

Abstract
Background
Neurodevelopmental disorders, a group of early‐onset neurological disorders with significant clinical and genetic heterogeneity, remain a diagnostic challenge for clinical genetic evaluation. Therefore, we assessed the diagnostic yield by combining standard phenotypes and whole‐exome sequencing in families with these disorders that were “not yet diagnosed” by the traditional testing methods.
Methods
Using a standardized vocabulary of phenotypic abnormalities from human phenotype ontology (HPO), we performed deep phenotyping for 45 “not yet diagnosed” pedigrees to characterize multiple clinical features extracted from Chinese electronic medical records (EMRs). By matching HPO terms with known human diseases and phenotypes from model organisms, together with whole‐exome sequencing data, we prioritized candidate mutations/genes. We made probable genetic diagnoses for the families.
Results
We obtained a diagnostic yield of 29% (13 out of 45) with probably genetic diagnosis, of which compound heterozygosity and de novo mutations accounted for 77% (10/13) of the diagnosis. Of note, these pedigrees are accompanied by a more significant number of non‐neurological features.
Conclusions
Deep phenotyping and whole‐exome sequencing improve the etiological evaluation for neurodevelopmental disorders in the clinical setting.
Keywords: deep phenotyping, genetic diagnosis, neurodevelopmental disorders, whole exome sequencing
We performed deep phenotyping for “not yet diagnosed” families with neurodevelopmental disorders. We conducted human phenotype ontology‐based standardization for phenotypes in neurodevelopmental disorders. Integrating standardized phenotypes with whole‐exome sequencing increased the diagnostic yield.
1. INTRODUCTION
Neurodevelopmental disorders, affecting more than 3% of children (Bellman et al., 2013), are classified into disorders of intellectual disability, communication, autism spectrum, attention‐deficit hyperactivity, specific learning, motor, and others (DSM‐5) (Regier et al., 2013). Neurodevelopmental disorders are common reasons for referrals to genetic counselors (Gahl et al., 2012), and there remain significant challenges in the genetic evaluation due to the heterogeneous clinical presentation (Soden et al., 2014).
Clinical laboratory investigations for neurodevelopmental disorders include neuroimaging, metabolic screening, traditional genetic testing (e.g., karyotype, chromosomal microarray analysis, and panel sequencing), and invasive tests (Shashi et al., 2014). However, >50% of such patients did not receive an etiologic diagnosis (Battaglia et al., 2013). Recently, the application of next‐generation sequencing for its diagnosis has been assessed. Recently, the application of next‐generation sequencing for genetic diagnosis has been assessed. A large family‐based study (n = 4293) showed that approximately 42% of patients with developmental disorders harbored de novo pathogenic mutations (McRae et al., 2017). A diagnosis rate of 36%–48% was obtained for patients with neurodevelopmental disorders (Evers et al., 2017; Nolan & Carlson, 2015; Thevenon et al., 2016). Furthermore, the diagnostic yield increased using an improved analysis pipeline, for example, increased from 27% to 40% by reanalyzing 1133 families with developmental disorders (Wright et al., 2018), and a 15.4% of additional diagnosis for 416 children with congenital anomalies or mental retardations was achieved (Nambot et al., 2018). However, there remains great interest in implementing a novel approach for increasing the diagnostic yield for “not yet diagnosed” patients in a clinical setting.
Deep phenotyping aims to provide the best clinical care for each patient based on disease stratification through the precise and comprehensive analysis of phenotypic abnormalities (Robinson, 2012). The human phenotype ontology (HPO), a standard vocabulary for describing the phenotypic abnormalities in human disease, offers the most comprehensive deep phenotyping resources (Robinson et al., 2008). Several tools based on the standardized phenotype ontology have been developed for clinical and genetic diagnosis. Phenolyzer discovers disease genes according to prior phenotype or disease information (Yang et al., 2015), and Exomiser prioritizes disease‐associated genes/mutations analyzing sequencing data with its matched phenotypes (Smedley et al., 2015). However, to the best of our knowledge, incorporating deep phenotyping with whole‐exome sequencing to assess the diagnostic yield for neurodevelopmental disorders in family‐based studies remains limited.
Here, we performed the phenotype‐driven diagnosis for “not yet diagnosed” pedigrees with neurodevelopmental disorders. Our analyses prioritized candidate pathogenic genes/mutations underlying these families.
2. MATERIALS AND METHODS
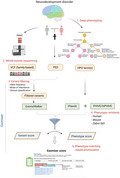
The conceptual framework for the phenotype‐driven diagnosis for a nuclear pedigree (i.e., parent‐offspring [s]) with neurodevelopmental disorders includes deep phenotyping, whole‐exome sequencing, variant filtering, and phenotype‐matching‐based prioritization (Figure 1).
FIGURE 1.
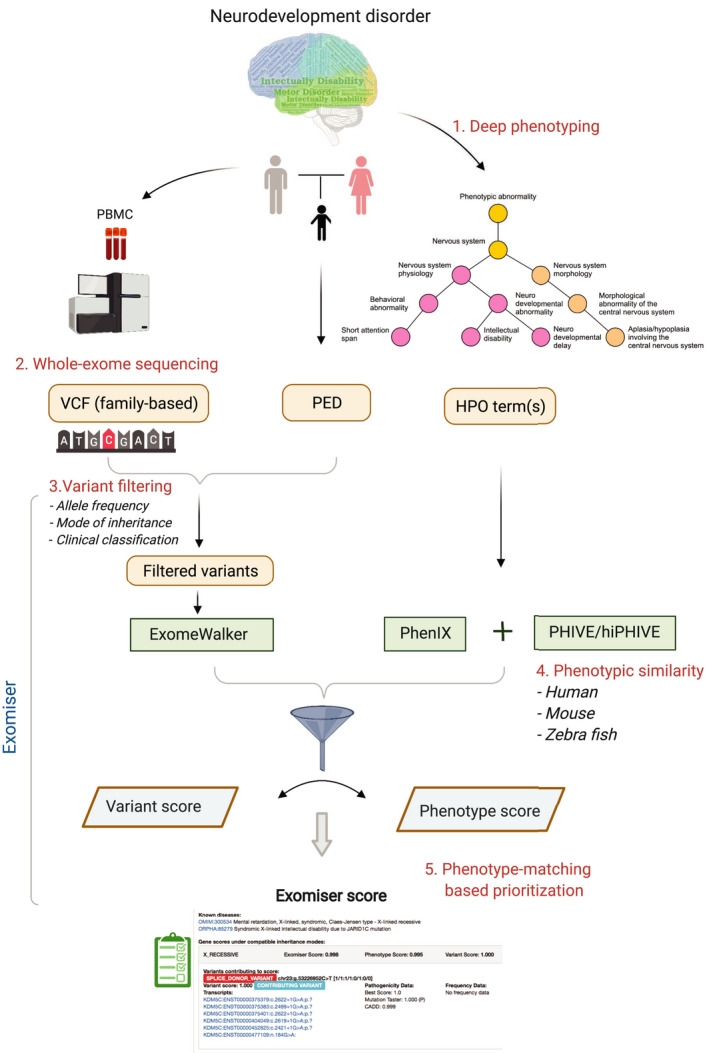
A framework of clinical genetic evaluation for “not yet diagnosed” nuclear pedigrees with neurodevelopmental disorders. VCF, variant calling format; PED, pedigree; and HPO, human phenotype ontology
2.1. Ethical compliance
The study was approved by the Institutional Research Board (IRB) at the Henan Provincial People's Hospital, and all participants or their guardians signed the informed consent.
2.2. The recruitment of pedigree with neurodevelopmental disorders
We recruited nuclear pedigrees with chief complaints of “developmental delay,” “intellectual disability,” or “seizure” (Table S1), who had genetic counseling visited our institute. Although epilepsy remains ostracized from the family of neurodevelopmental disorders, a recent editorial (Shankar et al., 2020) defined epilepsy as an orphan disorder within the neurodevelopmental family. Here, only individuals with a “conventional” neurodevelopmental disorder co‐existent with epilepsy were included. In addition, we excluded patients with known etiologies according to traditional genetic testing or metabolic screening (Table S2).
2.3. Deep phenotyping and phenotype standardization
Various clinical notes, including medical history, laboratory tests, and radiologic reports, were collected from the electronic medical records (EMRs). Deep phenotyping extracted clinical features of symptoms, signs, laboratory, and radiologic tests for each proband (and the affected siblings) (Chen et al., 2019; Tang et al., 2020). The extracted phenotypes were standardized by searching for encoded HPO terms in the Chinese Human Phenotypic Ontology browser (http://www.chinahpo.org). One of the most matched HPO terms was selected if multiple terms were noted (Table S3). The packages of “ontologyIndex” and “hpoPlot” in R were used to perform HPO‐based analyses (Greene et al., 2017).
2.4. Whole‐exome sequencing
Genomic DNA was extracted from the peripheral blood lymphocytes using the QIAamp DNA Blood Mini kit (Qiagen, Hilden Germany). The sequencing library was constructed using SureSelect Human All Exon V6 kit (Agilent, Santa Clara, CA), which was sequenced on the HiSeq Xten platform (Illumina, San Diego, CA) at the Beijing Genomics Center (Shenzhen, China). Sequencing data with a paired‐end length of 150 bp were obtained from 45 pedigrees (including 55 affected and 97 unaffected individuals), with an approximately mean sequencing depth of 49×.
2.5. Data analysis
Sequencing reads were aligned to the hg19 reference genome with BWA, applied GATK (v.4.1) for indel realignment, duplicate removal, and base score quality recalibration. Single nucleotide variants (SNVs) and small insertions and deletions (indels) across all individuals in a family were identified according to GATK Best Practice Guide. Following variant calling and hard filtering, we annotated and classified mutations into pathogenic, likely pathogenic, variants with uncertain significance (VUS), likely benign, and benign according to the ACMG/AMP guideline (Li & Wang, 2017; Richards et al., 2015), which were reviewed manually. The minor allele frequency (MAF) was ascertained from the 1000G (https://www.internationalgenome.org) and gnomAD database (Lek et al., 2016). Databases of ClinVar (Landrum et al., 2014), OMIM (omim.org), and HGMD (Stenson et al., 2014) were used to identify the known pathogenic variants.
To identify potentially causal genes/mutations underlying the affected pedigree and thus make a genetic diagnosis, we used Exomiser (Robinson et al., 2014) to prioritize mutations by integrating the assessment of phenotype similarity. Exomiser compares the standardized phenotypes (i.e., HPO‐coded phenotype terms) with the known Mendelian disease and phenotypes demonstrated in model organisms (e.g., mouse and zebrafish). A protocol for implementing Exomiser has been provided (Köhler et al., 2019), where a pedigree file, a variant calling format file, and a list of HPO terms were needed (Figure 1). The PHIVE (Phenotypic Interpretation of Variants in Exomes) (Robinson et al., 2014) used the wealth of genotype to phenotype data that already exists from model organism studies to assess the potential impact of these exome variants, and the hiPHIVE (Robinson et al., 2014) integrated phenotypic similarity calculation with zebrafish. In addition, ExomeWalker (Robinson et al., 2014) used the random walk method to identify new causal genes underlying Mendelian disease by identifying the vicinity between candidate genes in whole‐exome sequencing and phenotypically related genes in protein–protein association (PPA) networks. We also applied Phenolyzer (Yang et al., 2015) to discover genes implicated in neurodevelopmental disorders using HPO terms alone, leveraging prior biological knowledge and phenotype information.
To establish the genotype–phenotype correlation (i.e., clinical judgment) (Figure S1), two genetic counselors independently reviewed the mutations prioritized by Exomiser, including reassessment for mutations (MAF from 1000G/gnomAD, the known causal mutations in HGMD [Stenson et al., 2014] and Clinvar [Landrum et al., 2014]), and reviewing phenotypes. In addition, an experienced clinical geneticist examined the prioritized variants and their association with phenotypes. All identified potentially causal mutations were finally validated by Sanger sequencing.
3. RESULT
3.1. The recruited pedigrees
During 01‐2019 to 06‐2019, we recruited a total of 45 pedigrees with “not yet diagnosed” neurodevelopmental disorders (Table 1). The proband male/female ratio was approximately 2:1, and all probands had aged at symptom onset <8 years. The majority of the pedigrees were referred for prenatal genetic counseling, but five affected adults came for clinical genetic evaluation. These pedigrees included 31 parent–child trios, 12 parent–child quads, one parent–child quin, and one family with a second‐degree relative. Consanguinity was not documented for the parents of the proband.
TABLE 1.
Clinical characteristics of the recruited pedigrees
| Value | |
|---|---|
| Age (years) | Mean ± SD (range) |
| Symptom onset | 1.4 ± 1.4 (0–8) |
| Age group for proband (years) | Male (female) |
| Neonate [0,1) | 2 (0) |
| Infant [1,3) | 5 (3) |
| Child [3,13) | 21 (7) |
| Adolescent [13,18) | 0 (2) |
| Adult [18,) | 2 (3) |
| Size of pedigree | Number |
| s = 3 (trio) | 31 |
| s = 4 (quad a ) | 12 |
| s = 5 (quin a ) | 1 |
| s = 6 | 1 |
The “quads” and “quins” refer to two and three siblings, regardless of affected status.
3.2. HPO‐encoded phenotypes recapitulating significant clinical heterogeneity
Deep phenotyping compiled the clinical features extracted from the EMRs and ascertained a total of 121 HPO terms (Table S3 and Figure S2). An ontology plot showed the annotated HPO terms (i.e., as nodes indicated) as subgraphs of the full ontology, where a lineage represented a system hierarchy (Figure 2). For example, “HP:0001249 (intellectual disability)” ‘is‐a(n)’ abnormality of “HP:0012759 (neurodevelopmental abnormality).” Two branches of “nervous system physiology” and “nervous system morphology” under the lineage of “phenotypic abnormality of the nervous system” were noted. The plot also showed that the population frequency of HPO terms differed significantly among branches. Overall, more than half of HPO terms were neurologic features (n = 66, 55%), whereas the remaining were implicated in multiple nonnervous systems (Figure S3a). A median number of eight HPO terms per pedigree were annotated (Figure S3b). Nearly all families (n = 44) present with ≥ two phenotypes that such a multi‐morbidity has important clinical implications (Barnett et al., 2012). The phenotypes present in the affected individuals in the same pedigree varied, in part due to incomplete penetrance or later symptom onset (Table S4). One example in the undiagnosed pedigree (UDP) #7 showed that the proband (p701) exhibited “seizures,” “motor development delay,” and “language development delay” at 6 months of age, whereas his sibling (p702) only developed “seizures” at 4 years of age.
FIGURE 2.
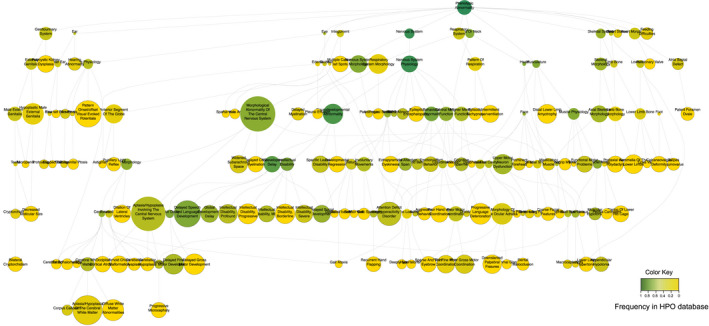
An ontology plot of HPO terms characterized in the recruited pedigrees with neurodevelopmental disorders. The relationship of phenotypes was indicated hierarchically. The color represents the frequency of a term in the HPO database (dark green: High; and yellow: Low). The solid circle represents the abnormal phenotype present in the recruited pedigrees. (A higher resolution for this plot was present at http://www.igenetics.org.cn/project/NDD/)
The most frequent neurologic phenotypes were “HP:0000750 (delayed speech and language development),” “HP:0001270 (motor delay),” “HP:0001249 (intellectual disability),” “HP:0012434 (delayed social development),” and “HP:0001263 (global developmental delay)” (Figure S3c). However, approximately half of these phenotypes were singleton (n = 35, 53%) or doubleton (n = 6, 9%), referring that the phenotype was noted in only one (or two) individuals in the cohort. According to DSM‐5, phenotypes under the lineage of “nervous system physiology” were grouped; each showed a predominant phenotype (e.g., “HP:0000750” in communication disorder, and “HP:0001270” in motor disorder). In contrast, phenotypes in attention‐deficit/hyperactivity disorder, autism spectrum disorder, and specific learning disorder were less present. Non‐neurologic features were also likely to be present (Figure S3d), indicative of syndromic features in a proportion of pedigrees (n = 29, 64%). The vast majority of these phenotypes were singleton or doubleton but “HP:0001252 (muscular hypotonia),” “HP:0012389 (appendicular hypotonia),” and “HP:0003808 (abnormal muscle tone)” had a frequency of 12.5%, 10%, and 7%, respectively.
3.3. An increased diagnostic yield by incorporating HPO‐encoded phenotypes and whole‐exome sequencing
We filtered the SNVs and indels by removing common variants (MAF > 1%) and then evaluating the remaining based on the predicted pathogenicity. Given the mode of inheritance, variants co‐segregated with the pedigree were selected. For example, the autosomal recessive inheritance mode required the homozygous or compound heterozygous mutation. We assigned a phenotypic score for genes based on comparison with known human diseases or animal models with mutations in ortholog and assigned the variant score based on allele frequency and pathogenicity (Smedley et al., 2015) to obtain the final ranking as the sum of the individual scores (Robinson et al., 2014). The prioritized variants were assigned according to the following criteria: (1) pathogenic variant (PV): a variant presented in HGMD, Clinvar or classified to be “pathogenic” based on ACMG guidelines with matched phenotypes to neurodevelopmental disorders; (2) likely pathogenic variant (LPV): a non‐HGMD or non‐ClinVar variant, but was classified as “pathogenic” based on ACMG guidelines in the gene for which previously reported patients had matched phenotypes to neurodevelopmental disorders, and (3) variant of unknown significance (VUS): the variants that do not fulfill the above criteria but the corresponding genes have matched phenotypes to neurodevelopmental disorders. We made a probable genetic diagnosis as a PV or LPV identified in a gene relevant to phenotypes in the patient (Table 2).
TABLE 2.
Probably genetic diagnosis for pedigrees with neurodevelopmental disorders
| Ped. | Gene | Location | Variant | Mutation pattern | Inheritance pattern | ACMG guideline | Syndrome (disease ID) |
|---|---|---|---|---|---|---|---|
| UDP1 | KDM5C (OMIM:314690) | X:53226952 | NM_004187.5: c.2517_2622del | – | XR | P (PVS1 + PM2 + PP1) | Mental retardation, X‐linked, syndromic, Claes‐Jensen type—X‐linked recessive (OMIM:300534) |
| UDP2 | CC2D2A (OMIM:612013) | 4:15534887 4:15575799 | NM_001080522.2: c.1538G > A NM_001080522.2: c.3626delC | CH | AR | P/P (PVS1 + PM2 + PP3/PVS1 + PM2) | Meckel syndrome (ORPHA:564) |
| UDP 9 | TSEN2 (OMIM:608753) | 3:12546725 3:12574176 | NM_025265.4: c.904G > A NM_025265.4: c.1354C > T | CH | AR | LP/P (PM1 + PM2 + PM5 + PP3/PVS1 + PM2 + PP3) | Pontocerebellar hypoplasia type 2B (OMIM:612389) |
| UDP 10 | MECP2 (OMIM:300005) | X:153296516 | NM_004992.3: c.763C > T | De novo | XD | P (PS2 + PM2 + PP3 + PP5) | Rett syndrome (OMIM:312750) |
| UDP 11 | BRWD3 (OMIM:300553) | X:79945476 | NM_153252.5: c.3718C > T | De novo | XR | P (PVS1 + PM2 + PP3 + PP5) | Mental retardation, X‐linked 93 (OMIM:300659) |
| UDP 14 | ARID1B (OMIM:614556) | 6:157222580 | NM_017519.2: c.1809delG | De novo | AD | P (PVS1 + PS2 + PM2) | Coffin‐Siris syndrome 1 (OMIM:135900) |
| UDP 19 | NFIX (OMIM:164005) | 19:13135823 | NM_001365985.2: c.13_14insAGCC | De novo | AD | LP (PVS1 + PS2) | Sotos syndrome 2 (OMIM:614753) |
| UDP 21 | NFIX (OMIM:164005) | 19:13186465 | NM_002501.4: c.935G > A | – | AD | P (PVS1 + PM2 + PP3) | Sotos syndrome 2 (OMIM:614753) |
| UDP 23 | KAT6A (OMIM:601408) | 8:41791815 | NM_006766.5: c.3921_3922delGA | De novo | AD | LP (PVS1 + PS2) | Mental retardation, autosomal dominant 32 (OMIM:616268) |
| UDP 30 | WWOX (OMIM:605131) |
16:79245511 16:78466441 |
NM_016373.3: c.1063G > C NM_016373.3: c.854delA |
CH | AR | LP/P (PM1 + PM2 + PM3 + PP3/PVS1 + PM2) | Epileptic encephalopathy, early infantile, 28 (OMIM:616211) |
| UDP 35 | BRAF (OMIM:164757) | 7:140477811 | NM_004333.6: c.1497A > C | De novo | AD | P (PS2 + PM1 + PM2 + PP3 + PP5) | Cardiofaciocutaneous syndrome (OMIM:115150) |
| UDP 44 | CHD2 (OMIM:602119) | 15:93487644 | NM_001271.4: c.1053‐1G > C | ‐ | AD | P (PVS1 + PM2 + PP3) | Myoclonic‐astatic epilepsy (ORPHA:1942) |
| UDP 49 | TPP1 (OMIM:607998) |
11:6636466 11:6636487 |
NM_000391.4: c.1361C > A NM_000391.4: c.1340G > A |
CH | AR | LP/P (PM1 + PM2 + PM3 + PP3/PM1 + PM2 + PP3 + PP5) | Ceroid lipofuscinosis, neuronal, 2 (OMIM:204500) |
Note: (See more details at http://www.igenetics.org.cn/project/NDD/). Mutation pattern: CH, Compound heterozygous; −, NA. Inheritance mode: XR, X‐lined recessive; AD, Autosomal dominant; AR, Autosomal recessive; XD, X‐linked dominant. ACMG guideline, P, Pathogenic; LP, Likely pathogenic; VUS, Variant with uncertain significance.
We hypothesized that replacing a given phenotype term (e.g., a term located at the low level of the ontology) with its ancestral term would affect phenotype‐matching. One example in UDP #9 highlighted such an effect on prioritization. Initially, “HP:0000252 (microcephaly)” was characterized, and further deep phenotyping updated with its descendent of “HP:0000253 (progressive microcephaly)” in the proband (p901). When “HP:0000252” was used, the compound heterozygous mutations in TSEN2 (OMIM:608753) were prioritized with a phenotype score of 0.878 and Exomiser score of 0.993, significantly greater than “HP:0000253” was used (0.000 and 0.015, respectively). Thus, we replaced 35 terms with their corresponding ancestral terms in 27 pedigrees (i.e., one for 19 and two for eight pedigrees) (Table S5).
Overall, we achieved 13 probably diagnoses (Table 2), leading to a diagnostic yield of 29%. Of the diagnosed pedigrees, six were inherited in an autosomal dominant (AD) manner, four in an autosomal recessive (AR) with compound heterozygous mutations, two in X‐linked recessive (XR), and one in X‐linked dominant (XD). In addition to the compound heterozygous mutation, the de novo mutation accounted for four pedigrees in AD manner, one in XD, and one in XR, respectively (Table 2). A detailed annotation for these mutations, including population frequency, ACMG‐guided clinical classification, and associated clinical syndromes, was provided in Table 2. The phenotype and Exomiser score (Figure 3c,d) increased slightly when the original HPO term was replaced with its ancestral term, obviously noted in UDP #9.
FIGURE 3.
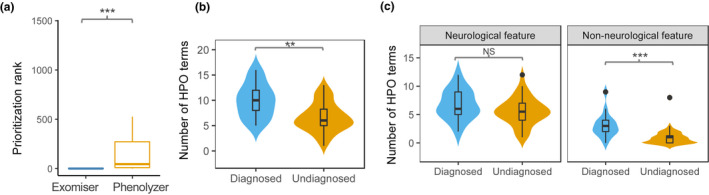
Genetic diagnosis for pedigrees with neurodevelopmental disorders. (a). The comparison of the rank of the prioritized genes identified in Exomiser and Phenolyzer; (b). The number of HPO terms was compared between diagnosed and undiagnosed pedigrees; and (c). The comparison of neurological and non‐neurological features in diagnosed and undiagnosed pedigrees. **, p < .01; ***, p < .001; NS, not significant
We also implemented Phenolyzer to identify the associated clinical syndromes underlying the pedigrees and their causal genes using HPO terms alone. The rank of the prioritized genes identified in Exomiser was compared with that of the genes seeded from Phenolyzer, indicating that incorporating rare mutations increased the ranking for the prioritized genes. In contrast, a broad set of seed genes generated by Phenolyzer created more difficulty in prioritization (Wilcoxon test, p = 6 × 10−5) (Figure 3a).
We next investigated whether the phenotypic structure differed between the “diagnosed” and the remaining pedigrees, as shown in the landscape of HPO‐encoded phenotypes for all pedigrees (Figure S1). The total number of phenotypes in the diagnosed pedigrees (range: 5 to 16; mean: 10) differed significantly from that in the remaining pedigrees (range: 1 to 13; mean: 6.63) (Wilcoxon test, p = .005) (Figure 3b). Although the number of neurologic features did not differ significantly between the diagnosed (mean: 6.76) and undiagnosed (mean: 5.63) families, the number of non‐neurologic features in the diagnosed families, was considerably greater than that in the undiagnosed families (Wilcoxon test, p = .0001) (Figure 3c). For the diagnosed 13 pedigrees, just one was “non‐syndromic” (i.e., only with neurological features). Still, for the undiagnosed 32 pedigrees, 15 were “non‐syndromic.” Diagnosis is more likely to be achieved in “syndromic” rather than “non‐syndromic” patients (χ 2 test, p = .03).
3.4. Case examples
For illustration, we summarized the analyses for two pedigrees accompanied by various phenotypes (Figures 4a and S4). The confirmation of Sanger sequencing for mutations prioritized for the remaining diagnosed pedigrees is shown in Figure S5.
FIGURE 4.
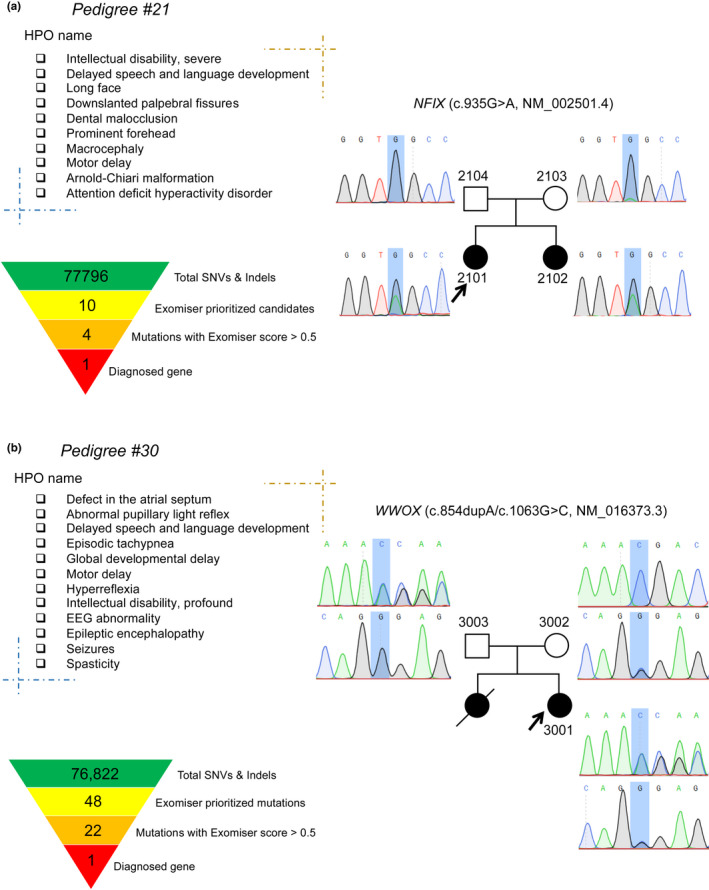
Case examples for UDP #21 (a) and #30 (b). A list of HPO terms present in the proband and the affected members were shown. The mutation filtering and prioritization process obtained genetics diagnosis, and Sanger sequencing confirmed the mutation
3.4.1. UDP #21
The pedigree demonstrated that whole‐exome sequencing in the parent‐offspring quad might increase the power to identify the causal mutation (Figure 4a). Both siblings were referred to a genetic counselor due to intellectual disability, accompanied by macrocephaly, postnatal overgrowth, delayed speech and language development, motor deterioration, and Chiari malformation. The proband (p2101) was delivered by Cesarean section at full term without hypoxia. At 3 years of age, his head circumference was 54 cm (>3 SD), and he received decompression surgery for subamygdala hernia in the cerebellum 6 months later. Her sibling (p2102) had a similar medical history and presented with delays in language and motor skills, poor coordination, and social adaptability at 19 months. She received decompression surgery at 2 years of age and developed intermittent seizures after that. She had a head circumference of 54 cm (>3 SD) at 28 months of age. Currently, she can speak and walk with support. Both siblings have facial abnormalities, including a prominent forehead, long face, short philtrum, dental malocclusion, everted lower lip vermilion, narrow mouth with open‐mouth appearance, and down slanted palpebral fissures. A heterozygous nonsense mutation (c.935G > A, p.Trp312*; NM 002501.4) in NFIX (OMIM:164005) was prioritized, which was associated with Sotos syndrome 2 (OMIM:614753) or Marshall–Smith syndrome (OMIM:602535). Sanger sequencing confirmed the same mutation in the mother but with a low mutation fraction, and the mechanism for the potentially gonadal mosaicism needs to be further investigated.
3.4.2. UDP #30
This pedigree showed that identifying pathogenic mutation might understand the pleiotropic effect of the causal gene (Figure 4b). The proband (a 2‐year‐old girl) was born naturally but developed epileptic seizures 10 days after giving birth with a frequency of 2 to 30 times per day, lasting for 5 s to 5 min for each occurrence, occasionally with status epilepticus and shortness of breath. She showed developmental delays at 09 months of age, for example, unable to raise his head, cry low, unwilling to laugh, abnormal gaze, without ocular pursuit, and pupillary light reflex disappeared. MRI scan showed a C‐shaped spine, thinning of the corpus callosum on the back, and dysplastic white matter. An electroencephalogram showed a periodic eruption‐suppression wave, and visual evoked potentials indicated severe abnormalities in bilateral visual pathways. Echocardiography reported an atrial septal defect (type II). Family history revealed that one of her siblings had similar symptoms and died 3 months after birth. A previous study reported that a pedigree with epileptic encephalopathy, early infantile, 28 (EIEE28, OMIM:616211) accompanying atrial septal defect resulted from a homozygous microdeletion involving WWOX (OMIM:605131) (Davids et al., 2019). In this case, compound heterozygosity in WWOX of a pathogenic frameshift mutation (c.854delA, p.N285fs*10) and a VUS missense mutation (c.1063G > C, p.G355R), derived from maternally and paternally, respectively, caused EIEE28.
3.5. Diagnostic potential of VUS
A VUS identified in the known genes associated with neurodevelopmental phenotypes in the patient may have diagnostic potential (Table S6 and Figure S6). In addition, another three genes (DSCAML1 [OMIM:611782], FOXO4 [OMIM:300033], and TREX2 [OMIM:300370]), prioritized by ExomeWalker implemented in Exomiser, may also have diagnostic potential. These genes closely interacted with the known genes associated with neurodevelopmental disorders, for example, DSCAML1, interactome to IQSEC2 (OMIM:300522); FOXO4, interactome to FOXG1 (OMIM:164874); and TREX2, interactome to ZEB2 (OMIM:605802). Studies have reported that DSCAML1 has an essential functional role in developing the nervous system (Barlow et al., 2002), and the expression of FOXO4 is related to the occurrence and outcome of epilepsy (Wang et al., 2017). Further investigation should be performed to confirm the causative roles of VUS in neurodevelopmental disorders.
4. DISCUSSION
In the present study, we increased the diagnostic yield for “not yet diagnosed” pedigrees with neurodevelopmental disorders through HPO‐based deep phenotyping, whole‐exome sequencing, and phenotype‐matching algorithm (Figures 1 and S1). We compiled multiple phenotypes from EMRs using HPO‐based deep phenotyping (Figures 2, S2 and S3). Together with the filtered rare mutations, we obtained an improved diagnostic yield of 29% and identified a large proportion of de novo and compound heterozygosity mutations underlying these pedigrees (Table 2). In clinical practice, the use of HPO in annotating phenotypic information remains unexplored (Aitken et al., 2019). However, using phenotype alone cannot obtain a satisfied genetic diagnosis; for example, Phenolyzer returned broad candidates of seed genes.
The current study may refine and extend the mutational and phenotypic spectrum of the known neurodevelopmental disorders. We reported at least 13 pathogenic mutations in known genes associated with neurodevelopmental disorders, providing a basis for further functional validation and the elucidation of molecular mechanisms. We also noted a cardiac phenotype in both affected siblings in UDP #30 diagnosed with EIEE28, caused by the compound heterozygous mutations (including a pathogenic and a VUS mutation) in WWOX. Although imprinting genes were not analyzed here, expanding the mutational spectrum in the causal genes and phenotypic spectrum of neurodevelopmental disorders could offer accurate and reliable genetic counseling and prenatal diagnosis to the patients and finally minimize the newly affected individuals in families (Katsanis & Katsanis, 2013).
Our results provide take‐home messages for health professionals in the clinical management of patients with neurodevelopmental disorders. The annotated phenotypes recapitulate syndromic features of neurodevelopmental disorders, which was unneglected in clinical diagnosis and genetics evaluation. Non‐neurologic features may have an increased power to make a genetic diagnosis, and precision phenotyping enables revealing causal genes with pleiotropic effects (Wang et al., 2019). Furthermore, the return of actionable genetic findings for genetic counselors will accelerate prenatal diagnosis, enabling parental choices (Bick et al., 2019).
Several issues should be considered in genetics evaluations for “not yet diagnosed” families. First, the diagnostic yields obtained by the proposed methods need to be further studied in a large cohort. Second, a diagnosed VUS may not meet good medical genetics practice (e.g., returning to the patient), and the interpretation of VUS remained challenging. Nevertheless, the diagnostic potential of VUS could create an opportunity for patients or family members to receive a timely diagnosis in response to the reclassification of VUS, or when more associated phenotypes emerge as the proband grows up. These VUSs may be a good reminder for the clinical practice to follow‐up since these families are still suffering, and a probable diagnosis is needed. Third, deep phenotyping may not extract all phenotypes from EMRs, such as non‐neurologic phenotypes. In addition, the potential bias of clinical features could result from different healthcare providers, which might affect the power to make genetic diagnoses. Fourth, the phenotype selection based on the hierarchical ontology may have a noticeable effect on phenotype‐matching (e.g., UDP #9). An algorithm by iterating all related ancestral terms may identify the most matched disorder. Finally, whole‐exome sequencing cannot capture genetic aberrations outside the exon regions, and a sequencing depth of approximately 49× may not accurately identify copy number variants (Zhang et al., 2019). However, whole‐genome sequencing may increase the power to identify noncoding causal variants and germline CNVs underlying the families.
5. CONCLUSIONS
The combined use of deep phenotyping and whole‐exome sequencing increased the diagnostic yield for nuclear pedigrees with neurodevelopmental disorders. Our analysis may provide an avenue for shortening the diagnostic challenge for such rare undiagnosed diseases in the clinical setting. An economical and practical approach will be widely applied to evaluate genetic etiology.
CONFLICT OF INTEREST
The authors disclose no conflict of interest.
AUTHORS CONTRIBUTIONS
Q.W., K.Y., X.H., and S.L. recruited pedigrees, Q.W. conducted deep phenotyping with guidance from K.D., X.T. and K.D. performed computation and data analysis, Q.W. established genotype–phenotype correlation with input from H.Z., K.D., and S.L., X.T. prepared the graphics and tables with input from Q.W., K.D., and Q.W. wrote the manuscript with input from X.T. and S.L., and K.D. and S.L. conceived and designed the project.
Supporting information
Supinfo
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 81650010, S.L.), the Science and Technology Cooperation Project of Henan Province (No.182106000058, S.L.), a grant from National Health Commission Key Laboratory of Birth Defects Prevention (ZD201907, K.D.), and a grant from Henan Provincial Key Laboratory of Genetic Diseases and Functional Genomics (HNSZD202003, X.T.).
Wang, Q. , Tang, X. , Yang, K. , Huo, X. , Zhang, H. , Ding, K. & Liao, S. (2022). Deep phenotyping and whole‐exome sequencing improved the diagnostic yield for nuclear pedigrees with neurodevelopmental disorders. Molecular Genetics & Genomic Medicine, 10, e1918. 10.1002/mgg3.1918
Qingqing Wang and Xia Tang contributed equally to this study.
Contributor Information
Keyue Ding, Email: ding.keyue@igenetics.org.cn.
Shixiu Liao, Email: ychslshx@henu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Aitken, S. , Firth, H. V. , McRae, J. , Halachev, M. , Kini, U. , Parker, M. J. , Lees, M. M. , Lachlan, K. , Sarkar, A. , Joss, S. , Splitt, M. , McKee, S. , Németh, A. H. , Scott, R. H. , Wright, C. F. , Marsh, J. A. , Hurles, M. E. , FitzPatrick, D. , & DDD Study . (2019). Finding diagnostically useful patterns in quantitative phenotypic data. American Journal of Human Genetics, 105(5), 933–946. 10.1016/j.ajhg.2019.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, G. M. , Micales, B. , Chen, X.‐N. , Lyons, G. E. , & Korenberg, J. R. (2002). Mammalian DSCAMs: Roles in the development of the spinal cord, cortex, and cerebellum? Biochemical and Biophysical Research Communications, 293(3), 881–891. 10.1016/s0006-291x(02)00307-8 [DOI] [PubMed] [Google Scholar]
- Barnett, K. , Mercer, S. W. , Norbury, M. , Watt, G. , Wyke, S. , & Guthrie, B. (2012). Epidemiology of multimorbidity and implications for health care, research, and medical education: A cross‐sectional study. The Lancet, 380(9836), 37–43. 10.1016/s0140-6736(12)60240-2 [DOI] [PubMed] [Google Scholar]
- Battaglia, A. , Doccini, V. , Bernardini, L. , Novelli, A. , Loddo, S. , Capalbo, A. , Filippi, T. , & Carey, J. C. (2013). Confirmation of chromosomal microarray as a first‐tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. European Journal of Paediatric Neurology, 17(6), 589–599. 10.1016/j.ejpn.2013.04.010 [DOI] [PubMed] [Google Scholar]
- Bellman, M. , Byrne, O. , & Sege, R. (2013). Developmental assessment of children. BMJ: British Medical Journal, 346, e8687, 10.1136/bmj.e8687 [DOI] [PubMed] [Google Scholar]
- Bick, D. , Jones, M. , Taylor, S. L. , Taft, R. J. , & Belmont, J. (2019). Case for genome sequencing in infants and children with rare, undiagnosed or genetic diseases. Journal of Medical Genetics, 56(12), 783–791. 10.1136/jmedgenet-2019-106111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Song, L. , Shao, Y. , Li, D. , & Ding, K. (2019). Using natural language processing to extract clinically useful information from Chinese electronic medical records. International Journal of Medical Informatics, 124, 6–12. 10.1016/j.ijmedinf.2019.01.004 [DOI] [PubMed] [Google Scholar]
- Davids, M. , Markello, T. , Wolfe, L. A. , Chepa‐Lotrea, X. , Tifft, C. J. , Gahl, W. A. , & Malicdan, M. C. V. (2019). Early infantile‐onset epileptic encephalopathy 28 due to a homozygous microdeletion involving the WWOX gene in a region of uniparental disomy. Human Mutation, 40(1), 42–47. 10.1002/humu.23675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers, C. , Staufner, C. , Granzow, M. , Paramasivam, N. , Hinderhofer, K. , Kaufmann, L. , Fischer, C. , Thiel, C. , Opladen, T. , Kotzaeridou, U. , Wiemann, S. , Schlesner, M. , Eils, R. , Kölker, S. , Bartram, C. R. , Hoffmann, G. F. , & Moog, U. (2017). Impact of clinical exomes in neurodevelopmental and neurometabolic disorders. Molecular Genetics and Metabolism, 121(4), 297–307. 10.1016/j.ymgme.2017.06.014 [DOI] [PubMed] [Google Scholar]
- Gahl, W. A. , Markello, T. C. , Toro, C. , Fajardo, K. F. , Sincan, M. , Gill, F. , Carlson‐Donohoe, H. , Gropman, A. , Pierson, T. M. , Golas, G. , Wolfe, L. , Groden, C. , Godfrey, R. , Nehrebecky, M. , Wahl, C. , Landis, D. M. , Yang, S. , Madeo, A. , Mullikin, J. C. , … Adams, D. (2012). The National Institutes of Health undiagnosed diseases program: Insights into rare diseases. Genetics in Medicine, 14(1), 51–59. 10.1038/gim.0b013e318232a005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, D. , Richardson, S. , & Turro, E. (2017). ontologyX: A suite of R packages for working with ontological data. Bioinformatics, 33(7), 1104–1106. 10.1093/bioinformatics/btw763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanis, S. H. , & Katsanis, N. (2013). Molecular genetic testing and the future of clinical genomics. Nature Reviews Genetics, 14, 415–426. 10.1038/nrg3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, S. , Øien, N. C. , Buske, O. J. , Groza, T. , Jacobsen, J. O. B. , McNamara, C. , Vasilevsky, N. , Carmody, L. C. , Gourdine, J. P. , Gargano, M. , McMurry, J. , Danis, D. , Mungall, C. J. , Smedley, D. , Haendel, M. , & Robinson, P. N. (2019). Encoding clinical data with the human phenotype ontology for computational differential diagnostics. Current Protocols in Human Genetics, 103(1), e92. 10.1002/cphg.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Riley, G. R. , Jang, W. , Rubinstein, W. S. , Church, D. M. , & Maglott, D. R. (2014). ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42(D1), D980–D985. 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , O'Donnell‐Luria, A. H. , Ware, J. S. , Hill, A. J. , Cummings, B. B. , Tukiainen, T. , Birnbaum, D. P. , Kosmicki, J. A. , Duncan, L. E. , Estrada, K. , Zhao, F. , Zou, J. , Pierce‐Hoffman, E. , Berghout, J. , … Exome Aggregation Consortium . (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , & Wang, K. (2017). InterVar: Clinical interpretation of genetic variants by the 2015 ACMG‐AMP guidelines. The American Journal of Human Genetics, 100(2), 267–280. 10.1016/j.ajhg.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae, J. F. , Clayton, S. , Fitzgerald, T. W. , Kaplanis, J. , Prigmore, E. , Rajan, D. , Sifrim, A. , Aitken, S. , Akawi, N. , Alvi, M. , Ambridge, K. , Barrett, D. M. , Bayzetinova, T. , Jones, P. , Jones, W. D. , King, D. , Krishnappa, N. , Mason, L. E. , Singh, T. , … Hurles, M. E. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambot, S. , Thevenon, J. , Kuentz, P. , Duffourd, Y. , Tisserant, E. , Bruel, A.‐L. , Mosca‐Boidron, A. L. , Masurel‐Paulet, A. , Lehalle, D. , Jean‐Marçais, N. , Lefebvre, M. , Vabres, P. , el Chehadeh‐Djebbar, S. , Philippe, C. , Tran Mau‐Them, F. , St‐Onge, J. , Jouan, T. , Chevarin, M. , Poé, C. , … Orphanomix Physicians' Group . (2018). Clinical whole‐exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: Substantial interest of prospective annual reanalysis. Genetics in Medicine, 20(6), 645–654. 10.1038/gim.2017.162 [DOI] [PubMed] [Google Scholar]
- Nolan, D. , & Carlson, M. (2015). Whole exome sequencing in pediatric neurology patients. Journal of Child Neurology, 31(7), 887–894. 10.1177/0883073815627880 [DOI] [PubMed] [Google Scholar]
- Regier, D. A. , Kuhl, E. A. , & Kupfer, D. J. (2013). The DSM‐5: Classification and criteria changes. World Psychiatry, 12(2), 92–98. 10.1002/wps.20050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, P. N. (2012). Deep phenotyping for precision medicine. Human Mutation, 33(5), 777–780. 10.1002/humu.22080 [DOI] [PubMed] [Google Scholar]
- Robinson, P. N. , Köhler, S. , Bauer, S. , Seelow, D. , Horn, D. , & Mundlos, S. (2008). The human phenotype ontology: A tool for annotating and analyzing human hereditary disease. The American Journal of Human Genetics, 83(5), 610–615. 10.1016/j.ajhg.2008.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, P. N. , Köhler, S. , Oellrich, A. , Sanger Mouse Genetics Project , Wang, K. , Mungall, C. J. , Lewis, S. E. , Washington, N. , Bauer, S. , Seelow, D. , Krawitz, P. , Gilissen, C. , Haendel, M. , & Smedley, D. (2014). Improved exome prioritization of disease genes through cross‐species phenotype comparison. Genome Research, 24(2), 340–348. 10.1101/gr.160325.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar, R. , Perera, B. , & Thomas, R. H. (2020). Epilepsy, an orphan disorder within the neurodevelopmental family. Journal of Neurology, Neurosurgery & Psychiatry, 91(12), 1245–1247. 10.1136/jnnp-2020-324660 [DOI] [PubMed] [Google Scholar]
- Shashi, V. , McConkie‐Rosell, A. , Rosell, B. , Schoch, K. , Vellore, K. , McDonald, M. , Jiang, Y. H. , Xie, P. , Need, A. , & Goldstein, D. B. (2014). The utility of the traditional medical genetics diagnostic evaluation in the context of next‐generation sequencing for undiagnosed genetic disorders. Genetics in Medicine, 16(2), 176–182. 10.1038/gim.2013.99 [DOI] [PubMed] [Google Scholar]
- Smedley, D. , Jacobsen, J. O. B. , Jäger, M. , Köhler, S. , Holtgrewe, M. , Schubach, M. , Siragusa, E. , Zemojtel, T. , Buske, O. J. , Washington, N. L. , Bone, W. P. , Haendel, M. A. , & Robinson, P. N. (2015). Next‐generation diagnostics and disease‐gene discovery with the exomiser. Nature Protocols, 10(12), 2004–2015. 10.1038/nprot.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soden, S. E. , Saunders, C. J. , Willig, L. K. , Farrow, E. G. , Smith, L. D. , Petrikin, J. E. , LePichon, J. , Miller, N. A. , Thiffault, I. , Dinwiddie, D. L. , Twist, G. , Noll, A. , Heese, B. A. , Zellmer, L. , Atherton, A. M. , Abdelmoity, A. T. , Safina, N. , Nyp, S. S. , Zuccarelli, B. , … Kingsmore, S. F. (2014). Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Science Translational Medicine, 6(265), 265ra168. 10.1126/scitranslmed.3010076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Shaw, K. , Phillips, A. D. , & Cooper, D. N. (2014). The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genetics, 133(1), 1–9. 10.1007/s00439-013-1358-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. , Chen, W. , Zeng, Z. , Ding, K. , & Zhou, Z. (2020). An ontology‐based classification of Ebstein's anomaly and its implications in clinical adverse outcomes. International Journal of Cardiology, 316, 79–86. 10.1016/j.ijcard.2020.04.073 [DOI] [PubMed] [Google Scholar]
- Thevenon, J. , Duffourd, Y. , Masurel‐Paulet, A. , Lefebvre, M. , Feillet, F. , el Chehadeh‐Djebbar, S. , St‐Onge, J. , Steinmetz, A. , Huet, F. , Chouchane, M. , Darmency‐Stamboul, V. , Callier, P. , Thauvin‐Robinet, C. , Faivre, L. , & Rivière, J. B. (2016). Diagnostic odyssey in severe neurodevelopmental disorders: Toward clinical whole‐exome sequencing as a first‐line diagnostic test. Clinical Genetics, 89(6), 700–707. 10.1111/cge.12732 [DOI] [PubMed] [Google Scholar]
- Wang, W. , Corominas, R. , & Lin, G. N. (2019). De novo mutations from whole exome sequencing in neurodevelopmental and psychiatric disorders: From discovery to application. Frontiers in Genetics, 10, 258. 10.3389/fgene.2019.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, K. , Zhao, J. , Liu, L. , & Feng, J. (2017). Expression is associated with the occurrence and outcome of seizures: An RNA‐sequencing analysis of low‐grade gliomas. Seizure, 52, 41–45. 10.1016/j.seizure.2017.09.012 [DOI] [PubMed] [Google Scholar]
- Wright, C. F. , McRae, J. F. , Clayton, S. , Gallone, G. , Aitken, S. , FitzGerald, T. W. , Jones, P. , Prigmore, E. , Rajan, D. , Lord, J. , Sifrim, A. , Kelsell, R. , Parker, M. J. , Barrett, J. C. , Hurles, M. E. , FitzPatrick, D. , Firth, H. V. , & DDD Study . (2018). Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome‐wide data in 1,133 families with developmental disorders. Genetics in Medicine, 20(10), 1216–1223. 10.1038/gim.2017.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Robinson, P. N. , & Wang, K. (2015). Phenolyzer: Phenotype‐based prioritization of candidate genes for human diseases. Nature Methods, 12(9), 841–843. 10.1038/nmeth.3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Bai, W. , Yuan, N. , & Du, Z. (2019). Comprehensively benchmarking applications for detecting copy number variation. PLoS Computational Biology, 15(5), e1007069. 10.1371/journal.pcbi.1007069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.