(III)-allenyl radical form C’, leading to the formation of γ-Co(III)-benzyl radical intermediate D and ε-Co(III)-benzyl radical intermediate D’, respectively. DFT calculations indicate radical addition of propargyl radical C is more favorable than allenyl radical C’ both kinetically (ΔG‡TS2 = 11.4 kcal/mol; ΔG‡TS2’ = 18.7 kcal/mol) and thermodynamically ΔG°D = −11.1 kcal/mol; ΔG°D’ = −7.2 kcal/mol). According to the DFT calculations, γ-Co(III)-alkyl radical D then undergoes radical substitution to produce alkynyl cyclopropane 3a while regenerating metalloradical catalyst [Co(P6)]. This final step of 3-exo-tet radical cyclization, which is exergonic by 15.2 kcal/mol, is found to be an almost barrierless process. The overall low activation barrier is in accordance with the experimental observation that the catalytic reaction could proceed effectively even at room temperature.
Scheme 3. Mechanistic Studies on Co(II)-Catalyzed Olefin Cyclopropanation with α-Alkynyldiazomethanes.
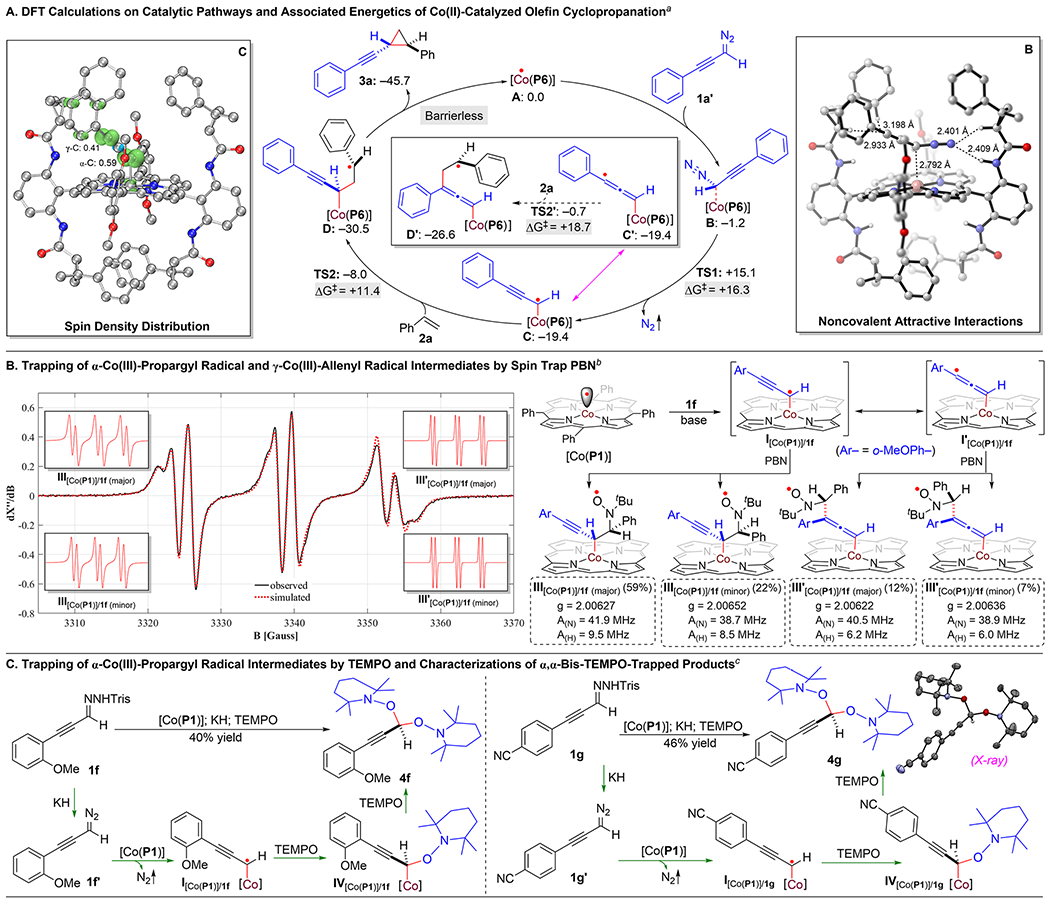
aApplied bp86/LANL2DZ for geometry optimization and B3LYP/def2-tzvp for calculations of single-point energies (kcal/mol) along with Grimme’s dispersion correction and SMD (ethyl acetate) solvation model. bCarried out with 1f (0.10 mmol), [Co(P1)] (2 mol %) and PBN (0.12 mmol) in the presence of Et3N (0.20 mmol) at 60 °C in benzene (1.0 mL) for 10 min; Ar = 2-methoxyphenyl. The simulation of the EPR spectrum was carried out by iteration of the isotropic g-values and line widths using the EPR simulation program SpinFit Xenon. cCarried out with 1 (0.10 mmol) and TEMPO (0.60 mmol) in the presence of KH (0.40 mmol) by [Co(P1)] (2 mol %) in ethyl acetate (0.6 mL) at 22 °C for 24 h; Tris = 2,4,6-triisopropylbenzene sulfonyl; isolated yield; structure determined by X-ray crystallography.
To provide direct evidence for the existence of the key α-Co(III)-propargyl radical intermediate, efforts were made to trap the Co-supported organic radicals for experimental detection and characterization. First, the spin trapping reagent N-tert-butyl-α-phenylnitrone (PBN) was added to the reaction mixture of alkynyldiazomethane 1f’ with metalloradical catalyst [Co(P1)] in the absence of olefin substrate and was then monitored by X-band electron paramagnetic resonance (EPR) spectroscopy at room temperature (Scheme 3B; see the Supporting Information for details). The observed isotropic EPR spectrum exhibits strong signals with the characteristic splitting pattern at g-value of ~2.00, which was taken as the evidence for formation of radical III[Co(P1)]/1f resulting from PBN trapping of the initially generated Co(III)-propargyl radical intermediate I[Co(P1)]/1f.21 In accordance with the spin density distribution from DFT calculations (Scheme 3A), the observed broad spectrum (in black) could be near perfectly simulated (in red) as four well-defined triplet of doublet signals (Scheme 3B) by involving four isomeric PBN-trapped radical species that are originated from the two resonance forms of α-Co(III)-propargyl radical intermediate I[Co(P1)]/1f and γ-Co(III)-allenyl radical intermediate I’[Co(P1)]/1f on the basis of the hyperfine coupling by 14N (I = 1) and 1H (I = 1/2): 81% of O-centered radicals III[co(P1)]/1f from I[Co(P1)]/1f as two diastereomers (59% of major diastereomer: g = 2.00627, A(N) = 41.9 MHz, A(H) = 9.5 MHz; 22% of minor diastereomer: g = 2.00652, A(N) = 38.7 MHz, A(H) = 8.5 MHz) and 19% of O-centered radicals III’[co(P1)]/1f from I’[Co(P1)]/1f as two diastereomers (12% of major isomer: g = 2.00622, A(N) = 40.5 MHz, A(H) = 6.2 MHz; 7% of minor isomer: g = 2.00636, A(N) = 38.9 MHz, A(H) = 6.0 MHz).
Besides the spectroscopic observation of the key radical intermediate by EPR, significant efforts were devoted to directly trap α-Co(III)-propargyl radical intermediate by stable TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) radical for structural characterization (Scheme 3c). When the metalloradical activation of alkynyldiazomethane 1f’ by [Co(P1)] was conducted in the presence of TEMPO without alkene substrates, we were able to isolate product 4f in 40% yield, which was shown to contain two geminal TEMPO units at the propargylic position. Formation of bis-TEMPO-trapped product 4f evidently implies the initial generation of α-Co(III)-propargyl radical I[Co(P1)]/1f, which was trapped by TEMPO through radical recombination to generate intermediate IV[Co(P1)]/1f. Subsequent radical substitution reaction of IV[Co(P1)]/1f with a second molecule of TEMPO was likely responsible for the final formation of bis-TEMPO-trapped product 4f upon the homolytic cleavage of the weak Co(III)–C bond. Analogous TEMPO-trapping experiment was carried out with alkynyldiazomethane 1g, resulting in the isolation of bis-TEMPO-trapped product 4g in 46% yield, the structure of which was further confirmed by the X-ray crystallography (Scheme 3C). In both reactions, α,α-bis-TEMPO-trapped products were exclusively generated from α-Co(III)-propargyl radical intermediate without the formation of α,γ-bis-TEMPO-trapped products from its γ-Co(III)-allenyl radical resonance form. The observed preference of reactivity at α-propargyl position over γ-allenyl position is in good accordance with the regioselectivity of Co(II)-based catalytic system for asymmetric cyclopropanation, which is also supported by the DFT computational studies.
Synthetic Applications.
Considering that the resulting enantioenriched alkynyl cyclopropanes contain both versatile C≡C triple bond as well as relatively acidic tertiary C—H bonds on the cyclopropane ring, they may serve as useful intermediates for stereoselective organic synthesis. To this end, we carried out several stereoselective transformations using enantioenriched alkynyl cyclopropanes 3f (95% ee) and 3a (96% ee) as the model compounds to demonstrate the synthetic utility of this methodology (Scheme 4).22 For example, it was shown that enantioenriched alkynyl cyclopropane 3f could undergo effective cis-hydrogenation with dihydrogen in the presence of Lindlar catalyst, generating vinyl cyclopropane 5f with (Z)-configuration exclusively in almost quantitative yield (99% yield) without any erosion of the original diastereoselectivity (100% ds) and enantioselectivity (100% es) (Scheme 4A). Furthermore, by taking advantage of the difference in acidity between the two tertiary C—H bonds on the cyclopropane ring, we demonstrated that the C—H bond neighboring at the alkynyl substituent in cyclopropane 3f could be preferentially deprotonated with n-BuLi over the C—H bond neighboring at the aryl substituent, leading to site-selective generation of cyclopropyl lithium intermediate 3f·Li (Scheme 4B).22a By adding different alkyl halides as the electrophiles, the in situ-generated cyclopropyl lithium intermediate 3f·Li could undergo subsequent nucleophilic substitution reactions, allowing for stereoselective synthesis of trisubstituted cyclopropanes with three different types of groups while creating an all-carbon quaternary stereogenic center without the formation of the potential allenylic products. For instance, the in situ reaction of 3f·Li with methyl iodide furnished corresponding 1-methyl-1-alkynyl-2-phenyl cyclopropane 6f in moderate yield (66%) with notable loss of stereopurities (50% ds and 85% es) presumably due to the small size of the electrophile (Scheme 4B). When the larger benzyl bromide was added as the electrophile, the reaction could deliver corresponding 1-benzyl-1-alkynyl-2-phenyl cyclopropane 7f in higher yield (72%) with complete retention of the diastereopurity (100% ds) and without significant loss of the original enantiopurity (98% es) (Scheme 4C). The reaction also worked similarly well with allylic bromide as the electrophilic partner, affording desired 1-allyl-1-alkynyl-2-phenyl cyclopropane 8f in high yield (92%) while maintaining the majority of the stereopurities (93% ds and 88% es) (Scheme 4D). Moreover, oxidative click reaction of cyclopropane 3f with sodium azide could be successfully achieved in the presence of PhI(OAc)2, producing 1,2,3-triazole-substituted cyclopropane 9f in high yield (81%) without any erosion of the original diastereoselectivity (100% ds) and enantioselectivity (100% es) (Scheme 4E).22b Presumably, the in situ-generated azidyl radical first underwent radical addition with the C≡ bond in cyclopropane 3f to generate vinyl radical intermediate A. Ensuing 5-endo-dig radical cyclization of intermediate A caused the formation of 5-membered aminyl radical intermediate B, which gave rise to final product 9f after hydrogen atom abstraction from the solvent.22b In addition to these demonstrated transformations for stereoselective synthesis of new cyclopropane derivatives, the three-membered cyclopropane ring in the resulting enantioenriched alkynyl cyclopropanes could be expanded to form 5-membered cyclic structure as exemplified by the reaction of enantioenriched alkynyl cyclopropane 3a with Na2S·9H2O in DMA at high temperature, affording 2-benzylidenetetrahydrothiophene 4a in high yield (73%) with good configuration control of the trisubstituted olefin (Z:E = 85:15) but with only moderate enantioselectivity (36% ee) (Scheme 4F). According to the proposed mechanism,22c the cyclopropane ring in 3a was first opened by in situ-generated trisulfur radical anion S3•− via radical substitution to generate propargylic radical intermediate C, which was protonated by H2O after one-electron reduction and followed by homolysis of the S—S bond to form thiyl radical intermediate D while releasing disulfur radical anion S2•−. The following 5-exo-dig radical cyclization of intermediate D led to the formation of vinyl radical intermediate E, which went through another sequence of reduction and protonation to give final product 4a.22c
Scheme 4. Synthetic Applications of Resulting Chiral Alkynyl Cyclopropanes from Co(II)-Catalyzed Olefin Cyclopropanation.
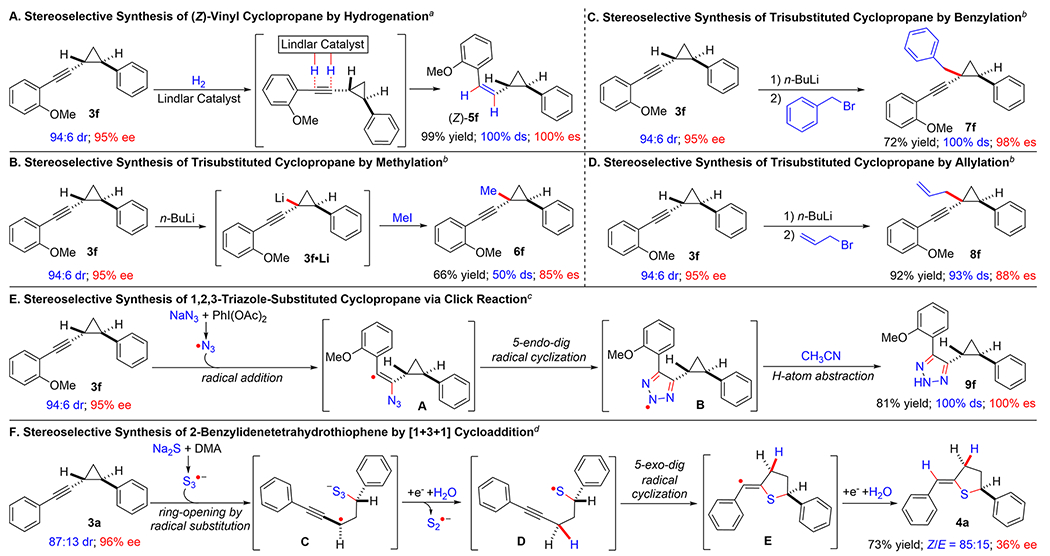
aCarried out with 3f (0.10 mmol) and quinoline (2.0 equiv) by Lindlar catalyst (1.2 equiv) in mixed solvent (2.0 mL; hexane:ethyl acetate = 1:1) under H2 atmosphere at 22 °C for 12 h. bCarried out with 3f (0.10 mmol) and n-BuLi (1.6 equiv) in THF (2.0 mL) under N2 atmosphere at −78 °C for 1 h, followed by addition of electrophile (1.6 equiv) and then stirred at 22 °C for 24 h. cCarried out with 3f (0.10 mmol), NaN3 (1.5 equiv), and PhI(OAc)2 (1.0 equiv) in MeCN (2.0 mL) under N2 atmosphere at 22 °C for 12 h. dCarried out with 3a (0.10 mmol) and Na2S·9H2O (6.0 equiv) in DMA (0.5 mL) at 150 °C for 12 h.
CONCLUSIONS
In summary, we have developed the first asymmetric catalytic system that can use in situ-generated α-alkynyldiazomethanes for direct cyclopropanation of alkenes via Co(II)-based metalloradical catalysis (MRC). On the basis of a remarkable ligand effect on the Co(II)-based catalytic system, D2-symmetric chiral amidoporphyrin 2,6-DiMeO-QingPhyrin has been identified as the optimal supporting ligand that offers suitable steric, electronic, and chiral environments surrounding the Co(II)-metalloradical center for engaging a network of noncovalent attractive interactions to facilitate the cyclopropanation process. The Co(II)-based metalloradical system is capable of activating different alkynyldiazomethanes under mild conditions for highly asymmetric cyclopropanation of diverse alkenes with varied electronic and steric properties, affording chiral alkynyl cyclopropanes in high yields with high both diastereoselectivity and enantioselectivity. The combined computational and experimental studies have shed light on the underlying stepwise radical mechanism of the Co(II)-based cyclopropanation system involving a unique α-metalloradical intermediate that is associated with two resonance forms of α-Co(III)-propargyl radical and γ-Co(III)-allenyl radical. In addition to rationalizing the unique profile of reactivity and selectivity, the established mechanism offers a convincing explanation of the regioselectivity toward the formation of alkynyl cyclopropanes via α-Co(III)-propargylic radical form without any complication from potential reaction via γ-Co(III)-allenyl radical form. The resulting enantioenriched alkynyl cyclopropanes, as showcased in several stereospecific transformations, may serve as useful intermediates for stereospecific organic synthesis. Considering the ubiquity of chiral alkynyl cyclopropanes, we believe this Co(II)-catalyzed asymmetric radical cyclopropanation process will find useful applications in organic synthesis.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support from the NIH (R01-GM102554) and in part by NSF (CHE-1900375).
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c13154
Accession Codes
CCDC 2128604 and 2128605 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c13154.
Experimental details and analytical data for all new compounds (PDF)
Contributor Information
Jing Ke, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
Wan-Chen Cindy Lee, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
Xiaoxu Wang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
Yong Wang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
Xin Wen, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
X. Peter Zhang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
REFERENCES
- (1).For selected books on radical reactions, see; (a) Zard SZ Radical Reactions in Organic Synthesis; Oxford University Press: Oxford, UK, 2003. [Google Scholar]; (b) Chatgilialoglu C; Studer A Encyclopedia of Radicals in Chemistry, Biology, and Materials. John Wiley & Sons: Chichester, U.K., 2012. [Google Scholar]; For selected reviews on radical reactions, see; (c) Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]; (d) Quiclet-Sire B; Zard SZ Fun with Radicals: Some New Perspectives for Organic Synthesis. Pure Appl. Chem 2010, 83, 519–551. [Google Scholar]; (e) Narayanam JM; Stephenson CR Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (f) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (2).For selected reviews on stereoselective radical reactions, see; (a) Bar G; Parsons AF Stereoselective Radical Reactions. Chem. Soc. Rev 2003, 32, 251–263. [DOI] [PubMed] [Google Scholar]; (b) Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3296. [DOI] [PubMed] [Google Scholar]; (c) Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed 2015, 54, 3872–3890 [DOI] [PubMed] [Google Scholar]; For selected examples of approaches to controlling radical reactivity and stereoselectivity, see.; (d) Du J; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (f) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kern N; Plesniak MP; McDouall JJ; Procter DJ Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem 2017, 9, 1198–1204. [DOI] [PubMed] [Google Scholar]; (i) Morrill C; Jensen C; Just-Baringo X; Grogan G; Turner NJ; Procter DJ Biocatalytic Conversion of Cyclic Ketones Bearing α-Quaternary Stereocenters into Lactones in an Enantioselective Radical Approach to Medium-Sized Carbocycles. Angew. Chem 2018, 130, 3754–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Proctor RS; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]; (k) Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereo-selective Radical Cyclization. Science 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Huang H-M; McDouall JJ; Procter DJ SmI2-Catalysed Cyclization Cascades by Radical Relay. Nat. Catal 2019, 2, 211–218. [Google Scholar]; (m) Cheng Y-F; Liu J-R; Gu Q-S; Yu Z-L; Wang J; Li Z-L; Bian J-Q; Wen H-T; Wang X-J; Hong X; Liu X-Y Catalytic Enantioselective Desymmetrizing Functionalization of Alkyl Radicals via Cu (i)/CPA Cooperative Catalysis. Nat. Catal 2020, 3, 401–410. [Google Scholar]; (n) Gu Q-S; Li Z-L; Liu X-Y Copper (i)-Catalyzed Asymmetric Reactions Involving Radicals. Acc. Chem. Res 2020, 53, 170–181. [DOI] [PubMed] [Google Scholar]; (o) Li Z-L; Fang G-C; Gu Q-S; Liu X-Y Recent Advances in Copper-Catalysed Radical-Involved Asymmetric 1,2-Difunctionalization of Alkenes. Chem. Soc. Rev 2020, 49, 32–48. [DOI] [PubMed] [Google Scholar]; (p) Nakafuku KM; Zhang Z; Wappes EA; Stateman LM; Chen AD; Nagib DA Enantioselective Radical C–H Amination for the Synthesis of β Amino Alcohols. Nat. Chem 2020, 12, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Wang Y; Carder HM; Wendlandt AE Synthesis of Rare Sugar Isomers through Site-Selective Epimerization. Nature 2020, 578, 403–408. [DOI] [PubMed] [Google Scholar]; (r) Yang C-J; Zhang C; Gu Q-S; Fang J-H; Su X-L; Ye L; Sun Y; Tian Y; Li Z-L; Liu X-Y Cu-Catalysed Intramolecular Radical Enantioconvergent Tertiary B-C(sp3)–H Amination of Racemic Ketones. Nat. Catal 2020, 3, 539–546. [Google Scholar]; (s) Zhang C; Li Z-L; Gu Q-S; Liu X-Y Catalytic Enantioselective C(sp3)–H Functionalization Involving Radical Intermediates. Nat. Commun 2021, 12, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Zhou H; Li Z-L; Gu Q-S; Liu X-Y Enabled Copper (i)-Catalyzed Asymmetric Radical C(sp3)–C CrossCoupling Reactions. ACS Catal 2021. 11, 7978–7986. [Google Scholar]
- (3).For selected reviews and highlights on Co(II)-based MRC, see; (a) Lu H; Zhang XP Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]; (b) Pellissier H; Clavier H Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (c) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (d) Huang H-M; Garduño-Castro MH; Morrill C; Procter DJ Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]; (e) Singh R; Mukherjee A Metalloporphyrin Catalyzed C–H Amination. ACS Catal. 2019, 9, 3604–3617. [Google Scholar]; (f) Roy S, Das SK; Khatua H; Das S; Chattopadhyay B. Road Map for the Construction of High-Valued N-Heterocycles via Denitrogenative Annulation. Acc. Chem. Res 2021, 54, 4395–4409. [DOI] [PubMed] [Google Scholar]
- (4).For selected examples of Ti(III)-based radical processes, see; (a) Nugent WA; RajanBabu T Transition-Metal-Centered Radicals in Organic Synthesis. Titanium (III)-Induced Cyclization of Epoxy Olefins. J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]; (b) RajanBabu a. T.; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (c) Gansäuer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers R Cationic Titanocene A III Complexes for Catalysis in Single-Electron Steps. Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (d) Gansäuer A; Hildebrandt S; Vogelsang E; Flowers R II Tuning the Redox Properties of the Titanocene (III)/(IV)-Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans. 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (e) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (f) Yao C; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]; (g) Ye K-Y; McCallum T, Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]
- (5).For selected examples of metalloradical-mediated radical processes, see; (a) Wayland BB; Poszmik G; Mukerjee SL; Fryd M Living Radical Polymerization of Acrylates by Organocobalt Porphyrin Complexes. J. Am. Chem. Soc 1994, 116, 7943–7944. [Google Scholar]; (b) Zhang X-X; Wayland BB Rhodium (II) Porphyrin Bimetalloradical Complexes: Preparation and Enhanced Reactivity with CH4 and H2. J. Am. Chem. Soc 1994, 116, 7897–7898. [Google Scholar]; (c) Chan KS; Li XZ; Dzik WI; de Bruin B Carbon-Carbon Bond Activation of 2,2,6,6-Tetramethyl-piperidine-1-oxyl by a RhII Metalloradical: A Combined Experimental and Theoretical Study. J. Am. Chem. Soc 2008, 130, 2051–2061. [DOI] [PubMed] [Google Scholar]; (d) Chan YW; Chan KS Metalloradical-Catalyzed Aliphatic Carbon-Carbon Activation of Cyclooctane. J. Am. Chem. Soc 2010, 132, 6920–6922. [DOI] [PubMed] [Google Scholar]; (e) Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co–H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (f) Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H• Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. [DOI] [PubMed] [Google Scholar]; (g) Roy S; Das SK; Chattopadhyay B Cobalt(II)-Based Metalloradical Activation of 2-(Diazomethyl) Pyridines for Radical Transannulation and Cyclopropanation. Angew. Chem., Int. Ed 2018, 57, 2238–2243. [DOI] [PubMed] [Google Scholar]; (h) Roy S; Khatua H; Das SK; Chattopadhyay B Iron (II)-Based Metalloradical Activation: Switch from Traditional Click Chemistry to Denitrogenative Annulation. Angew. Chem., Int. Ed 2019, 58, 11439–11443. [DOI] [PubMed] [Google Scholar]; (i) Das SK; Roy S; Khatua H; Chattopadhyay B Iron-Catalyzed Amination of Strong Aliphatic C(sp3)–H Bonds. J. Am. Chem. Soc 2020, 142, 16211–16217. [DOI] [PubMed] [Google Scholar]; (j) Zhang Z; Gevorgyan V Co-Catalyzed Transannulation of Pyridotriazoles with Isothiocyanates and Xanthate Esters. Org. Lett 2020, 22, 8500–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Roy S; Das SK; Khatua H; Das S; Singh KN; Chattopadhyay B. Iron-Catalyzed Radical Activation Mechanism for Denitrogenative Rearrangement over C(sp3)–H Amination. Angew. Chem., Int. Ed 2021, 60, 8772–8780. [DOI] [PubMed] [Google Scholar]
- (6).(a) Chen Y; Fields KB; Zhang XP Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]; (b) Caselli A; Gallo E; Ragaini F; Ricatto F; Abbiati G; Cenini S Chiral Porphyrin Complexes of Cobalt(II) and Ruthenium(II) in Catalytic Cyclopropanation and Amination Reactions. Inorg. Chim. Acta 2006, 359, 2924–2932. [Google Scholar]; (c) Chen Y; Ruppel JV; Zhang XP Cobalt-Catalyzed Asymmetric Cyclopropanation of Electron-Deficient Olefins. J. Am. Chem. Soc 2007, 129, 12074–12075. [DOI] [PubMed] [Google Scholar]; (d) Fantauzzi S; Gallo E; Rose E; Raoul N; Caselli A; Issa S; Ragaini F; Cenini S Asymmetric Cyclopropanation of Olefins Catalyzed by Chiral Cobalt (II)-Binaphthyl Porphyrins. Organometallics 2008, 27, 6143–6151. [Google Scholar]; (e) Zhu S; Perman JA; Zhang XP Acceptor/Acceptor-Substituted Diazo Reagents for Carbene Transfers: Cobalt-Catalyzed Asymmetric Z-Cyclopropanation of Alkenes with α-Nitrodiazoacetates. Angew. Chem., Int. Ed 2008, 47, 8460–8463. [DOI] [PubMed] [Google Scholar]; (f) Zhu S; Xu X; Perman JA; Zhang XP A General and Efficient Cobalt(II)-Based Catalytic System for Highly Stereoselective Cyclopropanation of Alkenes with α-Cyanodiazoacetates. J. Am. Chem. Soc 2010, 132, 12796–12799. [DOI] [PubMed] [Google Scholar]; (g) Xu X; Lu H; Ruppel JV; Cui X; Lopez de Mesa S; Wojtas L; Zhang XP Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]; (h) Paul ND; Mandal S; Otte M; Cui X; Zhang XP; de Bruin B Metalloradical Approach to 2H-Chromenes. J. Am. Chem. Soc 2014, 136, 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Reddy AR; Hao F; Wu K; Zhou CY; Che CM Cobalt(II) Porphyrin-Catalyzed Intramolecular Cyclopropanation of N-Alkyl Indoles/Pyrroles with Alkylcarbene: Efficient Synthesis of Polycyclic N-Heterocycles. Angew. Chem., Int. Ed 2016, 55, 1810–1815. [DOI] [PubMed] [Google Scholar]; (j) Chirila A; Gopal Das B; Paul ND; de Bruin B Diastereoselective Radical-Type Cyclopropanation of Electron-Deficient Alkenes Mediated by the Highly Active Cobalt(II) Tetramethyltetraaza[14]Annulene Catalyst. ChemCatChem 2017, 9, 1413. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wang J-Y; Xie J-J; Lee W-CC; Wang D-S; Zhang XP, Radical Differentiation of Two Esters in Unsymmetric Diazomalonates for Highly Asymmetric Olefin Cyclopropanation. Chem. Catal 2021, DOI: 10.1016/j.checat.2021.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Dzik WI; Xu X; Zhang XP; Reek JN; de Bruin B ‘Carbene Radicals’ in CoII(Por)-Catalyzed Olefin Cyclopropanation. J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]; (b) Belof JL; Cioce CR; Xu X; Zhang XP; Space B; Woodcock HL Characterization of Tunable Radical Metal–Carbenes: Key Intermediates in Catalytic Cyclopropanation. Organometallics 2011, 30, 2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu H; Dzik WI; Xu X; Wojtas L; de Bruin B; Zhang XP Experimental Evidence for Cobalt (III)-Carbene Radicals: Key Intermediates in Cobalt (II)-Based Metalloradical Cyclopropanation. J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- (8).(a) Wang Y; Wen X; Cui X; Wojtas L; Zhang XP Asymmetric Radical Cyclopropanation of Alkenes with in Situ-Generated Donor-Substituted Diazo Reagents via Co(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2017, 139, 1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee W-CC; Wang D-S; Zhang C; Xie J; Li B; Zhang XP Asymmetric Radical Cyclopropanation of Dehydroaminocarboxylates: Stereoselective Synthesis of Cyclopropyl α-Amino Acids. Chem 2021, 7, 1588–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang X; Ke J; Zhu Y; Deb A; Xu Y; Zhsang P Asymmetric Radical Process for General Synthesis of Chiral Heteroaryl Cyclopropanes. J. Am. Chem. Soc 2021, 143, 11121–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Noro M; Masuda T; Ichimura AS; Koga N; Iwamura H Carbene-Carbene Interconversion between 1-and 3-Phenyl-2-Propynylidenes. J. Am. Chem. Soc 1994, 116, 6179–6190. [Google Scholar]; (b) Bowling NP; Halter RJ; Hodges JA; Seburg RA; Thomas PS; Simmons CS; Stanton JF; McMahon RJ Reactive Carbon-Chain Molecules: Synthesis of 1-Diazo-2,4-Pentadiyne and Spectroscopic Characterization of Triplet Pentadiynylidene (H–C≡C–C≡C–H). J. Am. Chem. Soc 2006, 128, 3291–3302. [DOI] [PubMed] [Google Scholar]; (c) Wommack AJ; Moebius DC; Travis AL; Kingsbury JS Diverse Alkanones by Catalytic Carbon Insertion into the Formyl C–H Bond. Concise Access to the Natural Precursor of Achyrofuran. Org. Lett 2009, 11, 3202–3205. [DOI] [PubMed] [Google Scholar]; (d) Thomas PS; Bowling NP; Burrmann NJ; McMahon RJ Dialkynyl Carbene Derivatives: Generation and Characterization of Triplet tert-Butylpentadiynylidene (tBu–C≡C–C≡C–H) and Dimethylpentadiynylidene (Me–C≡C–C≡C–Me). J. Org. Chem 2010, 75, 6372–6381. [DOI] [PubMed] [Google Scholar]; (e) Kumar R; Turcaud S; Micouin L The Reaction of Dimethylalkynylaluminum Reagents with Trimethylsilyldiazomethane: Original Reactivity Leading to New α-Silylated Alkynyl Hydrazones. Org. Lett 2014, 16, 6192–6195. [DOI] [PubMed] [Google Scholar]
- (10).(a) Collin J; Lossing F Ionization and Dissociation of Allene, Propyne, 1-Butyne, and 1,2-and 1,3-Butadienes by Electron Impact; the C3H3+ Ion. J. Am. Chem. Soc 1957, 79, 5848–5853. [Google Scholar]; (b) Fantazier RM; Poutsma ML Reduction of Propargylic Chlorides with Tri-N-Butyltin Hydride. The Ambident Behavior of Propargylic Radicals. J. Am. Chem. Soc 1968, 90, 5490–5498. [Google Scholar]; (c) Kochi JK; Krusic PJ Electron Spin Resonance of Free Radicals from Acetylenes and Allenes. J. Am. Chem. Soc 1970, 92, 4110–4114. [Google Scholar]; (d) Kasai PH Electron Spin Resonance Studies of Vinyl, Propargyl, and Butatrienyl Radicals Isolated in Argon Matrices. J. Am. Chem. Soc 1972, 94, 5950–5956. [Google Scholar]; (e) Kuhnel W; Gey E; Ondruschka B Theoretical-Stydies of Characteristics of Unsaturated Hydrocarbon Radicals of the Allyl and Propargyl Types. Z. Phys. Chem 1987, 268, 805–814. [Google Scholar]; (f) Herges R; Mebel A Propargylene. J. Am. Chem. Soc 1994, 116, 8229–8237. [Google Scholar]; (g) Adam W; Ortega-Schulte CM Spin Delocalization by Triple-Bonded Functionalities in Propargyl and Heteropropargyl Radicals, Assessed from the EPR-Spectral D Parameter of 1,3 Cyclopentanediyl Triplet Diradicals. J. Org. Chem 2003, 68, 1007–1011. [DOI] [PubMed] [Google Scholar]; (h) Steinbauer M; Lang M; Fischer I; de Miranda BKC; Romanzin C; Alcaraz C The Photoionisation of Propargylene and Diazopropyne. Phys. Chem. Chem. Phys 2011, 13, 17956–17959. [DOI] [PubMed] [Google Scholar]; (i) Maury J; Jammi S; Vibert F; Marque SRA; Siri D; Feray L; Bertrand M EPR Investigation of Zinc/Iodine Exchange between Propargyl Iodides and Diethylzinc: Detection of Propargyl Radical by Spin Trapping. J. Org. Chem 2012, 77, 9081–9086. [DOI] [PubMed] [Google Scholar]; (j) Giegerich J; Petersen J; Mitrić R; Fischer I Photodissociation Dynamics of Propargylene, Hccch. Phys. Chem. Chem. Phys 2014, 16, 6294–6302. [DOI] [PubMed] [Google Scholar]; (k) Osborn DL; Vogelhuber KM; Wren SW; Miller EM; Lu Y-J; Case AS; Sheps L; McMahon RJ; Stanton JF; Harding LB; Ruscic B; Lineberger WC Electronic States of the Quasilinear Molecule Propargylene (HCCCH) from Negative Ion Photoelectron Spectroscopy. J. Am. Chem. Soc 2014, 136, 10361–10372. [DOI] [PubMed] [Google Scholar]; (l) Qiu S; Zhang Y; Huang X; Bao L; Hong Y; Zeng Z; Wu J 9-Ethynylfluoroenyl Radicals: Regioselective Dimerization and Post Ring-Cyclization Reactions. Org. Lett 2016, 18, 6018–6021. [DOI] [PubMed] [Google Scholar]; (m) Hansmann MM; Melaimi M; Bertrand G Crystalline Monomeric Allenyl/Propargyl Radical. J. Am. Chem. Soc 2017, 139, 15620–15623. [DOI] [PubMed] [Google Scholar]; (n) Reusch E; Kaiser D; Schleier D; Buschmann R; Krueger A; Hermann T; Engels B; Fischer I; Hemberger P Pentadiynylidene and Its Methyl-Substituted Derivates: Threshold Photoelectron Spectroscopy of R1-C5-R2 Triplet Carbon Chains. J. Phys. Chem. A 2019, 123, 2008–2017. [DOI] [PubMed] [Google Scholar]; (o) Dong X-Y; Zhan T-Y; Jiang S-P; Liu X-D; Ye L; Li Z-L; Gu Q-S; Liu X-Y Copper-Catalyzed Asymmetric Coupling of Allenyl Radicals with Terminal Alkynes to Access Tetrasubstituted Allenes. Angew. Chem., Int. Ed 2021, 60, 2160–2164. [DOI] [PubMed] [Google Scholar]
- (11).(a) Zampella A; D’Auria MV; Minale L; Debitus C; Roussakis C Callipeltoside A: A Cytotoxic Aminodeoxy Sugar Containing Macrolide of a New Type from the Marine Lithistida Sponge Callipelta sp. J. Am. Chem. Soc 1996, 118, 11085–11088. [Google Scholar]; (b) Zampella A; D’Auria MV; Minale L; Debitus C Callipeltosides B and C, Two Novel Cytotoxic Glycoside Macrolides from a Marine Lithistida Sponge Callipelta Sp. Tetrahedron 1997, 53, 3243–3248. [Google Scholar]; (c) Adkins JC; Noble S Efavirenz. Drugs 1998, 56, 1055–1064. [DOI] [PubMed] [Google Scholar]; (d) Evans DA; Hu E; Burch JD; Jaeschke G Enantioselective Total Synthesis of Callipeltoside A. J. Am. Chem. Soc 2002, 124, 5654–5655. [DOI] [PubMed] [Google Scholar]; (e) Trost BM; Gunzner JL; Dirat O; Rhee YH Callipeltoside A: Total Synthesis, Assignment of the Absolute and Relative Configuration, and Evaluation of Synthetic Analogues. J. Am. Chem. Soc 2002, 124, 10396–10415. [DOI] [PubMed] [Google Scholar]; (f) Carpenter J; Northrup AB; Chung d.; Wiener JJM; Kim SG; MacMillan DWC. Total Synthesis and Structural Revision of Callipeltoside C. Angew. Chem., Int. Ed 2008, 47, 3568–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Raddatz R; Tao M; Hudkins RL Histamine H3 Antagonists for Treatment of Cognitive Deficits in CNS Diseases. Curr. Top. Med. Chem 2010, 10, 153–169. [DOI] [PubMed] [Google Scholar]; (h) Rakhmanina NY; van den Anker JN Efavirenz in the Therapy of HIV Infection. Expert Opinion on Drug Metabolism & Toxicology 2010, 6, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) La Ferla B; Airoldi C; Zona C; Orsato A; Cardona F; Merlo S; Sironi E; D’Orazio G; Nicotra F Natural Glycoconjugates with Antitumor Activity. Nat. Prod. Rep 2011, 28, 630–648. [DOI] [PubMed] [Google Scholar]; (j) Frost JR; Pearson CM; Snaddon TN; Booth RA; Turner RM; Gold J; Shaw DM; Gaunt MJ; Ley SV Callipeltosides A, B and C: Total Syntheses and Structural Confirmation. Chem. - Eur. J 2015, 21, 13261–13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For general reviews on metal-catalyzed olefin cyclopropanation with diazo compounds, see; (a) Doyle MP Catalytic Methods for Metal Carbene Transformations. Chem. Rev 1986, 86, 919–939. [DOI] [PubMed] [Google Scholar]; (b) Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev 1998, 98, 911–936. [DOI] [PubMed] [Google Scholar]; (c) Intrieri D; Carminati DM; Gallo E The Ligand Influence in Stereoselective Carbene Transfer Reactions Promoted by Chiral Metal Porphyrin Catalysts. Dalton Trans 2016, 45, 15746–15761. [DOI] [PubMed] [Google Scholar]; (d) Mato M; Franchino A; Garcia-Morales C; Echavarren AM Gold-Catalyzed Synthesis of Small Rings. Chem. Rev 2021, 121, 8613–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For asymmetric synthesis of alkynylcyclopropanes from cyclopropanation of conjugated enynes, see ref 8 and; (a) Saha B; Uchida T; Katsuki T Highly Enantioselective Intramolecular Cyclopropanation of Alkenyl Diazo Ketones Using Ru(Salen) as Catalyst. Chem. Lett 2002, 31, 846–847. [Google Scholar]; (b) Du H; Long J; Shi Y Catalytic Asymmetric Simmons–Smith Cyclopropanation of Silyl Enol Ethers. Efficient Synthesis of Optically Active Cyclopropanol Derivatives. Org. Lett 2006, 8, 2827–2829. [DOI] [PubMed] [Google Scholar]; (c) Suematsu H; Kanchiku S; Uchida T; Katsuki T Construction of Aryliridium–Salen Complexes: Enantio- and Cis-Selective Cyclopropanation of Conjugated and Nonconjugated Olefins. J. Am. Chem. Soc 2008, 130, 10327–10337. [DOI] [PubMed] [Google Scholar]; (d) Singha S; Buchsteiner M; Bistoni G; Goddard R; Fürstner A A New Ligand Design Based on London Dispersion Empowers Chiral Bismuth–Rhodium Paddlewheel Catalysts. J. Am. Chem. Soc 2021, 143, 5666–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For asymmetric synthesis of alkynylcyclopropanes from direct functionalization of cyclopropenes, see; (a) Krámer K; Leong P; Lautens M Enantioselective Palladium-Catalyzed Carbozincation of Cyclopropenes. Org. Lett 2011, 13, 819–821. [DOI] [PubMed] [Google Scholar]; (b) Teng H-L; Ma Y; Zhan G; Nishiura M; Hou Z Asymmetric C(sp)–H Addition of Terminal Alkynes to Cyclopropenes by a Chiral Gadolinium Catalyst. ACS Catal 2018, 8, 4705–4709. [Google Scholar]; (c) Dian L; Marek I Pd-Catalyzed Enantioselective Hydroalkynylation of Cyclopropenes. ACS Catal 2020, 10, 1289–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Davies HML; Boebel TA Asymmetric Synthesis of 1-Alkynylcyclopropane-1-Carboxylates. Tetrahedron Lett 2000, 41, 8189–8192. [Google Scholar]
- (16).(a) Skell PS; Klebe J Structure and Properties of Propargylene C3H21. J. Am. Chem. Soc 1960, 82, 247–248. [Google Scholar]; (b) Padwa A; Gareau Y; Xu SL A Study of the Rearrangement Chemistry of Alkynyl Carbenes. Tetrahedron Lett 1991, 32, 983–986. [Google Scholar]; (c) Liu Z; Zhang X; Zanoni G; Bi X Silver-Catalyzed Cyclopropanation of Alkenes Using N-Nosylhydrazones as Diazo Surrogates. Org. Lett 2017, 19, 6646–6649. [DOI] [PubMed] [Google Scholar]
- (17).(a) Regitz M; Maas G Diazo Compounds: Properties and Synthesis; Academic Press: Orlando, FL, 1986; pp 407–426. [Google Scholar]; (b) Aggarwal VK; Alonso E; Bae I; Hynd G; Lydon KM; Palmer MJ; Patel M; Porcelloni M; Richardson J; Stenson RA; Studley JR; Vasse J-L; Winn CL A New Protocol for the in Situ Generation of Aromatic, Heteroaromatic, and Unsaturated Diazo Compounds and Its Application in Catalytic and Asymmetric Epoxidation of Carbonyl Compounds. Extensive Studies to Map out Scope and Limitations, and Rationalization of Diastereo- and Enantioselectivities. J. Am. Chem. Soc 2003, 125, 10926–10940. [DOI] [PubMed] [Google Scholar]; (c) Fulton JR; Aggarwal VK; de Vicente J The Use of Tosylhydrazone Salts as a Safe Alternative for Handling Diazo Compounds and Their Applications in Organic Synthesis. Eur. J. Org. Chem 2005, 2005, 1479–1492. [Google Scholar]; (d) Seburg RA; Hodges JA; McMahon RJ Propynal Equivalents and Diazopropyne: Synthesis of All Mono-13C Isotopomers. Helv. Chim. Acta 2009, 92, 1626–1643. [Google Scholar]; (e) Vuluga D; Legros J; Crousse B; Bonnet-Delpon D Synthesis of Pyrazoles Through Catalyst-Free Cycloaddition of Diazo Compounds to Alkynes. Green Chem 2009, 11, 156–159. [Google Scholar]; (f) Allouche EMD; Charette AB Cyclopropanation Reactions of Semi-Stabilized and Non-Stabilized Diazo Compounds. Synthesis 2019, 51, 3947–3963. [Google Scholar]
- (18).Mato M; Montesinos-Magraner M; Sugranyes AR; Echavarren AM Rh(II)-Catalyzed Alkynylcyclopropanation of Alkenes by Decarbenation of Alkynylcycloheptatrienes. J. Am. Chem. Soc 2021, 143, 10760–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ruppel JV; Jones JE; Huff CA; Kamble RM; Chen Y; Zhang XP A Highly Effective Cobalt Catalyst for Olefin Aziridination with Azides: Hydrogen Bonding Guided Catalyst Design. Org. Lett 2008, 10, 1995–1998. [DOI] [PubMed] [Google Scholar]
- (20).Zhu S; Ruppel JV; Lu H; Wojtas L; Zhang XP Cobalt-Catalyzed Asymmetric Cyclopropanation with Diazosulfones: Rigidification and Polarization of Ligand Chiral Environment via Hydrogen Bonding and Cyclization. J. Am. Chem. Soc 2008, 130, 5042–5043. [DOI] [PubMed] [Google Scholar]
- (21).(a) Wang Y; Wen X; Cui X; Zhang XP Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C–H Alkylation. J. Am. Chem. Soc 2018, 140, 4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wen X; Wang Y; Zhang XP Enantioselective Radical Process for Synthesis of Chiral Indolines by Metalloradical Alkylation of Diverse C(sp3)–H Bonds. Chem. Sci 2018, 9, 5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Ma S; He Q The Cyclopropyl Effect on the Regioselectivity of Coupling Reactions Involving the Lithiation of 1-Cyclopropyl-2-Arylacetylenes. Tetrahedron 2006, 62, 2769–2778. [Google Scholar]; (b) Hu L; Mück-Lichtenfeld C; Wang T; He G; Gao M; Zhao J Reaction between Azidyl Radicals and Alkynes: A Straightforward Approach to NH-1,2,3-Triazoles. Chem. - Eur. J 2016, 22, 911–915. [DOI] [PubMed] [Google Scholar]; (c) Li J-H; Huang Q; Wang S-Y; Ji S-J Trisulfur Radical Anion (S3•−) Involved [1 + 2 + 2] and [1 + 3 + 1] Cycloaddition with Aromatic Alkynes: Synthesis of Tetraphenylthiophene and 2-Benzylidenetetrahydrothiophene Derivatives. Org. Lett 2018, 20, 4704–4708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.