

Abstract
Arrhythmogenic cardiomyopathy (ACM) is a primary disease of the myocardium, predominantly caused by genetic defects in proteins of the cardiac intercalated disc, particularly desmosomes. Transmission is mostly autosomal dominant with incomplete penetrance, ACM also has wide phenotype variability, ranging from premature ventricular contractions to sudden cardiac death and heart failure. Among other drivers and modulators of phenotype, inflammation in response to viral infection and/or immune triggers, have been postulated to be an aggravator of cardiac myocyte damage and necrosis. This theory is supported by multiple pieces of evidence, including the presence of inflammatory infiltrates in more than two-thirds of ACM hearts, detection of different cardiotropic viruses in sporadic cases of ACM, the fact that ACM patients often fulfill the histological criteria of active myocarditis, and the abundance of anti-desmoglein-2, anti-heart and anti-intercalated disk autoantibodies in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC). In keeping with the frequent familial occurrence of ACM, it has been proposed that, in addition to genetic predisposition to progressive myocardial damage, a heritable susceptibility to viral infections and/or immune reactions may explain familial clustering of ACM. Moreover, considerable in vitro and in vivo evidence implicates activated inflammatory signaling in ACM. While the role of inflammation/immune response in ACM is not entirely clear, inflammation as a driver of phenotype and potential target for mechanism-based therapy warrants further research. This review discusses the present evidence supporting the role of inflammatory and immune responses in ACM pathogenesis and proposes opportunities for translational and clinical investigation.
Keywords: arrhythmogenic cardiomyopathy, genetics, ventricular arrhythmia, sudden cardiac death, inflammation, autoimmunity, myocarditis
Journal Subject Terms: Cardiomyopathy, Inflammation, Myocarditis, Sudden Cardiac Death
1. Inflammation – the missing piece in the pathogenesis of ACM?
Arrhythmogenic cardiomyopathy (ACM) is a genetically determined myocardial disease, characterized by fibrofatty myocardial infiltration and a high degree of electrical instability associated with significantly increased risk for sudden cardiac death (SCD).1, 2 Life-threatening ventricular arrhythmias, the cardinal manifestation of ACM, may occur at any disease stage, but typically precede ventricular dysfunction and pathological abnormalities.3 The classical form of ACM, arrhythmogenic right ventricular cardiomyopathy (ARVC), has been recognized for centuries and was formally defined in 1982.1 More recently, disease subtypes with predominantly left ventricular (LV) or biventricular involvement were described following wider implementation of post-mortem autopsy, increased use of contrast-enhancement cardiovascular magnetic resonance imaging (CMR) and detailed endocardial and epicardial voltage mapping, and improved understanding of genotype-phenotype associations.3–5 This warranted the shift of the nomenclature from ARVC to ACM, which collectively reflects ARVC, arrhythmogenic left ventricular cardiomyopathy (ALVC), and biventricular ACM.3, 6
ACM is a primary genetic disorder caused principally by desmosomal and infrequently by non-desmosomal gene variants.7 Remodeling of desmosomes leading to abnormal cardiac myocyte response to mechanical stress and subsequent myocyte loss is considered central to the disease pathogenesis (Figure 1, pathways A and B).8 However, the incomplete penetrance and the wide phenotypic variability among identical variant carriers has sparked numerous theories about the potential involvement of non-genetic factors in shaping the ultimate ACM phenotype.9 Endurance exercise has been shown to significantly increase the risk of ventricular tachyarrhythmias and disease penetrance in desmosomal pathogenic variant carriers, likely related to physical-stress-induced recurrent desmosomal damage.10 Additionally inflammation of infectious and/or immune etiology has been suggested to influence ACM pathophysiology. Initially, this concept was supported by the fact that patchy inflammatory infiltrates have been found in up to two thirds of ARVC hearts (Figure 2).11 With increased recognition of ACM as a distinct clinical entity, there have been reports of ACM cases misdiagnosed as myocarditis (lymphocytic and granulomatous), or vice versa, highlighting the close resemblance of their clinical presentation (Figure 3).12–15 These preliminary findings open the door to a discussion whether an inflammatory process secondary to infection or immune mechanism may trigger ACM disease initiation or progression in genetically predisposed subjects. The absence of manifest disease in ACM-associated pathogenic variant-positive patients, rare progressive degenerative/dysplastic process in pathogenic variant-positive patients and phenotypically similar disease process in some pathogenic variant-negative patients support inflammation as a driving force in ACM phenotype.16 The voltage mapping data (repeated at two different time periods) and excellent response to ablation during long-term follow-up provide evidence that ACM phenotype develops as a result of acute triggering event followed by longer periods of quiescence and/or very slow progression.5, 17
Figure 1. Schematic diagram illustrating mechanistic pathways involved in the pathogenesis of myocyte damage and replacement-type myocardial fibrosis in different genetic forms of arrhythmogenic cardiomyopathy.
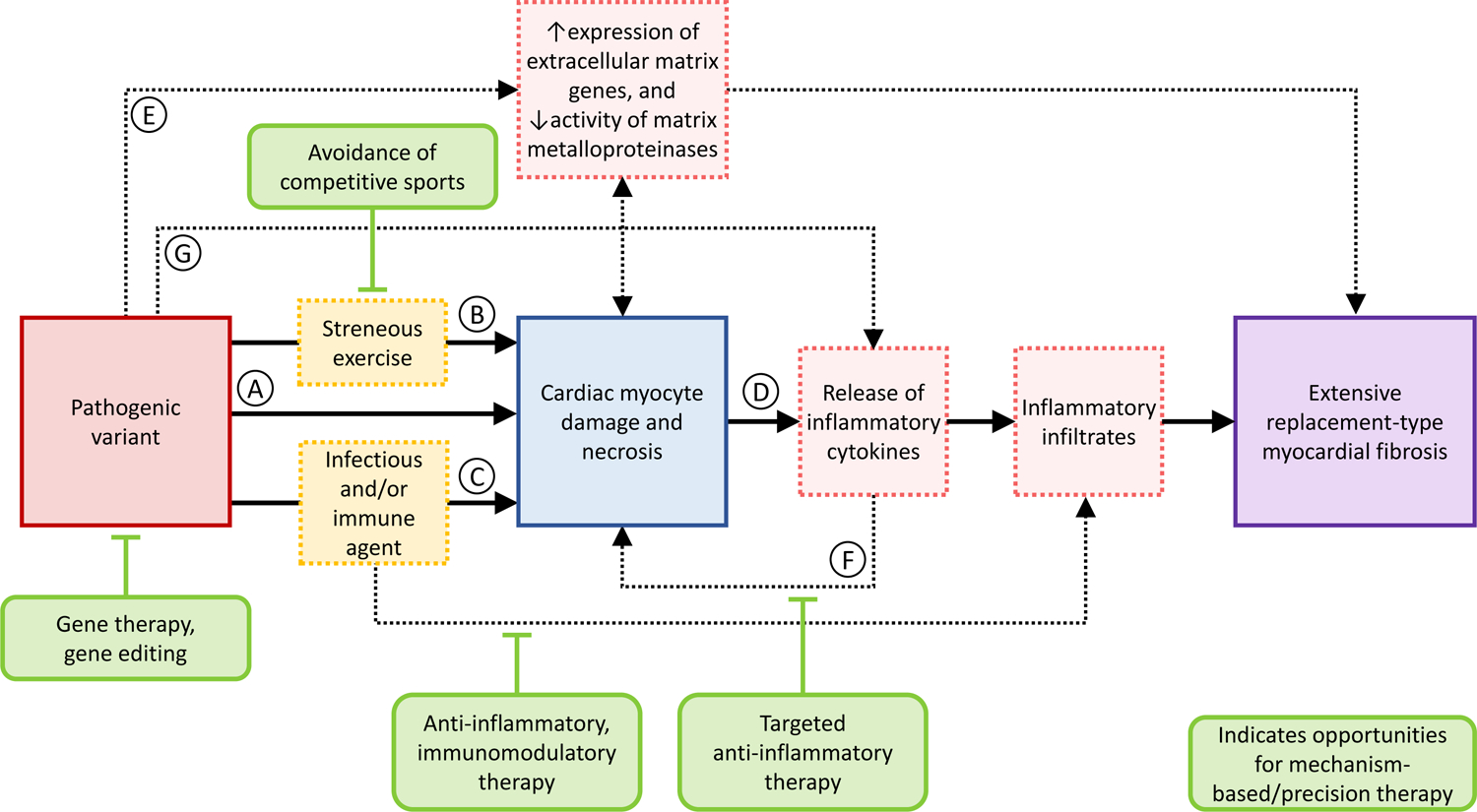
Please see text for details regarding the pathogenic cascade. Note, mechanisms involved in cardiac electrical susceptibility in the context of alterations in cardiac ion channels and intercalated discs, are not shown.
Figure 2. Histological evidence of myocardial inflammation in arrhythmogenic right ventricular cardiomyopathy.
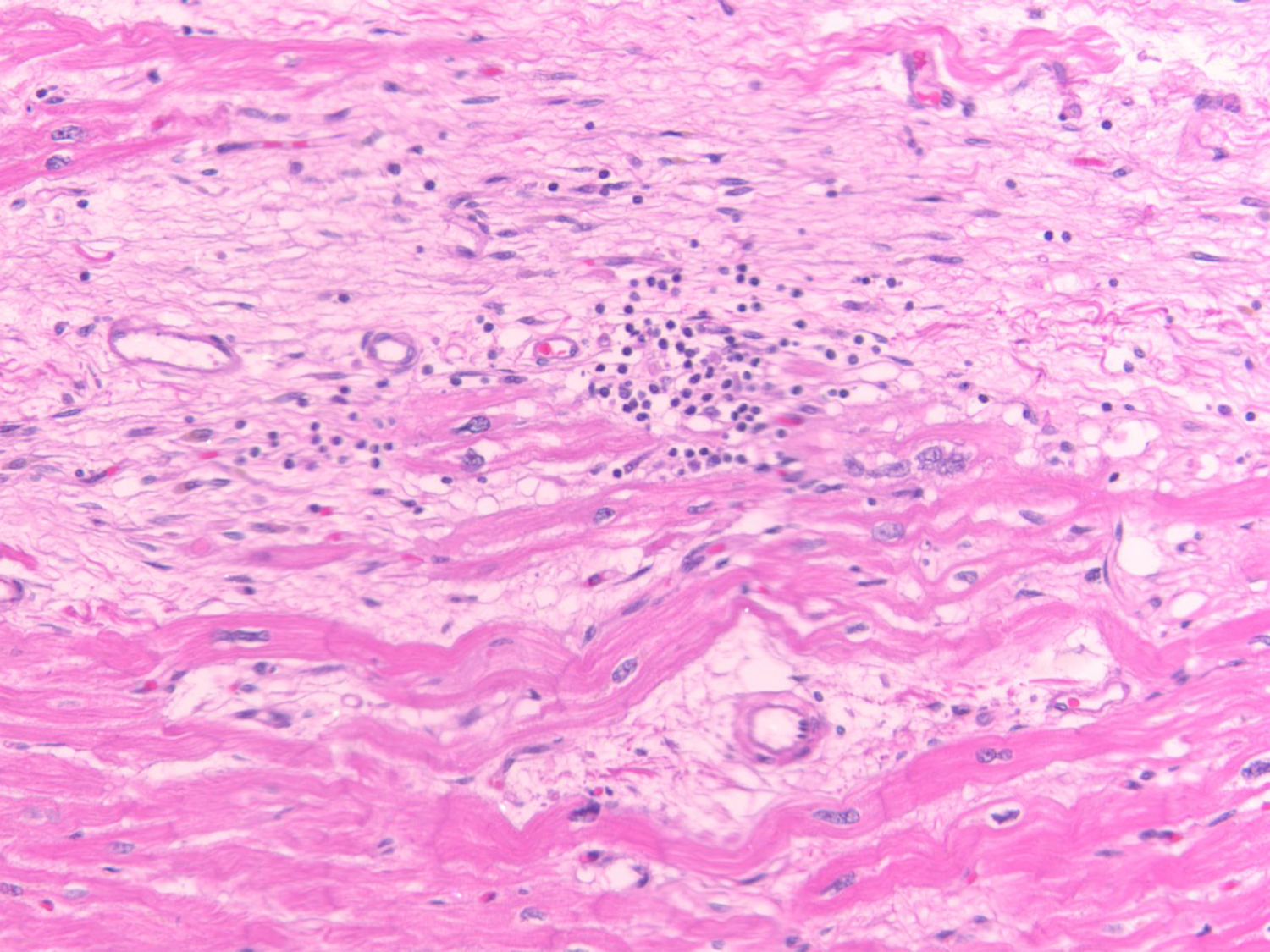
Histological specimen from the right ventricular myocardium of a 32-year old male patient with sudden cardiac death and post-mortem diagnosis of typical arrhythmogenic right ventricular cardiomyopathy showing extensive fibrosis, very few adipocytes and areas of focal inflammation in the, hematoxylin and eosin magnified 40 x. Given sudden death was the sentinel event, no in vivo ECG or CMR data were available. Two family members were subsequently diagnosed with ARVC. Genetic testing revealed a truncation pathogenic variant in the PKP2 gene, which segregated with phenotype in the family and was confirmed in the index patient.
Figure 3. Arrhythmogenic left ventricular cardiomyopathy and inflammation: case presentations.
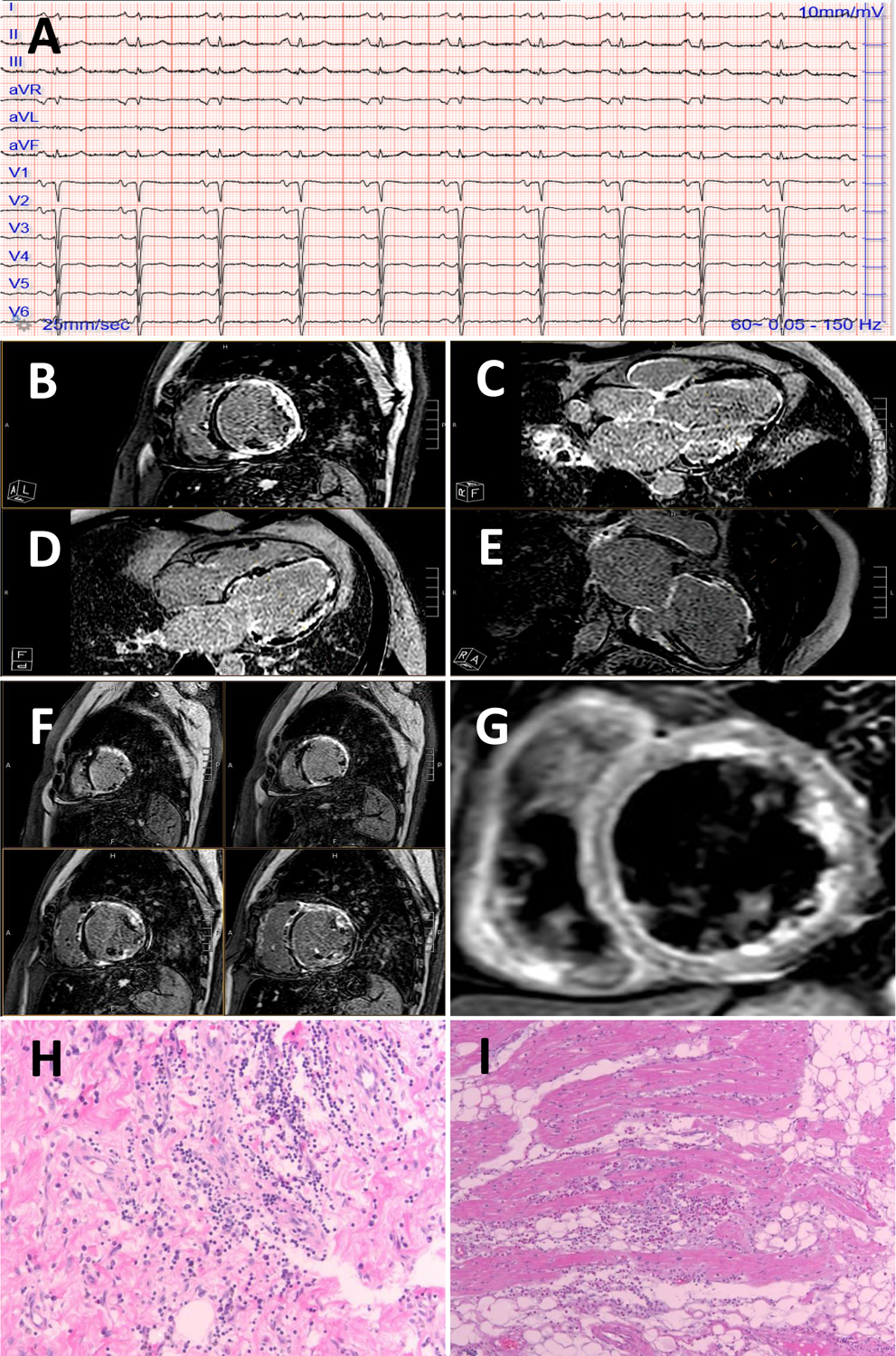
A 49-year-old male with a history of chest pain, troponin elevation, normal epicardial coronary arteries presented several weeks dyspnea, NTproBNP 1500 pg/ml. A 12-lead ECG showed sinus rhythm with T-wave inversion in the lateral leads (I, aVL, V5, V6) and poor R wave progression across the precordial leads (panel A).
A CMR showed extensive left ventricular late gadolinium enhancement with near circumferential ring-like pattern of the basal, mid and apical segments (panels B-F). T2 STIR imaging also showed edema (panel G). An arrhythmogenic cardiomyopathy and dilated cardiomyopathy panel was sent revealing a pathogenic DSP variant c.478C>T (p.Arg160Ter).
An unrelated patient who had died of a sudden cardiac death event on histological analysis showed lymphocytic infiltrates of the left ventricular myocardium (negative for viral PCR), hematoxylin and eosin 10 × panel (H) and magnified 20 × (I). This patient tested positive for a truncating pathogenic DSP variant.
2. The complex genetics and pathophysiology of ACM
Most genetic forms of ACM are transmitted as an autosomal dominant trait, with variable penetrance. Causative variants in desmosomal genes, namely plakophilin 2 (PKP2), desmoplakin (DSP), desmoglein 2 (DSG2), desmocolin 2 (DSC2), and plakoglobin (JUP), are the most prevalent and well validated of the genetic basis of ACM.18 Desmosomes are intercellular adhesion complexes, which in the heart reside within the intercalated disc. They anchor transmembrane cadherins to the desmin cytoskeleton via linker proteins, providing the heart with mechanical strength. Over the past decade, variants in genes encoding non-desmosomal proteins have been implicated in ACM, particularly in ALVC and biventricular ACM. These genes encode proteins with a diverse range of biological functions, including maintenance of cytoskeletal and nucleoskeletal architecture, nuclear envelope stability, calcium handling, sodium transport and cytokine signaling,9, 19 suggesting that ACM pathophysiology involves perturbation of multiple signaling pathways (Figure 4).
Figure 4. Genes associated with arrhythmogenic cardiomyopathy (ACM).
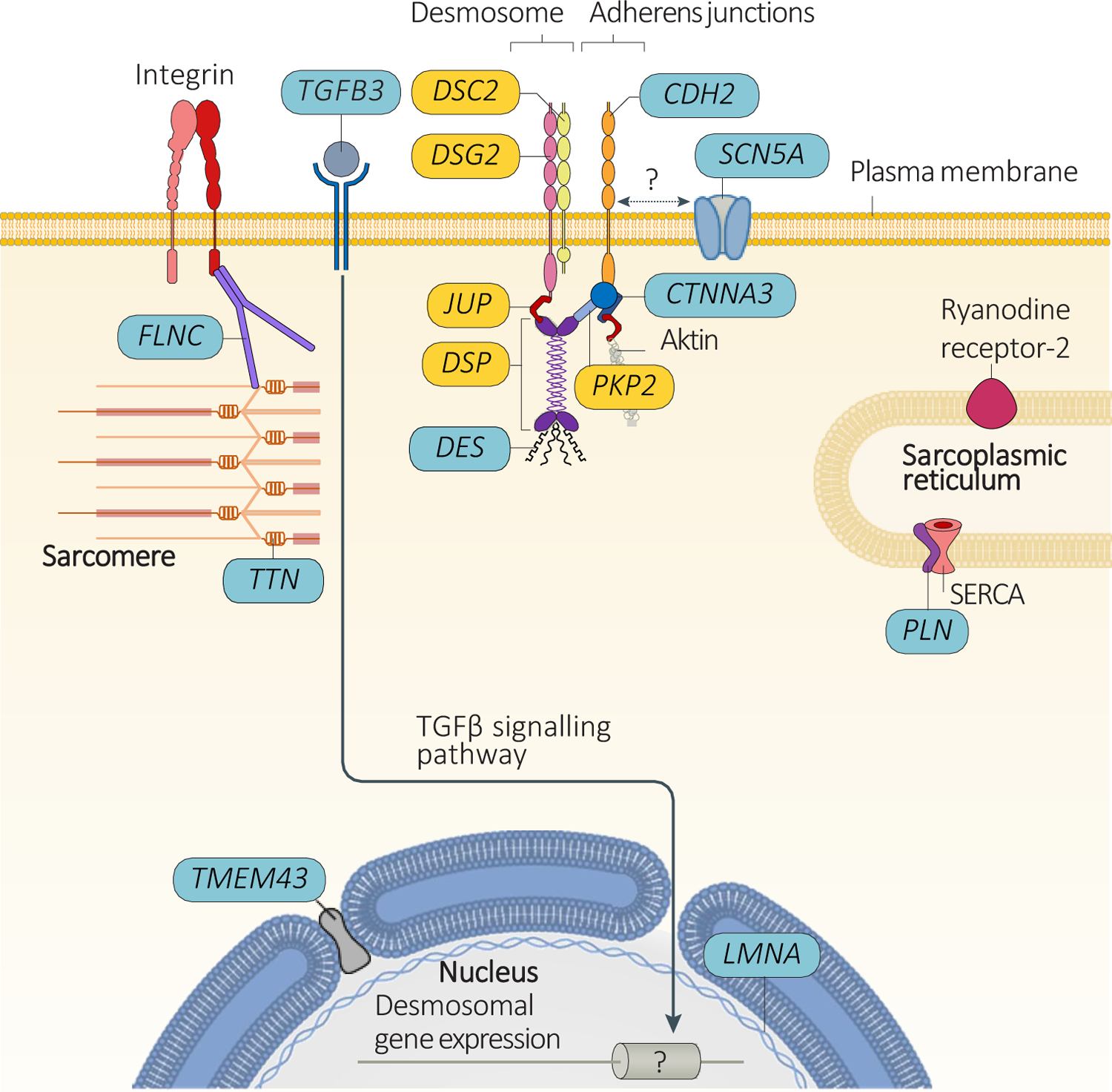
Pathogenic variants in desmosomal genes (shown in yellow) are very frequent among patients with ACM. Less frequently, pathogenic variants in ACM patients are found in genes encoding proteins that interact with the desmosome (CDH2, CTNNA3, and DES), in the genes potentially involved in expression of proteins (LMNA, TFGB3, and TMEM43), in the genes related to sarcoplasmic reticulum calcium homeostasis (PLN), or in the sarcomeric gene TTN (less frequently involved genes are shown in blue). Pathogenic variants in the SCN5A gene have been associated with defects in cadherin 2 (encoded by CDH2) at the intercalated disc, which might underlie the mechanism of ACM (although a direct interaction between these molecules has yet to be demonstrated). SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase 2; TGFβ, transforming growth factor-β.
The pathophysiology of ACM is complicated and insufficiently understood. Previous studies have identified four features that are often evident in ACM patients regardless of the underlying pathogenic variant, hence are likely part of a final common molecular pathway: 1) decreased immunoreactive signal for plakoglobin at cell-cell junctions, suggesting redistribution of plakoglobin from membrane to intracellular pools;20 2) gap junction remodeling indicated by decreased immunoreactive signal for the major ventricular gap junction protein, connexin 43 (Cx43), at cell-cell junctions;20 3) myocardial apoptosis;21 and 4) high circulating levels of proinflammatory cytokines and expression of cytokines by cardiac myocytes.13 The first three features have been reviewed elsewhere;9, 19 here we focus on inflammation and immune response in ACM.
3. Evidence from human ACM histopathological studies
In their original clinicopathological description of ARVC cases, Marcus et al.1 reported histiocytic and lymphocytic infiltrates in the interstitium of affected RV areas in ARVC biopsy samples. Following this report, sporadic cases of viral myocarditis facilitating arrhythmias or disease progression in ARVC patients were reported.22, 23 Yet, while inflammatory infiltrates are common, they are not universally present in ARVC hearts.24 In 1991, Thiene et al.24 reported on gross and pathohistological examinations and immunohistochemistry studies of 19 ARVC hearts. In six (32%) out of 19 cases, they found active inflammatory processes consisting of patchy CD45+ mononuclear infiltrates surrounding myocardial damage and necrosis, consistent with the histological definition of myocarditis. The authors drew parallels with an experimental model of perimyocarditis in BALB/c mice due to Coxsackie B3 virus, resulting in isolated RV wall thinning and aneurysms in later stages,25 and proposed that genetic factors may not only predispose to susceptibility for infection/inflammation but also play a role in the site of cardiac involvement – the epicardium of RV (Figure 1, pathway C).24 Another key study by Basso et al.11 described patchy inflammatory infiltrates, consisting of CD45+ and CD43+ lymphocytes, associated with cardiac myocyte death in 20 (67%) out of 30 ARVC hearts. Electron microscopy examination revealed lymphocytic infiltrates around capillary vessels, and cardiac myocyte debris within the fibrotic areas, attributable to recent cell death. Later, Corrado et al.26 showed that interstitial mononuclear infiltrates, located mainly in the subendocardial layers around necrotic or degenerative myocytes (Figure 1, pathway D), were more often found in cases with LV involvement (88%), as compared to those with isolated RV disease (30%), and infrequently showed features of focal giant cell myocarditis. More recently, Campuzano et al.27 identified ventricular multifocal inflammatory infiltrates in 14 (39%) out of 36 ARVC postmortem human heart samples. A subset of seven (19%) cases showed predominantly T cells, while in another seven cases, infiltrates were composed of mainly neutrophils, less of mast cells and macrophages, whilst T cells were absent. Infiltrates were more frequently identified in areas of fibrofatty replacement and when present, correlated with more severe structural cardiac abnormalities and biventricular involvement, implicating inflammatory triggers as a potential modulator of the ACM phenotype. Interestingly, the fibro-fatty pathology of ACM is not seen in viral myocarditis or autoimmune forms like giant cell or eosinophilic myocarditis.
4. ‘Hot phase’ clinical presentation in ACM
Several studies reported that inflammatory cells tend to be particularly abundant in the affected myocardial tissue during the so-called ‘hot phases of disease’, observed in some patients with ACM. At this stage, patients may present with signs of myocarditis, such as chest pain, elevated troponin levels, ECG abnormalities, or myocarditis pattern on CMR.28–31 Furthermore, various cardiotropic viruses, including enterovirus, adenovirus, cytomegalovirus, hepatitis C virus and parvovirus B19, have been detected in myocardial samples from sporadic ARVC cases.32–34 Whether or not viral infection has a pathogenic role in disease causality or progression, and whether these infections were superimposed on an already affected myocardium that is susceptible to viral infections, remains unknown.
An interesting study by Lopez-Ayala et al.35 examined the incidence and genetic basis of acute myocarditis in patients with ARVC or ALVC and their pathogenic variant-carrier family members. Over a median of 34 months follow-up, 6/131 (4.6%) patients, including one with ARVC, four with ALVC, and one with biventricular ACM, and 1/64 (1.6%) clinically healthy variant carrier developed acute chest pain, with associated increase of the troponin I values and progressive worsening of the LVEF, without an episode of fever or infection in the weeks prior to acute myocarditis.35 Notably, laboratory tests performed at the time of the episodes did not show elevations in C-reactive protein or leukocytes, nor did CMR demonstrate pericardial effusion or myocardial edema. Myocarditis clustered in families with the DSP-c.1339C>T (Glu447*) and LDB3-c.1051A>G (Thr351Ala) variants. Similarly, Bariani et al.36 have recently reported that among 560 ACM probands and family members only 23 (5%) presented with a ‘hot phase’, often at pediatric age. While only 10/23 (43%) patients met the diagnostic criteria for ARVC at first presentation, all but one individual fulfilled the 2010 Task Force criteria for ARVC after a mean follow-up of 17-years.
There are limited data on the presence of active myocarditis in living patients with ARVC using non-invasive imaging techniques. Campian et al.37 assessed regional myocardial inflammation with 67Ga scintigraphy in 8 ARVC patients and 9 controls. They found significantly higher 67Ga uptake in the RV wall in ARVC cases than in controls, while 67Ga uptake was not different between both groups in the interventricular septum and LV wall. Protonotarios et al.38 used 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) to assess the prevalence of myocardial inflammation in patients with ARVC. Among 16 definitive ARVC patients, 7 (44%) had positive FDG-PET scans, 2 of whom had cardiac sarcoidosis on endomyocardial biopsy. Of the remaining 5, 2 carried pathogenic DSP variants. FDG uptake was found in the LV myocardium in all cases, with only one patient having RV uptake additionally.
Recently, Poller et al.39 described a novel, recurrent myocarditis-like phenotype in two brothers heterozygous for the pathogenic DSP-p.Arg1458Ter variant. In both brothers, these episodes were characterized by chest pain, myocarditis-like ECG-changes, and massive increase of troponin T, and occurred after competitive sports activities. While neither brother fulfilled histological or CMR criteria for ARVC, both showed multifocal subepicardial late gadolinium enhancement (LGE) in the LV. Immunohistochemistry showed complete loss of desmoplakin protein from the myocardium of these patients despite their heterozygous desmoplakin defect at the genome level. Such acute myocardial injury occurs in approximately 15% of patients with pathogenic DSP variants, and is strongly associated with LV (90%) LGE even with normal LVEF.40 Childhood episodes of acute myocarditis have been described in those with homozygous variant carriers.14
5. Autoantibodies in ACM hearts
Circulating anti-DSG2 autoantibodies,41 anti-heart autoantibodies (AHAs) and anti-intercalated disk autoantibodies (AIDAs)42 were identified in patients with ACM, in keeping with the theory that autoimmune response against intercalated disk components and myosin is involved in ACM pathogenesis. Moreover, anti-DSG2 antibodies were present in Boxer dogs with ARVC, and absent in those without.41 In humans, the level of anti-DSG2 antibodies correlated with the burden of premature ventricular contractions, and antibodies caused gap junction dysfunction, a common feature of ARVC.41 Similarly, positive AHA status in patients with ARVC was associated with lower LVEF, higher frequency of cardiac symptoms and implantable cardioverter defibrillator implantation, and positive AIDA with lower biventricular ejection fraction.42 Remarkably, anti-DSG2 antibodies were present in ARVC subjects regardless of presence of or type of the underlying pathogenic variant, suggesting that a final common pathway can underlie gene-elusive ARVC cases.41 An increased frequency of AHAs and AIDAs in ARVC patients and relatives in comparison with controls was reported. Of note, AHA- and AIDA-positive status was more common in familial than in sporadic pedigrees (85% vs 45%). Overall, these primary studies implicate anti-DSG2 autoantibodies, AHAs, and AIDAs as sensitive and specific biomarkers and new therapeutic targets for ACM. However, the detection rate of anti-DSG2 antibodies was not investigated in disease entities overlapping with ACM, and the specificity of these antibodies requires further investigation.
6. The molecular crosslink between inflammatory signaling and ACM pathogenesis
6.1. TGFβ3 signaling and replacement-type fibrosis in ACM
The hypothesis that cytokine signaling plays a role in ACM pathogenesis first gained attention when the gene locus in one large ARVC family was mapped to 14q24.3, which includes the TGFB3 gene, encoding transforming growth factor-β3 (TGFβ3).43 After screening of the coding region of TGFB3 and of additional four genes within the critical region failed to reveal genetic alterations, Rampazzo et al.44 identified a rare, non-coding G>A nucleotide substitution (c.–30G>A) in the 5’ untranslated region (UTR) of TGFB3, which co-segregated with the disease in all affected family members.44 Investigation extended to 30 unrelated ARVC patients negative for pathogenic variants in the known ARVC genes, identified an additional substitution variant (c.*495C>T) in the 3’UTR of TGFB3 in one proband.44 Expression of UTR mutant TGFβ3 in murine myoblast cells significantly increased translation of luciferase reported gene, indicating gain-of-function. Interestingly, in keeping with the knowledge that TGFβ3 induces a fibrotic response by promoting expression of extracellular matrix genes and by suppressing the activity of matrix metalloproteinases, the authors suggested that UTR variants in the TGFB3 gene lead to increased myocardial fibrosis (Figure 1, pathway E).45 In accord with this theory, endomyocardial biopsies in the two probands with TGFB3-mediated ARVC showed extensive replacement-type fibrosis. Signal-averaged ECG, which strongly correlates with the extent of replacement fibrosis in ARVC hearts,46 revealed late potentials in six affected members of the 5’ UTR-TGFB3 family.44
Thus far, no pathogenic TGFB3 variants have been subsequently reported in ARVC. A recent ClinGen expert-curation of published studies revealed limited evidence to support the causal role of TGFB3 variants in monogenetic ARVC;18 however, given the regulatory role of TGFβ3 in cellular adhesion and extracellular matrix formation, its paracrine and autocrine signaling warrant further investigation with relation to ACM.
Interestingly, there is evidence of activation of the TGF-β profibrotic pathway in more cases of the ACM with different genetic backgrounds. Maione et al.47 observed higher levels of circulating TGF-β1 and higher levels of fibrotic markers in cardiac biopsies from desmosomal or gene-elusive ARVC patients than in healthy controls. ARVC-patient derived cardiac mesenchymal stromal cells were more responsive to TGF-β1 treatment, in terms of pro-fibrotic differentiation and higher activation of the SMAD2/3 signaling pathway, than those from healthy controls. Also, in a PKP2+/− mouse model and neonatal rat ventricular cardiomyocytes, loss of PKP2 was shown to promote TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiac myocytes.48 Notably, other pathways, including the Wnt pathway, have been postulated in promoting fibrosis in ACM hearts but there is thus far no clear evidence.
6.2. Proinflammatory cytokines and plakoglobin translocation
Another piece of evidence supporting the role of cytokine signaling proteins in the ACM pathogenesis is the key study by Asimaki et al,13 which showed marked reduction in the immunoreactive signal for plakoglobin at cardiac myocyte junctions in patients with granulomatous myocarditis, similar to findings in patients with ARVC. In contrast, plakoglobin signal was not depressed in non-granulomatous (lymphocytic) myocarditis. In vitro, brief exposure of cultured neonatal rat ventricular myocytes to low concentrations of interleukin (IL)-17, tumor necrosis factor-α (TNFα), and IL-6 – cytokines implicated in granulomatous myocarditis – caused translocation of plakoglobin from junctional to intracellular sites, whereas other potent cytokines implicated in non-granulomatous myocarditis had no effect on plakoglobin localization. Immunostaining of human ARVC myocardial samples revealed higher myocardial expression of IL-17 and TNF-α compared to controls. A sensitive immunoassay revealed elevated levels of multiple proinflammatory cytokines, including IL-6 receptor, IL-8, monocyte chemoattractant protein 1, macrophage inflammatory protein 1, TNF-α1 and TNF-αR2, and a significant reduction in the levels of IL-1R2, an anti-inflammatory protein, in the serum of ACM patients compared with controls. Remarkably, inflammatory mediators including IL-6, TNF-a, MIP-1a, and the chemokine RANTES, were also shown to be secreted by neonatal rat ventricular myocytes transfected to express JUP2157del2.49 These findings implicate cytokines, including those produced by the damaged cardiac myocytes, in redistribution of desmosomal proteins and pathogenesis of ARVC (Figure 1, pathway F).
6.3. Transcriptional link between PKP2 and host-response proteins in cardiac myocytes
Recently, Delmar and colleagues investigated the transcriptional link between PKP2 and the inflammatory/immune response in adult cardiac myocytes, and tested the hypothesis that PKP2 deficiency endogenously activates a transcriptional program that can lead to a ‘sterile myocarditis’.50 Cardiac-specific, tamoxifen-activated PKP2-knockout mice (PKP2cKO) were crossed with a RiboTag line to allow characterization of the ribosome-resident transcriptome of cardiac myocytes after PKP2 knockdown. In PKP2cKO/RiboTag adult murine cardiac myocytes, analysis of biological processes of upregulated transcripts showed over-representation of transcripts involved in platelet activation and chemokine signaling, viral response, inflammatory and immune pathways. The transcriptional changes in myocytes at this stage coincided with the presence of infiltrates positive for T-cell marker CD45 and for the neutrophil marker Ly-6G/Ly-6C. At an earlier stage, when apparent histological changes are still absent, the authors observed nonmyocyte cells and injured myocytes in the subepicardial region of the myocardium using serial block face scanning electron microscopy. Further, the authors assessed the correlation of PKP2 transcript with all others in the transcriptome of the left ventricle of 386 human decedents in the GTEx v8 database. Analysis of the transcripts that inversely correlated with PKP2, as in the case of PKP2cKO/RiboTag, revealed a predominance of functional pathways related to viral infection, platelet activation, inflammation, and immune response networks, indicating that the relation between PKP2 transcript levels and the abundance of transcripts involved in the inflammatory/immune response are independent of the presence of a cardiac disease. In a further step, the authors extracted transcripts significantly and inversely correlated with PKP2 in the PKP2cKO/RiboTag data and localized those in the GTEx data. Functional pathways from overlapping datasets were consistent with inflammation/immune response, in particular with pathways associated with viral infections. Collectively, these data support the concept that PKP2 might be transcriptionally linked, in cardiac myocytes, to genes coding for host-response molecules even in the absence of an exogenous trigger (Figure 1, pathway G). While there is no evidence for direct regulation of transcription by PKP2, it is known that PKP2 transcript abundance can affect the localization and function of intercalated disc proteins that affect transcription, particularly beta-catenin, plakoglobin, and protein kinase C-alpha.50
6.4. Activation of the NFκB signaling and GSK3β pathway in ACM
Considerable evidence implicates abnormal signaling by glycogen synthase kinase-3β (GSK3β) in ACM. SB216763, a small molecule annotated as an inhibitor of GSK3β, prevents and/or reverses the ACM phenotype (including arrhythmias, contractile dysfunction, myocyte injury and apoptosis, exercise-induced sudden cardiac death and re-distribution of junctional proteins) in ACM models in vitro,49 in vivo51 and ex vivo52.49 These studies demonstrate that arrhythmias and myocardial damage in ACM arise via a final common disease pathway that can be blocked by a single small molecule.51 Additionally, GSK3β distribution was found to be consistently altered in hearts of ACM patients regardless of the underlying pathogenic variant. Under physiological conditions, GSK3β resides in the so-called degradation complex in cytoplasm and phosphorylate substrate proteins targeting them for ubiquitination and proteasomal degradation. In ACM hearts, GSK3β translocates from the cytoplasm to the intercalated disk. This ‘reverse remodeling’ of the intercalated disk appeared to be specific to ACM as it did not occur in other forms of cardiomyopathies, viral or granulomatous myocarditis.51 Insightful as these studies were, effective mechanism-based drug therapy of ACM will require long-term administration and long-term inhibition of GSK3β, which could have unacceptable adverse consequences, including increased risk of carcinogenesis.
Accordingly, to identify potential new targets for drug therapy, Chelko et al.51 focused on signaling mediated by nuclear factor-κB (NFκB). A master regulator of cellular inflammatory responses, NFκB is closely linked to the GSK3β pathway. Activation of GSK3β promotes NFκB-mediated inflammation, whereas inhibition of GSK3β limits NFκB signaling. Activation of NFκB signaling, indicated by increased expression and nuclear accumulation of phospho-RelA/p65, occurs in both in vitro and in vivo mouse models of ACM (JUP2157del2, Dsg2mut/mut). Bay 11–7082, a small-molecule inhibitor of NFκB signaling, prevented the development of ACM features in vitro (redistribution of plakoglobin, Cx43, and GSK3β; myocyte apoptosis, release of inflammatory cytokines) and in vivo (myocardial necrosis and fibrosis, left ventricular contractile dysfunction, abnormalities in single-averaged ECG in Dsg2mut/mu mice). Hearts of Dsg2mut/mut mice expressed markedly increased levels of inflammatory cytokines and chemotactic molecules that were attenuated by Bay 11–7082. Beneficial effects of Bay 11–7082 correlated with the extent to which production of selected cytokines had been blocked. Treatment of Dsg2mut/mut mice with Bay 11–7082 also reduced the number of infiltrating inflammatory cells in the myocardium. The levels of LIX (CXCL5) and osteopontin (OPN) showed inverse correlation with ejection fraction and a positive correlation with myocardial fibrosis and apoptosis. NFκB signaling was also activated in hiPSC-cardiac myocytes derived from a patient with PKP2-mediated ACM. These cells produced and secreted abundant inflammatory cytokines under basal conditions, and this was also greatly reduced by Bay 11–7082. These results provide additional independent evidence of activation of an innate immune response in cardiac myocytes in ACM under the control of NFκB signaling.
Activation of the NFκB signaling was also reported in Tmem43 knock-in mouse model carrying the p.S358L mutation, which shows features of cardiomyopathy with fibro-fatty infiltration but preserved cardiac function.53 NFκB directly drove the expression of pro-fibrotic TGFβ1, and enhanced downstream signaling, indicating that TMEM43-S358L mutation up-regulates NFκB-TGFβ signal cascade during ARVC cardiac fibrosis. Overexpression of calcineurin splice variant calcineurin Aβ1 (which results in GSK3β inhibition) or chemical GSK3β inhibition improved cardiac function and increased mice life span.54 HiPSCs harboring the TMEM43-p.S358L variant also showed contractile dysfunction that was partially restored after GSK3β inhibition, providing solid evidence for targeting NFκB signaling in TMEM43-ARVC.
7. Conclusive remarks
Evidence accumulated over the past decade strongly suggests that inflammation – either reactive to internal influences or triggered by exogenous agents, such as viruses – is involved in the pathogenesis of ACM. Mechanisms by which inflammation and/or autoimmunity contribute to the ACM pathogenesis and the extent to which these factors shape the ultimate ACM phenotype may vary based on the causal gene variant. Most of the evidence on the role of inflammation and autoimmunity in ACM is based on desmosomal forms of disease; non-desmosomal disease subtypes require more research to explore possible phenotype modifiers. Current data suggests that inflammatory/immune factor might be more prevalent in ALVC than in typical ARVC, with DSP-cardiomyopathy typically manifesting as ALVC being increasingly recognized as distinct form of cardiomyopathy with inflammatory component. While patients with typical ARVC also often endorse episodes of myocarditis when asked, systematic evaluation with CMR and FDG CT-PET does not occur. It is, therefore, likely that we will see increased RV uptake as we systematically evaluate patients for it. Although speculative, it is plausible that inflammation may exacerbate exercise-induced desmosomal damage in genetically-predisposed patients and vice versa but determining the chain of causality is difficult in humans. Discerning whether there is a primary inflammatory susceptibility or a secondary inflammatory response in ACM will be critical to targeting these pathways; both primary and secondary inflammatory response mechanisms may be amenable to novel therapies. Whether targeting specific inflammatory pathways will help attenuate/prevent the phenotype in all disease forms or only be beneficial for a subgroup of patients, remains to be investigated. Despite the challenges ahead, pharmacological targeting of inflammatory signaling might represent a promising new avenue for treatment of ACM patients, particularly in the context of recent accomplishments in developing anti-interleukin medications (e.g. canakinumab, a monoclonal antibody against [IL-1β]) that is already safely and effectively used in clinical practice.
Acknowledgments
The authors acknowledge Dr. Salman Chatha for assistance with the figures.
Disclosures
Dr. Reichlin has received speaker/consulting honoraria or travel support from Abbott/SJM, Astra Zeneca, Brahms, Bayer, Biosense-Webster, Biotronik, Boston-Scientific, Daiichi Sankyo, Medtronic, Pfizer-BMS and Roche, all for work outside the submitted study. He has received support for his institution’s fellowship program from Abbott/SJM, Biosense-Webster, Biotronik, Boston-Scientific and Medtronic for work outside the submitted study. Dr. Owens consults for MyoKardia/Bristol Myers Squibb, Cytokinetics, PCM Scientific. The other authors report no potential conflicts of interest.
Source of Funding
Dr. Asimaki received funding from Rostrees Foundation and the British Heart foundation. Dr. Landstrom is supported by National Heart, Lung and Blood Institute (NHLBI) grants K08HL136839 and R01HL149870, Doris Duke Foundation, American Academy of Pediatrics, and the Children’s Cardiomyopathy Foundation. Dr. Gelzer, Chahal and Owens receive research funding from the Winkelman Family Fund for Cardiac Innovation and the ACVIM Cardiology Research Grant. Dr. Semsarian is the recipient of a National Health and Medical Research Council (NHMRC) Practitioner Fellowship (#1154992). Dr. Chahal receives funding from the Paul and Ruby Tsai Foundation.
Non-standard abbreviations and nonstandard acronyms
- ACM
arrhythmogenic cardiomyopathy
- AHA
anti-heart autoantibodies
- AIDA
anti-intercalated disk autoantibodies
- ALVC
arrhythmogenic left ventricular cardiomyopathy
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- FDG-PET
18F-fluorodeoxyglucose positron emission tomography
- SCD
sudden cardiac death
References
- 1.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C and Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. [DOI] [PubMed] [Google Scholar]
- 2.Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, Lazzari M, Migliore F, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol. 2020;319:106–114. [DOI] [PubMed] [Google Scholar]
- 4.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E and McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. [DOI] [PubMed] [Google Scholar]
- 5.Briceno DF, Liang JJ, Shirai Y, Markman TM, Chahal A, Tschabrunn C, Zado E, Hyman MC, Kumareswaran R, Arkles JS, et al. Characterization of Structural Changes in Arrhythmogenic Right Ventricular Cardiomyopathy With Recurrent Ventricular Tachycardia After Ablation: Insights From Repeat Electroanatomic Voltage Mapping. Circ Arrhythm Electrophysiol. 2020;13:e007611. [DOI] [PubMed] [Google Scholar]
- 6.Patel V, Asatryan B, Siripanthong B, Munroe PB, Tiku-Owens A, Lopes LR, Khanji MY, Protonotarios A, Santangeli P, Muser D, et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. Int J Mol Sci. 2020;21:6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat-Alarcon N, Abraham T, et al. Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hariharan V, Asimaki A, Michaelson JE, Plovie E, MacRae CA, Saffitz JE and Huang H. Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell-cell adhesion. Cardiovasc Res. 2014;104:280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Austin KM, Trembley MA, Chandler SF, Sanders SP, Saffitz JE, Abrams DJ and Pu WT. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2019;16:519–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP and Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basso C, Thiene G, Corrado D, Angelini A, Nava A and Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–991. [DOI] [PubMed] [Google Scholar]
- 12.Reichl K, Kreykes SE, Martin CM and Shenoy C. Desmoplakin Variant-Associated Arrhythmogenic Cardiomyopathy Presenting as Acute Myocarditis. Circ Genom Precis Med. 2018;11:e002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asimaki A, Tandri H, Duffy ER, Winterfield JR, Mackey-Bojack S, Picken MM, Cooper LT, Wilber DJ, Marcus FI, Basso C, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belkaya S, Kontorovich AR, Byun M, Mulero-Navarro S, Bajolle F, Cobat A, Josowitz R, Itan Y, Quint R, Lorenzo L, et al. Autosomal Recessive Cardiomyopathy Presenting as Acute Myocarditis. J Am Coll Cardiol. 2017;69:1653–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanawuttiwat T, Sager SJ, Hare JM and Myerburg RJ. Myocarditis and ARVC/D: variants or mimics? Heart Rhythm. 2013;10:1544–1548. [DOI] [PubMed] [Google Scholar]
- 16.Riley MP, Zado E, Bala R, Callans DJ, Cooper J, Dixit S, Garcia F, Gerstenfeld EP, Hutchinson MD, Lin D, et al. Lack of uniform progression of endocardial scar in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy and ventricular tachycardia. Circ Arrhythm Electrophysiol. 2010;3:332–338. [DOI] [PubMed] [Google Scholar]
- 17.Santangeli P, Zado ES, Supple GE, Haqqani HM, Garcia FC, Tschabrunn CM, Callans DJ, Lin D, Dixit S, Hutchinson MD, et al. Long-Term Outcome With Catheter Ablation of Ventricular Tachycardia in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Arrhythm Electrophysiol. 2015;8:1413–1421. [DOI] [PubMed] [Google Scholar]
- 18.James CA, Jongbloed JDH, Hershberger RE, Morales A, Judge DP, Syrris P, Pilichou K, Medeiros Domingo A, Murray B, Cadrin-Tourigny J, et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ Genom Precis Med. 2021;14:e003273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marian AJ, Asatryan B and Wehrens XHT. Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies. Cardiovasc Res. 2020;116:1600–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. [DOI] [PubMed] [Google Scholar]
- 21.Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M and Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 1996;335:1190–1196. [DOI] [PubMed] [Google Scholar]
- 22.Blomstrom-Lundqvist C, Sabel KG and Olsson SB. A long term follow up of 15 patients with arrhythmogenic right ventricular dysplasia. Br Heart J. 1987;58:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabel KG, Blomstrom-Lundqvist C, Olsson SB and Enestrom S. Arrhythmogenic right ventricular dysplasia in brother and sister: is it related to myocarditis? Pediatr Cardiol. 1990;11:113–116. [DOI] [PubMed] [Google Scholar]
- 24.Thiene G, Corrado D, Nava A, Rossi L, Poletti A, Boffa GM, Daliento L and Pennelli N. Right ventricular cardiomyopathy: is there evidence of an inflammatory aetiology? Eur Heart J. 1991;12 Suppl D:22–25. [DOI] [PubMed] [Google Scholar]
- 25.Matsumori A and Kawai C. Coxsackie virus B3 perimyocarditis in BALB/c mice: experimental model of chronic perimyocarditis in the right ventricle. J Pathol. 1980;131:97–106. [DOI] [PubMed] [Google Scholar]
- 26.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30:1512–1520. [DOI] [PubMed] [Google Scholar]
- 27.Campuzano O, Alcalde M, Iglesias A, Barahona-Dussault C, Sarquella-Brugada G, Benito B, Arzamendi D, Flores J, Leung TK, Talajic M, et al. Arrhythmogenic right ventricular cardiomyopathy: severe structural alterations are associated with inflammation. J Clin Pathol. 2012;65:1077–1083. [DOI] [PubMed] [Google Scholar]
- 28.Kostis WJ, Tedford RJ, Miller DL, Schulman SP and Tomaselli GF. Troponin-I elevation in a young man with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Interv Card Electrophysiol. 2008;22:49–53. [DOI] [PubMed] [Google Scholar]
- 29.Lazaros G, Anastasakis A, Tsiachris D, Dilaveris P, Protonotarios N and Stefanadis C. Naxos disease presenting with ventricular tachycardia and troponin elevation. Heart Vessels. 2009;24:63–65. [DOI] [PubMed] [Google Scholar]
- 30.Jordan AN, Lyne J, De Silva R and Wong T. Myocarditic appearance of arrhythmogenic right ventricular cardiomyopathy. Circulation. 2010;122:e556–557. [DOI] [PubMed] [Google Scholar]
- 31.Patrianakos AP, Protonotarios N, Nyktari E, Pagonidis K, Tsatsopoulou A, Parthenakis FI and Vardas PE. Arrhythmogenic right ventricular cardiomyopathy/dysplasia and troponin release. Myocarditis or the “hot phase” of the disease? Int J Cardiol. 2012;157:e26–28. [DOI] [PubMed] [Google Scholar]
- 32.Grumbach IM, Heim A, Vonhof S, Stille-Siegener M, Mall G, Gonska BD, Kreuzer H, Andreas S and Figulla HR. Coxsackievirus genome in myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiology. 1998;89:241–245. [DOI] [PubMed] [Google Scholar]
- 33.Bowles NE, Ni J, Marcus F and Towbin JA. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2002;39:892–895. [DOI] [PubMed] [Google Scholar]
- 34.Calabrese F, Basso C, Carturan E, Valente M and Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: is there a role for viruses? Cardiovasc Pathol. 2006;15:11–17. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Ayala JM, Pastor-Quirante F, Gonzalez-Carrillo J, Lopez-Cuenca D, Sanchez-Munoz JJ, Oliva-Sandoval MJ and Gimeno JR. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015;12:766–773. [DOI] [PubMed] [Google Scholar]
- 36.Bariani R, Cipriani A, Rizzo S, Celeghin R, Bueno Marinas M, Giorgi B, De Gaspari M, Rigato I, Leoni L, Zorzi A, et al. ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy. Europace. 2021;23:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campian ME, Verberne HJ, Hardziyenka M, de Groot EA, van Moerkerken AF, van Eck-Smit BL and Tan HL. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Nucl Med Mol Imaging. 2010;37:2079–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Protonotarios A, Wicks E, Ashworth M, Stephenson E, Guttmann O, Savvatis K, Sekhri N, Mohiddin SA, Syrris P, Menezes L, et al. Prevalence of (18)F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. 2019;284:99–104. [DOI] [PubMed] [Google Scholar]
- 39.Poller W, Haas J, Klingel K, Kuhnisch J, Gast M, Kaya Z, Escher F, Kayvanpour E, Degener F, Opgen-Rhein B, et al. Familial Recurrent Myocarditis Triggered by Exercise in Patients With a Truncating Variant of the Desmoplakin Gene. J Am Heart Assoc. 2020;9:e015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterjee D, Fatah M, Akdis D, Spears DA, Koopmann TT, Mittal K, Rafiq MA, Cattanach BM, Zhao Q, Healey JS, et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur Heart J. 2018;39:3932–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caforio ALP, Re F, Avella A, Marcolongo R, Baratta P, Seguso M, Gallo N, Plebani M, Izquierdo-Bajo A, Cheng CY, et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies With Disease Severity and Family History. Circulation. 2020;141:1238–1248. [DOI] [PubMed] [Google Scholar]
- 43.Rampazzo A, Beffagna G, Nava A, Occhi G, Bauce B, Noiato M, Basso C, Frigo G, Thiene G, Towbin J, et al. Arrhythmogenic right ventricular cardiomyopathy type 1 (ARVD1): confirmation of locus assignment and mutation screening of four candidate genes. Eur J Hum Genet. 2003;11:69–76. [DOI] [PubMed] [Google Scholar]
- 44.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. [DOI] [PubMed] [Google Scholar]
- 45.Leask A and Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. [DOI] [PubMed] [Google Scholar]
- 46.Turrini P, Angelini A, Thiene G, Buja G, Daliento L, Rizzoli G and Nava A. Late potentials and ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 1999;83:1214–1219. [DOI] [PubMed] [Google Scholar]
- 47.Maione AS, Stadiotti I, Pilato CA, Perrucci GL, Saverio V, Catto V, Vettor G, Casella M, Guarino A, Polvani G, et al. Excess TGF-beta1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int J Mol Sci. 2021;22:2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dubash AD, Kam CY, Aguado BA, Patel DM, Delmar M, Shea LD and Green KJ. Plakophilin-2 loss promotes TGF-beta1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J Cell Biol. 2016;212:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Hernandez M, Marron-Linares GM, Schlamp F, Heguy A, van Opbergen CJM, Mezzano V, Zhang M, Liang FX, Cerrone M and Delmar M. Transcriptomic Coupling of PKP2 With Inflammatory and Immune Pathways Endogenous to Adult Cardiac Myocytes. Front Physiol. 2020;11:623190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation. 2019;140:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asimaki A, Protonotarios A, James CA, Chelko SP, Tichnell C, Murray B, Tsatsopoulou A, Anastasakis A, te Riele A, Kleber AG, et al. Characterizing the Molecular Pathology of Arrhythmogenic Cardiomyopathy in Patient Buccal Mucosa Cells. Circ Arrhythm Electrophysiol. 2016;9:e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng G, Jiang C, Li Y, Yang D, Ma Y, Zhang B, Li X, Zhang P, Hu X, Zhao X, et al. TMEM43-S358L mutation enhances NF-kappaB-TGFbeta signal cascade in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Protein Cell. 2019;10:104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Padron-Barthe L, Villalba-Orero M, Gomez-Salinero JM, Dominguez F, Roman M, Larrasa-Alonso J, Ortiz-Sanchez P, Martinez F, Lopez-Olaneta M, Bonzon-Kulichenko E, et al. Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3beta. Circulation. 2019;140:1188–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]