

Abstract
Opioid tolerance (OT) leads to dose escalation and serious side effects, including opioid-induced hyperalgesia (OIH). We sought to better understand the mechanisms underlying this event in the gastrointestinal tract. Chronic in vivo administration of morphine by intraperitoneal injection in male C57BL/6 mice evoked tolerance and evidence of OIH in an assay of colonic afferent nerve mechanosensitivity; this was inhibited by the δ-opioid receptor (DOPr) antagonist naltrindole when intraperitoneally injected in previous morphine administration. Patch-clamp studies of DRG neurons following overnight incubation with high concentrations of morphine, the µ-opioid receptors (MOPr) agonist [D-Ala2, N-Me-Phe4, Gly5-ol]-Enkephalin (DAMGO) or the DOPr agonist [D-Ala2, D-Leu5]-Enkephalin evoked hyperexcitability. The pronociceptive actions of these opioids were blocked by the DOPr antagonist SDM25N but not the MOPr antagonist D-Pen-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2. The hyperexcitability induced by DAMGO was reversed after a 1 h washout, but reapplication of low concentrations of DAMGO or [D-Ala2, D-Leu5]-Enkephalin restored the hyperexcitability, an effect mediated by protein kinase C. DOPr-dependent DRG neuron hyperexcitability was blocked by the endocytosis inhibitor Pitstop 2, and the weakly internalizing DOPr agonist ARM390 did not cause hyperexcitability. Bioluminescence resonance energy transfer studies in HEK cells showed no evidence of switching of G-protein signaling from Gi to a Gs pathway in response to either high concentrations or overnight incubation of opioids. Thus, chronic high-dose opioid exposure leads to opioid tolerance and features of OIH in the colon. This action is mediated by DOPr signaling and is dependent on receptor endocytosis and downstream protein kinase C signaling.
SIGNIFICANCE STATEMENT Opioids are effective in the treatment of abdominal pain, but escalating doses can lead to opioid tolerance and potentially opioid-induced hyperalgesia. We found that δ-opioid receptor (DOPr) plays a central role in the development of opioid tolerance and opioid-induced hyperalgesia in colonic afferent nociceptors following prolonged exposure to high concentrations of MOPr or DOPr agonists. Furthermore, the role of DOPr was dependent on OPr internalization and activation of a protein kinase C signaling pathway. Thus, targeting DOPr or key components of the downstream signaling pathway could mitigate adverse side effects by opioids.
Keywords: colonic afferent nerves, delta-opioid receptor, dorsal root ganglia, mu-opioid receptor, opioid tolerance, opioid-induced hyperalgesia
Introduction
Chronic abdominal pain is a debilitating symptom in gastrointestinal disorders, such as irritable bowel syndrome and inflammatory bowel disease. While opioid drugs are effective, they cause serious side effects (Bielefeldt et al., 2009; Targownik et al., 2014). Opioid tolerance (OT), where increasing opioid doses are required to maintain the same level of analgesia, underlies escalating opioid dosing and the resulting risk of serious side effects (Allouche et al., 2014; Cahill et al., 2016; Stein, 2016, 2018). Moreover, there is evidence that increasing doses can lead to a paradoxical switch in signaling leading to enhanced pain (Mattioli et al., 2014), a condition called opioid-induced hyperalgesia (OIH), although this phenomenon is poorly understood.
Nociceptive signaling in the intestine is mediated by the small C-fiber and larger Aδ-fiber DRG neurons (Robinson and Gebhart, 2008; Kyloh et al., 2011) whose distal axons innervate the intestine and central axons project to second-order neurons in the dorsal horn of the spinal cord. Opioid drugs and endogenous opioids bind to G-protein-coupled μ-, δ-, or κ-opioid receptors (MOPr, DOPr, KOPr) on the plasma membrane of these neurons (Stein, 2016; Spahn et al., 2018). In addition to conventional G-protein-mediated signaling, recent evidence also revealed that β-arrestins (βarrs) couple receptors to clathrin and adaptor protein-2 and guide receptors and ligands to endosomes where further signaling occurs (Jensen et al., 2017; Jimenez-Vargas et al., 2018, 2020; Stoeber et al., 2018).
A large body of work has shown that the molecular mechanisms underlying OT are remarkably complex, and several mechanisms have been proposed, including opioid receptor interactions, receptor phosphorylation, G-protein uncoupling, and altered receptor internalization/recycling (Varga et al., 2004; Williams et al., 2013; Allouche et al., 2014; Galligan and Akbarali, 2014). Several studies have highlighted the importance of βarr2 in the development of morphine antinociceptive tolerance (Bohn et al., 1999; Raehal and Bohn, 2011; Kliewer et al., 2019; Grim et al., 2020; He et al., 2021). Interestingly, the mechanisms of tolerance may be tissue-dependent, further highlighting the need for studies in the intestine.
The analgesic actions of conventional opioid drugs in the intestine, such as morphine, are thought to be mediated primarily by MOPr activation, although DOPr signaling may also have important analgesic actions despite being less well studied (Pergolizzi et al., 2020). Recent clinical studies have shown that drugs targeting multiple opioid receptors, such as a novel agent with mixed effects (MOPr agonist/DOPr antagonist), in the GI tract may have sustained efficacy (Lacy, 2018), but their mechanism of action is poorly understood. To better understand the mechanisms of OT in nociceptive signaling in the intestine and the roles of MOPr and DOPr, we used a model of chronic morphine administration to induce OT and features of OIH. We found evidence of a significant functional MOPr–DOPr interaction underlying these phenomena and sought to understand the mechanisms involved.
Materials and Methods
Materials
SDM25N hydrochloride (SDM25N), D-Pen-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP), naloxone hydrochloride (NLX), AR-M1000390 hydrochloride (ARM390), H 89 hydrochloride (H89), and GF 109203× (GFX) were from Tocris Bioscience. Pitstop 2 (PS2) was from Abcam, and other reagents, including [D-Ala2, D-Leu5]-Enkephalin (DADLE), and [D-Ala2, N-Me-Phe4, Gly5-ol]-Enkephalin (DAMGO), and naltrindole hydrochloride (NTI), were from Sigma Aldrich, unless stated otherwise.
Animals
All studies were approved by the Queen's University animal care committees in accordance with guidelines from their National Council of Animal Care. C57BL/6 mice (males and in a small series, females; 8-12 weeks, Charles River Laboratories) were maintained under controlled temperature (22 ± 1°C) and light (12 h light/dark cycle) with free access to food and water.
Chronic morphine treatment
Morphine (Sandoz) was administered by intraperitoneal (i.p.) injection twice daily (9:00 A.M. and 6:00 P.M.) for 7 d, at increasing dose (10 mg/kg first day, 20 mg/kg second day, 30 mg/kg third day, 40 mg/kg fourth to seventh day; see Fig. 1A) (Mattioli et al., 2014). Control mice were injected with saline solution (NaCl 0.9% i.p.) twice daily for 7 d. On day 8, under general anesthetic (ketamine/xylazine), mice were killed, and the DRGs and colons were excised. Where indicated, mice were preinjected with 2.5 mg/kg i.p. NTI 20 min before morphine injections (Matthes et al., 1998). Mice weight was monitored each day during the protocol.
Figure 1.

Chronic morphine treatment induces tolerance in mouse DRG neurons. A, Schematic protocol of chronic morphine treatment. Dashed arrows indicate morning i.p. injections. Solid arrows indicate afternoon i.p. injections. Tail-flick assay (short ticks) was performed before and after morning injections. B, In vivo tail-flick test showed that morphine had an antinociceptive effect compared with saline (F(1,10) = 117.5, p < 0.0001, two-way repeated-measures ANOVA with Bonferroni's post hoc test); this effect declined after day 4 despite high doses suggesting OT. Results are expressed as the mean of % MPE ± SEM, saline mice = 6; morphine mice = 6. C, Effect of overnight 1 μm morphine on rheobase of DRG neurons measured by patch clamp from saline and morphine-treated mice shows tolerance to morphine in neurons from morphine-treated mice (F(1,49) = 5.57, p = 0.022, two-way ANOVA with Bonferroni's post hoc test). Number of cells appears in each bar. *p ≤ 0.05. **p ≤ 0.01. D, Representative recordings of the rheobase (minimal current to elicit an action potential) of DRG neurons from saline and morphine-treated mice exposed to vehicle (control) or morphine overnight.
Tail-flick assay
The in vivo antinociceptive effect of chronic morphine treatment was tested using the tail-flick assay. Mice were gently placed head first into a restrainer with air holes with both legs inside the restrainer and tail exposed, and the distal one-third of the tail was immersed in a water bath at 52°C. The latency to tail-flick was video recorded before (to determine the baseline) and 30 min after the morning injection with a cutoff time of 10 s to prevent tissue damage. Each assay was performed in triplicate. The latency was measured later in videos starting from the immersion of the tail in the water until the first tail-flick. The investigator analyzing videos was blinded to treatments. Data are presented as percent of maximum possible effect (% MPE) = [(post-drug latency − baseline latency) ÷ (10 − baseline latency)] × 100 (Mattioli et al., 2014; Kang et al., 2017). Attenuation of the maximal antinociceptive effect of morphine overtime was considered as OT.
Retrograde labeling of colon-projecting DRG neurons
To identify colon-projecting DRG neurons, surgeries were performed to inject the retrograde tracer Fast Blue (17740-1, Polysciences) into the colon wall. Female mice were anesthetized via isoflurane inhalation (1.5%-2.5% isoflurane; oxygen flow: 1 L/min), placed on a heating pad, and subjected to midline laparotomy. The colon was carefully exposed, and Fast Blue (1.7% w/v in sterile water) was injected in small volumes (1-2 µl) into multiple sites on the colon wall. The gut was wiped down after each injection to remove excess dye. Bupivacaine (2 mg/kg intradermally) and tramadol (20 mg/kg subcutaneously) were given before surgery, and tramadol (20 mg/kg subcutaneously) was given daily for 3 d as a postoperative analgesic. Mice were killed 7-14 d after surgery, and DRGs (T9-T13) were acutely dissociated for patch-clamp recordings.
Isolation of DRG neurons
DRG neurons were acutely dissociated, as previously described (Ibeakanma et al., 2011). Briefly, mice were killed after ketamine/xylazine i.p. injection, and DRGs from T9-T13 were dissected and collected on Hanks' balanced salt solution. Neurons were enzymatically and mechanically dissociated in F-12 supplemented media (10% FBS, penicillin, streptomycin), plated on Poly-D lysine/laminin-coated coverslips, and placed at 37°C (95% O2 and 5% CO2) for 2 h before being incubated overnight with F-12 supplemented media. Electrophysiological recordings were performed the following day. Each treatment was tested on DRG neurons from 6-8 male mice, with exception of the recordings on retrogradely labeled DRG neurons that were obtained from 2 female mice.
Patch-clamp recordings
All experiments were performed at room temperature as previously described (Valdez-Morales et al., 2013). Patch pipettes were pulled (PC-100 Puller, Narishige) and fire-polished (3-5 mΩ) from borosilicate glass capillaries (Warner Instruments). Voltage-clamp recordings were performed by using Multiclamp 700B amplifier, digitized by Digidata 1440A AD converter, and recorded using pClamp 10.5 software (all by Molecular Devices). Dissociated DRG neurons were incubated acutely (30 min) or overnight at 37°C with morphine, the selective MOPr agonist DAMGO (Yekkirala et al., 2010) or the selective DOPr agonists DADLE (Raynor et al., 1994) or ARM390 (Marie et al., 2003). The OPr antagonists NLX (Wang et al., 2008), CTOP (Raynor et al., 1994), and SDM25N (McLamore et al., 2001), PKA inhibitor H89 (Davies et al., 2000), PKC inhibitor GFX (Toullec et al., 1991), or the selective inhibitor of clathrin-mediated endocytosis PS2 (Robertson et al., 2014) were incubated 30 min before the application of agonists. All cells with a membrane capacitance ≤30 pF and a resting membrane potential more negative than −40 mV were analyzed (Moore et al., 2002; Stewart et al., 2003). Where indicated, colon-projecting DRG neurons were identified by the Fast Blue fluorescence emitted from labeled cell bodies under short exposure to UV light with a 365 nm filter.
Perforated patch-clamp recordings were obtained with amphotericin B (0.24 mg/ml) (Ibeakanma et al., 2011), and neuronal excitability was assessed by measuring the rheobase and the action potential discharge at twice rheobase. The extracellular solution composition was as follows (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 D-glucose, pH 7.4. The pipette solution composition was as follows (mm): 110 K-gluconate, 30 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, pH 7.25.
Ex vivo colonic afferent nerve recordings
Distal colon along with attached mesentery (containing the interior mesenteric ganglion) was removed intact and placed in an organ bath continuously superfused with carbogenated (5% CO2 and 95% O2) Krebs buffer (composition, in mm as follows: NaCl, 118.4; NaHCO3, 24.9; MgSO4, 1.2; KH2PO4, 1.2; glucose, 11.7; CaCl2, 1.9) at 34°C. Krebs contained the L-type calcium channel blocker nifedipine (3 μm) and the muscarinic acetylcholine receptor antagonist atropine (5 μm) to suppress smooth muscle activity (Zar et al., 1990), as well as the cyclooxygenase inhibitor indomethacin (3 μm) to suppress potential inhibitory actions of endogenous prostaglandins (Peiris et al., 2017). The preparations were opened longitudinally along the mesenteric border and pinned flat with the mucosal side up. Lumbar splanchnic nerve was identified in the neurovascular bundle, teased apart into 5-6 fibers, and individually drawn into a glass suction electrode attached to a Neurolog headstage (NL100, Digitimer). Afferent nerve signals were amplified ×10,000 (NL104), filtered (150-800 Hz, NL125 band pass filter), and recorded on a computer via a Micro 1401 interface and Spike 2 software (version 7, Cambridge Electronic Design).
Receptive fields were identified by systematically stroking the mucosal surface or the mesenteric attachment with a brush. Once identified, receptive fields were tested with three distinct mechanical stimuli to allow classification: probing (with 1 g von Frey filament), mucosal stroking (0.4 g von Frey filament), and circumferential stretch. The circumferential stretch was applied manually by holding the gut wall with forceps and stretching gently. Vascular afferents that only respond to probing on the gut wall or the mesenteric attachment were included in this study since they are thought to be nociceptors (Brookes et al., 2013). Following a 30 min equilibration period, control probing responses (1 g) of each unit were examined by probing 3 times, each for 3 s with 10 s intervals between probings. Morphine (1 μm) was superfused into the organ bath for 15 min before reassessment of the probing response. Single-unit analysis was performed offline using the spike sorting function of Spike2 to discriminate the afferent nerve activity of individual units. The afferent nerve response to probing was assessed as average firing frequency during a 3 s period using a custom-made script in Spike2. Baseline activity was assessed as average firing frequency during a 120 s period after equilibration and reassessed in the presence of morphine. The investigator recording from afferent nerves was blinded to the treatments.
cDNAs
DOPr and MOPr were from GenoCopoeia. GRK2 was from D. Jensen (Columbia University). RGFP-CAAX, Rluc2-Gαi/s/q, GFP10-Gγ2, Gβ1, and Rluc2-βarrestin-2 were from M. Bouvier (Université de Montréal) (Gales et al., 2006; Namkung et al., 2016).
Cell culture and transfection
For bioluminescence resonance energy transfer (BRET) assays of G protein activation, HEK293T cells were cultured and transfected, as described previously (Jimenez-Vargas et al., 2020), using JetPEI (Polyplus Transfection) with DOPr and MOPr (0.2 µg), GRK2 (0.2 μg) and Rluc2-Gαi/s/q (0.2 µg), and GFP10-Gγ2 (0.4 μg) with Gβ1 (0.1 μg). For ebBRET of βarr2 recruitment to the membrane, HEK293T cells were transfected with DOPr and MOPr (0.2 µg), GRK2 (0.2 μg), Rluc2-βarr2 (0.1 μg), and RGFP-CAAX (0.4 µg) for plasma membrane recruitment (Namkung et al., 2016).
BRET assays G protein activations and βarr recruitment to the plasma membrane
Transfected HEK293T cells were incubated overnight with vehicle, DADLE 10 μm, or DAMGO 10 μm. On the day of the assay, HEK293T cells were washed with Dulbecco's PBS, and then Tyrode's buffer was added, and cells were incubated for 30 min at 37°C. After the addition of Prolume Purple Coelenterazine (2.5 μm; NanoLight Technology), the cells were incubated for 5 min at 37°C. BRET was recorded for 22.5 min in a Synergy Neo2 Microplate reader (BioTek) (acceptor filter: 515 ± 30 nm; donor filter: 410 ± 80 nm). Cells were challenged with vehicle, DADLE 10 nm, DADLE 10 μm, DAMGO 10 nm, or DAMGO 10 μm after 2.5 min. Δ-BRET represents the BRET signal in the presence of agonist subtracted by the BRET signal in the presence of vehicle.
Statistical analyses
Data were analyzed using Prism 9 Software (GraphPad Software), and results are expressed as mean ± SEM. Normal distribution was assessed by parametric testing D'Agostino-Pearson. Differences between two groups were examined using unpaired t test, whereas multiple groups were examined using one-way ANOVA with Dunnett's or Tukey's post hoc test as indicated, or two-way ANOVA with Bonferroni's post hoc test for multiple comparisons. A p value ≤ 0.05 was considered significant. After classification of unit responses, data were loaded into a contingency table and then analyzed with the χ2 test for trend. A p value ≤ 0.05 was considered significant.
Results
OT and colonic OIH
Mice were treated with morphine for 7 d, using an established model of OIH (Fig. 1A) (Ferrini et al., 2013; Mattioli et al., 2014). Tolerance of somatic nerves was assessed using a tail-flick test in conscious mice. Morphine's antinociceptive effect was maximum on day 4 and declined over the following days despite high opioid doses (Fig. 1B; p = 0.002, two-way repeated-measures ANOVA, N = 6 saline-, 6 morphine-treated mice), suggesting development of OT (Mattioli et al., 2014; Kang et al., 2017). Patch-clamp recordings from DRG neurons isolated from morphine- and saline-treated mice demonstrated that overnight application of morphine (1 μm) inhibited neurons from saline-treated mice (rheobase increased by 49%; p = 0.006, two-way ANOVA, n = 9 control cells, 10 morphine-incubated cells/6 mice) but had no effect on neurons from mice treated with chronic morphine (Fig. 1C,D; p > 0.99, two-way ANOVA, n = 15 control cells, 19 morphine-incubated cells/6 mice).
To determine the effect of chronic morphine administration on excitability of afferent nerves innervating the colon, recordings were obtained from colons dissected from saline- and morphine-treated mice (Fig. 2A). Chronic morphine treatment decreased basal firing of afferent nerves (Fig. 2B; p = 0.028, two-way repeated-measures ANOVA, n = 12 units/6 saline-treated mice, 18 units/6 morphine-treated mice). However, while the acute application of morphine (1 μm) reduced the basal firing activity of afferent nerves from saline-treated mice (p = 0.16, two-way repeated-measures ANOVA, n = 12 units/6 saline-treated mice), it evoked a paradoxical increase in basal firing in afferent nerves from morphine-treated mice (Fig. 2B; p = 0.018, two-way repeated-measures ANOVA, n = 18 units/6 morphine-treated mice). Similarly, morphine also inhibited the mechanosensitive response (von Frey filament, 1 g) in saline-treated mice but did not alter mechanosensitive responses in the morphine-treated mice (Fig. 2D; p = 0.004, two-way repeated-measures ANOVA, n = 14 units/6 saline-treated mice). Based on responses to the superfusion of morphine, units were classified in three groups: excited (>20% increase over baseline or control probing response), inhibited (>20% decrease from baseline or control probing response), and unchanged (<20% change) (Yu et al., 2019). Compared with saline-treated mice in which acute morphine application inhibited both basal firing (Fig. 2C; χ2, df = 5.269, 1; p = 0.022) and mechanosensitivity (Fig. 2E; χ2, df = 8.283, 1; p = 0.004) in most afferent units, morphine-treated mice had significantly fewer units with decreased firing and a greater number with increased or unchanged excitability.
Figure 2.

Chronic morphine treatment induces tolerance and OIH in mouse colon. Effect of acute application of 1 μm morphine on the basal activity and probing response of colonic afferent axons from saline and morphine-treated mice. A, Representative recording of colonic nerve response to 1 g von Frey filament probing before and after administration of 1 μm morphine. B, Acute morphine application did not affect the basal firing in afferent axons from saline-treated mice, while it increased firing in afferent axons from morphine-treated mice (F(1,28) = 10.06, p = 0.0037, two-way repeated-measures ANOVA with Bonferroni's post hoc test). C, Analysis of basal nerve activity showed an increased number of excited units on morphine-treated mice, whereas most of the units were inhibited in saline-treated mice (χ2, df = 5.269, 1; p = 0.022). D, Acute application of 1 μm morphine inhibited the colonic afferent nerve response to probing in saline-treated mice but not in morphine-treated mice (F(1,30) = 6.35, p = 0.0173, two-way repeated-measures ANOVA with Bonferroni's post hoc test). E, Chronic exposure to morphine increased the number of units that were excited on probing response after acute morphine application (χ2, df = 8.283, 1; p = 0.004). B, D, Error bars indicate mean ± SEM. *p ≤ 0.05. **p ≤ 0.01.
Together, these studies demonstrate the development of OT in response to chronic morphine and provide evidence of a paradoxical increase in basal and mechanosensitive afferent nerve firing in response to the acute application of morphine, suggesting OIH.
Pronociceptive signaling evoked by morphine is mediated by DOPr
To examine mechanisms underlying the development of OT and morphine-induced hyperexcitability in colonic afferents neurons, DRG neurons from naive mice were treated with morphine at a low (10 nm) or a high (30 μm) concentration either acutely (30 min) or overnight, and patch-clamp recordings were then obtained. Concentrations in the nanomolar range activate the classical Gi-mediated inhibitory pathway (Roeckel et al., 2017), whereas concentrations in the micromolar to millimolar range have been used to induce a pronociceptive effect (Akaishi et al., 2000; Forster et al., 2009; Rowan et al., 2014). Acute application of low and high concentrations of morphine decreased neuron excitability compared with control; rheobase increased by 24% (p = 0.038, one-way ANOVA, n = 15-21) and 30% (p = 0.004, one-way ANOVA, n = 21 on each group), respectively (Fig. 3A). Overnight incubation with high concentrations of morphine induced an increase in neuron excitability compared with control; rheobase decreased by 18% (Fig. 3B; p = 0.039, one-way ANOVA, n = 22 on each group). These data parallel the findings in afferent nerve recordings (Fig. 2), demonstrating that prolonged exposure to high concentration of opioids induces tolerance and a paradoxical increase in excitability of nociceptive neurons. The pronociceptive effect evoked by chronic morphine exposure was blocked by SDM25N (DOPr antagonist, 100 nm) (McLamore et al., 2001) but not by CTOP (MOPr antagonist, 100 nm) (Raynor et al., 1994), suggesting that the increased excitability induced by prolonged in vitro exposure to a high concentration of morphine is mediated by DOPr (Fig. 3C; for SDM25N: p = 0.019; for CTOP: p > 0.99, two-way ANOVA, n = 10-15).
Figure 3.
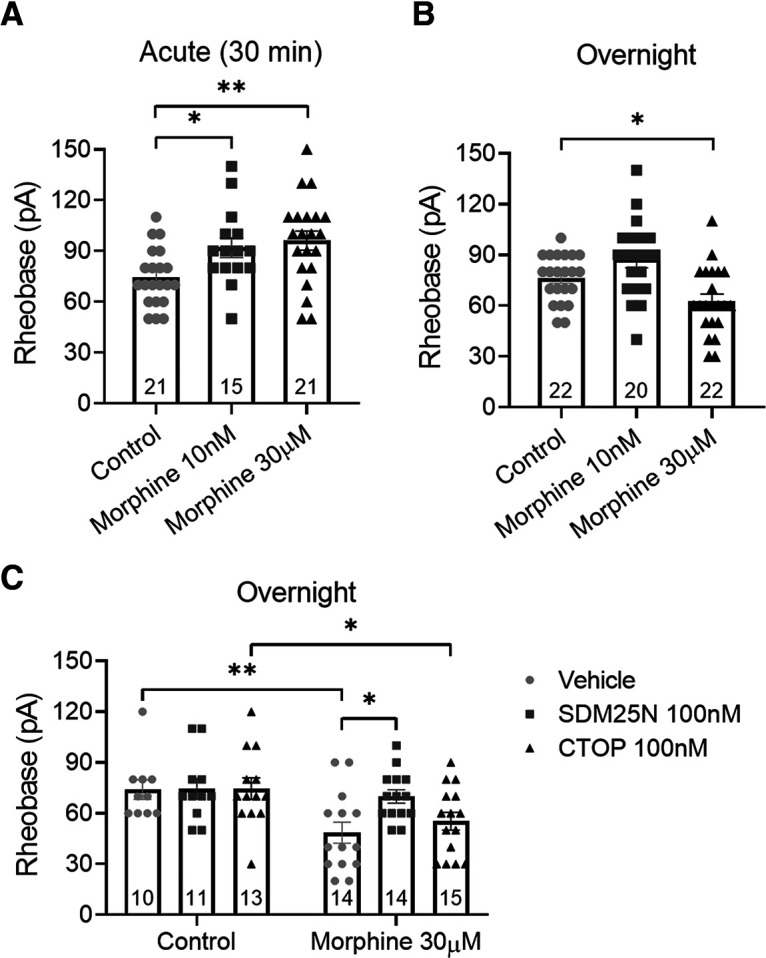
Overnight incubation with high concentrations of morphine has a pronociceptive effect on naive DRG neurons mediated by DOPr. A, Effect of the acute incubation of morphine on the excitability of DRG neurons measured by perforated patch clamp. Both low and high concentrations of morphine increased neuron rheobase compared with control (F(2,54) = 5.79, p = 0.0053, one-way ANOVA with Dunnett's post hoc test). B, Overnight incubation of low-concentration morphine had no effect on rheobase, while high concentration decreased neuron rheobase compared with control (F(2,61) = 8.93, p = 0.0004, one-way ANOVA with Dunnett's post hoc test). C, DOPr antagonist SDM25N but not MOPr antagonist CTOP prevented the pronociceptive effect induced by high-concentration morphine (F(1,71) = 12.45, p = 0.0007, two-way ANOVA with Bonferroni's post hoc test). Number of cells appears in each bar. Error bars indicate mean ± SEM. *p ≤ 0.05. **p ≤ 0.01.
To determine whether DOPr activation is also necessary for morphine-induced hyperalgesia in vivo, we examined the effect of the DOPr antagonist NTI (2.5 mg/kg) (Matthes et al., 1998). The DOPr antagonist was administered daily 20 min before morphine injections (Fig. 4A), and results from these mice were compared with those treated with morphine alone. The tail-flick test showed that NTI reduced the antinociceptive effect (MPE) of morphine at day 3, suggesting the involvement of DOPr in morphine-induced analgesia (Fig. 4B; p = 0.031, two-way repeated-measures ANOVA, N = 7 morphine-, 7 NTI + morphine-treated mice). Importantly, the analgesic effect of morphine was reduced on day 7 in morphine-treated mice (p = 0.003, two-way repeated-measures ANOVA, N = 7 mice), whereas NTI prevented this tolerance (p = 0.15, two-way repeated-measures ANOVA, N = 7 mice). In patch-clamp studies of DRG neurons, acute morphine application increased the excitability of nociceptive neurons from morphine-treated mice (rheobase decreased 26%, p = 0.049, two-way ANOVA, n = 15 control cells, 16 morphine-incubated cells/7 mice), but it did not affect the excitability of neurons from NTI + morphine mice (p > 0.99, two-way ANOVA, n = 17 control cells, 15 morphine-incubated cells/7 mice), demonstrating that blocking DOPr with NTI in vivo prevented the increase in DRG neuron excitability induced by chronic morphine (Fig. 4C). Ex vivo afferent nerve recordings in colons from mice receiving morphine alone showed that basal firing and mechanosensitivity were unaffected by the acute application of morphine (i.e., exhibited tolerance) (Fig. 4D,F; p > 0.99 and 0.25, respectively, two-way repeated-measures ANOVA, n = 15 units/7 morphine-treated mice), whereas in colons from NTI + morphine mice, the acute morphine application inhibited mechanosensitivity (Fig. 4F; p < 0.0001, two-way repeated-measures ANOVA, n = 16 units/7 NTI + morphine-treated mice). The number of afferent units with different responses to acute morphine application in terms of basal firing between morphine mice and NTI + morphine mice was similar (Fig. 4E; χ2, df = 2.280, 1; p = 0.13). However, acute morphine decreased the number of excited mechanosensitive units and increased the number of inhibited units in NTI + morphine mice (Fig. 4G; χ2, df = 14.06, 1; p = 0.0002), suggesting that blocking DOPr activation significantly reduced OIH in morphine-treated mice.
Figure 4.

The DOPr inhibitor NTI prevents tolerance and the pronociceptive effect induced by chronic morphine. A, Schematic protocol of chronic morphine treatment in the presence of DOPr antagonist NTI. B, In vivo tail-flick test on NTI + morphine-treated mice showed an inhibition on the maximum antinociceptive effect reached on day 3 by morphine-treated mice (F(1,12) = 5.52, p = 0.037, two-way repeated-measures ANOVA with Bonferroni's post hoc test). This maximum antinociceptive effect decreased on day 7 only in morphine-treated mice (F(1,12) = 17.67, p = 0.0012, two-way repeated-measures ANOVA with Bonferroni's post hoc test). Results are expressed as mean of % MPE ± SEM, morphine mice = 7; NTI + morphine mice = 7. C, Acute morphine incubation decreased rheobase of neurons from morphine-treated mice but did not have any effect on neurons from NTI + morphine-treated mice (F(1,59) = 4.163, p = 0.046, two-way ANOVA with Bonferroni's post hoc test). Number of cells appears in each bar. D-G, Effect of acute application of 1 μm morphine on the basal activity and probing response of colonic afferent axons from morphine (6 mice) and NTI + morphine-treated mice (5 mice). D, No changes were observed on basal firing in afferent nerves from morphine-or NTI + morphine-treated mice (F(1,29) = 1.53, p = 0.226, two-way repeated-measures ANOVA with Bonferroni's post hoc test). E, Quantification of units showed no difference between groups in terms of basal activity (χ2, df = 2.28, 1; p = 0.131). F, Acute application of morphine inhibited the afferent response to probing only in NTI + morphine mice (F(1,29) = 21.44, p < 0.0001, two-way repeated-measures ANOVA with Bonferroni's post hoc test). G, Most of the units were inhibited in response to probing after acute morphine exposure on NTI + morphine mice compared with those from morphine-treated mice, which showed a higher number of excited units (χ2, df = 14.06, 1; p = 0.0002). Error bars indicate mean ± SEM. *p ≤ 0.05. **p ≤ 0.01. ****p ≤ 0.0001.
The role of MOPr and DOPr signaling in the paradoxical increased DRG neuron excitability induced by opioids
We have previously shown that MOPr and DOPr are functionally expressed on colonic DRG neurons and coexpressed on a significant proportion of these neurons (Guerrero-Alba et al., 2018). To examine their specific roles in the development of opioid-induced neuronal hyperexcitability, the effects of the MOPr agonist DAMGO and the DOPr agonist DADLE were examined using acute application (30 min) and overnight incubation (Figs. 5 and 6, respectively) with low (10 nm) and high (10 μm) agonist concentrations. DAMGO and DADLE have similar binding affinities for their receptors (IC50 ∼ 2 nm), evoking inhibition at nanomolar concentrations (Raynor et al., 1994; Guerrero-Alba et al., 2018), and we sought to determine whether higher concentrations can evoke an excitatory effect. Following acute application of 10 nm or 10 μm DAMGO, neuronal excitability was decreased compared with control (Fig. 5A; p = 0.002 and 0.009, respectively, two-way ANOVA, n = 28-30). This decreased excitability was selectively blocked by 100 nm CTOP (Fig. 5A; p = 0.0012 and 0.02, respectively, two-way ANOVA, n = 18-30), but not by 100 nm SDM25N (Fig. 5B; p > 0.99, two-way ANOVA, n = 20-30). Acute application of 10 nm DADLE also decreased neuron excitability, whereas 10 μm DADLE increased neuron excitability (Fig. 5C; p = 0.026 and 0.018, two-way ANOVA, n = 23-26). Both effects were blocked by SDM25N (Fig. 5C; p = 0.003 and 0.046, two-way ANOVA, n = 23-26), whereas CTOP failed to block the excitatory effect by 10 μm DADLE (Fig. 5D; p > 0.99, two-way ANOVA, n = 14 on each group).
Figure 5.

Acute application of MOPr and DOPr agonists shows that pronociceptive actions are mediated by DOPr. A, B, Effect of low-concentration (10 nm) and high-concentration (10 μm) DAMGO applied acutely (30 min) on neuron excitability. Patch-clamp recordings show that low and high concentrations of DAMGO increased neuron rheobase. This antinociceptive effect was inhibited by MOPr antagonist CTOP (A: F(2,146) = 3.655, p = 0.028) but not by DOPr antagonist SDM25N (B: F(2,148) = 0.339, p = 0.713). C, D, A low concentration of DADLE increased neuron rheobase, whereas a high concentration decreased neuron rheobase. DOPr antagonist SDM25N blocked both effects induced by DADLE (C: F(1,134) = 8.35, p = 0.0004), whereas CTOP was unable to block the pronociceptive effect evoked by high-concentration DADLE (D: F(1,54) = 0.1555, p = 0.695). Number of cells appears in each bar. Error bars indicate mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; two-way ANOVA with Bonferroni's post hoc test.
Figure 6.

DOPr mediates the pronociceptive effect induced by high concentrations of MOPr and DOPr agonists applied overnight. A, B, Effect of low-concentration (10 nm) and high-concentration (10 μm) DAMGO overnight incubated on DRG neuron excitability. DRG neuron rheobase increased with low-concentration but decreased with high-concentration DAMGO. A, CTOP prevented the antinociceptive effect by 10 nm DAMGO but had no effect on the pronociceptive effect by 10 μm DAMGO (F(2,127) = 9.898, p < 0.0001). B, SDM25N inhibited only the pronociceptive effect by 10 μm DAMGO (F(2,71) = 8.144, p = 0.0007). C, D, Low-concentration DADLE had no effect on neuron rheobase, while high concentrations decreased neuron rheobase; this pronociceptive effect by high-concentration DADLE was inhibited by both SDM25N (C: F(2,161) = 5.022, p = 0.0077) and CTOP (D: F(1,72) = 5.605, p = 0.0206). E, Effect of 10 μm DADLE on DRG neurons innervating the colon of female mice. High-concentration DADLE decreased the rheobase of Fast Blue-labeled neurons (t(14) = 3.506, p = 0.0035, unpaired t test). Number of cells appears in each bar. Error bars indicate mean ± SEM. A–D, *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; two-way ANOVA with Bonferroni's post hoc test.
After overnight exposure, 10 nm DAMGO decreased neuron excitability compared with control, whereas 10 μm DAMGO increased excitability (Fig. 6A; p < 0.0001 and 0.03, respectively, two-way ANOVA, n = 23-25). Importantly, this excitatory effect of DAMGO was blocked by the DOPr antagonist SDM25N (Fig. 6B; p < 0.0001, two-way ANOVA, n = 10-13) but not by the MOPr antagonist CTOP (Fig. 6A; p > 0.99, two-way ANOVA, n = 22-23); 10 nm DADLE had no effect on the mean rheobase suggesting desensitization, whereas 10 μm DADLE increased neuron excitability (Fig. 6C; p = 0.296 and 0.004, respectively, two-way ANOVA, n = 23-28). This pronociceptive effect was also blocked by SDM25N (Fig. 6C; p = 0.006, two-way ANOVA, n = 23-33) but also by CTOP (Fig. 6D; p = 0.008, two-way ANOVA, n = 19 on each group), an unexpected finding given that CTOP did not block the pronociceptive action evoked by DADLE (Fig. 5D). Together, our data suggest that DOPr is involved in the paradoxical increased neuronal excitability induced by MOPr and DOPr agonists.
To confirm our fundamental observation in gut specific neurons, we performed patch-clamp recordings on colon-projecting DRG neurons (Fast Blue-labeled) from female mice. The DOPr agonist DADLE 10 μm overnight incubated increased DRG neuron excitability compared with vehicle (Fig. 6E; p = 0.0035, unpaired t test, n = 8 on each group).
The DOPr-mediated pronociceptive effect is endocytosis-dependent
To examine the intracellular signaling underlying hyperexcitability evoked by overnight incubation with high concentrations of DAMGO and DADLE, we used the opioid receptor antagonist NLX and the inhibitor of endocytosis, PS2. NLX is an alkaloid that can permeate the plasma membrane and also affect the intracellular signaling of opioid receptors (Wang et al., 2008), whereas PS2 is an inhibitor of clathrin-mediated endocytosis (Robertson et al., 2014). The pronociceptive effects of DAMGO and DADLE were blocked by 1 μm NLX (Fig. 7A; p < 0.0001 and 0.002 respectively, two-way ANOVA, n = 12-16) and also by 20 μm PS2 (Fig. 7B; p = 0.027 and 0.016 respectively, two-way ANOVA, n = 14-19). To further characterize the involvement of endocytic pathways in the increased neuronal excitability evoked by high concentrations of opioid agonists and mediated by DOPr, DRG neurons were incubated acutely (30 min) and overnight with high concentration of a weakly internalizing DOPr agonist ARM390 (10 μm) (Marie et al., 2003; Pradhan et al., 2009). We previously showed that DADLE and SNC80 but not ARM390 (100 nm) induced βarr recruitment as well as DOPr internalization (Jimenez-Vargas et al., 2020). Acute incubation with ARM390 did not alter the mean rheobase of DRG neurons, whereas overnight incubation decreased neuron excitability despite the high concentration (Fig. 7C; p = 0.039, one-way ANOVA, n = 20 or 21). Together, these data suggest that the neuronal hyperexcitability induced by opioids is dependent on DOPr internalization.
Figure 7.

Opioid-induced DRG neuronal hyperexcitability is dependent on receptor endocytosis. A, Patch-clamp recordings show that the OPr antagonist naloxone (NLX) prevented the decrease in the rheobase evoked by high-concentration DAMGO and DADLE applied overnight (F(2,78) = 8.143, p = 0.0006, two-way ANOVA with Bonferroni's post hoc test). B, The clathrin inhibitor PS2 also prevented the decrease on the rheobase evoked by high-concentration opioids (F(1,98) = 10.63, p = 0.0015, two-way ANOVA with Bonferroni's post hoc test). C, Acute application of a weakly internalizing DOPr agonist ARM390 at equally high concentration did not alter neuron rheobase, whereas overnight incubation increased neuron rheobase (F(2,58) = 3.064, p = 0.054, one-way ANOVA with Dunnett's post hoc test). Number of cells appears in each bar. Error bars indicate mean ± SEM. *p ≤ 0.05. **p ≤ 0.01. ***p ≤ 0.001.
Overnight exposure to a high concentration of opioids switches MOPr and DOPr signaling in response to acute opioid re-exposure to a pronociceptive pathway
To further examine the mechanisms underlying the switch of opioid signaling to a pronociceptive pathway, we studied the response to low-concentration (10 nm) MOPr and DOPr agonists following overnight incubation of DRG neurons with high concentrations (10 μm) of DAMGO or DADLE (Fig. 8A). As shown in Figure 6, sustained exposure to 10 μm DADLE increased neuron excitability. However, after 1 h washout of DADLE, neuron excitability recovered to control levels (Fig. 8B). Neurons were then re-exposed to acute application (30 min) of 10 nm of either a DOPr or MOPr agonist. DADLE 10 nm but not DAMGO increased neuron excitability rather than decreasing it (p = 0.002 and p > 0.99, respectively, one-way ANOVA, n = 16 or 17), as previously observed in naive neurons (Fig. 5), suggesting a switch in receptor downstream signaling pathways as a result of the prior overnight exposure to DADLE (Fig. 8B). Interestingly, following overnight incubation with 10 μm DAMGO, re-exposure to both 10 nm DAMGO or DADLE increased neuron excitability (Fig. 8C; p = 0.003 on both cases, one-way ANOVA, n = 10-12).
Figure 8.

Overnight exposure to high-concentration opioids switches low-concentration DOPr and MOPr agonist responses to a PKC-dependent pronociceptive pathway. A, Schematic protocol showing the paradigm used in B and C. Arrows indicate perforated patch-clamp recordings at different treatment points. B, The pronociceptive effect induced by high-concentration DADLE is lost after 1 h washout with control media, re-exposure to a low-concentration DADLE paradoxically decreased neuron rheobase (F(4,78) = 8.474, p < 0.0001). C, After washout of high-concentration DAMGO overnight, acute re-exposure to DAMGO or DADLE paradoxically decreased neuron rheobase (F(4,58) = 7.111, p < 0.0001). D, Schematic protocol showing the paradigm used in E–H. Arrows indicate perforated patch-clamp recordings after incubation with PKA or PKC inhibitors with or without opioid re-exposure. E, F, Following the washout of overnight high-concentration DADLE, PKC inhibitor GFX (F: F(2,34) = 0.382, p = 0.686) but not PKA inhibitor H89 (E: F(2,35) = 3.681, p = 0.035) prevented the paradoxical increased excitability induced by acute re-exposure to low-concentration DADLE. G, H, Following washout of overnight high-concentration DAMGO, PKA inhibitor H89 (G: F(2,53) = 4.1, p = 0.022) only prevented the pronociceptive effect induced by acute re-exposure to low-concentration DAMGO while PKC inhibitor GFX (H: F(2,50) = 2.246, p = 0.116) prevented the increased excitability induced by acute re-exposure to both low-concentration DAMGO or DADLE. Number of cells appears in each bar. Error bars indicate mean ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; one-way ANOVA with Dunnett's post hoc test.
PKC mediates the pronociceptive effect by opioids after overnight exposure to 10 μm DADLE
To further understand the intracellular mechanism underlying neuronal hyperexcitability after opioid re-exposure, we explored the role of PKA and PKC signaling. Opioid inhibition of neurons occurs via Gi-protein linked to PKA pathway, whereas PKCε signaling has been implicated in pronociceptive responses induced by opioids (Joseph et al., 2010). Following washout of the excitatory effect of overnight 10 μm DADLE or DAMGO incubation, PKA or PKC inhibitors were applied to DRG neurons 30 min before acute incubation with 10 nm opioids (Fig. 8D). H89 1 μm failed to block the pronociceptive effect of 10 nm DADLE re-exposure (Fig. 8E,G; rheobase decreased compared with H89 control; p = 0.02 and p = 0.012 respectively, one-way ANOVA, n = 12-16), but blocked the pronociceptive effect of 10 nm DAMGO re-exposure (Fig. 8G; rheobase not different compared with H89 control, p = 0.15, one-way ANOVA, n = 16 or 17). GFX 1 μm blocked the pronociceptive effect of 10 nm DADLE re-exposure following either overnight 10 μm DADLE or DAMGO incubation (Fig. 8F,H; rheobase not different from GFX control, p = 0.63 and p = 0.09, respectively, one-way ANOVA, n = 12-18), suggesting that PKC signaling plays a role in the pronociceptive responses mediated by DOPr.
Gαi signaling and βarr2 recruitment are lost after sustained exposure to opioids
BRET assays were performed in HEK293T cells to measure the activation of Gαi, Gαs, and Gαq proteins following acute opioid agonists re-exposure. Acute application of DADLE and DAMGO to cells not previously exposed to opioids activated Gαi subunits but minimal Gαq and not Gαs proteins. DADLE evoked a larger response than DAMGO (Fig. 9). Gαi subunit activation by acute opioids decreased when cells were previously exposed to a high concentration of DADLE (10 μm) or DAMGO (10 μm) (Fig. 9A–D; one-way ANOVA), suggesting possible desensitization of Gαi signaling after prolonged opioid exposure. We did not find evidence of Gαs or Gαq protein activation after opioid re-exposure (Fig. 9E–L; one-way ANOVA).
Figure 9.

Overnight incubation with high-concentration opioids decreased acute Gαi activation and βarr2 recruitment. Transfected HEK293T cells were treated overnight with vehicle, high-concentration (10 μm) DADLE or DAMGO, washed for 30 min, and then stimulated with vehicle, or agonist with low-concentration (10 nm) DAMGO or DADLE or high-concentration (10 μm) DAMGO or DADLE. BRET was recorded over time to measure activation of Gαi (A: F(5,24) = 45.25, p < 0.0001; B: F(5,24) = 99.2, p < 0.0001; C: F(5,23) = 0.1771, p = 0.9685; D: F(5,23) = 51.16, p < 0.0001), Gαs (E: F(5,24) = 0.8942, p = 0.5007; F: F(5,24) = 0.1545, p = 0.9766; G: F(5,23) = 0.9256, p = 0.4824; H: F(5,23) = 0.477, p = 0.7896), and Gαq (I: F(5,24) = 7.611, p < 0.0002; J: F(5,24) = 8.63, p < 0.0001; K: F(5,23) = 1.63, p = 0.1919; L: F(5,23) = 6.034, p = 0.001) by dissociation of Rluc2-Gα with GFP10-Gγ2 after agonist stimulation. Consequently, a decrease of BRET reflects an activation of Gα protein. Following the same cell treatment, recruitment of Rluc2-βarr2 to the plasma membrane (rGFP-CAAX) was also measured using ebBRET (M: F(5,24) = 8.966, p < 0.0001; N: F(5,24) = 24.1, p < 0.0001; O: F(5,23) = 0.7804, p = 0.574; P: F(5,23) = 13.28, p < 0.0001), an increase of ebBRET reflecting the recruitment of βarr2 at the plasma membrane. Data were collected from 3-7 independent experiments. Bars represent mean of the area under the curve ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.001; one-way ANOVA with multiple comparisons and Tukey's post hoc test.
βarr2 recruitment to the plasma membrane was also measured in HEK293T cells. Acute application of DADLE (Fig. 9M,N) or high concentration of DAMGO (Fig. 9P) evoked significant recruitment of βarr2 to the plasma membrane. This recruitment was absent when cells were re-exposed to opioids after overnight incubation with high concentration of DADLE or DAMGO (Fig. 9M–P; one-way ANOVA).
Discussion
The opioid crisis in the developed world has fueled ongoing interest in strategies to mitigate the serious and potentially lethal side effects of opioid drugs. These side effects are driven by escalating dosing as a result of OT and possibly the less well-described phenomenon of OIH, where opioids are thought to paradoxically increase nociceptive signaling following chronic exposure to high doses (Farmer et al., 2017). An important site of analgesia derived from systemic opioids is the peripheral nociceptive DRG neuron, such as those innervating the intestine (Guerrero-Alba et al., 2018). Here we focus on peripheral nociceptive signaling mechanisms in the colon using in vivo models of OT and corresponding in vitro models of OPr desensitization. We provide evidence of decreased antinociceptive and enhanced pronociceptive signaling from afferent nerves following chronic morphine exposure and corresponding changes in in vitro studies of isolated DRG neurons. Moreover, we found evidence of a functional MOPr–DOPr interaction where these events were driven by activation of DOPr in response to high doses of either MOPr or DOPr agonists.
Morphine is a common analgesic used to manage pain over days to weeks in multiple clinical settings. In our in vivo studies with morphine exposure for only 7 d, we observed decreased analgesia and enhanced pronociceptive signaling in colonic afferent nerves, when the nerves were challenged with an acute application of morphine. These findings were evident after quantification of the number of individual units differentially altered by acute morphine in basal afferent nerve firing and mechanical probing responses, suggesting that both basal firing and mechanosensitive properties are affected. In parallel, patch-clamp studies of individual DRG neurons from the same animals demonstrated receptor desensitization in response to the acute application of morphine. Furthermore, chronic incubation (overnight) with high-dose morphine recapitulates the switch from antinociceptive to pronociceptive signaling following an acute morphine challenge (Fig. 3C), a phenomenon observed by others (Khomula et al., 2019).
The analgesic actions of morphine and related compounds are mediated largely by MOPrs. Our studies, however, identified that the switch to pronociceptive pathways is mediated by DOPr signaling, based on several findings. Our in vitro studies of isolated DRG neurons demonstrated that the selective DOPr antagonists blocked the pronociceptive actions of overnight morphine incubation, whereas the MOPr antagonist had no effect. We observed a similar phenomenon in vivo where the pronociceptive actions of acute application of morphine following chronic exposure were blocked by the selective DOPr antagonist NTI, suggesting that DOPr plays a significant role in the development of hyperalgesia. We then used selective MOPr and DOPr agonists and their respective antagonists to recapitulate these findings. The higher concentrations we used may have exceeded concentrations reported to be selective (Raynor et al., 1994). Previous studies suggest that morphine and DAMGO at high concentrations can activate DOPr (Jordan et al., 2000; Zhao et al., 2003). Nonetheless, we found that the classical inhibition evoked by acute application at these high concentrations was blocked by their respective antagonists (Fig. 5).
Our studies may suggest a functional MOPr–DOPr interaction following sustained application of high-dose agonists. We previously showed that MOPr and DOPr are coexpressed on a significant proportion of nociceptive DRG neurons (Guerrero-Alba et al., 2018). The concept of MOPr–DOPr interaction underlying OPr-mediated inhibition is well described in previous studies (DiCello et al., 2020), although the mechanisms are poorly understood and may be multifactorial. Additionally, selectivity studies found that MOPr agonists morphine and DAMGO showed higher affinity to cells coexpressing MOPr-DOPr receptors compared with cells expressing only MOPr (Yekkirala et al., 2010). In our studies, where neurons were exposed to high concentrations of DADLE and DAMGO overnight and then excitability allowed to recover during a washout, we found diverging effects when we re-exposed these neurons to low concentrations of the agonists. DAMGO overnight exposure primed the neurons to a pronociceptive action in response to a subsequent challenge with both the MOPr and DOPr agonists, whereas overnight DADLE only resulted in a pronociceptive response to subsequent DADLE, but not DAMGO. These findings further support the concept that the pronociception evoked high-dose opioids are selectively mediated by activation of DOPr by either MOPr or DOPr agonists.
DOPr agonists are being developed for the treatment of chronic pain, as well as psychiatric disorders (Gendron et al., 2015). Like other GPCRs, DOPrs exhibit agonist-selective recruitment of arrestins, resulting in functional selectivity between agonists. Arrestins are multifaceted proteins that play an important role in DOPr trafficking and signaling. Recent studies suggest that weakly internalizing DOPr agonists have sustained signaling from the plasma membrane and exhibit sustained antinociception, whereas those that externalize and recruit arrestins exhibit rapid tolerance. OPr internalization has received considerable attention in studies of DOPr antinociceptive signaling and have shown that tolerance is induced by high internalizing DOPr agonist through βarr recruitment (Pradhan et al., 2010, 2016; Vicente-Sanchez et al., 2018), but the cellular mechanisms underlying DOPr hyperalgesia have not been studied. We found that the weakly internalizing DOPr agonist ARM390 did not evoke neuronal hyperexcitability, unlike DADLE, and that the actions of DADLE were blocked by the clathrin inhibitor PS2. Thus, our studies suggest that the DOPr-mediated pronociceptive signaling is also dependent on receptor internalization.
Opioid inhibitory effects are classically linked to Gαi and Gβγ protein signaling, and downstream inhibition of adenylate cyclase and PKA activity, although other signaling pathways have been implicated (Al-Hasani and Bruchas, 2011). We did not find a switch in Gαi to Gαs or Gαq signaling; however, our results suggest that DOPr-mediated pronociceptive signaling is PKC-dependent, which is similar to findings in other studies using MOPr selective agonists (Joseph et al., 2010). It has been reported that PKC activation can be downstream of Gαi pathway via Gβγ subunits or calcium entry (Camps et al., 1992; Nishizuka, 1992). Further studies are needed to determine the role of PKC in the mechanisms of phosphorylation and internalization of OR that might lead to OT and OIH.
There are several potential limitations of the current study. All opioid agonists are analgesic, yet selective activation of MOPr and DOPr exhibits different behavioral effects. DOPr agonists have anxiolytic and antidepressant activity (Valentino and Volkow, 2018), and MOPr agonists induce euphoria and stress coping. And while such actions could alter the outcome of behavioral studies, there is little evidence they have impacted the current study. Our electrophysiological measurements of tolerance and OIH were obtained using ex vivo peripheral afferent nerve preparations and in vitro patch-clamp studies of isolated neurons following chronic treatment and thus would not be directly influenced by the psychological impact of DOPr or MOPr signaling. While the tail flick test measures of tolerance could be influenced by psychological changes, our electrophysiological studies showed parallel correlates of tolerance and OIH in the same animals. Another potential limitation is the lack of in vivo dose ranging studies and correlation with the concentrations used in the in vitro studies. Further studies are needed in this regard, but the current studies show evidence of the observed in vitro correlates of tolerance and OIH in the in vivo studies, and the doses used were well tolerated by the animals and thus are likely comparable with those used in humans. A final issue relates to the identity of the DRG neurons studied. While the studies conducted using afferent recordings from the colon and patch-clamp studies of neurons retrogradely labeled from the colon of male and female mice demonstrate the pronociceptive actions of DOPr in gut-specific neurons, most of the patch-clamp studies were conducted on nonlabeled neurons; thus, many of these would be somatosensory neurons. Thus, it is likely that the findings described in this study relate not only to visceral neurons but also to the peripheral somatosensory system.
In conclusion, DOPr plays a major role in the development of OT and OIH in colonic afferent nociceptors. The development of OIH requires prolonged exposure to high opioid concentrations, leading to activation of mechanisms, such as OPr internalization and activation of PKC signaling pathway. Targeting key components on these identified pathways could mitigate the development of OT and OIH in the colon.
Footnotes
S.J.V., A.E.L., and D.E.R. were supported by Crohn's and Colitis Canada. N.W.B. was supported by National Institutes of Health Grants NS102722, DE026806, DK118971, and DE029951.
The authors declare no competing financial interests.
References
- Akaishi T, Saito H, Ito Y, Ishige K, Ikegaya Y (2000) Morphine augments excitatory synaptic transmission in the dentate gyrus through GABAergic disinhibition. Neurosci Res 38:357–363. 10.1016/s0168-0102(00)00177-2 [DOI] [PubMed] [Google Scholar]
- Al-Hasani R, Bruchas MR (2011) Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115:1363–1381. 10.1097/ALN.0b013e318238bba6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allouche S, Noble F, Marie N (2014) Opioid receptor desensitization: mechanisms and its link to tolerance. Front Pharmacol 5:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Davis B, Binion DG (2009) Pain and inflammatory bowel disease. Inflamm Bowel Dis 15:778–788. 10.1002/ibd.20848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498. 10.1126/science.286.5449.2495 [DOI] [PubMed] [Google Scholar]
- Brookes SJ, Spencer NJ, Costa M, Zagorodnyuk VP (2013) Extrinsic primary afferent signalling in the gut. Nat Rev Gastroenterol Hepatol 10:286–296. 10.1038/nrgastro.2013.29 [DOI] [PubMed] [Google Scholar]
- Cahill CM, Walwyn W, Taylor AM, Pradhan AA, Evans CJ (2016) Allostatic mechanisms of opioid tolerance beyond desensitization and downregulation. Trends Pharmacol Sci 37:963–976. 10.1016/j.tips.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P (1992) Isozyme-selective stimulation of phospholipase C-beta 2 by G protein beta gamma-subunits. Nature 360:684–686. 10.1038/360684a0 [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105. 10.1042/0264-6021:3510095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCello JJ, Carbone SE, Saito A, Rajasekhar P, Ceredig RA, Pham V, Valant C, Christopoulos A, Veldhuis NA, Canals M, Massotte D, Poole DP (2020) Mu and delta opioid receptors are coexpressed and functionally interact in the enteric nervous system of the mouse colon. Cell Mol Gastroenterol Hepatol 9:465–483. 10.1016/j.jcmgh.2019.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer AD, Gallagher J, Bruckner-Holt C, Aziz Q (2017) Narcotic bowel syndrome. Lancet Gastroenterol Hepatol 2:361–368. 10.1016/S2468-1253(16)30217-5 [DOI] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang WB, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De Koninck Y (2013) Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl– homeostasis. Nat Neurosci 16:183–192. 10.1038/nn.3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster AB, Reeh PW, Messlinger K, Fischer MJ (2009) High concentrations of morphine sensitize and activate mouse dorsal root ganglia via TRPV1 and TRPA1 receptors. Mol Pain 5:17. 10.1186/1744-8069-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gales C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol 13:778–786. 10.1038/nsmb1134 [DOI] [PubMed] [Google Scholar]
- Galligan JJ, Akbarali HI (2014) Molecular physiology of enteric opioid receptors. Am J Gastroenterol Suppl 2:17–21. 10.1038/ajgsup.2014.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Mittal N, Beaudry H, Walwyn W (2015) Recent advances on the delta opioid receptor: from trafficking to function. Br J Pharmacol 172:403–419. 10.1111/bph.12706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim TW, Schmid CL, Stahl EL, Pantouli F, Ho JH, Acevedo-Canabal A, Kennedy NM, Cameron MD, Bannister TD, Bohn LM (2020) A G protein signaling-biased agonist at the mu-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology 45:416–425. 10.1038/s41386-019-0491-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Alba R, Valdez-Morales EE, Jimenez-Vargas NN, Bron R, Poole D, Reed D, Castro J, Campaniello M, Hughes PA, Brierley SM, Bunnett N, Lomax AE, Vanner S (2018) Coexpression of mu and delta opioid receptors by mouse colonic nociceptors. Br J Pharmacol 175:2622–2634. 10.1111/bph.14222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Gooding SW, Lewis E, Felth LC, Gaur A, Whistler JL (2021) Pharmacological and genetic manipulations at the micro-opioid receptor reveal arrestin-3 engagement limits analgesic tolerance and does not exacerbate respiratory depression in mice. Neuropsychopharmacology 46:2241–2249. 10.1038/s41386-021-01054-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibeakanma C, Ochoa-Cortes F, Miranda-Morales M, McDonald T, Spreadbury I, Cenac N, Cattaruzza F, Hurlbut D, Vanner S, Bunnett N, Vergnolle N, Vanner S (2011) Brain-gut interactions increase peripheral nociceptive signaling in mice with postinfectious irritable bowel syndrome. Gastroenterology 141:2098–2108.e5. 10.1053/j.gastro.2011.08.006 [DOI] [PubMed] [Google Scholar]
- Jensen DD, Lieu TM, Halls ML, Veldhuis NA, Imlach WL, Mai QN, Poole DP, Quach T, Aurelio L, Conner J, Klein Herenbrink C, Barlow N, Simpson JS, Scanlon MJ, Graham B, McCluskey A, Robinson PJ, Escriou V, Nassini R, Materazzi S, et al. (2017) Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med 9:eaal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu TM, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, Klein Herenbrink C, Yeatman HR, Edgington-Mitchell L, Porter CJ, Halls ML, Canals M, Veldhuis NA, Poole DP, McLean P, Hicks GA, et al. (2018) Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci USA 115:E7438–E7447. 10.1073/pnas.1721891115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, Hegron A, Teng S, DiCello JJ, Rajasekhar P, Veldhuis NA, Carbone SE, Yu Y, Lopez-Lopez C, Jaramillo-Polanco J, Canals M, Reed DE, Lomax AE, Schmidt BL, Leong KW, Vanner SJ, et al. (2020) Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain. Proc Natl Acad Sci USA 117:15281–15292. 10.1073/pnas.2000500117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Cvejic S, Devi LA (2000) Opioids and their complicated receptor complexes. Neuropsychopharmacology 23:S5–S18. 10.1016/S0893-133X(00)00143-3 [DOI] [PubMed] [Google Scholar]
- Joseph EK, Reichling DB, Levine JD (2010) Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci 30:4660–4666. 10.1523/JNEUROSCI.5530-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Mischel RA, Bhave S, Komla E, Cho A, Huang C, Dewey WL, Akbarali HI (2017) The effect of gut microbiome on tolerance to morphine mediated antinociception in mice. Sci Rep 7:42658. 10.1038/srep42658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khomula EV, Araldi D, Levine JD (2019) In vitro nociceptor neuroplasticity associated with in vivo opioid-induced hyperalgesia. J Neurosci 39:7061–7073. 10.1523/JNEUROSCI.1191-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, Williams JT, Christie MJ, Schulz S (2019) Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun 10:367. 10.1038/s41467-018-08162-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyloh M, Nicholas S, Zagorodnyuk VP, Brookes SJ, Spencer NJ (2011) Identification of the visceral pain pathway activated by noxious colorectal distension in mice. Front Neurosci-Switz 5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy BE (2018) Review article: an analysis of safety profiles of treatments for diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther 48:817–830. 10.1111/apt.14948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie N, Landemore G, Debout C, Jauzac P, Allouche S (2003) Pharmacological characterization of AR-M1000390 at human delta opioid receptors. Life Sci 73:1691–1704. 10.1016/s0024-3205(03)00489-2 [DOI] [PubMed] [Google Scholar]
- Matthes HW, Smadja C, Valverde O, Vonesch JL, Foutz AS, Boudinot E, Denavit-Saubie M, Severini C, Negri L, Roques BP, Maldonado R, Kieffer BL (1998) Activity of the delta-opioid receptor is partially reduced, whereas activity of the kappa-receptor is maintained in mice lacking the mu-receptor. J Neurosci 18:7285–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattioli TA, Leduc-Pessah H, Skelhorne-Gross G, Nicol CJ, Milne B, Trang T, Cahill CM (2014) Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One 9:e97361. 10.1371/journal.pone.0097361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLamore S, Ullrich T, Rothman RB, Xu H, Dersch C, Coop A, Davis P, Porreca F, Jacobson AE, Rice KC (2001) Effect of N-alkyl and N-alkenyl substituents in noroxymorphindole, 17-substituted-6,7-dehydro-4,5alpha-epoxy-3,14-dihydroxy-6,7:2',3'-indolomorphina ns, on opioid receptor affinity, selectivity, and efficacy. J Med Chem 44:1471–1474. 10.1021/jm000511w [DOI] [PubMed] [Google Scholar]
- Moore BA, Stewart TM, Hill C, Vanner SJ (2002) TNBS ileitis evokes hyperexcitability and changes in ionic membrane properties of nociceptive DRG neurons. Am J Physiol Gastrointest Liver Physiol 282:G1045–G1051. 10.1152/ajpgi.00406.2001 [DOI] [PubMed] [Google Scholar]
- Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA (2016) Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7:12178. 10.1038/ncomms12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y (1992) Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258:607–614. 10.1126/science.1411571 [DOI] [PubMed] [Google Scholar]
- Peiris M, Hockley JR, Reed DE, Smith ES, Bulmer DC, Blackshaw LA (2017) Peripheral KV7 channels regulate visceral sensory function in mouse and human colon. Mol Pain 13:1744806917709371. 10.1177/1744806917709371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergolizzi JV Jr, Raffa RB, Rosenblatt MH (2020) Opioid withdrawal symptoms, a consequence of chronic opioid use and opioid use disorder: current understanding and approaches to management. J Clin Pharm Ther 45:892–903. 10.1111/jcpt.13114 [DOI] [PubMed] [Google Scholar]
- Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A, Evans C, Kieffer BL (2010) Ligand-directed trafficking of the delta-opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci 30:16459–16468. 10.1523/JNEUROSCI.3748-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Massotte D, Gaveriaux-Ruff C, Kieffer BL (2009) In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoS One 4:e5425. 10.1371/journal.pone.0005425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Perroy J, Walwyn WM, Smith ML, Vicente-Sanchez A, Segura L, Bana A, Kieffer BL, Evans CJ (2016) Agonist-specific recruitment of arrestin isoforms differentially modify delta opioid receptor function. J Neurosci 36:3541–3551. 10.1523/JNEUROSCI.4124-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM (2011) The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60:58–65. 10.1016/j.neuropharm.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T (1994) Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol 45:330–334. [PubMed] [Google Scholar]
- Robertson MJ, Deane FM, Stahlschmidt W, von Kleist L, Haucke V, Robinson PJ, McCluskey A (2014) Synthesis of the Pitstop family of clathrin inhibitors. Nat Protoc 9:1592–1606. 10.1038/nprot.2014.106 [DOI] [PubMed] [Google Scholar]
- Robinson DR, Gebhart GF (2008) Inside information: the unique features of visceral sensation. Mol Interv 8:242–253. 10.1124/mi.8.5.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeckel LA, Utard V, Reiss D, Mouheiche J, Maurin H, Robe A, Audouard E, Wood JN, Goumon Y, Simonin F, Gaveriaux-Ruff C (2017) Morphine-induced hyperalgesia involves mu opioid receptors and the metabolite morphine-3-glucuronide. Sci Rep 7:10406. 10.1038/s41598-017-11120-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MP, Bierbower SM, Eskander MA, Szteyn K, Por ED, Gomez R, Veldhuis N, Bunnett NW, Jeske NA (2014) Activation of mu opioid receptors sensitizes transient receptor potential vanilloid type 1 (TRPV1) via beta-arrestin-2-mediated cross-talk. PLoS One 9:e93688. 10.1371/journal.pone.0093688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn V, Del Vecchio G, Rodriguez-Gaztelumendi A, Temp J, Labuz D, Kloner M, Reidelbach M, Machelska H, Weber M, Stein C (2018) Opioid receptor signaling, analgesic and side effects induced by a computationally designed pH-dependent agonist. Sci Rep 8:8965. 10.1038/s41598-018-27313-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C (2016) Opioid receptors. Annu Rev Med 67:433–451. 10.1146/annurev-med-062613-093100 [DOI] [PubMed] [Google Scholar]
- Stein C (2018) New concepts in opioid analgesia. Expert Opin Investig Drugs 27:765–775. 10.1080/13543784.2018.1516204 [DOI] [PubMed] [Google Scholar]
- Stewart T, Beyak MJ, Vanner S (2003) Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons. J Physiol 552:797–807. 10.1113/jphysiol.2003.046409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeber M, Jullie D, Lobingier BT, Laeremans T, Steyaert J, Schiller PW, Manglik A, von Zastrow M (2018) A genetically encoded biosensor reveals location bias of opioid drug action. Neuron 98:963–976.e965. 10.1016/j.neuron.2018.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targownik LE, Nugent Z, Singh H, Bugden S, Bernstein CN (2014) The prevalence and predictors of opioid use in inflammatory bowel disease: a population-based analysis. Am J Gastroenterol 109:1613–1620. 10.1038/ajg.2014.230 [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F (1991) The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266:15771–15781. [PubMed] [Google Scholar]
- Valdez-Morales E, Guerrero-Alba R, Ochoa-Cortes F, Benson J, Spreadbury I, Hurlbut D, Miranda-Morales M, Lomax AE, Vanner S (2013) Release of endogenous opioids during a chronic IBD model suppresses the excitability of colonic DRG neurons. Neurogastroent Motil 25:39–46.e4. 10.1111/nmo.12008 [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Volkow ND (2018) Untangling the complexity of opioid receptor function. Neuropsychopharmacology 43:2514–2520. 10.1038/s41386-018-0225-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga EV, Navratilova E, Stropova D, Jambrosic J, Roeske WR, Yamamura HI (2004) Agonist-specific regulation of the delta-opioid receptor. Life Sci 76:599–612. 10.1016/j.lfs.2004.07.020 [DOI] [PubMed] [Google Scholar]
- Vicente-Sanchez A, Dripps IJ, Tipton AF, Akbari H, Akbari A, Jutkiewicz EM, Pradhan AA (2018) Tolerance to high-internalizing delta opioid receptor agonist is critically mediated by arrestin 2. Br J Pharmacol 175:3050–3059. 10.1111/bph.14353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Frankfurt M, Burns LH (2008) High-affinity naloxone binding to filamin a prevents mu opioid receptor-Gs coupling underlying opioid tolerance and dependence. PLoS One 3:e1554. 10.1371/journal.pone.0001554 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ (2013) Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65:223–254. 10.1124/pr.112.005942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Kalyuzhny AE, Portoghese PS (2010) Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective mu-delta agonists. ACS Chem Neurosci 1:146–154. 10.1021/cn9000236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Villalobos-Hernandez EC, Pradhananga S, Baker CC, Keating C, Grundy D, Lomax AE, Reed DE (2019) Deoxycholic acid activates colonic afferent nerves via 5-HT3 receptor-dependent and -independent mechanisms. Am J Physiol Gastrointest Liver Physiol 317:G275–G284. 10.1152/ajpgi.00016.2019 [DOI] [PubMed] [Google Scholar]
- Zar MA, Iravani MM, Luheshi GN (1990) Effect of nifedipine on the contractile responses of the isolated rat bladder. J Urol 143:835–839. 10.1016/s0022-5347(17)40112-1 [DOI] [PubMed] [Google Scholar]
- Zhao GM, Qian X, Schiller PW, Szeto HH (2003) Comparison of [Dmt1]DALDA and DAMGO in binding and G protein activation at mu, delta, and kappa opioid receptors. J Pharmacol Exp Ther 307:947–954. 10.1124/jpet.103.054775 [DOI] [PubMed] [Google Scholar]