

Abstract
Objective:
12/15-lipoxygenase (12/15LO) expression in the vessel wall is increased in animal models of metabolic syndrome and diabetes. Increased expression of 12/15LO enhances cultured vascular smooth muscle cell (VSMC) proliferation; an effect mediated by the helix-loop-helix factor Id3. Whether increased 12/15LO expression in vivo enhances neointimal formation (NIF) in response to injury is unknown.
Methods and Results:
Carotid endothelial denudation was performed on ApoE−/−, ApoE−/−/12/15LO−/− (DKO), C57BL/6 (BL-6), and 12/15LO-overexpressing transgenic mice (12/15LO-tg). DKO mice had attenuated and 12/15LO-tg mice had enhanced NIF compared with controls. 12/15LO-tg mice had greater post-injury carotid Id3 and Ki-67 expression, cell number, and fibronectin deposition compared with BL-6 mice. Loss of 12/15LO attenuated proliferation of cultured ApoE−/− VSMCs, while 12/15LO overexpression induced VSMC proliferation. Loss of Id3 enhanced ITF-2b binding to and activation of the p21cip1 promoter and abrogated 12/15LO-induced VSMC proliferation.
Conclusions:
These data are the first demonstration that increased expression of 12/15LO in the vessel wall enhances Id3-dependent cell proliferation, fibronectin deposition and NIF in response to injury. Results identify p21cip1 as a potential target of the 12/15LO-Id3 pathway and suggest that modulation of this pathway may have therapeutic implications for targeting the increased risk of restenosis in patients with diabetes.
Keywords: 12/15-Lipoxygenase, neointima, vascular injury, fibronectin, helix-loop-helix factors
Condensed abstract
Increased 12/15LO expression induces neointimal fibronectin deposition, Id3-mediated cell proliferation, and enhances neointimal formation in response to injury. These data suggest potential mechanisms for increased restenosis in states with elevated 12/15LO expression such as in metabolic syndrome and type 2 diabetes mellitus.
Individuals with type 2 diabetes have increased rates of restenosis after vascular interventional procedures.1, 2 Vascular smooth muscle cell (VSMC) proliferation and matrix production are key events in the process of neointimal formation (NIF) following percutaneous interventions.3, 4 Identifying the molecular mechanisms that mediate acceleration of these processes may provide important insight into strategies to attenuate restenosis in high risk populations, such as those with type 2 diabetes mellitus.
Previous studies have implicated 12/15-lipoxygenase enzyme (12/15LO) in the vascular response to injury. 12/15LO products of arachidonic acid such as 12S-HETE (hydroxyeicosatetraenoic acid), 15S-HETE, and 13S-HPODE (hydroperoxyoctadecadienoic acid) are produced in VSMCs and have hypertrophic effects.5, 6 In vitro, 12/15LO inhibition attenuates hypertrophic effects of angiotensin II in VSMCs, mitogenic effects of cytokines, and chemotactic effects of PDGF.5, 7, 8 Compared with VSMC from C57BL/6 (BL-6) mice, VSMCs from mice overexpressing 12/15LO (12/15LO-tg) grow faster, and VSMCs from 12/15LO−/− mice grow slower and display decreased S-phase entry in culture.9, 10 12/15LO and its products are increased in the vascular wall of animal models of atherosclerosis and injury-induced restenosis.11 Compared to uninjured carotids, 12/15LO expression is significantly increased in rat carotids on day 12 after injury.12 Moreover, pharmacologic or ribozyme-mediated inhibition of the 12/15LO gene in vascular injury models result in attenuated NIF,12, 13 yet, the effects of baseline elevations in 12/15LO on injury-induced NIF and the factors downstream of 12/15LO that mediate these effects in vivo are incompletely understood.
Many conditions that are implicated in accelerated vascular response to injury are associated with enhanced 12/15LO expression. In particular, there is evidence that activity and levels of 12/15LO are increased in diabetes. Patients with type-2 diabetes mellitus have increased levels of 12-HETE in the urine.14 Treatment of cultured VSMCs with high glucose increases the expression of 12- and 15-HETEs.15 Furthermore, 12/15LO expression is increased in the vascular wall in a porcine model of diabetes.11 Endothelial cells isolated from db/db diabetic mice have increased 12/15LO expression while the db/db mice also have increased levels of 12- and 15-S-HETE in vivo.16 The obese Zucker rat model of metabolic syndrome also displays increased carotid 12/15LO expression and NIF after balloon angioplasty relative to lean Zucker rats.17 Similarly, other candidates implicated in vascular response to injury including angiotensin II, growth factors such as PDGF, and inflammatory cytokines such as interleukin-1 are also known to be potent inducers of 12/15LO expression and activity in VSMCs. 11, 18–21
Inhibitor of differentiation 3 (Id3) is a helix-loop-helix (HLH) transcription factor that functions as an important regulator of cellular growth.22 Id3 expression is induced in response to mitogen stimulation, and inhibition of Id3 blocks mitogen-induced proliferation.23–25 Id3 expression in vivo is significantly increased at day 3 and 7 after vascular injury and returns to baseline by day 28.26, 27 Previous studies have demonstrated that 12/15LO induces Id3 expression and enhances growth in cultured VSMC. Importantly, overexpression of 12/15LO in culture increases growth in BL-6 VSMCs but not in Id3−/− VSMCs.10
The goal of the current study was to determine if the effects of increased 12/15LO on Id3 expression and proliferation in VSMC in culture, occurred in vivo and if increased expression of 12/15LO at baseline increases NIF in response to injury.
Methods
For full details of methods, please see http://atvb.ahajournals.org data supplemental material.
Real-time RT-PCR28, immunohistochemistry26, cell culture26, chromatin immunoprecipitation (ChIP)28 and promoter-reporter analysis10 were performed as previously described.
Animals
Studies were done in accordance with the institutional guidelines at the University of Virginia. C57BL/6 (BL-6), ApoE−/−, ApoE−/−/12/15LO−/−(DKO), 12/15LO-tg, 12/15LO-tg/Id3−/− mice were used for Left common carotid artery (LCCA) wire injury experiments. In an attempt to minimize variation, bias and the number of animals used, all wire injury in our study was performed by a single experienced individual blinded to the genotype of the mice.
Quantitative Histopathology
LCCA sections were stained using Russell’s modified Movat method. 29 Every injury section in all groups was reviewed by a panel of scientists experienced in vascular injury who were blinded to the genotypes of the injured mice. Any animal with visible disruption of the internal elastic lamina was excluded prior to unblinding. As such, every section that is included in the analysis has intact elastic lamina.
Statistical analysis
Statistical analyses were performed using PRISM 4 (GraphPad). Mann-Whitney U-test was used to compare continuous variables between groups and p-values <0.05 were considered significant. Analysis of Variance (ANOVA) was performed to evaluate the differences between multiple groups on continuous variables. Data is shown as Mean ± SD.
Results
Increased 12/15LO expression increases NIF in BL-6 mice, while 12/15LO deficiency attenuates NIF in ApoE−/− mice.
To evaluate whether12/15LO expression level at baseline influences NIF after vascular injury, 10-12 week old male BL-6 (n=8), 12/15LO-tg (n=11), ApoE−/− (n=5) and DKO (n=9) mice were fed Western diet for one week, after which they underwent LCCA endothelial denudation. We chose a 12/15LO knock-out model on ApoE−/− background to show the “loss of function” following loss of 12/15LO, because unlike ApoE−/−, BL-6 mice do not form robust neointima in response to injury. Twenty-eight days after wire injury, animals were euthanized and the LCCAs were harvested. Histomorphometric analysis revealed that 12/15LO-tg mice had significantly greater NIF compared with the BL-6 mouse (3427 ± 2017μm2 vs. 1469 ± 2162μm2, p=0.023), as shown in Figures 1A and B. Also consistent with prior inhibitor studies, the ApoE−/− control mice developed significant neointima in response to wire injury, while loss of 12/15LO resulted in a significant reduction in the lesion size (81824 ± 14151μm2 vs. 26461 ± 26529μm2, p=0.007), as shown in Figures 1C and D. There were no differences in glucose, cholesterol, or triglyceride levels between the groups (data not shown).
Figure 1. Increased 12/15LO expression enhances and 12/15LO deficiency attenuates injury-induced neointimal formation.
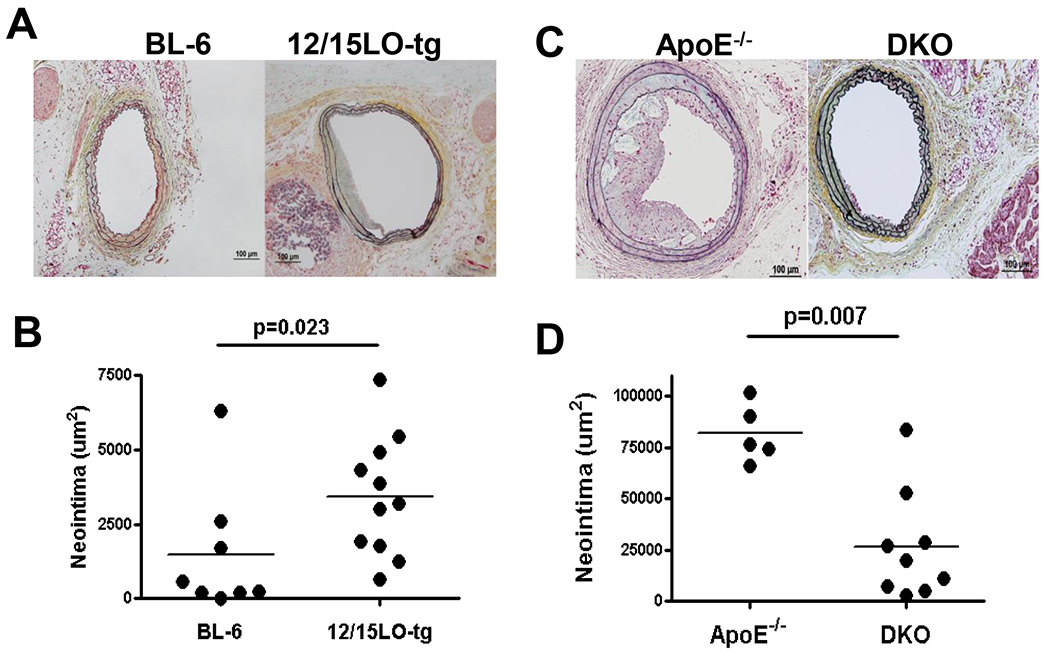
Male C57BL-6, 12/15LO-tg, ApoE−/−, and DKO mice were fed western diet starting at 10-12 weeks of age and underwent LCCA endothelial denudation a week later. Histomorphometric analysis was performed 28 days after the injury. 1A and C: Representative slides selected at 240μm proximal to the carotid bifurcation 1B: Compares mean neointimal formation in BL-6 and 12/15LO-tg mouse groups. 1D: Compares mean neointimal formation in ApoE−/− and DKO mouse groups. Each dot represents the mean of NIF in eight sections in one mouse.
Id3 expression in response to injury is greater in LCCA of 12/15LO-tg mice.
Prior studies demonstrated that cultured VSMCs from 12/15LO-tg animals had greater expression of Id3 than VSMCs from BL-6 mice and that 12LO-induced VSMC proliferation was mediated by Id3. Moreover, Id3 expression is increased in response to vascular injury in vivo.26, 27 To determine if 12/15LO-tg mice have enhanced Id3 expression in response to injury compared to BL-6, LCCAs were harvested at seven and 14 days after injury and were analyzed for 12/15LO and Id3 mRNA expressions. As expected, 12/15LO-tg mice had greater 12/15LO expression in LCCAs after injury compared with BL-6 (Figure 2A). Consistent with prior in vitro data, mice with increased 12/15LO expression had increased Id3 mRNA in the vessel wall compared with BL-6 mice (Figure 2B).
Figure 2. 12/15LO and Id3 expressions in post-injury LCCAs are greater in 12/15LO-tg compared with BL-6 mice.
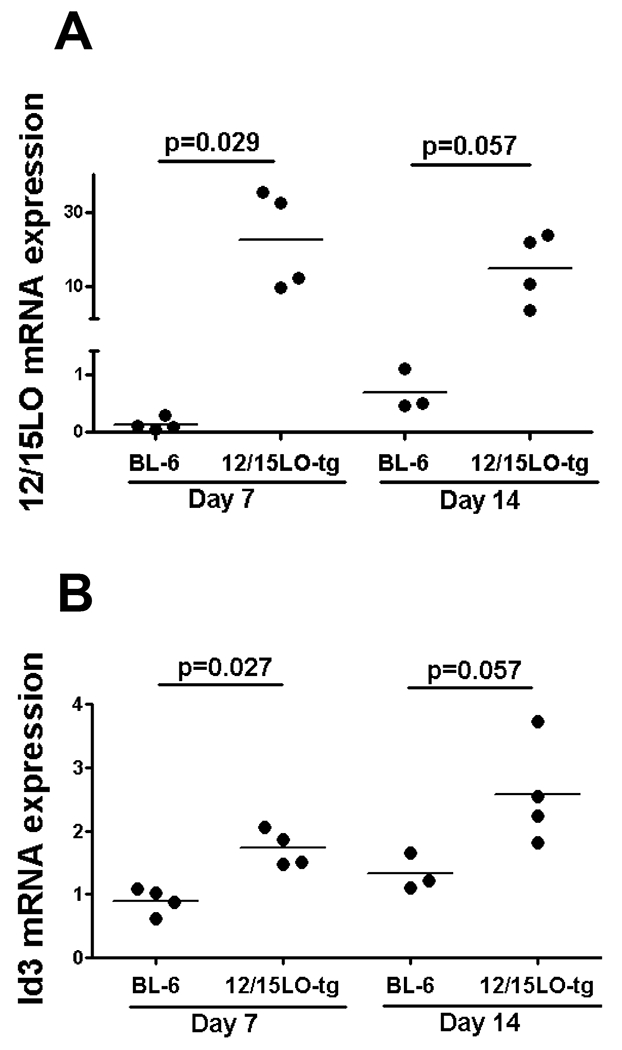
Male BL-6 and 12/15LO-tg mice were fed western diet starting at 10-12 weeks of age and underwent LCCA endothelial denudation one week later. LCCAs were harvested seven days and 14 days after the injury and 12/15LO (2A) and Id3 (2B) mRNA expressions were determined by real-time RT-PCR. Each dot represents mRNA expression of one mouse.
The 12/15LO-tg mouse has enhanced neointimal cell proliferation, an effect attenuated by loss of Id3.
Analysis of neointima 28 days after injury indicates that there is a greater cell number in the neointima of 12/15LO-tg mice compared with BL-6 mice (Supplemental Figure II). Ki-67 staining was performed seven days after injury to evaluate for in vivo differences in proliferation post-injury in 12/15LO-tg vs. BL-6 mice. Results demonstrate significantly more neointimal but not medial cell proliferation in 12/15LO-tg mice compared with BL-6 mice. Interestingly, cell proliferation was significantly attenuated in 12/15LO-tg mice null for Id3 compared with 12/15LO-tg mice (Figures 3A and B).
Figure 3. Increased Ki-67 in the neointima of 12/15LO-tg compared with BL-6 mice is abrogated by loss of Id3.
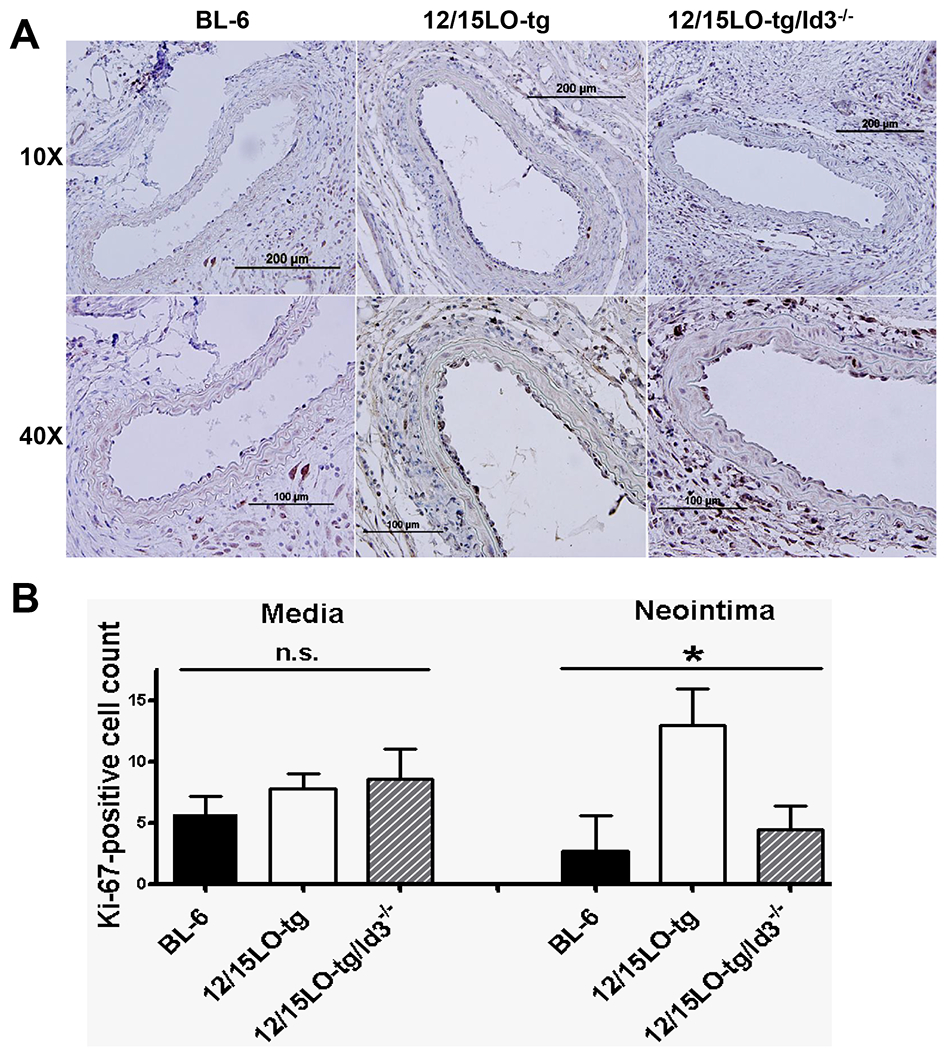
Male BL-6,12/15LO-tg, and 12/15LO-tg/Id3−/− mice were fed western diet starting at 10-12 weeks of age and underwent LCCA endothelial denudation one week later. LCCAs were harvested seven days after the injury 3A: Representative sections showing Ki-67 immunostaining in 7-day post-injury LCCAs at 10X (top row) and 40X (bottom row) magnifications 3B: Quantitation of Ki-67-positive cells seven days after injury (n=4 in each group), (*) represents significant difference in mean values of different groups by ANOVA. Error bars are SD.
12/15LO enhances VSMC proliferation in culture, an effect that is attenuated by Id3 deficiency.
We compared growth rates for matched passage (7-9) cultured VSMCs from BL-6, Id3−/−, 12/15LO-tg, and 12/15LO-tg/Id3−/− mice by direct cell counting. Similarly, we compared the growth rate between VSMCs from ApoE−/− and DKO mice. Consistent with our in vivo data, 12/15LO-tg VSMCs had accelerated proliferation compared with the BL-6 and loss of Id3 in 12/15LO-tg VSMCs resulted in a significant reduction in their proliferation (Figure 4A). Moreover, VSMCs from DKO mice had attenuated proliferation compared with VSMCs from ApoE−/− mice (Figure 4B). Similar growth rates were observed using a fluorometric DNA assay (data not shown).
Figure 4. Id3 deficiency inhibits accelerated proliferation observed in 12/15LO-tg VSMCs and loss of 12/15LO in cultured ApoE−/− VSMCs attenuates their proliferation.
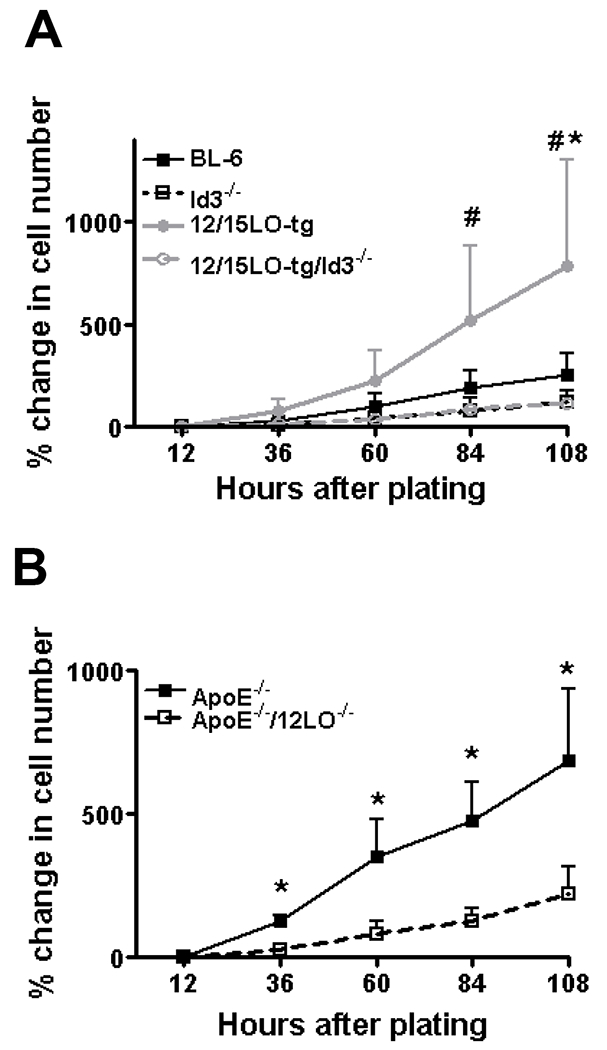
Passage 7-9 VSMCs from BL-6, 12/15LO-tg, Id3−/−, 12/15LO-tg/Id3−/−, ApoE−/−, and DKO mice were plated at the same density. By direct counting, the cell numbers quantified at twelve hours after plating (defined as baseline), and then every 24 hours x 4. Data is mean of four independent experiments. 4A: (#) p<0.05 for 12/15LO-tg vs. 12/15LO-tg/Id3−/−, (*) p<0.05 for 12/15LO-tg vs. BL-6. 4B: (*) p<0.05.
Loss of Id3 enhances ITF-2b binding to and activation of the p21cip1 promoter.
Id3 is a known growth factor-inducible gene that inhibits VSMC expression of the cyclin-dependent kinase inhibitor p21cip1. To explore a mechanism by which Id3 accelerates VSMC growth, we examined the effect of Id3 on the binding of HLH factor ITF-2b to the p21cip1 promoter by ChIP. Interestingly, ITF-2b binding to p21cip1 promoter was enhanced after loss of Id3 in 12/15LO-tg VSMCs (Figure 5A). In addition, by promoter-reporter assays, we demonstrated that ITF-2b expression potentiates p21cip1 promoter activity (Figure 5B).
Figure 5. Id3 deficiency enhances ITF-2b binding to p21cip1 promoter, and ITF-2b binding increases p21cip1 promoter activity.
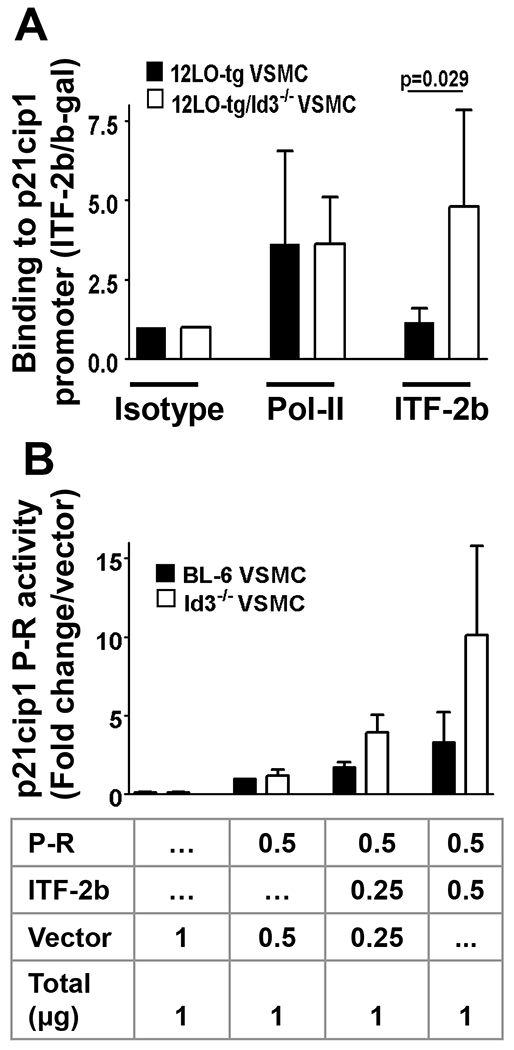
To explore a mechanism by which Id3 accelerates VSMC growth, we examined the effect of Id3 on the binding of helix-loop-helix factor ITF-2 to the p21cip1 promoter by ChIP. VSMCs of 12/15LO-tg and 12/15LO-tg/Id3−/− mice were cross-linked and precipitated with the indicated antibodies and immunoprecipitated DNA fragments were quantified by real time PCR and normalized to an internal β-galactosidase control for recovery. ChIP for RNA polymerase-II was used as positive control. Results are presented as fold increase in percent recovery relative to IP with isotype control and are average of triplicate PCR measurements from four independent ChIPs (5A). In addition, we demonstrated that ITF-2b biding to p21cip1 promoter enhances its transcription. BL-6 and Id3−/− VSMCS were cotransfected with a pEF4-ITF-2b expression vector and a pGL3 vector harboring 2.3Kb human p21cip1 promoter. Twenty-four hours after transfection, cell lystaes were assayed for luciferase activity. Values are p21cip1 promoter-repprter luciferase activity normalized to protein levels and are presented relative to the activity of promoter-reporter with only empty vector. Experiments were done three times in triplicate (5B).
12/15LO-tg mice have more neointimal fibronectin deposition compared with the BL-6 mice.
Reddy et al. previously demonstrated that 12(S)HETE, induces fibronectin promoter activity in VSMC.6 To determine if 12/15LO increases fibronectin production in vivo, potentially contributing to the increased NIF in 12/15LO-tg mice, we performed immunostaining for fibronectin on 28-day post-injury LCCA sections (eight animals in each group). When compared with the BL-6, 12/15LO-tg mice had significantly more fibronectin determined by both the absolute fibronectin-stained area and by the percent of the fibronectin-stained area relative to the neointimal lesion (Figure 6). Immunostaining for fibronectin in ApoE−/− and DKO demonstrated a trend to lower fibronectin deposition in DKO mice compared with ApoE−/− mice Supplemental Figure III). As 12S-HETE induction of fibronectin, has been shown to mediate monocyte adhesion30, 31, injured artery sections from BL-6, 12/15LO-tg, ApoE−/− and DKO mice were stained with a MAC-2 antibody(Supplemental figure IV). While there was increased MAC-2 staining in the mice on ApoE−/− compared to BL-6 background, there was no statistically significant differences in MAC-2 staining with increase or loss of 12/15LO expression (data not shown).
Figure 6. 12/15LO-tg mice have greater neointimal fibronectin (FN) compared with BL-6 in 28-day post-injury neointima.
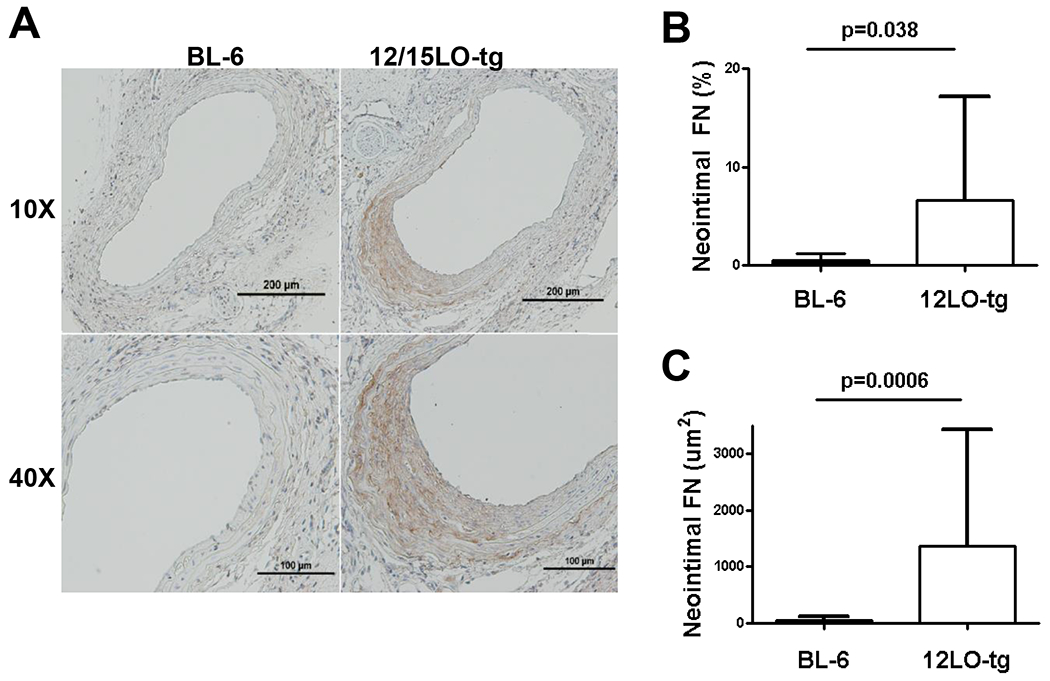
To examine whether enhanced fibronectin deposition contributed to the increased neointimal formation in the 12/15LO-tg mice, we stained the 28D post-injury LCCA sections for fibronectin. 6A: top row displays 10X magnification of the representative sections stained for fibronectin in BL-6 and 12/15LO-tg mice. The bottom row displays the same slides at 40X magnification. 6B and C: show that compared with the BL-6, neointima in the 12/15LO-tg mice had significantly more fibronectin determined by the absolute fibronectin-stained area and by the percent of the fibronectin-stained area to the neointimal lesion. Data is represented as mean ± SD of the mean of two LCCA sections obtained from equally distributed intervals from carotid bifurcation of eight animals in each group.
Discussion
In vivo studies have previously shown that 12/15LO expression is induced after vascular injury.12, 17 Ribozyme-mediated or pharmacologic inhibition studies have shown reduced NIF when injury-induced 12/15LO expression is blocked.12, 13 Our data demonstrating reduced neointimal formation in the ApoE−/−/12/15LO−/− compared to ApoE−/− mice provides further evidence that loss of 12/15LO attenuates the vascular response to injury. In addition to these loss of function studies, here we extend the existing literature, by providing the first data on the effect of increased baseline 12/15LO expression on NIF in response to injury. Relevance for determining the impact of baseline increase in 12/15LO on vascular lesion formation is provided by a number of studies demonstrating that conditions associated with accelerated vascular response to injury (such as increased oxidative stress and type 2 diabetes) are associated with enhanced 12/15LO expression.11, 18–21
12/15LO has been implicated in mediating monocyte adhesion to endothelial cells31, 32 and VSMCs33. In addition, 12/15LO is abundantly expressed in macrophages34 and regulates processes in macrophages implicated in atherogenesis31, 35. Indeed, disruption of the 12/15LO gene diminished atherosclerosis in the ApoE−/− mouse36, and macrophage 12/15LO has been implicated in this effect37. Results of the present study demonstrate that in addition to reduced atherosclerosis, ApoE−/−/12/15LO−/− mice have attenuated neointimal formation in response to injury. While loss of 12/15LO in macrophage may have contributed to the attenuated response to injury in ApoE−/−/12/15LO−/− mice, our findings that cultured VSMC from ApoE−/−/12/15LO−/− mice have attenuated proliferation compared to ApoE−/− controls suggest that reduced VSMC proliferation may be an additional mechanism contributing to attenuated neointimal formation in ApoE−/−/12/15LO−/− mice.
To address the role of 12/15LO in mediating VSMC proliferation in vivo, we performed additional injury studies utilizing gentle wire denudation of the carotid in a C57BL/6 mouse background. Utilizing this model limits the effects of marked hyperlipidemia and macrophage infiltration on neointimal formation. This model does not result in robust inflammatory lesions with macrophage infiltration38 like Western fed ApoE−/− mice39. Indeed, in contrast to ApoE−/− mice, MAC-2 staining revealed no immunoreactivity in the C57BL/6 control and 12/15LO-tg mice 28 days after injury (Supplemental Figure IV). Consistent with previously published data demonstrating enhanced proliferation in cultured C57BL/6 VSMC with increased 12/15LO expression9, 10, results in vivo provide evidence that animals with increased baseline 12/15LO expression have increased neointimal size in response to injury. While lesion size in this C57BL/6 model is small, results clearly demonstrate significant increases in Ki67 immunoreactivity and cell content. These data provide the first in vivo evidence suggesting that enhanced neointimal cell proliferation may be one mechanism whereby increased baseline 12/15LO promotes NIF.
Mechanisms whereby 12/15LO enhances VSMC proliferation are poorly understood. Previous studies in cultured VSMC provide evidence that 12/15LO-induced increase in VSMC proliferation is mediated by the helix-loop-helix factor, Id3. Overexpression of 12/15LO was shown to increase Id3 promoter activation suggesting that 12/15LO regulates expression of the Id3 gene at the level of transcription. Id3 is a known growth factor-inducible gene that had been shown to inhibit VSMC expression of the cyclin-dependent kinase inhibitor p21cip1, a key cell cycle factor that inhibits G1 to S progression and VSMC proliferation.22, 40 Id3 inhibits p21cip1 expression via dimerization with basic-HLH factors that activate p21cip1 transcription.41 Results on the present study are the first to identify ITF2b as a factor that activates the p21cip1 promoter in VSMC and determine that this activation is enhanced in the absence of Id3. Consistent with these findings, in 12/15LO-tg VSMC, ITF-2b binding to the p21cip1 promoter is significantly enhanced in the absence of Id3, suggesting one potential molecular mechanism whereby Id3 may mediated the growth promoting effects of 12/15LO.
Increased Id3 protein expression in VSMC with increased expression of 12/15LO and attenuated 12/15LO-induced VSMC proliferation in VSMC from mice null for Id310, provide evidence that Id3 is a downstream regulator of 12/15LO-induced VSMC proliferation in culture. In vivo, Id3 expression is induced during vascular lesion formation in response to injury 26, 27, 42. Here, we provide evidence that elevated 12/15LO expression at baseline resulted in increased post-injury Id3 expression. Moreover, loss of Id3 attenuated the increased neointimal Ki-67-positive staining in the injured 12/15LO-tg animals, providing evidence that Id3 is essential for 12/15LO-induced vascular wall proliferation.
In addition to VSMC proliferation, VSMC hypertrophy, migration or matrix production may contribute as mechanisms by which increased 12/15LO expression promotes NIF. 12/15LO promotes VSMC migration and 12/15LO products of arachidonic acid have hypertrophic effects and increase the expression of the fibronectin gene in VSMC in culture.5, 7, 8 Here, results extend these in vitro data, demonstrating a significant increase in fibronectin deposition in the neointima in response to injury in the 12/15LO-tg mice compared with the C57BL/6 controls.
Interestingly, while loss of Id3 limits neointimal proliferation in response to injury in the 12/15LO-tg mouse on a C57BL/6 background, loss of Id3 in ApoE−/− mice resulted in an increase in atherosclerosis43, underscoring the important differences in atherosclerosis and restenosis models. The pathophysiology of the response to percutaneous vascular interventions (restenosis) in humans and to endothelial denudation in animal models have distinct features from the pathophysiology of atherogenesis44. Factors that may play a key role in regulating the response to mechanical injury in the vessel wall, may have no or an opposite effect on atherogenesis45. Previously published studies have clearly demonstrated that the genetic determinants for injury-induced neointimal formation and diet-induced atherosclerosis in inbred mice are quite distinct38.
In summary, our in vivo study provides evidence that baseline elevated levels of 12/15LO as seen in metabolic syndrome and diabetes result in increased VSMC proliferation, fibronectin deposition, and NIF in response to injury. Moreover, the 12/15LO-induced proliferation is Id3-dependent, suggesting that 12/15LO and/or Id3 may be important targets for limiting restenosis.
Supplementary Material
Sources of Funding
This work was supported by National Institutes of Health (NIH) grants P01 HL55798 and RO1 HL-62522 (C.A.M.), a NIH Training Grant 5-T32 HL007355-29 (H.D. and R.M.), an American Heart Association Predoctoral Fellowship (A.C.D.), and an American Heart Association Postdoctoral Fellowship (H.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
Reference List
- 1.Elezi S, Kastrati A, Pache J, Wehinger A, Hadamitzky M, Dirschinger J, Neumann FJ, Schomig A. Diabetes mellitus and the clinical and angiographic outcome after coronary stent placement. J Am Coll Cardiol. 1998;32:1866–1873. [DOI] [PubMed] [Google Scholar]
- 2.Kastrati A, Schomig A, Elezi S, Schuhlen H, Dirschinger J, Hadamitzky M, Wehinger A, Hausleiter J, Walter H, Neumann FJ. Predictive factors of restenosis after coronary stent placement. J Am Coll Cardiol. 1997;30:1428–1436. [DOI] [PubMed] [Google Scholar]
- 3.Ross R The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. [DOI] [PubMed] [Google Scholar]
- 4.Thyberg J, Blomgren K, Hedin U, Dryjski M. Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: an electron-microscopic and stereological study. Cell Tissue Res. 1995;281:421–433. [DOI] [PubMed] [Google Scholar]
- 5.Natarajan R, Gonzales N, Lanting L, Nadler J. Role of the lipoxygenase pathway in angiotensin II-induced vascular smooth muscle cell hypertrophy. Hypertension. 1994;23:I142–147. [DOI] [PubMed] [Google Scholar]
- 6.Reddy MA, Thimmalapura PR, Lanting L, Nadler JL, Fatima S, Natarajan R. The oxidized lipid and lipoxygenase product 12(S)-hydroxyeicosatetraenoic acid induces hypertrophy and fibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMP response element-binding protein activation. Mediation of angiotensin II effects. J Biol Chem. 2002;277:9920–9928. [DOI] [PubMed] [Google Scholar]
- 7.Natarajan R, Bai W, Rangarajan V, Gonzales N, Gu JL, Lanting L, Nadler JL. Platelet-derived growth factor BB mediated regulation of 12-lipoxygenase in porcine aortic smooth muscle cells. J Cell Physiol. 1996;169:391–400. [DOI] [PubMed] [Google Scholar]
- 8.Natarajan R, Rosdahl J, Gonzales N, Bai W. Regulation of 12-lipoxygenase by cytokines in vascular smooth muscle cells. Hypertension. 1997;30:873–879. [DOI] [PubMed] [Google Scholar]
- 9.Reddy MA, Kim YS, Lanting L, Natarajan R. Reduced growth factor responses in vascular smooth muscle cells derived from 12/15-lipoxygenase-deficient mice. Hypertension. 2003;41:1294–1300. [DOI] [PubMed] [Google Scholar]
- 10.Taylor AM, Hanchett R, Natarajan R, Hedrick CC, Forrest S, Nadler JL, McNamara CA. The effects of leukocyte-type 12/15-lipoxygenase on Id3-mediated vascular smooth muscle cell growth. Arterioscler Thromb Vasc Biol. 2005;25:2069–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natarajan R, Gerrity RG, Gu JL, Lanting L, Thomas L, Nadler JL. Role of 12-lipoxygenase and oxidant stress in hyperglycaemia-induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia. 2002;45:125–133. [DOI] [PubMed] [Google Scholar]
- 12.Natarajan R, Pei H, Gu JL, Sarma JM, Nadler J. Evidence for 12-lipoxygenase induction in the vessel wall following balloon injury. Cardiovasc Res. 1999;41:489–499. [DOI] [PubMed] [Google Scholar]
- 13.Gu JL, Pei H, Thomas L, Nadler JL, Rossi JJ, Lanting L, Natarajan R. Ribozyme-mediated inhibition of rat leukocyte-type 12-lipoxygenase prevents intimal hyperplasia in balloon-injured rat carotid arteries. Circulation. 2001;103:1446–1452. [DOI] [PubMed] [Google Scholar]
- 14.Antonipillai I, Nadler J, Vu EJ, Bughi S, Natarajan R, Horton R. A 12-lipoxygenase product, 12-hydroxyeicosatetraenoic acid, is increased in diabetics with incipient and early renal disease. J Clin Endocrinol Metab. 1996;81:1940–1945. [DOI] [PubMed] [Google Scholar]
- 15.Natarajan R, Gu JL, Rossi J, Gonzales N, Lanting L, Xu L, Nadler J. Elevated glucose and angiotensin II increase 12-lipoxygenase activity and expression in porcine aortic smooth muscle cells. Proc Natl Acad Sci U S A. 1993;90:4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatley ME, Srinivasan S, Reilly KB, Bolick DT, Hedrick CC. Increased production of 12/15 lipoxygenase eicosanoids accelerates monocyte/endothelial interactions in diabetic db/db mice. J Biol Chem. 2003;278:25369–25375. [DOI] [PubMed] [Google Scholar]
- 17.Pei H, Gu J, Thimmalapura PR, Mison A, Nadler JL. Activation of the 12-lipoxygenase and signal transducer and activator of transcription pathway during neointima formation in a model of the metabolic syndrome. Am J Physiol Endocrinol Metab. 2006;290:E92–E102. [DOI] [PubMed] [Google Scholar]
- 18.Chamberlain J, Gunn J, Francis S, Holt C, Crossman D. Temporal and spatial distribution of interleukin-1 beta in balloon injured porcine coronary arteries. Cardiovasc Res. 1999;44:156–165. [DOI] [PubMed] [Google Scholar]
- 19.Kim JA, Gu JL, Natarajan R, Berliner JA, Nadler JL. A leukocyte type of 12-lipoxygenase is expressed in human vascular and mononuclear cells. Evidence for upregulation by angiotensin II. Arterioscler Thromb Vasc Biol. 1995;15:942–948. [DOI] [PubMed] [Google Scholar]
- 20.Scheidegger KJ, Butler S, Witztum JL. Angiotensin II increases macrophage-mediated modification of low density lipoprotein via a lipoxygenase-dependent pathway. J Biol Chem. 1997;272:21609–21615. [DOI] [PubMed] [Google Scholar]
- 21.Szabo A, Laki J, Madsen HO, Dosa E, Prohaszka Z, Rugonfalvi-Kiss S, Kokai M, Acsadi G, Karadi I, Entz L, Selmeci L, Romics L, Fust G, Garred P, Cervenak L. Early rise in serum VEGF and PDGF levels predisposes patients with a normal MBL2 genotype to restenosis after eversion endarterectomy. Stroke. 2007;38:2247–2253. [DOI] [PubMed] [Google Scholar]
- 22.Forrest ST, Taylor AM, Sarembock IJ, Perlegas D, McNamara CA. Phosphorylation regulates Id3 function in vascular smooth muscle cells. Circ Res. 2004;95:557–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. [DOI] [PubMed] [Google Scholar]
- 24.Christy BA, Sanders LK, Lau LF, Copeland NG, Jenkins NA, Nathans D. An Id-related helix-loop-helix protein encoded by a growth factor-inducible gene. Proc Natl Acad Sci U S A. 1991;88:1815–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deed RW, Hara E, Atherton GT, Peters G, Norton JD. Regulation of Id3 cell cycle function by Cdk-2-dependent phosphorylation. Mol Cell Biol. 1997;17:6815–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumura ME, Li F, Berthoux L, Wei B, Lobe DR, Jeon C, Hammarskjold ML, McNamara CA. Vascular injury induces posttranscriptional regulation of the Id3 gene: cloning of a novel Id3 isoform expressed during vascular lesion formation in rat and human atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:752–758. [DOI] [PubMed] [Google Scholar]
- 27.Nickenig G, Baudler S, Muller C, Werner C, Werner N, Welzel H, Strehlow K, Bohm M. Redox-sensitive vascular smooth muscle cell proliferation is mediated by GKLF and Id3 in vitro and in vivo. Faseb J. 2002;16:1077–1086. [DOI] [PubMed] [Google Scholar]
- 28.Doran AC, Meller N, Cutchins A, Deliri H, Slayton RP, Oldham SN, Kim JB, Keller SR, McNamara CA. The helix-loop-helix factors Id3 and E47 are novel regulators of adiponectin. Circ Res. 2008;103:624–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Movat HZ. Demonstration of all connective tissue elements in a single section; pentachrome stains. AMA Arch Pathol. 1955;60:289–295. [PubMed] [Google Scholar]
- 30.Guan JL, Hynes RO. Lymphoid cells recognize an alternatively spliced segment of fibronectin via the integrin receptor alpha 4 beta 1. Cell. 1990;60:53–61. [DOI] [PubMed] [Google Scholar]
- 31.Patricia MK, Kim JA, Harper CM, Shih PT, Berliner JA, Natarajan R, Nadler JL, Hedrick CC. Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19:2615–2622. [DOI] [PubMed] [Google Scholar]
- 32.Reilly KB, Srinivasan S, Hatley ME, Patricia MK, Lannigan J, Bolick DT, Vandenhoff G, Pei H, Natarajan R, Nadler JL, Hedrick CC. 12/15-Lipoxygenase Activity Mediates Inflammatory Monocyte/Endothelial Interactions and Atherosclerosis in Vivo. Journal of Biological Chemistry. 2004;279:9440–9450. [DOI] [PubMed] [Google Scholar]
- 33.Natarajan R, Nadler JL. Lipid Inflammatory Mediators in Diabetic Vascular Disease. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. [DOI] [PubMed] [Google Scholar]
- 34.Kim JA, Gu J-L, Natarajan R, Berliner JA, Nadler JL. A Leukocyte Type of 12-Lipoxygenase Is Expressed in Human Vascular and Mononuclear Cells : Evidence for Upregulation by Angiotensin II. Arterioscler Thromb Vasc Biol. 1995;15:942–948. [DOI] [PubMed] [Google Scholar]
- 35.Nagelin MH, Srinivasan S, Lee J, Nadler JL, Hedrick CC. 12/15-Lipoxygenase Activity Increases the Degradation of Macrophage ATP-Binding Cassette Transporter G1. Arterioscler Thromb Vasc Biol. 2008;28:1811–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E–deficient mice. The Journal of Clinical Investigation. 1999;103:1597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Pratico D, Witztum JL, Nadler JL, Funk CD, Ley K. Critical Role of Macrophage 12/15-Lipoxygenase for Atherosclerosis in Apolipoprotein E-Deficient Mice. Circulation. 2004;110:2024–2031. [DOI] [PubMed] [Google Scholar]
- 38.Kuhel DG, Zhu B, Witte DP, Hui DY. Distinction in Genetic Determinants for Injury-Induced Neointimal Hyperplasia and Diet-Induced Atherosclerosis in Inbred Mice. Arterioscler Thromb Vasc Biol. 2002;22:955–960. [DOI] [PubMed] [Google Scholar]
- 39.Hansson GrK. Inflammation, Atherosclerosis, and Coronary Artery Disease. New England Journal of Medicine. 2005;352:1685–1695. [DOI] [PubMed] [Google Scholar]
- 40.Yang ZY, Simari RD, Perkins ND, San H, Gordon D, Nabel GJ, Nabel EG. Role of the p21 cyclin-dependent kinase inhibitor in limiting intimal cell proliferation in response to arterial injury. Proc Natl Acad Sci U S A. 1996;93:7905–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prabhu S, Ignatova A, Park ST, Sun XH. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol Cell Biol. 1997;17:5888–5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forrest ST, Barringhaus KG, Perlegas D, Hammarskjold M-L, McNamara CA. Intron Retention Generates a Novel Id3 Isoform That Inhibits Vascular Lesion Formation. Journal of Biological Chemistry. 2004;279:32897–32903. [DOI] [PubMed] [Google Scholar]
- 43.Doran AC, Lehtinen AB, Meller N, Lipinski MJ, Slayton RP, Oldham SN, Skaflen MD, Yeboah J, Rich SS, Bowden DW, McNamara CA. Id3 Is a Novel Atheroprotective Factor Containing a Functionally Significant Single-Nucleotide Polymorphism Associated With Intima-Media Thickness in Humans. Circ Res;106:1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasterkamp G, de Kleijn DPV, Borst C. Arterial remodeling in atherosclerosis, restenosis and after alteration of blood flow: potential mechanisms and clinical implications. Cardiovascular Research. 2000;45:843–852. [DOI] [PubMed] [Google Scholar]
- 45.Sanz-González SM, BarquÃ-n L, GarcÃ-a-Cao I, Roque M, González JM, Fuster JJ, Castells MT, Flores JM, Serrano M, Andrés V. Increased p53 gene dosage reduces neointimal thickening induced by mechanical injury but has no effect on native atherosclerosis. Cardiovascular Research. 2007;75:803–812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.