

SUMMARY
To investigate changes in the tumor microenvironment (TME) during lung cancer progression, we interrogate tumors from two chest computed tomography (CT)-defined groups. Pure non-solid (pNS) CT density nodules contain preinvasive/minimally invasive cancers, and solid density nodules contain invasive cancers. Profiling data reveal a dynamic interaction between the tumor and its TME throughout progression. Alterations in genes regulating the extracellular matrix and genes regulating fibroblasts are central at the preinvasive state. T cell-mediated immune suppression is initiated in preinvasive nodules and sustained with rising intensity through progression to invasive tumors. Reduced T cell infiltration of the cancer cell nests is more frequently associated with preinvasive cancers, possibly until tumor evolution leads to a durable, viable invasive phenotype accompanied by more varied and robust immune suppression. Upregulation of immune checkpoints occurs only in the invasive nodules. Throughout progression, an effector immune response is present but is effectively thwarted by the immune-suppressive elements.
Graphical abstract
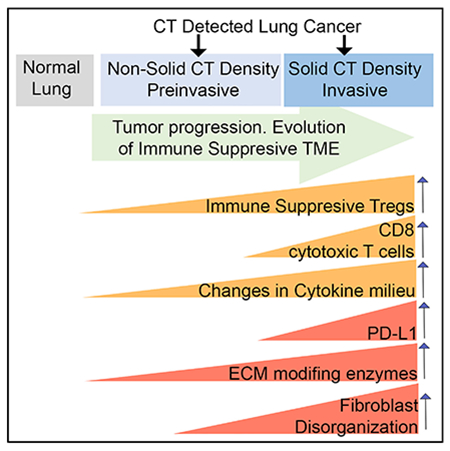
In brief
CT-scan-identified lung nodules present a significant clinical challenge. Altorki et al. define characteristics, both immune-context and microenvironment features, distinguishing normal lung, preinvasive, and invasive nodules. By capturing early features of disease progression, they inform future interception strategies and identify key questions to be investigated in mechanistic studies.
INTRODUCTION
The widespread use of chest computed tomography (CT), particularly for lung cancer screening purposes, has led to the detection of ground-glass or pure non-solid (pNS) nodules that are not visible on plain chest radiography (Izumchenko et al., 2015; Niho et al., 1999). In CT scans, pNS nodules appear as hazy opacities that do not obscure the underlying lung parenchyma or vasculature. pNS nodules between 1 and 3 cm in size typically harbor adenocarcinoma in situ (AIS) or minimally invasive adenocarcinoma (MIA), where the invasive component is 5 mm or less in size. The precise biological behavior of these lesions is unclear, as many remain unchanged in size and appearance for years. However, approximately 20%–40% of these nodules either exhibit continued growth or develop areas of increased CT density (a manifestation of more invasive malignancy) within a 4 year window (Kobayashi and Mitsudomi, 2013). Why some nodules retain an indolent behavior while others progress to invasive malignancy is unclear. The underlying pathobiology that drives some nodules to progress is incompletely understood, an uncertainty that commits patients to frequent, repeat imaging and, in some cases, potentially harmful biopsies.
A few recent studies have comprehensively characterized the genomic landscape of pulmonary preneoplasia and its progression to invasive carcinoma (Chen et al., 2019; Hu et al., 2019; Krysan et al., 2019; Qian et al., 2020). Histological progression was associated with progressive DNA alterations, including increased mutational burden with accumulation of single-nucleotide variants (SNV), somatic copy-number alterations, human leukocyte antigen (HLA) loss of heterozygosity, and mutations in canonical oncogenes such as EGFR, KRAS, and TP53.
In addition to genomic changes of cancer cells being linked to progression, the role of the tumor microenvironment (TME) in the initiation and progression of cancer is well recognized (Joyce and Fearon, 2015; Klemm and Joyce, 2015; Pyonteck et al., 2013; Turley et al., 2015). The TME is predominantly composed of immune cells, fibroblasts, and endothelial cells, as well as the extracellular matrix (ECM). There are a number of non-mutually exclusive hypotheses for how the TME contributes to tumorigenesis. For example, alterations in the immune microenvironment may result in a dominant immunosuppressive state within the tumor that drives progression of established cancer cells or favors proliferation and invasion of progenitor neoplastic cells. Similarly, dysregulation in the deposition and remodeling of the ECM, particularly enhanced desmoplasia, has been implicated in the development and progression of several solid tumors. Preexisting fibrotic disorders such as idiopathic pulmonary fibrosis, liver cirrhosis, and fibrotic breast disease have all been associated with increased incidence of cancers of the lung, liver, and breast, respectively (Ballester et al., 2019; Chung et al., 2018; Yaghjyan et al., 2015).
We posited that progression of lung cancer from preinvasive to invasive adenocarcinoma develops as a result of global alterations in the TME, including the emergence of an immunosuppressive phenotype as well as dysregulation of the ECM. To test this hypothesis, we used whole-exome sequencing, global gene expression profiling, and multiplex-image-based proteomic analyses to comprehensively characterize and contrast alterations in gene expression and cellular composition of the TME of nodules categorized into 2 groups based on CT-scanning radiographic characteristics. One group was nodules of pNS CT density, which are preinvasive/minimally invasive, and the other group was nodules of solid CT density, which are invasive adenocarcinoma.
RESULTS
The lung adenocarcinoma (LUAD) samples in this study were obtained from pNS nodules (n = 31), predominantly representing preinvasive and MIA histologies (n = 21: AIS = 5, and MIA = 16), and from solid nodules (n = 31) representing invasive adenocarcinoma. There were no statistically significant differences between the two groups in age, gender, radiographically determined tumor size, and smoking exposure (never smokers), although smoking pack years was increased the solid-nodule group (Table S1). The cancer cells in the more clinically advanced solid-nodule group were more proliferative than in the pNS-nodule group, as evidenced by Ki67 expression: 1.7% ± 0.28% and 8.1% ± 1.48% panCK-cell-positive for Ki67 in pNS nodules and in solid nodules, respectively (difference between nodule groups p < 0.001, Mann Whitney t test).
Mutation landscape
Whole-exome sequencing was done on all 31 pNS and 29 solid nodules. In the pNS nodules used for whole-exome sequencing, 19 were preinvasive/minimally invasive (4 AIS and 15 MIA). There was no difference between the two groups in mutational burden (Figure S1A). Common driver mutations were equally frequent in both groups, with KRAS and epidermal growth factor receptor (EGFR) mutations being the most common in both (Figure S1B). TP53 mutations were more frequent in the solid-nodule group, consistent with TP53 mutations occurring later in disease progression. Copy-number variations were significantly more frequent in the solid-nodule group (Figures S1C–S1E), consistent with previous findings of an increased prevalence of these genomic alterations in more advanced LUAD (Chen et al., 2019; Hu et al., 2019). The frequency of genome loss was increased in the solid group compared with the pNS group, whereas there was no difference in genome gains between the groups.
Differentially expressed genes and enriched molecular pathways in pNS nodules compared with normal lung
Principal-component analysis of RNA sequencing (RNA-seq) of normal lung (n = 10), pNS (n = 23), and solid (n = 21) nodules revealed clustering of the two tumor groups based on their CT density and a separate clustering of samples from normal lung (Figure S2). Lung tissue at least 5 cm from the tumor was used as normal lung. This distance was chosen to minimize any cancerization field effect of the tumor on surrounding tissue. Tumor-adjacent tissues were collected from 8 pNS nodules and 2 solid nodules. We refer to this group of 10 samples as normal lung. The principal-component analysis plot demonstrates that there is little variability among the 10 normal lung samples. In the pNS nodules used for RNA-seq analyses, there were 16 preinvasive/minimally invasive (2 AIS and 13 MIA) and 7 nodules of invasive histology.
The expressions of 924 genes were increased by at least 2-fold in pNS nodules versus normal lung tissue (Figure 1A; Table S2). Genes annotated to Gene Ontology biological pathways related to cell cycle, cell division, and cell growth were overrepresented among the upregulated genes in the pNS nodules, consistent with increased cell proliferation (Figure 1B). Genes annotated to ECM-related biological pathways were also overrepresented among the upregulated genes in the pNS nodules (Figure 1C). These genes encompass all aspects of ECM biology, including proteases that control the dynamics of the ECM and interactions of cells with the ECM (8 different matrix metalloproteinases [MMPs], HPN, TMPRSS6, ADAM12, and ADAMTS-14 and -16), regulators of those proteases (SPINK5, TIMP1, and TKT), intracellular proteins linking the ECM to the cytoskeleton (ITGA1, ITGB6, and FERMT1), structural components of the ECM (collagens, COMP, LAMA1, POST), and enzymes involved in the biosynthesis or post-translational modification of ECM proteins (HAS3, P4HA3, and SULF1). These data suggest that remodeling of the ECM is an early event in the development of preinvasive/minimally invasive LUAD.
Figure 1. Transcriptome differences between normal lung and pure non-solid nodules.
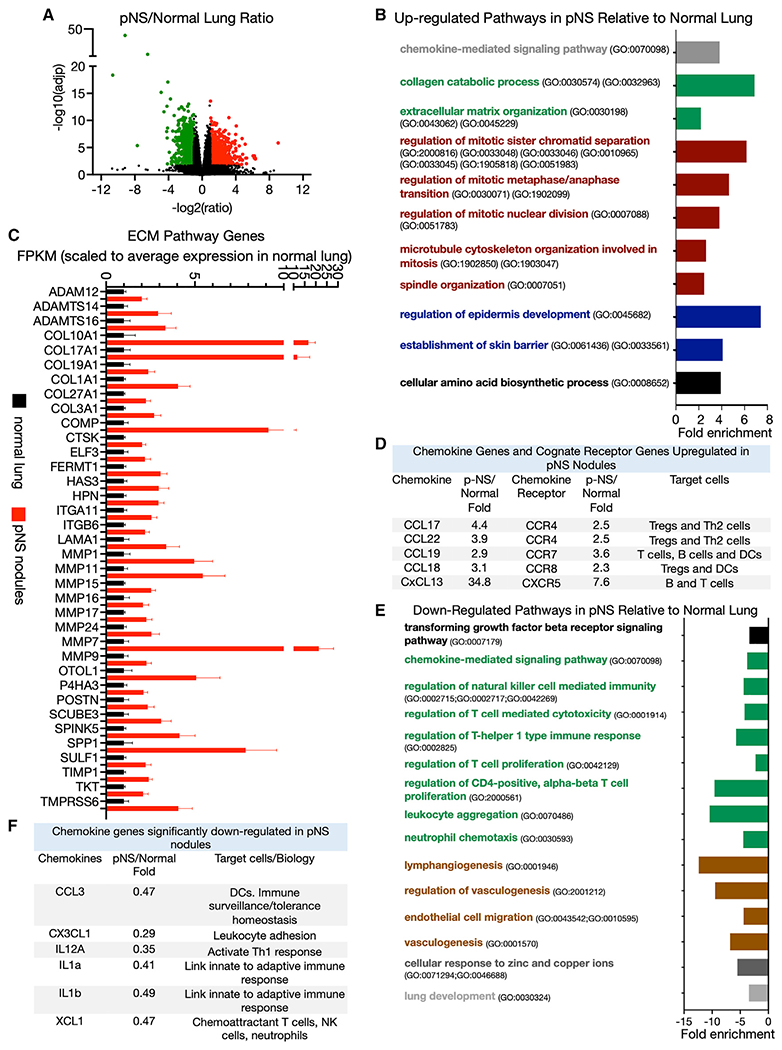
(A) Volcano plot of differentially expressed genes upregulated (red) and downregulated (green) in pure non-solid (pNS) nodules compared with normal lung with a ≥2-fold difference in expression and an FDR ≤0.05 (Benjamini-Hochberg correction). Comparison of 10 normal lung with 23 pNS nodules.
(B) Some biological pathways enriched among the significantly upregulated genes in pNS nodules compared with normal lung (FDR <0.05). The David Gene Ontology pathway identifiers (GO numbers) are noted.
(C) Significantly upregulated genes in pNS nodules relative to normal lung that are annotated to extracellular matrix (ECM) biological pathways. The FPKM of the individual genes are scaled to the average value in normal lung. Average values of the scaled fragments per kilobase of exon per million mapped fragments (FPKM) ±SEM for 10 normal lung samples and 23 pNS nodules.
(D) Chemokines and cognate receptors significantly upregulated in the pNS nodules compared with normal lung. The target cells (those cells that highly express the receptors) are noted.
(E) Some biological pathways enriched among the significantly downregulated genes in pNS nodules compared with normal lung (FDR <0.05). The David Gene Ontology pathway identifiers (GO numbers) are noted.
(F) Chemokine genes significantly downregulated in pNS nodules compared with normal lung. Target cells for these chemokines are noted.
Genes annotated to chemokine signaling pathway (GO: 0070098) were also overrepresented among the upregulated genes in the pNS nodules. Of note, the chemokine-signaling genes included five ligands and their cognate receptors (Figure 1D). These gene expression data suggest a cytokine profile of pNS nodules that is biased toward immune suppression due to enhanced recruitment of regulatory T cells (Tregs), T helper type 2 (Th2) cells, and myeloid-derived suppressive cells, an immune environment that perhaps contributes to a blunted anti-tumor immune response associated with the emergence of pNS nodules.
The expressions of 968 genes were reduced in the pNS compared with normal lung, representing the repression of a range of biological pathways in pNS nodules including immune-modulatory pathways and vasculogenesis pathways (Figure 1E; Table S2). The downregulated genes indicate the repression of a number of immune-modulatory pathways, including tumor growth factor beta (TGF-β) biology, natural killer (NK) cell biology, T cell proliferation, and immune modulation, suggesting a broadly immune-suppressive TME of pNS nodules that might contribute to immune escape. Of note is the downregulation of several chemokine genes linked to recruitment and/or activation of the innate and adaptive immune system (Figure 1F). The gene expression profile of the up- and downregulated immune-related pathways suggests a dominantly immune-suppressive microenvironment.
Differentially expressed genes and enriched molecular pathways in solid nodules compared with pNS nodules
There were 314 genes upregulated in solid nodules compared with pNS nodules (Figure 2A; Table S2). Over a quarter of these genes are annotated to the cell cycle (GO: 0007049), reflecting increases in many pathways involved in cell proliferation and indicative of the more advanced and aggressive nature of solid nodules (Figure 2B). Once again, ECM biology pathways were significantly enriched among genes upregulated in the solid-tumor group (Figure 2B). As was the case in the pNS nodules, these genes included a broad spectrum of functions in matrix deposition, degradation, and post-translational modification, suggesting that an ongoing remodeling of the ECM is a characteristic of tumor progression (Figure 2C).
Figure 2. Transcriptome differences between pNS and solid nodules.
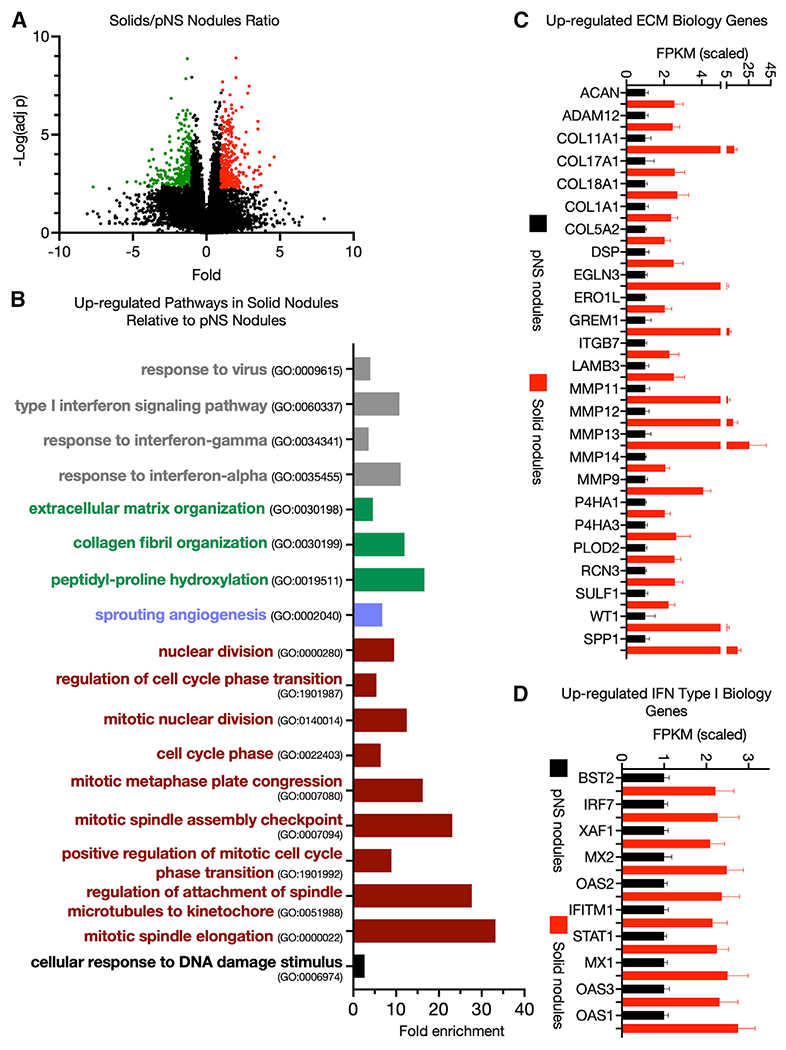
(A) Volcano plot of differentially expressed genes upregulated (red) and downregulated (green) in solid tumors compared with pNS tumors with a ≥2-fold difference in expression and an FDR <0.05 (Benjamini-Hochberg correction). Comparison of 23 pNS nodules with 21 solid nodules.
(B) Some biological pathways enriched among the significantly upregulated genes in solid tumors compared with pNS tumors (FDR <0.05). Gene Ontology pathway identifiers (GO numbers) are noted.
(C) ECM Gene Ontology pathway genes upregulated in solid tumors compared with pNS tumors. Average values of the scaled FPKM ±SEM for 23 pNS nodules and 21 solid nodules.
(D) Type I interferon response genes upregulated in solid tumors compared with pNS tumors. Average values of the scaled FPKM ±SEM for 23 pNS nodules and 21 solid nodules.
Type I interferon signaling and response to virus Gene Ontology pathways were overrepresented among the upregulated genes, driven by the expression of a core set of type I interferon-regulated genes (Figures 2B and 2D). The upregulated chemokines and chemokine receptor genes in the pNS-nodule group (i.e., Figure 1D) remained elevated in the solid-nodule group. Thus, the immune changes indicated by type I interferon activity are in addition to those identified in pNS nodules.
The gene expression differences between pNS and solid nodules suggest that the immune microenvironment of the solid nodules (i.e., invasive LUAD) is more complex, with activation of both immunostimulatory and immune-suppressive pathways in contrast to the preinvasive/minimally LUAD of the pNS-nodule group. Finally, in contrast to pNS nodules, pathways regulating angiogenesis and blood vessel morphogenesis were significantly enriched in solid nodules harboring invasive adenocarcinoma.
Among the 297 genes downregulated in solids compared with pNSs (Table S2), genes annotated to Gene Ontology pathways of epidermis development (GO: 0008544) and skin development (GO: 0043588) were significantly enriched (≤2-fold enrichment, false discovery rate [FDR] <0.05).
Upregulated genes paralleling development from normal lung to solid nodules
Of the over 900 genes whose expressions in pNS nodules were increased by greater than 2-fold, 90 were further increased by greater than 2-fold in solid tumors compared with pNS (Table S2). Not unexpectedly, most biological pathways overrepresented in the invasive and more advanced solid-tumor group were related to mitosis and nuclear division (Figure 3A).
Figure 3. Genes progressively upregulated from normal lung to solid nodules.
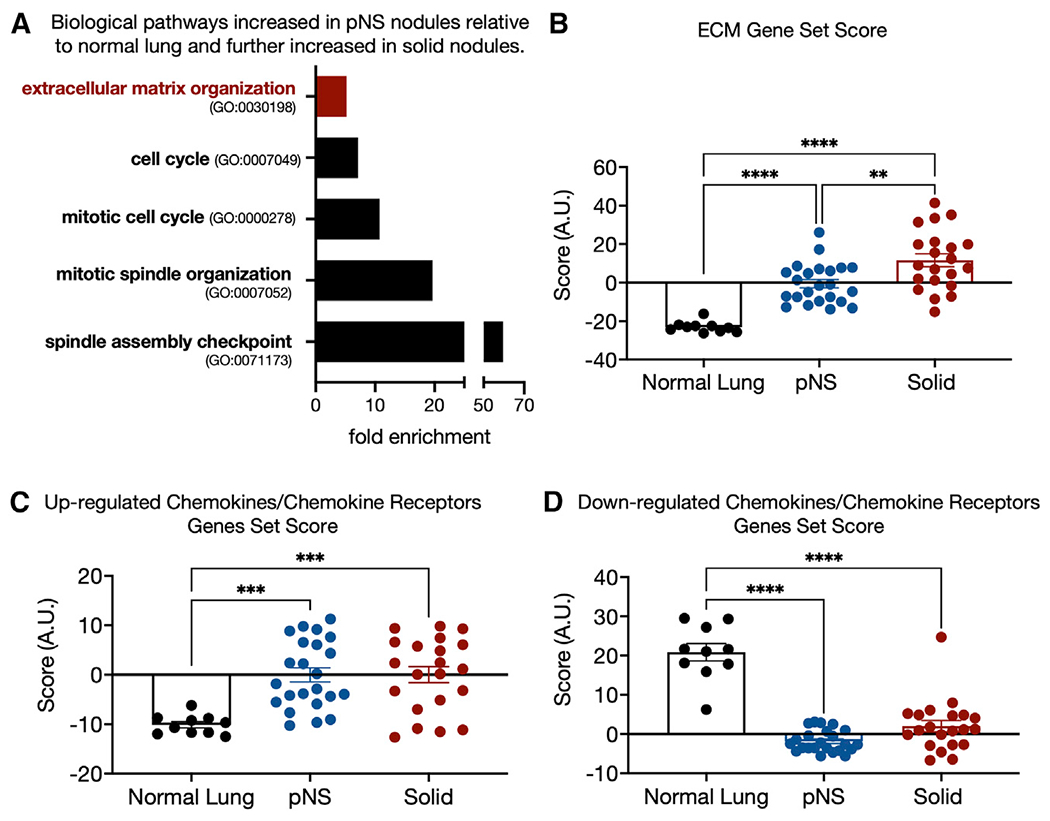
(A) Some biological pathways enriched among the 90 genes that are upregulated by ≥2-fold in pNS compared with normal lung and further upregulated by ≥2-fold in solid tumors relative to pNS tumors (FDR <0.05).
(B) ECM gene set score for each sample group generated as the sum of the Z scores of the 36 ECM pathway genes differential expressed between normal lung and pNS nodules (Figure 1C). Sidak’s p value for multiple comparison are shown. 10 normal lung, 23 pNS nodules, and 21 solid nodules.
(C) Gene set score for chemokine/chemokine receptor genes upregulated in pNS compared with normal lung generated as the sum of the Z scores of the individual genes (Figure 1D). Sidak’s p value for multiple comparison are shown. 10 normal lung, 23 pNS nodules, and 21 solid nodules.
(D) Gene set score for chemokine/chemokine receptor genes downregulated in pNS compared with normal lung generated as the sum of the Z scores of the individual genes (Figure 1F). Sidak’s p value for multiple comparison are shown. 10 normal lung, 23 pNS nodules, and 21 solid nodules.
In addition, genes for proteins that contribute to ECM biosynthesis, organization, and dynamics were enriched among these 90 genes (Figure 3A). To gain insights into how the expression of this set of ECM-related genes change during development from normal lung to pNS nodules to solid nodules, we generated a combined gene set score based on the sum of the Z scores of expressions of the 36 ECM biology genes increased in the pNS-nodule group (Figure 1B). These 36 genes are a subset of the 218 genes of the ECM Gene Ontology grouping (GO: 0030198). We restricted our analyses to the sum Z score of the 36 differentially expressed genes because we were specifically interested in how this subset of ECM-related genes changes during progression. There was a progressive increase in the ECM set Z score from normal lung to solid nodules, identifying progressive alterations in the ECM as a key early event in the development of preinvasive or minimally invasive LUAD and in progression to a more invasive phenotype (Figure 3B).
We also generated separate Z-score-based gene set scores for the chemokine/chemokine receptor genes upregulated and downregulated in pNS nodules versus normal lung. In contrast to the ECM gene set, neither the up- or downregulated chemokine/chemokine receptor gene set scores were further increased in the solid-nodule group compared with the pNS-nodule group (Figures 3C and 3D). We also tested these ECM and chemokines gene sets against nodules grouped by smoking status, EGFR versus KRAS mutational status, and tumor mutational burden (TMB) (Figures S3A–S3F). In none of those comparisons were any of the gene sets different between the comparison groups.
The expression of only 39 genes was significantly reduced in pNS compared with normal lung and was then further reduced in the solid-nodule group, too few for Gene Ontology pathway analyses (Table S2).
Evolution of immune microenvironment
We characterized the immune landscape in normal lung and both nodule groups using xCell deconvolution of the RNA-seq data (Figure 4A). Deconvolution by CIBERSORT (Chen et al., 2018) yielded qualitatively similar results. Compared with normal lung, pNS nodules were enriched for immune cells associated with both the innate and adaptive immune system. Immune cells increased in pNS nodules included B cells, plasma cells, CD4+ T cells, Tregs, NKT cells, and dendritic cells. In contrast, the immune landscape of the solid-nodule group was characterized by further increases in cells associated with an adaptive immune response including CD4+ T cells, Th1 and Th2 cells, and Tregs, as well as CD8+ cells. These data suggest a robust recruitment of diverse immune cell populations to the microenvironment of preinvasive/minimally invasive lesions (pNS nodules) that then progresses at pace with the development of a more invasive malignancy.
Figure 4. Immunofluorescence immune profiling.
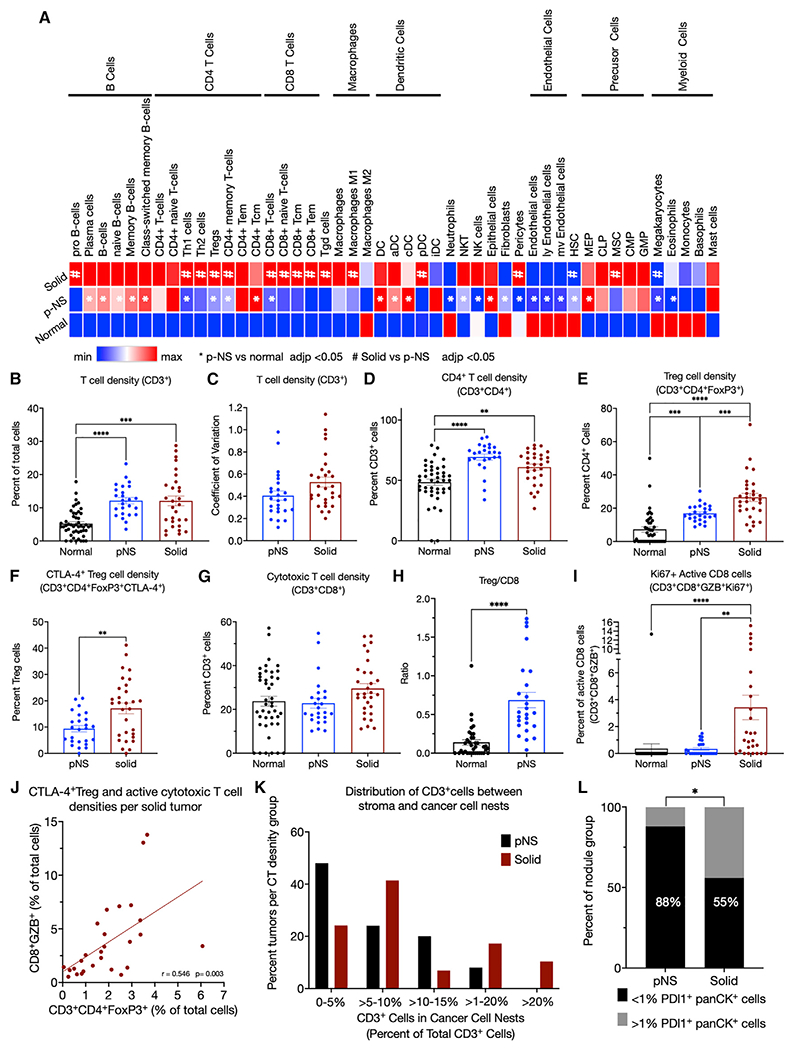
(A) Immune-cell compositions of normal, pNS tumors, and solid tumors determined by xCell deconvolution.
(B) Density of CD3+ cells determined as the percentage of total cells per ROI.
(C) Coefficient of variations of CD3+ cell densities of the 6 ROIs per tumor of the pNS and solid groups.
(D) Densities of CD4+ cells determined as percentage of CD3+ cells.
(E) Treg densities (CD3+CD4+FoxP3+) determined as the percentage of CD3+CD4+ cells.
(F) CTLA-4+ Treg densities (CD3+CD4+FoxP3+CTLA-4+) as the percentage of CD3+CD4+FoxP3+ cells.
(G) Cytotoxic T cell densities (CD3+CD8+) determined as the percentage of CD3+ cells.
(H) Ratios per tumor of Tregs (CD3+CD4+FoxP3+) to cytotoxic T cells (CD3+CD8+) in normal lung and pNS nodules.
(I) Active cytotoxic T cells densities (CD3+CD8+GZB+Ki67+) determined as the percentage of Ki67 + CD3+CD8+GZB+ cells. GZB, granzyme B.
(J) Correlation of CTLA-4+ Treg (CD3+CD4+FoxP3+CTLA-4+) and active cytotoxic T cell (CD3+CD8+GZB+) densities as percentages of total cells per ROI. The p value is for the difference in the slope of the correlation line from 0.
(K) T cells (CD3+) within the cancer cell nests as a percentage of total T cells in the 3 subcategories of solid tumors segregated by panCK+ PD-L1+ expression. (L) PD-L1-expressing panCK cells in pNS- and solid-nodule groups. The percent of nodules per group with <1% or ≥1% panCK+PD-L1+ cells. Fisher’s exact test. In all panels, the symbols for normal tissue are data from a single ROI per sample, whereas in the pNS and solid groups, each symbol is the mean of 3–6 ROIs per tumor. Total cells were determined by 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei per ROI. The group means ± SEM are shown for 10 normal lung, 25 pNS nodules, and 27 solid nodules. p values. ANOVA followed by Kruskal-Wallis test of p values for multiple comparisons. All significant differences are noted.
To characterize the spatial localization and activation states of immune cells within the TME, we performed multiplex immunofluorescence microscopy on 25 pNS, 27 solid nodules, and normal lung tissue 5 cm from the nodules using a panel of 16 antibodies identifying 12 cell types and distinct activation states (Table S3) with the Neogenomics imaging platform, an iterative multiplex method (Figure S4A [Xu-Monette et al., 2019)]). In the pNS group, there were 18 nodules with preinvasive/minimally invasive histologies (4 AIS and 14 MIA) and 7 with invasive histologies. To capture heterogeneity within individual tumors, multiple regions of interests (ROIs), between 3 and 6, were studied from each tumor. To broadly define the immune cell compositions of the microenvironment of the two groups, we determined the density of B cells (CD3−CD19+), macrophages (CD3−CD68+CD209−), and dendritic cells (CD3−CD209+CD68−). B cell density in the normal lung was low (0.06% of total cells) and significantly elevated in both pNS- and solid-nodule groups (2.7% and 2.1% of total cells, respectively) (Figure S4B). Increased B cells in the pNS- and solid-nodule groups relative to the normal lung is consistent with the RNA-seq deconvolution data. There was no difference in B cell density between the two tumor groups, identifying B cell infiltration as a possible early event. There was a small yet significant reduction in macrophage cell density in the solid-nodule groups compared with normal lung, whereas there was no difference between the pNS group and the normal lung (Figure S4C). There were also small increases in dendritic cell densities in pNS- and solid-nodule groups compared with normal lung (Figure S4D).
In line with the deconvolution of bulk RNA-seq profiles, T cell densities (CD3+ cells) were elevated in both the pNS- and solid-nodule groups compared with the normal lung group, whereas there was no significant difference in T cell density between pNS and solid nodules (Figure 4B). In addition, intra-tumor heterogeneity in T cell densities, assessed by the coefficient of variation of T cell density among 6 ROIs per tumor, was similar in the two nodule groups, demonstrating that intra-tumor heterogeneity in T cell density does not change significantly during the progression of pNS to solid nodules (Figure 4C).
T cell composition and activation states in pNS and solid nodules
CD4+ T cells, as a proportion of T cells, were significantly increased in the pNS nodule group compared with normal lung (Figure 4D). The subtype composition of the CD4+ T cells also varied between the normal lung and pNS-tumor groups. Immune-suppressive Tregs (FoxP3+) were significantly increased in pNS nodules at the expense of Th cells (FoxP3−) (Figure 4E). In addition, approximately 10% of Tregs in pNS nodules were CTLA-4+ (Figure 4F), of which about 2% were also Ki67+. No CTLA4+ Tregs were detected in normal lung. CTLA-4 and Ki67 expression are associated with enhanced Treg activation and proliferation (Pedros et al., 2017; Tang et al., 2008), suggesting an active immune-suppressive TME in the pNS-nodule group compared with normal lung.
The density of cytotoxic T (CD8+) cells, as the proportion of T cells, did not differ between normal lung and pNS-tumor groups (Figure 4G). The ratio of Tregs to cytotoxic T cells calculated for the individual nodules was significantly elevated in in pNS group compared with the normal lung (Figure 4H), further suggesting a predominantly Treg-mediated immune-suppressive microenvironment.
We next investigated differences in T cell composition and activation states between solid and pNS nodules. There was no difference in CD4+ cell density between the pNS- and solid-nodule groups (Figure 4D). In both groups, about 70% of the T cells were CD4+ cells. However, the percentage of Tregs was increased in the solid-nodule group, and the proportion of CTLA-4+ Tregs was significantly elevated in the solid-nodule group, consistent with a progressively immune-suppressive TME in solid nodules (Figures 4E and 4F). There was no significant difference in cytotoxic T cell density between the pNS-nodule and solid-nodule groups (Figure 4G). However, there was an increase in activated cytotoxic T cells (GZB+Ki67+) in the solid nodules, an unexpected finding considering that solid nodules harbored more invasive adenocarcinoma, whereas there was no increase in activated cytotoxic T cells in the pNS-nodule group compared with normal lung (Figure 4I). There was a positive correlation in the densities of Tregs and GZB+CD8+ T cells of individual tumors in the solid-nodule group, revealing an increase in immune-suppressive Tregs counterbalancing the increased cytotoxic cells (Figure 4J).
Intratumor location of CD3+ cells in pNS and solid nodules
We next investigated whether T cell localization to the tumor stroma or to cancer cell nests varied between the nodule groups. Despite there being no difference between the pNS- and solid-nodule groups in total intra-tumoral CD3+ density (Figure 3B), there was a difference between the nodule groups in intra-tumoral distribution of CD3+ cells. The frequency distribution of the CD3+ cells located to cancer cell nests, determined by identifying CD3+ T cells located within islands of panCK+ cells, revealed a shift toward increased infiltration in the solid-nodule group, thereby correlating infiltration of T cells among cancer cells with invasive LUAD (Figure 4K). Conversely, T cell exclusion from the cancer cell nests was more frequently observed in pNS, possibly contributing to immune evasion in preinvasive/minimally invasive neoplasia.
Immune-escape mechanisms during development of invasive LUAD nodules
Upregulation of immune checkpoints is an immune-escape mechanism in later stages non-small cell lung cancer (NSCLC). However, gene expression profiling revealed that out of a panel of 7 checkpoint genes, only CTLA-4 expression was increased in pNS nodules (Figures S4E–S4K). In addition, reductions of co-stimulatory immune checkpoints did not account for immune suppression. Co-stimulatory immune checkpoints CD137 and CD27 were increased with progression, whereas co-stimulatory OX40 and CD40 were unchanged (Figures S4L–S4O). Thus, based on expression in bulk tissue, we conclude that modulation of immune-checkpoint function genes does not account for escape of immune surveillance in the early progression of NSCLC captured in our sample groups.
Although there was no difference between the nodule groups in PD-L1 expression in bulk RNA-seq analysis, there was an increased prevalence of PD-L1+ cancer cells in the solid-nodule group (Figure 4L). Expression of PD-L1 is regulated both transcriptionally and post-transcriptionally, likely explaining the discordance in these findings (Sun et al., 2018; Yi et al., 2021). An advantage of the imaging analyses is that PD-L1 expression can be assigned to specific cell types within the tumor. In the pNS-nodule group, only 3 of 25 tumors had greater than 1% PD-L1+ cancer cells (i.e., panCK+ cells), whereas in the solid-nodule group, 13 of 29 tumors had greater than 1% PD-L1+ cancer cells. Thus, PD-L1 expression by cancer cells was not a main immune-escape mechanism among pNS nodules, and although it was more prevalent among solid tumors, it was not an immune-escape mechanism common to the more advanced tumors. There was neither an increase in the expression of immune modulatory IDO1 nor a decrease in major histocompatibility complex (MHC) class I expression (HLA-ABC+) in either nodule group compared with normal lung or between the nodule groups (Figures S4P–S4Q).
Correlation between fibroblast activation, tumor progression, and T cell localization
Cancer-associated fibroblasts (CAFs) are a major component of the TME and are the main contributors to the deposition, modification, and turnover of tumor ECM (Henke et al., 2019; Quintero-Fabian et al., 2019; Sahai et al., 2020). Because we found a strong association between the development and progression of LUAD with increased gene expression of proteins that control the ECM, we next investigated potential differences in fibroblasts between the nodule groups. We used alpha smooth muscle actin (αSMA) as a marker of fibroblasts, although it is also expressed in other cell types, most prominently myofibroblasts and pericytes (Yoshida et al., 2019). Anti-αSMA immunofluorescence analyses showed that there was no difference in the number of fibroblasts between pNS- and solid-nodule groups (Figure 5A). There were two prominent patterns of fibroblast organization represented in both nodule groups (Figure 5B). In one, αSMA was organized in an intact pattern circumscribing cancer cell nests. In the other, the αSMA pattern was discontinuous and fragmented. Although both of these patterns were observed in the two nodule groups, the discontinuous fibroblast pattern was significantly more prevalent in the solid-nodule group, revealing an association between the discontinuous fibroblast pattern and more invasive LUAD (Figure 5C).
Figure 5. TME morphology: SMA pattern.
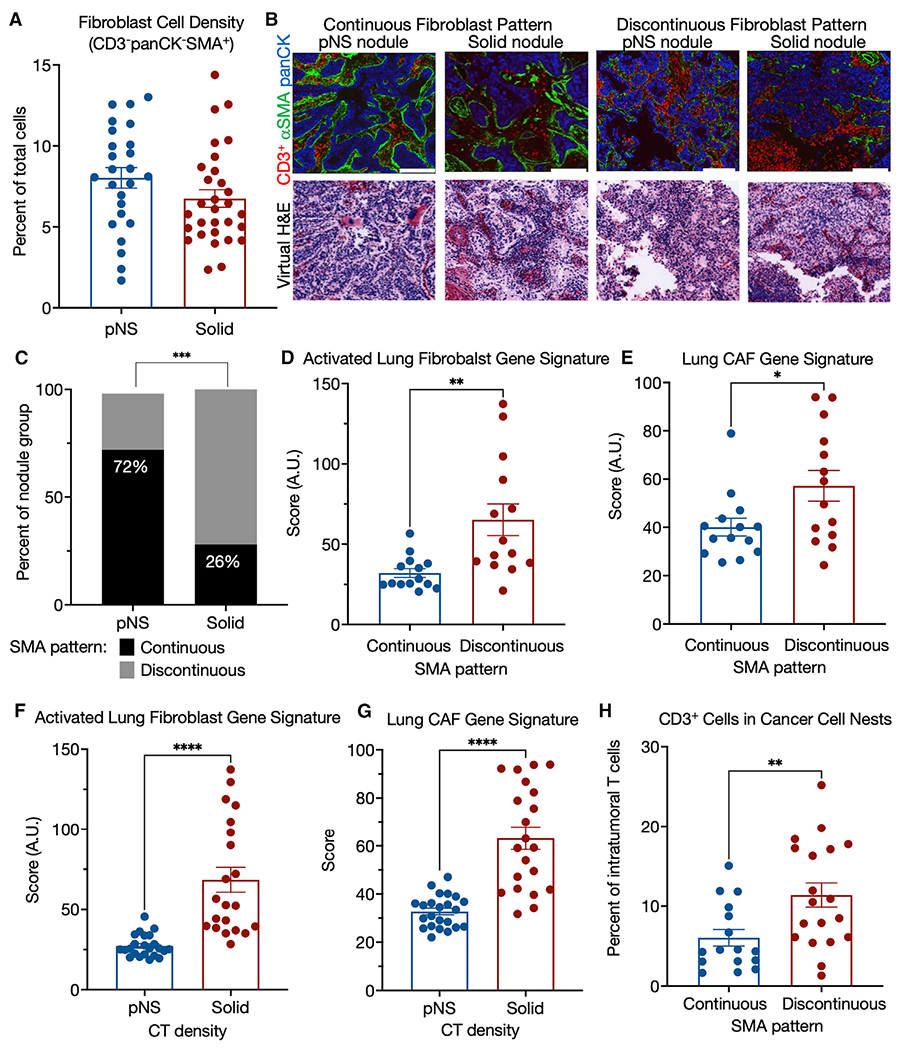
(A) Fibroblasts cell (CD3−panCK−SMA+) density in pNS- and solid-nodules groups.
(B) Representative pseudo-colored immunofluorescence and virtual H&E images of pNS and solid nodules with organized or disorganized αSMA barriers. panCK in red, αSMA in green, and DAPI in blue. Images acquired at 200× magnification. Scale bar, 200 μm.
(C) Predominant tumor αSMA morphology by tumor group. p value, Fischer’s exact test.
(D) Comparison of nodules, grouped by αSMA pattern, for an activated lung fibroblasts gene signature (Peyser et al., 2019) generated as sum Z score for the genes of the signature for each sample. Because the RNA-seq is from bulk tumor tissue, only those nodules that had either continuous or discontinuous morphologies, independent of CT density group, for each of the 3 ROIs were used for these analyses: n = 14 continuous and n = 14 discontinuous. Mann-Whitney nonparametric t test.
(E) Comparison of nodules, grouped by αSMA pattern, for a lung CAF gene signature (Navab et al., 2011) generated as sum Z score for the genes of the signature for each sample. Because the RNA-seq is from bulk tumor tissue, only those nodules that had either continuous or discontinuous morphologies, independent of CT density group, for each of the 3 ROIs were used for these analyses: n = 14 continuous and n = 14 discontinuous. Mann-Whitney nonparametric t test.
(F) Comparison of nodules, grouped by CT density, for an activated lung fibroblasts gene signature (Peyser et al., 2019) generated as sum Z score for the genes of the signature for each sample. Mann-Whitney nonparametric t test. Means ± SEM for 23 pNS and 21 solid nodules.
(G) Comparison of nodules, grouped by CT density, for a lung CAF gene signature (Navab et al., 2011) generated as sum Z score for the genes of the signature for each sample. Mann-Whitney nonparametric t test. Means ± SEM for 23 pNS and 21 solid nodules.
(H) Comparison of nodules, by αSMA pattern, for CD3+ cell infiltration into cancer cell nests as the percentage of T cells in the ROI (cancer cell nests plus stroma). Mann-Whitney nonparametric t test. Means ± SEM for 16 continuous and 19 discontinuous nodules with the same fibroblast morphology pattern in all three ROIs.
We next investigated if the two αSMA patterns are associated with fibroblast activation. We grouped samples based on their αSMA pattern (continuous versus discontinuous), irrespective of CT density, and compared these groups using a previously reported gene signature of activated lung fibroblasts (Peyser et al., 2019) (list of genes in Table S4). We restricted this analysis to those tumors that had the same αSMA pattern (continuous or discontinuous) in all 3 ROIs because the RNA-seq data were necessarily not generated from the identical region of the tumor as the imaging, and we wanted to control for intra-tumor heterogeneity in αSMA patterns. This analysis revealed a significant association of the activated fibroblast signature with nodules that had a discontinuous αSMA pattern (Figure 5D). A second, independently derived signature for activated fibroblasts, this one derived for human lung CAFs (Navab et al., 2011) (list of genes in Table S4), was also positively correlated with the discontinuous αSMA pattern (Figure 5E). This association was not driven by differences in the number of fibroblasts, as there was no correlation between the number of fibroblasts and the αSMA pattern (Figure S5) nor between the activated fibroblast signature and the number of fibroblasts (Pearson’s r = −0.025).
These two gene set signatures of activated fibroblasts and of lung CAFs were also associated with progression from preinvasive to invasive, as determined by CT density grouping of the nodules (Figures 5F and 5G). Together, these results support a strong association between activation of fibroblasts, discontinuous αSMA morphology, and tumor progression.
We next asked whether the two αSMA+ staining patterns were also associated with T cell localization within the tumor. Nodules with a discontinuous αSMA pattern had significantly higher infiltration of T cells into cancer cell nests compared with nodules with a continuous αSMA pattern (Figure 5H). These data directly associate T cell distribution between the stroma and cancer cells to an alteration in fibroblast organization, which, in turn, is associated with published activated fibroblast signatures, suggesting a positive association between fibroblast activation and T cell trafficking within the tumor nodules.
DISCUSSION
The principal objective of this work was to address gaps in our understanding of the global evolution of the TME associated with the development of early preinvasive and minimally invasive pulmonary adenocarcinoma manifesting radiographically as pNS nodules in CT scanning. To accomplish this objective, we used genomic, transcriptomic, and multiplex immunofluorescence to compare pNS nodules, which primarily harbor pre/minimally invasive LUAD, with normal lung as well as solid nodules containing more invasive adenocarcinoma. The results of our profiling analyses demonstrate distinct biologies associated with these nodule classifications, validating CT density classification, a clinically actionable diagnostic parameter, in the exploration of tumor progression.
One of our main findings is the significant upregulation of genes involved in remodeling the ECM in pNS nodules harboring preinvasive adenocarcinoma and MIA. Although remodeling of the ECM has been previously implicated in cancer progression and metastases (Henke et al., 2019; Huang et al., 2021; Winkler et al., 2020), our results demonstrate alteration in matrix biology as an early event in the development of LUAD. The ECM, a major component of the TME, possesses potent pro-tumorigenic properties mediated by various remodeling mechanisms including matrix deposition, post-translational modification of matrix components, proteolytic degradation, and force-mediated mechanical remodeling. Matrix remodeling can drive tumorigenesis through various mechanisms such as release of bound growth factors, changes in mechanical forces of the tissue impacting signal transduction, alterations in cell migration, and support of sprouting angiogenesis (Belhabib et al., 2021; Henke et al., 2019; Quintero-Fabian et al., 2019; Sahai et al., 2020). Our study supports the hypothesis that ECM changes precede acquisition of invasive properties, thereby identifying ECM biology as a reasonable therapeutic target to intercept early disease progression. Although our profiling data do not provide a possible hypothesis as to what triggers alteration in the ECM in early neoplasia, it is of interest that ECM gene enrichment was not associated with smoking history, KRAS or EGFR mutations, or TMB.
The expressions of ECM biology genes are further increased in the solid-nodule group, indicating that tumor ECM modification and reorganization continue to evolve over time. As previously stated, activated CAFs are the major producers of matrix in solid tumors and thus have a prominent role in ECM biology. We found that two independently derived signatures of activated lung fibroblasts (one from lung CAFs [Peyser et al., 2019] and another from bleomycin-activated fibroblasts [Navab et al., 2011]) were progressively enriched in the transitions from normal lung to pNS to solid nodules, associating fibroblast activation with CT-scan image as well as with early development and progression of LUAD.
The same activated fibroblast gene signatures were enriched in tumors with the discontinuous αSMA pattern, correlating fibroblast activation with fibroblast organization within the TME. We also found that the continuous fibroblasts pattern was significantly associated with a reduced infiltration of T cells into the cancer cell nest. Although our analyses do not establish a direct functional link between T cell exclusion from cancer cell nests and CAFs, previous studies do support this concept. For example, it has been shown in human lung tumors that a dense collagen matrix inhibits immune cells from migrating into the cancer cell nests (Salmon et al., 2012). However, the shielding effect of CAFs does not have to be a physical barrier since several CAF-mediated mechanisms for modulating immune responses have been proposed (Sahai et al., 2020). Nonetheless, our data reveal a strong association between the organization of CAFs and T cell infiltration into the cancer cell nests, possibly as a result of cancer cell and/or fibroblast-driven ECM remodeling with disruption of the fibroblastic barrier.
Provocatively, genes regulating angiogenesis and the broader activation of the adaptive immune response were also enriched in solid nodules compared with pNS nodules, perhaps associated with the continued remodeling of the ECM. The solid tumors, due to increased cell density, are likely under greater nutrient and hypoxic stress, requiring enhanced vascularization, and the further increases in genes of proteins that control ECM biology in solid nodules might reflect a role of ECM reorganization in promoting angiogenesis.
Our transcriptomic analyses also revealed significant enrichment in the expression of chemokine/cytokine genes and their cognate receptors in the pNS-nodule group compared with normal lung. The upregulated chemokine profile is biased toward an immune-suppressive milieu, favoring recruitment of Tregs and Th2 cells, as well as B cells. Our deconvolution and immunofluorescence data confirm increases of these cell types in the pNS nodules. Interestingly, these chemokine and chemokine receptors genes are not further increased in the solid nodules. However, in the more advanced nodule group there was an increase in genes involved in type I interferon biology as well as genes generally associated with an adaptive immune response. As a result, the immune TME of solid nodules containing invasive LUAD is more complex and characterized by increased frequency of activated cytotoxic T cells whose antitumor effect we propose is neutralized by enhanced immune suppression mediated by an increased frequency of CTLA-4+ Tregs as well as enhanced expression of PD-L1 in the cancer cells. PD-L1 is the checkpoint ligand for PD1, signifying a more robust immune-suppressive environment in the solid nodules. Interferon-gamma, which is secreted predominantly by activated T cells, is one of the most potent activators of PD-L1 expression (Garcia-Diaz et al., 2019). Therefore, the increased expression of PD-L1 in the invasive solid nodules provide additional support for an ongoing yet ineffective anti-tumor immune response.
In contrast, the immune TME of the pNS nodules is noteworthy for how few immune-suppressive mechanisms are upregulated in the preinvasive and minimally invasive stages, the principal immune-suppressive mechanism being increased density of Tregs. This finding agrees with two recent studies of lung tumors (Dejima et al., 2021; Lavin et al., 2017). A second potential mechanism of immune escape in pNS nodules is T cell exclusion from the cancer cell nests that seems mediated by an organized fibroblastic pattern. The phenomenon of T cell exclusion from the tumor mass has been reported as a mechanism of immune evasion in multiple cancers including pancreatic, ovarian, and colorectal (Beatty et al., 2015; Sanchez-Paulete et al., 2017; Tauriello et al., 2018). A similar finding was observed in ductal carcinoma in situ of the breast, where cancer cells are separated from tumor infiltrating T cells by a continuous layer of myoepithelial cells and myofibroblasts (Gil Del Alcazar et al., 2017). T cell exclusion from the entire tumor mass was not observed in any of our cases. The T cell density of the pNS-nodule group is significantly increased compared with normal lung, and it is similar to that of the solid-nodule group, data that demonstrate that both nodule groups are equally T cell inflamed. Despite the same density of intratumor T cells in both nodule groups, as discussed above, the nodule groups differ in the disposition of T cells between the stroma and the cancer cells nest. While reduced T cells within cancer cell nests were frequently observed in pNS nodules and are associated with the fibroblast pattern, we do not know at this time the functional consequence of this association.
Our data associate increased T cell infiltration of cancer cell nests with the invasive nodules, linking an intermingling of T cells and cancer cells to a more varied and robust immune-escape mechanisms as well as to signs of an enhanced immune response, the latter demonstrated by an increase in active cytotoxic CD8+ cells. This concurrent increase in immune response and suppression in the solid nodules likely signifies a shift from successful immune surveillance to more complete escape that occurs early in invasion.
There have been recent reports on the genomic and transcriptomic landscapes of preinvasive squamous cell cancer (Mascaux et al., 2019; Teixeira et al., 2019), and a recent report identified a number of potential immune-escape mechanisms, including the impairment of antigen presentation, upregulation of CCL27-CCR10 ligand receptor expression, and downregulation of the immune modulator TNSF9 (Pennycuick et al., 2020). There are only a few studies of preinvasive pulmonary adenocarcinoma, most of which have focused on genomic alterations within the neoplastic cells, such as mutational burden and somatic copy-number alterations, rather than on cellular and molecular evolution within the TME, as we have in this report (Chen et al., 2019; Hu et al., 2019; Krysan et al., 2019). Our genomic analyses confirm those previous reports of preinvasive LUAD. We did not find a difference in TMB between the pNS and solid nodules in our study. However, it is important to note that the TMB values of our samples, which are clinical stage 1A LUAD tumors, are considerably lower than those reported for more clinically advanced human LUAD. We observed an increase in copy-number variations (CNVs) associated with progression, an observation consistent with the recently reported findings of a more comprehensive study of the association of genomic alterations and immune response in early LUAD. The results of that study support the hypothesis that genomic alterations in cancer cells initiate the changes in immune response and thereby trigger progression to more advanced disease (Chen et al., 2019).
In summary, our data suggest a complex dynamic interaction between the tumor and its immune microenvironment in the very early stages of development. Alterations in the ECM and CAFs are central at the preinvasive and early invasive states of clinically early stage 1A disease. A dominant regulatory T cell-mediated immune suppression is initiated at the precursor level and is sustained with rising intensity throughout malignant progression. T cell exclusion from the cancer cell nests appears to be an additional mechanism of immune evasion deployed more frequently in preinvasive lung cancers, possibly until tumor evolution leads to a durable, viable invasive phenotype accompanied by more varied and robust immune suppression. Throughout the entire process, a nascent effector immune response is present but is effectively thwarted by the immune-suppressive elements, suggesting that different interception strategies need to be employed at different stages of tumor evolution.
Limitations of the study
Although our data represent a detailed mapping of the cellular and transcriptional landscape of the TME in preinvasive/minimally invasive adenocarcinoma, it is pertinent to note that the pNS tumors studied here, for clinical reasons, had been surgically removed rather than relegated to continued clinical surveillance. Consequently, our study group of pNS nodules might be biased toward nodules that are progressing rather than remaining indolent. Additionally, our findings represent a snapshot in time of the TME of a group of these early lesions contrasted with a group of more advanced cancers. It is not possible in human studies to molecularly characterize individual nodules before and after progression.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Tim McGraw (temcgraw@med.cornell.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the Lead contact upon request.
The RNAseq data are available at Gene Expression Omnibus (GEO), accession number GSE19375. The DNA seq data are available at the database of Genotypes and Phenotypes (dbGaP), accession number phs002818.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gapprev/gap/cgi-bin/study.cgi?study_id=phs002818.v1.p1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Tissue samples for this study were obtained from archived surgically resected material obtained from two groups of patients who had surgical resection of their nodules at New York-Presbyterian-Weill Cornell Medicine. One group were 31 patients with pure non-solid nodules, predominantly representing AIS and MIA histologies. We grouped these nodules of preinvasive (AIS) and invasive (MIA) histologies together because they are known to have similar indolent clinical and biological behaviors. The second group included 31 patients with solid nodules representing invasive adenocarcinoma. CT scan images were re-reviewed by 2 observers and classified as pure non-solid or solid nodules based on CT attenuation. The corresponding resected tissues were re-reviewed by an expert pulmonary pathologist and the extent of invasion was measured using Aperio whole scanned slides. The study was approved by the New York-Presbyterian-Weill Cornell Medicine institutional review board and patient consent was waived. Subjects with p-NS nodules ranged in age from 48 to 84 with a mean age of 69 years, and 22 were female and 9 male (Table S1). Subjects with solid nodules ranged in age from 54 to 86 with a mean age of 73 years, and 17 were female and 14 male (Table S1).
METHOD DETAILS
RNAseq
RNA was extracted from FFPE samples with RNeasy FFPE kit from Qiagen, Part# 73504. Tissue from tumor adjacent lung were used as controls. NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB #E7760, New England BioLabs, Ipswich, MA) was used for sample library preparation according to the manufacturer’s instruction. PCR enrichment of adaptor ligated cDNA were pooled and hybridized with probes from Twist Exome Kit #2 (Twist Bioscience, San Francisco, CA). After post capture PCR amplification and purification, the quality of final libraries was checked and loaded to Illumina NovaSeq6000 for sequencing at PE2x100 cycles. The raw sequencing reads in BCL format were processed through bcl2fastq 2.19 (Illumina) for FASTQ conversion and demultiplexing.
All reads were independently aligned with STAR_2.4.0f1 for sequence alignment against the human genome build hg19, downloaded via the UCSC genome browser, and SAMTOOLS v0.1.19 for sorting and indexing reads. HTSeq was used to quantity counts. Cufflinks (2.0.2) was used to get the expression values (FPKMs), and Gencode v19 GTF file for annotation. Differential gene expression was determined using Benjamini-Hochberg correction for multiple testing with an alpha of ≤0.05 and an expression difference of 2 fold or greater. Gene ontology pathway enrichment analyses were performed using Panther online software (Ashburner et al., 2000; Gene Ontology, 2021; Mi et al., 2019). Gene set scores (signatures) were constructed as the sum of the z-scores of the individual genes of the gene set. Cell compositions of the nodules were investigated by deconvolution of the RNAseq data. Two methods, xCell (Aran et al., 2017) and CIBERSORT (Chen et al., 2018) were used.
Whole-exome variant calling
DNA was extracted with Maxwell 16 FFPE Plus LEV DNA Purification Kit from Promega, Part# AS1135. DNA extracted from tumor adjacent normal lung served as reference genome for each tumor.
Less than 200ng of FFPE gDNA sample based on Qubit quantification was enzymatically fragmented using Agilent Enzymatic DNA Fragmentation Protocol (Agilent Technologies, Santa Clara, CA). Fragmented FFPE gDNA were used to perform end repair, A-tailing and adapter ligation with Agilent SureSelect XT Low Input library preparation kit following the manufacturer instructions. Then, the indexed libraries were captured by EXaCT-2 Target Enrichment Human All Exon probes that was custom-designed by Agilent Technologies, Inc. (SureSelect XT Low Input Target Enrichment) and amplified. The quality and quantities of the enriched indexed NGS libraries were checked by Agilent 4200 TapeStation System and Invitrogen Qubit Fluorometer (Thermo Fisher, Waltham, MA). The final libraries were pooled and sequenced on an Illumina NovaSeq6000 sequencer (Illumina Inc, San Diego, CA) at PE 2 × 150 cycles. The raw sequencing reads in BCL format were processed through bcl2fastq 2.19 (Illumina) for FASTQ conversion and demultiplexing.
Whole exome sequencing data was obtained from the Exome Cancer Test v2.0 (EXaCT-2), a whole exome sequencing approach adapted from Agilent’s SureSelect Human All Exon V6 (Agilent Technologies, Santa Clara, CA) that combines exome sequencing and deep sequencing of cancer hotspots. Sequencing data reads, after removal of adaptor sequences using fastp, were aligned to the reference human genome (hg19) using the Burrows-Wheeler Alignment tool (bwa mem). After alignment, removal of PCR duplicates, realignment around InDels, fixing mates and base quality score recalibration was performed using PICARD or the Genome Analysis Toolkit (GATK) according to GATK best practices and PICARD. For somatic variant calling, a consensus mutation calling approach was used which is a variation of the pipeline reported by the TCGA’s Multi-Center Mutation Calling in Multiple Cancers (MC3) project. Our pipeline includes 6 mutation callers (VarScan, SomaticSniper, MuTect, Strelka, RADIA, and Muse) to identify SNVs and 4 mutation callers (VarScan, MuTect, Strelka, Pindel) to identify insertion/deletions (InDels). Each caller is run individually on a paired tumor and its matched normal sample; resulting variants from each tool are then merged and a final set of variants is selected based on consensus calls made by ≥ 2 tools (except for if a call is made by Pindel due to its sensitivity). The variants were filtered based on their read depths (Germline: min read depth = 10; min alternate allele depth = 1, Tumor: min read depth = 10; min alternate allele depth = 3) and VAfs (Germline <1%; Tumor >5%). The variants are then annotated using Oncotator. The dbSNPs which were not found in COSMIC were also excluded from further analysis. TMB was calculated for each sample based on the total number of non-synonymous mutations (including truncating mutations) normalized to the number of bases in the exonic regions per million.
Multiplex imaging
Tissue microarrays were constructed for multiplex immunofluorescence staining. Tissue microarrays slides of 3 ROIs (1 mm diameter punch cores) per samples were generated. Tumor areas rich in stroma were selected to insure profiling of the TME. Normal tissue was obtained from separate paraffin blocks of tissue taken away from the tumor mass. The manual tissue arrayer (Beecher Instruments, Sun Prairie, WI) was used to generate tissue microarray (TMA) blocks.
The immunofluorescence imaging was performed using the Neogenomics platform as previously described (Sood et al., 2016; Xu-Monette et al., 2019). Briefly, formalin-fixed paraffin-embedded tissue arrays were baked at 65°C for 1 h. Slides were deparaffinized with xylene, rehydrated by decreasing ethanol concentration washes, and then processed for antigen retrieval. A two-step antigen retrieval was adopted to allow antibodies with different antigen retrieval conditions to be used together on the same samples (Gerdes et al., 2013). Samples were then blocked against nonspecific binding with 10% (wt/vol) donkey serum and 3% (wt/vol) BSA in phosphate-buffered solution (PBS) for 1 h at room temperature and stained with DAPI for 15 min. Directly conjugated primary antibodies were diluted in PBS supplied with 3% (wt/vol) BSA to optimized concentrations and applied for 1 h at room temperature on a Leica Bond III Stainer. In the case of primary-secondary antibody staining, samples were incubated with primary antibody, followed by incubation with species-specific secondary antibodies conjugated to either Cyanine 3 (cy3) or cyanine 5 (cy5).
Stained images were collected on INCell analyzer 2200 microscope (GE Healthcare Life Sciences) equipped with high-efficiency fluorochrome specific filter sets for DAPI, cy3 and cy5 (Xu-Monette et al., 2019). For multiplexed staining where co-localization was desired, the regions of interest (~0.4–0.6 mm2 tissue area) were imaged, and stage coordinates were saved. The coordinates of each image region were then recalled for each subsequent round after minor readjustment using reference points from the first-round DAPI image and determining the appropriate offset. The exposure times were set at a fixed value for all images of a given marker. For image analyses, microscopy images were exported as full-resolution TIFF images in grayscale for each individual channel collected.
MultiOmyx image analytics
The acquired images from sequential rounds were registered using DAPI images acquired in the first round of staining via a rigid registration algorithm for each region of interest. The parameters of transformation were then applied to the subsequent rounds, which ensured that the pixel coordinates across all the imaging rounds corresponded to the same physical locations on the tissue. Classification and co-expression analysis were performed in multiple stages. First, a nuclear segmentation algorithm was applied on the DAPI image to delineate and identify individual cells. Location information and expression of all the markers were computed for every cell identified. Then, morphologic image analysis and shape detection were performed using proprietary algorithms (Neogenomics Laboratories; https://neogenomics.com/pharma-services/lab-services/multiomyx). These algorithms detect and classify cells as positive or negative for each marker depending on their subcellular localization and morphology. A tissue-quality algorithm was also applied to the images to ensure image artifacts that arose owing to tissue folding or tear did not affect cell classification. Coexpression analysis and phenotype identification were performed by combining individual marker classification results.
αSMA morphology
The αSMA morphology of 3 ROIs per tumor were independently scored by 2 investigators (NKA and TEM) as either organized (value of 1) or disorganized (value of 0). Representative images of these 2 classes of morphology are shown in Figure 5. Each tumor was assigned a predominant αSMA morphology based on the sum score: disorganized ≤ 1; organized ≥2.
QUANTIFICATION AND STATISTICAL ANALYSES
Prism, GraphPad Software (version 9.1.2) and Morpheus online software (https://software.broadinstitute.org/morpheus) were used for statistical analyses. Mean group values ±SEM are reported. N’s are numbers of nodules or normal lung tissue per group. Unpaired student’s T test, Mann-Whitney test for non-parametric data or ANOVA with Tukey’s correction for multiple testing or Sidak’s p value for multiple comparison were used for comparisons, as appropriate. Statistical details of experiments are reported in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 [F7.2.38] | Dako | Catalog# M7254 |
| CD4 [EPR6855] | Abcam | Catalog# ab181724 |
| CD8 [C8/144B] | Dako | Catalog# M7103 |
| CD68 [KP1] | BioLegend | Catalog# 916104 |
| CD103 [EPR4166(2)] | Abcam | Catalog# ab221210 |
| CD209 / DC-SIGN [D7F5C] | Cell Signaling | Catalog# 13193 |
| CTLA-4 [CAL49] | Abcam | Catalog# ab251599 |
| FOXP3 [206D] | BioLegend | Catalog# 320114 |
| Granzyme B [GrB-7] | Dako | Catalog# M7235 |
| HLA-ABC [EMR8-5] | Abcam | Catalog# ab70328 |
| HLA-DR [WR18] | Novus | Catalog# NB100-64358PBSonly |
| Ki67 [SP6] | Abcam | Catalog# ab16667 |
| Pan Cytokeratin [PCK-26] | Sigma-Aldrich | Catalog# C5992 |
| Cytokeratin Pan Type I [AE1] | eBioscience | Catalog# 14-9001-82/cust02300 |
| PD-1 [EPR4877(2)] | Abcam | Catalog# ab186928 |
| PD-L1 [SP142] | Abcam | Catalog# ab236238 |
| SMA [1A4] | Sigma | Catalog# A5228 |
| Biological samples | ||
| 31 Archival (FFPE) human nodules, pure non-solid CT density | This study | N/A |
| 31 archival (FFPE) human nodules, solid CT density | This Study | N/A |
| Critical commercial assays | ||
| Neo Genomics Multiplex Fluorescence Imaging | NeoGenomiucs | N/A |
| Deposited data | ||
| DNA sequence data | dbGaP #phs002818.v1.p1. | accession number phs002818.v1.p1 |
| RNAseq data | GEO #GSE19375 | accession number GSE19375 |
| Software and algorithms | ||
| Prisim 9 | https://www.graphpad.com | https://www.graphpad.com/ |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| CIBERSORT | https://cibersort.stanford.edu/ | N/A |
| DESeq2 | https://bioconductor.org/packages/DESeq2 | N/A |
| Gene Ontology Resource | http://geneontology.org | N/A |
| Morpheus | https://software.broadinstitute.org/morpheus/ | N/A |
| xCell | https://xcell.ucsf.edu | N/A |
Highlights.
Non-solid CT density lung adenocarcinomas are predominantly pre/minimally invasive
Immune-suppressive Tregs are increased in non-solid nodules
ECM genes are increased in non-solid nodules, implicating fibroblasts activation
Increased immune suppression parallels progression to invasion
ACKNOWLEDGMENTS
The project was supported in part by NCI UG3 CA244697 (N.K.A., A.C.B., V.M., O.E., and T.E.M.), the Yoram Cohen family foundation (N.K.A.), the Vicky and Jay Furhman family fund (N.K.A.), and the Weill Cornell Medicine Meyer Cancer Center (N.K.A. and T.E.M.). O.E. is also supported by UL1TR002384, R01CA194547, and LLS SCOR grants 180078–02 and 7021–20. T.E.M. is also supported by DOD LC180227. We thank Gary Koretzky (Cornell University), Niroshana Anandasabapathy (WCM), and Juan Cubillos-Ruiz (WCM) for helpful discussions and critical reading of the manuscript. We thank Murtaza Malbari for expert oversite of the project and Abu Nasar, J. Nathan Mynard, Cathy Spinelli, and Joyce Gakuria for clinical database management. The WCM genomics core facility was used for RNA and DNA sequencing. The multiplex immunofluorescence (IF) was performed by Neogenomics Laboratories, Inc. (Fort Meyers, FL, USA) on a fee-for-service basis.
DECLARATION OF INTERESTS
N.K.A. has equity in Angiocrine Bioscience, TMRW, and View Point Medical. O.E. is supported by Janssen, J&J, Astra-Zeneca, Volastra, and Eli Lilly research grants. He is a scientific advisor and an equity holder in Freenome, Owkin, Volastra Therapeutics, and One Three Biotech and a paid scientific advisor to Champions Oncology. T.E.M. receives research funding from Janssen and from Pfizer, Inc. All other authors declare no competing interests.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110639.
REFERENCES
- Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester B, Milara J, and Cortijo J (2019). Idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int. J. Mol. Sci 20, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guirnalda PD, et al. (2015). Exclusion of T Cells from pancreatic carcinomas in mice is regulated by Ly6-C(low) F4/80(+) extratumoral macrophages. Gastroenterology 140, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhabib I, Zaghdoudi S, Lac C, Bousquet C, and Jean C (2021). Extracellular matrices and cancer-associated fibroblasts: targets for cancer diagnosis and therapy? Cancers (Basel) 13, 3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Khodadoust MS, Liu CL, Newman AM, and Alizadeh AA (2018). Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol 1711, 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Carrot-Zhang J, Zhao Y, Hu H, Freeman SS, Yu S, Ha G, Taylor AM, Berger AC, Westlake L, et al. (2019). Genomic and immune profiling of pre-invasive lung adenocarcinoma. Nat. Commun 10, 5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W, Jo C, Chung WJ, and Kim DJ (2018). Liver cirrhosis and cancer: comparison of mortality. Hepatol. Int 12, 269–276. [DOI] [PubMed] [Google Scholar]
- Dejima H, Hu X, Chen R, Zhang J, Fujimoto J, Parra ER, Haymaker C, Hubert SM, Duose D, Solis LM, et al. (2021). Immune evolution from preneoplasia to invasive lung adenocarcinomas and underlying molecular features. Nat. Commun 12, 2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2019). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 29, 3766. [DOI] [PubMed] [Google Scholar]
- Gene Ontology C (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, et al. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. U S A. 110, 11982–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Del Alcazar CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, Zi X, Kwak M, Bergholtz H, Su Y, et al. (2017). Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov. 7, 1098–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke E, Nandigama R, and Ergun S (2019). Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci 6, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Fujimoto J, Ying L, Fukuoka J, Ashizawa K, Sun W, Reuben A, Chow CW, McGranahan N, Chen R, et al. (2019). Multi-region exome sequencing reveals genomic evolution from preneoplasia to lung adenocarcinoma. Nat. Commun 10, 2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, and Qiao Y (2021). Extracellular matrix and its therapeutic potential for cancer treatment. Signal. Transduct. Target Ther 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumchenko E, Chang X, Brait M, Fertig E, Kagohara LT, Bedi A, Marchionni L, Agrawal N, Ravi R, Jones S, et al. (2015). Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat. Commun 6, 8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JA, and Fearon DT (2015). T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80. [DOI] [PubMed] [Google Scholar]
- Klemm F, and Joyce JA (2015). Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 25, 198–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, and Mitsudomi T (2013). Management of ground-glass opacities: should all pulmonary lesions with ground-glass opacity be surgically resected? Transl. Lung Cancer Res. 2, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysan K, Tran LM, Grimes BS, Fishbein GA, Seki A, Gardner BK, Walser TC, Salehi-Rad R, Yanagawa J, Lee JM, et al. (2019). The immune contexture associates with the genomic landscape in lung adenomatous premalignancy. Cancer Res. 79, 5022–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. (2017). Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascaux C, Angelova M, Vasaturo A, Beane J, Hijazi K, Anthoine G, Buttard B, Rothe F, Willard-Gallo K, Haller A, et al. (2019). Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 571, 570–575. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navab R, Strumpf D, Bandarchi B, Zhu CQ, Pintilie M, Ramnarine VR, Ibrahimov E, Radulovich N, Leung L, Barczyk M, et al. (2011). Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 108, 7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niho S, Yokose T, Suzuki K, Kodama T, Nishiwaki Y, and Mukai K (1999). Monoclonality of atypical adenomatous hyperplasia of the lung. Am. J. Pathol 154, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedros C, Canonigo-Balancio AJ, Kong KF, and Altman A (2017). Requirement of Treg-intrinsic CTLA4/PKCeta signaling pathway for suppressing tumor immunity. JCI Insight 2, e95692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennycuick A, Teixeira VH, AbdulJabbar K, Raza SEA, Lund T, Akarca AU, Rosenthal R, Kalinke L, Chandrasekharan DP, Pipinikas CP, et al. (2020). Immune surveillance in clinical regression of preinvasive squamous cell lung cancer. Cancer Discov. 10, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, et al. (2019). Defining the activated fibroblast population in lung fibrosis using single-cell sequencing. Am. J. Respir. Cell Mol. Biol 61, 74–85. [DOI] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 19, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Zhao S, Zou Y, Rahman SMJ, Senosain MF, Stricker T, Chen H, Powell CA, Borczuk AC, and Massion PP (2020). Genomic underpinnings of tumor behavior in in situ and early lung adenocarcinoma. Am. J. Respir. Crit. Care Med 201, 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero-Fabian S, Arreola R, Becerril-Villanueva E, Torres-Romero JC, Arana-Argaez V, Lara-Riegos J, Ramirez-Camacho MA, and Alvarez-Sanchez ME (2019). Role of matrix metalloproteinases in angiogenesis and cancer. Front. Oncol 9, 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F, and Donnadieu E (2012). Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest 122, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Perez-Gracia JL, Sanchez-Arraez A, Sancho D, and Melero I (2017). Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol 28, xii74. [DOI] [PubMed] [Google Scholar]
- Sood A, Miller AM, Brogi E, Sui Y, Armenia J, McDonough E, Santamaria-Pang A, Carlin S, Stamper A, Campos C, et al. (2016). Multiplexed immunofluorescence delineates proteomic cancer cell states associated with metabolism. JCI Insight 1, e87030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Mezzadra R, and Schumacher TN (2018). Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AL, Teijaro JR, Njau MN, Chandran SS, Azimzadeh A, Nadler SG, Rothstein DM, and Farber DL (2008). CTLA4 expression is an indicator and regulator of steady-state CD4+ FoxP3+ T cell homeostasis. J. Immunol 181, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, et al. (2018). TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543. [DOI] [PubMed] [Google Scholar]
- Teixeira VH, Pipinikas CP, Pennycuick A, Lee-Six H, Chandrasekharan D, Beane J, Morris TJ, Karpathakis A, Feber A, Breeze CE, et al. (2019). Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat. Med 25, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley SJ, Cremasco V, and Astarita JL (2015). Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol 15, 669–682. [DOI] [PubMed] [Google Scholar]
- Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, and Werb Z (2020). Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun 11, 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Monette ZY, Xiao M, Au Q, Padmanabhan R, Xu B, Hoe N, Rodriguez-Perales S, Torres-Ruiz R, Manyam GC, Visco C, et al. (2019). Immune profiling and quantitative analysis decipher the clinical role of immune-checkpoint expression in the tumor immune microenvironment of DLBCL. Cancer Immunol. Res 7, 644–657. [DOI] [PubMed] [Google Scholar]
- Yaghjyan L, Colditz GA, Rosner B, and Tamimi RM (2015). Mammographic breast density and breast cancer risk: interactions of percent density, absolute dense, and non-dense areas with breast cancer risk factors. Breast Cancer Res. Treat 150, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Niu M, Xu L, Luo S, and Wu K (2021). Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida GJ, Azuma A, Miura Y, and Orimo A (2019). Activated fibroblast program orchestrates tumor initiation and progression; molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci 20, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the Lead contact upon request.
The RNAseq data are available at Gene Expression Omnibus (GEO), accession number GSE19375. The DNA seq data are available at the database of Genotypes and Phenotypes (dbGaP), accession number phs002818.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gapprev/gap/cgi-bin/study.cgi?study_id=phs002818.v1.p1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.