

Abstract
Developing efficient and inexpensive CO2 capture technologies is a significant way to reduce carbon emissions. In this work, a novel chitosan/lithium sulfonate double network high strength hydrogel is synthesized by electron beam radiation. Due to the electron beam having a wide radiation area and certain penetrating power, the free radical polymerization can be initiated more uniformly and quickly in the hydrogel. The network structure of the hydrogel prepared by radiation-initiated polymerization is more uniform than that prepared by conventional chemical initiator-initiated polymerization. Meanwhile, the introduction of the second network to construct the double network structure does not reduce the surface area of the aerogel, which is different from the conventional method of grafting or impregnation modified porous materials. Moreover, the synthesized aerogels have good physical and chemical stability. The freeze-dried aerogels possess a porous structure and CO2 capture ability due to the CO2-philic double network structure. Because of the inexpensive raw material and convenient radiation process, this work can reduce the cost of CO2 adsorbents and has prospects of application in the field of CO2 solid adsorbents.
Chitosan hydrogel is regenerated from alkali/urea aqueous solution and the lithium sulfonate second network is introduced by electron beam radiation-initiated in situ free radical polymerization. The freeze-dried aerogel has CO2 capture capacity.
1. Introduction
Carbon dioxide (CO2) is a common greenhouse gas emitted by industrial manufacture and human activities. It is generally believed that the massive emission of CO2 is the main cause of global climate change.1 Most CO2 emissions came from burning fossil fuels in industrial production and transportation.2 Developing Carbon Capture, Utilization and Sequestration (CCUS) is critical to reducing CO2 emissions and mitigating severe climate change.3
Amine scrubbing technology is a traditional method of CO2 absorption in industry, but inherent disadvantages are serious corrosion of equipment and high cost of regeneration.4,5 Compared with liquid amine scrubbing strategy, solid adsorption technology is more desirable due to its low energy consumption, simple operation, easy regeneration, and so on.6 All the time, researchers have developed various porous CO2 solid adsorbents such as zeolites, metal–organic frameworks (MOFs),7 carbon-based materials8 and porous organic polymers (POPs).9 Porous polymer is considered as a promising solid adsorption material because of its high specific surface area, adjustable pore size and stable chemical structure.10,11 In addition to physical and chemical stability, high CO2 absorption capacity and selectivity of porous materials under ambient conditions are also essential.12 However, the physical adsorbents require large pressure of CO2 and exhibit relatively low selectivity toward CO2 in the presence of other gases.13 Theoretically, increasing the isosteric heat of CO2 adsorption via the introduction of CO2-philic groups should have a great effect on the capture capacity and selectivity.14 However, the conventional methods of grafting and impregnation to introduce CO2-philic groups usually has negative influences on surface area and may result in a decrease in CO2 absorption capacity.15 Possible strategy to overcome the shortcoming is utilizing porous polymer with high specific surface area and physicochemical stability as the matrix and then introducing CO2-philic groups.16
Hydrogels are a series of functional materials with high water content and three dimensional crosslinked hydrophilic networks. Due to the characteristics of high water content, porosity and typical cell compatibility, hydrogels can be applied in drug release, pollution adsorption, sensors and tissue engineering by regulating the chemical composition and structure of hydrogels.17–20 However, due to the inherent structural heterogeneity or lack of effective energy dissipation mechanism, hydrogels often do not have sufficient mechanical strength, which limits their applications in many fields.21 Researchers designed tough hydrogels by introducing effective energy dissipation mechanisms or unique structures, such as double-network hydrogels,22 nanocomposite hydrogels23 and so on. Aerogel is highly porous and lightweight material produced by sublimating the liquid components of conventional gel, with an average pore diameter of tens of nanometers.24 The traditional preparation methods of aerogels are freeze-drying (low temperature, low pressure) or critical point drying (CPD, high temperature, high pressure) of precursor materials or gels, in which the aqueous phase is replaced with gas (air) by transitioning through either a solid (freeze-drying) or supercritical (CPD) regime.25 As a kind of porous material, aerogels have been widely investigated in the fields of thermal insulation, catalysis and adsorption in recent years due to their excellent performance such as low volume density, high porosity, large specific surface area and low thermal conductivity.26–29
Chitosan is a natural biopolymer prepared by deacetylation of chitin.30 Chitosan has excellent characteristics including biocompatibility, biodegradability and low immunogenicity, which make it an ideal candidate for the construction of microcapsules, films, hydrogels and so on.31 In the past decades, researchers have designed numerous methods for synthesis of chitosan hydrogels. Most chitosan-based hydrogels were constructed based on chitosan acidic solvent system and the mechanical properties are relatively fragile, which limits its application in some fields.32,33 Some strategy such as chemical cross-linking, nanoparticle reinforcement and blending with other polymers have been used to enhance their mechanical strength. However, these methods resulted in only a moderate improvement and sometimes even sacrificed the inherent properties of chitosan.34–36 Zhang and co-workers broke through these limitations of traditional method of acid dissolving chitosan, using a new solvent system (4.5 wt% LiOH/7 wt% KOH/8 wt% urea aqueous solution) to successfully dissolve chitosan at low temperature. The chitosan alkali/urea solution was immersed into hot water (T ≥ 40 °C). Heating and poor solvents can destroy the alkali/urea hydrogen-bonded chitosan complexes, resulting in the aggregation of the chitosan chain in parallel to form regenerated chitosan nanofibers and then construct chitosan physical hydrogels.37
Ionizing radiation has long been considered as a very suitable method of forming hydrogels.38 The advantage of this method is that free radicals are generated directly on the monomer, thus avoiding the use of any chemical initiator. Polymerization can take place at any temperature and the polymerization time can be as short as a few minutes.39 In addition, the radiation method is relatively simple and convenient, and the degree of cross-linking or grafting—which strongly impacts the properties of hydrogels—can be controlled by changing the radiation dose.40 Szafulera et al. synthesized derivatives of dextran by coupling of methacrylated glycidyl to the structure of polysaccharide, and constructed chemically crosslinked hydrogels by electron-beam irradiation of in aqueous solution. The swelling property and cell compatibility of the hydrogels were evaluated, implying possibility of biomedical applications especially in the field of soft tissue regenerative medicine of the dextran-based hydrogels.41
Herein is reported the chitosan hydrogel regenerated from alkali/urea aqueous solution and the introduction of lithium sulfonate second network by electron beam radiation-initiated in situ free radical polymerization. The freeze-dried aerogel can be used as a solid adsorbent for CO2 capture due to the porous structure and CO2-philic polymer double network. Firstly, the chitosan alkali/urea aqueous solution is immersed into hot water and the chitosan physical hydrogel is prepared. Then, the hydrogel is immersed in a solution containing lithium sulfonate monomer and the concentration difference impels the monomer molecules into the hydrogel. Finally, under the electron beam radiation, the in situ free radical polymerization of the monomer is initiated in the chitosan hydrogel and the double-network hydrogel containing second network is constructed. Due to the physically/chemically crosslinked double network, the hydrogel exhibits great mechanical properties. Through a series of characterization of FTIR, XRD, SEM, TG, N2 adsorption–desorption and so on, the composition, structure, microtopography, stability and porous properties of the hydrogel and freeze-dried aerogel are researched. The impacts of different radiation dose on the surface area of aerogels are also studied. In addition, the properties of traditional chemical initiator-initiated and radiation-initiated hydrogel and aerogel are discussed by these characterizations. Moreover, the CO2 capture capacity of radiation-initiated and chemical initiator-initiated aerogels is measured and studied.
2. Experimental section
2.1. Materials
Chitosan (degree of deacetylation ≥90%) was purchased from Shanghai Macklin Biochemical Co., Ltd. Lithium hydroxide (LiOH) (98%), 2-acrylamide-2-methylpropanesulfonic acid (AMPS) (98%), acrylamide (AAm) (99%), N,N′-methylenebisacrylamide (MBA) (99%) were purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd. Potassium hydroxide (KOH) (AR), urea (AR) and potassium persulfate (KPS) (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. All the reagents were not further purified.
2.2. Preparation of chitosan hydrogels
An aqueous solution containing 4.5 wt% LiOH/7 wt% KOH/8 wt% urea was prepared and precooled to −18 °C. Weighing 96 g of alkali/urea aqueous solution into a beaker and then 4 g chitosan powder was added to it and stirring fully under ice water bath for 10 min. The suspension was placed at −50 °C for 24 h and then taken out to melt in ice water bath with mechanical stirring. After stirred evenly, 4 wt% chitosan alkali/urea aqueous solution was prepared. 20 g of the solution was centrifuged to eliminate bubbles and then transferred into a Petri dish to form a ∼3 mm thick disk by tape casting. The Petri dish was immersed in hot water at 45 °C for 30 min and then the chitosan physical hydrogel was prepared. The hydrogel was swelled in deionized water for 72 h to remove residual alkali/urea molecules. The original physical hydrogel was coded as O-gel.
2.3. Synthesis of chitosan/P(AMPSL-co-AAm) hydrogels initiated by chemical initiator
Firstly, a series of prepolymer water solutions (20 mL) consisting of different amounts of AMPS, LiOH, AAm, MBA and KPS were configured under the ice water bath (Table S1†). The prepared O-gel was immersed into the prepolymer solution and stood at 5 °C for 72 h. The concentration difference inside and outside the hydrogels impelled the monomer, crosslinker and initiator molecules into the hydrogels. The hydrogels were taken out from the solution, then sealed and transferred to a 65 °C oven. The in situ free radical polymerization of the hydrogels was initiated by chemical initiator for 10 h. Typically, the prepolymer water solution (20 mL) consisted AMPS (4.0 g), LiOH (0.462 g), AAm (0.412), MBA (0.0077 g) and 5% KPS (136 μL) and the hydrogel synthesized by this raw material ratio was coded as I-gel.
2.4. Synthesis of chitosan/P(AMPSL-co-AAm) hydrogels initiated by electron beam radiation
A prepolymer water solutions (20 mL) containing AMPS (4.0 g), LiOH (0.462 g) and different amounts of AAm and MBA were configured under the ice water bath (Table S1†). The O-gel was immersed into the prepolymer solution and stood at 5 °C for 72 h. The concentration difference inside and outside the hydrogels impelled the monomer and crosslinker molecules into the hydrogels. The hydrogels were taken out from the solution and then sealed. The hydrogels were irradiated by a 1 MeV electron accelerator (Wasik Associates, USA) at ambient temperature with different total dose (kGy) and dose rate of 10 kGy/pass. The in situ free radical polymerization of the hydrogels was initiated under electron beam radiation. Typically, the prepolymer water solution (20 mL) consisted AMPS (4.0 g), LiOH (0.462 g), AAm (0.412) and MBA (0.0077 g) and the hydrogel synthesized by a total dose of 40 kGy was coded as R-gel.
In addition, the P(AMPSL-co-AAm) hydrogel was synthesized for comparison. Typically, AMPS (4.0 g), LiOH (0.462 g), AAm (0.412) and MBA (0.0077 g) were dissolved in 10 mL deionized water. The solution was poured into a Petri dish. The hydrogel was synthesized with a total dose of 40 kGy and dose rate of 10 kGy/pass. The P(AMPSL-co-AAm) hydrogel was coded as P-gel.
Fig. 1 is the schematic diagram of preparation of hydrogels by electron beam radiation. A tray containing the hydrogel sample follows the track into the room, where is equipped with an electron accelerator. Each time the tray enters the room, the sample is exposed to one dose rate (10 kGy/pass) of radiation. The total radiation dose to the sample is equal to the product of the dose rate and the number of circles.
Fig. 1. Schematic diagram of preparation of hydrogels by electron beam radiation.

2.5. Characterization
The chemical composition, micromorphology and structure of the aerogel samples were characterized by Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM) and X-ray diffraction (XRD). The chemical composition of the aerogel samples was characterized by FTIR recorded on a Nicolet iS50 instrument. ATR mode was used for FTIR characterization. The transmittance of the samples with wave number in the range of 400–4000 cm−1 was recorded. SEM observation of the inner morphology of the aerogel was made on a Hitachi SU8010 scanning electron microscopy. The samples were plated with Au before SEM characterization. XRD measurement was carried out on an XRD diffractometer (Bruker D8 Advance). The XRD patterns with Cu Kα radiation (λ = 0.15406 nm) at 30 kV and 15 mA were recorded in the region of 2θ from 5° to 45°.
The mechanical properties of the hydrogel samples were characterized by tension tests, which were performed on a universal testing machine (SANS CMT) at a speed of 2 mm min−1. The hydrogels were cut into a dumbbell shape (gauge length = 50 mm, thickness = 3 mm and narrow part width = 4 mm) for tensile test.
The swelling behavior of the hydrogels in deionized water was characterized by gravimetric method. The hydrogels were immersed in 200 mL of deionized water, and then every other day the hydrogel was placed in a fresh 200 mL of deionized water. After 3 d, the swelling ratio (SR, g g−1) was calculated by the eqn (1).
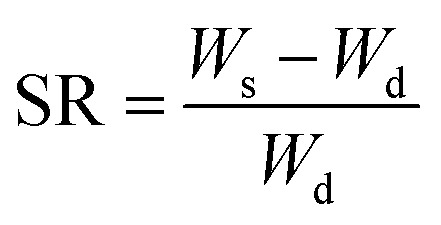 |
1 |
where Ws (g) is hydrogel weight after swelling for 3 d, and Wd (g) is the sample weight of freeze-dried hydrogel.
Porosity analyses were performed using Vsorb 2800P analyzer using high purity and high pressure N2 and He. The pressure of N2 and He was controlled to 0.1 MPa by the pressure reducing valve. Before characterization the samples were subjected to vacuum heat treatment for 3 h to remove the adsorbed impurity molecules. The N2 adsorption–desorption isotherms were recorded at liquid nitrogen temperature (77 K) and a relative pressure (P/P0) range of 0 to 0.1. The surface area and pore volume and size were calculated from N2 adsorption–desorption isotherm by means of Brunauer–Emmett–Teller (BET) model and Barrett–Joyner–Halenda (BJH) model.
The thermal properties of aerogels were measured by thermogravimetric analysis (TGA, Netzsch TG-209) at a programmed heating rate of 10 °C min−1 from room temperature up to 790 °C in N2 atmosphere. The TG curves recorded the relationship between the weight loss and temperature rise. The DTG curves is the first derivative of the TG curves with respect to temperature, representing the relationship between the rate of weight loss and temperature.
2.6. CO2 capture
The CO2 capture capacity of the aerogel samples was measured by volumetric method in a gas adsorption instrument (Vsorb 2800P, China). High purity and pressure CO2 was used in the experiment, and the outlet pressure was controlled at 0.1 MPa using the pressure reducing valve. Before the CO2 adsorption experiment, the aerogels were pretreated in a vacuum environment at 60 °C for 3 h to remove the adsorbed impurity molecules and then cooled to room temperature. The sample tube was placed in a dewar filled with water (25, 50 and 75 °C, respectively) and the CO2 was gradually fed into the sample tube. The instrument automatically calculated the CO2 adsorption volume under different pressures and converted it to the value in the standard temperature and pressure (STP), thus obtaining the adsorption isotherm. The capture capacity (CC, mg g−1) was calculated by the eqn (2).
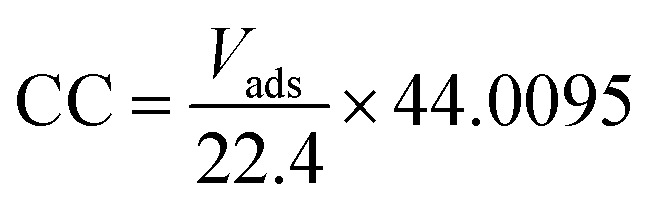 |
2 |
where Vads is the adsorption volume (cm3 g−1, STP), 22.4 and 44.0095 is the molar volume of gas under STP (L mol−1) and CO2 molar mass (g mol−1), respectively.
3. Results and discussion
3.1. Characterization
3.1.1. FTIR analysis
FT-IR spectra were carried out to clarify the chemical composition of chitosan and P(AMPSL-co-AAm) in R-gel and I-gel (Fig. 2). The peak (1028, 1059 cm−1) assigned to the saccharide structure42 (C–O–C, C–OH stretching vibration) is observed in O-gel, which both appear in the spectrum of R-gel (1039 cm−1) and I-gel (1038 cm−1), indicating the presence of chitosan macromolecules in R-gel and I-gel. The peaks (1588, 1655 cm−1) attributed to chitosan –NH2 bending vibration also appear in the R-gel (1545, 1660 cm−1) and I-gel (1545, 1661 cm−1) spectrum but the peak intensity is obviously enhanced.43 That is resulted from the superposition of absorption peaks (–NH bending vibration and C O stretching vibration), indicating the presence of acylamino groups in P(AMPSL-co-AAm).44 It is worth noting that the peaks ascribed to –OH (3362 cm−1) of O-gel shifted to 3320 cm−1 (R-gel) and 3327 cm−1 (I-gel) respectively, revealing that the introduction of P(AMPSL-co-AAm) allows the hydrogel to form a stronger hydrogen bond network.42 The strong peaks (1182, 1159, 623 cm−1) in R-gel and (1183, 1159, 624 cm−1) in I-gel corresponding to the –SO3Li characteristic peak, also indicating the successful introduction of the second network.16
Fig. 2. FTIR spectra of O-gel, R-gel and I-gel.

3.1.2. SEM analysis
Fig. 3 shows the SEM images of O-gel, P-gel, R-gel and I-gel. Intuitively, each sample has its unique microtopography. There is a specific range of pore size distribution in the SEM images of each samples, confirming the porous structure of aerogels. In Fig. 3a, all pore sizes seem to be relatively close. The approximate diameter of a typical and obvious pore is marked and compared it with the scale (30 μm). The diameter of the pore is about 5 μm can be calculated. That indicates the chitosan macromolecules aggregate in parallel to form regenerated nanofibers and form physical entanglement and cross-linking. The SEM image of P-gel (Fig. 3b) shows the pores are denser than O-gel, and most of the pores are distributed in about 5 μm (although containing a few pores of larger diameter). Actually, although both the O-gel and P-gel all have porous microtopography, their pore characteristics are different. There is a combination of these two pore characteristics in Fig. 3c, representing the complex of the CTS and P(AMPSL-co-AAm). However, the I-gel double network structure (Fig. 3d) is not as uniform as that of R-gel. The reason may be that KPS in the prepolymer solution inevitably initiates the polymerization (Scheme 1), leading to some oligomers in I-gel, further resulting in uneven dispersion of monomer and crosslinker molecules in the hydrogel. However, the prepolymer solution of R-gel does not contain KPS, which allows the monomer and crosslinker molecules evenly dispersed in the hydrogel. In addition, because the electron beam has wide radiation area and a certain penetrating power, the free radical polymerization can be initiated more uniformly and quickly. Therefore, the double-network structure of R-gel is firmer and more uniform, further resulting in the more uniform pore structure.
Fig. 3. SEM images of freeze-dried hydrogels. (a) O-gel; (b) P-gel; (c) R-gel; (d) I-gel.

Scheme 1. Schematic illustration of the synthesis of electron beam radiation initiated chitosan/P(AMPSL-co-AAm) hydrogel and aerogel.

3.1.3. XRD analysis
In Fig. 4, two diffraction peaks appear at 2θ = 11° and 20° in pattern (a), corresponding to the diffraction peaks of chitosan (020) and (110) crystal face, respectively. Pattern (b) and (c) likewise have two diffraction peaks at 2θ = 11° and 20°, but the peak intensity is decreased. This may be due to the introduction of the second network, which reduces the degree of order in chitosan macromolecules. In addition, the peak intensity in pattern (b) is lower than (c), indicating that electron beam radiation is more likely to cause the degradation of chitosan than high temperature.45
Fig. 4. XRD patterns of freeze-dried hydrogels. (a) O-gel; (b) R-gel; (c) I-gel.

3.1.4. Mechanical properties
The mechanical properties of hydrogels are essential for their applications. Fig. 5 shows the typical tensile stress–strain curves of O-gel, R-gel, I-gel and P-gel, and the tensile fracture stress and strain of O-gel, R-gel, I-gel and P-gel are (82 kPa, 57.5%) (98 kPa, 71.3%), (88 kPa, 65.9%) and (73 kPa, 98.8%), respectively. The results show that the mechanical properties especially the tensile strength of the double-network hydrogel I-gel and R-gel are improved compared with O-gel and P-gel. Because the double-network hydrogel contains effective energy dissipation mechanism compared with the single-network hydrogel, and there are more sacrificial bonds paly significant role during stretching.46 Furthermore, the polymer chain is slightly degraded and depolymerized due to the electron beam radiation, yet the tensile strength of R-gel is higher than I-gel. That is probably because prepolymer solution of I-gel contains initiator KPS, which inevitably initiates polymerization, further leading to the uneven and inadequate dispersion of monomers and crosslinkers in hydrogel during immersion. However, the prepolymer solution of R-gel contains monomers and crosslinkers exclusively, making the even and adequate dispersion of monomers and crosslinkers in hydrogel. Moreover, the electron beam radiation is faster and more even than the heat conduction during the polymerization process, hence the network structure of R-gel is more uniform,47 which corresponding to the analysis of SEM pictures. Interestingly, it can be intuitively seen from the hydrogel pictures that the light transmittance of R-gel is lower than O-gel (Fig. 6a), and the R-gel can be severely knotted, twisted and stretched (Fig. 6b–d).
Fig. 5. Tensile stress–strain curves of the O-gel, P-gel, R-gel and I-gel at room temperature.

Fig. 6. Pictures of (a) transmittance of O-gel and R-gel; (b) knotted R-gel; (c) twisted R-gel; (d) stretched R-gel.

3.1.5. Analysis of textural properties
Information related to porous properties of the aerogels is obtained from their respective nitrogen adsorption–desorption isotherms recorded at 77 K (Fig. 7a). In the relative pressure range of P/P0 = 0–0.01, the aerogels exhibit a sharp absorption of N2 molecules. The steep absorption of adsorbent at low P/P0 is due to the enhanced adsorbent–adsorptive interactions in the narrow micropores, causing the micropores to be filled at low P/P0. That indicates the presence of micropores in the aerogels.48,49 In the higher relative pressure interval (P/P0 = 0.1–0.9), N2 gas absorption increases gradually. In the relative pressure range of P/P0 = 0.9–1.0, the N2 gas absorption is particularly rapid, and the hysteresis appears in the desorption stage. This adsorption behaviour is depended on the adsorbent–adsorptive interactions and also the interactions between the molecules in the condensed state. Capillary condensation accompanied with hysteresis often occurs when the mesopore width exceeds a certain critical width.49 That indicates the presence of mesopores in aerogels. These characterization results show the presence of particular pores in the polymer network. In other words, the pore structure of aerogels is essentially hierarchical, with pore sizes ranging from microporous to mesoporous.50 BJH adsorption pore size distribution (PSD) curves of aerogels (Fig. 7b) are estimated from N2 adsorption data at 77 K. According to the PSD profile, aerogels contains micropores (<2 nm) and mesopores (2–50 nm). The BET surface area, Langmuir surface area, BJH adsorption cumulative volume and BJH and SF median pore width of aerogels are listed in Table 1. The SABET of O-gel is 85.22 m2 g−1. With the introduction of the second network, the surface area of I-gel and R-gel increased to 102.43 m2 g−1 and 114.18 m2 g−1, respectively. And the pore size of aerogels decreases with the increase of surface area. Compared with I-gel, R-gel has larger surface area and smaller pore width. This may be because the monomer and crosslinker molecules evenly dispersed in the hydrogel before radiation process. Moreover, due to the wide radiation area and good penetrating power of electron beam, the free radical polymerization can be initiated more uniformly and quickly. Therefore, the double-network structure of R-gel is more uniform and stable, further resulting in the more uniform pore structure. This result also corresponds to the SEM and mechanical properties characterization.
Fig. 7. (a) N2 adsorption–desorption isotherm of aerogels at 77 K, (b) distribution of pore sizes in aerogels.

Pore properties of aerogels.
| Sample | SABET (m2 g−1) | SALang (m2 g−1) | V total (cm3 g−1) | W BJH median (nm) | W SF median (nm) |
|---|---|---|---|---|---|
| O-gel | 85.22 | 121.16 | 0.213 | 3.91 | 1.30 |
| I-gel | 102.43 | 150.20 | 0.238 | 2.84 | 1.05 |
| R-gel | 114.18 | 157.05 | 0.261 | 2.05 | 0.99 |
3.1.6. Thermal properties
Thermogravimetric analysis is an important method to evaluate the thermal stability of polymer composites, also can reflect the interaction of components in the composite system. The good stability of solid adsorbents used for CO2 capture is often emphasized. Especially in the flue gas environment, the thermal stability is essential for the adsorption capacity.8,51 In order to investigate the thermal properties of aerogels, the TG and DTG data of O-gel, R-gel and I-gel is obtained during the heating program of 25–790 °C with a heating rate 10 °C min−1 (Fig. 8). O-gel loses adsorbed free water obviously in the range of 70–100 °C. At 290 °C, the chitosan macromolecules undergo severe thermal degradation, corresponding to a rapidly decreasing weight curve.42 The mass decreases slowly in the range of 350–790 °C. For the R-gel and I-gel, the severe mass loss also occurs at 290 °C, but the rate of mass loss is slower. Moreover, the mass loss of R-gel and I-gel compared to that of O-gel is reduced at high temperatures. These indicate that there is a strong interaction between P(AMPSL-co-AAm) and CTS hydrogel matrix.42 The mass loss of R-gel is significantly decreased than I-gel at higher temperature, implying that the interaction between double networks of R-gel is stronger. The peaks about 320, 370 and 700 °C in DTG curves (Fig. 8b) of I-gel and R-gel are attributed to the thermal decomposition of P(AMPSL-co-AAm). In summary, the results indicate that the thermal stability of R-gel is better than that of O-gel and I-gel. R-gel CO2 adsorbent will be stable enough to function in flue gas environments (generally up to around 120 °C) due to its good thermal properties.52,53
Fig. 8. (a) TGA curves and (b) DTG curves of freeze-dried O-gel, R-gel and I-gel (25–790 °C, 10 °C min−1).

3.2. CO2 capture properties
Establishing strong interaction between the polymer network and CO2 is a crucial method to enhance its CO2 capture capacity. In addition to the inherently high surface area, if its surface is CO2-Philic, such a polymer porous material will exhibit more efficient CO2 absorption capacity.9 The chemical functionalization of porous material with polar groups (such as nitrogen-rich groups, oxygen-rich groups and inorganic ions) can enhance the average dipole–quadruple interactions with CO2, thus improving the CO2 capture capacity.54 Besides, ensuring the presence of nitrogen-based structural motifs in porous materials can also promote the Lewis base–acid interaction. Nitrogen-rich entities (such as amine, imine, azo and cyano) on the polymer skeleton act as Lewis-base sites can bind to acidic CO2 molecules, thus improving the CO2-philic properties of porous surfaces.9,55–57 The chitosan skeleton matrix of the aerogels herein reported is actually amino-rich macromolecules. The CO2 capture capacity of O-gel at 298 K is investigated primarily. CO2 sorption isotherms are measured at 298 K with the pressure range from 0 to 0.1 MPa (Fig. 9a), and the gravimetric capture of CO2 by O-gel is 38.6 mg g−1. Theoretically, an electric field can be generated on the surface of a functionalized carbon-based polymer network, which can enhance the affinity of the network to CO2 through high quadrupole moment.16 There are up to three open coordination sites after Li+ cation in –SO3Li is fully activated, leading to stronger interaction with CO2 molecules.58 Sulfonate-functionalized porous materials will enhance its CO2 capture capacity. Indeed, lithium sulfonate-functionalized I-gel showed conspicuous increases in gravimetric CO2 capture with the value of 63.3 mg g−1 at 298 K and 0.1 MPa. Moreover, lithium sulfonate-functionalized R-gel exhibits a gravimetric CO2 capture of 67.9 mg g−1 at 298 K and 0.1 MPa. Compared with I-gel, the CO2 capture capacity of R-gel is more prominent. That is probably because the network of R-gel is more uniform and the pores are firmer and more stable, which indicates the superiority of radiation method over traditional chemical initiation method, and this also corresponds to the above characterization. The capture capacity is better than that of the previously reported quaternized chitosan/PVA aerogels (0.18 mmol g−1),59 and also superior to the reported chitosan grafted graphene oxide aerogel (0.257 mmol g−1).60 As the operating temperature increases from 298 K to 348 K, the CO2 capture capacity of the R-gel decreases from 67.9 mg g−1 to 43.9 mg g−1 (Fig. 9b). The adsorbing capacity of physical adsorption is usually lower at high temperatures because of the desorption of CO2 at high temperatures. The increased motion of CO2 molecular at high temperature reduces the surface interaction of the adsorbent, resulting in a decrease in adsorption capacity.61,62
Fig. 9. (a) CO2 adsorption isotherm of three aerogels at 298 K, (b) CO2 adsorption isotherm of R-gel at different temperatures.

It is generally understood that CO2 capture capacity is determined by numerous factors such as surface area, pore size and pore function, etc., each of which carries different weights at different pressures and temperatures. At low pressures, the interaction between CO2 and pore surface plays a leading role in CO2 capture. The CO2 adsorption isotherm of the aerogels rises sharply in low pressure range, indicating that the amino and lithium sulfonate present in aerogels have favorable binding energy with CO2 molecules. Researchers have investigated the mechanism of the absorption of CO2 by amino groups and proposed two possible explanations. One mechanism shows that two amines react with one CO2 to produce carbamate, which can be further converted to urea and followed by CO2 regeneration. The other mechanism is that one amine group reacts with one CO2 and one H2O to form bicarbonate or two amine groups reacts with one CO2 and one H2O to form carbonate (Fig. 10a).54,63,64 The FTIR spectra before and after CO2 capture of R-gel are compared (Fig. 10b). After CO2 capture, R-gel reacted with CO2 to produce bicarbonate species (1453 cm−1). In addition, the bending vibration peak (1377 cm−1) of –OH turns into two peaks (1387 cm−1, 1371 cm−1) in the R-gel spectrum after CO2 capture, indicating the presence of carbonate species. This may indicate that R-gel captured CO2 in the presence of moisture. Because the hydrophilic polymer network inevitably adsorbs some moisture, which may be not completely removed in the pretreatment stage before CO2 capture. When the pressure increases to 0.1 MPa, the effect of functionalization on CO2 capture gradually weakened, simultaneously the effect of surface area gradually increased. Therefore, the CO2 capture capacity of R-gel is larger than I-gel due to its higher surface area.16
Fig. 10. (a) Mechanism of CO2 capture by amino in the absence and presence of moisture; (b) FTIR spectra of R-gel before and after CO2 capture in the range of 1300–1600 cm−1.

4. Conclusion
Herein is reported a novel chitosan/lithium sulfonate hydrogel fabricated by electron beam radiation-initiated in situ free radical polymerization. Because the electron beam has wide radiation area and a certain penetrating power, the polymerization can be initiated more uniformly and quickly. Due to the introduction of the radiation-initiated second network, the double-network structure of R-gel is firmer and more uniform, further resulting in the more uniform pore structure of aerogel. This is also reflected in SEM, mechanical testing, TG, surface area and pore size analysis and other characterization results. Different from the traditional grafting or impregnation modified porous materials, the surface area can be improved by introducing a second network to construct a double network structure. N2 adsorption–desorption shows that aerogels have a specific surface area of 114.18 m2 g−1 and hierarchical porous structure from microporous to mesoporous. Because of the porous structure and presence of CO2-philic amino and lithium sulfonate, the R-gel can capture CO2 efficiently (67.9 mg g−1 at 298 K, 0.1 MPa). Since the aerogel have physical and chemical stability, practical applications in capturing CO2 are entirely possible in flue gas environment. Due to the raw materials of chitosan are sustainable and widely available, and the radiation process is simple and convenient, we hope to provide reference for the design of inexpensive and efficient CO2 adsorbents.
Conflicts of interest
There is no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This work is in memory of Prof. Lina Zhang, and it is supported by grants from the National Natural Science Foundation of China (51903080), Natural Science Foundation of Hubei Province (2018CFC854), Scientific Research Foundation of Hubei University of Science and Technology (2019-20KZ06), the Open Project of Key Laboratory of Green Chemical Engineering Process of Ministry of Education (GCP20200210).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra03041h
References
- Solomon S. Plattner G.-K. Knutti R. Friedlingstein P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. U. S. A. 2009;106(6):1704–1709. doi: 10.1073/pnas.0812721106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueres C. Le Quéré C. Mahindra A. Bäte O. Whiteman G. Peters G. et al., Emissions are still rising: ramp up the cuts. Nature. 2018;564:27–30. doi: 10.1038/d41586-018-07585-6. [DOI] [PubMed] [Google Scholar]
- Vega F. Baena-Moreno F. Fernández L. M. G. Portillo E. Navarrete B. Zhang Z. Current status of CO2 chemical absorption research applied to CCS: towards full deployment at industrial scale. Appl. Energy. 2020;260:114313. doi: 10.1016/j.apenergy.2019.114313. [DOI] [Google Scholar]
- Liang L. Liu C. Jiang F. Chen Q. Zhang L. Xue H. et al., Carbon dioxide capture and conversion by an acid-base resistant metal-organic framework. Nat. Commun. 2017;8(1):1–10. doi: 10.1038/s41467-016-0009-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-R. Ma Y. McCarthy M. C. Sculley J. Yu J. Jeong H.-K. et al., Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord. Chem. Rev. 2011;255(15–16):1791–1823. doi: 10.1016/j.ccr.2011.02.012. [DOI] [Google Scholar]
- Choi S. Drese J. H. Jones C. W. Adsorbent materials for carbon dioxide capture from large anthropogenic point sources. ChemSusChem. 2009;2(9):796–854. doi: 10.1002/cssc.200900036. [DOI] [PubMed] [Google Scholar]
- Cai Z. Bien C. E. Liu Q. Wade C. R. Insights into CO2 Adsorption in M–OH Functionalized MOFs. Chem. Mater. 2020;32(10):4257–4264. doi: 10.1021/acs.chemmater.0c00746. [DOI] [Google Scholar]
- Zhang Z. Luo D. Lui G. Li G. Jiang G. Cano Z. P. et al., In-situ ion-activated carbon nanospheres with tunable ultramicroporosity for superior CO2 capture. Carbon. 2019;143:531–541. doi: 10.1016/j.carbon.2018.10.096. [DOI] [Google Scholar]
- Bera R. Ansari M. Alam A. Das N. Triptycene, phenolic-OH, and Azo-functionalized porous organic polymers: efficient and selective CO2 capture. ACS Appl. Polym. Mater. 2019;1(5):959–968. doi: 10.1021/acsapm.8b00264. [DOI] [Google Scholar]
- Luo R. Chen M. Liu X. Xu W. Li J. Liu B. et al., Recent advances in CO2 capture and simultaneous conversion into cyclic carbonates over porous organic polymers having accessible metal sites. J. Mater. Chem. A. 2020;8:18408–18424. doi: 10.1039/D0TA06142E. [DOI] [Google Scholar]
- Das S. Heasman P. Ben T. Qiu S. Porous organic materials: strategic design and structure–function correlation. Chem. Rev. 2017;117(3):1515–1563. doi: 10.1021/acs.chemrev.6b00439. [DOI] [PubMed] [Google Scholar]
- Manyà J. J. González B. Azuara M. Arner G. Ultra-microporous adsorbents prepared from vine shoots-derived biochar with high CO2 uptake and CO2/N2 selectivity. Chem. Eng. J. 2018;345:631–639. doi: 10.1016/j.cej.2018.01.092. [DOI] [Google Scholar]
- Ma R. Hu P. Xu L. Fan J. Wang Y. Niu M. et al., Nanostructured polyethylenimine decorated palygorskite for reversible CO2 capture. Mater. Express. 2017;7(4):253–260. doi: 10.1166/mex.2017.1374. [DOI] [Google Scholar]
- Dawson R. Adams D. J. Cooper A. I. Chemical tuning of CO2 sorption in robust nanoporous organic polymers. Chem. Sci. 2011;2(6):1173–1177. doi: 10.1039/C1SC00100K. [DOI] [Google Scholar]
- Rao N. Wang M. Shang Z. Hou Y. Fan G. Li J. CO2 adsorption by amine-functionalized MCM-41: a comparison between impregnation and grafting modification methods. Energy Fuels. 2018;32(1):670–677. doi: 10.1021/acs.energyfuels.7b02906. [DOI] [Google Scholar]
- Lu W. Yuan D. Sculley J. Zhao D. Krishna R. Zhou H.-C. Sulfonate-grafted porous polymer networks for preferential CO2 adsorption at low pressure. J. Am. Chem. Soc. 2011;133(45):18126–18129. doi: 10.1021/ja2087773. [DOI] [PubMed] [Google Scholar]
- Fu J. Hydrogel properties and applications. J. Mater. Chem. B. 2019;7(10):1523–1525. doi: 10.1039/C9TB90023C. [DOI] [PubMed] [Google Scholar]
- Gaharwar A. K. Peppas N. A. Khademhosseini A. Nanocomposite hydrogels for biomedical applications. Biotechnol. Bioeng. 2014;111(3):441–453. doi: 10.1002/bit.25160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman A. S. Hydrogels for biomedical applications. Adv. Drug Delivery Rev. 2012;64:18–23. doi: 10.1016/j.addr.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Vieira R. M. Vilela P. B. Becegato V. A. Paulino A. T. Chitosan-based hydrogel and chitosan/acid-activated montmorillonite composite hydrogel for the adsorption and removal of Pb+2 and Ni+2 ions accommodated in aqueous solutions. J. Environ. Chem. Eng. 2018;6(2):2713–2723. doi: 10.1016/j.jece.2018.04.018. [DOI] [Google Scholar]
- Gong J. P. Why are double network hydrogels so tough? Soft Matter. 2010;6(12):2583–2590. doi: 10.1039/B924290B. [DOI] [Google Scholar]
- Gong J. P. Katsuyama Y. Kurokawa T. Osada Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003;15(14):1155–1158. doi: 10.1002/adma.200304907. [DOI] [Google Scholar]
- Wang J. Lin L. Cheng Q. Jiang L. A strong bio-inspired layered PNIPAM–clay nanocomposite hydrogel. Angew. Chem., Int. Ed. 2012;51(19):4676–4680. doi: 10.1002/anie.201200267. [DOI] [PubMed] [Google Scholar]
- Akimov Y. K. Fields of application of aerogels. Instrum. Exp. Tech. 2003;46(3):287–299. doi: 10.1023/A:1024401803057. [DOI] [Google Scholar]
- De France K. J. Hoare T. Cranston E. D. Review of hydrogels and aerogels containing nanocellulose. Chem. Mater. 2017;29(11):4609–4631. doi: 10.1021/acs.chemmater.7b00531. [DOI] [Google Scholar]
- Hüsing N. Schubert U. Aerogels—airy materials: chemistry, structure, and properties. Angew. Chem., Int. Ed. 1998;37(1-2):22–45. doi: 10.1002/(SICI)1521-3773(19980202)37:1/2<22::AID-ANIE22>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Zu G. Kanamori K. Nakanishi K. Lu X. Yu K. Huang J. et al., Superelastic multifunctional aminosilane-crosslinked graphene aerogels for high thermal insulation, three-component separation, and strain/pressure-sensing arrays. ACS Appl. Mater. Interfaces. 2019;11(46):43533–43542. doi: 10.1021/acsami.9b16746. [DOI] [PubMed] [Google Scholar]
- Yuan H. Yang G. Luo Q. Xiao T. Zuo Y. Guo X. et al., A 3D net-like structured fluorescent aerogel based on carboxy-methylated cellulose nanofibrils and carbon dots as a highly effective adsorbent and sensitive optical sensor of Cr (VI) Environ. Sci.: Nano. 2020;7(3):773–781. doi: 10.1039/C9EN01394F. [DOI] [Google Scholar]
- Peydayesh M. Suter M. K. Bolisetty S. Boulos S. Handschin S. Nyström L. et al., Amyloid fibrils aerogel for sustainable removal of organic contaminants from water. Adv. Mater. 2020;32(12):1907932. doi: 10.1002/adma.201907932. [DOI] [PubMed] [Google Scholar]
- Zhong Q.-Q. Yue Q.-Y. Gao B.-Y. Li Q. Xu X. A novel amphoteric adsorbent derived from biomass materials: Synthesis and adsorption for Cu (II)/Cr (VI) in single and binary systems. Chem. Eng. J. 2013;229:90–98. doi: 10.1016/j.cej.2013.05.083. [DOI] [Google Scholar]
- Dash M. Chiellini F. Ottenbrite R. M. Chiellini E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011;36(8):981–1014. doi: 10.1016/j.progpolymsci.2011.02.001. [DOI] [Google Scholar]
- Hua S. Yang H. Wang W. Wang A. Controlled release of ofloxacin from chitosan–montmorillonite hydrogel. Appl. Clay Sci. 2010;50(1):112–117. doi: 10.1016/j.clay.2010.07.012. [DOI] [Google Scholar]
- Chatterjee S. Lee M. W. Woo S. H. Adsorption of congo red by chitosan hydrogel beads impregnated with carbon nanotubes. Bioresour. Technol. 2010;101(6):1800–1806. doi: 10.1016/j.biortech.2009.10.051. [DOI] [PubMed] [Google Scholar]
- Zhang H. Qadeer A. Chen W. In situ gelable interpenetrating double network hydrogel formulated from binary components: thiolated chitosan and oxidized dextran. Biomacromolecules. 2011;12(5):1428–1437. doi: 10.1021/bm101192b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnol C. Rodrigues F. H. Pereira A. G. Fajardo A. R. Rubira A. F. Muniz E. C. Superabsorbent hydrogel composite made of cellulose nanofibrils and chitosan-graft-poly (acrylic acid) Carbohydr. Polym. 2012;87(3):2038–2045. doi: 10.1016/j.carbpol.2011.10.017. [DOI] [Google Scholar]
- Chen Y. Chen L. Bai H. Li L. Graphene oxide–chitosan composite hydrogels as broad-spectrum adsorbents for water purification. J. Mater. Chem. A. 2013;1(6):1992–2001. doi: 10.1039/C2TA00406B. [DOI] [Google Scholar]
- Duan J. Liang X. Cao Y. Wang S. Zhang L. High strength chitosan hydrogels with biocompatibility via new avenue based on constructing nanofibrous architecture. Macromolecules. 2015;48(8):2706–2714. doi: 10.1021/acs.macromol.5b00117. [DOI] [Google Scholar]
- Rosiak J. Ulański P. Synthesis of hydrogels by irradiation of polymers in aqueous solution. Radiat. Phys. Chem. 1999;55(2):139–151. doi: 10.1016/S0969-806X(98)00319-3. [DOI] [Google Scholar]
- Safrany A. Beiler B. László K. Svec F. Control of pore formation in macroporous polymers synthesized by single-step γ-radiation-initiated polymerization and cross-linking. Polymer. 2005;46(9):2862–2871. doi: 10.1016/j.polymer.2005.02.024. [DOI] [Google Scholar]
- Nho Y. C. Lim Y. M. Lee Y. M. Preparation, properties and biological application of pH-sensitive poly (ethylene oxide)(PEO) hydrogels grafted with acrylic acid (AAc) using gamma-ray irradiation. Radiat. Phys. Chem. 2004;71(1–2):239–242. doi: 10.1016/j.radphyschem.2004.03.046. [DOI] [Google Scholar]
- Szafulera K. Wach R. A. Olejnik A. K. Rosiak J. M. Ulański P. Radiation synthesis of biocompatible hydrogels of dextran methacrylate. Radiat. Phys. Chem. 2018;142:115–120. doi: 10.1016/j.radphyschem.2017.01.004. [DOI] [Google Scholar]
- Duan J. Liang X. Guo J. Zhu K. Zhang L. Ultra-stretchable and force-sensitive hydrogels reinforced with chitosan microspheres embedded in polymer networks. Adv. Mater. 2016;28(36):8037–8044. doi: 10.1002/adma.201602126. [DOI] [PubMed] [Google Scholar]
- Zhang C. Wang Y. Guo J. Liu Y. Zhou Y. Chitosan nanoparticle carrier based on surface molecularly imprinted polymers for the recognition and separation of proteins. RSC Adv. 2015;5(128):106197–106205. doi: 10.1039/C5RA14088A. [DOI] [Google Scholar]
- Tang X. Huang T. Zhang S. Wang W. Zheng H. The role of sulfonated chitosan-based flocculant in the treatment of hematite wastewater containing heavy metals. Colloids Surf., A. 2020;585:124070. doi: 10.1016/j.colsurfa.2019.124070. [DOI] [Google Scholar]
- Zhao W.-W. Yu L. Zhong X.-G. Zhang Y.-F. Sun J.-Z. Wang Q. Studies on Radiation Effect of Chitosan (II)–Some of the Chemical Changes Induced by Irradiation. Chem. Res. Chin. Univ. 1993;06:868–871. [Google Scholar]
- Lin T. Bai Q. Peng J. Xu L. Li J. Zhai M. One-step radiation synthesis of agarose/polyacrylamide double-network hydrogel with extremely excellent mechanical properties. Carbohydr. Polym. 2018;200:72–81. doi: 10.1016/j.carbpol.2018.07.070. [DOI] [PubMed] [Google Scholar]
- Sokker H. El-Sawy N. M. Hassan M. El-Anadouli B. E. Adsorption of crude oil from aqueous solution by hydrogel of chitosan based polyacrylamide prepared by radiation induced graft polymerization. J. Hazard. Mater. 2011;190(1–3):359–365. doi: 10.1016/j.jhazmat.2011.03.055. [DOI] [PubMed] [Google Scholar]
- Qi S.-C. Yu G.-X. Xue D.-M. Liu X. Liu X.-Q. Sun L.-B. Rigid supramolecular structures based on flexible covalent bonds: A fabrication mechanism of porous organic polymers and their CO2 capture properties. Chem. Eng. J. 2020;385:123978. doi: 10.1016/j.cej.2019.123978. [DOI] [Google Scholar]
- Thommes M. Kaneko K. Neimark A. V. Olivier J. P. Rodriguez-Reinoso F. Rouquerol J. et al., Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report) Pure Appl. Chem. 2015;87(9–10):1051–1069. [Google Scholar]
- Patel H. A. Je S. H. Park J. Jung Y. Coskun A. Yavuz C. T. Directing the structural features of N2-phobic nanoporous covalent organic polymers for CO2 capture and separation. Chem.–Eur. J. 2014;20(3):772–780. doi: 10.1002/chem.201303493. [DOI] [PubMed] [Google Scholar]
- Yang M. Ma C. Xu M. Wang S. Xu L. Recent Advances in CO2 Adsorption from Air: a Review. Curr. Pollut. Rep. 2019;5(4):272–293. doi: 10.1007/s40726-019-00128-1. [DOI] [Google Scholar]
- Aaron D. Tsouris C. Separation of CO2 from flue gas: a review. Sep. Sci. Technol. 2005;40(1–3):321–348. doi: 10.1081/SS-200042244. [DOI] [Google Scholar]
- He F. Wang T. Fang M. Wang Z. Yu H. Ma Q. Screening test of amino acid salts for CO2 absorption at flue gas temperature in a membrane contactor. Energy Fuels. 2017;31(1):770–777. doi: 10.1021/acs.energyfuels.6b02578. [DOI] [Google Scholar]
- Zou L. Sun Y. Che S. Yang X. Wang X. Bosch M. et al., Porous organic polymers for post-combustion carbon capture. Adv. Mater. 2017;29(37):1700229. doi: 10.1002/adma.201700229. [DOI] [PubMed] [Google Scholar]
- Zhu Y. Long H. Zhang W. Imine-linked porous polymer frameworks with high small gas (H2, CO2, CH4, C2H2) uptake and CO2/N2 selectivity. Chem. Mater. 2013;25(9):1630–1635. doi: 10.1021/cm400019f. [DOI] [Google Scholar]
- Arab P. Parrish E. İslamoğlu T. El-Kaderi H. M. Synthesis and evaluation of porous azo-linked polymers for carbon dioxide capture and separation. J. Mater. Chem. A. 2015;3(41):20586–20594. doi: 10.1039/C5TA04308E. [DOI] [Google Scholar]
- Deng G. Wang Z. Triptycene-based microporous cyanate resins for adsorption/separations of benzene/cyclohexane and carbon dioxide gas. ACS Appl. Mater. Interfaces. 2017;9(47):41618–41627. doi: 10.1021/acsami.7b15050. [DOI] [PubMed] [Google Scholar]
- Xu Q. Liu D. Yang Q. Zhong C. Mi J. Li-modified metal–organic frameworks for CO2/CH4 separation: a route to achieving high adsorption selectivity. J. Mater. Chem. 2010;20(4):706–714. doi: 10.1039/B912407A. [DOI] [Google Scholar]
- Song J. Liu J. Zhao W. Chen Y. Xiao H. Shi X. et al., Quaternized chitosan/PVA aerogels for reversible CO2 capture from ambient air. Ind. Eng. Chem. Res. 2018;57(14):4941–4948. doi: 10.1021/acs.iecr.8b00064. [DOI] [Google Scholar]
- Hsan N. Dutta P. Kumar S. Bera R. Das N. Chitosan grafted graphene oxide aerogel: Synthesis, characterization and carbon dioxide capture study. Int. J. Biol. Macromol. 2019;125:300–306. doi: 10.1016/j.ijbiomac.2018.12.071. [DOI] [PubMed] [Google Scholar]
- Sevilla M. Fuertes A. B. Sustainable porous carbons with a superior performance for CO2 capture. Energy Environ. Sci. 2011;4(5):1765–1771. doi: 10.1039/C0EE00784F. [DOI] [Google Scholar]
- Alhwaige A. A. Agag T. Ishida H. Qutubuddin S. Biobased chitosan hybrid aerogels with superior adsorption: role of graphene oxide in CO2 capture. RSC Adv. 2013;3(36):16011–16020. doi: 10.1039/C3RA42022A. [DOI] [Google Scholar]
- Planas N. Dzubak A. L. Poloni R. Lin L.-C. McManus A. McDonald T. M. et al., The mechanism of carbon dioxide adsorption in an alkylamine-functionalized metal–organic framework. J. Am. Chem. Soc. 2013;135(20):7402–7405. doi: 10.1021/ja4004766. [DOI] [PubMed] [Google Scholar]
- Sayari A. Belmabkhout Y. Stabilization of amine-containing CO2 adsorbents: dramatic effect of water vapor. J. Am. Chem. Soc. 2010;132(18):6312–6314. doi: 10.1021/ja1013773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.