

Abstract
Systemic sclerosis is the rheumatic disease with the highest individual mortality. The severity of the disease is determined by the extent of fibrotic changes to cutaneous and internal organ tissues, the most life-threatening visceral manifestations being interstitial lung disease, SSc-associated-pulmonary arterial hypertension and myocardial involvement. The heterogeneity of the disease has initially hindered the design of successful clinical trials, but considerations on classification criteria have improved patient selection in trials, allowing the identification of more homogeneous groups of patients based on progressive visceral manifestations or the extent of skin involvement with a focus of patients with early disease. Two major subsets of systemic sclerosis are classically described: limited cutaneous systemic sclerosis characterized by distal skin fibrosis and the diffuse subset with distal and proximal skin thickening. Beyond this dichotomic subgrouping of systemic sclerosis, new phenotypic considerations based on antibody subtypes have provided a better understanding of the heterogeneity of the disease, anti-Scl70 antibodies being associated with progressive interstitial lung disease regardless of cutaneous involvement. Two targeted therapies, tocilizumab (a monoclonal antibody targeting interleukin-6 receptors (IL-6R)) and nintedanib (a tyrosine kinase inhibitor), have recently been approved by the American Food & Drug Administration to limit the decline of lung function in patients with SSc-associated interstitial lung disease, demonstrating that such better understanding of the disease pathogenesis with the identification of key targets can lead to therapeutic advances in the management of some visceral manifestations of the disease. This review will provide a brief overview of the pathogenesis of SSc and will present a selection of therapies recently approved or evaluated in this context. Therapies evaluated and approved in SSc-ILD will be emphasized and a review of recent phase II trials in diffuse cutaneous systemic sclerosis will be proposed. We will also discuss selected therapeutic pathways currently under investigation in systemic sclerosis that still lack clinical data in this context but that may show promising results in the future based on preclinical data.
Keywords: Systemic sclerosis, scleroderma, interstitial lung disease, fibrosis, tocilizumab, nintedanib
1. Introduction
Systemic sclerosis (SSc or scleroderma) is the rheumatic disease with the highest individual mortality[1]. SSc is characterized by the presence of vascular damage associated with fibrotic and inflammatory manifestations[2, 3]. The severity of the disease is determined by the extent of fibrotic changes to cutaneous and internal organ tissues, the most life-threatening visceral manifestations including interstitial lung disease (ILD), pulmonary arterial hypertension (PAH), SSc-associated cardiomyopathy or scleroderma renal crisis (SRC)[4, 5]. The prevalence of these manifestations is highly variable; SSc is a heterogeneous disorder in terms of severity and clinical presentations[6]. Two main subsets of the disease are classically described based on the extent of skin fibrosis: the limited cutaneous subset (limited cutaneous SSc, or lcSSc) characterized by skin thickening distal to the knees and elbows, and the diffuse cutaneous subset (dcSSc) defined by the association of distal and proximal skin changes including the chest, torso, abdomen, and thighs[7]. Although visceral manifestations can occur in both subgroups, dcSSc is considered as a more severe form of SSc, with a higher prevalence of ILD or SRC and subsequent lower survival[8]. The initial presentation of dcSSc patients is usually characterized by early inflammatory manifestations such as synovitis and skin edema, whereas vascular manifestations are more prominent in early lcSSc[9]. Anti-nuclear antibodies are almost always detected in the serum of SSc patients, the most common specificities being anti-centromere, anti-topoisomerase I (anti-Scl70) or anti-RNA polymerase III antibodies[5]. SSc-autoantibodies are classically mutually exclusive and recent studies suggest that they may help to predict the onset of visceral manifestations and guide treatment strategy[10]. Anti-topoisomerase I antibodies are associated with clinical ILD in both cutaneous subsets and anti-RNA-polymerase III antibodies are a risk factor of SRC[5]. Distinct molecular basis may participated in explaining clinical diversity between autoantibody subsets, notably in dcSSc[11].
Early diagnosis and common intervention with immunomodulatory agents such as cyclophosphamide (CYC) or mycophenolate mofetil (MMF) have led to substantial progress in the management of SSc, although there is still no disease modifying-agent that has demonstrated benefit to all SSc patients[12, 13]. The identification and evaluation of new therapeutic approaches is thus still needed[14]. The heterogeneity of the disease as well as the unpredictable trajectory of some SSc-related manifestations, including spontaneous regression of skin fibrosis in placebo arms, have participated in hindering drug development and have limited the success of clinical trials [15]. Nonetheless, progress in the clinical characterization of the patients as well as in outcome measure selection and a better understanding of the molecular basis of the clinical heterogeneity have led to recent approvals. The American College of Rheumatology and European League Against Rheumatisms (ACR-EULAR) classification criteria for SSc revised in 2013 allow the inclusion of patients with earlier disease in comparison with the 1980 ACR criteria[16]. Early SSc, before the onset of irreversible fibrotic damages, has been identified has a window of opportunity for therapeutic intervention especially considering skin involvement in dcSSc or ILD in both subsets. The identification of auto-antibodies such as Scl70 as a risk factor for ILD progression has also improved patient stratification in international trials[17]. Two targeted therapies, tocilizumab (a monoclonal antibody targeting interleukin-6 receptors (IL-6R)) and nintedanib (a tyrosine kinase inhibitor), have recently been approved by the American Food & Drug Administration (FDA) to limit the decline of lung function in patients with SSc-ILD, demonstrating that a better understanding of the disease pathogenesis with the identification of key targets can lead to therapeutic advances in the management of some visceral manifestations of the disease[17–22].
This review will provide a brief overview of the pathogenesis of SSc, will present a selection of therapies recently approved or evaluated in this context, and will discuss selected therapeutic pathways currently under investigation in SSc.
2. Overview of the pathogenesis of SSc and key targeted cell types
SSc pathogenesis includes a triad of pathological processes (Figure 1): 1) endothelial dysfunction promoting occlusive vasculopathy with vascular rarefaction[23, 24]; 2) immune dysregulation involving both innate and adaptive immunity, associated with markers of autoimmunity[25–27]; 3) uncontrolled production of extracellular matrix (ECM) by activated myofibroblasts with collagen deposits leading to increase stiffness in fibrotic tissues such as skin or lung[28, 29].
Figure 1: Main pathogenic mechanisms and approved or recently evaluated therapies in Phase II/III trials with published results in SSc.
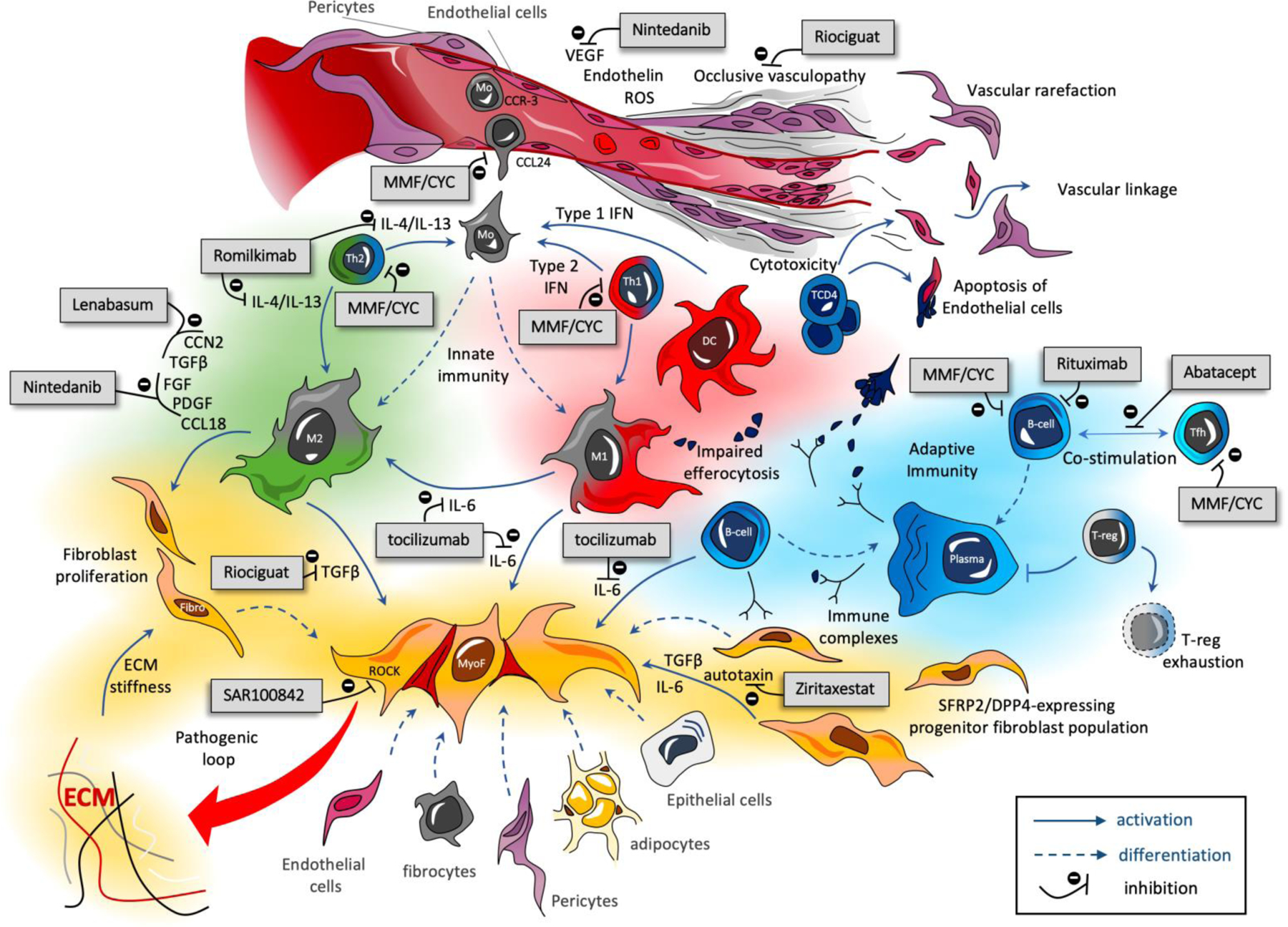
The pathogenesis of SSc involves three main mechanisms: occlusive vasculopathy with endothelial dysfunction, early inflammatory processes and uncontrolled extra-cellular matrix (ECM) production with fibrosis. T-CD4 cells may directly participate to the endothelial damage by inducing cytotoxic dependent-apoptosis of endothelial cells; the impaired capacities of macrophages to phagocyte apoptotic cells (altered efferocytosis); the impact of immune complexes composed by autoantibodies and intra-nuclear proteins (topoisomerase, centromere proteins) that participate to macrophage and fibroblast activation; the over-activation of both M1 (pro-inflammatory) and M2 (pro-fibrotic) macrophages at the early stages of the disease. ECM stiffness also activate fibroblast and TGF-β signaling in a TLR4 and/or Integrin αV dependent manner, leading to a sustain autoactivation loop of myofibroblasts. Only the main mechanisms of action of the therapies recently evaluated are presented here.
Apoptosis of endothelial cells triggered by auto-reactive clonal CD4+ T cells suggest a direct link between vasculopathy and immune dysregulation in SSc[30, 31]. Uncontrolled adaptive immunity also leads to the production of autoantibodies targeting intra-nuclear components[32]. Immune complexes composed of auto-antibodies with auto-antigens, such as the topoisomerase I, could activate fibroblasts[33, 34]. B-cells are also responsible for the production of interleukin 6 (IL-6), another key cytokine activating fibroblasts[35, 36]. IL-6 is produced by activated myofibroblasts in an autocrine manner[37]. Innate immunity participates in SSc-associated tissue damages, as macrophage associated-genes are amongst the most represented cellular signatures in the skin of patients with early diffuse SSc[38]. At the earliest phase of the disease, pro-inflammatory M1 macrophages and Th1 signaling notably including interferon type I and II are the most prominent[39, 40]. Impaired pro-resolving properties of macrophages, such as reduced capacities for performing efferocytosis, i.e. the phagocytosis of apoptotic debris, may also participate in chronic inflammation and persistence of autoantigens in SSc[41, 42]. Secondly but still early in the disease process, a Th2 microenvironment, including IL-4 and IL-13, favors the differentiation of recruited blood-monocytes into pro-fibrotic M2 macrophages[43]. These alternatively activated M2 macrophages have pro-fibrotic properties notably mediated by the secretion of transforming growth factor-β (TGF-β), fibroblast growth factor (FGF), connective tissue growth factor (CTGF also called CCN2), platelet-derived growth factor (PDGF) and CCL18[44]. Such pro-fibrotic mediators participate in the trans-differentiation of fibroblasts into activated myofibroblasts[29]. Although many cellular subtypes are potential precursors of myofibroblasts in SSc, recent single cell RNA-seq studies have shown that dermal myofibroblasts mainly derive from a subpopulation of dermal fibroblasts[45]. SSc-associated myofibroblasts acquire a mesenchymal phenotype with apoptosis resistance and are responsible for an uncontrolled production of ECM components[46]. In return, ECM stiffness of fibrotic tissues can further activate myofibroblasts notably through Toll-like receptors (TLR) and integrin-dependent signals[47–49].
SSc-associated vasculopathy and immune dysregulation are considered as being amongst the earliest events of SSc pathogenesis whereas myofibroblasts are the final integrators of proinflammatory and profibrotic pathways, notably through the over-expression of STAT-3 dependent signals[50]. The interaction between the different intracellular pathways in SSc has been recently reviewed elsewhere[51]. Therapeutic approaches designed to limit life-threatening fibrotic manifestations of the disease, such as ILD, focus on the direct inhibition of these key activating pathways or target cells indirectly responsible for fibroblast activation[52].
3. Therapeutic options recently approved in SSc-ILD and key up-coming trials
SSc-ILD is amongst the leading cause of SSc-related death[53]. About 50% of SSc patients have or will develop ILD during the course of their disease[54, 55]. SSc-ILD onset classically occurs in the 5 first years and this manifestation affects both patients with lcSSc and dcSSc, although ILD is more frequent in the diffuse subset[56]. Other risk factors for SSc-ILD are positivity for anti-topoisomerase I antibodies, male gender and Afro-Caribbean ethnicity[55]. Considering the impact of SSc-ILD on survival, therapeutic research has particularly focused on this manifestation in the past two decades. The first demonstration that conventional immunomodulatory agents could limit the decline of lung function in patients with SSc-ILD was based on the results from Scleroderma Lung Study I (SLS-I)[12]. This RCT evaluated the effects of oral CYC versus placebo on lung function in SSc-ILD and demonstrated that the mean absolute difference in adjusted 12 month forced vital capacity (FVC) expressed as the percent predicted (%pred), was 2.53% favoring CYC (p<0.03). Scleroderma Lung Study II (SLS-II) demonstrated that oral CYC and MMF in SSc-ILD had a similar impact on lung function with significant improvement of FVC(%predicted) in both arms after 24 months, with no between-arm difference (P=0.24)[13]. Considering its better safety profile, MMF became the standard of care for the treatment of SSc-ILD[57]. Phase II and III trials conducted in the 5 past years have led to the FDA approval of two targeted therapies in SSc-ILD: tocilizumab and nintedanib. Both drugs are now indicated to slow the rate of decline of pulmonary functions in patients with SSc-ILD[58].
3.1. Tocilizumab
As pro-inflammatory signals may precede and favor irreversible fibrotic tissue damages in SSc, early disease has been identified as a window of opportunity for therapeutic measures, especially in dcSSc[52]. IL-6 is over-expressed in the serum of patients with early dcSSc, and inflammatory features of the disease such as joint involvement or high IL-6 dependent C-reactive protein (CRP) serum levels are associated with the onset of more severe visceral manifestations, notably SSc-ILD[59]. IL-6 is produced by B-cells, macrophages and myofibroblasts[36]. IL-6 can directly participate in the activation of myofibroblasts and favor M2 polarization, through a potent upregulation of IL-4 and IL-13 receptor membrane expression on macrophages[60, 61]. IL-6 and immune complexes synergistically favor the expression of SPP1 (or osteopontin) in macrophages, and these SPP1-high macrophages may have specific proliferating properties in fibrotic diseases, as recently identified through single-cell RNA-sequencing in lung tissues from patients with ILD[62, 63]. In the inflammatory bleomycin mouse model, targeting IL-6 signaling with monoclonal anti-IL-6 receptor antibody prevented skin fibrosis[59].
Tocilizumab is a therapeutic monoclonal antibody targeting the human IL-6 receptor. The phase II faSScinate randomized controlled trial (RCT) evaluated the efficacy of tocilizumab in patients with early dcSSc in comparison with placebo[18]. There was a numerical difference in the evolution of skin fibrosis evaluated by change in mRSS favoring tocilizumab at week 24 and 48: the least square mean decrease in mRSS at 24 weeks was –3.92 and –1.22 in the tocilizumab and placebo groups, respectively, (difference –2.70, 95% CI –5.85 to 0.45; p=0.0915). At week 48, between treatment difference was −3.55, (95% CI −7.23 to 0.12; p=0.0579), in favor of tocilizumab. Tocilizumab treatment was also associated with sharply decreased serum CCL18 levels and decreased skin biomarker gene expression, as well as decreased expression of genes associated with IL-6 induced macrophage polarization, including CCL18. Active therapy also had a significant impact on the trajectory of lung involvement, as a smaller decrease in lung function assessed through FVC was observed in the tocilizumab arm in comparison with placebo at week 24 (tocilizumab –34 mL versus placebo –171 mL; least square mean difference 136 mL, 95% CI 9 to 264; p=0.0368). Fewer patients in the tocilizumab group than in the placebo group had worsening of FVC (%pred) at week 24 (p=0.009) and 48 (p=0.037). These promising results on SSc-ILD were confirmed in the phase III focuSSced trial[19]. In this international RCT, 212 patients with early active dcSSc were included (105 and 107 in the tocilizumab and placebo arm, respectively); amongst them 67% had SSc-ILD at baseline in the tocilizumab arm and 65% in the placebo arm. In these patients with SSc-ILD, the least square-mean of FVC (%pred) change from baseline was −6.4 in the placebo group and 0.1 in the tocilizumab arm with a least-square mean difference between treatment groups of 6.5 (95%CI 3.4–9.5; p<0.0001). There was also a shift in the distribution of change from baseline in FVC (%pred) at week 48 favoring tocilizumab (van Elteren nominal p=0.002 versus placebo). These clinically meaningful results on FVC were also consistent with the effects of active therapy on the evolution of quantitative evaluation of ILD and lung fibrosis on high-resolution computed tomography (HRCT) that significantly favored tocilizumab. Post-hoc analyses of focuSSced also demonstrated that the effects of tocilizumab on FVC were observed in all subgroups of patients after stratification on SSc-ILD extent. In patients with mild (ILD involving 5–10% of the whole lung surface), moderate (10–20%), and severe (>20%) ILD based on HRCT quantification, the mean decline in the FVC (%pred) in the active therapy group at 48 weeks were −4.1, 0.7, and 2.1, and in the placebo group were −10.0, −5.7, and −6.7, respectively[64]. These results suggest that tocilizumab could equally benefit to patients with SSc-ILD regardless of the initial ILD severity and regardless of the prior rate of progression since the history of FVC decline or the presence of ILD were not amongst the inclusion criteria for focuSSced. Based on the results of faSScinate and focuSSced, the FDA approved tocilizumab to slow the rate of decline in pulmonary function in patients with SSc-ILD, regardless of the cutaneous subset[65]. FocuSSced highlights the need for early treatment, especially in patients at high risk of progressive ILD, even when initial ILD presentation is mild, to prevent the development of irreversible fibrotic damages[20]. This trials also demonstrated that phenotyping of SSc based on systemic and extra-pulmonary manifestations of the disease can help to successfully identify patients at high risk of progressive ILD that would benefit the most from active therapy. This approach suggests a shift of paradigm in SSc-ILD, towards early introduction of treatments before the progression of the disease occurs, instead of identifying patients that have already experienced effective progression and/or FVC decline[66]. Recent results from real world experience also suggest that patients with anti-Scl70/anti-topoisomerase would especially benefit from the effects of tocilizumab on lung function[67].
3.2. Nintedanib
Fibrosis is the end-stage pathogenic process of SSc-ILD, as the excess of ECM in the pulmonary interstitium leads to increase tissue stiffness with reduction of pulmonary compliance and volumes[28, 68]. The sustained and uncontrolled activation of pulmonary myofibroblasts notably relies on mediators from the FGF family produced by myofibroblasts in an autocrine manner or by M2 macrophages with paracrine effects[69]. Amongst other macrophage mediators, PDGF participates to the activation of myofibroblasts[70]. SSc-associated vascular damages may also contribute to fibrotic changes in SSc-ILD and key mediators of the occlusive vasculopathy such as vascular endothelial growth factor (VEGF) participate in lung fibrosis[23, 71].
Nintedanib is a tyrosine kinase inhibitor targeting the common domain of the receptors of PDGF, FGF (1 to 3) and VEGF (1 to 3) through competitive binding to ATP–binding pocket of these receptors, stopping intracellular signaling[72]. Nintedanib can directly inhibit the activation of myofibroblasts mediated by these pathways. Nintedanib also prevents the differentiation of monocytes into macrophages in vitro[44]. In mouse models of SSc, nintedanib also inhibits macrophages activation and ameliorates vascular and fibrotic manifestations of the disease[73]. Nintedanib was initially approved in 2014 for the treatment of idiopathic pulmonary fibrosis (IPF)[74, 75].
The SENSCIS trial evaluated the efficacy and safety of nintedanib in patients with SSc-ILD[17]. This international phase III trial is the largest RCT ever conducted in SSc. 580 patients were included (288 in the nintedanib and placebo groups, 3 patients randomized despite non-eligibility and one withdrawal). Patients with diffuse or limited cutaneous subsets were eligible if they had SSc-ILD with CT showing fibrosis affecting at least 10% of the lungs and FVC(%pred) higher than 40%. The primary end-point was the annual rate of decline in FVC (milliliters per year), assessed over a 52-week period. The adjusted annual rate of change in FVC was −52.4mL per year in the nintedanib group and −93mL per year in the placebo group (difference, 41.0 mL per year; (95%CI 2.9 to 79.0, p=0.04)). The most common side effect was diarrhea, reported in 76% of the patients in the nintedanib group and in 32% in the placebo arm. Nintedanib had no effect on the extra-pulmonary manifestations of SSc. The INBUILD trial also evaluated the efficacy of nintedanib compared to placebo to limit the annual rate of decline in the FVC in patients with progressive fibrosing ILD of various etiologies[66]. In this trial, 23 (6.9%) and 16 (4.8%) had SSc-ILD in the nintedanib and placebo arms respectively. INBUILD demonstrated the efficacy of nintedanib to limit the annual rate of FVC decline in patients with progressive fibrosing ILD (between-group difference 107.0 mL per year (95%CI 65.4 to 148.5 (p<0.001)). In 2019, nintedanib was approved by the FDA to slow the rate of decline of pulmonary functions in patients with SSc-ILD.
3.3. Perspectives for the treatment of SSc-ILD considering these recent approvals: potential role of combination therapies and other anti-fibrotic agents inherited from IPF.
In SLS-II, MMF showed improvement of pulmonary function over time in the majority of patients and was similarly active with respect to evolution over time of mRSS as well as dyspnea and health-related quality of life evaluated by patient reported outcomes (PROs)[76–78]. Tocilizumab in focuSSced showed a numerical impact on skin fibrosis but had no statistically significant effect on dyspnea[19]. In SENSCIS, nintedanib showed no benefit on dyspnea and health-related quality of life evaluated by PROs, and did not impact extra-pulmonary manifestations of the disease[17]. Based on these results, nintedanib is not considered disease-modifying on SSc as a whole. Both tocilizumab and nintedanib, nonetheless, showed biological effects that could be considered specifically disease-modifying in SSc-ILD[20].
In focuSSced no background immunomodulatory therapy was allowed, whereas patients with stable doses of MMF for at least 6 months were permitted to enroll in SENSCIS[17, 19]. This may have participated in limiting intergroup-difference between placebo and nintedanib, notably on extra-pulmonary manifestations and PROs potentially improved by MMF. The treatment effect of nintedanib on the annual rate of change in FVC as primary outcome was numerically lower in participants who were taking MMF at baseline than in those not taking MMF (difference of nintedanib versus placebo of 26.3 mL per year (95%CI −27.9 to 80.6) and 55.4 mL per year (95%CI 2.3–108.5) in the groups receiving and not receiving MMF, respectively)[79]. Nonetheless, with a relative reduction of 40% and 46% of annual rate of change in FVC in the groups with and without MMF respectively, statistical testing did not indicate significant heterogeneity in the treatment effect of nintedanib between the subgroups defined by MMF use. Nevertheless, this result suggests potential additive effects of MMF and nintedanib. Although this question needs further investigations to be fully explored in dedicated RCTs, these post-hoc sub-group analyses of SENSCIS pave the way for future evaluations of combination therapies in SSc[15]. The mechanisms of action of MMF, nintedanib, and tocilizumab show complementary effects with impact on distinct pathways that would support their combined use (Table 1). The combination of tocilizumab with either nintedanib or MMF may also appear relevant based on their respective targets. The safety profile of combining a tyrosine kinase inhibitor with an anti-IL6 monoclonal antibody is still to be determined in SSc, which precludes the use of such combinations in daily practice to date.
Table 1:
mechanisms of action of the main drugs used in SSc-ILD
| Cellular target/pathway | MMF (MFA)inhibitor of inosinemonophosphate dehydrogenase / inhibition of novo synthesis of guanosine nucleotides | Tocilizumab Anti-IL6-R |
Nintedanib Anti-PDGF, VEGF and FGF-R |
|---|---|---|---|
| T-cells (Th1,2,17) | Inhibition of T cell-proliferation But limited effects of the production of Th2 cytokine in vitro (pro-fibrotic IL-4) |
Increased T-reg frequency and a blunted T-effector cytokine response compared to controls in renal transplant when TCZ is used as add-on therapy with MMF. | Limits lymphocytic interstitial infiltration |
| Macrophages | Participates to the resolution of inflammation through a switch from M1 to M2 in kidney (but upregulation of M2 macrophages that may exert profibrotic properties) MPA increased the expression of M2 surface markers, including CD163 and CD200R, on M1 macrophages MMF reduces macrophage infiltrates in the skin through a potential impact of monocyte recruitment via a down-regulation of chemokines such as CCL2 |
Limited/no effects on M1 polarization markers Down-regulation of M2 markers notably through a potential effect on the IL-6 dependent expression of the receptors of Th2 cytokines IL-4 and IL-13. |
Limits the secretion of M1 cytokines (IL-1b, IL-8, CXCL13) Limits M2 polarization markers (CD206, CD209, CD200R) |
| Endothelial cells and/or vascular smooth muscle cells (VSMC) | Inhibition of the proliferation of endothelial cell and VSMC Limits PAH in rat models Decreases VEGF serum levels |
Scarce data on effects of TCZ on SSc-associated endothelial dysfunction | Blocks the effects of VEGF and limits disturbance of vessel morphology and occlusive vasculopathy Reduced apoptosis of endothelial cells Decrease the number of proliferating VSMC |
| Endothelial to mesenchymal transition /epithelial to mesenchymal transition | Potential inhibitory effects of endoMT induced by TGFβ+IL-6 or IL-1β via its impact on TGFβ | Potential inhibitory effects of endoMT induced by TGFb+IL-6 via its impact on IL-6 | Controversial data on EMT in alveolar epithelial cells |
| Fibroblasts / Myofibroblasts | Inhibition of Fibroblast proliferation Down-regulation of STAT3 signaling, Down-regulation of IL-6 secretion. |
Normalize functional and phenotypical properties of dermal fibroblast (lower CTGF/CCN2 production, decreased aSMA)Inhibition of IL-6/STAT3 dependent autocrine loop activation of myofibroblasts | Limits the proliferation, migration and survival Inhibits transdifferenciation Limits the proliferation, migration and survival Reduced TIMP-2 levels, together with increased pro-MMP-2 with potential reduction of collagen deposits |
| TGFβ signaling | TGF beta-induced cell motility, collagenmatrix contraction and cell morphology in vitro (kidney epithelial cells) | Contradictory effects, but limited direct effects on TGFβ signaling Reverses gene expression profiles dominated by TGFβ-regulated genes and molecular pathways in dermal fibroblasts through potent indirect effects |
Reduces TGFβ-stimulated collagen secretion |
| IL-6/IL-6R | Down regulation of IL-6 production in various in vitro model, including autocrine production by myofibroblasts | Main mechanisms of action | Potential indirect reduction of IL-6 levels |
Mechanisms of actions and drug-related effects considered in this table are derived from clinical or preclinical data from in vivo or in vitro models of inflammatory or fibrotic diseases sharing some similarities with systemic sclerosis (SSc), or directly based on effects demonstrated in SSc models.
The Scleroderma Lung Study III (SLS-III) will provide specific insights on the relevance of combining immunomodulatory agents with anti-fibrotic drugs, as this ongoing phase III trial is evaluating the efficacy of pirfenidone versus placebo as add-on therapy with MMF in SSc-ILD (NCT03221257). Pirfenidone is an oral anti-fibrotic therapy approved in the treatment of IPF[80]. Although its precise mechanism of action is still to be determined, pirfenidone may impact TGF-β signaling with subsequent inhibition of myofibroblast trans-differentiation[81]. The LOTUS study evaluated the safety of pirfenidone in SSc-ILD, showing a good tolerance profile notably in combination with MMF[82]. The efficacy of pirfenidone to limit FVC decline in SSc-ILD was also evaluated in a pilot study including 34 patients[83]. Although pirfenidone failed to improve/stabilize FVC in comparison with placebo over 6 months, the limited sample size precludes firm conclusions. The efficacy and safety of pirfenidone in patients with non-IPF progressive fibrotic ILDs (ILD patterns on HRCT or lung biopsy) was also recently assessed in a phase IIb multicenter German RCTs (the RELIEF study)[84]. Patients were excluded in case of a pre-existent steroid and/or immunosuppressant therapy modified within the last 3 months and/or if such therapy would need to be changed during the study period. The presence of progressive ILD prior enrollment was a mandatory inclusion criterion and was defined by an annual FVC decline of at least 5% predicted, based on at least three FVC measurements within 6–24 months before enrolment. Eight patients with SSc-ILD were included in this study. An interim analysis undertaken due to the slow recruitment rate has resulted in early termination of this trial. Considering this premature termination and missing data secondary to withdrawals, the results have to be interpreted with caution. 187 patients per group (i.e. 374 patients in total) were initially expected but only 127 were enrolled before study early termination (64 and 63 in the pirfenidone and placebo arm respectively). In the overall population of 127 patients at week 48, rank ANCOVA with diagnostic group included as a factor showed a significantly lower decline in FVC % predicted in the pirfenidone group as compared to placebo (p=0.043); the result was similar when the model was stratified by underlying diagnostic group (p=0.042)[84]. The most frequent underlying diagnosis was hypersensitivity pneumonitis (45%). Amongst CTD-ILD patients (n=37), 46% had rheumatoid arthritis whereas 22% of CTD-ILD patients had SSc. This low number of SSc patients precludes conclusion regarding specific effects of pirfenidone on this subgroup in this trial. Considering the first preliminary encouraging results in RELIEF, the awaited SLS-III study will help to determine the potential place of pirfenidone in the treatment of SSc-ILD.
4. Therapeutic approaches recently evaluated in phase II trials in early dcSSc.
Beyond SSc-ILD, recent trials in the field of SSc have especially focused on the diffuse cutaneous subset since it is considered as the most severe subgroup, although only concerning 20 to 30% of all SSc-patients[85]. In the majority of these recent phase II trials including early dcSSc patients, mRSS was used as the primary outcome[86–89]. Although trends were observed favoring active therapy, many of these trials have failed to reach statistical significance on mRSS, whereas secondary outcomes such as FVC progression were able to discriminate placebo from active treatment[18, 86]. In some of these trials, a combined response index, the American College of Rheumatology Composite Response Index for Clinical Trials in Early Diffuse Cutaneous Systemic Sclerosis (ACR-CRISS), notably including FVC progression and mRSS evolution, successfully discriminated active therapy from placebo[19, 90, 91]. These results suggest that mRSS alone as a primary outcome measure in dcSSc may be insufficient to characterize meaningful change in response to treatment [15]. Nonetheless, considering the statistical trend favoring active therapy in many of these trials and the significant differences in others, mRSS in dcSSc Phase II trials is informative for a go-no-go decision guiding the design of future phase III trials[88]. Its inclusion in the ARC-CRISS also informs on overall disease improvement and on the potential disease-modifying effects of the considered therapy. The results of the following phase II trials should be interpreted in the light of these considerations on outcomes measure in dcSSc (Table 2).
Table 2:
Therapies recently evaluated in phase II trials dedicated to patients with dcSSc
| Compound | Structure /IUPAC/Chemical Name | Mechanisms of action and cellular targets | Phase II trial in dcSSc (clinicaltrial.gov) |
|---|---|---|---|
| Romilkimab / SAR156597 | Engineered, humanized, bispecific IgG4 antibody that neutralizes IL-4 and IL-13 | Neutralizes IL-4 and IL-13, two Th2 cytokines directly responsible for profibrotic M2 polarization of macrophages and for the direct activation of fibroblasts. | NCT02921971 |
| Abatacept | Fusion protein composed of the Fc region of the immunoglobulin IgG1 fused to the extracellular domain of CTLA-4 | Binds to CD80 and CD86 with subsequent inhibition of co-stimulation and prevention of mutual B-cell/T-cell co-activation. The impact of abatacept on T-cell activation may directly down-regulates Th2 dependent activation of macrophages and fibroblasts. Abatacept also limits in vivo B-cell infiltrates in damaged tissues. | NCT02161406 |
| Riociguat | Carbamate ester that is the methyl ester of {4,6-diamino-2-[1-(2-fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl}methylcarbamic acid | Stimulator of the soluble guanylate cyclase either through a synergistic NO-dependent stimulation or through a direct NO-independent pathway. This stimulation of the soluble guanylate cyclase leads to the production of cGMP with vasorelaxation and anti-remodeling properties as well as inhibitory effects on TGF-β dependent activation of fibroblasts. | NCT02283762 |
| Lenabasum | Ajulemic acid, a synthetic cannabinoid derivative of the Tetrahydrocannabinol metabolite 11-nor-9-carboxy-THC : (6aR,10aR)-1-hydroxy-6,6-dimethyl-3-(2-methyloctan-2-yl)-6a,7,10,10a-tetrahydrobenzo[c]chromene-9-carboxylic acid | Agonist of the cannabinoid receptors CB2,reducing extra-cellular-matrix deposition in vitro and inhibition of the trans-differentiation of fibroblasts into myofibroblasts. CB2 agonists decreases expression of CTGF/CCN2 and TGF-β1, through a down-regulation of canonical TGF-β signaling in fibrotic tissues. | NCT02465437 |
| Ziritaxestat (GLPG1690) | 2-((2-ethyl-6-(4-(2-(3-hydroxyazetidin-1-yl)-2-oxoethyl)piperazin-1-yl)-8-methylimidazo[1,2-a]pyridin-3-yl)(methyl)amino)-4-(4-fluorophenyl)thiazole-5-carbonitrile | Inhibits autotaxin dependent-production of Lysophosphatidic acid (LPA) with subsequent down-regulation of LPA-related signaling and inhibition of myofibroblast activation as well as potential improvement of macrophage pro-resolving and anti-inflammatory properties. | NCT03798366 |
| SAR100842 | 2-{4-methoxy-3-[2-(3-methylphenyl)ethoxy]benzamido}−2,3-dihydro-1H-indene-2-carboxylic acid | Selective oral antagonist of LPA1 with subsequent inhibition of myofibroblast activation and potential improvement of macrophage pro-resolving and anti-inflammatory properties. | NCT01651143 |
4.1. The proof of concept 24-week phase II trial evaluating the efficacy and safety of Romilkimab (SAR156597) versus placebo in early dcSSc
IL-4 and IL-13 play an important role in the pathogenesis of SSc. Elevated serum levels of IL-4 have been measured in the serum of SSc patients[92]. IL-13 has a similar and redundant impact since IL-4 and IL-13 receptors share common subunits and downstream activating pathways, notably including the transcription factor STAT-6[93]. These Th2 cytokines participate in the polarization of macrophages into pro-fibrotic alternatively activated M2 macrophages[94]. IL-4 also favors the proliferation of fibroblasts and increases their production of pro-fibrotic markers such as TGF-β or CTGF/CCN2[95]. The redundancy of IL-4 and IL-13 on downstream regulating pathways supports the concomitant inhibition of both cytokines to obtain substantial biological effects[96].
Romilkimab (SAR156597) is an engineered, humanized, bispecific immunoglobulin-G4 antibody that neutralizes IL-4 and IL-13[88]. The efficacy and safety of romilkimab was recently evaluated in a phase II RCT, including 48 and 49 patients in the active therapy and placebo arms respectively. The change from baseline to week 24 in mRSS was chosen as the primary outcome. Romilkimab showed efficacy over placebo as mean change in mRSS was −4.76 (0.86) versus −2.45 (0.85) for romilkimab and placebo respectively[88]. The mean difference in change from baseline to week 24 in mRSS was −2.31 (90% CI −4.32 to −0.31; p=0.0291, one-sided) in favor of active therapy. The change in diffusing lung capacity of carbon monoxide (DLco) and FVC also tend to favor romilkimab. Romilkimab significantly improved the European Quality of Life-5 Dimension-5 Level, as change from baseline to week 24 was 0.07 (0.03) for romilkimab versus 0.00 (0.03) for placebo with a mean difference (95% CI) of 0.07 (SEM=0.04) (–0.01 to 0.15; p=0.0363). There was no difference regarding the probability of improvement based on the ACR-CRISS (0.3811 (0.4372) and 0.4245 (0.4266) in the placebo and romilkimab arms respectively, p= 0.27).
The result on mRSS in this proof of concept trial highlighted that targeting pro-fibrotic Th2 signaling in early diffuse SSc may constitute a relevant approach, suggesting that these patients may also benefit from early anti-fibrotic therapy and not only from early anti-inflammatory drugs[97]. This result may support the hypothesis that early up-front combination of immunomodulatory agents with anti-fibrotic properties, such as romilkimab, with agents with anti-inflammatory effects, such as MMF, could be a relevant therapeutic approach in dcSSc. The design of this proof of concept trial precludes conclusions regarding this issue, as different background immunomodulatory agents were allowed including methotrexate (43% and 25% of the patients in the placebo and romilkimab arms respectively), MMF (14 and 21%) and Azathioprine (2 and 8%). Only a dedicated RCT, including a stratification based on background therapy or a unique background immunomodulatory agent with an anti-fibrotic agent or placebo as add-on therapy would validate this hypothesis in early dcSSc.
4.2. Abatacept trials: targeting co-stimulation in early dcSSc.
Beyond the secretion of Th2 cytokines, CD4+ T cells also play a direct role in SSc-associated tissue damages. The perivascular infiltrate in the skin of early dcSSc is characterized by the presence of CD4+ T cells that may directly participate to the endothelial damage by inducing cytotoxic dependent-apoptosis of endothelial cells (Figure 1)[30]. This oligoclonal CD4+ T-cell subpopulation positive for CD319 (SLAM-F7) is also expanded in the blood of patients with early dcSSc in comparison with healthy controls[98]. Recent transcriptomic analyses of the skin in dcSSc also support an elevated type II interferon signature which is classically related to Th1 lymphocytes [38]. Through the process of CD80/CD86-CD28 dependent-mutual co-stimulation, CD4+ T cells also participate in the activation of B-cells and are directly involved in the autoimmune features of SSc. Abatacept is a fusion protein that binds to CD80 and CD86 molecules preventing the co-stimulatory signals activating both CD4+ T-cells and B-cells[27]. Abatacept is approved in the treatment of rheumatoid arthritis and was able to prevent experimental dermal fibrosis and to reduce established inflammation-driven fibrosis in mouse models of SSc[99]. Treatment with abatacept limited B-cell infiltrates in bleomycin-induced skin fibrosis[27]. Concomitantly, this decrease of B-cell count was associated with improved dermal thickness, decreased collagen deposit and reduced myofibroblast skin infiltrate both in preventive and curative protocols. Observational data from the European Scleroderma Trials and Research group (EUSTAR) in 27 patients suggest that abatacept may have some effectiveness on joint involvement and related disability in SSc[100].
The Abatacept Systemic SclErosis Trial (ASSET trial) was a phase II RCT evaluating the safety and efficacy of abatacept in patients with early dcSSc[87]. The inclusion criteria in this trial focused on recruiting patients with early, active skin disease based on disease duration and mRSS progression. The primary outcome was the change from baseline in mRSS at 12 months. Although there was no significant difference between placebo and active therapy for this primary endpoint, numerical change in mRSS favored active therapy (−6.24 ± 1.14 and −4.49 ± 1.14, for abatacept and placebo respectively with a treatment difference of −1.75 [95% CI −4.93, 1.43]). The probability of improvement based on the ACR CRISS at 12 months also favored abatacept (probability of 0.02 (IQR=0.75) and 0.72 (0.99) in the placebo and abatacept groups respectively; p=0.03 by Van Elteren test with adjustment for disease duration). Health assessment questionnaire-quality index (HAQ-DI) evolution also demonstrated higher improvement of quality of life with active therapy as compared to placebo. The open-label data at 18 months showed clinically meaningful improvements in mRSS and FVC in both the abatacept and initial placebo groups when patients transitioned to open-label treatment with abatacept[101]. These results, in addition to previous reports, support the relevance and need for further assessment of the abatacept efficacy in early dcSSc[102].
In ASSET, patients were also sub-classified based on the initial transcriptomic signature identified on baseline skin biopsy. Three subgroups were identified: an inflammatory pattern, a proliferative pattern and a normal-like pattern[87]. These intrinsic skin gene expression subsets were consistent with previous reports in dcSSc[103–105]. Interestingly, such classification may help to stratify patients based on their potential treatment response, as the least square mean change in mRSS over 12 months was significantly different between the abatacept and placebo groups for the inflammatory and normal-like subsets (P < 0.001 and P = 0.03, respectively) whereas there was no difference for the proliferative group. Similar results on treatment response were obtained in a previous exploratory RCT including 7 patients in the abatacept arm and 3 in the placebo arm[102]. The data from these abatacept trials made the proof of concept that intrinsic gene expression could help to refine inclusion criteria for a future abatacept trial and that this approach could lay a path towards personalized medicine in dcSSc[106].
4.3. Soluble Guanylate cyclase pathway: Exploring the anti-fibrotic effects of Riociguat in dcSSc.
Soluble guanylate cyclase (sGC) is one of the main catalysts in the production of cyclic guanosine monophosphate (cGMP). Cyclic GMP induces vasorelaxation and shows anti-proliferative properties. Impairment of the nitric-oxide (NO.)- sGC-cGMP pathway is involved in the pathogenesis of SSc-PAH and participates in SSc-associated occlusive vasculopathy[107]. Riociguat is a sGC stimulator with a dual mode of action: it can stimulate cGMP production by sGC either through a synergistic NO-dependent stimulation or through a direct NO-independent pathway[108]. In preclinical models of PAH, riociguat show vasorelaxation and anti-remodeling properties, suggesting its relevance in this context. The phase III PATENT-1 trial demonstrated that riociguat increased the 6-minute walk distance, improved vascular resistance, NT-proBNP levels, dyspnea and time-to-clinical worsening in patients with PAH, notably including SSc-PAH[108, 109]. Beyond these effects on vascular remodeling, riociguat also shows anti-fibrotic properties. Soluble GC stimulators could prevent and induce regression of fibrosis in inflammatory and non-inflammatory mouse models of SSc, through the inhibition of TGF-β dependent activation of fibroblasts[110]. These anti-fibrotic effects of riociguat were notably driven by the protein kinase G (PKG), a downstream mediator of the sGC-cGMP pathway. PKG specifically interferes with the TGF-β-induced activation of ERK, that participates in fibroblast activation in SSc[111, 112]. Other TGF-β non-canonical and canonical SMAD dependent signals are not impacted by riociguat.
The RIociguat Safety and Efficacy in patients with diffuse cutaneous Systemic Sclerosis (RISE-SSc) study was an international randomized, double-blind, placebo-controlled, parallel-group clinical trial evaluating the impact of riociguat on change in mRSS from baseline to week 52 in patients with early dcSSc at high risk of progression[86]. The primary endpoint was not met, as change from baseline in mRSS was –2.09 and –0.77 in the riociguat (n=57) and placebo (n=52) arms respectively, (difference of least squares means –2.34 (95% CI –4.99 to 0.30; p=0.08)) at week 52. However, skin fibrosis was also analyzed by prespecified exploratory analyses of mRSS progression ; it was observed that 11 (18.6%)/59 patients with riociguat and 22 (36.7%)/60 patients with placebo exhibited skin progression (Mantel-Haenszel estimate of difference: –17.99% (95% CI –33.57% to –2.40%; nominal p=0.0237). There was no significant between-arm difference in the probability of improvement based on the ACR-CRISS. This result could be expected as RISE-SSc was designed to detect prevention of progression and not improvement. Change in FVC(%pred) and prevention of new digital ulcers tended to favor active therapy. The open-label section of the trial showed similar results regarding skin evolution, with mRSS regression in both arms without statistical difference between placebo and active therapy[113]. Riociguat increased cGMP levels and decreased the serum levels of SSc-associated severity biomarkers such as sPECAM-1 and CXCL-4 at week 14[114]. The limited impact of riociguat on SMAD dependent TGF-β signaling and on other non-canonical pathways may explain, in part, the disappointing anti-fibrotic effects of riociguat in RISE-SSc. Despite the clear benefit of riociguat in SSc-PAH, its relevance for the treatment of other manifestations of the disease and its potential disease modifying-effects are not supported by the results of RISE-SSc.
4.4. Cannabinoid agonists and PPAR-γ agonists
The endocannabinoid system includes two cannabinoid receptors, CB1 and CB2. CB2 may exert anti-fibrotic effects as CB2−/− mice are more sensitive to bleomycin-induced dermal fibrosis [115]. Both CB1 and CB2 receptors are over-expressed in dcSSc fibroblasts. Cannabinoid receptor agonists could reduce ECM deposition in vitro, could limit the trans-differentiation of fibroblasts into myofibroblasts and could counter-act their resistance to apoptosis [116]. In vivo, cannabinoid receptor agonists significantly prevented experimental bleomycin-induced dermal fibrosis and could limit fibrosis progression in a curative model [117]. In these in vivo mouse models, the anti-fibrotic effects of cannabinoid receptor agonists in the skin were PPAR-γ-dependent, as co-treatment with PPAR-γ antagonist alleviated the effects of cannabinoid receptor agonists. In a mouse model of ILD induced by trans-oral instillation of bleomycin, cannabinoid receptor agonists could reduce lung fibrosis as well [118]. These pulmonary effects were associated with a decreased expression of CTGF/CCN2 and TGF-β1, through a down-regulation of canonical TGF-β signaling as suggested by reduced phosphorylation of SMAD2/3 and a decreased in αSMA expression. On the contrary, cannabinoid receptor agonists induced an increased pulmonary expression of PPAR-γ. Considering these anti-fibrotic effects on skin and lung, cannabinoid receptor agonists appear to be relevant therapeutic agents that may show disease-modifying effects in SSc.
Lenabasum is a synthetic agonist of CB2 that was evaluated in a phase II RCT in early dcSSc[119, 120]. The primary efficacy outcome measure was the probability of improvement based on the ACR-CRISS at week 12. Twenty-seven and 15 patients were randomized to receive lenabasum and placebo, respectively. Improvement in median ACR-CRISS score was observed in the lenabasum arm starting at week 8 and increased over time, with a maximum of 0.33 (IQR 0.01–0.82), in comparison with 0.00 (IQR 0.000–0.16) in the placebo arm, at week 16 (p=0.04 by 1-sided mixed-effects model repeated-measures analysis and p=0.07 by 2-sided mixed-effects model repeated-measures analysis). Lenabasum also tended to improve change in mRSS with a mean difference of −2.6 (SEM=1.9) at week 16 in favor of lenabasum (p=0.09 by 1-sided mixed-effects model repeated-measures analysis and p=0.17 by 2-sided mixed-effects model repeated-measures analysis). Lenabasum was well tolerated in this trial. These encouraging results led to the phase III RESOLVE-1 trial (NCT03398837) using ACR-CRISS at 52 week as primary outcome. The publication of the results in a peer-reviewed journal is still awaited, but the press announcement (September 2020) specified that the primary outcome was not met as ACR-CRISS scores at week 52 were 0.887 in the placebo arm and 0.888 in the lenabasum 20 mg twice daily arm[121]. Publication of the final results and subgroup analyses may help to identify if a subset of patients had shown specific treatment response.
An increased expression of PPAR-γ may participate in the effects of cannabinoid receptor agonists. Regarding PPAR, the pan-PPAR agonist Lanifibranor (IVA337) showed promising results in preclinical models of SSc. In the bleomycin mouse model, IVA337 induced decreased ECM deposition and decreased skin expression of phosphorylated SMAD2/3[122]. The phase 2 trial FASST evaluating IVA337 versus placebo in dcSSc included 145 patients using mean change of mRSS from baseline to week 48 as primary outcome. Although the results of this trial have not been published in a peer-review journal, press-release announced the absence of efficacy of active therapy in comparison with placebo on the primary outcome and on secondary outcomes. This could be partly explained by a decrease of mRSS in the placebo arm potentially due to concomitant use of immunosuppressive therapies. Lanifibranor showed a favorable trend in patients’ global assessment of disease activity (p=0.08). Based on the results of this phase II trial, the development of IVA337 in SSc was discontinued.
4.5. Rho/ROCK, LPA1 pathway and Autotaxin inhibitors
The RhoA/ROCK signal is a system of intracellular kinases comprising signaling G proteins from the Rho GTPase family subsequently activating the Rho-associated protein kinase ROCK[123]. The RhoA/ROCK pathway contributes to the contractile phenotype of activated myofibroblasts and ECM stiffness induces STAT-3 phosphorylation in a RhoA/ROCK-dependent manner[124]. RhoA/ROCK is also involved in immune regulation and impacts inflammation resolution as RhoA/ROCK limits pro-resolving properties of macrophages such as efferocytosis (Figure 1)[42]. TGF-β, CTGF/CCN2 and integrins are amongst the upstream activators of this kinase cascade[51]. Lysophosphatidic acid (LPA) receptor family can also activate this pathway and LPA1 (LPA receptor 1) activation can induce fibroblast trans-differentiation via RhoA/ROCK activation[125]. LPA is notably produced in pro-inflammatory conditions and derived from lysophospholipids such as lysophosphatidylcholine through the action of lysophospholipase D, also called autotaxin[126]. A pathogenic loop including IL-6 and autotaxin may participate in LPA1 activation in SSc. Therefore, targeting autotaxin-dependent production of LPA, LPA receptors or downstream RhoA/ROCK pathway may constitute a relevant approach to limit fibrotic manifestations of SSc. The inhibition of LPA/LPA-receptor pathway and/or RhoA/ROCK prevents or improves fibrosis in various mouse models of SSc, strengthening the relevance of investigating these targets in this fibrotic disorder.
Ziritaxestat (GLPG1690) is an orally administrated autotaxin inhibitor that was recently evaluated in a phase 2a randomized, double-blind, placebo-controlled trial for the treatment of early dcSSc[89]. This RCT included 21 and 12 patients in the treatment and placebo arm respectively, and utilized change from baseline mRSS at 24 weeks as primary efficacy endpoint. Between-group difference for this primary endpoint was –2.8 (IC95% –5.6, –0.1) favoring ziritaxestat (p=0.0411). The ACR-CRISS also showed numerically higher probability of improvement with active therapy highlighting that the LPA/Autotaxin pathway could constitute a relevant approach for a disease-modifying strategy. The open-label extension of this phase 2 trial has nonetheless been terminated, and reason for this termination as stated on clinicaltrial.gov is: “the benefit-risk profile no longer supports continuing the studies”.
The safety and efficacy of SAR100842, a potent selective oral antagonist of LPA1 was evaluated in an 8-week double-blind, randomized, placebo-controlled study followed by a 16-week open-label extension in patients with early dcSSc[127]. Although safety profile evaluation was the main objective, mRSS served as a surrogate marker for efficacy at week 8. Between-group difference for this exploratory endpoint favored active therapy (−3.57 (4.18) in the SAR100842 (n=17) versus −2.76 (4.85) for the placebo (n=15) p=0.46) and SAR100842 was well tolerated. Two RCTs are registered to evaluate the safety and efficacy of ROCK-2 inhibition by KD025 (belumosudil) in dcSSc (NCT04680975 and NCT03919799). The announced primary endpoint in these trials is the ACR-CRISS after 24 weeks of therapy. The current discussion on FDA approval of Belumosudil (KD025) in chronic graft-versus-host disease (cGVHD) strengthens the relevance of targeting this pathway in dcSSc, as the pathogenesis of cGVHD shows similarities with SSc[128].
4.6. Stem cell transplantation
The widespread involvement of adaptive and innate immunity in the pathogenesis of severe manifestations associated with dcSSc and the efficacy of cyclophosphamide in SSc-ILD and associated PRO suggest that intensive immunosuppression through myeloablative chemotherapy followed by rescuing hematopoietic stem cell transplantation (HSCT) could constitute a disease-modifying approach in dcSSc. The ASTIS and SCOT trials have both demonstrated the efficacy of HSCT to improve survival in patients with severe SSc [129, 130]. Only highly selected patients with -or at high risk of- life-threatening manifestations of the disease were included in these trials. Although both trials showed a statistically significant impact on survival at the end of the follow-up, with differences in PRO and quality of life indices favoring HSCT procedure, myeloablative chemotherapy was associated with early morbidity and mortality. To improve these outcomes in clinical practice, the inclusion criteria of candidates for HSCT have been narrowed in some centers, notably by excluding patients with severe heart involvement[131]. Therefore, although HSCT is a disease-modifying approach in severe dcSSc in expert centers, this intervention is restricted to a very limited number of highly selected patients. More accessible therapeutic strategies adapted to a wider population of SSc patients are thus still needed.
Immunomodulatory properties of allogeneic or autologous mesenchymal stem cell transplantation is currently being evaluated in SSc and may show promising results in the future[132, 133]. In mouse models of SSc, mesenchymal stem/stromal cell transplantation had immunosuppressive, tissue remodeling and anti-oxidative properties[134]. Similarly, adipose-derived stem cells and autologous stromal vascular fraction from adipose tissue showed pro-angiogenic and anti-fibrotic effects in vitro[135]. An open-label clinical trial assessing subcutaneous injections of autologous adipose-derived stromal vascular fraction into the fingers of SSc patients has shown improvement in hand functioning and quality of life, suggesting that such procedure could be promising in the future[136, 137].
5. A selection of treatments or pathways under investigation in a clinical or preclinical stage
Beyond the phase II and phase III trials already discussed in SSc-ILD and dcSSc, other pathways are under-investigation in a less advanced stage of development. Although some of these pathways are old concepts in the field of SSc, new therapeutic approaches based on a better understanding of such signals may show promising results.
5.1. Revisiting the inhibition of TGF-β signaling,
TGF-β plays a central role in SSc-related fibrosis. TGF-β family comprises three isoforms (TGF-β1, 2 and 3) that are all increased in the skin and serum of SSc patients, although a large proportion remains in an inactive form[29]. This latent form can be activated by various signals including increased ECM stiffness, actions of integrins or thrombospondins. TGF-β signaling is up-regulated in the fibrotic tissues of all main mouse models of SSc, including genetic and induced models with inflammatory or non-inflammatory related fibrosis[28]. There are two main TGF-β-dependent downstream signaling pathways: a canonical pathway involving SMAD 2/3 and 4; and a non-canonical pathway, SMAD independent, that notably involves RhoA/ROCK, c-Abl, TAK1, p38, JNK, SRC and JAK2-STAT-3[138–141]. These pathways are redundant and the concomitant inhibition of several of them is needed to ensure significant biological effects. The different isoforms of TGF-β have pleiotropic activities and TGF-β2 notably participates in hematopoiesis and myocardial functioning[29]. The key roles of different TGF-β isoforms during development and carcinogenesis along with adverse effects of TGFβ receptor kinase inhibitors on heart valves in pre-clinical murine studies led to safety concerns regarding TGF-β inhibition. Despite these concerns, non-selected targeting of all TGF-β isoforms in a short open-label study of fresolimumab in dcSSc was associated with striking a decrease in skin TGF-β-regulated gene expression biomarkers, as well as in the MRSS[142]. Adverse events, including epistaxis, gum bleeding, subconjunctival eye hemorrhage and anemia were reported amongst the 15 dcSSc patients, suggesting that pan-TGF-β inhibition might be associated with vascular complications[142]. Considering the involvement of TGF-β2 in hematopoiesis, selective inhibition of TGF-β1 and TGF-β3 may have a better safety profile. AVID200 results from the fusion of a TGF-β-Receptor ectodomain with an IgG Fc region, acting as a potent TGF-β trap and showing high selectivity against TGF-β1 and TGF-β3 with limited impact on TGF-β2-associated pathways[143]. The safety and preliminary efficacy of AVID200 was evaluated in a phase I trial in dcSSc, showing that AVID200 at the dose of 1 and 3 mg/kg was well-tolerated[144]. Further investigation of AVID200 in dcSSc, and other therapeutics targeting TGF-β under development in oncoimmunology, hold promise that TGF-β inhibition will eventually become available[145].
Integrins from the αV class participate in the activation of latent TGF-β and may represent a relevant therapeutic target in fibrotic diseases. Abituzumab is a humanized monoclonal antibody inhibiting the activity of αV integrins[146]. The safety and efficacy of abituzumab as add-on therapy to MMF in SSc-ILD were evaluated in a phase II, double-blind, parallel-group, multicenter trial (NCT02745145). The primary efficacy endpoint was the annual rate of change in absolute FVC. Patients were randomized (2:2:1) to receive abituzumab 1500mg, abituzumab 500mg or placebo every 4 weeks for 104 weeks but the trial was prematurely terminated due to slow enrollment[146]. There were no concerns regarding safety; no conclusion regarding efficacy could be drawn due to premature ending of the trial. Other therapies targeting the TGF-β non-canonical pathway (TAK1 inhibitor) or canonical pathway (ALK5 inhibitor, NOX1/4 inhibitor) may have some role in SSc, although clinical data are still awaited for these candidate drugs[147–149].
5.2. Targeting JAK/STAT pathways
Janus Kinases (JAK) are a class of intracellular kinase mediating the signal of numerous receptors of growth factors and major cytokines notably through the phosphorylation of transcription factors from the STAT (Signal Transducer and Activator of Transcription) family[150]. JAK/STAT signaling is involved in several pro-inflammatory and pro-fibrotic pathways in SSc[151]. TGF-β non-canonical pathway involves JAK2-STAT-3; IL-6R mediates signal through JAK1/2/TYK2-STAT-1/3; IL-13 and IL-4 receptors notably through JAK1/3-STAT 6; and IFN type I and II through JAK1/TYK2 and (predominantly) STAT-1[93]. JAK inhibitors are orally administrated tyrosine kinase inhibitors (TKIs) targeting one or more of the 4 members of the JAK family. Various JAK inhibitors have been approved in the treatment of rheumatic diseases such as rheumatoid arthritis (tofacitinib, baricitinib, upadacitinib, filgotinib) or chronic inflammatory skin diseases such as moderate to severe atopic dermatitis (baricitinib)[152–154]. In mouse models of SSc, JAK inhibitors such as Ruxolitinib or Tofacitinib, considered as pan-JAK inhibitors, could prevent the onset of skin and lung fibrosis, notably through their impact on macrophage polarization and fibroblast activation[96, 155–157]. Tofacitinib nonetheless failed to reverse lung and skin fibrosis in the bleomycin mouse model[155].
The safety profile of tofacitinib was evaluated in a Phase I/II double-blind, randomized, placebo-controlled trial including 15 patients with dcSSc (2:1, 10 with active therapy and 5 with placebo)[158]. Change in mRSS and probability of improvement evaluated with the ACR-CRISS favored active therapy and tofacitinib was well tolerated. These encouraging results of a pan-JAK inhibitor in dcSSc are also supported by the recent data of ruxolitinib in cGVHD, showing efficacy in a phase III trial dedicated to this scleroderma-like disorder[159, 160]. Assessment of the impact of a more selective inhibition, such as the targeting of JAK1 with itacitinib, is announced in early dcSSc with mRSS change as primary endpoint (NCT04789850). The recent long-term safety concerns regarding tofacitinib in RA may nonetheless temper the initial enthusiasm surrounding JAK inhibitors in rheumatic diseases[161].
5.3. inhibition of B-cell activation and adaptive immunity
Auto-antibodies are amongst the hallmarks of immune dysregulation in SSc[5]. Immune-complexes composed of SSc-associated anti-nuclear antibodies and their specific nuclear antigen, such as anti-Scl70 antibodies/topoisomerase I complexes, could directly participate in fibroblast activation and endothelial dysfunction [33, 34]. SSc auto-antibodies could also have functional properties as demonstrated by the potential pro-fibrotic effects of anti-PDGF-R antibodies through direct stimulation of PDGF-R with subsequent fibroblast activation[162]. Similar effects of anti-endothelin receptor antibodies on endothelial cells have also been suggested and may participate in endothelial dysfunction[163]. These data strengthen the relevance of targeting adaptive immunity in SSc. B-cells could also directly participate in the immuno-inflammatory process of the disease through the secretion of cytokines such as IL-6[36]. This IL-6 secretion could especially rely on memory B cells expressing CD19 and CD95. Serum levels of BAFF (B cell-activating factor, also known as BLyS for B Lymphocyte Stimulator)), a mediator facilitating survival and maturation of B cells, are elevated in SSc patients[164]. In mouse models of SSc, B-cell depletion with an anti-CD20 antibody prevents the fibrotic manifestations of the disease[165]. Inhibiting BAFF attenuates skin and lung fibrosis in the bleomycin mouse model as well[164]. The effects of abatacept on co-stimulation as previously described also support the relevance of targeting adaptive immunity in SSc[99].
Using a matching strategy based on a propensity score, recent analyses of 254 patients receiving rituximab (Anti-CD20 antibody) in the observational EUSTAR cohort suggested that this therapeutic approach could improve skin fibrosis in comparison with 9575 matched-SSc controls who did not receive rituximab[166]. Meta-analyses suggest that rituximab could also improve lung function in the first year of treatment, as demonstrated by the evolution of FVC and DLco after rituximab initiation[167, 168]. In EUSTAR analyses, PFTs changes were not different between the 2 groups, although a subset receiving MMF and rituximab seemed to have some benefit raising one more time the interest of combination therapies. In a randomized, double-blind, placebo-controlled, phase II multicenter trial, including 57 patients with SSc-PAH, rituximab failed to improve six-minute walk distance (6MWD) at week 24. Nonetheless a secondary analysis model including 6MWD data out to week 48 as a secondary outcome favored rituximab[169]. A Japanese double-blind RCT including 56 patients (dc or lcSSc patients) recently demonstrated the efficacy of rituximab on skin involvement. In this trial, the absolute change in mRSS at week 24 was lower in the rituximab group as compared to the placebo group (−6.30 versus 2.14 in the rituximab and placebo group respectively; difference −8.44 [95% CI −11.00 to −5.88] favoring rituximab; p<0.0001). Rituximab also showed promising result on ILD as the change in FVC (% predicted) from baseline to week 24 was 0.09% in the rituximab group compared with –2.87% in the placebo group (difference 2.96% [95% CI 0.08–5.84]; p=0.044)[170]. These results reinforce the need for investigating rituximab in international RCTs in SSc[171].
The relevance of BAFF inhibition with belimumab (monoclonal antibody targeting BAFF) has been evaluated in a recent single-center, double-blind, placebo-controlled, pilot study including 20 patients with dcSSc and recently started on MMF[172]. Belimumab was well tolerated. An improvement of mRSS was observed in both arms but the comparison of change from baseline favored belimumab (−10 (IQR −13, −9) and −3.0 (IQR −15, −1) in the belimumab and placebo groups respectively; p=0.411). A phase II trial evaluating the combined effect of rituximab and belimumab as an add-on therapy with MMF in early dcSSc is currently ongoing (NCT03844061). The primary outcome will be the probability of improvement based on the ACR-CRISS. This will be the first RCT evaluating the combination of two monoclonal antibodies in SSc. The results of this pivotal trial may pave the way for other combination approaches in the future.
Ibrutinib is a small orally administrated drug that inhibits the function of Bruton’s tyrosine kinase (BTK), an enzyme participating in the B-cell receptor-associated signaling and B-cell survival[173]. A phase Ib/II trial evaluating Ibrutinib in cGVHD showed promising results leading to its FDA-approval for this scleroderma-like condition[174]. To date, there is no registered clinical trial evaluating ibrutinib in SSc, but pre-clinical data on B-cells and peripheral blood mononuclear cells from SSc patients suggest that ibrutinib in vitro could notably limit the production of IL-6[173]. Clinical evidence is nonetheless needed to determine the potential effects of BTK inhibitors in SSc.
5.4. α-MSH and Melanocortin-1 receptor agonists
Alpha-Melanocyte-stimulating hormone (α-MSH) is locally produced in the skin where it could exert physiological anti-fibrotic and anti-inflammatory effects[175]. Melanocortin receptors (MCR) have a pleiotropic distribution and melanocytes, skin fibroblasts, monocytes, macrophages, lymphocytes as well as neutrophils notably expressed MC receptor-1 (MC1R), one of the five identified α-MSH receptors[176]. α-MSH exerts anti-inflammatory and pro-resolving effects notably by enhancing efferocytosis capacities of macrophages and down-regulating the production of IL-6 and IFN-γ. The anti-fibrotic effects of MC1R signaling are notably mediated through the suppression of TGF-β1-dependent collagen production by human skin fibroblasts[177]. Interestingly, patients with dcSSc may experience skin hyperpigmentation, potentially linked to an over-expression of α-MSH that could represent a compensating physiological anti-fibrotic mechanism[178]. In the inflammatory bleomycin-induced mouse model of SSc, α-MSH reduced skin fibrosis and dermal collagen deposit. These effects were partially mediated by the up-regulation of the antioxidant superoxide dismutase 2 and heme-oxygenase 1 in this model including oxidative stress in its pathogenesis[177]. In the animal model of ILD induced by the transoral instillation of bleomycin, α-MSH analogs also decreased IL-6 and TGF-β expression in fibrotic lung tissues[179].
The efficacy, safety, and tolerability of MT-7117, a new synthetic, orally-administered selective agonist of MC1R, will be evaluated in a phase II, multicenter, randomized, double-blind, placebo-controlled, trial in patients with early dcSSc. ACR-CRISS at week 52 will be the primary endpoint and inclusion criteria will select patients with active inflammatory disease through a recruitment strategy similar to the faSScinate and focuSSced trials. Seventy-two patients will be included in this study which is currently recruiting (NCT04440592).
5.5. Eotaxin-2 and its receptor CCR3
Because they participate in cell trafficking, migration and activation, chemokines are thought to play a role in the pathogenesis of SSc. Chemokine c-c motif ligand 24 also called eotaxin-2 may participate in Th2 signaling and associated M2 macrophage activation through its receptor CCR3 (C-C chemokine receptor type 3)[180]. CCR3 is over-expressed at the surface blood monocytes from patients with SSc[181]. Eotaxin-2 is also involved in fibroblasts migration and CCR3 is notably expressed on dermal fibroblasts participating in tissue remodeling and wound healing processes[182, 183]. Eotaxin-2 promotes the production of collagen by lung fibroblasts and is over-expressed in lung tissues and broncho-alveolar lavage of patients with fibrotic lung diseases such as IPF[184]. Eotaxin-2 knockout mice had reduced skin fibrosis after dermal injections of bleomycin in comparison with wild-type littermates. The blockade of this pathway by CM-101, a therapeutic monoclonal antibody against CCL24, led to similar results on bleomycin-induced skin fibrosis and on lung inflammation after trans-oral instillation of bleomycin[182]. In vitro, CM-101 significantly reduced the activation of dermal fibroblasts and their trans-differentiation into myofibroblasts when exposed to serum from SSc patients. CM-101 could also prevent the activation of endothelial cells after exposure to serum from SSc patients, as demonstrated by a down-regulation of VCAM-1, an adhesion molecule classically expressed by activated endothelial cells. Since CM-101 could impact fibrosis as well as vascular manifestations of SSc in pre-clinical models, a multicenter phase II RCT testing the efficacy and safety of CM-101 is planned in early dcSSc and lcSSc patients.
5.6. Therapeutic intravenous Immunoglobulins (IVIg)
IVIg have immunomodulatory properties notably driven by their F(ab) portion that can target and neutralize auto-antibodies. IVIg can thus prevent the effects of immune complexes on fibroblasts, monocytes and endothelial cells[185]. They could also neutralize auto-antibodies with functional properties such as anti-PDGF-receptor or anti-endothelin receptor antibodies with agonist properties[186]. Through their Fc portion, IVIg could also prevent monocyte activation and disrupt the differentiation of monocyte-derived macrophages into anti-and pro-inflammatory macrophages[187]. IVIg could directly impact fibroblast activation as after treatment with IVIg, skin fibroblast showed reduced expression of pro-collagen I, α-SMA and TGF-β receptor I and II[188]. Preventive and therapeutic use of IVIg limits collagen content and macrophage dermal infiltrate in the bleomycin mouse model.
Observational cohort studies suggest an effect of IVIg on GI involvement assessed with the University of California Los Angeles Scleroderma Clinical Trials Consortium gastrointestinal tract 2.0 (UCLA GIT 2.0) and muscle weakness[189, 190]. IVIg could also have a steroid-sparing effect[189]. The effect of IVIg on mRSS change was evaluated in a double-blind, placebo-controlled, multicenter trial including 63 patients with dcSSc, showing no efficacy 12 weeks after administration or at discontinuation of active therapy. The different protocols used for IVIg administration is one of the limitations of this trial[191]. Two phase II trials evaluating IVIg are currently referenced on clinical trial in dcSSc (NCT04137224 and NCT04138485). Safety has been announced as the primary outcome in NCT04137224. NCT04138485 was designed to evaluate safety and efficacy based on the ACR-CRISS after 48 weeks, but this trial appeared to be withdrawn due to business reasons as stated on clinical-trial.gov.
5.7. Other pathways: Epigenetic reprogramming, senescence, modulation of regulatory T-cells and the CD30 pathway.
There is growing interest for epigenetic reprogramming in SSc, notably regarding the regulation of myofibroblast activation[192]. The transcription factor PU.1 has been recently identified as a key regulator of pro-fibrotic activation of fibroblasts and its expression and effects are controlled by epigenetic mechanisms[193]. The pharmacological or genetic inactivation of PU.1 induced the regression of the fibrotic features notably in bleomycin-induced skin fibrosis, suggesting that such an approach may be a promising therapeutic option in SSc.
Cellular senescence and aging could participate in the pathogenesis of SSc[194]. Efferocytosis is impaired during aging in a P38-MAPK-dependent manner and the premature or sustained accumulation of senescent cells; notably senescent fibroblasts; in fibrotic tissues can have a detrimental impact on fibrosis[195]. Senescent fibroblasts can secrete pro-fibrotic mediators such as IL-6 and TGF-ß (a secretome called senescence-associated secretory pattern, SASP) leading to a sustain profibrotic activation loop[196]. Senolytic agents targeting senescent fibroblasts such as dasatinib, could be especially relevant in dcSSc patients with high skin expression of senescence-associated genes, as suggested in a recent pilot study[197].
Regulatory T-cells (Tregs) participate in the maintenance of immunological self-tolerance and circulatory Tregs have decreased functional abilities in SSc[198]. Low-dose of IL-2 could expand and activate Tregs, showing promising results on several systemic autoimmune diseases in a phase I-IIa open label study[199]. The specific impact of such Treg modulating approach in SSc is still to be determined but may constitute a relevant strategy especially in patients with inflammatory manifestations of the disease such as early dcSSc. Among other pathways under-investigation in SSc, the CD30 pathway may also appear especially relevant. CD30 is expressed on activated T and B lymphocytes as well as NK cells[200]. CD30+ B-cells may be especially involved in the pathogenesis of SSc[201]. Activated monocytes and eosinophils may also express low levels of CD30[200]. Brentuximab Vedotin (BV) is a mouse-human chimeric anti-CD30 antibody conjugated to an anti-mitotic agent (monomethylauristatin). BV especially targets CD30+ cells and the anti-mitotic agent released in the intracellular medium interferes with microtubule formation inducing cell cycle arrest[202]. BV may thus show widespread immunomodulatory properties on activated immune cells in SSc. The BRAVOS study is a phase I/II trial exploring the tolerance of BV in dcSSc (NCT0322249). BRAVOS will also evaluate exploratory efficacy endpoints such as mRSS, ACR-CRISS and FVC change. Study completion date is expected in October 2023.
6. Conclusion and prospects
A better understanding of the pathogenesis of key manifestations of SSc, such as ILD, has led to substantial progress in the management of the disease, as demonstrated by the FDA approval of two targeted therapies in this indication, tocilizumab and nintedanib. These medications initially showed efficacy in diseases sharing common pathogenic aspects with SSc, and were secondly successfully repurposed to treat SSc-ILD. The results of phase III trials in other fibrotic diseases may therefore pave the way for future trials in SSc. To that extent, the phase III trials that evaluates CTGF/CCN2 targeting with the monoclonal antibody Pamrevlumab in IPF (NCT039551346 and NCT04419558) could be of specific interest. CTGF/CCN2 is a pro-fibrotic mediator common to the pathogenesis of IPF and SSc-ILD and Pamrevlumab has shown promising results in pre-clinical models of SSc [203]. In the field of systemic diseases, therapeutic advances in systemic lupus erythematosus may also offer new opportunities for SSc. As interferon type I signature has been identified in several studies as a severity marker of SSc, anifrolumab, a human monoclonal antibody to type I interferon receptor subunit 1, may also be considered in the future for the treatment of SSc[39, 204]. In a phase III trial in systemic lupus erythematosus, anifrolumab resulted in a higher percentage of patients showing therapeutic response as defined by a composite end-point at week 52 in comparison with placebo, in contrast to the findings of a previous phase III trial using a different responder index as primary outcome[205, 206]. This suggests that the choice of the most relevant primary endpoint remains a challenge in systemic diseases. In SSc-ILD, FVC which is a quantitative measurement with good reproducibility and sensitivity to change have allowed the demonstration of efficacy of 2 drugs. For a more global disease assessment, reflecting potential disease modifying properties, the broader use of the composite index ACR-CRISS for dcSSc in phase II and III trials may constitute a turn of the tide in the design of SSc RCTs, especially considering that ACR-CRISS could discriminate placebo from active therapy in phase II and III trials that did not met their primary outcome. An emphasis on therapies that have the potential of having clinically meaningful effects on PROs will also be mandatory in the future as we still lack therapeutic strategies that could be proposed both to lcSSc and dcSSc with a long-lasting impact on the way patients feel, function, and survive[207].
Funding:
Dr. Khanna was supported by the NIH/NIAMS K24-AR063120 and R01- AR-070470
Dr. Lescoat was funded by the French network of the University Hospitals HUGO (Hôpitaux Universitaires du Grand Ouest) (AAP JCM2020) and a grant from Rennes University Hospital (CORECT Visiting Grant 2020).
Dr. Roofeh was funded by the NIH/NIAMS T32-AR007080
Dr. Lafyatis was funded by NIH/NIAMS P50 AR060780
Abbreviations
- α-MSH
Alpha-Melanocyte-stimulating hormone
- ACR-CRISS
American College of Rheumatology Composite Response Index for Clinical Trials in Early Diffuse Cutaneous Systemic Sclerosis
- FDA
American Food & Drug Administration
- BAFF
B cell-activating factor
- BLyS
B Lymphocyte Stimulator
- BTK
Bruton’s tyrosine kinase
- CRP
C-reactive protein
- CCR3
C-C chemokine receptor type 3
- CTGF
connective tissue growth factor
- cGMP
cyclic guanosine monophosphate
- CYC
cyclophosphamide
- dcSSc
diffuse cutaneous systemic sclerosis
- DLco
diffusing lung capacity of carbon monoxide
- EndoMT
Endothelial to mesenchymal transition
- EMT
epithelial to mesenchymal transition
- EUSTAR
European Scleroderma Trials and Research group
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- FVC
forced vital capacity
- HAQ-DI
Health assessment questionnaire-quality index
- IL-6
interleukin 6
- ILD
interstitial lung disease
- IVIg
intravenous Immunoglobulins
- JAK
Janus Kinase
- LcSSc
limited cutaneous systemic sclerosis
- mRSS
modified Rodnan total Skin thickness Score
- MMF
mycophenolate mofetil
- PROs
patient reported outcomes
- PDGF
Platelet-derived growth factor
- PKG
protein kinase G
- PAH
pulmonary arterial hypertension
- RCT
Randomized controlled trial
- SLS-I
Scleroderma Lung Study I
- SLS-II
Scleroderma Lung Study II
- SRC
scleroderma renal crisis
- sGC
Soluble guanylate cyclase
- SSc
Systemic sclerosis
- TLR
Toll-like receptor
- TGF-β
transforming growth factor-β
- UCLA GIT 2.0
University of California Los Angeles Scleroderma Clinical Trials Consortium gastrointestinal tract 2.0
- VEGF
vascular endothelial growth factor
Footnotes
Conflicts of interest/Competing interests
AL: Nothing to disclose
DR: Nothing to disclose
MK: Grant support from Boehringer-Ingelheim, Ono Pharmaceuticals, Consultant: AbbVie, Boehringer-Ingelheim, Corbus, Mochida, Kissei, Galapagos NV, Speaker bureau: Asahi Kasei Pharma, Astellas, Boehringer-Ingelheim, Chugai. GSK, Janssen. Mitsubishi-Tanabe, Nippon Shinyaku.
RL: has served as a consultant for Pfizer, Bristol Myers Squibb, Boehringer-Ingleheim, Formation, Sanofi, Boehringer-Mannheim, Merck and Genentech/Roche; holds or recently had research grants from Corbus, Formation, Moderna, Regeneron, Pfizer and Kiniksa and holds equity in Thirona Bio.
YA: Grant support from Alpine ImmunoSciences, Roche and Sanofi.. Consultant: Bayer, Boehringer Ingelheim, Genentech/Roche, Medsenic, Sanofi-Aventis
DK: Grant support from NIH, Immune Tolerance Network, Bayer, BMS, Horizon, Pfizer. Consultant: Acceleron, Actelion, Abbvie, Amgen, Bayer, Boehringer Ingelheim, CSL Behring, Corbus, Gilead, Galapagos, Genentech/Roche, GSK, Horizon, Merck, Mitsubishi Tanabe Pharma, Sanofi-Aventis, and United Therapeutics. Stocks: Eicos Sciences, Inc (less than 5%). Leadership/Equity position – Chief Medical Officer, CiviBioPharma/Eicos Sciences, Inc
Availability of data and material
Not applicable
Code availability
Not applicable
All authors agreed with the content of this review and gave explicit consent to submit
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
REFERENCES
- 1.Denton CP, Khanna D (2017) Systemic sclerosis. The Lancet 390:1685–1699. 10.1016/S0140-6736(17)30933-9 [DOI] [PubMed] [Google Scholar]
- 2.Allanore Y, Simms R, Distler O, et al. (2015) Systemic sclerosis. Nat Rev Dis Primers 1:15002. 10.1038/nrdp.2015.2 [DOI] [PubMed] [Google Scholar]
- 3.Lescoat A, Coiffier G, de Carlan M, et al. (2018) Combination of Capillaroscopic and Ultrasonographic Evaluations in Systemic Sclerosis: Results of a Cross-Sectional Study. Arthritis Care Res (Hoboken) 70:938–943. 10.1002/acr.23413 [DOI] [PubMed] [Google Scholar]
- 4.Denton CP, Lapadula G, Mouthon L, Müller-Ladner U (2009) Renal complications and scleroderma renal crisis. Rheumatology (Oxford) 48 Suppl 3:iii32–35. 10.1093/rheumatology/ken483 [DOI] [PubMed] [Google Scholar]
- 5.Nihtyanova SI, Sari A, Harvey JC, et al. (2020) Using Autoantibodies and Cutaneous Subset to Develop Outcome‐Based Disease Classification in Systemic Sclerosis. Arthritis Rheumatol 72:465–476. 10.1002/art.41153 [DOI] [PubMed] [Google Scholar]
- 6.Lescoat A, Cavalin C, Ehrlich R, et al. (2019) The nosology of systemic sclerosis: how lessons from the past offer new challenges in reframing an idiopathic rheumatological disorder. The Lancet Rheumatology 1:e257–e264. 10.1016/S2665-9913(19)30038-4 [DOI] [PubMed] [Google Scholar]
- 7.LeRoy EC, Black C, Fleischmajer R, et al. (1988) Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 15:202–205 [PubMed] [Google Scholar]
- 8.Frantz C, Avouac J, Distler O, et al. (2016) Impaired quality of life in systemic sclerosis and patient perception of the disease: A large international survey. Seminars in Arthritis and Rheumatism 46:115–123. 10.1016/j.semarthrit.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 9.Lescoat A, Ballerie A, Belhomme N, et al. (2018) Synovial involvement assessed by power Doppler ultra-sonography in systemic sclerosis: results of a cross-sectional study. Rheumatology (Oxford) 57:2012–2021. 10.1093/rheumatology/key214 [DOI] [PubMed] [Google Scholar]
- 10.Heijnen IAFM, Foocharoen C, Bannert B, et al. (2013) Clinical significance of coexisting antitopoisomerase I and anticentromere antibodies in patients with systemic sclerosis: a EUSTAR group-based study. Clin Exp Rheumatol 31:96–102 [PubMed] [Google Scholar]
- 11.Clark KEN, Campochiaro C, Csomor E, et al. (2021) Molecular basis for clinical diversity between autoantibody subsets in diffuse cutaneous systemic sclerosis. Ann Rheum Dis annrheumdis-2021–220402 10.1136/annrheumdis-2021-220402 [DOI] [PubMed] [Google Scholar]
- 12.Tashkin DP, Elashoff R, Clements PJ, et al. (2006) Cyclophosphamide versus Placebo in Scleroderma Lung Disease. New England Journal of Medicine 354:2655–2666. 10.1056/NEJMoa055120 [DOI] [PubMed] [Google Scholar]
- 13.Tashkin DP, Roth MD, Clements PJ, et al. (2016) Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 4:708–719. 10.1016/S2213-2600(16)30152-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roofeh D, Lescoat A, Khanna D (2020) Emerging drugs for the treatment of scleroderma: a review of recent phase 2 and 3 trials. Expert Opin Emerg Drugs 1–12. 10.1080/14728214.2020.1836156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pope JE (2020) The future of treatment in systemic sclerosis: can we design better trials? The Lancet Rheumatology 2:e185–e194. 10.1016/S2665-9913(20)30010-2 [DOI] [PubMed] [Google Scholar]
- 16.van den Hoogen F, Khanna D, Fransen J, et al. (2013) 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative: ACR/EULAR Classification Criteria for SSc. Arthritis & Rheumatism 65:2737–2747. 10.1002/art.38098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Distler O, Highland KB, Gahlemann M, et al. (2019) Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. New England Journal of Medicine 380:2518–2528. 10.1056/NEJMoa1903076 [DOI] [PubMed] [Google Scholar]
- 18.Khanna D, Denton CP, Jahreis A, et al. (2016) Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 387:2630–2640. 10.1016/S0140-6736(16)00232-4 [DOI] [PubMed] [Google Scholar]
- 19.Khanna D, Lin CJF, Furst DE, et al. (2020) Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 8:963–974. 10.1016/S2213-2600(20)30318-0 [DOI] [PubMed] [Google Scholar]
- 20.Roofeh D, Lescoat A, Khanna D (2021) Treatment for systemic sclerosis-associated interstitial lung disease. Curr Opin Rheumatol 33:240–248. 10.1097/BOR.0000000000000795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuwana M, Distler O (2020) Recent progress and missing gaps to achieve goal in the care of systemic sclerosis–associated interstitial lung disease. Journal of Scleroderma and Related Disorders 5:3–5. 10.1177/2397198320902551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khanna D, Lescoat A, Roofeh D, et al. (2021) SYSTEMIC SCLEROSIS-ASSOCIATED INTERSTITIAL LUNG DISEASE: How to incorporate two Food and Drug Administration-approved therapies in clinical practice Arthritis Rheumatol. 10.1002/art.41933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuwana M, Okazaki Y (2012) Quantification of circulating endothelial progenitor cells in systemic sclerosis: a direct comparison of protocols. Ann Rheum Dis 71:617–620. 10.1136/annrheumdis-2011-200713 [DOI] [PubMed] [Google Scholar]
- 24.Shirai Y, Okazaki Y, Inoue Y, et al. (2015) Elevated levels of pentraxin 3 in systemic sclerosis: associations with vascular manifestations and defective vasculogenesis. Arthritis Rheumatol 67:498–507. 10.1002/art.38953 [DOI] [PubMed] [Google Scholar]
- 25.Lescoat A, Ballerie A, Jouneau S, et al. (2019) M1/M2 polarisation state of M-CSF blood-derived macrophages in systemic sclerosis. Ann Rheum Dis 78:e127. 10.1136/annrheumdis-2018-214333 [DOI] [PubMed] [Google Scholar]
- 26.Lescoat A, Lecureur V, Roussel M, et al. (2017) CD16-positive circulating monocytes and fibrotic manifestations of systemic sclerosis. Clin Rheumatol 36:1649–1654. 10.1007/s10067-017-3597-6 [DOI] [PubMed] [Google Scholar]
- 27.Boleto G, Guignabert C, Pezet S, et al. (2018) T-cell costimulation blockade is effective in experimental digestive and lung tissue fibrosis. Arthritis Res Ther 20:197. 10.1186/s13075-018-1694-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varga J, Abraham D (2007) Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117:557–567. 10.1172/JCI31139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lafyatis R (2014) Transforming growth factor β--at the centre of systemic sclerosis. Nat Rev Rheumatol 10:706–719. 10.1038/nrrheum.2014.137 [DOI] [PubMed] [Google Scholar]
- 30.Maehara T, Kaneko N, Perugino CA, et al. (2020) Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J Clin Invest 130:2451–2464. 10.1172/JCI131700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lescoat A, Yelnik CM, Coiffier G, et al. (2019) Ulnar Artery Occlusion and Severity Markers of Vasculopathy in Systemic Sclerosis: A Multicenter Cross-Sectional Study. Arthritis & Rheumatology (Hoboken, NJ) 71:983–990. 10.1002/art.40799 [DOI] [PubMed] [Google Scholar]
- 32.Harris ML, Rosen A (2003) Autoimmunity in scleroderma: the origin, pathogenetic role, and clinical significance of autoantibodies. Curr Opin Rheumatol 15:778–784. 10.1097/00002281-200311000-00016 [DOI] [PubMed] [Google Scholar]
- 33.Raschi E, Privitera D, Bodio C, et al. (2020) Scleroderma-specific autoantibodies embedded in immune complexes mediate endothelial damage: an early event in the pathogenesis of systemic sclerosis. Arthritis Res Ther 22:265. 10.1186/s13075-020-02360-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raschi E, Chighizola CB, Cesana L, et al. (2018) Immune complexes containing scleroderma-specific autoantibodies induce a profibrotic and proinflammatory phenotype in skin fibroblasts. Arthritis Res Ther 20:187. 10.1186/s13075-018-1689-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denton CP, Ong VH, Xu S, et al. (2018) Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: insights from the faSScinate clinical trial in systemic sclerosis. Ann Rheum Dis 77:1362–1371. 10.1136/annrheumdis-2018-213031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taher TE, Ong VH, Bystrom J, et al. (2018) Association of Defective Regulation of Autoreactive Interleukin-6-Producing Transitional B Lymphocytes With Disease in Patients With Systemic Sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 70:450–461. 10.1002/art.40390 [DOI] [PubMed] [Google Scholar]
- 37.Juhl P, Bondesen S, Hawkins CL, et al. (2020) Dermal fibroblasts have different extracellular matrix profiles induced by TGF-β, PDGF and IL-6 in a model for skin fibrosis. Sci Rep 10:17300. 10.1038/s41598-020-74179-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skaug B, Khanna D, Swindell WR, et al. (2020) Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis 79:379–386. 10.1136/annrheumdis-2019-215894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skaug B, Assassi S (2020) Type I interferon dysregulation in Systemic Sclerosis. Cytokine 132:154635. 10.1016/j.cyto.2018.12.018 [DOI] [PubMed] [Google Scholar]
- 40.Assassi S, Li N, Volkmann ER, et al. (2020) Predictive Significance of Serum Interferon-Inducible Protein Score for Response to Treatment in Systemic Sclerosis-Related Interstitial Lung Disease. Arthritis Rheumatol 10.1002/art.41627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ballerie A, Lescoat A, Augagneur Y, et al. (2019) Efferocytosis capacities of blood monocyte-derived macrophages in systemic sclerosis. Immunol Cell Biol 97:340–347. 10.1111/imcb.12217 [DOI] [PubMed] [Google Scholar]
- 42.Lescoat A, Ballerie A, Lelong M, et al. (2020) Crystalline Silica Impairs Efferocytosis Abilities of Human and Mouse Macrophages: Implication for Silica-Associated Systemic Sclerosis. Front Immunol 11:219. 10.3389/fimmu.2020.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhandari R, Ball MS, Martyanov V, et al. Pro-fibrotic Activation of Human Macrophages in Systemic Sclerosis. Arthritis & Rheumatology n/a: 10.1002/art.41243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bellamri N, Morzadec C, Joannes A, et al. (2019) Alteration of human macrophage phenotypes by the anti-fibrotic drug nintedanib. Int Immunopharmacol 72:112–123. 10.1016/j.intimp.2019.03.061 [DOI] [PubMed] [Google Scholar]
- 45.Tabib T, Huang M, Morse N, et al. (2021) Myofibroblast transcriptome indicates SFRP2+ fibroblast progenitors in systemic sclerosis skin. bioRxiv 2021.04.30.442148. 10.1101/2021.04.30.442148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinz B, Lagares D (2020) Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol 16:11–31. 10.1038/s41584-019-0324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya S, Wang W, Morales-Nebreda L, et al. (2016) Tenascin-C drives persistence of organ fibrosis. Nat Commun 7:11703. 10.1038/ncomms11703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y, Huang X, Hecker L, et al. (2013) Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest 123:1096–1108. 10.1172/JCI66700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lagares D, Busnadiego O, García-Fernández RA, et al. (2012) Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum 64:1653–1664. 10.1002/art.33482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakraborty D, Šumová B, Mallano T, et al. (2017) Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun 8:1130. 10.1038/s41467-017-01236-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lescoat A, Varga J, Matucci-Cerinic M, Khanna D (2021) New promising drugs for the treatment of systemic sclerosis: Pathogenic considerations, enhanced classifications, and personalized medicine. Expert Opin Investig Drugs 10.1080/13543784.2021.1923693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roofeh D, Khanna D (2020) Management of systemic sclerosis: the first five years. Curr Opin Rheumatol 32:228–237. 10.1097/BOR.0000000000000711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elhai M, Meune C, Boubaya M, et al. (2017) Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis 76:1897–1905. 10.1136/annrheumdis-2017-211448 [DOI] [PubMed] [Google Scholar]
- 54.Bernstein EJ, Jaafar S, Assassi S, et al. (2020) Performance Characteristics of Pulmonary Function Tests for the Detection of Interstitial Lung Disease in Adults with Early Diffuse Cutaneous Systemic Sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 10.1002/art.41415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Distler O, Assassi S, Cottin V, et al. (2020) Predictors of progression in systemic sclerosis patients with interstitial lung disease. Eur Respir J 55:. 10.1183/13993003.02026-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cottin V, Brown KK (2019) Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir Res 20:13. 10.1186/s12931-019-0980-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann-Vold A-M, Maher TM, Philpot EE, et al. (2021) Assessment of recent evidence for the management of patients with systemic sclerosis-associated interstitial lung disease: a systematic review. ERJ Open Res 7:. 10.1183/23120541.00235-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roofeh D, Distler O, Allanore Y, et al. (2020) Treatment of systemic sclerosis–associated interstitial lung disease: Lessons from clinical trials. Journal of Scleroderma and Related Disorders 5:61–71. 10.1177/2397198320903208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Desallais L, Avouac J, Fréchet M, et al. (2014) Targeting IL-6 by both passive or active immunization strategies prevents bleomycin-induced skin fibrosis. Arthritis Res Ther 16:R157. 10.1186/ar4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mauer J, Chaurasia B, Goldau J, et al. (2014) Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 15:423–430. 10.1038/ni.2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Denton CP, Ong VH, Xu S, et al. (2018) Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: insights from the faSScinate clinical trial in systemic sclerosis. Ann Rheum Dis 77:1362–1371. 10.1136/annrheumdis-2018-213031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morse C, Tabib T, Sembrat J, et al. (2019) Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. European Respiratory Journal 54:. 10.1183/13993003.02441-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao X, Jia G, Guttman A, et al. (2020) Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis. Cell Rep Med 1:100140. 10.1016/j.xcrm.2020.100140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roofeh D, Lin CJF, Goldin J, et al. (2021) Tocilizumab Prevents Progression of Early Systemic Sclerosis Associated Interstitial Lung Disease. Arthritis Rheumatol 10.1002/art.41668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Denton CP, Khanna D (2021) Rational repurposing of tocilizumab for treatment of lung fibrosis in systemic sclerosis. The Lancet Rheumatology 3:e321–e323. 10.1016/S2665-9913(21)00111-9 [DOI] [PubMed] [Google Scholar]
- 66.Flaherty KR, Wells AU, Cottin V, et al. (2019) Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med 381:1718–1727. 10.1056/NEJMoa1908681 [DOI] [PubMed] [Google Scholar]
- 67.Suleman Y, Clark KEN, Cole AR, et al. (2021) Real-world experience of tocilizumab in systemic sclerosis: potential benefit on lung function for anti-topoisomerase-positive patients. Rheumatology (Oxford) 60:3945–3946. 10.1093/rheumatology/keab273 [DOI] [PubMed] [Google Scholar]
- 68.Volkmann ER (2020) Natural history of systemic sclerosis–related interstitial lung disease: How to identify a progressive fibrosing phenotype. Journal of Scleroderma and Related Disorders 5:31–40. 10.1177/2397198319889549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lescoat A, Ballerie A, Augagneur Y, et al. (2018) Distinct Properties of Human M-CSF and GM-CSF Monocyte-Derived Macrophages to Simulate Pathological Lung Conditions In Vitro: Application to Systemic and Inflammatory Disorders with Pulmonary Involvement. Int J Mol Sci 19:. 10.3390/ijms19030894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jaguin M, Fardel O, Lecureur V (2015) AhR-dependent secretion of PDGF-BB by human classically activated macrophages exposed to DEP extracts stimulates lung fibroblast proliferation. Toxicol Appl Pharmacol 285:170–178. 10.1016/j.taap.2015.04.007 [DOI] [PubMed] [Google Scholar]
- 71.Matucci-Cerinic M, Kahaleh B, Wigley FM (2013) Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum 65:1953–1962. 10.1002/art.37988 [DOI] [PubMed] [Google Scholar]
- 72.Wollin L, Wex E, Pautsch A, et al. (2015) Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 45:1434–1445. 10.1183/09031936.00174914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang J, Maier C, Zhang Y, et al. (2017) Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Annals of the Rheumatic Diseases 76:1941–1948. 10.1136/annrheumdis-2016-210823 [DOI] [PubMed] [Google Scholar]
- 74.Richeldi L, Costabel U, Selman M, et al. (2011) Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 365:1079–1087. 10.1056/NEJMoa1103690 [DOI] [PubMed] [Google Scholar]
- 75.Richeldi L, du Bois RM, Raghu G, et al. (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082. 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 76.Volkmann ER, Tashkin DP, LeClair H, et al. (2020) Treatment With Mycophenolate and Cyclophosphamide Leads to Clinically Meaningful Improvements in Patient-Reported Outcomes in Scleroderma Lung Disease: Results of Scleroderma Lung Study II. ACR Open Rheumatol 2:362–370. 10.1002/acr2.11125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Namas R, Tashkin DP, Furst DE, et al. (2018) Efficacy of Mycophenolate Mofetil and Oral Cyclophosphamide on Skin Thickness: Post Hoc Analyses From Two Randomized Placebo-Controlled Trials. Arthritis Care Res (Hoboken) 70:439–444. 10.1002/acr.23282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goldin JG, Kim GHJ, Tseng C-H, et al. (2018) Longitudinal Changes in Quantitative Interstitial Lung Disease on Computed Tomography after Immunosuppression in the Scleroderma Lung Study II. Ann Am Thorac Soc 15:1286–1295. 10.1513/AnnalsATS.201802-079OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Highland KB, Distler O, Kuwana M, et al. (2021) Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: a subgroup analysis of the SENSCIS trial. Lancet Respir Med 9:96–106. 10.1016/S2213-2600(20)30330-1 [DOI] [PubMed] [Google Scholar]
- 80.King TE, Bradford WZ, Castro-Bernardini S, et al. (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370:2083–2092. 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 81.Hall CL, Wells AR, Leung KP (2018) Pirfenidone reduces profibrotic responses in human dermal myofibroblasts, in vitro. Lab Invest 98:640–655. 10.1038/s41374-017-0014-3 [DOI] [PubMed] [Google Scholar]
- 82.Khanna D, Albera C, Fischer A, et al. (2016) An Open-label, Phase II Study of the Safety and Tolerability of Pirfenidone in Patients with Scleroderma-associated Interstitial Lung Disease: the LOTUSS Trial. J Rheumatol 43:1672–1679. 10.3899/jrheum.151322 [DOI] [PubMed] [Google Scholar]
- 83.Acharya N, Sharma SK, Mishra D, et al. (2020) Efficacy and safety of pirfenidone in systemic sclerosis-related interstitial lung disease-a randomised controlled trial. Rheumatol Int 40:703–710. 10.1007/s00296-020-04565-w [DOI] [PubMed] [Google Scholar]
- 84.Behr J, Prasse A, Kreuter M, et al. (2021) Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 9:476–486. 10.1016/S2213-2600(20)30554-3 [DOI] [PubMed] [Google Scholar]
- 85.Fretheim H, Halse A-K, Seip M, et al. (2020) Multidimensional tracking of phenotypes and organ involvement in a complete nationwide systemic sclerosis cohort. Rheumatology keaa026. 10.1093/rheumatology/keaa026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khanna D, Allanore Y, Denton CP, et al. (2020) Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): randomised, double-blind, placebo-controlled multicentre trial. Ann Rheum Dis 79:618–625. 10.1136/annrheumdis-2019-216823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khanna D, Spino C, Johnson S, et al. (2020) Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis & Rheumatology 72:125–136. 10.1002/art.41055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Allanore Y, Wung P, Soubrane C, et al. (2020) A randomised, double-blind, placebo-controlled, 24-week, phase II, proof-of-concept study of romilkimab (SAR156597) in early diffuse cutaneous systemic sclerosis. Annals of the Rheumatic Diseases 79:1600–1607. 10.1136/annrheumdis-2020-218447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khanna D, Denton C, Furst D, et al. (2020) A Phase 2a Randomized, Double-blind, Placebo-controlled Study of Ziritaxestat in Early Diffuse Cutaneous Systemic Sclerosis (NOVESA). Arthritis & Rheumatology 72 (suppl 10): [DOI] [PubMed] [Google Scholar]
- 90.Khanna D, Berrocal VJ, Giannini EH, et al. (2016) The American College of Rheumatology Provisional Composite Response Index for Clinical Trials in Early Diffuse Cutaneous Systemic Sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 68:299–311. 10.1002/art.39501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khanna D, Huang S, Lin CJF, Spino C (2020) New composite endpoint in early diffuse cutaneous systemic sclerosis: revisiting the provisional American College of Rheumatology Composite Response Index in Systemic Sclerosis. Ann Rheum Dis 10.1136/annrheumdis-2020-219100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hasegawa M, Fujimoto M, Kikuchi K, Takehara K (1997) Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol 24:328–332 [PubMed] [Google Scholar]
- 93.Alim K, Bruyère A, Lescoat A, et al. (2021) Interactions of janus kinase inhibitors with drug transporters and consequences for pharmacokinetics and toxicity. Expert Opin Drug Metab Toxicol 1–13. 10.1080/17425255.2021.1862084 [DOI] [PubMed] [Google Scholar]
- 94.Jaguin M, Houlbert N, Fardel O, Lecureur V (2013) Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol 281:51–61. 10.1016/j.cellimm.2013.01.010 [DOI] [PubMed] [Google Scholar]
- 95.Murray PJ, Allen JE, Biswas SK, et al. (2014) Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20. 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lescoat A, Lelong M, Jeljeli M, et al. (2020) Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem Pharmacol 178:114103. 10.1016/j.bcp.2020.114103 [DOI] [PubMed] [Google Scholar]
- 97.Galdo FD, Hartley C, Allanore Y (2020) Randomised controlled trials in systemic sclerosis: patient selection and endpoints for next generation trials. The Lancet Rheumatology 2:e173–e184. 10.1016/S2665-9913(20)30007-2 [DOI] [PubMed] [Google Scholar]
- 98.Fox DA, Lundy SK, Whitfield ML, et al. (2021) Lymphocyte subset abnormalities in early diffuse cutaneous systemic sclerosis. Arthritis Res Ther 23:10. 10.1186/s13075-020-02383-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ponsoye M, Frantz C, Ruzehaji N, et al. (2016) Treatment with abatacept prevents experimental dermal fibrosis and induces regression of established inflammation-driven fibrosis. Ann Rheum Dis 75:2142–2149. 10.1136/annrheumdis-2015-208213 [DOI] [PubMed] [Google Scholar]
- 100.Castellví I, Elhai M, Bruni C, et al. (2020) Safety and effectiveness of abatacept in systemic sclerosis: The EUSTAR experience. Semin Arthritis Rheum 50:1489–1493. 10.1016/j.semarthrit.2019.12.004 [DOI] [PubMed] [Google Scholar]
- 101.Chung L, Spino C, McLain R, et al. (2020) Safety and efficacy of abatacept in early diffuse cutaneous systemic sclerosis (ASSET): open-label extension of a phase 2, double-blind randomised trial. The Lancet Rheumatology 2:e743–e753. 10.1016/S2665-9913(20)30237-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chakravarty EF, Martyanov V, Fiorentino D, et al. (2015) Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res Ther 17:1–14. 10.1186/s13075-015-0669-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hinchcliff M, Huang C-C, Wood TA, et al. (2013) Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 133:1979–1989. 10.1038/jid.2013.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Franks JM, Martyanov V, Cai G, et al. (2019) A Machine Learning Classifier for Assigning Individual Patients With Systemic Sclerosis to Intrinsic Molecular Subsets. Arthritis & Rheumatology 71:1701–1710. 10.1002/art.40898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pendergrass SA, Lemaire R, Francis IP, et al. (2012) Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol 132:1363–1373. 10.1038/jid.2011.472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hinchcliff M, Mahoney JM (2019) Towards a new classification of systemic sclerosis. Nature Reviews Rheumatology 34:1–2. 10.1038/s41584-019-0257-z [DOI] [PubMed] [Google Scholar]
- 107.Stasch J-P, Pacher P, Evgenov OV (2011) Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123:2263–2273. 10.1161/CIRCULATIONAHA.110.981738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ghofrani H-A, Galiè N, Grimminger F, et al. (2013) Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369:330–340. 10.1056/NEJMoa1209655 [DOI] [PubMed] [Google Scholar]
- 109.Humbert M, Coghlan JG, Ghofrani H-A, et al. (2017) Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: results from PATENT-1 and PATENT-2. Ann Rheum Dis 76:422–426. 10.1136/annrheumdis-2015-209087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dees C, Beyer C, Distler A, et al. (2015) Stimulators of soluble guanylate cyclase (sGC) inhibit experimental skin fibrosis of different aetiologies. Ann Rheum Dis 74:1621–1625. 10.1136/annrheumdis-2014-206809 [DOI] [PubMed] [Google Scholar]
- 111.Matei A-E, Beyer C, Györfi A-H, et al. (2018) Protein kinases G are essential downstream mediators of the antifibrotic effects of sGC stimulators. Ann Rheum Dis 77:459. 10.1136/annrheumdis-2017-212489 [DOI] [PubMed] [Google Scholar]
- 112.Sandner P, Stasch JP (2017) Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir Med 122 Suppl 1:S1–S9. 10.1016/j.rmed.2016.08.022 [DOI] [PubMed] [Google Scholar]
- 113.Khanna D, Pope J, Matucci-Cerinic M, et al. (2020) Op0249 Long-Term Extension Results of Rise-Ssc, a Randomized Trial of Riociguat in Patients with Early Diffuse Cutaneous Systemic Sclerosis (dcssc). Annals of the Rheumatic Diseases 79:156–157. 10.1136/annrheumdis-2020-eular.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Distler O, Kramer F, Höfler J, et al. (2020) Fri0575 Biomarker Analysis from the Rise-Ssc Study of Riociguat in Early Diffuse Cutaneous Systemic Sclerosis (dcssc). Annals of the Rheumatic Diseases 79:890–891. 10.1136/annrheumdis-2020-eular.3138 [DOI] [Google Scholar]
- 115.Akhmetshina A, Dees C, Busch N, et al. (2009) The cannabinoid receptor CB2 exerts antifibrotic effects in experimental dermal fibrosis. Arthritis Rheum 60:1129–1136. 10.1002/art.24395 [DOI] [PubMed] [Google Scholar]
- 116.Garcia-Gonzalez E, Selvi E, Balistreri E, et al. (2009) Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology (Oxford) 48:1050–1056. 10.1093/rheumatology/kep189 [DOI] [PubMed] [Google Scholar]
- 117.Gonzalez EG, Selvi E, Balistreri E, et al. (2012) Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann Rheum Dis 71:1545–1551. 10.1136/annrheumdis-2011-200314 [DOI] [PubMed] [Google Scholar]
- 118.Lucattelli M, Fineschi S, Selvi E, et al. (2016) Ajulemic acid exerts potent anti-fibrotic effect during the fibrogenic phase of bleomycin lung. Respir Res 17:49. 10.1186/s12931-016-0373-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Spiera R, Hummers L, Chung L, et al. (2020) Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with systemic sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 10.1002/art.41294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hinchcliff M (2020) Lenabasum for Skin Disease in Patients With Diffuse Cutaneous Systemic Sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 10.1002/art.41302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Inc CPH (2020) Corbus Pharmaceuticals Announces Topline Results from RESOLVE-1 Phase 3 Study of Lenabasum for Treatment of Systemic Sclerosis. In: GlobeNewswire News Room https://www.globenewswire.com/news-release/2020/09/08/2089940/0/en/Corbus-Pharmaceuticals-Announces-Topline-Results-from-RESOLVE-1-Phase-3-Study-of-Lenabasum-for-Treatment-of-Systemic-Sclerosis.html. Accessed 6 May 2021 [Google Scholar]
- 122.Ruzehaji N, Frantz C, Ponsoye M, et al. (2016) Pan PPAR agonist IVA337 is effective in prevention and treatment of experimental skin fibrosis. Ann Rheum Dis 75:2175–2183. 10.1136/annrheumdis-2015-208029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bros M, Haas K, Moll L, Grabbe S (2019) RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 8:733. 10.3390/cells8070733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oh RS, Haak AJ, Smith KMJ, et al. (2018) RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J Cell Sci 131:. 10.1242/jcs.209932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao Y, Natarajan V (2013) Lysophosphatidic acid (LPA) and its receptors: role in airway inflammation and remodeling. Biochim Biophys Acta 1831:86–92. 10.1016/j.bbalip.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Castelino FV, Bain G, Pace VA, et al. (2016) An Autotaxin/Lysophosphatidic Acid/Interleukin-6 Amplification Loop Drives Scleroderma Fibrosis. Arthritis Rheumatol 68:2964–2974. 10.1002/art.39797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Allanore Y, Distler O, Jagerschmidt A, et al. (2018) Lysophosphatidic Acid Receptor 1 Antagonist SAR100842 for Patients With Diffuse Cutaneous Systemic Sclerosis: A Double-Blind, Randomized, Eight-Week Placebo-Controlled Study Followed by a Sixteen-Week Open-Label Extension Study. Arthritis Rheumatol 70:1634–1643. 10.1002/art.40547 [DOI] [PubMed] [Google Scholar]
- 128.Jagasia M, Lazaryan A, Bachier CR, et al. (2021) ROCK2 Inhibition With Belumosudil (KD025) for the Treatment of Chronic Graft-Versus-Host Disease. J Clin Oncol JCO2002754 10.1200/JCO.20.02754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. (2018) Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N Engl J Med 378:35–47. 10.1056/nejmoa1703327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van Laar JM, Farge D, Sont JK, et al. (2014) Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 311:2490–2498. 10.1001/jama.2014.6368 [DOI] [PubMed] [Google Scholar]
- 131.Di Benedetto P, Ruscitti P, Cipriani P, Giacomelli R (2020) Haematopoietic stem cell transplantation in systemic sclerosis: Challenges and perspectives. Autoimmun Rev 19:102662. 10.1016/j.autrev.2020.102662 [DOI] [PubMed] [Google Scholar]
- 132.Maria ATJ, Maumus M, Le Quellec A, et al. (2017) Adipose-Derived Mesenchymal Stem Cells in Autoimmune Disorders: State of the Art and Perspectives for Systemic Sclerosis. Clin Rev Allergy Immunol 52:234–259. 10.1007/s12016-016-8552-9 [DOI] [PubMed] [Google Scholar]
- 133.Rozier P, Maria A, Goulabchand R, et al. (2018) Mesenchymal Stem Cells in Systemic Sclerosis: Allogenic or Autologous Approaches for Therapeutic Use? Front Immunol 9:2938. 10.3389/fimmu.2018.02938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maria ATJ, Toupet K, Bony C, et al. (2016) Antifibrotic, Antioxidant, and Immunomodulatory Effects of Mesenchymal Stem Cells in HOCl-Induced Systemic Sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 68:1013–1025. 10.1002/art.39477 [DOI] [PubMed] [Google Scholar]
- 135.Magalon J, Velier M, Simoncini S, et al. (2019) Molecular profile and proangiogenic activity of the adipose-derived stromal vascular fraction used as an autologous innovative medicinal product in patients with systemic sclerosis. Ann Rheum Dis 78:391–398. 10.1136/annrheumdis-2018-214218 [DOI] [PubMed] [Google Scholar]
- 136.Granel B, Daumas A, Jouve E, et al. (2015) Safety, tolerability and potential efficacy of injection of autologous adipose-derived stromal vascular fraction in the fingers of patients with systemic sclerosis: an open-label phase I trial. Ann Rheum Dis 74:2175–2182. 10.1136/annrheumdis-2014-205681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Guillaume-Jugnot P, Daumas A, Magalon J, et al. (2016) Autologous adipose-derived stromal vascular fraction in patients with systemic sclerosis: 12-month follow-up. Rheumatology (Oxford) 55:301–306. 10.1093/rheumatology/kev323 [DOI] [PubMed] [Google Scholar]
- 138.Dees C, Tomcik M, Palumbo-Zerr K, et al. (2012) JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor β in systemic sclerosis. Arthritis Rheum 64:3006–3015. 10.1002/art.34500 [DOI] [PubMed] [Google Scholar]
- 139.Thannickal VJ, Lee DY, White ES, et al. (2003) Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 278:12384–12389. 10.1074/jbc.M208544200 [DOI] [PubMed] [Google Scholar]
- 140.Roberts AB, Sporn MB, Assoian RK, et al. (1986) Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 83:4167–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Varga J, Rosenbloom J, Jimenez SA (1987) Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J 247:597–604. 10.1042/bj2470597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rice LM, Padilla CM, McLaughlin SR, et al. (2015) Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest 125:2795–2807. 10.1172/JCI77958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim B-G, Malek E, Choi SH, et al. (2021) Novel therapies emerging in oncology to target the TGF-β pathway. J Hematol Oncol 14:55. 10.1186/s13045-021-01053-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lafyatis R, Spiera R, Domsic R, et al. (2020) Thu0329 Safety, Target Engagement, and Initial Efficacy of Avid200, a First-in-Class Potent and Isoform-Selective Inhibitor of Tgf-Beta 1 and 3, in Patients with Diffuse Cutaneous Systemic Sclerosis (dcssc): A Phase 1 Dose Escalation Study. Annals of the Rheumatic Diseases 79:394–395. 10.1136/annrheumdis-2020-eular.1753 [DOI] [Google Scholar]
- 145.Huynh LK, Hipolito CJ, ten Dijke P (2019) A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 9:. 10.3390/biom9110743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Khanna D, Tashkin DP, Wells AU, et al. (2021) STRATUS: A Phase II Study of Abituzumab in Patients With Systemic Sclerosis–associated Interstitial Lung Disease. The Journal of Rheumatology in press: [DOI] [PubMed] [Google Scholar]
- 147.Murphy-Marshman H, Quensel K, Shi-Wen X, et al. (2017) Antioxidants and NOX1/NOX4 inhibition blocks TGFβ1-induced CCN2 and α-SMA expression in dermal and gingival fibroblasts. PLoS One 12:e0186740. 10.1371/journal.pone.0186740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen Y, Shi-wen X, Eastwood M, et al. (2006) Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum 54:1309–1316. 10.1002/art.21725 [DOI] [PubMed] [Google Scholar]
- 149.Da Q, Yan Z, Li Z, et al. (2020) TAK1 is involved in sodium L-lactate-stimulated p38 signaling and promotes apoptosis. Mol Cell Biochem 10.1007/s11010-020-03952-y [DOI] [PubMed] [Google Scholar]
- 150.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW (2002) Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285:1–24. 10.1016/s0378-1119(02)00398-0 [DOI] [PubMed] [Google Scholar]
- 151.Deverapalli SC, Rosmarin D (2018) The use of JAK inhibitors in the treatment of progressive systemic sclerosis. J Eur Acad Dermatol Venereol 32:e328. 10.1111/jdv.14876 [DOI] [PubMed] [Google Scholar]
- 152.Lee EB, Fleischmann R, Hall S, et al. (2014) Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 370:2377–2386. 10.1056/NEJMoa1310476 [DOI] [PubMed] [Google Scholar]
- 153.Genovese MC, Kremer J, Zamani O, et al. (2016) Baricitinib in Patients with Refractory Rheumatoid Arthritis. N Engl J Med 374:1243–1252. 10.1056/NEJMoa1507247 [DOI] [PubMed] [Google Scholar]
- 154.Reich K, Kabashima K, Peris K, et al. (2020) Efficacy and Safety of Baricitinib Combined With Topical Corticosteroids for Treatment of Moderate to Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol 156:1333–1343. 10.1001/jamadermatol.2020.3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang W, Bhattacharyya S, Marangoni RG, et al. (2020) The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. Journal of Scleroderma and Related Disorders 5:40–50. 10.1177/2397198319865367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Aung WW, Wang C, Xibei J, et al. (2020) Immunomodulating role of the JAKs inhibitor tofacitinib in a mouse model of bleomycin-induced scleroderma. J Dermatol Sci 10.1016/j.jdermsci.2020.12.007 [DOI] [PubMed] [Google Scholar]
- 157.Talotta R (2020) The rationale for targeting the JAK/STAT pathway in scleroderma-associated interstitial lung disease. Immunotherapy 13:241–256. 10.2217/imt-2020-0270 [DOI] [PubMed] [Google Scholar]
- 158.Khanna D, Nagaraja V, Koenig A, et al. Tofacitinib in Early Diffuse Cutaneous Systemic Sclerosis— Results of Phase I/II Investigator-Initiated, Double-Blind Randomized Placebo-Controlled Trial. ACR Meeting Abstracts [Google Scholar]
- 159.Jagasia M, Zeiser R, Arbushites M, et al. (2018) Ruxolitinib for the treatment of patients with steroid-refractory GVHD: an introduction to the REACH trials. Immunotherapy 10:391–402. 10.2217/imt-2017-0156 [DOI] [PubMed] [Google Scholar]
- 160.Zeiser R (2020) Ruxolitinib (RUX) Vs Best Available Therapy (BAT) in Patients with Steroid-Refractory/Steroid-Dependent Chronic Graft-Vs-Host Disease (cGVHD): Primary Findings from the Phase 3, Randomized REACH3 Study. ASH [Google Scholar]
- 161.Rheumatology TL (2021) JAK inhibitors: fate in doubt for rheumatoid arthritis? The Lancet Rheumatology 3:e161. 10.1016/S2665-9913(21)00041-2 [DOI] [PubMed] [Google Scholar]
- 162.Baroni SS, Santillo M, Bevilacqua F, et al. (2006) Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med 354:2667–2676. 10.1056/NEJMoa052955 [DOI] [PubMed] [Google Scholar]
- 163.Becker MO, Kill A, Kutsche M, et al. (2014) Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med 190:808–817. 10.1164/rccm.201403-0442OC [DOI] [PubMed] [Google Scholar]
- 164.Matsushita T, Kobayashi T, Mizumaki K, et al. (2018) BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci Adv 4:eaas9944. 10.1126/sciadv.aas9944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hasegawa M, Hamaguchi Y, Yanaba K, et al. (2006) B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am J Pathol 169:954–966. 10.2353/ajpath.2006.060205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Elhai M, Boubaya M, Distler O, et al. (2019) Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: a prospective cohort study. Ann Rheum Dis 78:979–987. 10.1136/annrheumdis-2018-214816 [DOI] [PubMed] [Google Scholar]
- 167.Goswami RP, Ray A, Chatterjee M, et al. (2020) Rituximab in the treatment of systemic sclerosis-related interstitial lung disease: a systematic review and meta-analysis. Rheumatology (Oxford) 10.1093/rheumatology/keaa550 [DOI] [PubMed] [Google Scholar]
- 168.Tang R, Yu J, Shi Y, et al. (2020) Safety and efficacy of Rituximab in systemic sclerosis: A systematic review and meta-analysis. Int Immunopharmacol 83:106389. 10.1016/j.intimp.2020.106389 [DOI] [PubMed] [Google Scholar]
- 169.Zamanian RT, Badesch D, Chung L, et al. (2021) Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis Associated Pulmonary Arterial Hypertension: A Multi-center, Double-blind, Randomized, Placebo-controlled Trial. Am J Respir Crit Care Med 10.1164/rccm.202009-3481OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ebata S, Yoshizaki A, Oba K, et al. (2021) Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. The Lancet Rheumatology 0: 10.1016/S2665-9913(21)00107-7 [DOI] [PubMed] [Google Scholar]
- 171.Hughes M, Khanna D (2021) Rituximab for the treatment of systemic sclerosis: urgent need for an international randomised controlled trial. The Lancet Rheumatology 0: 10.1016/S2665-9913(21)00149-1 [DOI] [PubMed] [Google Scholar]
- 172.Gordon JK, Martyanov V, Franks JM, et al. (2018) Belimumab for the Treatment of Early Diffuse Systemic Sclerosis: Results of a Randomized, Double-Blind, Placebo-Controlled, Pilot Trial. Arthritis Rheumatol 70:308–316. 10.1002/art.40358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Einhaus J, Pecher A-C, Asteriti E, et al. (2020) Inhibition of effector B cells by ibrutinib in systemic sclerosis. Arthritis Res Ther 22:66. 10.1186/s13075-020-02153-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Waller EK, Miklos D, Cutler C, et al. (2019) Ibrutinib for Chronic Graft-versus-Host Disease After Failure of Prior Therapy: 1-Year Update of a Phase 1b/2 Study. Biol Blood Marrow Transplant 25:2002–2007. 10.1016/j.bbmt.2019.06.023 [DOI] [PubMed] [Google Scholar]
- 175.Dinparastisaleh R, Mirsaeidi M (2021) Antifibrotic and Anti-Inflammatory Actions of α-Melanocytic Hormone: New Roles for an Old Player. Pharmaceuticals (Basel) 14:. 10.3390/ph14010045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Spana C, Taylor AW, Yee DG, et al. (2018) Probing the Role of Melanocortin Type 1 Receptor Agonists in Diverse Immunological Diseases. Front Pharmacol 9:1535. 10.3389/fphar.2018.01535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kokot A, Sindrilaru A, Schiller M, et al. (2009) alpha-melanocyte-stimulating hormone suppresses bleomycin-induced collagen synthesis and reduces tissue fibrosis in a mouse model of scleroderma: melanocortin peptides as a novel treatment strategy for scleroderma? Arthritis Rheum 60:592–603. 10.1002/art.24228 [DOI] [PubMed] [Google Scholar]
- 178.Leroy V, Henrot P, Barnetche T, et al. (2019) Association of skin hyperpigmentation disorders with digital ulcers in systemic sclerosis: Analysis of a cohort of 239 patients. J Am Acad Dermatol 80:478–484. 10.1016/j.jaad.2018.07.033 [DOI] [PubMed] [Google Scholar]
- 179.Colombo G, Gatti S, Sordi A, et al. (2007) Production and effects of alpha-melanocyte-stimulating hormone during acute lung injury. Shock 27:326–333. 10.1097/01.shk.0000239764.80033.7e [DOI] [PubMed] [Google Scholar]
- 180.Forssmann U, Uguccioni M, Loetscher P, et al. (1997) Eotaxin-2, a novel CC chemokine that is selective for the chemokine receptor CCR3, and acts like eotaxin on human eosinophil and basophil leukocytes. J Exp Med 185:2171–2176. 10.1084/jem.185.12.2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee R, Reese C, Perry B, et al. (2015) Enhanced chemokine-receptor expression, function, and signaling in healthy African American and scleroderma-patient monocytes are regulated by caveolin-1. Fibrogenesis Tissue Repair 8:11. 10.1186/s13069-015-0028-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mor A, Salto MS, Katav A, et al. (2019) Blockade of CCL24 with a monoclonal antibody ameliorates experimental dermal and pulmonary fibrosis. Annals of the Rheumatic Diseases 78:1260–1268. 10.1136/annrheumdis-2019-215119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Buskermolen JK, Roffel S, Gibbs S (2017) Stimulation of oral fibroblast chemokine receptors identifies CCR3 and CCR4 as potential wound healing targets. J Cell Physiol 232:2996–3005. 10.1002/jcp.25946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Foster MW, Morrison LD, Todd JL, et al. (2015) Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res 14:1238–1249. 10.1021/pr501149m [DOI] [PubMed] [Google Scholar]
- 185.Domínguez-Soto Á, Simón-Fuentes M, de Las Casas-Engel M, et al. (2018) IVIg Promote Cross-Tolerance against Inflammatory Stimuli In Vitro and In Vivo. J Immunol 201:41–52. 10.4049/jimmunol.1701093 [DOI] [PubMed] [Google Scholar]
- 186.Schwab I, Nimmerjahn F (2013) Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol 13:176–189. 10.1038/nri3401 [DOI] [PubMed] [Google Scholar]
- 187.Kozicky LK, Zhao ZY, Menzies SC, et al. (2015) Intravenous immunoglobulin skews macrophages to an anti-inflammatory, IL-10-producing activation state. J Leukoc Biol 98:983–994. 10.1189/jlb.3VMA0315-078R [DOI] [PubMed] [Google Scholar]
- 188.Asano Y, Ihn H, Asashima N, et al. (2005) A case of diffuse scleroderma successfully treated with high-dose intravenous immune globulin infusion. Rheumatology (Oxford) 44:824–826. 10.1093/rheumatology/keh600 [DOI] [PubMed] [Google Scholar]
- 189.Chaigne B, Rodeia S, Benmostefa N, et al. (2020) Corticosteroid-sparing benefit of intravenous immunoglobulin in systemic sclerosis-associated myopathy: A comparative study in 52 patients. Autoimmun Rev 19:102431. 10.1016/j.autrev.2019.102431 [DOI] [PubMed] [Google Scholar]
- 190.Sanges S, Rivière S, Mekinian A, et al. (2017) Intravenous immunoglobulins in systemic sclerosis: Data from a French nationwide cohort of 46 patients and review of the literature. Autoimmun Rev 16:377–384. 10.1016/j.autrev.2017.02.008 [DOI] [PubMed] [Google Scholar]
- 191.Takehara K, Ihn H, Sato S (2013) A randomized, double-blind, placebo-controlled trial: intravenous immunoglobulin treatment in patients with diffuse cutaneous systemic sclerosis. Clin Exp Rheumatol 31:151–156 [PubMed] [Google Scholar]
- 192.Broen JCA, Radstake TRDJ, Rossato M (2014) The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat Rev Rheumatol 10:671–681. 10.1038/nrrheum.2014.128 [DOI] [PubMed] [Google Scholar]
- 193.Wohlfahrt T, Rauber S, Uebe S, et al. (2019) PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566:344–349. 10.1038/s41586-019-0896-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Martyanov V, Whitfield ML, Varga J (2019) Senescence Signature in Skin Biopsies From Systemic Sclerosis Patients Treated With Senolytic Therapy: Potential Predictor of Clinical Response? Arthritis & Rheumatology 71:1766–1767. 10.1002/art.40934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.De Maeyer RPH, van de Merwe RC, Louie R, et al. (2020) Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol 21:615–625. 10.1038/s41590-020-0646-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Volkmann ER, Varga J (2019) Emerging targets of disease-modifying therapy for systemic sclerosis. Nat Rev Rheumatol 15:208–224. 10.1038/s41584-019-0184-z [DOI] [PubMed] [Google Scholar]
- 197.Kanemaru R, Takahashi F, Kato M, et al. (2018) Dasatinib Suppresses TGFβ-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Inhibits Pulmonary Fibrosis. Lung 196:531–541. 10.1007/s00408-018-0134-6 [DOI] [PubMed] [Google Scholar]
- 198.Frantz C, Auffray C, Avouac J, Allanore Y (2018) Regulatory T Cells in Systemic Sclerosis. Front Immunol 9:2356. 10.3389/fimmu.2018.02356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rosenzwajg M, Lorenzon R, Cacoub P, et al. (2019) Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Annals of the Rheumatic Diseases 78:209–217. 10.1136/annrheumdis-2018-214229 [DOI] [PubMed] [Google Scholar]
- 200.Barbieri A, Dolcino M, Tinazzi E, et al. (2015) Characterization of CD30/CD30L+ Cells in Peripheral Blood and Synovial Fluid of Patients with Rheumatoid Arthritis. J Immunol Res 2015:. 10.1155/2015/729654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Higashioka K, Kikushige Y, Ayano M, et al. (2020) Generation of a novel CD30+ B cell subset producing GM-CSF and its possible link to the pathogenesis of systemic sclerosis. Clinical & Experimental Immunology 201:233–243. 10.1111/cei.13477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shea L, Mehta-Shah N (2020) Brentuximab Vedotin in the Treatment of Peripheral T Cell Lymphoma and Cutaneous T Cell Lymphoma. Curr Hematol Malig Rep 15:9–19. 10.1007/s11899-020-00561-w [DOI] [PubMed] [Google Scholar]
- 203.Makino K, Makino T, Stawski L, et al. (2017) Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res Ther 19:134. 10.1186/s13075-017-1356-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guo X, Higgs BW, Bay-Jensen AC, et al. (2015) Suppression of T Cell Activation and Collagen Accumulation by an Anti-IFNAR1 mAb, Anifrolumab, in Adult Patients with Systemic Sclerosis. The Journal of Investigative Dermatology 135:2402–2409. 10.1038/jid.2015.188 [DOI] [PubMed] [Google Scholar]
- 205.Morand EF, Furie R, Tanaka Y, et al. (2020) Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med 382:211–221. 10.1056/NEJMoa1912196 [DOI] [PubMed] [Google Scholar]
- 206.Bruce IN, Nami A, Schwetje E, et al. (2021) Pharmacokinetics, pharmacodynamics, and safety of subcutaneous anifrolumab in patients with systemic lupus erythematosus, active skin disease, and high type I interferon gene signature: a multicentre, randomised, double-blind, placebo-controlled, phase 2 study. The Lancet Rheumatology 3:e101–e110. 10.1016/S2665-9913(20)30342-8 [DOI] [PubMed] [Google Scholar]
- 207.Lescoat A, Murphy SL, Roofeh D, et al. (2020) Considerations for a combined index for limited cutaneous systemic sclerosis to support drug development and improve outcomes. Journal of Scleroderma and Related Disorders 2397198320961967. 10.1177/2397198320961967 [DOI] [PMC free article] [PubMed] [Google Scholar]