

Abstract
Phage, the most prevalent creature on the planet, serves a variety of critical roles. Phage's primary role is to facilitate gene-to-gene communication. The phage proteins can be defined as the virion proteins and the nonvirion ones. Nowadays, experimental identification is a difficult process that necessitates a significant amount of laboratory time and expense. Considering such situation, it is critical to design practical calculating techniques and develop well-performance tools. In this work, the Phage_UniR_LGBM has been proposed to classify the virion proteins. In detailed, such model utilizes the UniRep as the feature and the LightGBM algorithm as the classification model. And then, the training data train the model, and the testing data test the model with the cross-validation. The Phage_UniR_LGBM was compared with the several state-of-the-art features and classification algorithms. The performances of the Phage_UniR_LGBM are 88.51% in Sp,89.89% in Sn, 89.18% in Acc, 0.7873 in MCC, and 0.8925 in F1 score.
1. Introduction
Phage, which can be treated as the most abundant organism, has many important functions on earth [1, 2]. The major function of phage is to promote gene-to-gene communication [3–5]. The second function of phages is to maintain microbial diversity [6]. If the number of a bacterial species increases rapidly in a bacterial population, the corresponding bacteriophage will specifically infect this type of bacteria and kill them, so that the entire bacterial population returns to a balanced state [7, 8]. In addition, phages also participate in the Earth's material cycle [9, 10]. Blue bacteria are a kind of very important bacteria in the ocean, which can absorb carbon dioxide and convert it into glucose through photosynthesis [11]. About half of the blue bacteria will eventually be lysed by its corresponding phage and released to the entire marine environment, providing nutrients for the surrounding biological system [12–14]. Phages are also an important part of the human microbial community. Each human gut contains about 1014 bacteria, while the number of phages is 1015-16, 10 times more than bacteria. The function of phages is far beyond the above-mentioned issues. Nowadays, phage-targeted therapy has become a hot topic. However, phages can survive only by relying on bacterial hosts, and phages are difficult to culture. Therefore, it is particularly important to predict the interaction between phages and bacteria through bioinformatics methods [15–17].
Phage-host interaction is an effective means of studying adaptive evolution of bacteria and plays an important role in human health and disease, which may contribute to new therapeutic agents, such as phage therapy against multidrug-resistant infections [8, 18]. The continuous evolution of pathogenic bacteria and the resistance to new antibiotics may cause as many as 10 million people to lose their lives each year [19–22]. Antibiotics are usually small molecules that inhibit bacterial growth in some way. Because of their abuse and selective pressure on bacterial communities, bacteria have produced many mechanisms of resistance to these molecules over the years, such as their metabolism or excretion that render antibiotics ineffective, and the discovery of new antibiotics has become increasingly difficult.
Because bacteria are closely related to human health and the environment, ecologists and microbiologists have been studying bacterial communities to discover potential laws that are beneficial to humans and the environment. According to research, human microbial communities have been influenced by phages, and some phages will change the composition of microbial communities, resulting in changes in these communities. Phages play a key role in maintaining the microbial community structure of human and environment and provide potential tools for accurately manipulating specific microorganisms. Recent studies have further shown that interactions between phages and microbes can affect mammalian health and disease.
It is worth noted that a great deal of features included amino acid composition [23], atomic composition (ATC) [24], chain-transition distribution (CTD) [25], pseudo amino acid composition [23], and amino acid pair [26] in the sequence level. What is more, several feature selection methods have been taken into account in this field. These approaches mainly focus on giving an increasingly effective and detailed modeling feature as the input of the classification model. On the other hand, the artificial intelligence technologies develop with the rocket speed. The machine learning algorithms, including neural network [27–29], random forest, support vector machine [30, 31], k-nearest neighbor [32], logistic regression [33, 34], and some deep learning models [35–37], have become one of the hottest topics among this field. In this work, we propose the Phage_UniR_LGBM. Such model utilizes the UniRep as the feature and the LightGBM algorithm as the classification model. In detailed, the dataset of phage proteins has been initially divided into 2 parts, including both the training ones and the testing ones. And then, the training data train the model, and the testing data test the model with the cross-validation. In order to demonstrate the performances of this method, the above-mentioned classification models and the features have been compared with the Phage_UniR_LGBM. We demonstrate the outline with these steps of the Phage_UniR_LGBM in Figure 1.
Figure 1.
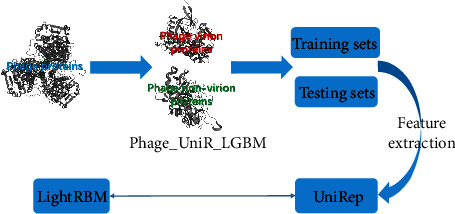
The outline of the Phage_UniR_LGBM.
2. Methods and Materials
2.1. Dataset
In order to classify the phase proteins, the data employed the Ding's effort, which mainly focus on phage virion proteins researches. Such dataset is a reliable dataset, which selected with many filtering schemes [38]. Meanwhile, such dataset can hardly be treated the redundant data. The protein sequences can pairwise with any other one in low homologous. Last but not the least, several state-of-the-art methods have tested the performances with such dataset. Therefore, such dataset can be treated as a typical benchmark dataset in this field. Considering such situation, we employed such dataset as the identified data in this work. The detailed information of the employed data should be demonstrated in Table 1.
Table 1.
The information of phage virion proteins.
| Protein type | Phage virion proteins | Nonphage virion proteins |
|---|---|---|
| Scale | 99 | 208 |
From Table 1, we can easily find that there are two types of these proteins, the 99 sequences of phage virion proteins and the 208 ones of nonphage virion proteins. The whole number of proteins is 307. According to this situation, we can define the phage virion proteins as the positive samples. So the nonphage virion proteins can be treated as the negative ones. With this information, such work can be abstracted as the typical two-type classification issue in the machine learning area. A necessary step should be taken into account. The 90% of both the positive and negative samples should be treated as the training and testing data, and the rest of the 10% of the whole dataset can be treated as the independent data in this work. It was pointed out that the training and testing dataset can hardly overlap with the independent ones. The training and testing dataset utilized the 2, 3, 4, 5, 6, 8, 10-fold cross-validation to demonstrate the stability of the Phage_UniR_LGBM.
2.2. LightGBM Algorithm
The gradient boosting decision tree (GBDT) [39, 40], which has the ability to learn the performances of learners, is continuously improving with several computational iterations. During the iteration of such special algorithm, several parameters of this algorithm should be listed. The current iteration of model achievement can be defined as the Fc (x). In detail, the c means the current iteration. With a similar theory, the Fc−n (x) means the last n iterations' model achievement, and the Fc+n (x) means the next n iterations' model ones. What is more, the loss function of the current iteration can be defined as the Loss (y, Fc (x)). Such algorithm can focus on searching and dropping out the weak learner hc (x) with the minimization of the loss function in the current round. And then, the loss function's negative gradient can be calculated on the current iteration's loss function. It was pointed out that the square difference plays a significant role during this algorithm. So, such parameter can be measured with the method of the fitting hc (x) in equation (1), and the loss function can be evaluated by equation (2).
| (1) |
| (2) |
In the final step, the most potential learner can be selected with the method, shown in equation (3), in the current iteration.
| (3) |
When it comes to the Light Gradient Boosting Machine, such algorithm, can be abbreviated LightGBM [41, 42], is a special type of the above-mentioned algorithm. In detail, the GBDT algorithm mainly relies on the gradient one-side sampling and exclusive feature bundling. Such two characteristics can be treated as the main contributions of effective and high-performance. However, the shortcomings can hardly be neglected that the GBDT may speed huge computation resources during the algorithm operation. So as to overcome such deficiency, the LightGBM algorithm, which has the ability to achieve the same accuracy with the 5% time-consuming, is proposed. The majority of LightGBM algorithm may follow the next four steps. Initially, the input data can be transformed with the histogram form. After such transformation, a histogram can be constructed with the same size of input integers. The constructed histogram has the ability to capture the optimal cutting point. With such approach, several unnecessary calculations can hardly be operated. So, the computation resources may save to some degree. The second step focuses on constructing a histogram's leaf nodes. With the method of a histogram for subtraction, the computational time can cut half of the traditional method. The next step utilizes the leaf-wise growth method, which is limited to the depth of tree construction. With this approach, the performances may be further accelerated than the GBDT ones. The final step of the LightGBM should not be neglected. The parallel computation can further enhance the speed without losing accuracy.
2.3. UniRep Feature
The features of protein description can be utilized by the UniRep [43, 44], which is a novel approach to demonstrate the protein information at various levels. With the further researches, it can be found that the amino-acid embedding approaches are learned by the UniRep. The UniRep method contains several properties in the protein level. For instance, the UniRep selected physicochemical feature with the method of amino acid residues' cluster. In detail, the whole 20 types of amino acid residues own their properties, including hydrophobic aliphatic, charged basic, charged acidic, polar neutral, unique, and hydrophobic aromatic. Such feature sets also separated protein from a huge number of structural classifications of proteins. These types can be classified with the crystallographic information. In order to evaluate the feature of identified proteins, we employ the whole protein peptide sequences from the identified phage virion proteins. So, the protein sequence can be transformed into a numeric vector. And then, we apply the LightGBM algorithm to identify the phage virion proteins numeric vectors and the nonphage ones. The input of the LightGBM is the identified numeric vector from each sample, and the output of the LightGBM is the calculated results from these samples.
2.4. Measurements of Performance
In this classification problem, samples can be defined as two types, including the phage virion proteins sequences and the nonphage virion proteins sequences. Defined positive samples mean the phage virion proteins sequences. On the contrary, the defined negative samples mean the nonphage virion proteins sequences. According to the definition of the classified samples, they can cause the four results in a common situation. We can easily obtain these formulations, including sensitivity, specificity, accuracy, F1 scores, and MCC. And the detailed information is shown in the following equations:
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
where P is the scale of positive samples and N is the scale of negative ones. T is a set of the true predicted result, and F is a set of the false predicted result.
When it comes to the F1 score, such performance can be treated as an index utilized to evaluate the positive and negative samples' distribution in the field of the two-type issue. Such performance should take into account several parameters, including the four basic parameters, which are TP, FP, TN, and FN. Such performance can be treated as a harmonic average of model accuracy and recall.
Another important performance is the MCC, which is abbreviated by Matthews correlation coefficient. Such performance's value ranges from -1 to 1. It means the relationship between the outputs and computational results. Considering the true results, false-positive ones, and true-negative ones, the MCC has the ability to demonstrate the balance of the above-mentioned three parameters. The area under receiver operating characteristic, which can be shorted as the AUC, is a significant evaluation metric. Such performance shows the relationship between the label and computational result in each sample, respectively.
3. Results and Discussions
To understand the classification issue of the phage virion proteins sequences and the nonphage virion proteins sequences, we define the label of the phage virion protein as 1 and the label of nonphage virion protein as 0. In other words, a phage virion one is treated as a positive sample and a nonphage virion one is treated as a negative sample. Therefore, the UniRep features of each protein sequence sample as the input of the LightGBM model and the output of each sample should be compared with their own label, respectively. In order to demonstrate the stability and reliability, we utilize the 2-, 3-, 4-, 5-, 6-, 8-, and 10-fold cross-validation. After this operation, we utilize the constructed model to test the performance in the independence dataset. So, in the following part, we demonstrate the detailed processions of the Phage_UniR_LGBM.
3.1. Performances of Different Classification Algorithms
The above-mentioned seven cross-validation test ways are utilized to validate the stability and reliability of the Phage_UniR_LGBM. And then, five state-of-the-art classification algorithms, including k-nearest neighbors, logistic regression, Gauss naive Bayes, support vector machine, and random forest, are utilized to classify the phage virion proteins and the nonphage virion proteins with the UniRep features. In order to show the stability and generality of the model, we employed the 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 8-fold and 10-fold cross validation methods.Table S1-S6 show the detailed information of Sn, Sp, Acc, MCC, and F1 scores of the LightGBM model and other state-of-the-art machine learning algorithms using various cross-validation methods. During the 5 cross-validation, we can easily find that the logistic regression algorithm can get the well performances in the Sn and the employed LightGBM model can get the well performances in the Sp. Meanwhile, the logistic regression can hardly work well in Sp. Therefore, it may cause such models own the low-performances in other three measurements, including accuracy, MCC, and F1 score. During the 2, 3, 4, 5, 6, and 8 cross-validation, the k-nearest neighbors model replaces the logistic regression model in Sp. It is noted that the LightGBM model's Sn is better than other compared algorithms. So, the LGBM can get the available results in the key classification performances, including Acc, MCC, and F1 score. From Figures 2–8 and Table S1-S7, we can get the conclusion that the LightGBM model can get the available effectiveness and stability during the cross-validation. On the other hand, the support vector machine and random forest model demonstrate their advantages during six parameters cross-validations.
Figure 2.
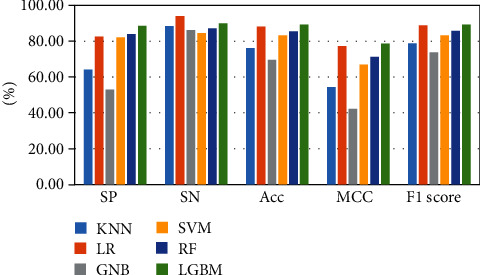
Seven classification algorithms comparisons in 2-fold validation.
Figure 3.
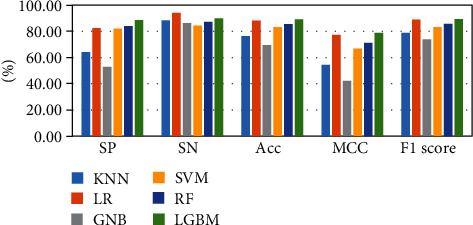
Seven classification algorithms comparisons in 3-fold validation.
Figure 4.
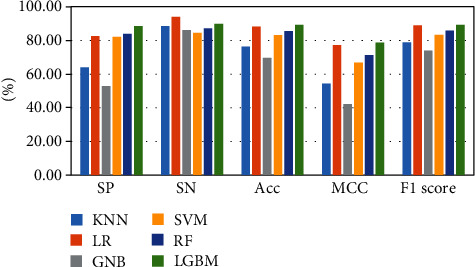
Seven classification algorithms comparisons in 4-fold validation.
Figure 5.
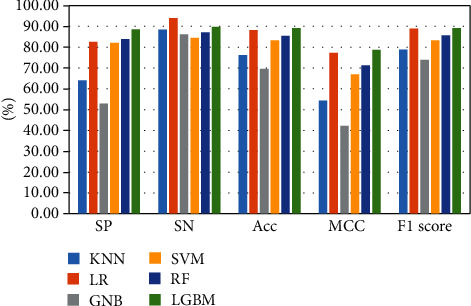
Seven classification algorithms comparisons in 5-fold validation.
Figure 6.
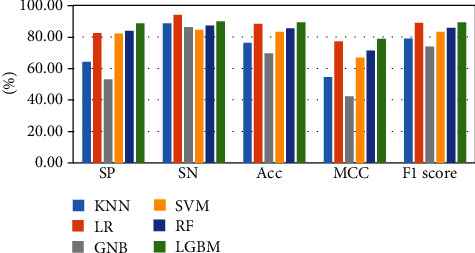
Seven classification algorithms comparisons in 6-fold validation.
Figure 7.
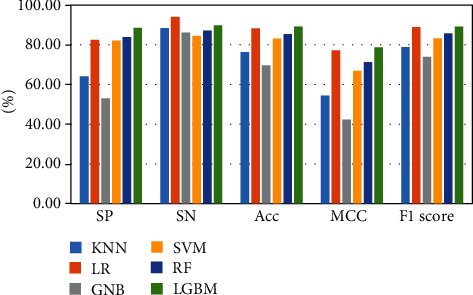
Seven classification algorithms comparisons in 8-fold validation.
Figure 8.
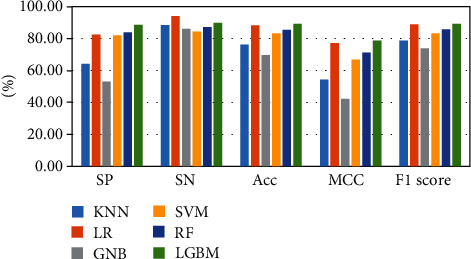
Seven classification algorithms comparisons in 10-fold validation.
In order to demonstrate the performances of this imbalance classification issue, the AUC has been employed to evaluate each classification algorithm in this work. With the 2-, 3-, 4-, 5-, 6-, 8-, and 10-fold cross-validations, we find that the employed 7 cross-validations follow a similar trend in this work. So, we evaluate the AUCs of each classification algorithm, including KNN, LR, GNB, SVM, RF, and LGBM. From these results, it could be seen that LGBM has the best AUC values among seven single classifiers. Figure 9 shows the AUC values of each algorithm in 10-fold cross-validation and the detailed values in Table S8.
Figure 9.
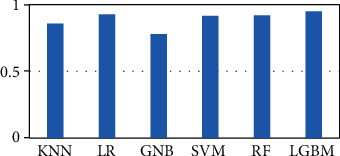
The AUCs of seven algorithms in 10-fold validation.
3.2. Performances of Different Features
In this work, the UniRep features compare with several state-of-the-art features, which include amino acid composition (AAC), atomic composition (ATC), chain-transition distribution (CTD), pseudo amino acid composition (PseAAC), and amino acid pair (AAP), in the protein sequence level. To compare the performances of each feature, we utilize the above-mentioned five machine learning algorithms and the LightGBM model to test these features, respectively. With the cross-validation test, we select the 10-fold one to demonstrate their performances. And the performances of different features in these classification algorithms are shown in Tables 2– 7.
Table 2.
The performances of different features in KNN model.
| SP | SN | Acc | MCC | F1 score | |
|---|---|---|---|---|---|
| AAC | 45.50% | 82.27% | 63.89% | 0.2986 | 0.6949 |
| ATC | 55.76% | 75.19% | 65.47% | 0.3155 | 0.6853 |
| CTD | 63.45% | 78.73% | 71.09% | 0.4268 | 0.7314 |
| PseAAC | 66.65% | 89.51% | 73.58% | 0.4761 | 0.7529 |
| AAP | 62.81% | 84.04% | 73.42% | 0.4794 | 0.7597 |
| UniRep | 64.09% | 88.46% | 76.26% | 0.5440 | 0.7885 |
Table 7.
The performances of different features in LGBM model.
| SP | SN | Acc | MCC | F1 score | |
|---|---|---|---|---|---|
| AAC | 16.75% | 48.54% | 32.64% | -0.3661 | 0.4188 |
| ATC | 50.25% | 93.49% | 71.87% | 0.4850 | 0.7687 |
| CTD | 48.48% | 43.15% | 45.81% | -0.0838 | 0.4433 |
| PseAAC | 84.62% | 85.65% | 85.14% | 0.7027 | 0.8506 |
| AAP | 60.82% | 71.91% | 66.37% | 0.3294 | 0.6813 |
| UniRep | 88.51% | 89.89% | 89.18% | 0.7873 | 0.8925 |
When it comes to evaluating the performances of each feature, the five employed performances should be compared, respectively. Nevertheless, the performances, such as the Acc, MCC, and F1 score, could be computed by the two basic performances, including the Sp and Sn. In order to more easily compare the performances of each feature, we initially compared the Sp and Sn for each one. From the performances of each feature in kNN model, we can easily find that the PseAAC can achieve the 66.65% in Sp and such performances are higher than the other five features. Meanwhile, such feature also gets the 89.51% in Sn, and such performance is better than other ones. It was noted that the features, including AAC, AAP, and the UniRep feature, can get the Sn more than 80% and the CTD, AAP, and UniRep can get the Sp more than 60% with the method of kNN classification model. Considering such phenomenon in KNN model, we propose a threshold, which is named well performance. The well performance means the evaluated performance can be higher than 70%. If the evaluated performance is 70.5%, it can be defined as the well performance. If the evaluated one is 69.99%, it can hardly be defined as the well performance. Therefore, the topic well performances are PseAAC, UniRep, and CTD in Sp, and the topic three well performances are AAP, UniRep, and CTD in sn with the LR classification algorithm. In the GNB model, the top three well performances are PseAAC, UniRep, and ATC in Sp and UniRep, PseAAC, and AAP in sn. In the SVM model, the well-performances are UniRep and PseAAC in Sp and PseAAC, UniRep, CTD, and AAP in Sn. In the RF model, the well performances are ATC, CTD, UniRep, and PseAAC in Sp and PseAAC, UniRep, AAP, and CTD in Sn. In the employed model, the well performances are UniRep and PseAAC in Sp and ATC, UniRep, PseAAC, and AAP in Sn.
After the above features comparison, we can find that the UniR_LGBM method can get the available performances than other ones. It can hardly be neglected that the PseAAC work well in several classification models.
4. Conclusions
In this work, a novel model, which is named Phage_UniR_LGBM, was proposed to deal with the phage virion proteins classification issue. This classification can be treated as a typical imbalance binary classification issue in the field of machine learning. In order to utilize the effective feature of the protein sequence, we employed the UniRep feature to quantitate the identified phage virion protein sequences. And then, the LightGBM algorithm was employed to evaluate the protein numeric vectors with the UniRep processing. In order to demonstrate the Phage_UniR_LGBM's stability and robustness, the 2, 3, 4, 5, 6, 8, and 10 cross-validation methods have been utilized in this work. Moreover, several typical machine learning algorithms include k-nearest neighbors, logistic regression, Gauss naive Bayes, support vector machine, and random forest. And then, the UniRep features were compared with several state-of-the-art features, which include amino acid composition (AAC), atomic composition (ATC), chain-transition distribution (CTD), pseudo amino acid composition (PseAAC), and amino acid pair (AAP), in the protein sequence level. From these comparisons, we find that the Phage_UniR_LGBM can be treated as an effective model to classify the phage virion protein.
From the Phage_UniR_LGBM, we find some interesting points in this classification issue. The scale of identified samples can hardly meet the need of deep learning algorithms. So, how to utilize the deep learning tools to deal with this issue? Meanwhile, the scale of negative samples and the scale of positive ones do not follow the 1 : 1 ratio. So, which strategy can be employed to deal with the typical imbalance binary classification issue in the machine learning area? The current efforts of protein sequences feature focus on the whole sequence of protein. Meanwhile, the reduction of useless feature information should be taken into account in this work. Considering such situations, we will focus on effectively solving these problems in the future efforts.
Table 3.
The performances of different features in LR model.
| SP | SN | Acc | MCC | F1 score | |
|---|---|---|---|---|---|
| AAC | 60.23% | 78.99% | 69.61% | 0.3993 | 0.7221 |
| ATC | 51.16% | 76.16% | 63.66% | 0.2822 | 0.6770 |
| CTD | 66.83% | 88.39% | 77.61% | 0.5655 | 0.7979 |
| PseAAC | 89.11% | 82.11% | 85.61% | 0.7140 | 0.8610 |
| AAP | 56.93% | 95.91% | 76.42% | 0.5738 | 0.8027 |
| UniRep | 82.51% | 94.03% | 88.23% | 0.7721 | 0.8893 |
Table 4.
The performances of different features in GNB model.
| SP | SN | Acc | MCC | F1 score | |
|---|---|---|---|---|---|
| AAC | 31.26% | 69.80% | 50.53% | 0.0115 | 0.5852 |
| ATC | 48.22% | 39.64% | 43.93% | -0.1219 | 0.4142 |
| CTD | 41.33% | 78.41% | 59.87% | 0.2126 | 0.6615 |
| PseAAC | 69.95% | 80.89% | 75.42% | 0.5115 | 0.7400 |
| AAP | 47.16% | 80.14% | 63.65% | 0.2892 | 0.6879 |
| UniRep | 52.99% | 86.17% | 69.56% | 0.4225 | 0.7390 |
Table 5.
The performances of different features in SVM model.
| SP | SN | Acc | MCC | F1 score | |
|---|---|---|---|---|---|
| AAC | 26.26% | 54.04% | 40.15% | -0.2051 | 0.4745 |
| ATC | 23.80% | 40.53% | 32.16% | -0.3618 | 0.3740 |
| CTD | 32.82% | 79.37% | 56.10% | 0.1378 | 0.6439 |
| PseAAC | 80.42% | 84.82% | 82.62% | 0.6530 | 0.8223 |
| AAP | 49.24% | 63.33% | 56.28% | 0.1269 | 0.5916 |
| UniRep | 82.06% | 84.44% | 83.22% | 0.6690 | 0.8327 |
Table 6.
The performances of different features in RF model.
| SP | SN | Acc | MCC | F1 score | |
|---|---|---|---|---|---|
| AAC | 76.32% | 59.27% | 67.80% | 0.3612 | 0.6479 |
| ATC | 91.48% | 67.11% | 79.30% | 0.6042 | 0.7643 |
| CTD | 84.71% | 71.47% | 78.09% | 0.5668 | 0.7654 |
| PseAAC | 78.84% | 88.49% | 83.67% | 0.6765 | 0.8284 |
| AAP | 66.59% | 72.34% | 69.47% | 0.3900 | 0.7032 |
| UniRep | 83.87% | 87.16% | 85.48% | 0.7121 | 0.8572 |
Acknowledgments
Wenzheng Bao and Qingyu Cui have the same role of first authors. Baitong Chen and Bin Yang have the same role of corresponding authors. This work was supported by the Natural Science Foundation of China (No. 61902337), Fundamental Research Funds for the Central Universities, 2020QN89, Xuzhou Science and Technology Plan Project KC21047 and KC19142, Shandong Provincial Natural Science Foundation, China (No. ZR2015PF007), the PhD Research Startup Foundation of Zaozhuang University (No.2014BS13), Zaozhuang University Foundation (No. 2015YY02), Jiangsu Provincial Natural Science Foundation (No. SBK2019040953), Natural Science Fund for Colleges and Universities in Jiangsu Province (No. 19KJB520016), and Young Talents of Science and Technology in Jiangsu.
Data Availability
The data used to support the findings of this study are available within the manuscript and the supplementary files.
Conflicts of Interest
The authors declare that they have no competing interests.
Authors' Contributions
Wenzheng Bao and Qingyu Cui, who designed the experiments of this work, contributed equally to this work. The two authors are treated as the co-first authors. Baitong Chen and Bin Yang constructed the classification model and edited the manuscript of this work. The two authors are treated as the co-corresponding author.
Supplementary Materials
The file of Supplemental Data is the computational results of this classification issue. The sna means the sensitivity, the spa means the specificity, the acc1 means the accuracy, the mcca1 means the Matthews correlation coefficient, and the f1 means the f1 score in the classification issue. The other parameters are the procession parameters of this work.
References
- 1.Ladinsky M. S., Mastronarde D. N., Mcintosh J. R., Howell K. E., Staehelin L. A. Golgi structure in three dimensions: functional insights from the normal rat kidney cell. The Journal of Cell Biology . 1999;144(6):1135–1149. doi: 10.1083/jcb.144.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Su L. J., Auluck P. K., Outeiro T. F., et al. Compounds from an unbiased chemical screen reverse both Er-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Disease Models & Mechanisms . 2010;3(3-4):194–208. doi: 10.1242/dmm.004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alipanahi B., Delong A., Weirauch M. T., Frey B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nature Biotechnology . 2015;33(8):831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 4.Salekin S., Zhang J., Huang Y. Base-pair resolution detection of transcription factor binding site by deep deconvolutional network. Bioinformatics . 2018;34(20):3446–3453. doi: 10.1093/bioinformatics/bty383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habeler M., Redl B. Phage-display reveals interaction of lipocalin allergen Can f 1 with a peptide resembling the antigen binding region of a human γδT-cell receptor. Biological Chemistry . 2021;402(4) doi: 10.1515/hsz-2020-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durmaz E., Higgins D. L., Klaenhammer T. R. Molecular characterization of a second abortive phage resistance gene present in Lactococcus lactis subsp. lactis ME2. Journal of Bacteriology . 2019;174(22):7463–7469. doi: 10.1128/jb.174.22.7463-7469.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandenburger A., Godson G. N., Radman M., Glickman B. W., Sluis C. A. Radiation-induced base substitution mutagenesis in single-stranded DNA phage M13. Nature . 1981;294(5837):180–182. doi: 10.1038/294180a0. [DOI] [PubMed] [Google Scholar]
- 8.Thomas D. Identification of phage SP01 proteins coded by regulatory genes 33 and 34. Nature . 1976;262(5571):748–753. doi: 10.1038/262748a0. [DOI] [PubMed] [Google Scholar]
- 9.Herskovits A. A. A Metzincin and TIMP-like protein pair of a phage origin sensitize Listeria monocytogenes to phage lysins and other cell wall targeting agents. Microorganisms . 2021;9(6):p. 1323. doi: 10.3390/microorganisms9061323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin D. M., Koskella B., Lin H. C., Gastroenterology S. O., System N., Biology D. Phage therapy: an alternative to antibiotics in the age of multi-drug resistance. World Journal of Gastrointestinal Pharmacology and Therapeutics . 2017;8(3):p. 162. doi: 10.4292/wjgpt.v8.i3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roach D. R., Leung C. Y., Henry M., Morello E., Debarbieux L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host & Microbe . 2017;22(1):38–47.e4. doi: 10.1016/j.chom.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 12.Orekhova M., Koreshova A., Artamonova T., Khodorkovskii M., Yakunina M. The study of the phiKZ phage non-canonical non-virion RNA polymerase. Biochemical and Biophysical Research Communications . 2019;511(4):759–764. doi: 10.1016/j.bbrc.2019.02.132. [DOI] [PubMed] [Google Scholar]
- 13.Wei C. Recent advances of computational methods for identifying bacteriophage virion proteins. Protein and Peptide Letters . 2020;27(4):259–264. doi: 10.2174/0929866526666190410124642. [DOI] [PubMed] [Google Scholar]
- 14.Koonin E. V., Yutin N. The crAss-like phage group: how metagenomics reshaped the human virome. Trends in Microbiology . 2020;28(5):349–359. doi: 10.1016/j.tim.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Clarkson S. G., Smith H. O., Walter S., Gross K. W., Birnstiel M. L. Integration of eukaryotic genes for 5S RNA and histone proteins into a phage lambda receptor. Nucleic Acids Research . 1976;3(10):2617–2632. doi: 10.1093/nar/3.10.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pereboeva L. A., Pereboev A. V., Morris G. E. Identification of antigenic sites on three hepatitis C virus proteins using phage-displayed peptide libraries. Journal of Medical Virology . 1998;56(2):105–111. doi: 10.1002/(SICI)1096-9071(199810)56:2<105::AID-JMV1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 17.Drulis-Kawa Z., Majkowska-Skrobek G., Maciejewska B. Bacteriophages and phage-derived proteins – application approaches. Current Medicinal Chemistry . 2015;22(14):1757–1773. doi: 10.2174/0929867322666150209152851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodríguez-Rubio L., Gutiérrez D., Donovan D. M., Martínez B., Rodríguez A., García P. Phage lytic proteins: biotechnological applications beyond clinical antimicrobials. Critical Reviews in Biotechnology . 2016:1–11. doi: 10.3109/07388551.2014.993587. [DOI] [PubMed] [Google Scholar]
- 19.Seguritan V., Alves N., Arnoult M., Raymond A., Segall A. M. Artificial neural networks trained to detect viral and phage structural proteins. PLoS Computational Biology . 2012;8(8, article e1002657) doi: 10.1371/journal.pcbi.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chibli H., Ghali H., Park S., Peter Y. A., Nadeau J. L. Immobilized phage proteins for specific detection of staphylococci. Analyst . 2014;139(1):179–186. doi: 10.1039/C3AN01608K. [DOI] [PubMed] [Google Scholar]
- 21.Gross A. L., Gillespie J. W., Petrenko V. A. Promiscuous tumor targeting phage proteins. Protein Engineering Design and Selection . 2016;29(3):93–103. doi: 10.1093/protein/gzv064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutiérrez D., Fernández L., Rodríguez A., García P., Gilmore M. S. Are phage lytic proteins the secret weapon to kill Staphylococcus aureus? MBio . 2018;9(1) doi: 10.1128/mBio.01923-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou K.-C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Structure Function & Bioinformatics . 2001;43(3):246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 24.Rao H. B., Zhu F., Yang G. B., Li Z. R., Chen Y. Z. Update of PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Research . 2011;39(suppl_2):W385–W390. doi: 10.1093/nar/gkr284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balachandran M., Shin T. H., Kim M. O., Gwang L. AIPpred: sequence-based prediction of anti-inflammatory peptides using random forest. Frontiers in Pharmacology . 2018;9 doi: 10.3389/fphar.2018.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habib T., Zhang C., Yang J. Y., Deng Y. Y. Supervised learning method for the prediction of subcellular localization of proteins using amino acid and amino acid pair composition. BMC Genomics . 2008;9(S1) doi: 10.1186/1471-2164-9-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan L., Zhu L., Guo W. L., et al. Nonconvex penalty based low-rank representation and sparse regression for eQTL mapping. IEEE/ACM Transactions on Computational Biology & Bioinformatics . 2017;14(5):1154–1164. doi: 10.1109/TCBB.2016.2609420. [DOI] [PubMed] [Google Scholar]
- 28.Ji Z., Wu D., Zhao W., et al. Systemic modeling myeloma-osteoclast interactions under normoxic/hypoxic condition using a novel computational approach. Scientific Reports . 2015;5(1):p. 13291. doi: 10.1038/srep13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bao W., Wang D., Chen Y. Classification of protein structure classes on flexible neutral tree. IEEE/ACM Transactions on Computational Biology and Bioinformatics . 2017;14(5):1122–1133. doi: 10.1109/TCBB.2016.2610967. [DOI] [PubMed] [Google Scholar]
- 30.Yuan L., Guo L. H., Yuan C. A., Zhang Y. H., Huang D. S. Integration of multi-omics data for gene regulatory network inference and application to breast cancer. IEEE/ACM Transactions on Computational Biology and Bioinformatics . 2019;16 doi: 10.1109/tcbb.2018.2866836. [DOI] [PubMed] [Google Scholar]
- 31.Yuan L., Zhao J., Sun T., Shen Z. A machine learning framework that integrates multi-omics data predicts cancer-related LncRNAs. BMC Bioinformatics . 2021;22(1):p. 332. doi: 10.1186/s12859-021-04256-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bao W., Yuan C.-A., Zhang Y., et al. Mutli-features prediction of protein translational modification sites. IEEE/ACM Transactions on Computational Biology & Bioinformatics . 2018;15 doi: 10.1109/tcbb.2017.2752703. [DOI] [PubMed] [Google Scholar]
- 33.Bao W., Yang B., Chen B. 2-hydr_Ensemble: lysine 2-hydroxyisobutyrylation identification with ensemble method. Chemometrics and Intelligent Laboratory Systems . 2021;215:p. 104351. doi: 10.1016/j.chemolab.2021.104351. [DOI] [Google Scholar]
- 34.Bao W., Yang B., Li D., Li Z., Zhou Y., Bao R. CMSENN: computational modification sites with ensemble neural network. Chemometrics and Intelligent Laboratory Systems . 2019;185:65–72. doi: 10.1016/j.chemolab.2018.12.009. [DOI] [Google Scholar]
- 35.Hu H., Qiu G., Chen S., Ji Z., Lin Y. Detection and recognition for life state of cell cancer using two-stage cascade CNNs. IEEE/ACM Transactions on Computational Biology and Bioinformatics . 2017;17 doi: 10.1109/tcbb.2017.2780842. [DOI] [PubMed] [Google Scholar]
- 36.Ji Z., Jing S., Dan W., Peng H., Zhao W. Predicting the impact of combined therapies on myeloma cell growth using a hybrid multi-scale agent-based model. Oncotarget . 2017;8 doi: 10.18632/oncotarget.13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang B., Bao W., Wang J. Active disease-related compound identification based on capsule network. Briefings in Bioinformatics . 2022;23(1) doi: 10.1093/bib/bbab462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng C., Zhang J., Ye X., Guo F., Zou Q. Review and comparative analysis of machine learning-based phage virion protein identification methods. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics . 2020;1868(6, article 140406) doi: 10.1016/j.bbapap.2020.140406. [DOI] [PubMed] [Google Scholar]
- 39.Zhou C., Yu H., Ding Y., Guo F., Gong X. J., Liu B. Multi-scale encoding of amino acid sequences for predicting protein interactions using gradient boosting decision tree. PLoS One . 2017;12(8, article e0181426) doi: 10.1371/journal.pone.0181426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zulfiqar H., Yuan S. S., Huang Q. L., Sun Z. J., Lin H. Identification of cyclin protein using gradient boost decision tree algorithm. Computational and Structural Biotechnology Journal . 2021;19:4123–4131. doi: 10.1016/j.csbj.2021.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D., Yang Z., Yi Z. LightGBM: an effective miRNA classification method in breast cancer patients. Proceedings of the 2017 International Conference on Computational Biology and Bioinformatics - ICCBB 2017; 2017; [DOI] [Google Scholar]
- 42.Zhao Y., Khushi M. Wavelet Denoised-ResNet CNN and LightGBM Method to Predict Forex Rate of Change . 2021. Papers.
- 43.Jan M. UNIREP: a microcomputer program to find unique and repetitive nucleotide sequences in genomes. Computer Applications in the Biosciences . 1993;9(3):355–360. doi: 10.1093/bioinformatics/9.3.355. [DOI] [PubMed] [Google Scholar]
- 44.Mrázek J., Kypr J. UNIREP: a microcomputer program to find unique and repetitive nucleotide sequences in genomes. Bioinformatics . 1993;9(3):355–360. doi: 10.1093/bioinformatics/9.3.355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The file of Supplemental Data is the computational results of this classification issue. The sna means the sensitivity, the spa means the specificity, the acc1 means the accuracy, the mcca1 means the Matthews correlation coefficient, and the f1 means the f1 score in the classification issue. The other parameters are the procession parameters of this work.
Data Availability Statement
The data used to support the findings of this study are available within the manuscript and the supplementary files.