

Abstract
Coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), resulting in a contagious respiratory tract infection that has become a global burden since the end of 2019. Notably, fewer patients infected with SARS-CoV-2 progress from acute disease onset to death compared with the progression rate associated with two other coronaviruses, SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV). Several research organizations and pharmaceutical industries have attempted to develop successful vaccine candidates for the prevention of COVID-19. However, increasing evidence indicates that the SARS-CoV-2 genome undergoes frequent mutation; thus, an adequate analysis of the viral strain remains necessary to construct effective vaccines. The current study attempted to design a multi-epitope vaccine by utilizing an approach based on the SARS-CoV-2 structural proteins. We predicted the antigenic T- and B-lymphocyte responses to four structural proteins after screening all structural proteins according to specific characteristics. The predicted epitopes were combined using suitable adjuvants and linkers, and a secondary structure profile indicated that the vaccine shared similar properties with the native protein. Importantly, the molecular docking analysis and molecular dynamics simulations revealed that the constructed vaccine possessed a high affinity for toll-like receptor 4 (TLR4). In addition, multiple descriptors were obtained from the simulation trajectories, including the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), solvent-accessible surface area (SASA), and radius of gyration (Rg), demonstrating the rigid nature and inflexibility of the vaccine and receptor molecules. In addition, codon optimization, based on Escherichia coli K12, was used to determine the GC content and the codon adaptation index (CAI) value, which further followed for the incorporation into the cloning vector pET28+(a). Collectively, these findings suggested that the constructed vaccine could be used to modulate the immune reaction against SARS-CoV-2.
COVID-19 is caused by SARS-CoV-2, resulting in a contagious respiratory tract infection. For designing a multi-epitope vaccine, we utilized the four structural proteins from the SARS-CoV-2 by using bioinformatics and immunoinformatics analysis.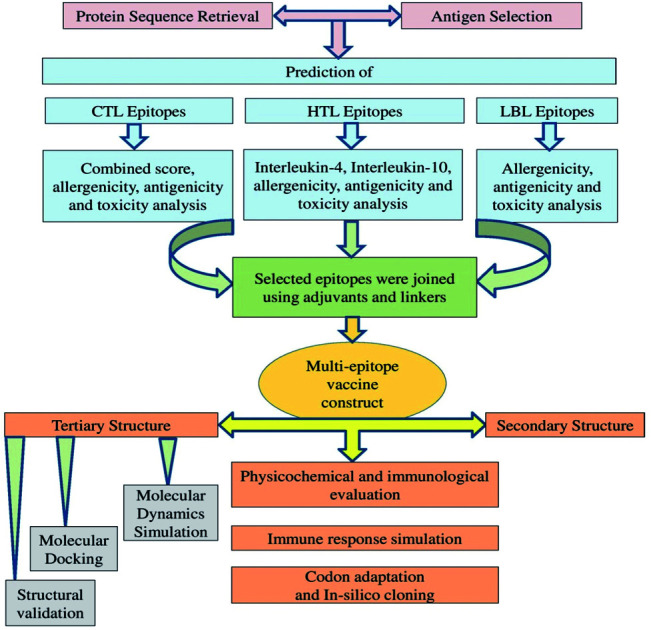
1. Introduction
A cluster of febrile respiratory cases that greatly resembled viral pneumonia with unknown etiology was reported by the Chinese authorities in Wuhan City of Hubei Province in late December 2019. The investigation of isolated bronchoalveolar lavage fluid samples and the metagenomic RNA sequencing of the causative agent associated with this illness resulted in the identification of a new strain of human beta-coronavirus, which was designated severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The clinical condition caused by SARS-CoV-2 is referred to as coronavirus disease 2019 (COVID-19).1 SARS-CoV-2 belongs to an emergent group of 2B coronavirus within the Coronaviridae family and represents the third such zoonotic coronavirus to be identified during the present century.2,3 In China, the first death due to COVID-19 infection was reported on January 11, 2020. The first case of infection reported outside of China was recorded in Thailand on January 13, 2020.4 Since then, this highly transmissible virus has spread rapidly across the globe during a short period of time, forcing the World Health Organization (WHO) to declare this outbreak to be a pandemic. The mortality rate continues to increase worldwide, and, as of January 19, 2021, over 93 million infected cases and over 2 million deaths have been reported globally since the start of the pandemic.5
Coronaviruses (CoVs) are enveloped, positive-sense RNA viruses that are widely distributed among birds, humans, and other mammals, including bats. Human-animal interface activities enhance the likelihood of cross-species infections and spillover events, and zoonotic CoV infections have been associated with respiratory, enteric, hepatic, and neurologic disorders.6 Before the emergence of SARS-CoV-2, six strains of pathogenic human coronaviruses had been identified. Among these genetically diverse species, four species (229E, OC43, NL63, and HKU1) typically cause the common cold and mild fever symptoms in immunocompromised patients. The other two coronaviruses, SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV), are zoonotic pathogenic species that are known to cause fatal respiratory illness and death.7,8 The present SARS-CoV-2 virus demonstrates genetic similarity with previously identified zoonotic CoVs, sharing 79% and 50% genome sequence identity with SARS-CoV and MERS-CoV, respectively.9 However, SARS-CoV-2 infections have been associated with significantly higher mutation rates and mortality rates lower than those associated with SARS-CoV and MERS-CoV infections.10
The human-to-human transmission of COVID-19 occurs via direct close contact with infected individuals (i.e., the spraying of droplets through coughs and sneezes) or indirect surface contact (fomite transmission). COVID-19 infections may occur without presenting any clinical symptoms, or the patient may exhibit mild, moderate, severe, or critical symptoms. These symptoms are typically a combination of the acute upper respiratory tract or digestive symptoms, including fever, fatigue, myalgia, cough, sore throat, sneezing, pneumonia with hypoxemia, nausea, vomiting, abdominal pain, and diarrhea. Patients with mild conditions, excluding febrile symptoms, may recover within 1 to 4 weeks without receiving major treatment. This group is considered to be responsible for the community transmission of the infection. Additionally, older individuals and those with pre-existing medical conditions (diabetes, asthma, and heart disease) have the worst prognosis. Critical cases have been associated with the occurrence of acute respiratory distress syndrome (ARDS), neurological complications, myocardial injury, heart attack, coagulation dysfunction, and acute kidney injury, which can lead to death.10–12
The prediction of the host immune response against SARS-CoV-2 is based on the accumulated clinical and experimental knowledge of the immune response to previous coronaviruses. The genomic RNA of SARS-CoV-2 encodes several non-structural polyproteins (nsp) and four vital structural proteins, including spike (S), nucleocapsid (N), membrane (M), and envelope (E) proteins. The trimeric S glycoprotein is cleaved into two subunits (S1 and S2) during viral infection, and this cleavage event plays a crucial role in virus pathogenesis. Organ tropism occurs because the S protein mediates the entry of the virus through receptor recognition and membrane fusion. The S protein binds to host angiotensin-converting enzyme 2 (ACE2) receptors through the receptor-binding domain (RBD) in type 2 lung alveolar cells, which increases virus transmissibility. Viral particle entry into the host cell depends on the S1 subunit, whereas the integration of the viral and host cell membranes depends on the S2 subunit, making the S protein highly antigenic. These functions of the S protein make it one of the most promising antigen formulations for COVID-19 vaccine development.13–16 The E protein is the smallest of the structural proteins that are essential for the life cycle of the virus, involved in assembly, budding, envelope formation, and interactions with other structural and host cell proteins.17,18 The N protein is a highly immunogenic RNA-binding protein containing two highly conserved domains that bind to viral genomic RNA and regulate viral RNA transcription, replication, and folding. This makes the N protein a potential target for vaccine development and diagnostic methods.19–21 The M protein is involved in determining the virion shape, facilitating membrane curvature, and binding to the N protein.12
Increased CD4+ and CD8+ T cell counts in the peripheral blood and the high concentrations of cytotoxic granules have been reported in infected patients. The disease severity can increase due to T cell overactivation, which can cause injury to the immune systems of COVID-19 patients. By contrast, less effective T-cell responses may allow the progression of viral pathology, resulting in an increased fatality rate. CD4+ and CD8+ T cell responses provide long-lasting protection against this deadly virus. Additionally, the antibody-mediated immune response, together with cellular immunity, plays a vital role in the protection against viral infections.22 Vaccine-mediated protection is provided by virus-specific T cells, known as effector T cells, and the production of antiviral cytokines also increases.23 The viral epitopes presented by the major histocompatibility complex (MHC) class I and MHC class II proteins are recognized by CD4+ and CD8+ T cells, respectively. The heterogeneity in T cells responses to this novel coronavirus may be partly correlated with the capacity of the MHC proteins to recognize the viral antigens.24,25
Vaccine administration has been established as the most useful way to control and eliminate diseases associated with pathogenic organisms by triggering a specific immune response against a foreign particle within the host body.26,27 The modulation of the immune system prepares the body to fight against contagious viruses and can save millions of lives. An effective vaccine must provide a robust and diverse immune response that activates both cell-mediated immune responses and B-cell mediated humoral immunity to elicit immunogenic responses.28 B-cell receptors can reactivate memory B cells, which protect against later re-infection.29
Traditional vaccine development based on biochemical trials can be costly, allergenic, time-consuming, and requires the in vitro culture of pathogenic viruses to ensure safety.17 Vaccine candidates that rely on high-molecular-weight antigenic proteins can be difficult to develop because high-molecular-weight proteins are challenging to express and can interfere with normal complement system function. However, a multi-epitope vaccine may lack these limitations and also possess significant advantages, including increased safety and enhanced immunogenicity,30 and several previous studies have described the design of multi-epitope vaccines for a variety of pathogens.31–33 Recently, various immunoinformatics approaches have been described to predict and assess the antigenicity, allergenicity, toxicity, and immunogenicity of various viral epitopes. The application of computational biology tools can greatly reduce the number of experiments required through the appropriate application of in silico predictions and additional methodologies, making computational methods both time- and cost-effective.12,34 Most of the recent vaccine candidates that have been developed using an immunoinformatics approach have been based on SARS-CoV-2 S glycoprotein variants. However, the immune responses that have been generated through the use of a single SARS-CoV-2 protein have thus far been insufficient to warrant use for effective prophylactic tool development. Multi-epitope vaccine candidates have previously been designed, and their efficacies have been reported against several viruses, including previous coronaviruses, such as SARS-CoV and MERS-CoV. Multi-epitope vaccines present reduced biohazard risks compared with other types of immunizations.35 In our two previous studies, we utilized the SARS-CoV-2 N protein and S protein to predict several epitopes.36,37 In our present study, we utilized an immunoinformatics approach to predict the potential of T-cell and B-cell epitopes within the two other SARS-CoV-2 structural proteins, the E and M proteins. Combined with the findings from previous studies, we identified potential MHC class I, class II and B-cell epitopes that could be combined through the addition of sufficient adjuvants and linkers into a multi-epitope antigen Fig. 1. We expect our current findings to facilitate COVID-19 vaccine development and the performance of experimental laboratory work, which remain necessary to validate our result further.
Fig. 1. Schematic representation of the overall workflow applied for the multi-epitope-based vaccine design using epitopes identified within the SARS-CoV-2 structural proteins.

2. Materials and methods
2.1. Protein sequence retrieval and sequence analysis
The SARS-CoV-2 structural (S, E, N, and M) protein sequences were used as targets for epitope screening. We previously published two articles in which we incorporated cytotoxic T-lymphocytes (CTL) and linear B-lymphocyte (LBL) epitopes from the N protein and CTL, helper T-lymphocytes (HTL), and LBL epitopes from the S protein of the SARS-CoV-2.36,37 In the present study, all available sequences for the E protein were retrieved from the UniProt database, those for the M protein were retrieved from the NCBI database, and the amino acid sequences for both proteins were downloaded in FASTA format.38,39 The antigenicity of the selected structural proteins was anticipated using the VaxiJen v.2.0 (http://www.ddg-pharmfac.net/vaxijen/) server,40 and a default threshold value of 0.4 for the virus was used.41 We chose the structural protein sequences with the highest antigenic score for the next step of investigations for the E protein and M protein.
2.2. Prediction and assessment of cytotoxic T-lymphocyte epitopes
CTLs are one of several cell types in the immune system that can directly interact with infectious cells and are capable of killing infectious cells.42 CTLs can enter pathogenic cells and play an essential role in the host defense mechanism. To predict CTL epitopes, the selected SARS-CoV-2 E and M protein sequences were submitted to the NetCTL v1.2 server, which is available at http://www.cbs.dtu.dk/services/NetCTL/.43 The predicted epitopes were further assessed through the following servers using the default parameters: VaxiJen v2.0 40 to predict antigenicity and MHC class I immunogenicity (http://tools.iedb.org/immunogenicity/);44 ToxinPred, (http://crdd.osdd.net/raghava/toxinpred/)45 to predict toxicity; and AllerTop v2.0 (https://ddg-pharmfac.net/AllerTOP/)46 to predict allergenicity.
2.3. Prediction and evaluation of helper T-lymphocyte epitopes
HTLs are essential for inducing adaptive immunity because they recognize foreign antigens and activate B cells and cytotoxic T cells, resulting in the elimination of infectious pathogens.42 We used the MHC class II binding allele prediction tool IEDB to determine the HTL epitopes (http://tools.iedb.org/mhcii/) and selected HTL epitopes using the CONSENSUS method, based on a percentile rank of 5%.47 Previously, we did not perform HTL epitope prediction for the N protein. Therefore, in this study, in addition to the SARS-CoV-2 E and M proteins, we also performed HTL epitope prediction for the N protein. We further evaluated the predicted epitopes based on their antigenicity and cytokine stimulating ability for the induction of interleukin-4 (IL4) and interleukin-10 (IL10). Antigenicity was determined using the VaxiJen v2.0 server, whereas IL4 and IL10 characteristics were predicted using the default parameters in the IL4pred (http://crdd.osdd.net/raghava/il4pred/)48 and IL10pred (http://crdd.osdd.net/raghava/IL-10pred/)49 servers, respectively.
2.4. Prediction and assessment of linear B-lymphocyte epitopes
B-cell epitopes play vital roles in inducing humoral or antibody-mediated immunity. B cells destroy pathogenic organisms by interacting with secreted antibodies and activating the immune system.50 We predicted the LBL epitopes in the E and M proteins using the ABCpred server.51 We further evaluated the predicted LBL epitopes for the SARS-CoV-2 E and M proteins and the previously predicted LBL epitopes from the S and N proteins using the VaxiJen v2.0,40 ToxinPred,45 and AllerTop v2.046 servers.
2.5. Peptide modeling and molecular docking
The molecular docking analysis was performed using the methodologies described in our previous studies.36,37,52 For modeling, the targeted CTL epitopes were submitted to the PEP-FOLD v3.0 server.53 The energy of each structure was determined by the SWISS-PDB VIEWER, and the structure with the lowest energy was chosen for further analysis.54 For molecular docking simulations, HLA-B*15:01 and HLA-B*35:01 were selected as MHC class I alleles. The crystal structures of HLA-B*15:01 and HLA-B*35:01 were downloaded from the Protein Data Bank (PDB) database.55 Molecular docking analysis was performed using AutoDock Vina in PyRx 0.8, defining the HLA-B*15:01 and HLA-B*35:01 molecules as the receptor proteins and the identified epitopes as the ligand molecules.56 By utilizing a sophisticated gradient optimization method, AutoDock Vina effectively provides an optimization algorithm from a single evaluation.57 First, we used the protein preparation wizard UCSF Chimera (Version 1.11.2) to prepare the protein for docking analysis by deleting the attached ligand and adding hydrogens and Gasteiger–Marsili charges. The prepared file was then added to the AutoDock wizard of PyRx 0.8 and converted into the .pdbqt format. The energy form of the ligand was minimized and converted to the .pdbqt format by OpenBabel.58 The parameters used for the docking simulation were set to default parameters. The size of the grid box in AutoDock Vina was maintained at 78.701 Å × 65.296 Å × 90.806 Å, respectively, for the X-, Y-, and Z-axes. AutoDock Vina was implemented via the shell script offered by AutoDock Vina developers. Docking results are reported as negative scores in kcal mol−1, as the binding affinities of ligands are depicted in negative energies.57 In addition, for the validation of the docking approach, we selected the epitopes that were associated with the respective PDB IDs to serve as positive controls and performed molecular docking analysis for these epitopes using the same parameters. The molecular docking analyses were visualized using Discovery Studio (DS) version 4.5, and figures were generated using Adobe Illustrator CC 18.
2.6. Physicochemical and immunological evaluation
The physiochemical features describe the basic properties of a protein. We used the ProtParam server (https://web.expasy.org/protparam/) to anticipate the physicochemical features and understand the fundamental nature of the designed vaccine.59 We further evaluated the immunological properties using the VaxiJen v2.0,40 AllerTop,46 and SOLpro60 servers.
2.7. Secondary structure prediction
We used the improved self-optimized prediction method (SOPMA) server (https://npsa-prabi.ibcp.fr/NPSA/npsa_seccons.html) and PSIPRED v4.0 server (http://bioinf.cs.ucl.ac.uk/psipred/), with default parameters, to identify the two-dimensional (2D) structural protein features of the vaccine construct, such as the formation of alpha-helices, beta-turns, and random coils.61,62 SOPMA has been reported to return greater than 80% prediction accuracy.61 We retrieved and evaluated the 2D structural features to understand the composition of the constructed vaccine.
2.8. Homology modeling, 3D structure refinement, and validation
The three-dimensional (3D) model of the constructed vaccine was generated using the homology modeling tool Raptor-X server.63 The refinement of the model was performed using the Galaxy Refine server.64 The validation of the constructed vaccine was performed using the ProSA-web and Procheck web servers. ProSA-web predicted the Z-score, which indicates the overall quality of the constructed vaccine,65 and a Ramachandran plot was analyzed by the Procheck server to determine the overall quality.66
2.9. Construction of the multi-epitope vaccine candidate
The vaccine construct was designed using the targeted CTL, HTL, and LBL epitopes from the structural proteins of SARS-CoV-2. In addition, a suitable adjuvant was added using appropriate linkers during vaccine construction.67,68 In the current experiment, we used a TLR4 agonist as an adjuvant because the viral glycoproteins adapt it. In addition, the inclusion of this adjuvant is necessary to maximize the production of the target vaccine candidate for optimal translation.69 The 50 S ribosomal protein L7/L12 (NCBI ID: P9WHE3) was considered as an adjuvant to increase the constructed vaccine's immunogenicity. The adjuvant was linked with the front portion of the vaccine using an EAAAK linker, which is bi-functional and has the capability of several lengths of helix-forming peptides to separate two weakly interacting β-domains. In addition, the CTL epitopes were linked together using AAY linkers, which represents a proteasomal cleavage site that increases the stability of proteins.24,70 Further, the HTL epitopes were linked through GPGPG linkers to prevent junctional epitopes and enable immunological processing. Finally, the LBLs were linked by incorporating KK linkers,67,68 also known as bi-lysine linkers, and are primarily associated with the independent immunoactivities of a vaccine.
2.10. Molecular docking
TLR4 (PDB: 4G8A) was extracted from PDB to assess the interaction pattern. The protein structure was optimized and prepared in DS version 4.5 and the PyMOL software package. Initially, the bound ligands from TLR4 were deleted and saved in.pdb format using DS version 4.5. Afterward, the .pdb file was loaded into the PyMOL software package. The AutoDock tool was run in the PyMOL software package, hydrogen and charges were assigned, and water molecules were removed. The docking study was performed in PatchDock server,71 and further refinement was performed in Firedock web-server.72 The docking interaction was visualized in PyMOL73 and DS.74
2.11. Molecular dynamics simulation
To analyze the structural stability and variations in the vaccine and receptor protein compounds, a molecular dynamics simulation was conducted in YASARA version 20.1.1.75 The AMBER14 force field76 was used for this study, and the protein complex was initially cleaned and optimized, followed by hydrogen bond orientation. A cubic simulation cell was created in which the system was neutralized by the addition of 0.9% NaCl at a temperature of 310 K temperature and pH 7.4. The system temperature was maintained using a Berendsen thermostat.77 The long-range electrostatic interactions were calculated by the particle mesh Ewald method.78 The simulation time step was set to 1.25 fs, and the simulation trajectory was saved after every 100 ps. Finally, the simulation study was performed for 150 ns to analyze the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), the radius of gyration (Rg), the solvent-accessible surface area (SASA), and hydrogen bond formation.79–86 In addition, we have used the peptide QYIKWPWYI as a control in this study, based on evidence in the literature and wet lab results.87 Currently, these peptide molecules are being examined in clinical trials (EudraCT 2020-002502-75, EudraCT 2020-002519-23), and we compared their dynamic behavior with that of the vaccine complex.
2.12. Immune response simulation
The normal mode analysis (NMA) of a vaccine complex was assessed to understand the stability and flexibility of the vaccine complex.88 This tool may serve as an alternative solution for more costly molecular dynamics simulations.89 The motif stiffness of the vaccine complex was evaluated using eigenvalues in which the main chain deformity was predicted by measuring the biological target's efficacy. The elastic network model and covariance matrix were also determined for the vaccine complex.90
2.13. Codon adaptation and in silico cloning
Codon adaptation and in silico cloning are two important steps during the process of vaccine design. Amino acids can be encoded by more than one codon in different organisms. In this workflow, codon optimization was conducted to identify specific codons that can be used to encode a specific amino acid more efficiently in a particular organismal system.91 Codon adaptation was implemented in the Jcat server for translation in suitable expression vectors.92 The vaccine was expressed in the Escherichia coli strain K12 host system, and the generated cDNA sequences were analyzed based on the percent CG content and the codon adaptation index (CAI). Rho-independent transcription terminators and prokaryotic ribosome binding sites were avoided. Finally, the optimized vaccine sequence was incorporated into the pET28(+) vector through the addition of XhoI and NdeI restriction sites at the N- and C-terminus, respectively, using the SnapGene tool.93
3. Results
3.1. Protein selection
We downloaded all relevant E protein sequences from the UniProt database and all relevant M protein sequences from the NCBI database in the present study. We analyzed the antigenicity of each sequence by inputting the sequences into the VaxiJen database. We selected the protein sequences with the highest scores, which were UniProt entry no. A0A6M8FIC5 for E protein and NCBI accession no. QIC53216.1 for the M protein, with antigenicity scores of 0.6908 for E protein and 0.5102 for M protein.
3.2. Predicted potential CTL epitopes
In our previously published articles, we determined potential CTLs for the S and N protein. In addition, we screened potential CTL epitopes from the E and M protein in the current experiment, based on those with the highest antigenicity and the results of toxicity and allergenicity analyses. In addition, the NetCTL 1.2 server provides a combinatorial score based on proteasomal cleavage efficiency and MHC class I binding. After the appropriate screening, we predicted one CTL epitope from the E protein and 3 CTLs from the M protein. The CTL epitopes used in the experiment for the multi-epitope vaccine construct are indicated in Table 1.
The selected CTL epitopes for the final vaccine construction.
| Epitope | Protein | C-score | Antigenicity | Toxicity | Allergenicity |
|---|---|---|---|---|---|
| VSLVKPSFY | E | 3.1860 | 0.7476 (probable antigen) | Non toxin | Non-allergen |
| LVGLMWLSY | M | 2.8970 | 1.0633 (probable antigen) | Non toxin | Non-allergen |
| AGDSGFAAY | M | 2.6730 | 0.9095 (probable antigen) | Non toxin | Non-allergen |
| ATSRTLSYY | M | 3.0900 | 0.6108 (probable antigen) | Non toxin | Non-allergen |
| NTASWFTAL | N | 0.9521 | 0.5192 (probable antigen) | Non toxin | Non-allergen |
| WTAGAAAYY | S | 3.1128 | 0.6306 (probable antigen) | Non toxin | Non-allergen |
| GAAAYYVGY | S | 1.2194 | 0.6604 (probable antigen) | Non toxin | Non-allergen |
3.3. Predicted potential HTL epitopes
In our previous studies, we predicted HTL epitopes for the SARS-CoV-2 S protein. However, we did not perform HTL epitope prediction for the N protein. Thus, the present study also included the prediction of HTL epitopes for the E, M, and N proteins. The selection of HTL epitopes was primarily based on the analyses of antigenicity, IL4- and IL10-inducing capability, toxicity, and allergenicity. According to the selection criteria, the HTL epitopes with the best profiles were selected for further analysis (Table 2).
The selected HTL epitopes for the final vaccine construction.
| Epitope | Protein | Antigenicity | IL4 | IL10 | Toxicity | Allergenicity |
|---|---|---|---|---|---|---|
| VKPSFYVYSRVKNLN | E | 1.2319 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| KPSFYVYSRVKNLNS | E | 0.8229 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| PSFYVYSRVKNLNSS | E | 0.7986 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| VSLVKPSFYVYSRVK | E | 0.7974 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| LAAVYRINWITGGIA | M | 1.0581 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| RWYFYYLGTGPEAGL | N | 0.7505 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| AQFAPSASAFFGMSR | N | 0.5266 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
| AALQIPFAMQMAYRF | S | 0.9108 (probable antigen) | Inducer | Inducer | Non toxin | Non-allergen |
3.4. Predicted potential LBL epitopes
In addition to the best LBL epitopes from the previous two studies, we incorporated LBL epitopes from the E and M proteins of SARS-CoV-2, based on the most antigenic, non-toxic profiles. The selected epitopes were also non-allergenic in nature. The LBL epitopes selected for incorporation into the multi-epitope vaccine are shown in Table 3.
The selected LBL epitopes for the final vaccine construction.
| Epitope | Protein | Antigenicity | Toxicity | Allergenicity |
|---|---|---|---|---|
| RTQLPPAYTNS | N | 0.5605 (probable antigen) | Non toxin | Non-allergen |
| QRQKKQQ | N | 0.4606 (probable antigen) | Non toxin | Non-allergen |
| LEGKQGN | S | 1.8367 (probable antigen) | Non toxin | Non-allergen |
| KNHTSPDVDLG | S | 1.4039 (probable antigen) | Non toxin | Non-allergen |
| KFLPFQQ | S | 1.0416 (probable antigen) | Non toxin | Non-allergen |
| DQLTPTWRVY | S | 0.6489 (probable antigen) | Non toxin | Non-allergen |
| NVSLVKPSFYVYSRVK | E | 0.7865 (probable antigen) | Non toxin | Non-allergen |
| YVYSRVKNLNSSRVPD | E | 0.5457 (probable antigen) | Non toxin | Non-allergen |
| LTWICLLQFAYANRNR | M | 1.1530 (probable antigen) | Non toxin | Non-allergen |
| ACFVLAAVYRINWITG | M | 0.9714 (probable antigen) | Non toxin | Non-allergen |
| SFRLFARTRSMWSFNP | M | 0.9510 (probable antigen) | Non toxin | Non-allergen |
| GIALAMACLVGLMWLS | M | 0.8896 (probable antigen) | Non toxin | Non-allergen |
| LWPVTLACFVLAAVYR | M | 0.8961 (probable antigen) | Non toxin | Non-allergen |
3.5. Molecular docking studies of the CTL epitopes and alleles
We used the molecular docking simulation to delineate the interactions between the targeted CTL epitopes and their respective HLA alleles. We selected the HLA-B*15:01 allele for the E protein, which was paired with the CTL epitopes LVKPSFYVY and VSLVKPSFY. Additionally, the HLA-B*35:01 allele was selected for the analysis of the M protein interactions, and two selected CTL epitopes from the M protein were AGDSGFAAY and LVGLMWLSY. We also obtained the epitopes that were bound to each HLA allele in the PDB IDs for the validation of the docking studies. The result from the molecular docking studies revealed that our selected epitopes and the positive controls all interacted with their respective HLA alleles. Although the positive controls bound with more negative energies than the targeted epitopes, the interactions with the targeted epitopes were similar to the positive controls, except for the epitope AGDSGFAAY, which only formed a single hydrogen bond with HLA-B*35:01. Although VSLVKPSFY possessed a lower docking score than the other epitope and the positive control, VSLVKPSFY formed nine hydrogen bonds with HLA-B*15:01, which was more bonds than were formed by either LVKPSFYVY or the positive control. Moreover, all of the selected epitopes and the positive controls interacted with the receptors with hydrophobic interactions (Table 4, Fig. 2 and 3).
Binding affinities and interaction between selected epitopes and HLA alleles.
| Protein | Allele | Epitope | Docking score (kcal mol−1) | Hydrogen bond interaction | Hydrophobic bond interaction | Unfavorable bumps | Attractive charges |
|---|---|---|---|---|---|---|---|
| E | HLA-B*15:01 | LVKPSFYVY | −8.4 | Arg62, Asn80, Arg97, Gln155 | Tyr7 (pi–alkyl), Asn70 (carbon–hydrogen bond), Leu95 (pi–sigma), Trp147 (pi–pi T-shaped), Tyr159 (carbon–hydrogen bond) | — | Lys146 |
| VSLVKPSFY | −7.5 | Arg62, Asn70, Tyr74, Asn80, Tyr84, Lys146, Ala150, Glu152, Gln155 | Ile66 (pi–alkyl), Ser77 (carbon–hydrogen bond), Tyr99 (pi–pi stacked), Tyr159 (pi–pi stacked) | Trp147 | — | ||
| Control | −8.8 | Glu63, Arg97, Trp147, Glu152, Gln155 | Tyr7 (pi–sigma), Met45 (pi–alkyl), Ile66 (pi–alkyl), Lys146 (pi–alkyl), Trp147 (pi–alkyl), Trp167 (pi–sigma) | Arg62, Tyr99, Tyr159 | Glu63 | ||
| M | HLA-B*35:01 | AGDSGFAAY | −7.7 | Tyr7 | Tyr99 (pi–pi stacked), Val152 (pi–alkyl), Leu156 (pi–alkyl), Trp167 (pi–sigma) | — | Arg62, Arg97 |
| LVGLMWLSY | −8.5 | Tyr7, Asn70, Trp147, Gln155, Trp167 | Tyr7 (pi–pi stacked), Ile66 (pi–alkyl), Tyr99 (pi–pi stacked), Lys146 (pi–alkyl) | Lys146, Trp147 | Trp167 | ||
| Control | −9.3 | Asn70, Asn80, Tyr84, Thr143, Lys146, Trp147, Tyr171 | Tyr59 (pi–alkyl), Asn63 (carbon–hydrogen bond), Ile95 (pi–alkyl), Trp147 (pi–pi T-shaped), Tyr159 (pi–alkyl), Trp167 (pi–alkyl) | — | Tyr7 |
Fig. 2. Three-dimensional representations of the molecular docking analysis showing the predicted epitopes of the SARS-CoV-2 E protein, (A) LVKPSFYVY and (B) VSLVKPSFY, and (C) the positive control bound to the groove of the HLA-B*15:01, in which the hydrogen bonds are displayed as green ball and stick images, attractive charges are displayed as gold ball and stick images, hydrophobic (pi–pi/pi–alkyl stacking) bonds are displayed as pink ball and stick images, and carbon–hydrogen bonds are displayed as white ball and stick images.

Fig. 3. Three-dimensional representation of the molecular docking analysis showing the predicted epitopes of the SARS-CoV-2 M protein, (A) LVGLMWLSY and (B) AGDSGFAAY, and (C) the positive control bound to the groove of the HLA-B*35:01, in which hydrogen bonds are displayed as green ball and stick images, attractive charges are displayed as gold ball and stick images, hydrophobic (pi–pi/pi–alkyl stacking) bonds are displayed as pink ball and stick images, carbon–hydrogen bonds are displayed as white ball and stick images.

3.6. Formulation of a vaccine construct
The formulation of the vaccine construct was performed by compiling the CTL, HTL, and LBL epitopes together, separated by the described linkers. In the current experiment, 3 CTLs were selected from the M protein, two CTLs were selected from the S protein, and a single CTL was selected from each of the E and N proteins. Among HTL epitopes, four were selected from the E protein, two were selected from the N protein, and a single epitope was selected from each of the S and M proteins. Moreover, 12 LBL epitopes were chosen from among the 4 SARS-CoV-2 structural proteins for the multi-epitope vaccine construct. The constructed vaccine consisted of 575 amino acid residues. The sequence of the final vaccine construct was as follows: MAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVAVAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVSGLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTVKEAAAKNTASWFTALAAYVSLVKPSFYAAYWTAGAAAYYAAYGAAAYYVGYAAYLVGLMWLSYAAYAGDSGFAAYAAYATSRTLSYYAAYGPGPGRWYFYYLGTGPEAGLGPGPGAQFAPSASAFFGMSRGPGPGVKPSFYVYSRVKNLNGPGPGKPSFYVYSRVKNLNSGPGPGPSFYVYSRVKNLNSSGPGPGVSLVKPSFYVYSRVKGPGPGAALQIPFAMQMAYRFGPGPGLAAVYRINWITGGIAGPGPGKKRTQLPPAYTNSKKQRQKKQQKKNVSLVKPSFYVYSRVKKKYVYSRVKNLNSSRVPDKKLEGKQGNKKKNHTSPDVDLGKKKFLPFQQKKDQLTPTWRVYKKLTWICLLQFAYANRNRKKACFVLAAVYRINWITGKKSFRLFARTRSMWSFNPKKGIAIAMACLVGLMWLSKKLWPVTLACFVLAAVYR (Fig. 4).
Fig. 4. Graphical map of the formulated multi-epitope vaccine construct. The vaccine constructs included (left to right) an adjuvant, CTL, HTL, and LBL epitopes, which are shown in the dark blue, red, green, and blue rectangular boxes, respectively. The adjuvant and the first CTL epitope were linked using an EAAAK linker (blue), the CTL epitopes were joined using AYY linkers (off-white), the HTL epitopes were linked using GPGPG linkers (dark yellow), and the LBL epitopes were joined using KK linkers (black).

3.7. Physicochemical properties and immunological evaluation
The physicochemical properties of the vaccine construct were evaluated, as displayed in Table 5. The molecular weight of the vaccine construct was 62 355.33 Da. The other determined properties were as follows: the theoretical isoelectric point (pI) was calculated at 9.91, indicating a basic nature; the chemical formula was determined to be C2883H4445N747O773S13; the instability index value was 31.26; the aliphatic index value was 76.47; and the grand average of hydropathicity (GRAVY) value was −0.139, indicating the hydrophilic nature of the construct. We further assessed the antigenicity and allergenicity of the vaccine construct. The construct was antigenic, with a score of 0.6153, and non-allergenic. Moreover, the vaccine construct was soluble, exhibiting a score of 0.701681 on a scale of 1 (Table 5).
Antigenic, allergenic and physicochemical characteristics of the construct.
| Characteristics | Finding | Remarks |
|---|---|---|
| Number of amino acids | 575 | Suitable |
| Molecular weight | 62 355.33 Da | High |
| Theoretical pI | 9.91 | Basic |
| Chemical formula | C2883H4445N747O773S13 | — |
| Extinction coefficient (at 280 nm in H2O) | 118 860 M−1 cm−1 | — |
| Estimated half-life (mammalian reticulocytes, in vitro) | 30 hours | — |
| Estimated half-life (yeast-cells, in vivo) | >20 hours | — |
| Estimated half-life (E. coli, in vivo) | >10 hours | — |
| Instability index of vaccine | 31.26 | Stable |
| Aliphatic index of vaccine | 76.47 | Thermostable |
| Grand average of hydropathicity (GRAVY) | −0.139 | Hydrophilic |
| Antigenicity | 0.6153 | Antigenic |
| Allergenicity | No | Non-allergen |
| Solubility | 0.701681 | Soluble |
The evaluation of secondary structural features, including α-helices, β-strands, and random coils, was performed using two servers. The SOPMA server anticipated 40.00% α-helices, 19.30% β-strands, and 40.70% random coils in the secondary structure (Table 6 and ESI Fig. S1†). By contrast, the PSIPRED server predicted 45.74% α-helices, 15.30% β-strands, and 38.96% random coils in the construct (Table 6 and ESI Fig. S1†).
The secondary structural features of the vaccine construct.
| Features | SOPMA server | PSIPRED server | ||
|---|---|---|---|---|
| Amino acid | Percentage | Amino acid | Percentage | |
| Alpha helix | 230 | 40.00% | 263 | 45.74% |
| Beta strand | 111 | 19.30% | 88 | 15.30% |
| Random coil | 234 | 40.70% | 224 | 38.96% |
3.8. Tertiary structure, refinement, and validation
The 3D structure of the constructed vaccine was generated using the Raptor-X online server. A total of 5 models were generated by the Raptor-X server, which was further evaluated by ProSA and Procheck web servers. In this study, model 1 returned the highest Z-score of −7.68, and the percentage of Ramachandran favored regions for model 1 was 90.6%. Although model 3 showed a similar percentage of Ramachandran favored regions as that for model 1, the Z-score for model 3 was −6.60. Thus, we selected model 1 for further analysis in the present study (Fig. 5).
Fig. 5. (A) 3D structure of the vaccine construct, (B) Z-score of the vaccine construct, as predicted by the ProSA server, and (C) Ramachandran plot analysis of the vaccine construct.

3.9. Molecular docking studies
The molecular interaction between the vaccine molecule and the immune cell requires a stable immune response, which depends on the geometry of the protein surface and the electrostatic interactions between the protein and the cell. Patchdock tools were used to rank the top ten interaction models between the receptor and the immune cell. The best complexes were sent for refinement in Firedock. The vaccine and TLR4 complex had better binding interactions in solution eight, in which the global energy was −33.27 kJ mol−1, van der Walls energy (vdW) was −47.06, repulsive vdW was 25.91, atomic contact energy (ACE) was 11.41, hydrogen bond energy was −3.07 (Fig. 6).
Fig. 6. The vaccine-TLR4 complex predicted by molecular docking.

3.10. Molecular dynamics simulation
The RMSD of the c-alpha atoms in the protein complex was evaluated to understand the structural deviations across the simulation trajectory. As shown in Fig. 7, the complex exhibited a sharp increase in the RMSD value at the beginning of the simulation. The protein complex then stabilized until the last phase of the simulation. The vaccine complex showed relatively less RMSD compared with the control, which indicated the stable nature of the vaccine candidate compared with the control. The RMSD value of the complex ranged from 2.5 to 2.7 Å, which indicated structural stability and less flexibility.
Fig. 7. The molecular dynamics simulation of the complexes. (A) Hydrogen bond analysis from the simulation system. (B) Root-mean-square fluctuations of the amino acid residues. (C) Root-mean-square deviation of the complexes. (D) Radius of gyration. (E) Solvent-accessible surface area.

The protein complex's SASA was also evaluated to determine the change in surface area. The vaccine construct possessed a higher SASA trend, which demonstrated an increase in the surface volume. However, this complex did not demonstrate a high level of SASA deviations, which indicated that no significant changes were occurring to the protein's surface area.
The Rg descriptor for a protein system defines the compactness of the complex. The protein complex displayed a steady Rg trend from the very beginning of the simulation. After a 30 ns simulation, the complex had a higher Rg value, which is responsible for the protein's labile nature. This increase in Rg might be due to the folding or unfolding of the protein complex. The hydrogen bonds in a biological system define the stability of the system. Fig. 7 indicates that the vaccine complex's hydrogen bonds did not fluctuate compared with those in the control, which indicated structural integrity.
The RMSF of the vaccine complex and the control were evaluated to determine the amino acid residue flexibility. Fig. 7 demonstrated that most residues exhibited RMSF values below 2.5 Å. This RMSF profile defines a less flexible and more rigid vaccine complex compared with the complex formed by the control.
3.11. Immune simulation
The stability of the vaccine complex was evaluated by deformability, eigenvalue, elastic network model, covariance map, and B factor analyses. Fig. 8 indicates that the hinge region represented a high deformability region; the average RMS was also present for the B factor. The eigenvalue was higher; 4.131509 × 10−6, The elastic network model and correlation matrix are depicted in Fig. 8. These results correlate with the lower chance of deformation among the vaccine complex molecules.
Fig. 8. The normal mode analysis of vaccine protein. (A) Covariance map. (B) Elastic network map. (C) B factor. (D) Deformability. (E) Eigenvalues. (F) Variance.

3.12. In silico cloning
To determine whether the vaccine sequence can be expressed in E. coli host cells, in silico cloning was performed. Codon optimization is essential for protein expression. To generate a cDNA sequence, the Jcat tool was used, which exhibits the 1725-nucleotide sequence used for vaccine expression. The CAI score and GC content of our optimized vaccine sequences were 0.9485 and 50.49%, respectively, denoting an efficient and potentially stable expression by the E. coli K-12 strain (Fig. 9). Generally, a GC content of 30%–70% and a CAI value greater than 0.8 are considered to denote good protein expression conditions in a host. The designed nucleotide sequence was inserted into the pET28(+) plasmid vector by inserting the appropriate restriction sites through the SnapGene tool.
Fig. 9. Restriction digestion and in silico cloning of the gene sequence of the final construct into pET28a(+) expression vector.

4. Discussion
Currently, the whole world is experiencing a pandemic caused by SARS-CoV-2. Initially, some significant events, including a cruise to Japan, attendance at ski resorts in Austria and Italy, a mass gathering of people in South Korea, and travel to a well-known pilgrimage city in Iran, contributed to the rapid and global spread of this deadly virus. Since then, the global dissemination of SARS-CoV-2 has increased, and this pathogenic organism has resulted in many deaths across numerous countries.
Genetically, SARS-CoV-2 is closely related to SARS-CoV, which was highly lethal and caused several deaths in late 2002; however, after intensive health measures, the SARS-CoV virus was faded from the public domain. In addition, another deadly coronavirus, MERS-CoV, showed an even higher fatality rate than SARS-CoV. Although the novel coronavirus (SARS-CoV-2) has reduced mortality compared with the two previous coronaviruses, the transmission rate of SARS-CoV-2 is greatly higher than that of either SARS-CoV or MERS-CoV.94 Additionally, SARS-CoV-2 possesses a longer incubation period than other viruses, such as the influenza virus. Both SARS-CoV and MERS-CoV were found at low levels in the upper respiratory tract (URT), whereas the viral load of SARS-CoV-2 is the opposite. For SARS-CoV-2, a high viral load can be detected in the URT, which declines after 5–6 days; the presence of the virus in the URT requires the isolation and quarantine of patients to prevent disease spread.95,96 Conversely, SARS-CoV loads peaked within 6–11 days after the onset of symptoms. These differences can explain the scenario that has resulted in the pandemic situation.97,98 Recently, a new variant of SARS-CoV-2 has been reported in England, which is estimated to be approximately 70% more transmissible than the previous strain. Importantly, evidence has suggested that the new strain might be associated with a higher mortality rate compared with other variants.99 The newly identified strain contains eight mutations in the S protein of SARS-CoV-2, one of which increases the chance of interaction with ACE2.100 Additionally, in South Africa, another SARS-CoV-2 variant has emerged, which is associated with an increased rate of infection and a higher mortality rate. Further, the South Africa strain is less effectively neutralized by convalescent patients' plasma. Another novel variant that has emerged in the USA features a mutated lysine residue at position 452, which affects the binding of the S protein to certain monoclonal antibodies.100
The development of vaccines for coronaviruses has generally been considered a low priority because most coronaviruses cause mild diseases. Although several vaccine candidates were tested pre-clinically to address SARS-CoV infections, vaccine development was halted after the virus was exterminated from the human population, and no cases of SARS-CoV in humans have been reported since 2004.101,102 In addition, vaccines against MERS-CoV are currently under development. Previous studies reported the lucidity of the antigenic target for both SARS-CoV and MERS-CoV.103,104 Both SARS-CoV and MERS-CoV, in addition to other coronaviruses, encode an S protein, which is responsible not only for binding with the host receptor but also for the fusion of the virus with the cell membrane.105 The S protein of SARS-CoV-2 was identified as being antigenic and was targeted for the early development of vaccines against SARS-CoV-2. However, a study by Zheng et al. reported that SARS-CoV-2 possessed 24.5% amino acid residues that are not conserved in comparison with the sequence of the S protein from SARS-CoV, which might be responsible for the antigenic differences between the two strains.106
The N protein of SARS-CoV-2 was previously documented as being responsible for viral replication and the pathogenesis of SARS-CoV-2.107 Another study predicted that the E protein of SARS-CoV-2 was more antigenic than other structural proteins.108 Furthermore, the N protein interacted with the M protein in the infected cell lipid membrane, forming a vesicular bilayer structure, which further interacts with the host proteins.109,110 These findings have led to the hypothesis that the SARS-CoV-2 structural proteins might be responsible for various immunogenic responses. Thus, in the current study, we sought to predict a peptide-based vaccine candidate against SARS-CoV-2 by utilizing the organism's structural proteins. In our previous two studies, we successfully predicted epitopes from two structural proteins, the S and N proteins.36,37 In this study, we also considered the other two structural proteins, the E and M proteins, to further predict antigenic peptides. The present study focused on designing a multi-epitope vaccine, which has greater immunogenic potential compared with classical and single-epitope-based vaccines. Furthermore, multi-epitope vaccines possess several unique characteristics, including the availability to present multiple HTL, CTL, and B-cell epitopes, allowing for the induction of both cellular and humoral immune responses. Likewise, it consists of multiple HLA epitopes, which can be easily recognized by several T-cell receptors and consists of epitopes from different immunogenic proteins, increasing the target organism's range.
First, we downloaded the SARS-CoV-2 E protein sequences from the UniProt database and the M protein sequences from the NCBI protein database. The sequences were then inputted into the VaxiJen server, and the sequences of both the E and M proteins with the highest antigenicity scores were used for further analysis. Based on the highest combinatorial scores and MHC class I binding scores, four epitopes were chosen from the E protein, and ten epitopes were selected from the M protein, and the antigenicity, allergenicity, and toxicity of these selected epitopes were determined. After screening out, one CTL epitope, VSLVKPSFY, and three additional epitopes, including LVGLMWLSY, AGDSGFAAY, and ATSRTLSYY, were selected for further multi-epitope study. The results from the MHC I interaction showed that two epitopes from the E protein sequence interacted with two MHC I alleles, HLA-B*15:25 and HLA-B*15:01, with greater interaction. In the M protein, two of the selected CTL epitopes interacted with HLA-B*35:01 with greater affinities. Although both M protein epitopes interacted with HLA-A*29:02, we only considered those HLA molecules available on the PDB database. Therefore, for the molecular docking studies, HLA-B*15:25 was selected for docking with the epitopes from the E protein, and HLA-B*35:01 was selected for the epitopes from the M protein. Although the molecular docking simulation results showed that the chosen epitopes were able to significantly interact with the targeted MHC I alleles, the binding affinities were less than those of the positive controls selected for the study. However, the CTL epitopes selected from the E protein formed significantly more hydrogen bonds with HLA-B*15:01 than the control epitope. Only AGDSGFAAY from the M protein showed less interaction toward HLA-B*35:01, forming only a single hydrogen bond with Tyr7, which was fewer bonds than were formed by another CTL epitope from the M protein or the positive control.
We also predicted HTL epitopes from both the E and M proteins of SARS-CoV-2. Importantly, in our previous study of the SARS-CoV-2 N protein, we did not analyze HTL epitopes from the N protein. We, therefore, also performed the prediction of HTL epitopes from the N protein of SARS-CoV-2, in addition to the prediction of HTL epitopes from the E and M proteins. We also analyzed the HTL epitopes obtained from our previous study of the SARS-CoV-2 S protein. We considered the antigenicity, allergenicity, toxicity, and IL-4- and IL-10-inducing capabilities during the selection of HTL epitopes from these four structural SARS-CoV-2 proteins. The selected epitopes were screened for further analysis.
We predicted B-cell epitopes from the SARS-CoV-2 E and M proteins and considered selected B-cell epitopes from the previous two studies. In the current study, we analyzed the antigenicity, allergenicity, and toxicity of the selected B-cell epitopes. The selected epitopes were further considered for the multi-epitope vaccine.
The selection of pertinent antigenic epitopes from the targeted proteins for inclusion in a multi-epitope vaccine is crucial for the design of such vaccines. In the current study, the antigenicity, allergenicity, toxicity, and degree of conservation of the epitopes were determined. Afterward, all targeted CTL, HTL, and B-cell epitopes were attached using the desired linkers, which were integrated to increase the stability, patterns of the expression, and folding capacity of the vaccine candidate.111 The EAAAK linker was used to attach the adjuvant to the CTL epitopes, which is associated with inducing a higher degree of cellular and immunogenic humoral responses.112 The attachment of the EAAAK linker to the adjuvant increases the stability and longevity of the vaccine construct.113 After connecting the CTL, HTL, and B-cell epitopes with their respective linkers, we developed a final vaccine construct that contained 575 amino acid residues. The antigenicity and allergenicity of the final vaccine construct were determined, which showed that the final vaccine construct was antigenic and non-allergenic, indicating that it could serve as a potential vaccine. The molecular weight was determined to be approximately 62 kDa, which was an average molecular weight. The theoretical pI was calculated as 9.91, which demonstrated that the vaccine construct was basic in nature. Solubility is considered to be an important characteristic of a recombinant vaccine construct.114 We predicted the solubility of the constructed vaccine when expressed by a host E. coli strain and found that the vaccine construct was soluble inside the host E. coli.
After predicting the 3D structure of the constructed vaccine, we identified a significant Z-score, and most amino acid residues in the Ramachandran plot were in the favored region. Molecular studies were used to predict the interaction between the vaccine constructs and the viral glycoprotein-binding TLR4. The analysis resulted in a global binding score of −33.27 kJ mol−1, indicated that the vaccine construct interacted well with TLR4 on the cell surface. TLRs belong to a family of conserved pattern recognition receptors (PPRs) that function to recognize the specific pattern of pathogens, including viruses, bacteria, or fungi, and are capable of distinguishing between self and non-self-materials.115 As many as 10 TLRs have been found in the human gene database, primarily characterized as transmembrane proteins with leucine-rich repeats in the extracellular domain. The activation of TLRs by specific ligands eventually results in the production of cytokines and the upregulation of MHC molecules, which ultimately links the innate immune response with adaptive immune cells.116 The cytoplasmic region of TLRs similar to the IL-1 receptor family; however, the extracellular portions of TLRs are structurally different. A critical functional of TLR4 is the recognition of microbial lipopolysaccharide (LPS), which is a potential immune activator.117 In addition, TLR2 recognizes LPS from non-enterobacterial origins, which are structurally different from the LPS that is recognized by TLR4.118
Additionally, we performed a molecular dynamics simulation, which is a powerful tool for assessing a protein's physical structure and the functional analysis of large macromolecules. To perform dynamics studies of the vaccine construct, 150 ns molecular dynamic simulations were performed. The results were analyzed based on the RMSD and RMSF scores, the Rg score, the SASA profile, and the hydrogen bond analysis. The RMSD value primarily denotes different conformations of the targeted molecular system. In the present study, the RMSD value indicated the structural stability of the vaccine construct. Despite a sharp increase at the start of the simulation, the RMSD value stabilized continued to present stability. Additionally, the RMSF value indicated the rigidity of the vaccine construct. The vaccine candidate's robustness and stability were also evaluated using the Rg score, hydrogen bond analysis, and SASA profile. We also evaluated a control in this study, which is currently under clinical investigation. The molecular dynamics simulation performed for the control indicated that the control had less uniformity than our predicted vaccine construct.
When designing a multi-epitope candidate, the successful cloning and expression in a suitable vector is a crucial step. In silico cloning is considered to represent a tremendously useful protocol in the field of biotechnology that can be applied prior to performing in vitro experiments using a vaccine construct. The in silico protocols allow for the reduction of human-introduced errors and are less time-consuming and more cost-effective than other methods.119 Several recent studies have already reported that the in silico cloning approach has significant applications for the fields of microbiology, molecular biology, and biotechnology and can yield a full or partial complementary DNA (cDNA) sequence.120,121 In addition, the secondary structure of RNA, particularly in the untranslated regions of the genome, has been demonstrated to be involved in various processes throughout the life cycle of the pathogen. Understanding the specific information that viruses use for replication will increase the chances of identifying the basic biological features that are necessary for pathogenic proliferation, which are crucial for designing vaccines.122 Therefore, in the current study, in silico cloning was performed to validate the vaccine construct's expression and translation in the expression vector pET-28a(+). Because the vaccine construct consisted of CTL, HTL, and B-cell epitopes, it could play a crucial role in inducing host immune responses, which may pave the way for the activation of several immune cells through complex signaling.
5. Conclusions
The development of multiple immunoinformatics tools has paved the way for designing and developing an epitope-based vaccine in a cost-effective manner within a short period of time. Because viruses can activate both cellular and humoral immunity, we combined a potential set of T-cell and B-cell epitopes from the major structural proteins of SARS-CoV-2 to construct a multi-epitope vaccine in this study. After evaluating the antigenicity, immunogenicity, and allergenicity of these epitopes, different linkers were efficiently applied to join the selected epitopes and present them to T-cell and B-cell receptors. Importantly, our vaccine construct strongly bound to TLR4, suggesting a robust immune response can be activated upon novel coronavirus infection. The vaccine construct demonstrated structural stability, less flexibility, and more rigidity in the molecular dynamics simulation with a lower chance of deformation in the immune simulation study than the positive control. The vaccine construct also revealed high protein expression levels in the E. coli host and was able to be successfully inserted into the pET28(+) plasmid vector, which indicates that our construct may serve as a potential vaccine candidate. Despite showing significant results in the in silico analyses, our preliminary design requires further experimental verification to assess the engineered vaccine's effectiveness.
Availability of data and material
The datasets supporting the conclusions of this study are included within the article (and its additional files).
Author contributions
Conceptualization: Ahmad J. Obaidullah, Mohammed M. Alanazi, Nawaf A. Alsaif, Hussam Albassam, Abdulrahman A. Al-Mehizia. Formal analysis: Ahmad J. Obaidullah, Mohammed M. Alanazi. Funding acquisition: Ahmad J. Obaidullah, Mohammed M. Alanazi. Investigation: Ahmad J. Obiadullah, Mohammed M. Alanazi, Nawaf A. Alsaif, Hussam Albassam, Abdulrahman A. Al-Mehizia, Ali M. Alqahtani, Shafi Mahmud, Saad Ahmed Sami, Talha Bin Emran. Methodology: Ahmad J. Obaidullah, Shafi Mahmud. Project administration: Talha Bin Emran. Resources: Ahmad J. Obaidullah, Mohammed M. Alanazi. Software: Ahmad J. Obaidullah, Mohammed M. Alanazi, Shafi Mahmud. Supervision: Ahmad J. Obaidullah, Mohammed M. Alanazi, Talha Bin Emran. Writing – original draft: Ahmad J. Obaidullah, Mohammed M. Alanazi. Writing – review & editing: Ahmad J. Obaidullah, Mohammed M. Alanazi, Ali M. Alqahtani, Talha Bin Emran.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no. (RG-1441-398).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02885e
References
- Hu B. Guo H. Zhou P. Shi Z.-L. Nat. Rev. Microbiol. 2020:1–14. [Google Scholar]
- Huang C. Wang Y. Li X. Ren L. Zhao J. Hu Y. Zhang L. Fan G. Xu J. Gu X. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski L. E. Menachery V. D. Viruses. 2020;12:135. doi: 10.3390/v12020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Horby P. W. Hayden F. G. Gao G. F. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- W. H. Organization, Weekly epidemiological update, 19 January 2021, https://www.who.int/publications/m/item/weekly-epidemiological-update---19-january-2021
- Zhu N. Zhang D. Wang W. Li X. Yang B. Song J. Zhao X. Huang B. Shi W. Lu R. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. Wong G. Shi W. Liu J. Lai A. C. Zhou J. Liu W. Bi Y. Gao G. F. Trends Microbiol. 2016;24:490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. Li F. Shi Z.-L. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R. Zhao X. Li J. Niu P. Yang B. Wu H. Wang W. Song H. Huang B. Zhu N. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S. Navid A. Farid R. Abbas G. Ahmad F. Zaman N. Parvaiz N. Azam S. S. Eur. J. Pharm. Sci. 2020;151:105387. doi: 10.1016/j.ejps.2020.105387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki K. Fujiogi M. Koutsogiannaki S. Clin. Immunol. 2020:108427. doi: 10.1016/j.clim.2020.108427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R. Chu Z. Yu F. Zha Y. Front. Immunol. 2020;11:1784. doi: 10.3389/fimmu.2020.01784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairkhah N. Aghasadeghi M. R. Namvar A. Bolhassani A. PLoS One. 2020;15:e0240577. doi: 10.1371/journal.pone.0240577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. Infect., Genet. Evol. 2020;79:104211. doi: 10.1016/j.meegid.2020.104211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T. M. Buchmeier M. J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. Xiao G. Chen Y. He Y. Niu J. Escalante C. R. Xiong H. Farmar J. Debnath A. K. Tien P. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmageed M. I. Abdelmoneim A. H. Mustafa M. I. Elfadol N. M. Murshed N. S. Shantier S. W. Makhawi A. M. BioMed Res. Int. 2020:2020. doi: 10.1155/2020/2683286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño-Rodriguez C. Honrubia J. M. Gutiérrez-Álvarez J. DeDiego M. L. Nieto-Torres J. L. Jimenez-Guardeño J. M. Regla-Nava J. A. Fernandez-Delgado R. Verdia-Báguena C. Queralt-Martín M. MBio. 2018;9 doi: 10.1128/mBio.02325-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-k. Lo S.-C. Wang Y.-S. Hou M.-H. Drug Discovery Today. 2016;21:562–572. doi: 10.1016/j.drudis.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S. F. Quadeer A. A. McKay M. R. Viruses. 2020;12:254. doi: 10.3390/v12030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. Liu L. Kou G. Zheng Y. Ding Y. Ni W. Wang Q. Tan L. Wu W. Tang S. J. Clin. Microbiol. 2020:58. doi: 10.1128/JCM.00461-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samad A. Ahammad F. Nain Z. Alam R. Imon R. R. Hasan M. Rahman M. S. J. Biomol. Struct. Dyn. 2020:1–17. doi: 10.1080/07391102.2020.1792347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F. Geginat J. Lanzavecchia A. Annu. Rev. Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- Abdellrazeq G. S. Fry L. M. Elnaggar M. M. Bannantine J. P. Schneider D. A. Chamberlin W. M. Mahmoud A. H. Park K.-T. Hulubei V. Davis W. C. Vaccine. 2020;38:2016–2025. doi: 10.1016/j.vaccine.2019.12.052. [DOI] [PubMed] [Google Scholar]
- Astuti I. Diabetes Metab. Syndr. 2020;14:407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood B. Philos. Trans. R. Soc., B. 2014;369:20130433. doi: 10.1098/rstb.2013.0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky A. A. Life Sciences. 2020;248:117477. doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanna I. J. Slifka M. K. Virology. 2011;411:206–215. doi: 10.1016/j.virol.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanna I. J. Carlson N. E. Slifka M. K. N. Engl. J. Med. 2007;357:1903–1915. doi: 10.1056/NEJMoa066092. [DOI] [PubMed] [Google Scholar]
- Gu Y. Sun X. Li B. Huang J. Zhan B. Zhu X. Front. Microbiol. 2017;8:1475. doi: 10.3389/fmicb.2017.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. Sood D. van der Spek P. J. Sharma H. S. Chandra R. J. Proteome Res. 2020;19:4678–4689. doi: 10.1021/acs.jproteome.0c00367. [DOI] [PubMed] [Google Scholar]
- Kumar N. Sood D. Sharma N. Chandra R. J. Chem. Inf. Model. 2019;60:421–433. doi: 10.1021/acs.jcim.9b01051. [DOI] [PubMed] [Google Scholar]
- Kumar N. Sood D. Chandra R. ACS Pharmacol. Transl. Sci. 2020 doi: 10.1021/acsptsci.0c00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ul Qamar M. T. Shahid F. Aslam S. Ashfaq U. A. Aslam S. Fatima I. Fareed M. M. Zohaib A. Chen L.-L. Infect. Dis. Poverty. 2020;9:1–14. doi: 10.1186/s40249-019-0617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M. S. Hoque M. N. Islam M. R. Akter S. Alam A. R. U. Siddique M. A. Saha O. Rahaman M. M. Sultana M. Crandall K. A. PeerJ. 2020;8:e9572. doi: 10.7717/peerj.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakib A. Sami S. A. Mimi N. J. Chowdhury M. M. Eva T. A. Nainu F. Paul A. Shahriar A. Tareq A. M. Emon N. U. Comput. Biol. Med. 2020;124:103967. doi: 10.1016/j.compbiomed.2020.103967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakib A. Sami S. A. Islam M. Ahmed S. Faiz F. B. Khanam B. H. Marma K. K. S. Rahman M. Uddin M. M. N. Nainu F. Molecules. 2020;25:5088. doi: 10.3390/molecules25215088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R. Bairoch A. Wu C. H. Barker W. C. Boeckmann B. Ferro S. Gasteiger E. Huang H. Lopez R. Magrane M. Martin M. J. Natale D. A. O’Donovan C. Redaschi N. Yeh L. S. L. Nucleic Acids Research. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. Zaretskaya I. Raytselis Y. Merezhuk Y. McGinnis S. Madden T. L. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I. A. Flower D. R. BMC Bioinf. 2007;8:1–7. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N. Zeller M. Kayala M. A. Vigil A. Randall A. Felgner P. L. Baldi P. Bioinformatics. 2010;26:2936–2943. doi: 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. Chen P. Wang J. Feng J. Zhou H. Li X. Zhong W. Hao P. Sci. China: Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. V. Lundegaard C. Lamberth K. Buus S. Lund O. Nielsen M. BMC Bioinf. 2007;8:424. doi: 10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calis J. J. Maybeno M. Greenbaum J. A. Weiskopf D. De Silva A. D. Sette A. Keşmir C. Peters B. PLoS Comput. Biol. 2013;9:e1003266. doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. Kapoor P. Chaudhary K. Gautam A. Kumar R. Raghava G. P. PLoS One. 2013;8:e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I. Flower D. R. Doytchinova I. BMC Bioinf. 2013;14(suppl. 6):S4. doi: 10.1186/1471-2105-14-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. Sidney J. Kim Y. Sette A. Lund O. Nielsen M. Peters B. BMC Bioinf. 2010;11:568. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda S. K. Gupta S. Vir P. Raghava G. P. Clin. Dev. Immunol. 2013;2013:263952. doi: 10.1155/2013/263952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal G. Usmani S. S. Dhanda S. K. Kaur H. Singh S. Sharma M. Raghava G. P. Sci. Rep. 2017;7:42851. doi: 10.1038/srep42851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nain Z. Karim M. M. Sen M. K. Adhikari U. K. Mol. Immunol. 2020;120:146–163. doi: 10.1016/j.molimm.2020.02.009. [DOI] [PubMed] [Google Scholar]
- Saha S. Raghava G. P. Methods Mol. Biol. 2007;409:387–394. doi: 10.1007/978-1-60327-118-9_29. [DOI] [PubMed] [Google Scholar]
- Rakib A. Nain Z. Sami S. A. Mahmud S. Islam A. Ahmed S. Siddiqui A. B. F. Babu S. O. F. Hossain P. Shahriar A. Briefings Bioinf. 2021;22:1476–1498. doi: 10.1093/bib/bbab045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maupetit J. Derreumaux P. Tuffery P. Nucleic Acids Res. 2009;37:W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N. Peitsch M. C. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Berman H. M. Westbrook J. Feng Z. Gilliland G. Bhat T. N. Weissig H. Shindyalov I. N. Bourne P. E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S., PyRx-Python Prescription v.0.8, The Scripps Research Institute, 2008, p. 2010 [Google Scholar]
- Trott O. Olson A. J. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Boyle N. M. Banck M. James C. A. Morley C. Vandermeersch T. Hutchison G. R. J. Cheminf. 2011;3:1–14. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Wilkins M. R., Appel R. D. and Bairoch A., The proteomics protocols handbook, 2005, pp. 571–607 [Google Scholar]
- Magnan C. N. Randall A. Baldi P. Bioinformatics. 2009;25:2200–2207. doi: 10.1093/bioinformatics/btp386. [DOI] [PubMed] [Google Scholar]
- Geourjon C. Deleage G. Bioinformatics. 1995;11:681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- Buchan D. W. Minneci F. Nugent T. C. Bryson K. Jones D. T. Nucleic Acids Res. 2013;41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. Li W. Liu S. Xu J. Nucleic Acids Res. 2016;44:W430–W435. doi: 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T. Cozzetto D. Jones D. T. Proteins: Struct., Funct., Bioinf. 2014;82:98–111. doi: 10.1002/prot.24377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M. Sippl M. J. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A. MacArthur M. W. Moss D. S. Thornton J. M. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Dorosti H. Eslami M. Negahdaripour M. Ghoshoon M. B. Gholami A. Heidari R. Dehshahri A. Erfani N. Nezafat N. Ghasemi Y. J. Biomol. Struct. Dyn. 2019 doi: 10.1080/07391102.2018.1519460. [DOI] [PubMed] [Google Scholar]
- Nain Z. Abdulla F. Rahman M. M. Karim M. M. Khan M. S. A. Sayed S. B. Mahmud S. Rahman S. R. Sheam M. M. Haque Z. J. Biomol. Struct. Dyn. 2020;38:4850–4867. doi: 10.1080/07391102.2019.1692072. [DOI] [PubMed] [Google Scholar]
- Olejnik J. Hume A. J. Mühlberger E. PLoS Pathog. 2018;14:e1007390. doi: 10.1371/journal.ppat.1007390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthwick N. Silva-Arrieta S. Llano A. Takiguchi M. Brander C. Hanke T. Vaccines. 2020;8:28. doi: 10.3390/vaccines8010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D. Inbar Y. Nussinov R. Wolfson H. J. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach E. Schneidman-Duhovny D. Andrusier N. Nussinov R. Wolfson H. J. Nucleic Acids Res. 2008;36:W229–W232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. CCP4 Newsletter on Protein Crystallography. 2002;40:82–92. [Google Scholar]
- A. S. Inc., Accelrys Discovery Studio. Accelrys Software Inc., San Diego, 2012 [Google Scholar]
- Krieger E., Vriend G. and Spronk C., http://YASARA.org, 2013, vol. 993
- Dickson C. J. Madej B. D. Skjevik Å. A. Betz R. M. Teigen K. Gould I. R. Walker R. C. J. Chem. Theory Comput. 2014;10:865–879. doi: 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E. Vriend G. J. Comput. Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E. Nielsen J. E. Spronk C. A. Vriend G. J. Mol. Graphics Modell. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Kumar N. Sood D. Tomar R. Chandra R. ACS Omega. 2019;4:21370–21380. doi: 10.1021/acsomega.9b03035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. Sood D. Singh S. Kumar S. Chandra R. Eur. J. Pharm. Sci. 2021;156:105572. doi: 10.1016/j.ejps.2020.105572. [DOI] [PubMed] [Google Scholar]
- Kumar N. Sood D. Gupta A. Jha N. K. Jain P. Chandra R. Biosci. Rep. 2020:40. doi: 10.1042/BSR20202349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N. Singh A. Grover S. Kumari A. Kumar Dhar P. Chandra R. Grover A. J. Biomol. Struct. Dyn. 2019;37:2098–2109. doi: 10.1080/07391102.2018.1477620. [DOI] [PubMed] [Google Scholar]
- Bappy S. S. Sultana S. Adhikari J. Mahmud S. Khan M. A. Kibria K. K. Rahman M. M. Shibly A. Z. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1726815. [DOI] [PubMed] [Google Scholar]
- Khan M. A. Mahmud S. Alam A. R. U. Rahman M. E. Ahmed F. Rahmatullah M. J. Biomol. Struct. Dyn. 2020:1–7. doi: 10.1080/07391102.2020.1796813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. S. Mahmud S. Sultana R. Dong W. Arabian J. Chem. 2020;13:8600–8612. doi: 10.1016/j.arabjc.2020.09.044. [DOI] [Google Scholar]
- Swargiary A. Mahmud S. Saleh M. A. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1835729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelde A. Bilich T. Heitmann J. S. Maringer Y. Salih H. R. Roerden M. Lübke M. Bauer J. Rieth J. Wacker M. Nat. Immunol. 2021;22:74–85. doi: 10.1038/s41590-020-00808-x. [DOI] [PubMed] [Google Scholar]
- Van Aalten D. M. De Groot B. L. Findlay J. B. Berendsen H. J. Amadei A. J. Comput. Chem. 1997;18:169–181. doi: 10.1002/(SICI)1096-987X(19970130)18:2<169::AID-JCC3>3.0.CO;2-T. [DOI] [Google Scholar]
- Tama F. Brooks III C. L. Annu. Rev. Biophys. Biomol. Struct. 2006;35:115–133. doi: 10.1146/annurev.biophys.35.040405.102010. [DOI] [PubMed] [Google Scholar]
- Prabhakar P. K. Srivastava A. Rao K. K. Balaji P. V. J. Biomol. Struct. Dyn. 2016;34:778–791. doi: 10.1080/07391102.2015.1054430. [DOI] [PubMed] [Google Scholar]
- Sarkar B., Ullah A., Araf Y., Islam N. N. and Zohora U. S., Expert Review of Vaccines, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote A. Hiller K. Scheer M. Münch R. Nörtemann B. Hempel D. C. Jahn D. Nucleic Acids Res. 2005;33:W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M. F. Roeske E. K. Ward L. N. Pengo T. Dileepan T. Kotov D. I. Jenkins M. K. Immunity. 2018;49:1090–1102. doi: 10.1016/j.immuni.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; , e1097
- Petersen E. Koopmans M. Go U. Hamer D. H. Petrosillo N. Castelli F. Storgaard M. Al Khalili S. Simonsen L. Lancet Infect. Dis. 2020;20:e238–e244. doi: 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To K. K.-W. Tsang O. T.-Y. Yip C. C.-Y. Chan K.-H. Wu T.-C. Chan J. M.-C. Leung W.-S. Chik T. S.-H. Choi C. Y.-C. Kandamby D. H. Clin. Infect. Dis. 2020;71:841–843. doi: 10.1093/cid/ciaa149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.-S. Zhang C. Lancet. 2020;395:391–393. doi: 10.1016/S0140-6736(20)30300-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P. K. Wong D. A. Tong L. K. Ip S.-M. Lo A. C. Lau C.-S. Yeung E. Y. Lim W. W. Lancet. 2004;363:1699–1700. doi: 10.1016/S0140-6736(04)16255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y. Zhang D. Yang P. Poon L. L. Wang Q. Lancet Infect. Dis. 2020;20:411–412. doi: 10.1016/S1473-3099(20)30113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horby P., Huntley C., Davies N., Edmunds J., Ferguson N., Medley G., Hayward A., Cevik M. and Semple C., NERVTAG, https://assets.publishing.service.gov, 2021, vol. 1 [Google Scholar]
- Mascola J. R. Graham B. S. Fauci A. S. JAMA. 2021;325:1261–1262. doi: 10.1001/jama.2021.2088. [DOI] [PubMed] [Google Scholar]
- Martin J. E. Louder M. K. Holman L. A. Gordon I. J. Enama M. E. Larkin B. D. Andrews C. A. Vogel L. Koup R. A. Roederer M. Vaccine. 2008;26:6338–6343. doi: 10.1016/j.vaccine.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. Zhang J.-S. Su N. Xu J.-G. Wang N. Chen J.-T. Chen X. Liu Y.-X. Gao H. Jia Y.-P. Antiviral Ther. 2007;12:1107. [PubMed] [Google Scholar]
- Yong C. Y. Ong H. K. Yeap S. K. Ho K. L. Tan W. S. Front. Microbiol. 2019;10:1781. doi: 10.3389/fmicb.2019.01781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R. L. Donaldson E. F. Baric R. S. Nat. Rev. Microbiol. 2013;11:836–848. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M. A. and Veesler D., in Advances in virus research, Elsevier, 2019, vol. 105, pp. 93–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M. Song L. Cell. Mol. Immunol. 2020;17:536–538. doi: 10.1038/s41423-020-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. A. Gorelick R. J. Virus Res. 2008;134:39–63. doi: 10.1016/j.virusres.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong E. Wong M. U. Huffman A. He Y. Front. Immunol. 2020;11:1581. doi: 10.3389/fimmu.2020.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L. Hurst-Hess K. R. Koetzner C. A. Masters P. S. J. Virol. 2016;90:4357–4368. doi: 10.1128/JVI.03212-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya M. Sharma A. R. Patra P. Ghosh P. Sharma G. Patra B. C. Lee S. S. Chakraborty C. J. Med. Virol. 2020;92:618–631. doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamriz S. Ofoghi H. Moazami N. Comput. Biol. Med. 2016;76:24–29. doi: 10.1016/j.compbiomed.2016.06.015. [DOI] [PubMed] [Google Scholar]
- Bonam S. R. Partidos C. D. Halmuthur S. K. M. Muller S. Trends Pharmacol. Sci. 2017;38:771–793. doi: 10.1016/j.tips.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Lee S. Nguyen M. T. Immune Netw. 2015;15:51. doi: 10.4110/in.2015.15.2.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon N. Pandey R. K. Prajapati V. K. Sci. Rep. 2017;7:1–12. doi: 10.1038/s41598-016-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri A. Das P. Chakravortty D. Vaccine. 2008;26:6777–6783. doi: 10.1016/j.vaccine.2008.09.045. [DOI] [PubMed] [Google Scholar]
- Akira S. Takeda K. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Takeda K. Akira S. Int. Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Netea M. G. van Deuren M. Kullberg B. J. Cavaillon J.-M. Van der Meer J. W. Trends Immunol. 2002;23:135–139. doi: 10.1016/S1471-4906(01)02169-X. [DOI] [PubMed] [Google Scholar]
- Mahmud S. Uddin M. A. R. Paul G. K. Shimu M. S. S. Islam S. Rahman E. Islam A. Islam M. S. Promi M. M. Emran T. B. Saleh M. A. Brief Bioinform. 2021;22:1402–1414. doi: 10.1093/bib/bbaa428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. Fu Y. Xie J. Cheng J. Ghabrial S. A. Li G. Yi X. Jiang D. PLoS One. 2012;7:e42147. doi: 10.1371/journal.pone.0042147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brassac J. Blattner F. R. Syst. Biol. 2015;64:792–808. doi: 10.1093/sysbio/syv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyde K. Harris E. J. Virol. 2006;80:2170–2182. doi: 10.1128/JVI.80.5.2170-2182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this study are included within the article (and its additional files).