

Abstract
MHC‐related protein 1 (MR1) presents microbial riboflavin metabolites to mucosal‐associated invariant T (MAIT) cells for surveillance of microbial presence. MAIT cells express a semi‐invariant T‐cell receptor (TCR), which recognizes MR1–antigen complexes in a pattern‐recognition‐like manner. Recently, diverse populations of MR1‐restricted T cells have been described that exhibit broad recognition of tumor cells and appear to recognize MR1 in association with tumor‐derived self‐antigens, though the identity of these antigens remains unclear. Here, we have used TCR gene transfer and engineered MR1‐expressing antigen‐presenting cells to probe the MR1 restriction and antigen reactivity of a range of MR1‐restricted TCRs, including model tumor‐reactive TCRs. We confirm MR1 reactivity by these TCRs, show differential dependence on lysine at position 43 of MR1 (K43) and demonstrate competitive inhibition by the MR1 ligand 6‐formylpterin. TCR‐expressing reporter lines, however, failed to recapitulate the robust tumor specificity previously reported, suggesting an importance of accessory molecules for MR1‐dependent tumor reactivity. Finally, MR1‐mutant cell lines showed that distinct residues on the α1/α2 helices were required for TCR binding by different MR1‐restricted T cells and suggested central but distinct docking modes by the broad family of MR1‐restricted αβ TCRs. Collectively, these data are consistent with recognition of distinct antigens by diverse MR1‐restricted T cells.
Keywords: Antigen processing and presentation, MAIT, MR1, T‐cell receptor
The study provides evidence validating the self‐reactivity of tumor‐reactive MHC‐related protein 1 (MR1)‐restricted T cells. Furthermore, we show that these T‐cell receptors exhibit differential antigen recognition and utilize diverse docking modes to recognize MR1–antigen complexes.

INTRODUCTION
Major histocompatibility complex (MHC) molecules are highly polymorphic and present peptide antigens to T cells. Beyond classical MHC molecules, there exists a series of monomorphic MHC class I‐like molecules that present conserved, nonpeptidic antigens for T‐cell surveillance. 1 One such molecule, MHC‐related protein 1 (MR1), captures small metabolites derived from microbial riboflavin biosynthesis. 2 Here, the riboflavin intermediate 5‐amino‐6‐D‐ribitylaminouracil (5‐A‐RU), the enzymatic formation of which is conserved across microbial species, 3 acts as a precursor for a series of aromatic compounds that bind MR1. 4 , 5 These MR1–antigen complexes are in turn recognized by a specialized T‐cell subset called mucosal‐associated invariant T (MAIT) cells, 4 which orchestrate an antimicrobial immune response. 5 , 6 Central to MAIT cell MR1–antigen reactivity is the semi‐invariant MAIT T‐cell receptor (TCR), which consists of a restricted TCR‐α chain, encoded by TRAV1‐2 rearranged with either TRAJ12, TRAJ20 or TRAJ33 to form a fixed complementarity‐determining region 3α (CDR3α) loop, 7 , 8 , 9 combined with a TCR‐β chain enriched for TRBV20‐1 and TRBV6 family genes. 8 This forms a TCR capable of docking MR1 and probing the antigen‐binding cleft for the presence of the ribityl moiety of the antigen. 10 , 11 , 12 Accordingly, the key function of the MR1–MAIT TCR axis is to monitor microbial infection via the riboflavin synthesis pathway, in a manner akin to a pattern‐recognition receptor–ligand interaction.
That the MAIT TCR and MR1 genes are highly conserved across mammalian evolution suggests a strong selective pressure to maintain this immunological process at the molecular level, centered around presentation of 5‐A‐RU‐based antigens. 8 , 13 , 14 The MR1‐binding cleft can nonetheless accommodate diversity in these ribityl compounds, 2 , 10 , 12 , 15 , 16 the most potent of which are the pyrimidine neoantigens, such as 5‐(2‐oxopropylideneamino)‐6‐D‐ribitylaminouracil (5‐OP‐RU). 12 Central to the potency of these pyrimidines is their formation of a covalent bond with a lysine at position 43 of MR1 (K43). 12 Generation of this so‐called Schiff base occurs in the endoplasmic reticulum, anchors the antigen to the MR1‐binding cleft and triggers egression of MR1 to the cell surface. 17 , 18 The MR1‐binding cleft can also accommodate a range of nonribityl compounds, for example, folate‐derivative 6‐formylpterin (6‐FP) 2 and its synthetic analog acetyl (Ac)‐6‐FP, 19 among others. 16 , 20 , 21 The antigenicity and physiological roles of these nonribityl compounds remain unclear. While all MAIT TCRs recognize the pyrimidine 5‐OP‐RU, select clones appear capable of differentiating between other ribityl or nonribityl antigens in vitro in a CDR3β‐dependent manner. 16 , 21 , 22 Whether this extends to polyclonal primary MAIT cells in vivo, however, is unknown.
Beyond TRAV1‐2+ MAIT cells, there also exists populations of TRAV1‐2− T cells that exhibit MR1 reactivity. 23 In the first description of these cells, MR1–antigen tetramers furnishing archetypal ligands 5‐OP‐RU, 6‐FP and Ac‐6‐FP were used to isolate diverse TRAV1‐2− populations that recognize ribityl and/or nonribityl antigens. 22 Another study isolated TRAV1‐2− MR1‐reactive cells that recognized microbially infected antigen‐presenting cells (APCs). 24 This included an MR1‐5‐OP‐RU‐reactive clone that also reacted to APCs infected with Streptococcus pyogenes, a species lacking riboflavin synthesis enzymes, implying recognition of a nonribityl microbial antigen. A population of TRAV1‐2−, MR1‐5‐OP‐RU‐reactive cells that maintains the innate‐like features of MAIT cells has also been described, 25 and this subset is enriched for a public, invariant TRAV36+/TRBV28+ TCR. More recently, a subset of γδ T cells that recognizes MR1 in antigen‐dependent and antigen‐independent manners was reported, extending MR1‐mediated immunity beyond the αβ repertoire. 26 In contrast to the pattern‐recognition receptor‐like recognition of MR1 by the MAIT TCR, structural studies have revealed that TRAV1‐2− TCRs employ diverse docking modes to mediate MR1 recognition, 22 , 26 , 27 offering a potential mechanism for detection of antigenic variation.
The physiological role of the broader MR1‐restricted T‐cell family, however, remains unknown, and indeed a role for MR1‐mediated immunity beyond microbial surveillance is yet to be ascribed. Given the diversity of MR1‐binding ligands, however, it seems reasonable that MR1 may capture and present a range of small molecules, akin to MHC presentation of diverse peptides and CD1 presentation of diverse lipids. 1 Thus, for example, perturbed metabolism resulting from cellular transformation in the setting of cancer may generate tumor‐associated MR1 antigens for T‐cell surveillance. To this end, Lepore et al. 28 isolated TRAV1‐2− T cells that responded to tumor cell lines engineered to overexpress MR1‐β2m complexes. Intriguingly, this reactivity was independent of K43, and moreover, distinct clones detected different fractions from tumor lysates. More recently, Crowther et al. 29 isolated a TRAV1‐2− clone (clone MC.7.G5) with broad tumor specificity and ignorance toward healthy tissues. Genome‐wide clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein 9 (CRISPR/Cas9) screening revealed that this reactivity was MR1 dependent. Moreover, these cells could mediate tumor cell killing in vivo and the TCR could be transferred to patient‐derived T cells to mediate in vitro killing of autologous and nonautologous tumor cells. In both studies, 6‐FP or Ac‐6‐FP could block activation, suggesting displacement of tumor‐associated antigen. However, unlike the study by Lepore et al., 28 MC.7.G5 reactivity was K43‐dependent. Collectively, these studies suggest MR1‐dependent recognition of at least three distinct tumor‐associated antigens by diverse TRAV1‐2− MR1‐restricted T cells. Accordingly, presentation of tumor‐associated antigens by MR1 may be an important and underappreciated mode of T cell‐mediated tumor surveillance, and more studies on this axis are needed. Here, we use TCR‐gene transfer, novel MR1‐expressing cell lines and model antigen‐presentation assays to confirm MR1 reactivity by the MR1‐restricted TCRs from the studies by Lepore et al. 28 and Crowther et al. 29 We show differential dependence on K43 by these clones and that this reactivity is inhibited by 6‐FP. We also perform alanine‐scan‐based assays to map the molecular hotspot of MR1 utilized by these and other MR1‐restricted TCRs.
Results
MR1‐reactivity by tumor‐reactive MR1‐restricted T‐cell clones
To interrogate the MR1 reactivity of tumor‐reactive MR1‐restricted T‐cell clones, we tested a panel of MR1‐restricted TCRs, including the MC.7.G5 TCR from the study by Crowther et al. 29 and two TCRs from the study by Lepore et al., 28 DGB129 and DGB70, that demonstrated recognition of distinct tumor lysate fractions. These TCRs were then studied in comparison with previously characterized TCRs, including TRAV1‐2+ MAIT TCRs (M33‐64 9 and MBV28 22 ), a TRAV1‐2− nonclassical MAIT TCR (MAV36 22 ) and a TRAV1‐2− 6‐FP‐reactive TCR (MAV21 22 ; Figure 1a). To first assess the MR1–antigen restriction of the MR1‐restricted TCRs, TCR and CD3 genes were transiently expressed in HEK293T cells, and the cells stained with MR1 tetramers loaded with 5‐OP‐RU or Ac‐6‐FP (Figure 1b). As expected, the MAIT clones M33‐64 and MBV28 stained brightly with human MR1‐5‐OP‐RU tetramers and showed strong xenoreactivity with mouse MR1‐5‐OP‐RU tetramers. Furthermore, the MR1‐autoreactive MAIT clone M33‐64 demonstrated weak staining with MR1‐6‐FP tetramers as previously reported. 22 The tumor‐reactive clones, however, failed to stain with either of the tetramers in a TCR‐dependent manner, suggesting that 5‐OP‐RU and Ac‐6‐FP are not agonists for these TCRs, as previously demonstrated. 28 , 29 To formally assess and compare the distinct TCRs in a cell‐based assay, we next generated SKW‐3 reporter lines that stably expressed these TCRs. Unlike primary T‐cell clones, in this system the matched SKW‐3 background allows direct comparison of the distinct TCRs in isolation from factors that may otherwise affect activation when using primary T cells, such as differential expression of costimulatory molecules. As a readout for MR1 reactivity, the SKW‐3 lines were cocultured with C1R APC lines and CD69 measured on the SKW‐3 cells (Figure 1c). All clones responded to anti‐CD3 and anti‐CD28‐conjugated beads, confirming that their TCR‐signaling apparatus was intact. In line with previous reports, MBV28 neither responded to wild‐type C1R cells (C1R.WT) nor to C1R cells genetically modified to overexpress MR1 (C1R.MR1), whereas M33‐64 showed an MR1‐dependent autoreactivity. 22 These two MAIT cell clones responded robustly in the presence of exogenous 5‐OP‐RU. Both MC.7.G5 and DGB129 responded to C1R.MR1 cells and this response was not enhanced in the presence of 5‐OP‐RU. By contrast, DGB70 failed to respond robustly to either APC line. To validate the MR1 reactivity of MC.7.G5 and DGB129, cocultures were repeated in the presence or absence of anti‐MR1 antibodies. M33‐64 was included as a control for known MR1 reactivity (Figure 1d). Activation of all three SKW‐3 lines was blocked in the presence of anti‐MR1 clone 26.5, an antibody known to block MAIT activation. 30 To assess whether a fully folded form of MR1 was required for activation, an antibody that only binds an unfolded form of MR1, clone 8G3, was also included. 18 However, this antibody failed to block activation. While this suggests that fully folded MR1 is likely required for activation, we cannot rule out that the antibody simply binds a noncompeting epitope and therefore does not have blocking capacity. Collectively, these data validate that MC.7.G5 and DGB129 TCRs are indeed MR1‐reactive and can detect MR1 in the absence of exogenous antigen. However, DGB70 failed to respond robustly in this setting.
Figure 1.
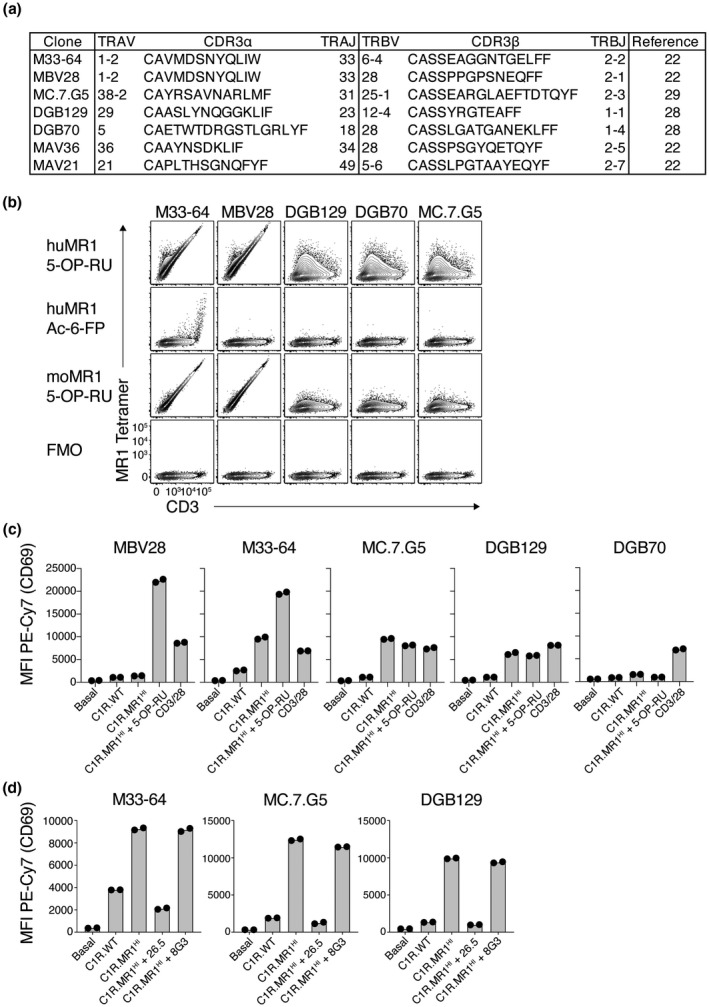
MHC‐related protein 1 (MR1) reactivity by diverse T‐cell receptors (TCRs). (a) Table showing TCR sequences of clones used in this study. (b) Flow cytometric contour plots showing MR1 tetramer staining on 293T cells transiently transfected to express diverse TCRs. Plots are representative of two independent experiments. huMR1, human MR1; moMR1, mouse MR1. (c) Bar graphs showing activation of TCR‐expressing SKW‐3 cells, as measured by CD69, when cultured alone (basal) or together with C1R.WT cells, C1R.MR1HI cells, C1R.MR1HI cells plus synthetic 5‐(2‐oxopropylideneamino)‐6‐D‐ribitylaminouracil (5‐OP‐RU) ligand or CD3/CD28‐coated beads. (d) Bar graphs showing activation of TCR‐expressing SKW‐3 cells, as measured by CD69, when cultured alone or together with C1R.WT cells, C1R.MR1HI cells or C1R.MR1HI cells in the presence of MR1‐specific monoclonal antibodies 26.5 and 8G3. Data points in b and c are duplicate wells, and data are representative of two independent experiments. MFI, mean fluorescent intensity; PE, phycoerythrin; WT, wild type.
Differential dependence on K43 by MR1‐restricted T‐cell clones
To assess the role of K43 in the MR1 reactivity of these distinct clones, we generated novel APC lines including lines in which K43 was mutated to alanine (MR1K43A). To generate an APC solely expressing MR1K43A, we reasoned that knockout of endogenous MR1 was required to ensure that no wild‐type MR1 could confound our analysis. We also opted to generate APCs from distinct lines including 293T cells that robustly activated the original MC.7.G5 clone 29 as well as A375 cells that were used in the study by Lepore et al. 28 We first used CRISPR/Cas9 to knock out MR1 from each of the three lines and generated single‐cell clones that were then screened for MR1 gene deletion. Unlike their wild‐type counterparts, downselected clones knocked out for MR1 (MR1KO) failed to stain with anti‐MR1 after pulsing with Ac‐6‐FP (Figure 2a) and furthermore failed to activate SKW‐3 cells furnishing a MAIT TCR (clone MBV28) when cultured in the presence of 5‐OP‐RU (Figure 2b). Complementary DNA from these lines were sequenced, confirming that the region within exon 3 of the MR1 gene targeted by the CRISPR single guide RNA exhibited excision of an 11‐bp stretch of sequence which resulted in a frameshift in the MR1 gene, rendering MR1 nonfunctional (Supplementary figure 1). These MR1KO lines were then retrovirally transduced to overexpress wild‐type MR1 (MR1HI) or MR1K43A and fluorescence‐activated cell sorting (FACS) sorted to purity for high MR1 expression. Relative to MR1KO lines, A375.WT cells had low endogenous expression of MR1, whereas 293T.WT cells had undetectable MR1 expression (Figure 2c). Overexpression of wild‐type MR1 resulted in a marked increase in basal MR1 levels on both lines, though A375 cells expressed substantially higher levels than did 293T cells. Overexpression of MR1K43A resulted in an even greater boost in MR1 levels, consistent with reduced endoplasmic reticulum (ER) retention as a result of the mutation 17 (Figure 2c). Thus, 293T.MR1K43A cells expressed similar amounts of MR1 as did A375.MR1HI cells. These cell lines were then used in activation‐based assays with tumor‐reactive SKW‐3 lines as in Figure 1 (Figure 2d). Cocultures with C1R cell lines gave similar results to Figure 1, and cocultures with A375.WT and A375.MR1HI lines largely mirrored the activation induced by their C1R counterparts, with M33‐64, MC.7.G5 and DGB129 responding to overexpression of wild‐type MR1. M33‐64 and DGB129 responded to A375.MR1K43A, indicating that their activation was independent of K43. Indeed, this response was greater than that toward A375.MR1HI, which may be a result of the higher expression of surface MR1 in the MR1K43A line. By contrast, despite this higher surface expression, MC.7.G5 failed to respond to A375.MR1K43A cells, suggesting a critical dependence on K43. 293T‐based APCs were very poor at activating the SKW‐3 lines (Figure 2d). Accordingly, in line with published data, MC.7.G5 and DGB129 demonstrate differential dependence on K43, suggesting the possibility of distinct antigen reactivity. Of note, DGB70 again failed to mount a robust response to either of the APCs, suggesting that this TCR maybe MR1 reactive but that that reactivity is not as strong relative to the other TCRs in this model system. We therefore opted not to use this line in subsequent experiments. To further compare the robustness of the MR1‐mediated response by the three MR1‐reactive TCRs, dose titrations with A375.MR1 and C1R.MR1 APCs were performed (Figure 2e). All three SKW‐3 lines responded more strongly to C1R.MR1 cells than they did to A375.MR1 cells, suggesting that in this assay C1R.MR1 cells are stronger APCs, possibly a reflection of the lack of adherence by this cell line. Furthermore, MC.7.G5 gave the strongest response toward both APC lines across the dose range, suggesting that a stronger level of TCR‐mediated signaling, and by inference, MR1 binding, 32 , 33 by this TCR.
Figure 2.
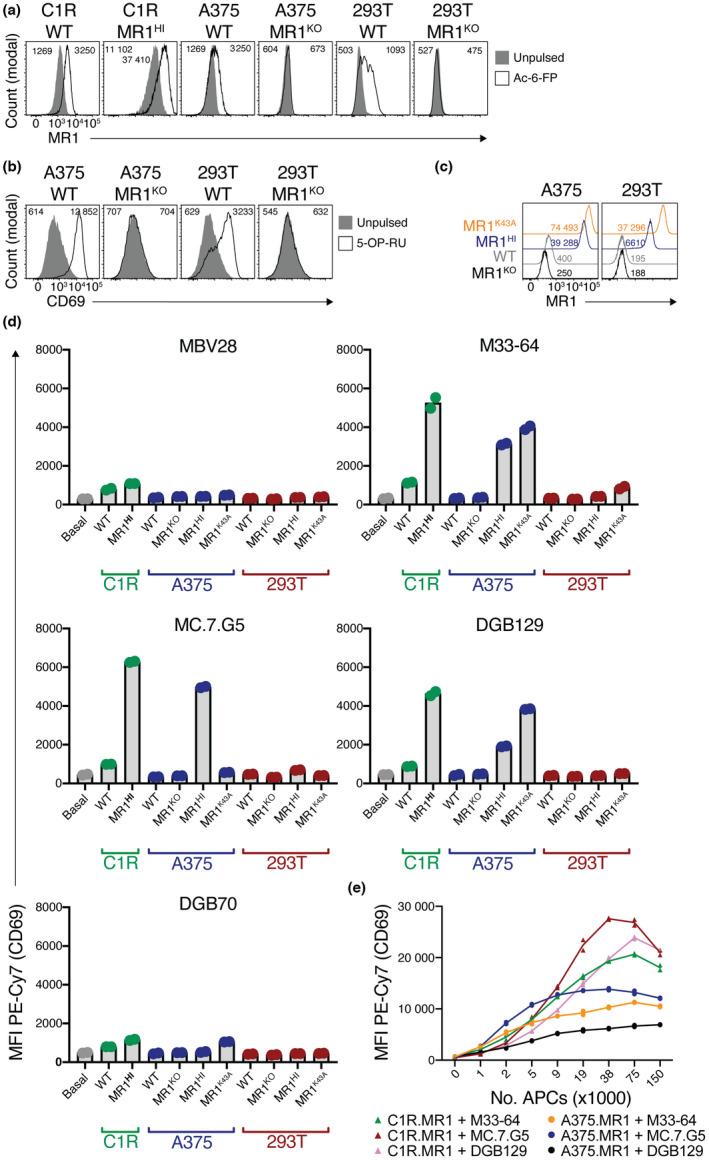
Differential dependence on K43 by different MHC‐related protein 1 (MR1)‐restricted T‐cell receptors (TCRs). (a) Histogram overlays showing MR1 expression on different antigen‐presenting cell lines when pulsed or not with 500 μm acetyl‐6‐formylpterin (Ac‐6‐FP). Numbers on plots indicate mean fluorescent intensity (MFI) of MR1 staining for unpulsed (left) or pulsed (right) cells. (b) Histogram overlays showing CD69 expression on SKW‐3.MBV28 cells when cocultured with different antigen‐presenting cell lines when pulsed or not with synthetic 5‐(2‐oxopropylideneamino)‐6‐D‐ribitylaminouracil (5‐OP‐RU). Numbers on plots indicate MFIs of CD69 as per a. (c) Histogram overlays showing MR1 expression on variant A375 or 293T cells. Numbers on plots depict MFIs of MR1. (d) Bars graphs showing activation of TCR‐expressing SKW‐3 cells, as measured by CD69, after coculture with a panel of antigen‐presenting cell lines. (e) Line graphs showing activation of SKW‐3 cell lines in response to titrated numbers of A375.MR1 or C1R.MR1 cells. a–e are representative of two independent experiments each. Data points in d and e are replicate wells and graphs are representative of three and two independent experiments, respectively. MFI, mean fluorescent intensity; PE, phycoerythrin; WT, wild type.
Blockade of activation with exogenous 6‐FP
The dependence of MC.7.G5 on K43 suggests the possibility that this TCR recognizes MR1 presenting a K43‐bound antigen. Accordingly, addition of K43‐binding ligands such as 6‐FP or Ac‐6‐FP may displace any exogenous ligand(s) and inhibit activation of this line. Indeed, both 6‐FP and Ac‐6‐FP can outcompete 5‐OP‐RU to inhibit MAIT cell activation. 19 A confounding feature of this system, however, is that ligands that neutralize K43 and strongly stabilize MR1 result in enhanced MR1 surface expression. 17 , 19 In the context of TCR clones that exhibit MR1 autoreactivity, addition of exogenous 6‐FP or Ac‐6‐FP may displace endogenous ligand, but the increased MR1 surface expression may counteract this effect. We therefore first measured MR1 surface expression on A375.MR1HI lines in response to titrated doses of 6‐FP and Ac‐6‐FP. While both 6‐FP and Ac‐6‐FP induced MR1 upregulation, Ac‐6‐FP induced far higher levels of surface MR1 (Figure 3a). Activation of MR1‐restricted SKW‐3 lines in response to A375.MR1HI cells cultured with 6‐FP or Ac‐6‐FP was then assessed (Figure 3b). The TRAV1‐2+ MAIT clone M33‐64 exhibited enhanced activation in response to the top dose of 6‐FP and a dose‐dependent response to Ac‐6‐FP. This reactivity aligned with surface MR1 levels in response to these ligands, likely reflecting the MR1‐autoreactive nature of the M33‐64 TCR. 22 By contrast, activation of both MC.7.G5 and DGB129 was reduced with the highest doses of 6‐FP, despite the moderate increases in surface MR1, suggesting the possibility that 6‐FP was displacing an endogenous agonist ligand. This effect was not seen, however, for Ac‐6‐FP cultures, whereby DGB129 exhibited a similar dose‐dependent activation to that of M33‐64, and activation of MC.7.G5 was largely unaffected in response to Ac‐6‐FP. This is possibly a result of the dramatic increase in surface MR1 in response to Ac‐6‐FP, relative to 6‐FP, potentially confounding the effect of the ligand in isolation. Similar results were obtained when C1R.MR1HI cells were used as APCs (Supplementary figure 2a). Given the enhanced MR1‐surface expression is a result of the stable binding of these ligands to the MR1 ligand‐binding groove via K43, 17 we next assessed how 6‐FP and Ac‐6‐FP affected activation in response to A375.MR1K43A APCs. Although the basal surface MR1 levels are moderately higher on A375.MR1K43A cells compared with A375.MR1HI cells, while A375.MR1HI cells upregulated MR1 moderately and strongly in response to 6‐FP and Ac‐6‐FP, respectively, A375.MR1K43A cells did not increase surface MR1 levels in response to 6‐FP, and only a slight increase in response the top dose of Ac‐6‐FP (Supplementary figure 2b). When cocultured with A375.MR1K43A cells, addition of 6‐FP did not affect M33‐64; however, there was a dose‐dependent increase in response to Ac‐6‐FP (Figure 3c). The K43‐dependent clone MC.7.G5, by contrast, exhibited activation in response to the highest dose of 6‐FP, but not Ac‐6‐FP. K43‐independent clone DGB129 also exhibited an increase in activation in response to 6‐FP, and this was also not observed in response to Ac‐6‐FP. This raises the possibility that 6‐FP is weakly agonistic to these TCRs, and this aligns with the observation that 6‐FP did not fully knockdown activation when A375.MR1HI cells were used (Figure 3b). Our initial tetramer staining experiments utilized MR1‐Ac‐6‐FP tetramers (Figure 1b). We therefore repeated this staining using MR1‐6‐FP tetramers; however, while these tetramers weakly bound the M33‐64 line as expected,22 they failed to stain the MC.7.G5, DGB129 and DGB70 lines (Supplementary figure 3). We also noted that 6‐FP derived from folate in the Roswell Park Memorial Institute‐1640 (RPMI‐1640) culture media may be contributing to the apparent MR1 autoreactivity of these clones; however, culturing these cells in folate‐free RPMI‐1640‐based media prior to, and during, coculture experiments did not affect activation of the MR1‐restricted lines, though we cannot rule out fetal bovine serum as a source of folate in these experiments (Supplementary figure 4). We next reasoned that tetramers produced from soluble TCRs may stain the MR1‐expressing APCs that are agonistic to those TCRs, as previously reported for TRAV1‐2+ MAIT TCRs. 17 We thus produced biotinylated soluble MBV28, M33‐64, MC.7.G5 and DGB129 TCRs for use as tetramers. These TCRs were detected with anti‐αβ TCR antibody in an ELISA assay, supporting the appropriate folding of the TCRs (Figure 2c). These TCR tetramers, however, failed to stain MR1‐overexpressing C1R and A375 cells (Supplementary figure 5b, c). Nonetheless, when A375.MR1HI cells were cultured with 5‐OP‐RU, both MAIT TCR tetramers derived from clones MBV28 and M33‐64 bound strongly to the cells. When pulsed with Ac‐6‐FP, the MR1‐autoreactive clone M33‐64 also bound. This, however, was not observed when cells were pulsed with 6‐FP, suggesting the possibility that very high levels of MR1 are required to facilitate TCR tetramer binding to a weak agonist. MC.7.G5 and DGB129 failed to bind both Ac‐6‐FP‐ and 6‐FP‐pulsed cells (Supplementary figure 5d).
Figure 3.
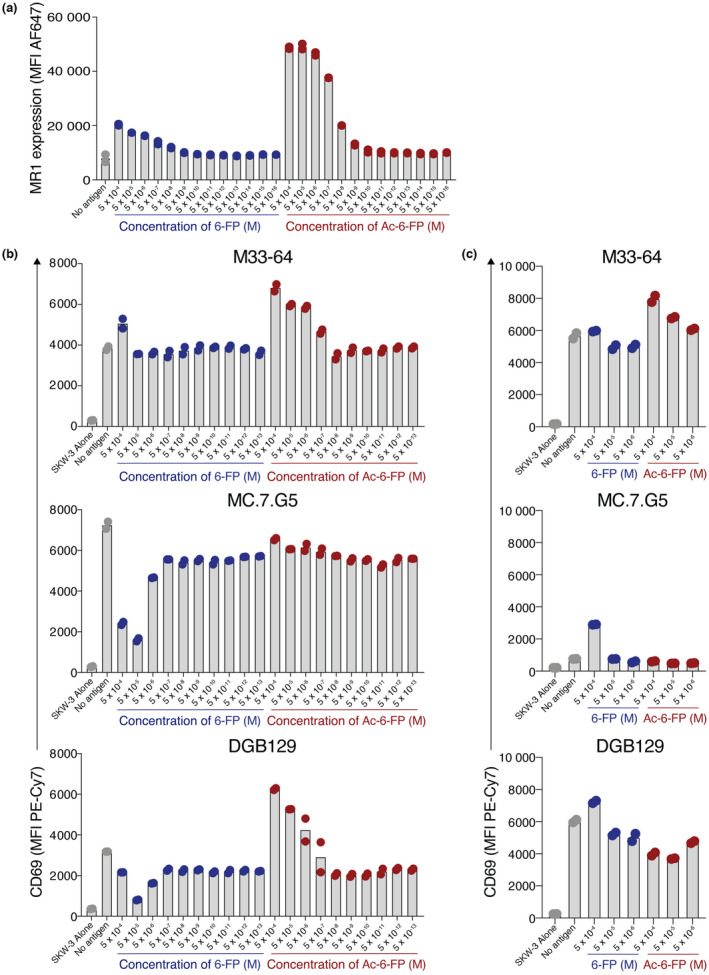
Differential effects on activation by 6‐formylpterin (6‐FP) and acetyl‐6‐FP (Ac‐6‐FP). (a) Bar graphs showing the mean fluorescence intensity (MFI) of MHC‐related protein 1 (MR1) expression on A375.MR1HI cells when pulsed with titrating concentrations of 6‐FP or Ac‐6‐FP. (b, c) Bar graphs showing activation of T‐cell receptor (TCR)‐expressing SKW‐3 cells as measured by CD69, after coculture with, (b) A375.MR1HI cells or (c) A375.MR1K43A cells, with titrating doses of 6‐FP or Ac‐6‐FP. Points on all plots are replicate wells, and data are representative of two independent experiments. PE, phycoerythrin.
Limited MR1 reactivity of MR1‐restricted SKW‐3 lines to diverse tumor lines
Crowther et al. 29 showed a remarkable ability of the primary MC.7.G5 clone to respond to diverse tumor‐derived cell lines, while remaining unresponsive to healthy tissues. To determine whether these distinct MR1‐restricted SKW‐3 lines could recapitulate this observation, their activation was assessed after coculture with a diverse panel of tumor‐derived cell lines (Figure 4a). In contrast to the primary cells used by Crowther et al., 29 for almost all APC lines, unless MR1 was overexpressed (right panel), very limited activation above basal CD69 levels was observed (left panel). To confirm that these APC lines possessed the correct antigen presentation machinery, surface MR1 expression was measured with and without incubation with Ac‐6‐FP. Most cell lines, except LM‐MEL‐19, showed an increase in MR1 surface expression in response to Ac‐6‐FP, suggesting the ability of these cells to capture and present antigen (Figure 4b). All cell lines, except LM‐MEL‐19 again, were also capable of activating SKW‐3 cells expressing a MAIT TCR when 5‐OP‐RU was added to cultures, albeit to varying degrees (Figure 4c). Thus, in contrast to the use of primary cells, when using SKW‐3 reporter cell lines, these TCRs show limited tumor‐cell reactivity despite the ability of the tumor cell lines to present antigen.
Figure 4.
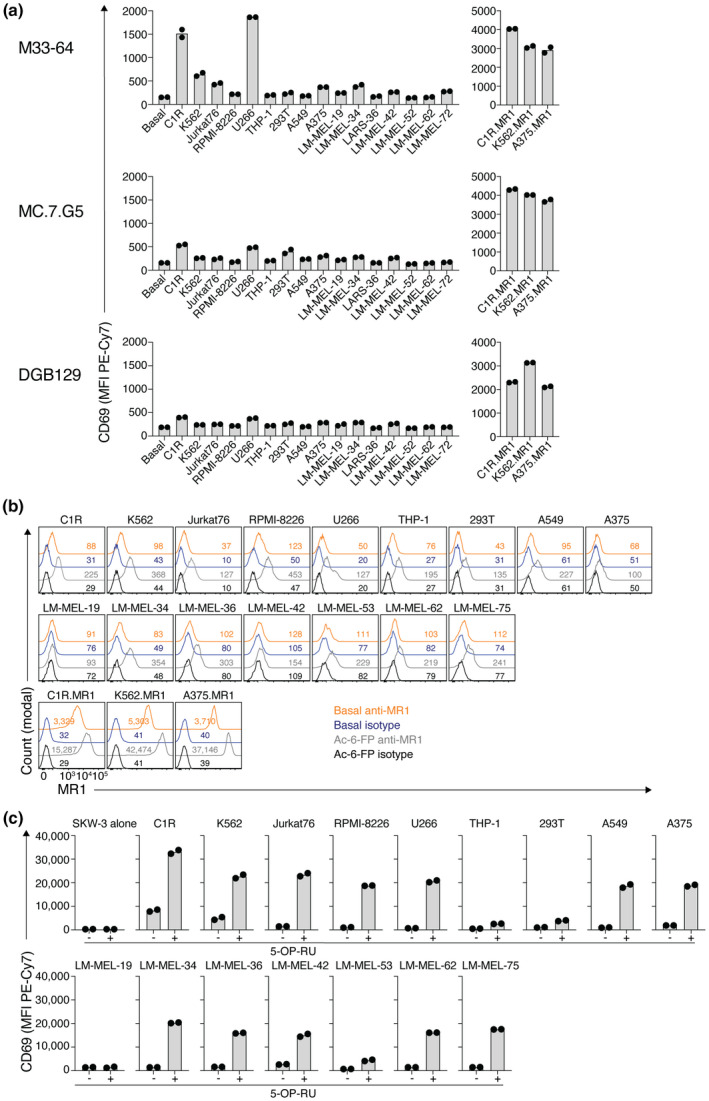
Poor recognition of diverse cell lines by MHC‐related protein 1 (MR1)‐restricted T‐cell receptor (TCR)‐expressing SKW‐3 lines. (a) Bar graphs showing activation of TCR‐expressing SKW‐3 cells as measured by CD69, after coculture with a panel of tumor‐derived cell lines (left), including those transduced to overexpress MR1 (right). Points on plots are replicate wells, and data are representative of two independent experiments. (b) Histogram overlays showing MR1 expression on different antigen‐presenting cell lines when pulsed or not with 500 μm acetyl‐6‐formylpterin (Ac‐6‐FP). Numbers on plots indicate mean fluorescent intensity of MR1 staining. (c) Bar graphs showing activation of mucosal‐associated invariant T TCR‐expressing SKW‐3 cells as measured by CD69, after coculture with a panel of tumor‐derived cell lines in the presence (+) or absence (−) of 5‐(2‐oxopropylideneamino)‐6‐D‐ribitylaminouracil (5‐OP‐RU). Points on plots are replicate wells, and data are representative of two independent experiments. PE, phycoerythrin.
Diversity of MR1 docking modes by MR1‐restricted TCRs
To investigate the diversity of docking modes employed by distinct MR1‐restricted TCRs, we utilized a panel of C1R APC lines, each engineered to overexpress MR1 with single alanine substitutions at solvent‐exposed amino acid residues along the α1 or α2 helix, allowing us to map an energetic hotspot on MR1 for each TCR 33 (Figure 5). A range of TCRs were assessed, including the autoreactive TRAV1‐2+ MAIT TCR clone M33‐64; tumor‐reactive TCRs MC.7.G5 and DGB129; folate‐derivative‐reactive clone MAV21 and nonclassical TRAV36+ MAIT TCR MAV36. The MAV36 TCR is MR1‐5‐OP‐RU restricted, thus exogenous 5‐OP‐RU was added to cultures, whereas no antigen was added to cocultures with the remaining TCRs that relied on their inherent MR1 autoreactivity for activation. As per MAIT TCRs previously assessed with this same panel of mutants, activation of M33‐64 was abrogated in response to L65A and E158A, whereas L151A drove enhanced activation. 33 Activation of the MAV36 TCR was also impaired with the L65A and E158A mutations, as well as in response to N146A. Mutations for both TCRs mapped to relatively centric energetic hotspots, shifted slightly toward the antigen‐binding A′ pocket. This aligns with crystal structures of MR1–antigen–TCR ternary complexes, which demonstrated central docking modes above the A′ pocket for these two TCRs. 22 Results from MC.7.G5 and DGB129 TCRs also suggested central hotspots, albeit with dependence on a different array of residues. Activation of MC.7.G5 was knocked down in response to L65A, M72A and N155A, whereas DGB129 depended on M72, N146, H148 and E158. Conversely, MAV21 had the greatest number of mutants that affected binding, including R61A, L65A, M72A, R79A, Q141A, N146A and H148A. These mutations spanned the length of the α1 helix and the F′ end of the α2 helix, suggesting a docking mode shifted closer toward the F′ pocket. Accordingly, MR1‐restricted TCRs employ distinct docking modes that rely on a range of amino acids across the helical jaws of MR1, spanning both the A′ and F′ pockets.
Figure 5.
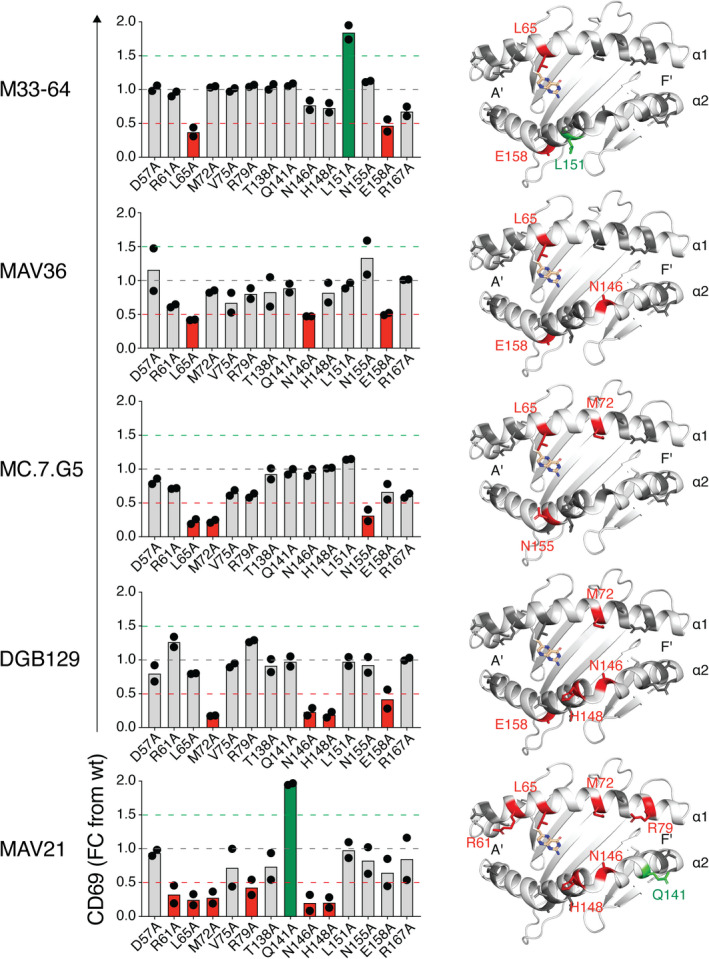
Analysis of docking modes by diverse MHC‐related protein 1 (MR1)‐restricted T‐cell receptors (TCRs). Left: Bar graphs showing activation of TCR‐expressing SKW‐3 cells as measured by CD69, after coculture with a panel of C1R cell lines expressing MR1 alanine point mutants. Points on plots are the mean of replicates from two independent experiments. Right: Cartoon representations of the top–down view of the α1 and α2 domains of MR1 (PDB: 4GUP), showing the amino acid residues that impact binding.
Discussion
Here, we used TCR gene transfer to generate a model system to probe the specificity of previously isolated MR1‐restricted TCRs. We have confirmed some of the salient features of the MC.7.G5 and DGB129 TCRs, from Crowther et al. 29 and Lepore et al. 29 respectively. Namely, these TCRs recognize MR1, have differential dependence on K43 and their activation can be blocked with 6‐FP. Moreover, our ala‐scan data suggest classical docking modes atop the α1–α2 helices. Collectively, these data are in line with the recognition of undefined antigens by these two TCRs, and indeed suggest that they recognize distinct antigens.
That the MC.7.G5 TCR had a strong dependence on K43 for activation suggests an antigen is required. Given that K43 is positioned deep in the base of the A′ pocket and is likely to be inaccessible to the reach of the TCR, it seems unlikely that it is being sensed directly. Although a γδ TCR has previously been shown to extend its CDR3δ loop deep into the cleft of an MHC I‐like molecule to mediate recognition, 34 this relied on an exceptionally long CDR3δ loop and an open‐ended antigen‐binding cleft, and this is in contrast to the CDR loops of the MC.7.G5 TCR and the enclosed A′ pocket of MR1. Indeed, previous studies have demonstrated that the K43A mutation does not result in any dramatic structural changes to the overall conformation of the α1/α2 helices and β‐sheet platform, nor to the antigen‐binding pocket, and is still amenable to recognition by MAIT TCRs as validated here with the M33‐64 TCR. 9 K43 has, however, emerged as a key molecular feature of the MR1 antigen‐binding pocket via formation of a Schiff base between K43 and antigen, stabilizing the MR1–antigen complex and triggering egress of MR1 to the cell surface. 2 , 17 , 19 Thus, the most likely explanation seems to be that the MR1K43A mutant can no longer efficiently capture a K43‐dependent antigen. This is also in line with the blocking of activation with 6‐FP, which binds MR1 via K43, 2 suggesting that at high doses, 6‐FP displaces or outcompetes an undefined antigen, as it does for ribityl antigens that activate TRAV1‐2+ MAIT cells. 19 These results also rule out that 6‐FP derived from folate in the RPMI media 25 is responsible for activation and aligns with our experiments performed with folate‐free media. We cannot, however, rule out that another, as‐yet‐undefined, component of the culture media is providing a source of antigen or antigen precursor. Notably, unlike 6‐FP, Ac‐6‐FP did not block activation of the MC.7.G5 TCR, and this contrasts with the paper by Crowther et al. 29 in which Ac‐6‐FP blocked killing of a melanoma line by the primary MC.7.G5 clone. The discrepancy here may be a result of differences in the experimental approaches; in our study, the MR1‐overexpressing cell lines have unphysiologically high translation of MR1 and thus exogenous Ac‐6‐FP induces exceptionally high levels of surface MR1. That the TCR tetramers failed to bind these MR1‐overexpressing cells suggests that any activating ligand(s) that is present is either in low abundance or engaged TCR with weak affinity or both. As these TCR‐transduced SKW‐3 lines are highly sensitive to TCR‐stimulation, they may have a much lower threshold of activation relative to primary cells and thus exhibit a degree of MR1 autoreactivity in the absence of a bona fide physiological antigen. Thus, any potential displacement of endogenous antigen may be countered by responsiveness to the increased surface MR1 levels, confounding interpretation of these results. That 6‐FP did not fully block activation, and high doses of 6‐FP in the A375.MR1K43A cultures resulted in low‐level activation, suggests that 6‐FP may be mildly agonistic to the MC.7.G5 TCR. This contrasts with Ac‐6‐FP, which failed to activate in the A375.MR1K43A cultures. Although these two compounds only differ by an acetyl group, this moiety is positioned at the opening of the cleft and is theoretically accessible to docked TCRs. 19 The acetyl group also causes localized conformational remodeling of amino acid side chains lining the antigen‐binding pocket and may thus either directly or indirectly inhibit TCR binding. 15 Indeed, TRAV1‐2+ MAIT TCRs that can differentiate between 6‐FP and Ac‐6‐FP on account of CDR3β variability have previously been reported. 22 In contrast to MC.7.G5, activation of the DGB129 TCR was not affected by the K43A mutation. It was, however, blocked with 6‐FP, suggesting displacement of a ligand from the A′ pocket as per MC.7.G5, although the independence from K43 suggests that this ligand is distinct from that recognized by MC.7.G5 and does not require a covalent bond with K43. While K43‐independent MR1 ligands have been described, their precise antigen capture and processing pathway and antigenicity relative to K43‐binding antigens remain unclear. Elucidation of their identity should shed much needed light on this axis.
In the study by Crowther et al., 29 MC.7.G5 responded robustly to the relatively low surface levels of MR1, including toward cell lines used in this study such as HEK293T, Jurkat and K562. 29 We found, however, that MR1 overexpression was required to elicit a response from the MC.7.G5 cells, and furthermore 293T cells were extremely poor activators even when MR1 was overexpressed. These discrepancies may be explained by the different origin of the T cells used between the two studies. Our system is specifically designed to interrogate TCR‐mediated signaling events. It does not consider the contribution of accessory molecules expressed by primary cells, nor the threshold of activation set in these cells during thymic development. While it is clear that the MC.7.G5 TCR is MR1 reactive, it seems likely that other interactions between the T cells and tumor cells are required to elicit an antitumor response. For example, unlike the SKW‐3 cells, the original MC.7.G5 clone expressed high levels of the CD8 coreceptor, 29 which is known to directly interact with MR1 and contribute to MR1‐restricted T‐cell agonism. 35 , 36 Costimulatory molecules such as CD27 and CD28, and activating receptors such as NKG2D, among many others, may also be critical in mediating tumor cell recognition in the context of expression of the MC.7.G5 TCR. Moreover, the threshold of TCR signaling required for activation, set in the thymus during development and involving proteins such as Themis, may contribute to this process. 37 Accordingly, along with the critical questions pertaining to the identification of the tumor‐derived ligands, unravelling the non‐TCR‐mediated mechanisms of tumor‐cell reactivity by MR1‐restricted T cells is highly pertinent and may offer novel avenues of therapeutic relevance. Therapeutics that genetically transfer the specificity of these TCRs, such as CAR‐T cells, also offer great potential as demonstrated in the study by Crowther et al. 29 Further interrogation of the TCR repertoire of tumor‐reactive MR1‐restricted TCRs, and their modes of antigen recognition is required. Moreover, the role of these cells in cancer development and progression is entirely unknown and more studies assessing their in vivo function are critical.
Collectively, this study supports the suggestion of the presentation by MR1 of distinct tumor‐associated antigens to diverse TRAV1‐2− MR1‐restricted T cells. The notion that MR1 may sample tumor metabolites for T‐cell surveillance is intriguing, and identification of these ligands will be a major step forward not only in understanding MR1 and MR1‐restricted T cells, but also in our understanding of T cell‐mediated tumor immunity. Further studies on this axis should provide great insight into the fascinating biology of MR1 and the T cells that recognize it.
METHODS
Generation of MR1‐knockout cell lines by CRISPR/Cas9
lentiCRISPRv2GFP plasmid 38 was a gift from David Feldser (Addgene plasmid #82416; http://n2t.net/addgene:82416; RRID:Addgene_82416). A single guide RNA (AGTGATTGTAGTGCCTCTGT) targeting a sequence in exon 3 of MR1 was designed using the Benchling CRISPR Guide RNA Design platform and cloned into lentiCRISPRv2GFP as described by the GeCKO Lentiviral CRISPR toolbox (Zheng Laboratory, Broad Institute, MIT, Cambridge, MA). Plasmid was then transiently transfected into HEK293T and A375 cells using FuGENE 6 transfection reagent (Promega, Madison, WI). After 24 h, cells with high GFP expression were FACS sorted at one cell per well into 96‐well plates. After 10 days, clones were screened for MR1 expression, and downselected for cells with negligible MR1 staining (phycoerythrin, clone 26.5; BioLegend, San Diego, CA). Clones were then assessed for MR1 upregulation in response to a 4‐h culture with 500 μm Ac‐6‐FP, followed by staining with anti‐MR1 as above, and those clones that failed to upregulate MR1 were further downselected. Clones were then assessed for their ability to stimulate SKW‐3.MBV28 cells (described below) when cocultured overnight in the presence of 100 nm 5‐OP‐RU (provided by David Fairlie, University of Queensland) and clones further downselected. RNA was subsequently extracted from downselected clones using Isolate II RNA Mini Kit (Bioline, London, UK). Complementary DNA was generated using SuperScript VILO cDNA synthesis kit (Thermo Fisher Scientific). Complementary DNA encoding exon 3 of MR1 was then amplified using KAPA‐HiFi‐HotStart ReadyMix PCR kit (Roche, Basel) using the following primers: forward, CCTCCTTTCCAGGGACGCAC; reverse, CGGATACAGAGATGGGGAAGAGTG. PCR products were separated by agarose gel electrophoresis and DNA excised from gel and purified using a Gel DNA Extraction Kit (Zymo Research, Irvine, CA). Purified DNA was sequenced via Sanger Sequencing (AGRF) and CRISPR‐mediated excision of target DNA confirmed. A single clone for each of A375 and 293T cells was taken forward and henceforth referred to as A375.MR1KO and 293T.MR1KO, respectively.
Generation of TCR or MR1 overexpressing cell lines by retroviral transduction
C1R.MR1HI 22 cells were described previously. C1R.MR1 alanine mutants 33 were described previously, but were stained with anti‐MR1 (phycoerythrin, clone 26.5, BioLegend) and further FACS sorted for high and matched expression of MR1. For SKW‐3.TCR lines, genes encoding the full‐length TCR‐α and TCR‐β chains linked by a P2A linker for M33‐64, MBV28, MAV21, MC.7.G5, DGB70 and DGB129 TCRs were synthesized as double‐stranded DNA fragments (Integrated DNA Technologies, Coralville, IA) and cloned into the pMSCV‐IRES‐GFP II (pMIG II) expression vector 39 ; a gift from Dario Vignali (Addgene plasmid # 52107; http://n2t.net/addgene:52107; RRID:Addgene_52107). pMIG.TCR plasmids along with a pMIG II plasmid encoding human CD3ε, δ, γ and ξ subunits linked via p2A linkers (pMIG.huCD3) were used to retrovirally transduce SKW‐3.β2mKO cells 18 as previously described, 39 to generate SKW‐3.M33‐64, SKW‐3.MBV28, SKW‐3.MAV21, SKW‐3.MC.7.G5, SKW‐3.DGB129 and SKW‐3.DGB70. Transduced cells were then stained with anti‐CD3 (BV421, clone UCHT1, Becton Dickinson, Franklin Lakes, NJ) and FACS sorted for GFPHICD3HI cells. A375.MR1KO and 293T.MR1KO cells were retrovirally transduced as previously described 33 with the pMIG II plasmid encoding full‐length human MR1 or MR1.K43A mutant genes. pMIG.MR1 plasmid was generated previously, 22 and pMIG.MR1‐K43A plasmid was generated by synthesizing double‐stranded DNA fragments encoding full‐length MR1 (Integrated DNA Technologies) and cloning into pMIG II. After transduction, cells were stained with anti‐MR1 (phycoerythrin, clone 26.5; BioLegend) and FACS sorted for GFPHIMR1HI cells. These resulted in A375.MR1HI, A375.MR1K43A, 293T.MR1HI and 293T.MR1K43A lines.
Transient transfections
HEK293T cells were transiently co‐transfected with pMIG.TCR and pMIG.huCD3 plasmids using FuGENE 6 transfection reagent (Promega). Cells were cultured for 48–72 h, harvested by mechanical disruption and stained with anti‐CD3 (BV421, clone UCHT1; BD), LIVE/DEAD near‐infrared viability dye (Thermo Fisher Scientific) and MR1 tetramers. Cells were then fixed with 2% paraformaldehyde and analyzed by flow cytometry.
Flow cytometry
All flow cytometry experiments were performed on an LSRFortessa (Becton Dickinson) equipped with a yellow–green laser. Cell sorting was performed using a FACSAria III cell sorter (Becton Dickinson). Data were analyzed using FlowJo version 10.7.1 (Becton Dickinson).
SKW‐3 coculture assays
Coculture assays were performed in RF10 media, consisting of an RPMI‐1640 base (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific), penicillin (100 U mL−1), streptomycin (100 mg mL−1), GlutaMAX (2 mmol L−1), sodium pyruvate (1 mmol L−1), non‐essential amino acids (0.1 mmol L−1), HEPES [4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid] buffer (15 mmol L−1), pH 7.2–7.5 (all from Thermo Fisher Scientific, Waltham) and 2‐mercaptoethanol (50 mmol L−1, Sigma). A375 cells were maintained in the same media but using a Dulbecco’s Modified Eagle’s Medium base (Thermo Fisher Scientific) in place of RPMI‐1640. SKW‐3 and APC lines were cocultured in 96‐well plates for approximately 18 h before cells were harvested. Adherent APC lines were plated 12–24 h before the addition of SKW‐3 lines, whereas nonadherent lines were added to SKW‐3 cells on the same day. Adherent lines were plated at 36 000 cells per well, which resulted in full confluency at the time of SKW‐3 addition. APCs were labeled with Cell Trace Violet (Thermo Fisher Scientific) prior to plating. For assays involving the addition of exogenous 5‐OP‐RU or blocking antibodies, antigen or blocking antibodies were added when the cells were combined and included for the entirety of the coculture. Blocking antibodies were used at 10 μg mL−1. Purified anti‐MR1 clone 26.5 was obtained from BioLegend, whereas clone 8G3 was produced in‐house. 18 For assays with 6‐FP and Ac‐6‐FP, ligands were added approximately 12 h after the cells were plated and incubated for 4 h before SKW‐3 cells were added directly to the APCs without removing the media. For assays involving folate‐free media, SKW‐3 and APC lines were cultured for 48 h prior to the experiment in RF10 media made with a folate‐free RPMI‐1640 base (Thermo Fisher Scientific) and this media was used during the course of the experiment. Upon harvest, cells were stained with LIVE/DEAD near‐infrared viability dye (Thermo Fisher Scientific), anti‐CD69 (PE‐Cy7, clone FN50; BD), fixed in 2% paraformaldehyde and acquired by flow cytometry. For analysis, APCs were removed by excluding Cell Trace Violet‐positive cells; SKW‐3 cells were then gated based on forward scatter‐area and side scatter‐area after dead cell and doublet removal. GFPHI cells were then gated, and CD69 mean fluorescence intensity was assessed.
MR1 expression assays
For antigen‐induced MR1‐upregulation assays, adherent APC lines were plated the day prior to the addition of antigen, whereas nonadherent lines were plated on the same day as the addition of antigen as above. APC lines were incubated with antigen for 4 h before being harvested. Adherent cell lines were mechanically disrupted. Cells were then stained with LIVE/DEAD near‐infrared viability dye (Thermo Fisher Scientific) and anti‐MR1‐PE antibody (Clone 26.5, BioLegend), fixed in 2% paraformaldehyde and acquired by flow cytometry. For analysis, APCs were gated based on forward scatter‐area and side scatter‐area after dead cell and doublet removal.
APC lines
HEK293T and A375 cells were obtained from ATCC. C1R cells were obtained from the McCluskey Laboratory (University of Melbourne). Jurkat cells were supplied by Mirjam HM Heemskerk. 40 RPMI‐8226, U266, K562, MEG‐01 and THP‐1 cells were obtained from the Neeson Laboratory (Peter MacCallum Cancer Centre). Melanoma lines were provided by Andreas Behren 41 (Olivia Newton‐John Cancer Research Institute).
Production of recombinant MR1 and TCR tetramers
TCR tetramers: TCR‐α and TCR‐β variable domain genes were synthesized (Integrated DNA Technologies) and cloned into the pET‐30 expression vector containing engineered mouse TCR‐α and TCR‐β constant domains for enhanced stability. 42 The TCR‐α chain included a C‐terminal (G3S)2 linker followed by an AVI tag for biotinylation. Individual TCR chains were expressed in BL21 E. coli and extracted as inclusion bodies. Matched TCR‐α and TCR‐β chains were then refolded by oxidative refolding as previously described. 43 Refolded TCRs were purified by weak anion exchange (DEAE Sepharose; Cytiva, Marlborough, MA), size‐exclusion chromatography (Superdex‐75; Cytiva) and strong anion exchange (Mono Q). TCR monomers were then biotinylated using BirA enzyme (synthesized in‐house) and further purified from biotinylation reagents using size‐exclusion chromatography (Superdex‐75; Cytiva). Biotinylation was confirmed by co‐incubating denatured TCR protein with streptavidin and running on sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Typically, more than 95% of protein ran at a higher apparent molecular weight when incubated with streptavidin, confirming efficient biotinylation. Appropriate conformation of TCR protein was confirmed via ELISA, using anti‐αβTCR monoclonal antibody clone 12H8 (produced in‐house), anti‐γδ TCR monoclonal antibody clone B1 (Becton Dickinson, Franklin Lakes) and anti‐Vα7.2 monoclonal antibody clone 3C10 (BioLegend). The previously described G115 γδ TCR was used as a control. 31 MR1 tetramers were produced with AVI‐tagged MR1.C262S constructs as previously described. 25 In brief, pET‐30 expression vectors encoding the truncated extracellular domains of human and mouse MR1.C262S with C‐terminal AVI‐tag and 6xHIS‐tag, or full‐length β2m were expressed as inclusion bodies as above. MR1 and β2m were then refolded by oxidative refolding in the presence of 6‐FP, Ac‐6‐FP (Schircks Laboratories, Buechstrasse, Switzerland) or 5‐amino‐6‐D‐ribitylaminouracil (provided by David Fairlie, University of Queensland) together with methylglyoxal (Millipore–Sigma, St. Louis, MO) for 5‐OP‐RU as previously described. 2 , 12 Refolded MR1 was purified by Ni‐NTA (Thermo Fisher Scientific), and biotinylated as above. Tetramers were formed by serial addition of streptavidin–phycoerythrin (Becton Dickinson) in 10 equal additions, every 10 min to a final molar ratio of 4:1 monomers to streptavidin–phycoerythrin.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTION
Rebecca Seneviratna: Investigation; Writing – review & editing. Samuel J Redmond: Investigation; Writing – review & editing. Hamish E McWilliam: Resources; Writing – review & editing. Rangsima Reantragoon: Resources; Writing – review & editing. Jose A Villadangos: Resources; Writing – review & editing. James McCluskey: Resources; Writing – review & editing. Dale I Godfrey: Conceptualization; Funding acquisition; Resources; Supervision; Writing – review & editing. Nicholas A Gherardin: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing.
Supporting information
ACKNOWLEDGMENTS
We thank staff from the flow cytometry facilities at the Department of Microbiology and Immunology at the Peter Doherty Institute. We thank Dr Lars Kjer‐Nielsen for his role in the provision of C1R.MR1 mutants used in TCR ala‐scan experiments, Associate Professor Andreas Behren (Olivia Newton‐John Cancer Research Institute, Australia) for provision of human melanoma “LM‐MEL” cell lines and Professor David Fairlie (University of Queensland, Australia) for the provision of 5‐OP‐RU and 5‐amino‐6‐D‐ribitylaminouracil. This work was supported by the Australian Research Council (ARC; CE140100011 and DP170102471) and the National Health and Medical Research Council, Australia (NHMRC; 1113293, 1159932 and 2003192). NAG and HEGM were supported by ARC DECRA Fellowships (DE210100705 and DE170100575). DIG was supported by an NHMRC Senior Principal Research Fellowship (1117766). JAV was supported by an NHMRC Principal Research Fellowship (1154502). Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
DATA AVAILABILITY STATEMENT
The authors declare that the data supporting the findings of this study are available within the article, supplementary information and source data or are available upon reasonable requests to the authors.
REFERENCES
- 1. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen‐presenting molecules. Annu Rev Immunol 2015; 33: 169–200. [DOI] [PubMed] [Google Scholar]
- 2. Kjer‐Nielsen L, Patel O, Corbett AJ, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012; 491: 717–723. [DOI] [PubMed] [Google Scholar]
- 3. Bacher A, Eberhardt S, Fischer M, Kis K, Richter G. Biosynthesis of vitamin b2 (riboflavin). Annu Rev Nutr 2000; 20: 153–167. [DOI] [PubMed] [Google Scholar]
- 4. Treiner E, Duban L, Bahram S, et al. Selection of evolutionarily conserved mucosal‐associated invariant T cells by MR1. Nature 2003; 422: 164–169. [DOI] [PubMed] [Google Scholar]
- 5. Le Bourhis L, Martin E, Peguillet I, et al. Antimicrobial activity of mucosal‐associated invariant T cells. Nat Immunol 2010; 11: 701–708. [DOI] [PubMed] [Google Scholar]
- 6. Gold MC, Cerri S, Smyk‐Pearson S, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol 2010; 8: e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4‐8‐ α/β T cells demonstrates preferential use of several Vβ genes and an invariant TCR α chain. J Exp Med 1993; 178: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tilloy F, Treiner E, Park SH, et al. An invariant T cell receptor α chain defines a novel TAP‐independent major histocompatibility complex class Ib‐restricted α/β T cell subpopulation in mammals. J Exp Med 1999; 189: 1907–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reantragoon R, Corbett AJ, Sakala IG, et al. Antigen‐loaded MR1 tetramers define T cell receptor heterogeneity in mucosal‐associated invariant T cells. J Exp Med 2013; 210: 2305–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patel O, Kjer‐Nielsen L, Le Nours J, et al. Recognition of vitamin B metabolites by mucosal‐associated invariant T cells. Nat Commun 2013; 4: 2142. [DOI] [PubMed] [Google Scholar]
- 11. Lopez‐Sagaseta J, Dulberger CL, McFedries A, Cushman M, Saghatelian A, Adams EJ. MAIT recognition of a stimulatory bacterial antigen bound to MR1. J Immunol 2013; 191: 5268–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corbett AJ, Eckle SB, Birkinshaw RW, et al. T‐cell activation by transitory neo‐antigens derived from distinct microbial pathways. Nature 2014; 509: 361–365. [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto K, Hirai M, Kurosawa Y. A gene outside the human MHC related to classical HLA class I genes. Science 1995; 269: 693–695. [DOI] [PubMed] [Google Scholar]
- 14. Riegert P, Wanner V, Bahram S. Genomics, isoforms, expression, and phylogeny of the MHC class I‐related MR1 gene. J Immunol 1998; 161: 4066–4077. [PubMed] [Google Scholar]
- 15. Awad W, Le Nours J, Kjer‐Nielsen L, McCluskey J, Rossjohn J. Mucosal‐associated invariant T cell receptor recognition of small molecules presented by MR1. Immunol Cell Biol 2018; 96: 588–597. [DOI] [PubMed] [Google Scholar]
- 16. Harriff MJ, McMurtrey C, Froyd CA, et al. MR1 displays the microbial metabolome driving selective MR1‐restricted T cell receptor usage. Sci Immunol 2018; 3: eaao2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McWilliam HE, Eckle SB, Theodossis A, et al. The intracellular pathway for the presentation of vitamin B‐related antigens by the antigen‐presenting molecule MR1. Nat Immunol 2016; 17: 531–537. [DOI] [PubMed] [Google Scholar]
- 18. McWilliam HEG, Mak JYW, Awad W, et al. Endoplasmic reticulum chaperones stabilize ligand‐receptive MR1 molecules for efficient presentation of metabolite antigens. Proc Natl Acad Sci USA 2020; 117: 24974–24985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eckle SB, Birkinshaw RW, Kostenko L, et al. A molecular basis underpinning the T cell receptor heterogeneity of mucosal‐associated invariant T cells. J Exp Med 2014; 211: 1585–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salio M, Awad W, Veerapen N, et al. Ligand‐dependent downregulation of MR1 cell surface expression. Proc Natl Acad Sci USA 2020; 117: 10465–10475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keller AN, Eckle SB, Xu W, et al. Drugs and drug‐like molecules can modulate the function of mucosal‐associated invariant T cells. Nat Immunol 2017; 18: 402–411. [DOI] [PubMed] [Google Scholar]
- 22. Gherardin NA, Keller AN, Woolley RE, et al. Diversity of T cells restricted by the MHC class I‐related molecule MR1 facilitates differential antigen recognition. Immunity 2016; 44: 32–45. [DOI] [PubMed] [Google Scholar]
- 23. Gherardin NA, McCluskey J, Rossjohn J, Godfrey DI. The diverse family of MR1‐restricted T cells. J Immunol 2018; 201: 2862–2871. [DOI] [PubMed] [Google Scholar]
- 24. Meermeier EW, Laugel BF, Sewell AK, et al. Human TRAV1‐2‐negative MR1‐restricted T cells detect S. pyogenes and alternatives to MAIT riboflavin‐based antigens. Nat Commun 2016; 7: 12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koay HF, Gherardin NA, Xu C, et al. Diverse MR1‐restricted T cells in mice and humans. Nat Commun 2019; 10: 2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le Nours J, Gherardin NA, Ramarathinam SH, et al. A class of γδ T cell receptors recognize the underside of the antigen‐presenting molecule MR1. Science 2019; 366: 1522–1527. [DOI] [PubMed] [Google Scholar]
- 27. Awad W, Meermeier EW, Sandoval‐Romero ML, et al. Atypical TRAV1‐2‐ T cell receptor recognition of the antigen‐presenting molecule MR1. J Biol Chem 2020; 295: 14445–14457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lepore M, Kalinichenko A, Calogero S, et al. Functionally diverse human T cells recognize non‐microbial antigens presented by MR1. Elife 2017; 6: e24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crowther MD, Dolton G, Legut M, et al. Genome‐wide CRISPR‐Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I‐related protein MR1. Nat Immunol 2020; 21: 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang S, Gilfillan S, Cella M, et al. Evidence for MR1 antigen presentation to mucosal‐associated invariant T cells. J Biol Chem 2005; 280: 21183–21193. [DOI] [PubMed] [Google Scholar]
- 31. Rigau M, Ostrouska S, Fulford TS, et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 2020; 367. [DOI] [PubMed] [Google Scholar]
- 32. Gras S, Chen Z, Miles JJ, et al. Allelic polymorphism in the T cell receptor and its impact on immune responses. J Exp Med 2010; 207: 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reantragoon R, Kjer‐Nielsen L, Patel O, et al. Structural insight into MR1‐mediated recognition of the mucosal associated invariant T cell receptor. J Exp Med 2012; 209: 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams EJ, Chien YH, Garcia KC. Structure of a γδ T cell receptor in complex with the nonclassical MHC T22. Science 2005; 308: 227–231. [DOI] [PubMed] [Google Scholar]
- 35. Kurioka A, Jahun AS, Hannaway RF, et al. Shared and distinct phenotypes and functions of human CD161++ Vα7.2+ T cell subsets. Front Immunol. 2017; 8: e1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gold MC, Eid T, Smyk‐Pearson S, et al. Human thymic MR1‐restricted MAIT cells are innate pathogen‐reactive effectors that adapt following thymic egress. Mucosal Immunol 2013; 6: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fu G, Casas J, Rigaud S, et al. Themis sets the signal threshold for positive and negative selection in T‐cell development. Nature 2013; 504: 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walter DM, Venancio OS, Buza EL, et al. Systematic In Vivo inactivation of chromatin‐regulating enzymes identifies setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res 2017; 77: 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holst J, Szymczak‐Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T‐cell receptor retrogenic mice. Nat Protoc 2006; 1: 406–417. [DOI] [PubMed] [Google Scholar]
- 40. Heemskerk MH, Hoogeboom M, de Paus RA, et al. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA‐2‐specific T‐cell receptor complexes expressing a conserved alpha joining region. Blood 2003; 102: 3530–3540. [DOI] [PubMed] [Google Scholar]
- 41. Behren A, Anaka M, Lo PH, et al. The Ludwig institute for cancer research Melbourne melanoma cell line panel. Pigment Cell Melanoma Res 2013; 26: 597–600. [DOI] [PubMed] [Google Scholar]
- 42. Boulter JM, Glick M, Todorov PT, et al. Stable, soluble T‐cell receptor molecules for crystallization and therapeutics. Protein Eng 2003; 16: 707–711. [DOI] [PubMed] [Google Scholar]
- 43. Garboczi DN, Utz U, Ghosh P, et al. Assembly, specific binding, and crystallization of a human TCR‐αβ with an antigenic Tax peptide from human T lymphotropic virus type 1 and the class I MHC molecule HLA‐A2. J Immunol 1996; 157: 5403–5410. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article, supplementary information and source data or are available upon reasonable requests to the authors.