

Abstract
Background:
Modern intrathecal drug delivery systems (IDDS) are technologically advanced to deliver medication through various automated and patient-controlled programs. They also are associated with unique complications ranging from postoperative complications, medication-related adverse events (AE), device malfunction, to refill associated AE.
Objectives:
To systematically analyze real-world complications and AE reported on the Food and Drug Administration’s Manufacturer and User Facility Device Experience database (MAUDE) associated with IDDS among patients predominantly with chronic pain disorders.
Materials and Methods:
MAUDE database was sampled for a month four times a year during the study period, February 2018 to February 2019. The database was resampled every six months till August 2020 to evaluate for any additional reported cases during the index months. The two FDA approved IDDS, were included. AE were broadly classified into causes related to catheter malfunction, pump malfunction, biologic, and medication-related AE.
Results:
A total of 1001 reports were included in the final analysis. The top three reasons for adverse report are infection/erosion (15.7%, n = 157), motor stall (12.4%, n = 125) and adverse medication reactions (11.8%, n = 119), respectively. There were five deaths among patients with IDDS. Epidural hematoma (n = 3) after IDDS surgery resulted in a death and residual neurological deficits after surgical evacuation. Programming errors, medication concentration discrepancy, and failure to turn on the pump after reprogramming are various preventable causes of medication-related IDDS AEs.
Conclusions:
Analysis of AE associated with IDDS from the MAUDE database provided a real-world perspective different from reported registry complications. Awareness and vigilance of preventable IDDS-related complications is the first step toward mitigating risks to provide safe and effective intrathecal drug delivery for chronic pain management.
Keywords: Chronic pain, complications, FDA, implantable devices, intrathecal drug delivery system
INTRODUCTION
Implantable intrathecal drug delivery systems (IDDS) are utilized for the treatment of severe refractory pain conditions. Systematic reviews of published trials and expert consensus statements report that patients with IDDS had better pain control, fewer medication-related side effects, and increased life expectancy among cancer patients (1-3). Since the introduction of intrathecal opioids for treating chronic pain, significant strides were made with the design of implantable devices, surgical techniques, and management of these devices. Modern intrathecal drug delivery systems (IDDS) are technologically advanced to deliver medication through various automated and patient-controlled programs. They are also associated with unique complications ranging from those occurring during the immediate post-operative period to medication-related adverse events (AE), device malfunction, and follow-up-related AE. These complications and AE associated with IDDS were reported from small, single-center retrospective reviews, observational studies, and systematic reviews (4). Clinical trials often enroll a narrowly defined group of patients undergoing care from experts in the field, and the follow-up is limited to the study duration (3). AE reports of clinical registries often include only one particular type of device getting their case from the experts (5). These events are often not generalizable to the real-world population. Passive surveillance systems like Manufacturer And User Facility Device Experience (MAUDE) provide unique insights into real-world occurrence of AE (6). In this study, we attempt to analyze real-world complications, and AE systematically reported on the MAUDE database associated with IDDS among patients with chronic pain disorders.
MATERIALS AND METHODS
Protocol Development
The protocol was developed by two authors (V.G. and S.H.). Due to the deidentified nature of publicly available data, the study is deemed to be exempted from Institutional Review Board review.
Data Source
The Food and Drug Administration’s (FDA) Manufacturer And User Facility Device Experience (MAUDE) database is a reporting system of adverse events associated with medical devices. The FDA guides manufacturers, importers, and device user facilities to report device-related adverse events. The guidance is stipulated in Medical Device Reporting (MDR) regulation (21 CFR part 803). The manufacturers and importers must report any device malfunction that would likely cause death or contribute to serious injury. The device user facilities, which include all health care facilities, excluding a physician’s office, are also required to report any device malfunction. Voluntary reports from patients, consumers, and health care professionals regarding device malfunctions are included in the database. The database is searchable and contains reports for the last 10 years. The data are reported to the end of the previous month.
The results of the database are available for a query on a publicly accessible website (https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfMAUDE/search.CFM). MDR data cannot be used to examine trends or to compare events between devices.
Sampling Strategy
We postulated that the reports of adverse device-related events remain stable after a significant period of entry into the market. As a passive surveillance system, MAUDE is designed to provide representative real-world effects of device-related malfunctions. To account for seasonal trends, we sampled the database for one month period four times a year. The study period involved February 2018 to February 2019. The database was resampled every six months till August 2020 to evaluate for any additional reported cases during the index months. There are currently two devices for IDDS: SynchroMed (Medtronic, Minneapolis, MN, USA) and Prometra (Flowonix, NJ, USA), both included in the study. There was a single report of discontinued Codman 3000 series implantable infusion pump (Johnson & Johnson Medical Devices, NJ, USA), which was not included in our analysis. The study design is summarized in Figure 1. A separate analysis was performed for deaths during the entire study period.
Figure 1.

Summary of the study design. The database was resampled every six months until August 2020 to evaluate any additional cases during the index months. SynchroMed (Medtronic) and Prometra (Flowonix).
Development of AE Classification Scheme
We reviewed reports from February 2018 to develop a malfunction classification scheme. The AE scheme broadly divides complications into those related to catheter malfunction, pump malfunction, biologic, or medication-related AE (Table 1).
Table 1.
Complication and AE Categories Associated With IDDS.
| Catheter-related AE | |
| C1 | Catheter severed/damaged/nicked/broken/fractured |
| C2 | Catheter kink |
| C3 | Catheter migration |
| C4 | Catheter occlusion |
| C5 | Fluid collection around the catheter |
| Pump-related AE | |
| P1 | Motor stall |
| P2 | Pump flipped |
| P3 | Pump empty/low volume |
| P4 | High residual volume |
| P5 | ERI/EOS |
| Biologic causes | |
| G | Granuloma |
| I | Infection/erosion |
| H | Spinal headache |
| A | Allergic reaction |
| He | Hematoma |
| PD | Pump site discomfort |
| Medication-related Reports | |
| M | Medication-related adverse events |
EOS, end of service; ERI, elective replacement indicator.
Data Manipulation
The monthly data of malfunction reports were downloaded as comma-separated value (.csv) files and were reviewed in Microsoft Excel (Microsoft Office 2010). Two study authors reviewed each report to exclude duplicates. Each report was analyzed for patient symptoms, type of malfunction, the pharmacologic agent used in the IDDS, indication for therapy (cancer and non-cancer-related chronic pain), and subsequent management. We suspect that most of these patients were treated for chronic pain (vs. spasticity, hypertension, and other rare indications). Reports associated with the infusion of the drugs related to spasticity (e.g., baclofen) and pulmonary hypertension (e.g., treprostinil) were removed to focus on patients with chronic pain disorders. Reports with inadequate or insufficient information for analysis of adverse events were also excluded from the study.
Analysis
Strategies for management of malfunction were reviewed and reported as a nonsurgical treatment or requiring surgical revision.
RESULTS
The inclusion criteria for the cohort of individuals with reports included for final analysis are summarized in Figure 2. A total of 1001 reports were included for the final analysis. Among these reports, 932 (93.1%) AE and 69 (6.9%) AE were associated with SynchroMed (Medtronic) and Prometra (Flowonix) pumps, respectively. The details of individual reports are summarized in Table 2. The top three reasons for adverse report are infection/erosion (15.7%, n = 157), motor stall (12.4%, n = 125) and adverse medication reactions (11.8%, n = 119), respectively. The overall number of reports per month per each AE category seems to vary during the study period.
Figure 2.
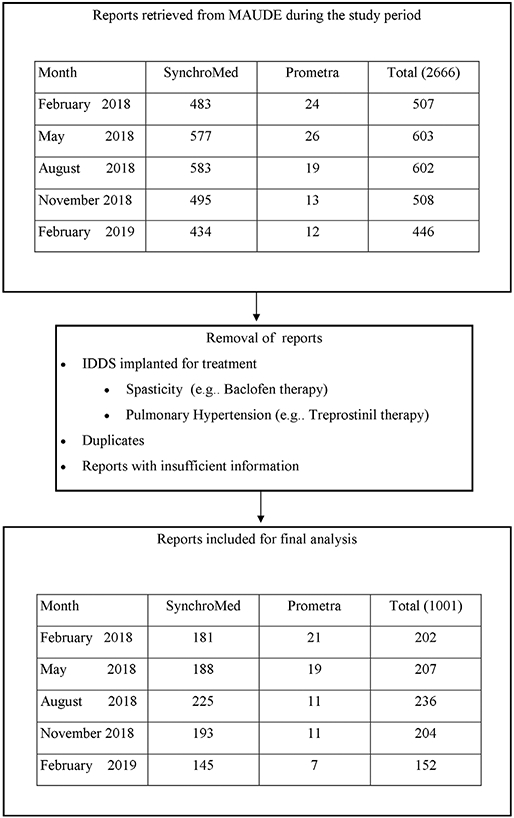
CONSORT style flow chart of cohort derivation. MAUDE, Manufacturer and User Facility Device Experience.
Table 2.
Summary of Complications and AE Associated With IDDS.
| Total | Feb 2018 | May 2018 | Aug 2018 | Nov 2018 | Feb 2019 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catheter-related AE | |||||||||||
| Catheter severed/damaged/nicked/broken/fractured | 82 | 19 | 2 | 16 | NR | 19 | NR | 13 | 2 | 11 | NR |
| Catheter kink | 53 | 10 | 1 | 13 | 1 | 6 | NR | 12 | 1 | 9 | NR |
| Catheter migration | 37 | 7 | NR | 4 | 1 | 10 | NR | 11 | NR | 4 | NR |
| Catheter occlusion | 51 | 8 | 3 | 3 | 1 | 10 | 1 | 17 | NR | 8 | NR |
| Fluid collection around catheter | 26 | 3 | NR | 1 | NR | 8 | NR | 6 | 1 | 7 | NR |
| Pump-related AE | |||||||||||
| Motor stall | 125 | 17 | NR | 27 | NR | 24 | NR | 31 | NR | 26 | NR |
| Pump movement in the pocket | 72 | 17 | NR | 11 | NR | 21 | 1 | 12 | NR | 10 | NR |
| Pump empty/low volume | 86 | 25 | 1 | 18 | NR | 38 | 1 | 1 | NR | 2 | NR |
| High residual volume | 50 | 5 | 8 | 6 | 4 | 6 | 2 | 8 | 5 | 0 | 6 |
| ERI/EOS | 24 | 8 | NR | 1 | NR | 3 | NR | 8 | NR | 4 | NR |
| Biologic AE | |||||||||||
| Granuloma | 19 | 2 | NR | 2 | NR | 1 | NR | 6 | NR | 8 | NR |
| Infection/Erosion | 157 | 25 | 4 | 26 | 6 | 34 | 2 | 32 | 1 | 27 | NR |
| Spinal headache | 18 | 3 | NR | 5 | NR | NR | 1 | 2 | NR | 7 | NR |
| Allergic reaction | 6 | NR | NR | 3 | 1 | 1 | NR | NR | NR | 1 | NR |
| Hematoma | 3 | NR | NR | 1 | NR | NR | NR | 2 | NR | NR | NR |
| Pump site discomfort | 68 | 8 | 1 | 11 | 3 | 16 | 2 | 15 | 1 | 11 | NR |
| Medication-related AE | |||||||||||
| Medication-related AE | 119 | 24 | 1 | 37 | 2 | 28 | 1 | 16 | NR | 10 | NR |
Gray columns indicate adverse events associated with Prometra (Flowonix, NJ, USA). EOS, end of service; ERI, elective replacement indicator; NR, not reported.
DEATHS
There were a total of five deaths (0.5%) during the study period. One patient had bacterial meningitis after the IDDS implant, and the patient never received intrathecal medications through the pump. The second patient had a spinal epidural hematoma one week after the implant procedure. The patient subsequently died of complications resulting from spinal epidural hematoma. The flow through the catheter was noted to be low due to the pressure effect from the hematoma. There was a patient with chronic regional pain syndrome (CRPS) with IDDS who underwent hip surgery who died of an opioid overdose after an escalation of oral opioids in the post-operative period. There was one report of a patient who died of sepsis after device implant. There was a report of a patient with a Prometra pump (Flowonix) who underwent MRI without emptying the pump. The death was thought to be secondary to morphine overdose.
When the search was expanded to include the entire period of February 2018 to February 2019, three additional reports were found. There is a report of a patient with blunt trauma secondary to a fall after discontinuation of IDDS therapy following marijuana use. There were two other reports of opioid overdose-related deaths, one with continued use of oral opioids after initiation of IDDS therapy and another due to a programming error.
DEVICE COMPONENT AE
Damage to the Catheter
There were a total of 82 (8.2%) case reports in which the catheter was damaged, severed, nicked, broken, or fractured. Clinical presentation varied, and the most common manifestation was worsening pain or withdrawal symptoms from intrathecal therapies (n = 20). Nine patients had medication leak into the pump pocket site during follow-up, resulting in overdose symptoms (n = 3), numbness (n = 1), and fluid at the pocket site (n = 3). Two patients presented with a post-dural puncture headache because of CSF leak from the damaged catheter. Catheter damage was diagnosed in one patient during the workup of a pump that was mobile and flipping inside the pocket. There were eight cases of catheter damage during planned spine surgery. During one of the spine surgeries, the catheter was disconnected due to technical reasons. The majority (n = 59) of the patients with catheter damage underwent catheter replacement, revision surgery, or the replacement of the entire system. Few patients (n = 3) underwent the explantation of the whole system.
Kinking of the Catheter
There were a total of 53 (2.2%) cases of catheter kinking during the study period. The catheter kink was attributed to the Dacron pouch in one report. The most common presentation (n = 32) was worsening pain, withdrawal symptoms, or loss of efficacy of therapy. A few patients had high residual volume during device refills (n = 3). The majority of the patients (n = 40) underwent surgery to revise or replace the catheter. A few (n = 2) patients underwent explantation of the IDDS.
Catheter Migration
There were a total of 37 (3.7%) reports with catheter migration. The most common presentation (n = 22) was worsening pain, loss of efficacy of IDDS therapy, or withdrawal symptoms. One patient reported worsening pain during attempted aspiration from the catheter access port. During workup for migration in one patient, the catheter was found in the epidural space. The majority of patients (n = 32) had catheter revision, and one patient had the entire system explantation.
Catheter Occlusion
There were 51 (0.5%) cases with catheter occlusion. Complete catheter occlusion was classified as a catheter occlusion event. The majority (n = 26) had symptoms of worsening pain, loss of efficacy of IDDS therapy, or withdrawal symptoms. Most of the patients were treated with revision surgery (n = 39). Some patients had their device filled with saline (n = 2) or underwent device explantation (n = 3).
Fluid Collection Around the Catheter and the Pump
There were a total of 26 (2.6%) reports of patients with fluid collection around the spinal incision site and or the pocket. The fluid collection collections could be due to CSF leaks, infectious complications, or serosanguinous collections. Patients were treated with observation, drainage (n = 3), purse-string suture at the catheter entry site (n = 3), revision (n = 10), and explantation (n = 4). After a fall with resultant CSF leak at the spinal incision site, one patient had a fluid collection, which was treated with a purse-string suture placement and fibrin glue application.
Pump Motor Stall
There were a total of 125 (12.4%) cases of pump motor stall during the study period. The patients presented with worsening pain and or withdrawal symptoms (n = 40), device alarms (n = 30), or were found during incidentally device interrogation (n = 28). Several patients had motor stall after MRI scanning (n = 26). The patients were treated with device reprogramming (n = 11), replacement (n = 61) or explantation procedures (n = 6). The motor stall problem was resolved without surgical intervention in some patients (n = 23). Return product analysis frequently found the following defects: electrical shorting due to electrochemical migration across the electrical feed-through insulator, motor gear train, or a shaft bearing anomaly due to wear and tear.
Pump Movement in the Pocket
There were a total of 72 (7.2%) cases associated with pump movement in the pocket. Difficulty in refilling the pump was the most common clinical presentation in these patients. The majority of the patients required corrective surgery (n = 50) or underwent an explantation procedure (n = 4). The device was manipulated over the skin in one patient to have access to the catheter port. In one patient, a pouch was used to minimize motion in the pocket. There were reports (n = 2) of difficulties using personal therapy manager (PTM, Medtronic), likely due to communication problems with the pump.
Empty or Low Residual Pump Volume
There were a total of 86 (8.6%) patients who presented with pump alarm (n = 41) or worsening pain/withdrawal symptoms (n = 21). Patients with empty or low residual volumes were treated with pump refill (n = 32), replacement (n = 2), explant (n = 4), dose reduction (n = 1), and turning off the pump (n = 2). Two patients were noted to have low volumes after missing refill appointments. Patients with empty or low residual pump volumes were hospitalized due to opioid overdose symptoms associated with volume discrepancy, withdrawal symptoms, and uncontrolled hypertension.
High Residual Pump Volume
There were a total of 50 (4.9%) cases of incidental high pump residual volumes during device refills. A few patients described uncontrolled pain or withdrawal symptoms (n = 12). The patients were treated with catheter replacement (n = 2), reduction in refill volumes (n = 3), device explantation (n = 17), and replacement of the system (n = 4).
Premature Battery Depletion (ERI/EOS)
There were a total of 24 (2.4%) patients with pumps with premature battery depletion. Patients presented with device alarm (n = 10) or worsening pain or withdrawal symptoms (n = 10). Most patients underwent replacement (n = 17) or pump explantation (n = 1).
BIOLOGIC AE
Granuloma
There were a total of 19 (1.9%) cases of granuloma noted in the study. The presentation ranged from asymptomatic incidental diagnosis on back MRI to new back pain, lack of efficacy, reservoir volume discrepancy, worsening leg weakness, and paralysis-like symptoms. Patients were treated with device explant (n = 6), catheter repositioning (n = 3), catheter replacement (n = 2) and unclear surgical intervention (n = 2). One report of a patient was noted to have a granuloma on MR imaging of the back, and his device was filled with saline. There was also a report of a patient who was followed up with serial imaging tests.
Infection/Erosion
There were a total of 157 (15.7%) cases of infection/erosion noted in the study. One hundred and twenty-three patients underwent device explantation, and four patients were treated conservatively with antibiotics only. There were two patients with sepsis requiring admission to the intensive care unit (ICU). There was one report of a patient developing a fungal infection of the IDDS. One patient was reported to have developed an infection after an abdominal massage. There was one report of device infection that was recognized during pump refill as pus was aspirated. One patient had evidence of abscess on MR imaging. There was one report of a patient who developed subdural hematoma after device surgery, who subsequently developed device infection.
Spinal Headache
There were 18 (1.8%) reports of spinal headache complications during the study period. There were reports of patients who had spinal headaches after device implant that were conservatively treated with hydration and bed rest. The majority of the patients had typical positional symptoms, and they improved with the epidural blood patch. There was one report of a patient who presented with headaches, delusional symptoms, and CSF accumulation around the pump site. The patient was successfully treated with catheter revision. There was a report of a patient with IDDS who had headaches after implant that did not improve with blood patches, and he was eventually diagnosed with sagittal sinus thrombosis. There was a report of a patient who had symptoms of spinal headaches and weakness had to undergo explantation of the system eventually.
Allergic Reactions
There were six (0.6%) reported cases of allergic reactions to IDDS. The database does not document the findings for the suspicion of allergic reaction nor any tests performed for its diagnosis. One patient underwent device explantation after adequate workup. The report describes a patient who developed a rash around a newly implanted device attributed to an allergic reaction. There was an accumulation of fluid around the catheter site. The patient was managed conservatively. There were three cases of skin changes around the pump site attributed to allergic reactions and changes resolved with extraction. Additionally, there was a report of a patient with discomfort at the pump site that settled with the removal of Dacron pouch and pocket revision surgery.
Spinal Epidural Hematoma
There were three reports of hematoma during our study period, with one of them resulting in death. The second reported a cauda equina syndrome 36 hours after IDDS implantation. Imaging tests showed epidural hematoma in the thoracolumbar spine, and the patient underwent T10-L1 laminectomies with the extraction of the system. Unfortunately, the patient developed residual neurological deficits in the lower extremities. The third patient developed lower extremity sensory deficits soon after surgery. The epidural hematoma was diagnosed and was surgically evacuated.
Pump Site Discomfort
There were a total of 68 (6.8%) cases associated with site discomfort. After appropriate workup, many patients underwent revision surgery (n = 45) or explantation (n = 9). There was a report of a patient who developed site discomfort after an accident and another after an MRI, and both were managed conservatively. A smaller volume reservoir pump was required in another patient with site discomfort. The use of abdominal binder and aspiration of pocket hematoma were other strategies used to manage pump site discomfort.
MEDICATION-RELATED AE
There were 119 (11.8%) reports of AE attributed to medication-related effects. There were four cases of medication-related adverse events among patients with the Prometra pump. The rest of the events were reported in patients with SynchroMed (Medtronic) pump. The cases are summarized in Table 3. Medication overdose events were the most common category, followed by events associated with withdrawal symptoms. Programming errors, failure to turn back the pump after MRI, and the concentration discrepancy of the medication are some of the preventable causes of medication-related AE reported in the database. Patients with medication-related pocket fill required visits to the emergency room or hospitalization for monitoring.
Table 3.
Medication-Related Adverse Events.
| Type of event | Number of reports (n = 119) |
|---|---|
| Overdose symptoms | |
| Medication synergy | 10 |
| Concentration discrepancy | 3 |
| Programming error | 8 |
| Pocket fill | 7 |
| Unclear reason | 48 |
| Change of catheter position | 1 |
| Withdrawal symptoms | 20 |
| IDDS was not turned back on after MRI | 1 |
| Urinary retention | 2 |
| Altered mental status | 3 |
| Urinary retention and altered mental status | 1 |
| Persistent rash | 1 |
| Adverse reaction to Ziconotide | 3 |
| Priapism | 1 |
| Pain and lymphedema | 1 |
| Unspecified reaction | 9 |
IDDS, intrathecal drug delivery system, MRI, magnetic resonance imaging.
DISCUSSION
Our study is a systemic analysis of AE associated with IDDS among patients with chronic pain disorders from the MAUDE reporting system. The study’s main findings are that the top three reasons for adverse reports after IDDS in the MAUDE database are infection, motor stall, and medication-related AE. Patients with pump motor stalls had medication withdrawal symptoms, and patients with an empty pump or low residual volumes were admitted with opioid overdose or withdrawal symptoms. Spinal epidural hematoma after IDDS surgery and programming errors can result in the most severe events, including death. Some of the deaths noted in the study are preventable with strict protocols for device management during the magnetic resonance imaging, prevention of programming errors, and judicious use of oral opioids among patients with IDDS. The AE seen in our study may reflect a heterogeneous group of patients with varying severity of underlying illness and providers with varying expertise.
AE and Complications
An adverse event is defined as an injury caused by medical management rather than the natural course of the disease (7). Adverse events happening immediately after a surgical procedure are referred to as postoperative complications. All pain management physicians must be aware of medical errors, near-miss events, and serious medical errors. A continuous quality improvement process is essential to identify such events, track outcomes, and make systematic changes to avoid recurrences.
Reporting of AE Associated With IDDS
There are several ways of reporting AE in the literature. They include case reports, case series, results from clinical trials, large registries to passive surveillance systems like MAUDE. The accuracy and generalizability of these reports vary based on the source of data. The key features of these reports are highlighted in Table 4. AE from case reports and case series report cases reveal unusual presentations and are more likely to represent severe cases (8) (Fig. 3).
Table 4.
Various Study Designs for Evaluating AE.
| Reports | Accuracy | Generalizability | Special attributes |
|---|---|---|---|
| Case reports | + | Poor | Highlights rare sensational attributes |
| Case series | ++ | Poor | Institutional bias |
| Clinical trials | +++++ | ++ | Small samples Expert care Sicker patients not represented AE are usually lower |
| Clinical registries | ++++ | +++++ | Often expensive Specific purposes Access for analysis is limited selection bias |
| Passive surveillance | +++ | +++++ | Inexpensive Publicly available Cross-sectional snapshot Reporting may not be accurate |
Figure 3.

The figure illustrates AE from various studies. Panel a shows how we would like to make inferences to a target population from a study population that was performed on a source population that can be enumerated. Panels b and c reveal case reports and case series from patients with IDDS. Notice that they often highlight cases with more severe AE (red circles) than milder AE (green circles). Panel d shows AE from clinical registries, which are a subset of all the IDDS patients. There may be a selection bias associated with clinical registries. Panel e shows AE from clinical trials. Often the AE rates are low and are not representative of the real-world practice. Panel f shows AE from passive surveillance systems like MAUDE. They represent real-world data, and the limitations are concerns with follow-up and lack of standardization of data reporting.
Several small studies and registries had shed light on AE associated with IDDS. Implantable systems performance registry (ISPR) was a voluntary product performance registry of Medtronic implantable devices that had reported AE among 6093 patients with IDDS (5). AE occurred in about 11% of the patients in this registry and are mostly related to catheter malfunction. Motor stalls occurred in 0.9% of the patients in the registry. Motor stalls were reported among patients exposed to magnetic fields from household appliances such as laptop speakers (9,10). Prometra (Flowonix) pumps have flow activated valves and do not have a motor that has potential for stalls, as seen in SynchroMed (Medtronic) pumps. The most common AE in the ISPR registry was catheter kink/ occlusion, which was noted in around 3.5% of the patients. Off label compounded drug formulations are associated with corrosion and premature pump failure (11). The rate of surgical site infection in one reported study was 6.5% (12). Catheter disconnection, leakage, programming errors, and bacterial meningitis were the reported AE in another small study (4). Post dural puncture headache (PDPH) after IDDS implantation was noted in 23% of the patients, and most patients were treated with conservative therapy, blood or fibrin glue patch (13). The low number of PDPH events (1.8%) seen in our study raises the possibility of underreporting to the MAUDE database.
In our study, there were seven cases of IDDS pocket fill during refill. A recent study showed that patients preferred ultrasound-guided refill as they were associated with less pain than template guided procedures (14). Ultrasound-guided refills took longer, and the safety events were similar in both the groups. Volume discrepancy between injected and sensor measured volumes should cue the providers for closer monitoring to prevent late AEs (15). Ultrasound guidance helps localize difficult to access reservoir port (16). Retained foreign bodies (Tuohy needle) leading to arachnoiditis was reported in the literature after IDDS implantation and was not noted in our study (17). Several studies have shown that adherence to infection control practices reduces the infection risk associated with IDDS surgery (18). Several patients with intrathecal inflammatory mass (granuloma) related to high dose morphine delivery through the IDDS reported in the literature (19).
Future Directions
A review of reports during the study period revealed more than 1000 adverse events associated with IDDS therapy. We anticipate that with improvements in computing, artificial intelligence, natural language processing, we may have a system to track AE’s, summarize them, and provide real-time feedback to the practitioners (20). Currently, most of the reports are submitted by the manufacturer, nurses, and patients. Physicians reports account for only 0.09% of the total MAUDE reports (11). Physician awareness and direct reporting may improve the quality of the reports.
Strengths and Limitations
Our study is a retrospective database analysis, and it has the limitations of any retrospective study. The study cannot provide rates of AE or compare AE between the two commercially available pumps. A total of 11,132 SynchroMed (Medtronic) targeted delivery pumps were implanted in 8997 patients till October 31, 2019, in the United States (21). A total of 6670 Prometra (Flowonix) programmable infusion pumps were implanted in the United States by December 24, 2019 (22). The number of pumps in service among patients with pain disorders during the study period could not be reliably obtained due to the lack of a centralized registry. Therefore, we are unable to evaluate the incidence rates of adverse events. It is also likely that an increased number of AE among patients with SynchroMed (Medtronic) pumps may reflect a higher rate of use in the general population. The unique patient characteristics (cancer pain vs. non-cancer pain indications) and long-term follow-up details are unavailable from the database. The possibility of increased AE among cancer-related pain patients due to associated disorders (immunosuppression, cachexia, etc.) and use of concomitant treatments (steroids, chemotherapeutic agents, and radiation therapy) could not be confirmed from our study. Lack of standardization of reporting resulted in significant variability in the reports. The causal relationship of AE to outcomes could not be adjudicated in our study. The strengths and weaknesses of our MAUDE database based study are summarized in Table 5.
Table 5.
Strengths and Weaknesses of a MAUDE Database-Based AE Study.
| Strengths | Limitations |
|
|
CONCLUSIONS
Analysis of AE associated with IDDS from the MAUDE database provided a real-world perspective different from reported registry complications. The study confirmed that even though the individual risk of AEs from IDDS for every single patient is low, the probability of encountering severe complications from IDDS is not negligible, as seen by the occurrence of spinal epidural hematoma and even mortality after IDDS surgery. The study emphasizes the need for extra vigilance during IDDS surgery and during follow-up to avoid complications and provide safe and effective intrathecal drug delivery in chronic pain management.
Supplementary Material
COMMENTS.
Goel et al. have published an important piece of work that adds to our understanding of IDDS therapy and the complications that can be seen. It is not surprising that complication rates are different than the ISPR as the reporting communities are different and likely the implanting community is different between the two groups. This information allows more targeted educational campaigns (e.g. around preventing motor stalls and preventing device infections for example) as well as better description to patients of the landscape of complications associated with this therapy. As neither are a true national or global registry that can capture the full incidence of complications our information is necessarily incomplete. We should work towards filling in the missing parts of the puzzle that remain. With the strong proviso of interpretation, I see the MAUDE database as a useful enquiry in the pursuit of neuromodulation safety.
Marc Russo, MBBS
Broadmeadow, NSW Australia
***
This article clearly shows how Database and registries could be important tools to evaluate in the future Adverse effects and complications of implanted devices.
Denis Dupoiron, MD
Angers, France
***
Much needed clarity and warning of continued vigilance with the use of IDDS.
Yeshvant Navalgund, MD
Greensburg, PA USA
***
I think this is an interesting overview of the potential adverse outcomes after placing an intrathecal pump. It is prudent for the readers to be refreshed on common pitfalls.
Mourad Shehebar, MD
New York, NY USA
Source(s) of financial support:
No funding was available for this study. Their respective departments support the investigators.
Footnotes
For more information on author guidelines, an explanation of our peer review process, and conflict of interest informed consent policies, please go to http://www.wiley.com/WileyCDA/Section/id-301854.html
Conflict of Interest: Dr. Patwardhan reports serving as a site PI for a Boston Scientific trial. Dr. Shankar has received honorarium from Medtronic for a focus group on acute pain. The remaining authors declare no relevant conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the supporting information tab for this article.
REFERENCES
- 1.Bottros MM, Christo PJ. Current perspectives on intrathecal drug delivery. J Pain Res 2014;7:615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deer TR, Pope JE, Hayek SM et al. The Polyanalgesic consensus conference (PACC): recommendations for intrathecal drug delivery: Guidance for improving safety and mitigating risks. Neuromodulation. 2017;20:155–176. [DOI] [PubMed] [Google Scholar]
- 3.Smith TJ, Coyne PJ, Staats PS et al. An implantable drug delivery system (IDDS) for refractory cancer pain provides sustained pain control, less drug-related toxicity, and possibly better survival compared with comprehensive medical management (CMM). Ann Oncol 2005;16:825–833. [DOI] [PubMed] [Google Scholar]
- 4.Kamran S, Wright BD. Complications of intrathecal drug delivery systems. Neuromodulation 2001;4:111–115. [DOI] [PubMed] [Google Scholar]
- 5.Konrad PE, Huffman JM, Stearns LM et al. Intrathecal drug delivery systems (IDDS): the implantable systems performance registry (ISPR). Neuromodulation 2016;19:848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doran J, Ward M, Ward B, Paskhover B, Umanoff M, Mammis A. Investigating complications associated with occipital nerve stimulation: a MAUDE study. Neuromodulation 2018;21:296–301. [DOI] [PubMed] [Google Scholar]
- 7.Sitzman BT. Adverse event protocol for interventional pain medicine: the importance of an organized response. Pain Med 2008;9:S108–S112. [Google Scholar]
- 8.Srinivasan SK, Patil AA. Axial (central) downward herniation of the brain as a complication of intra-thecal drug delivery system (IDDS) implant - a case report. Clin Neurol Neurosurg 2014;125:78–80. [DOI] [PubMed] [Google Scholar]
- 9.Huh B, Roldan CJ. Magnetic fields and intrathecal pump malfunction. Am J Emerg Med 2016;34:115 e115–115 e116. [DOI] [PubMed] [Google Scholar]
- 10.Kosturakis A, Gebhardt R. SynchroMed II intrathecal pump memory errors due to repeated magnetic resonance imaging. Pain Physician 2012;15:475–477. [PubMed] [Google Scholar]
- 11.Prager J, Deer T, Levy R et al. Best practices for intrathecal drug delivery for pain. Neuromodulation 2014;17:354–372. discussion 372. [DOI] [PubMed] [Google Scholar]
- 12.Scanlon MM, Gazelka HM, Moeschler SM et al. Surgical site infections in cancer patients with intrathecal drug delivery devices. Pain Med 2017;18:520–525. [DOI] [PubMed] [Google Scholar]
- 13.Neuman SA, Eldrige JS, Qu W, Freeman ED, Hoelzer BC. Post dural puncture headache following intrathecal drug delivery system placement. Pain Physician 2013;16:101–107. [PubMed] [Google Scholar]
- 14.Singa RM, Buvanendran A, McCarthy RJ. A comparison of refill procedures and patient outcomes following ultrasound-guided and template-guided intrathecal drug delivery systems with recessed ports. Neuromodulation. 2019;23:938–943. [DOI] [PubMed] [Google Scholar]
- 15.Maino P, Perez R, Koetsier E. Intrathecal pump refills, pocket fills, and symptoms of drug overdose: A prospective, observational study comparing the injected drug volume vs. the drug volume effectively measured inside the pump. Neuromodulation 2017;20:733–739. [DOI] [PubMed] [Google Scholar]
- 16.Shankar H. Ultrasound-guided localization of difficult-to-access refill port of the intrathecal pump reservoir. Neuromodulation. 2009;12:215–218. [DOI] [PubMed] [Google Scholar]
- 17.Kochany JZ, Tran ND, Sarria JE. Increasing back and radicular pain 2 years following intrathecal pump implantation with review of arachnoiditis. Pain Med 2013;14:1658–1663. [DOI] [PubMed] [Google Scholar]
- 18.Burgher AH, Barnett CF, Obray JB, Mauck WD. Introduction of infection control measures to reduce infection associated with implantable pain therapy devices. Pain Pract 2007;7:279–284. [DOI] [PubMed] [Google Scholar]
- 19.Ramsey CN, Owen RD, Witt WO, Grider JS. Intrathecal granuloma in a patient receiving high dose hydromorphone. Pain Physician 2008;11:369–373. [PubMed] [Google Scholar]
- 20.Crowson MG, Hamour A, Lin V, Chen JM, Chan TCY. Machine learning for pattern detection in cochlear implant FDA adverse event reports. Cochlear Implants Int 2020;21:313–322. [DOI] [PubMed] [Google Scholar]
- 21.SynchroMed Product Performance Report. https://www.medtronic.com/content/dam/medtronic-com/products/product-performance/ppr-reports/2019-product-performance-report-combined.pdf
- 22.Class 2 Device Recall Prometra Programmable Infusion Pump. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRES/res.cfm?id=178926
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.