

Abstract
Research progress from mainly over the last five years is described for a multidisciplinary collaborative program project directed toward the discovery of potential anticancer agents from a broad range of taxonomically defined organisms. Selected lead compounds with potential as new antitumor agents that are representative of considerable structural diversity have continued to be obtained from each of tropical plants, terrestrial and aquatic cyanobacteria, and filamentous fungi. Recently, a new focus has been on the investigation of the constituents of U.S. lichens and their fungal mycobionts. A medicinal chemistry and pharmacokinetics component of the project has optimized structurally selected lead natural products, leading to enhanced cytotoxic potencies against selected cancer cell lines. Biological testing has shown several compounds to have in vivo activity, and relevant preliminary structure-activity relationship and mechanism of action studies have been performed. Several promising lead compounds worthy of further investigation have been identified from the most recent collaborative work performed.
Graphical Abstract
INTRODUCTION
Cancer remains a formidable threat to public health in the United States and elsewhere, despite there being some room for optimism as a result of current methods of diagnosis and treatment. Thus, Seigel et al. reported recently that there has been a 31% decrease in the mortality rate due to cancer in the United States, during the period 1981–2018.1 In spite of this, it has been estimated in 2021, about 1.9 million people in the U.S. will be diagnosed with cancer, and that close to 600,000 persons will die of this disease.1 Globally, the International Agency for Research on Cancer (IARC) of the World Health Organization has projected that in 2020 the incidence of cancer was 19.3 million people, with the corresponding mortality figure being 10 million.2
While there are several effective approaches to the treatment of cancer, including radiation, surgery, immunotherapy, and targeted therapy, natural products have been of particular interest over the last 60 years in terms of providing a broad range of cancer chemotherapeutic agents with varied mechanisms of action. Indeed, of 259 small-molecule antitumor agents approved in western medicine from 1946–2019, 100 (38.6%) were either unaltered natural products or natural product synthetic derivatives, while other such drugs were inspired by natural product structures.3 Two examples of recently FDA-approved antitumor natural product derivatives are midostaurin and lurbinectedin, for which the source organisms of the lead compounds are of terrestrial and marine origin, respectively. Midostaurin, a semi-synthetic derivative of the indolocarbazole alkaloid, staurosporine, from a soil-derived microbe, was approved in 2017 for the chemotherapy of adult patients with advanced systemic mastocytosis associated with mast-cell leukemia.4 In turn, lurbinectedin received accelerated approval in 2020 to treat metastatic small-cell lung cancer in association with or after platinum-based chemotherapy, and its lead compound, the complex indole alkaloid, trabectedin, was sourced from a Caribbean Sea tunicate species,5 although the actual biosynthetic source is a bacterial endosymbiont.6 Many diverse types of marine and terrestrial organisms have afforded useful anticancer agents,3 and, for example, our group has recently reviewed new developments in the discovery of antitumor agents from higher plants.7 Others have documented that a wide range of endophytic and epiphytic bacteria and fungi are now known to produce well-known plant constituents such as camptothecin, homoharringtonine, paclitaxel, and podophyllotoxin.8,9 A major development that has accelerated over the last decade has been the approval of “antibody-drug conjugates” (ADCs), in which a highly cytotoxic natural product-derived “warhead” or “payload” is complexed often after considerable synthetic modification through a linker to a monoclonal antibody.7,10 The natural products lead compounds used in FDA-approved ADCs for cancer therapy are mainly of terrestrial microbial and marine organism origin,e.g., 11 but recently, trastuzumab deruxtecan, based on a potent semi-synthetic derivative of camptothecin, was approved to treat HER2-positive breast cancer.12
Since 2007, our collaborative team has been very fortunate to have been funded through the program project (P01) mechanism by the U.S. National Cancer Institute (P01CA125066; “Discovery of Anticancer Agents of Diverse Natural Origin)”. The present main objectives of the project are the discovery of new natural product lead compounds with antitumor activity from tropical plants, U.S. lichens and their mycobionts, aquatic and freshwater cyanobacteria, and filamentous fungi. In order to do this in an effective manner, the project has organism collection, taxonomic, isolation chemistry, synthetic chemistry and pharmacokinetics, biological evaluation, and biostatistics technical components. Members of three universities [the Ohio State University (OSU), the University of Illinois at Chicago (UIC), and the University of North Carolina at Greensboro (UNCG)] and a fungus research company (Mycosynthetix, Inc., Hillsborough, NC) constitute the primary members of the current project team. We have previously published summaries of the overall group technical progress of this P01 project,13,14 as well as of the tropical plant-focused lead compound discovery component of this collaboration.15,16
An organizational scheme for our P01 project was illustrated in our last comprehensive review of the program project published in 2016.14 This is still largely in force, with current Projects 1–3 (at OSU, UIC, and UNCG, respectively) focusing, in turn, on the isolation chemistry of bioactive compounds from tropical plants and U.S. lichens and their mycobionts, aquatic and terrestrial cyanobacteria, and filamentous fungi, respectively. Over the last five years, tropical plants for investigation in Project 1 have continued to be sourced by Project 2 from southeast Asia, while fungi for evaluation in Project 3 have been provided by Mycosynthetix, Inc. There is a biological testing component in both Project 1 and at Columbia University for Project 3. However, the main biological screening work for the P01 program as a whole has been carried out at UIC in Core 1 (previously Core A), using a panel of selected cancer cell lines, and more recently an in vitro autophagy bioassay. In addition, highly promising compounds have been evaluated in vivo using high-grade serous ovarian cancer murine xenograft models. The work in Core 2 (previously Core B) constitutes medicinal chemistry optimization of certain lead natural product molecules obtained from Projects 1–3, in addition to a preliminary pharmacokinetics component. Biostatistics under the direction of Dr. Xiaoli Zhang is carried out for the entire project in Core A (formerly Core C), and this unit also provides overall project administrative services. Active collaborations are sought with external institutions and colleagues, as will be mentioned in the paragraphs below. The P01 project continues to be overseen by an NCI Program Official (Dr. Yali Fu) and is advised by both an External Advisory Committee and an Internal Ohio State University Advisory Committee. We are grateful, in particular, to Drs. William Gerwick (University of California-San Diego), Susan Horwitz (Albert Einstein College of Medicine), the late G. Robert Pettit (Arizona State University), William C. Rose (formerly of Bristol-Myers Squibb), and Stephen M. Swanson (University of Wisconsin-Madison) for kindly serving as external advisors in recent years.
EXAMPLES OF RECENTLY OBTAINED BIOACTIVE COMPOUNDS FROM THE PROGRAM PROJECT RESEARCH
Metabolites from Tropical Plants and U.S. Lichens and Their Mycobionts.
Project 1 at the College of Pharmacy, OSU, currently has a dual focus, and works on both potential antitumor agents from tropical plants collected in southeast Asia and also from lichens and their fungal mycobionts obtained from coastal regions of the United States. Recent progress made from both of these classes of organisms are summarized in the following paragraphs.
Tropical Plant-derived Compounds.
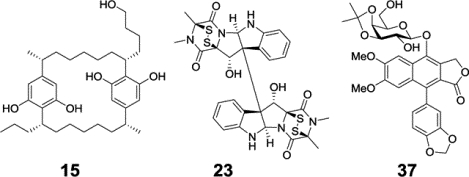
The structures (1-6) of six representative higher plant-derived lead compounds that have been investigated in our recent program project work are shown in Figure 1. Not all of these are new compounds, with some being previously known but for which additional biological and mechanistic investigations have been carried out. As one of the previously known compounds investigated in greater detail, the rocaglate derivative silvestrol (1) has been shown to occur in additional Aglaia species and also has been further evaluated biologically. The structure and absolute configuration of this rare dioxanyl-ring containing cyclopenta[b]benzofuran were reported initially in 2004 as a constituent of Aglaia foveolata (Meliaceae) from our work conducted at UIC, and then it was shown active in several cancer-related in vivo test systems at OSU.17 Recently, Greger has reviewed in detail the comparative phytochemistry of the rocaglate (flavagline) derivatives isolated from the genus Aglaia.18 Crucially, at McGill University in Montreal, silvestrol was determined to act mechanistically as a protein translation inhibitor by acting on eIF4A, which is an RNA helicase subunit of the eIF4F complex.19 In 2016, silvestrol was reported as a constituent of two further Aglaia species, namely, from the leaves of A. perviridis collected in Yunnan Province of the People’s Republic of China,20 and from the stems of A. stellatopilosa obtained from Sarawak, Malaysia.21 In work performed by Project 1 on a sample of A. perviridis collected in Vietnam, four new analogues of silvestrol were reported based on a new carbon skeleton, having a dihydrofuran ring fused to dioxanyl and aryl rings, as exemplified by compound 2. Unlike silvestrol, compound 2 and its three epimeric analogs, are substituted by a carboxamide group at C-2, rather than a carbomethoxy group. However, compound 2 was not cytotoxic when tested against the HT-29 human colon cancer and PC-3 human prostate cancer cell lines (IC50 values >10 μM).22 Rocaglate derivatives containing a dioxanyl ring like silvestrol (1) appear to be very rare, and, to date, have been found in only three of the ca. 120 species of the genus Aglaia.23
Figure 1.
Structures of compounds Isolated from tropical plants (1-6) and a mycobiont of a U.S. lichen (7 and 8).
In 2013, in earlier work performed by Project 1 on a Vietnamese collection of Aglaia perviridis, the known compound (−)-didesmethylrocaglamide (3) was purified as a trace constituent of a mixture of the dried leaves, twigs, and fruits.24 Compound 3 was originally isolated by Dumontet and associates from the seeds and leaves of Aglaia argentea, and found to display potent growth inhibitory effects against the KB cell line.25 In our hands, this rocaglate derivative was determined as being potently cytotoxic for the HT-29 colon cancer cell line (IC50 21 nM), and showed no discernible cytotoxicity for a normal colon cell line (CCD-112CoN) at a concentration level of 50 μM.24 We have been fortunate to be able to establish a collaboration with Dr. Long-Sheng Chang and his colleagues at the Center for Childhood Cancer and Blood Diseases, Nationwide Children’s Hospital, Columbus, OH, in which their group has tested several cyclopenta[b]benzofuran derivatives in neurofibromatosis-related tumor and pediatric sarcoma bioassay systems. When a small panel of 10 of these compounds were evaluated against four schwannoma, meningioma, and malignant peripheral nerve sheath tumor (MPNST) cell lines, (−)-didesmethylrocaglamide (3) was found to be the most highly cytotoxic, showing IC50 values in the 5–10 nM range for each cell line.26 Silvestrol (1) was somewhat less active than compound 3 when tested against these cell lines (IC50 range 10–70 nM).26 Prior work at Nationwide Children’s Hospital using 1 as a test compound showed that the eIF4F complex is a potential therapeutic target for MPNST, vestibular schwannomas, and meningiomas, and it was found to decrease the levels of several pertinent cell cycle proteins and upstream signaling kinases.27,28 Unfortunately, when it was evaluated in a standard toxicology test, 1 was determined to cause pulmonary toxicity in dogs.23,26 Therefore, despite previously being selected for evaluation through the NCI Experimental Therapeutics NExT program,14 further development of this compound as a potential cancer chemotherapeutic agent has been suspended.26 However, silvestrol is currently available commercially as a potent standard protein translation inhibitor, for use in laboratory studies.23 (−)-Didesmethylrocaglamide (3) as an eIF4A inhibitor has been shown to have good oral bioavailability, and to exhibit tumor growth in vivo in multiple models of pediatric sarcoma, and, accordingly, recently it has been chosen along with another rocaglate derivative, rocaglamide, for further development as a potential sarcoma treatment through the U.S. NCI Experimental Therapeutics NExT program.29 Rocaglamide was the initial cyclopenta[b]benzofuran derivative discovered in 1981 from an Aglaia species, and was reported as an antileukemic agent from A. elliptifolia by King et al.30
Three plant-derived compounds that are members of the cardiac glycoside class (4-6) have been investigated over the last few years in our program project. In an initial study, (+)-strebloside (4) was isolated along with several other cardiac glycosides from the stem bark of a medicinal plant, Streblus asper (Moraceae), collected in Vietnam.31 Also, six new analogues and one known compound were synthesized from compound 4.31 (+)-Strebloside (4) was characterized structurally from S. asper by Khare et al. in 1962,32 and was found to be a cytotoxic component of this same species, when collected in Thailand, for the KB cell line.33 In our further work, compound 4 was determined as being potently growth inhibitory for human breast, melanoma, and ovarian cell lines (IC50 range 44–134 nM).31 In addition, it was shown that the C-5 and C-14 hydroxy groups, the C-10 formyl group, and the sugar unit are all important for the mediation of the cytotoxicity of (+)-strebloside (4) against HT-29 human colon cells.31 Moreover, using an in vivo hollow fiber model (reviewed in Ref. 34), (+)-strebloside (4) demonstrated cancer cell growth inhibitory effects in NCr nu/nu mice implanted intraperitoneally (i.p.) with MDA-MB-231 human breast and OVCAR3 human ovarian cancer cells, at doses of 5 and 10 mg/kg, respectively. Moreover, this test compound did not show any obvious toxicity to mice at the i.p. doses used up to 30 mg/kg in this study.31
In a follow-up mechanistic study of (+)-strebloside (4) using both an in vitro assay and molecular docking, it was indicated that this compound binds to and inhibits the enzyme Na+/K+-ATPase in a similar manner to more well-known cardenolides such as digitoxin and ouabain.35 In a side-by-side comparison investigation, (+)-strebloside (4) displayed potent growth inhibition against a small panel of high grade serous ovarian cancer cell lines, but it was less active than digitoxin for most of the cell lines used. In OVCAR3 cells, (+)-strebloside caused apoptosis, and blocked cell cycle progression at the G2 phase, and resulted in PARP cleavage. It also inhibited NF-κB in human ovarian cells, and inhibited mutant p53 expression through the induction of ERK pathways.35 A further discussion on the cellular mechanism of action of (+)-strebloside is provided in the section on biological evaluation later in this review article.
In an additional phytochemical examination on the combined flowers, leaves, and twigs of S. asper, also collected in Vietnam, a new non-cytotoxic derivative, (+)-17β-hydroxystrebloside (5) was isolated along with (+)-strebloside (4). Compound 5 was inactive (IC50 >10 μM) against three human cancer cell lines (HT-29 colon; MDA-MB-435 melanoma; and OVCAR3 ovarian).36 Molecular docking profiles demonstrated that compounds 4 and 5 bind differentially to Na+/K+-ATPase, since the latter cardenolide fits the cation binding site of this enzyme with at least three different poses and as a result this tends to depotentiate its binding.36 In an additional docking profile study, (+)-strebloside (4) was postulated as targeting HIF-1, PI3K, and Nrf2 and p53 protein-protein interactions, in addition to Na+/K+-ATPase.37
Another cardenolide, the diglycoside corchorusoside C (6), has been used as a test compound in the development of a zebrafish (Danio rerio) assay of potential use for the evaluation of natural product candidate anticancer agents, in the laboratory of Dr. Esperanza Carcache de Blanco.38 Compound 6 was isolated for this study from the stems of a further Vietnamese plant, Streptocaulon juventas (Apocynaceae). Corchorusoside C (6) was tested against six human cancer cell lines, and shown to be most potently active against DU-145 prostate cells, with an IC50 value of 80 nM. It also induced cell shrinkage and detachment using this cell line. When evaluated against a non-tumorigenic cell line (CCD-112CoN colon cells), somewhat reduced cytotoxic potency (IC50 2.7 μM) was observed.38 A preliminary mechanistic investigation showed that compound 6 decreased the protein expression of NF-κB, IKK, ICAM-1, and BCl-2 and increased the expression of PARP-1 and the levels of caspases 3 and 7, leading to the induction of apoptosis in DU-145 cells. It was found also that cochorusoside C (6) modulated NF-κB and caspase levels in vivo in zebrafish.38 It is noteworthy that corchorusoside C did not display a significant impact on induction of cellular calcium concentration, in comparison with a positive control used, digoxin. Corchorusoside C (6) did not affect the development of zebrafish in the manner shown by digoxin at the same concentration. Accordingly, compound 6 may exhibit a lesser toxic effect than reported for digoxin and other cardiac glycosides on human myocytes, making this cardenolide worthy of consideration for further studies.38
Over the last few years, higher plants have been collected for our program project from Vietnam and more recently from Laos (Lao PDR) by a team from UIC headed by Dr. Djaja D. Soejarto (Project 2).15,39,40 The plant collection work has been conducted in accordance with requirements of international treaties, and has involved the formulation of a detailed Memorandum of Agreement between UIC and each of the Institute of Ecology and Biological Research (IEBR), Hanoi, Vietnam and the Institute of Traditional Medicine (ITM), Vientiane, Laos. Aspects involved with plant collection, such as species selection, field note data requirements, collection strategies, processing of collected samples, and exporting from southeast Asia and importing to United States of plants for our P01 project have been documented.15 A very important consideration for the accurate taxonomic identification of each species is the preparation of herbarium voucher specimens. For our recent collections, these have been deposited at IEBR (Vietnamese plants) and ITM (Lao plants) herbaria, respectively, with a second set deposited in each case at the John G. Searle Herbarium of the Field Museum, Chicago, IL. Further herbarium voucher specimens were also sent to appropriate taxonomic specialists at recognized institutions worldwide, when necessary to help with final taxonomic determinations of the plant species.15
Compounds from Lichens and Their Mycobionts.
In an effort to expand the range of organisms evaluated in our project and the potential for new bioactive compound discovery, a number of lichen specimens have been collected from the coastal areas of the western United States. This work was initiated through a two-year “Research Supplement to Promote Diversity in Health-related Research” to the overall P01 project, to promote the research program of Dr. Liva Rakotondraibe. Lichens are organisms composed of combinations of photobionts (cyanobacteria and/or microalgae) and mycobionts (fungi) that occur symbiotically, and have long been of interest for their bioactive constituents present.41 It was hypothesized that presently unexplored lichens and their photobionts and mycobionts, since they are highly stressed in their natural growth environment, may produce biologically active compounds representing unusual chemotypes that may serve as lead antineoplastic compounds, and then merit further development.
In work of this type that has been conducted to date, the component of the work on the taxonomic identification of lichens has been conducted by Dr. Richard W. Spjut, of World Botanical Associates, Bakersfield, CA, and the fungal identification conducted by Dr. Chad A. Rappleye, Department of Microbiology, Ohio State University.42,43 The chemical profile and the antiproliferative activity of U.S. endemic lichens from the genus Niebla collected from coastal areas in Marin County, California, and their mycobionts have been investigated. Although new and known triterpenes as well as previously identified depsides and depsidones were isolated and characterized from a lichen sample identified as Niebla homalea, only the known compound usnic acid among these was found to display antiproliferative activity against A2780 human ovarian and MCF-7 human breast cancer cells, with IC50 values of 3.8 and 6.8 μM, respectively.43 From one of the microbial associates isolated, an extract obtained from a fungus identified as Penicillium aurantiacobrunneum (Trichocomaceae), which exhibited moderate activity against both the A2780 and MCF-7 cancer cell lines, was subjected to chromatographic fractionation. A yellow α-pyrone constituent that was isolated and characterized spectroscopically as 4-epi-citreoviridin (7) (Figure 1) displayed cytotoxic activities against these same two cell lines (A2780, IC50 8.2±2.7 μM; MCF-7 IC50, 6.0±1.6 μM).42 It is worth noting that this compound did not show antiproliferative activities against the HT-29 colon adenocarcinoma and DU-145 prostate cancer cell lines. A metabolite named auransterol (8) (Figure 1) was found to be the most active among the sterol compounds isolated and exhibited a selective inhibition toward the colon adenocarcinoma (HT-29) cell line while lacking activity against MCF-7, A2780, and DU-145 cells. Due to this observation, the antiproliferative mechanism of 8 was investigated and it was shown to inhibit cell proliferation by inducing apoptosis with a mechanism independent of the tumor suppressor p53. This was evidenced by the upregulation of the apoptotic regulators such as BAX, cytochrome complex (Cyt-c), PARP-1, p21 and procaspase-3 proteins and downregulation of Bcl-2, with no modifications in procaspase-7 and p53.44
Metabolites from Cultured Cyanobacteria.
The main focus of Project 2, located at UIC and headed by Dr. Jimmy Orjala, is the investigation of bioactive metabolites from cultured freshwater cyanobacteria. The UIC collection of cultured cyanobacteria has become one of the largest in the U.S., and presently contains over 1,200 strains. Cyanobacteria need no source of organic carbon to grow under laboratory culture conditions. As photosynthetic organisms, they require exposure to light-dark cycles and a bicarbonate-carbonate source, which can be provided in the form of a buffer or simply as CO2 from an air flow. In addition, widely used culture media contain varying levels of inexpensive ingredients, such as nitrate, phosphate, potassium, magnesium, calcium, sodium, sulfate, chloride, trace metals, a chelating agent, and vitamins. Extra salts are added for marine and brackish water media.45 In our work, cyanobacterial strains are inoculated in four 2.8 L Fernbach flasks with 2 L of medium each (total volume: 8 L), grown under illumination (18/6-hour light/dark cycle) at 22 °C, and aerated with sterile filtered air for 6–8 weeks. The freeze-dried cell material then is extracted with dichloromethane-methanol (1:1). Extracts are next fractionated by vacuum-liquid chromatography using a Diaion HP-20SS column and a step gradient of isopropanol-water, to give six major fractions. Both laboratory grown and field-collected cyanobacteria have been shown to yield valuable secondary metabolites.46–53 From a biotechnological perspective, unialgal laboratory cultures have important benefits for natural product discovery. First, they allow for a more accurate identification of the source organism, which can sometimes be challenging for compounds isolated from field-collected cyanobacterial assemblages. Second, reproducible results can be ensured by the use of standardized culture conditions. Third, sufficient biomass can be obtained for cyanobacteria that do not grow in high densities in the natural environment.47
Extracts and fractions from the cyanobacterial strains are evaluated against a panel of human cancer cell lines to detect cytotoxic activity (see Biological Evaluation Core section, below). This workflow includes the large-scale cultivation of each strain in 8 L of medium under standard culture conditions, followed by extraction, fractionation, and cytotoxicity testing, along with HPLC-MS-NMR dereplication of active fractions for strain prioritization.54 In the following paragraphs, selected compounds from cyanobacterial sources obtained recently in the program project work will be featured.
[7,7]Paracyclophanes belong to a group of bioactive compounds produced by members of the Nostocaceae family, and several Nostoc spp. from the UIC cyanobacterial library have been identified to produce these compounds.55–59 These macrocyclic polyketides were first identified by Moore et al. in 1991.60,61 Since then, additional classes of [7,7]paracyclophanes have been discovered with different moieties attached at the alpha position of the resorcinol core and the terminal carbon of the aliphatic chain.55–59,62,63
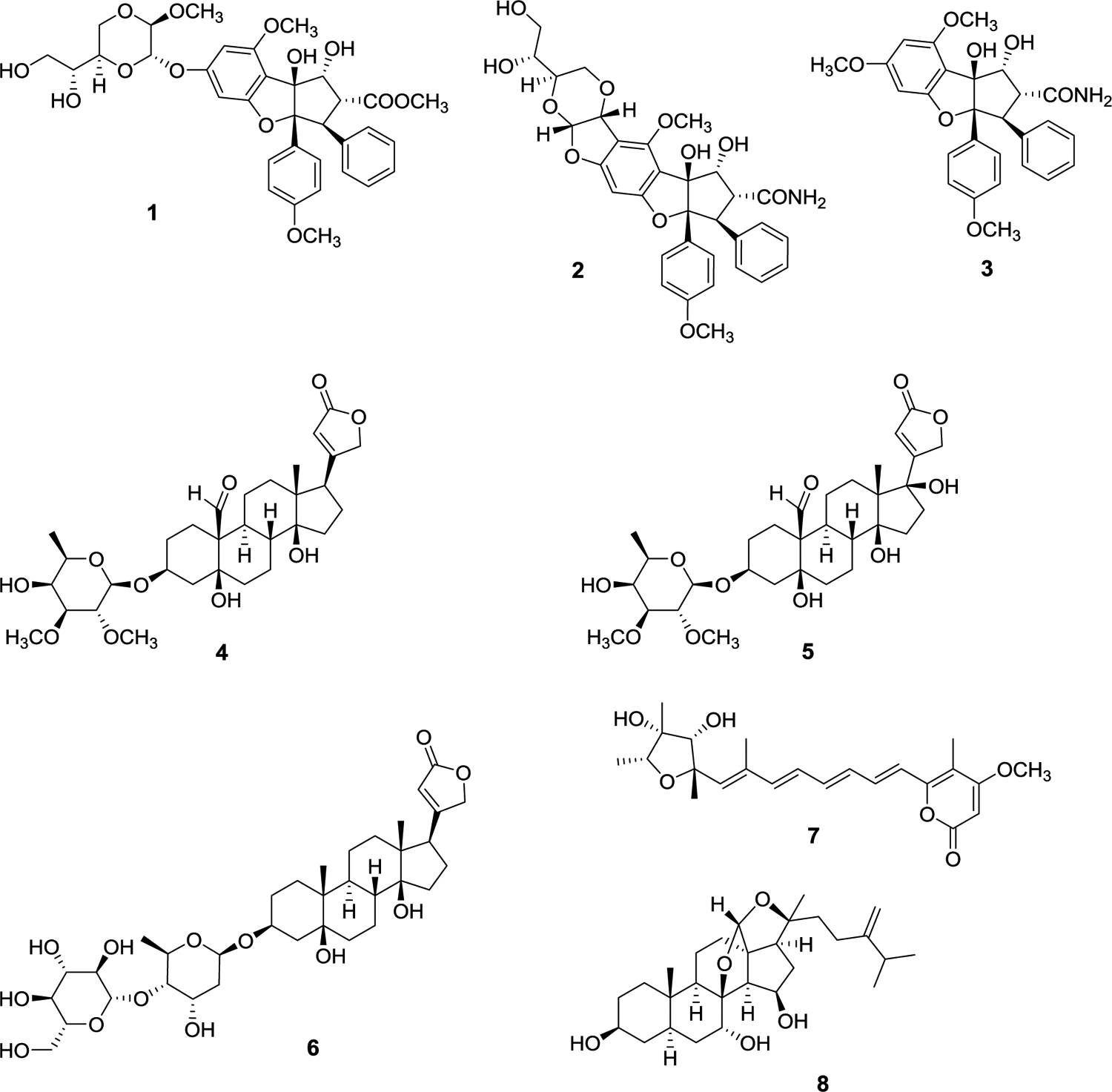
A fraction of a cultured Nostoc sp. (UIC 10279), obtained from a sample collected in the southwest suburbs of Chicago, displayed antiproliferative activity against the MDA-MB-435 human melanoma cancer cell line. A second Nostoc sp. (UIC 10366), obtained from a collection in the northwestern suburbs of Chicago, also displayed antiproliferative activity against this same cell line.59 LC-MS and 1H NMR-based dereplication suggested the presence of glycosylated cyclophanes in both strains (Figure 2). Bioassay-guided fractionation led to the isolation of five glycosylated cylindrocyclophanes, named ribocyclophanes A-E (9-13). Ribocyclophanes A-D (9-12) are glycosylated analogues of the cylindrocyclophanes, whereas the structurally related ribocyclophane E (13) is a glycosylated analog of the cylindrofridins, and a likely biosynthetic intermediate of 12.
Figure 2.
Structures of cyclophane derivatives obtained from cyanobacterial species.
A notable structural feature of compounds 9-12 is the presence of a β-d-ribopyranosyl glycone moiety linked to the benzylic carbon of the cylindrocyclophane aglycone. The only previously reported glycosylated [7,7]paracyclophanes from cyanobacteria are nostocyclophanes A and B, with glycosylation on the phenolic moieties by β-d-glucose.60 Interestingly, ribocyclophane A (9) was found in both the UIC 10279 and UIC 10366 samples, while ribocyclophanes B (10) and C (11) were found only in UIC 10279 and ribocyclophanes D (12) and E (13) only in UIC 10366. Ribocyclophanes A-E were tested for their antiproliferative activity against MDA-MB-231 (breast cancer) and MDA-MB-435 (melanoma) cancer cells and displayed IC50 values from 0.6 to 6.4 μM, with ribocyclophane D (12) being the most potent with IC50 values of less than 1 μM for both cell lines (IC50 0.8 μM and 0.6 μM, respectively). Ribocyclophane E (13) showed no discernible antiproliferative activity against either cell line at 25 μM, corroborating the findings of Preisitsch et al. that an unclosed [7,7]paracyclophane core structure does not retain antiproliferative activity.64
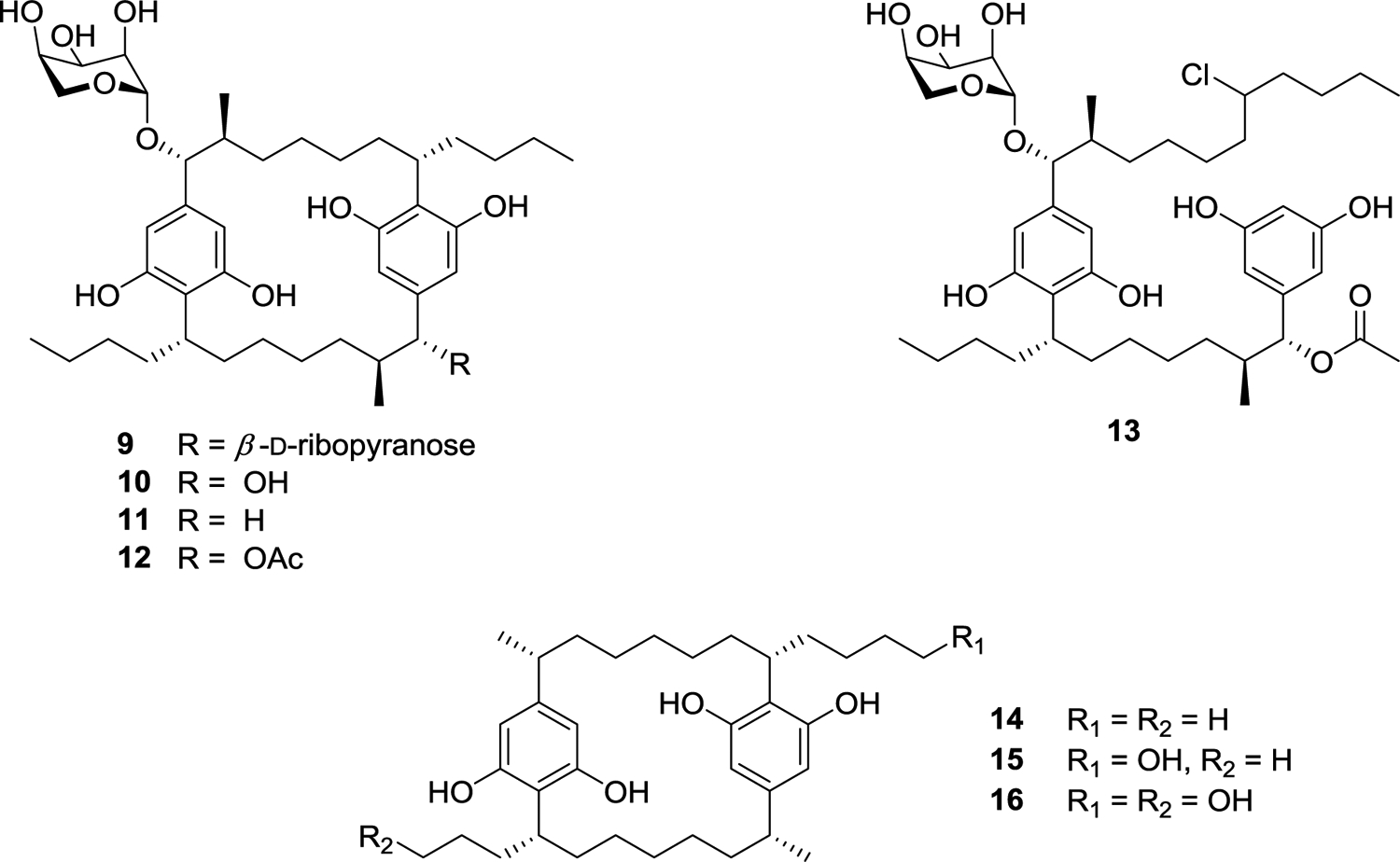
Another Nostoc sp. (UIC 10110) from our cyanobacterial library was found to produce a different set of [7,7]paracyclophanes, namely, merocyclophane A (14) as well as merocyclophanes C and D (15 and 16).58 The merocyclophane core structure is characterized by an α-branched methyl group at C-1/C-14. Merocyclophanes A, C, and D were evaluated for their antiproliferative activity against MDA-MB-231 (breast cancer) as well as other cancer cells and displayed IC50 values from 6.2 to 1.0 μM. A hollow fiber tumor assay was performed to evaluate the in vivo efficacy of the merocyclophanes. Merocyclophane C (15) was chosen for this assay due to its larger abundance in the cyanobacterial cell extract and its comparative ease of isolation. Hollow fibers containing cultured MDA-MB-231 cancer cells were inserted intraperitoneally (i.p.) in NCr nu/nu mice.34 The mice were treated with the merocyclophane C formulation for four days at 10 mg/kg or 15 mg/kg before being sacrificed. The hollow fibers were recovered, and the cancer cells were assessed for viability in an antiproliferation assay. No statistically significant growth inhibition was observed at either concentration; however, the MDA-MB-231 cancer cells showed inhibited growth with a p value of 0.051 at 15 mg/kg when compared to the negative control. This prompted a repeat of the experiment against the MDA-MB-231 cells at 17 mg/kg. However, during this laboratory work, the mice lost significant weight and the treatment proved too toxic, so the experiment was terminated.
The Orjala laboratory at UIC is the only group thus far to have reported the isolation of the merocyclophanes. This has provided a unique opportunity to identify the biosynthetic gene cluster that produces these compounds, and specifically to find a biosynthetic source for the α-methyl group, the major structural difference seen in the merocyclophanes as compared to other [7,7]paracyclophanes that all have a branched β-methyl group. To identify the biosynthetic origin of the α-methyl branching of the merocyclophanes, genomic DNA isolated from Nostoc sp. UIC 10110 was sequenced by Illumina MiSeq. The resulting assembly was analyzed by AntiSMASH 3.0.65 A gene cluster designated as a type I PKS-type III PKS hybrid by AntiSMASH, with close homology to the previously reported cylindrocyclophane and carbamidocyclophane gene clusters, was identified.64,66–69 This putative merocyclophane gene cluster contained many genes similar to cylindrocyclophane biosynthetic gene clusters. However, comparison of the gene clusters also indicated the genetic basis for the observed branching differences in the cylindrocyclophanes and the merocyclophanes. MerC (Type I PKS) installs a malonyl-CoA unit, performs a full reduction of the carbonyl group to the methylene, and installs the α-methyl group via a C-methyl transferase domain (Figure 3).
Figure 3.
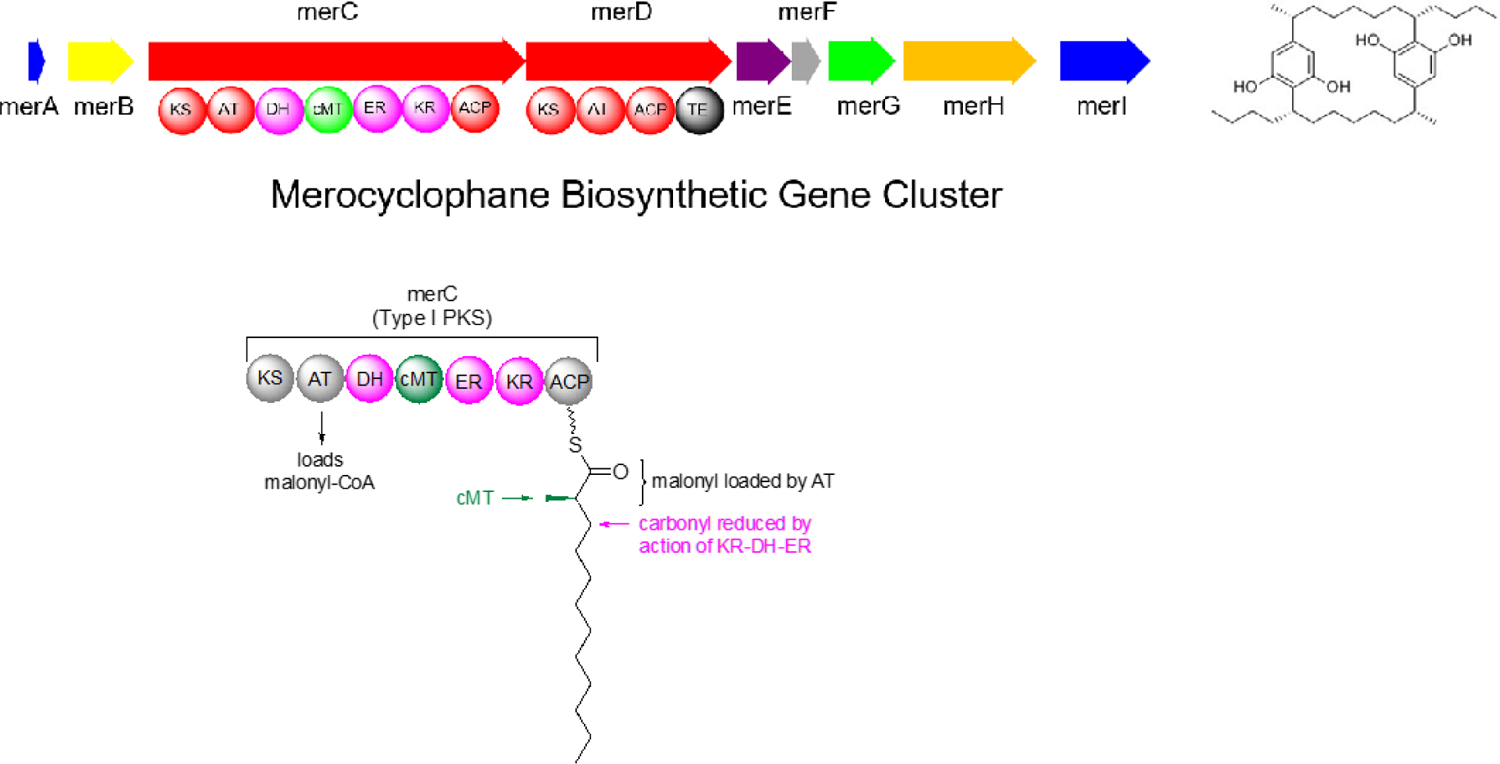
Merocyclophane biosynthetic gene cluster with zoom in on merC to show the proposed biosynthetic origin of the α-methyl group.
Phylogenetic analysis of [7,7]paracyclophane-producing strains using 16S rRNA identified three distinct clades that are known to produce [7,7]paracyclophanes: Nostoc group III, Nostoc group I, and Cylindrospermum (Figure 4).59 Interestingly, the two α-methyl group [7,7]paracyclophane core (merocyclophane) producers, UIC 10062 and UIC 10110, both clade in Nostoc group I, while the β-methyl group [7,7] paracyclophane cores (cylindrocyclophanes, carbamidocyclophanes, and ribocyclophanes) all clade in Nostoc group III, thus providing potential insight into the evolutionary relationships of different [7,7]paracyclophane-producing strains.
Figure 4.
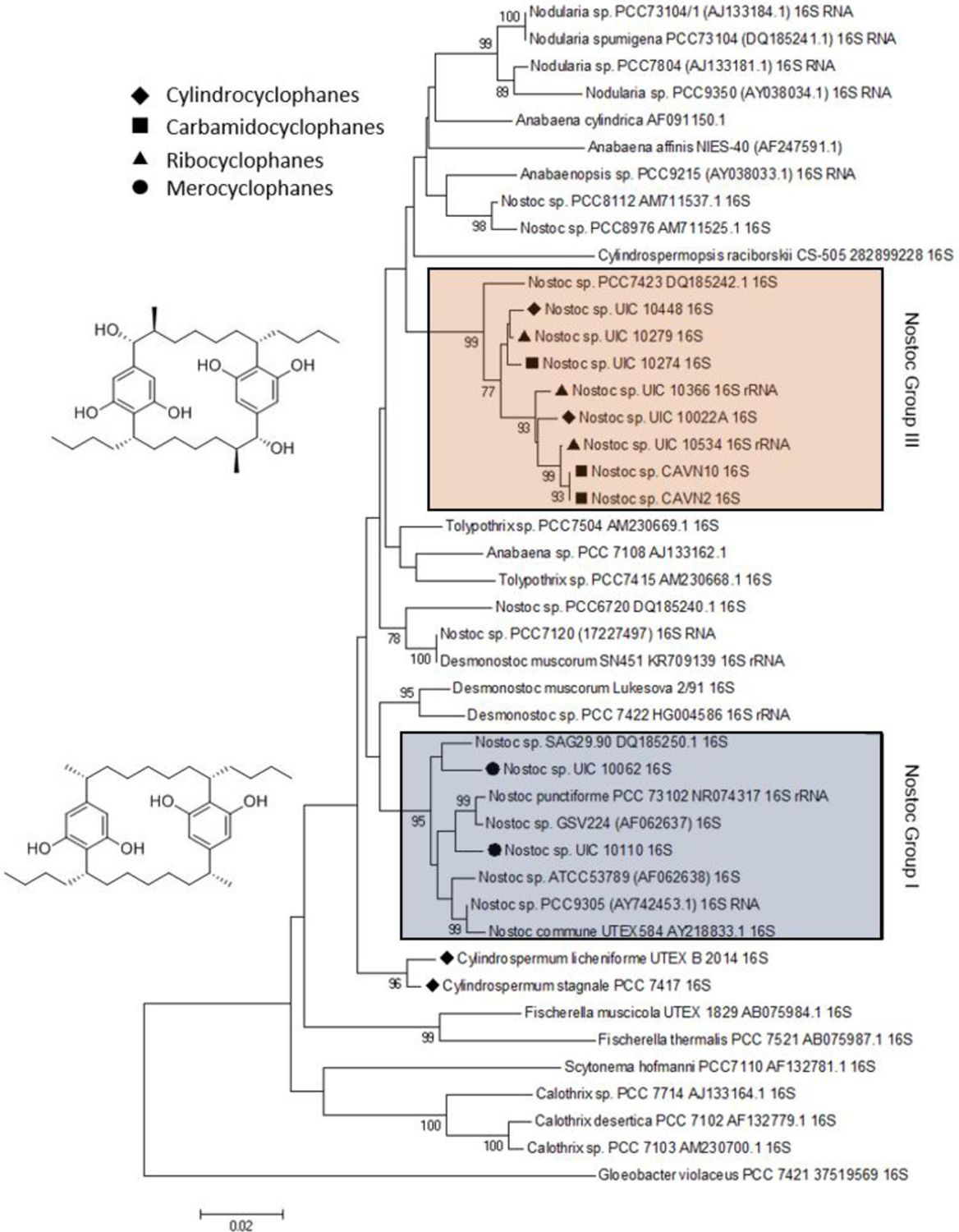
Taxonomic distribution of [7,7]paracyclophanes in cyanobacterial species.
As genome mining becomes a more widely used approach to identify bacterial natural products, the challenge of matching biosynthetic gene clusters to their cognate secondary metabolites has become more apparent. Bioinformatic platforms such as AntiSMASH have enabled great progress to be made in predicting chemical structures from genetic information. However, the predicted structures are often incomplete, which complicates identifying such predicted compounds by mass spectrometry. Secondary metabolites produced by cyanobacteria represent an excellent opportunity for bridging this gap. Cyanobacteria are known to produce biologically active metabolites and are able to encode numerous BGCs in their genomes.58,70–73 Cultured cyanobacteria incorporate inorganic nitrogen provided in chemically defined media into all nitrogen-containing secondary metabolites. Thus, stable isotope labeling with 15N labeled nitrate and subsequent comparative metabolomics can be used to match biosynthetic gene clusters to their cognate compounds in cell extracts (Figure 5).
Figure 5.
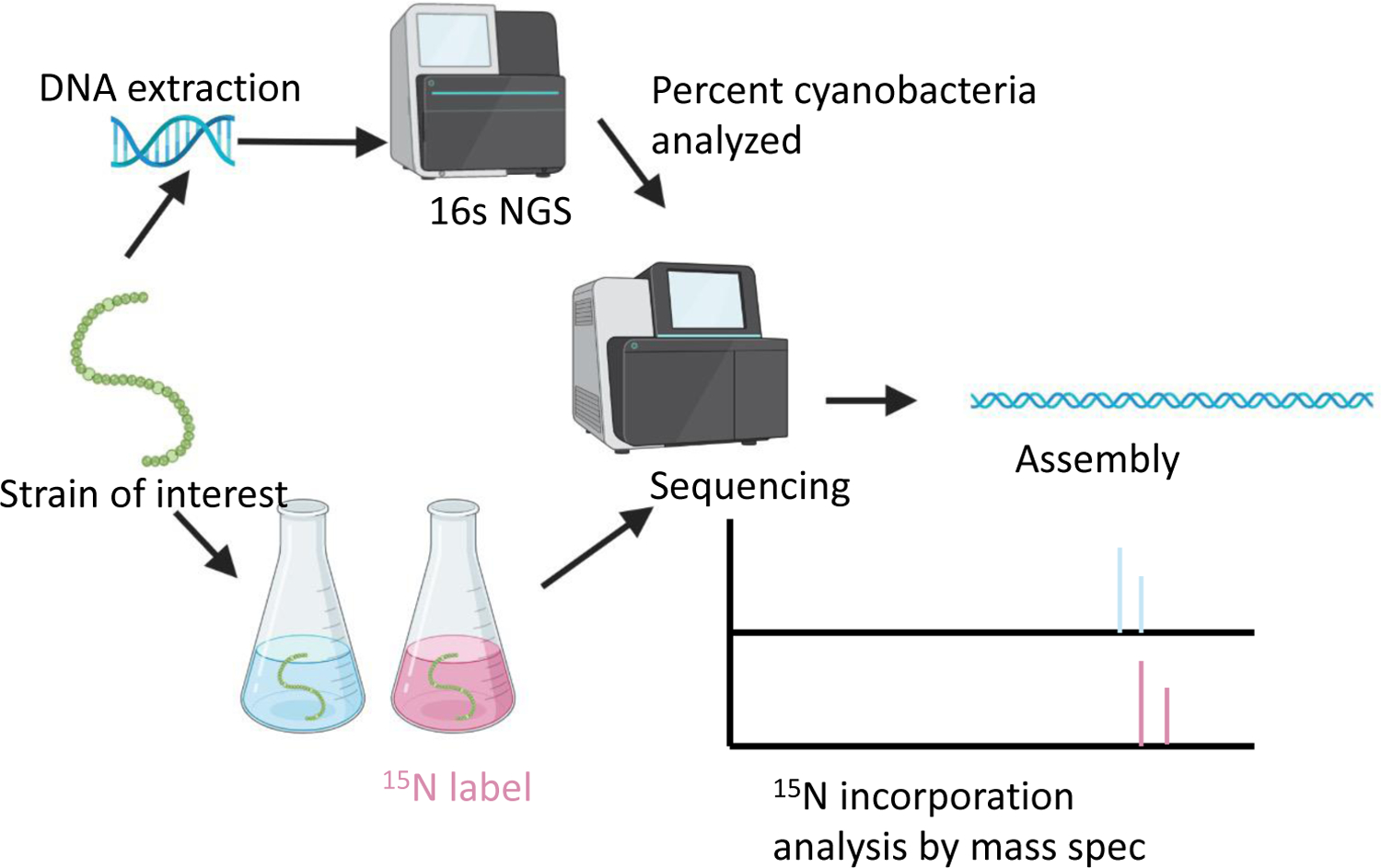
Diagram of the genome mining and comparative metabolomic approach used to match cyanobacterial BGCs to their respective natural products.
The feasibility of this approach was investigated using the cultured Nostoc sp. strain UIC 10630 from the UIC Cyanobacteria Culture Collection.74 AntiSMASH analysis of the sequenced genome was used to identify six biosynthetic gene clusters (BGCs) predicted to encode the production of secondary metabolites with at least one nitrogen atom (Table 1). Comparative metabolomic analysis of the 15N labeled and 14N unlabeled cell extracts revealed four nitrogen-containing compounds that contained the same number of nitrogen atoms as were predicted in four different biosynthetic gene clusters.74
Table 1.
List of Biosynthetic Gene Clusters (BCGs) Containing Nitrogen Identified by AntiSMASH on Cyanobacterial Acquisition UIC 10630 (Nostoc sp.)
| Cluster number | BGC type | % Similarity to known BGC | Adenylation domain predicted number of nitrogens | Genbank accession number |
|---|---|---|---|---|
| BGC4 | NRPS-PKS Hybrid | Nostopeptolide (100%) | 10 | MN701090 |
| BGC5 | NRPS | Anabaenopeptin (87%) | 7 | MN701092 |
| BGC6 | NRPS | Aeruginoside (41%) | 6 | MN701094 |
| BGC7 | NRPS-PKS Hybrid | None | 3 | MN701091 |
| BGC8 | NRPS-PKS Hybrid | None | 11 | MN701089 |
| BGC9 | NRPS-PKS Hybrid | None | 2 | MN701093 |
When using this approach, it was possible to verify the presence of the known biosynthetic gene cluster (BGC) for BGC4 and its product, nostopeptolide A1 (17), containing 10 nitrogen atoms.75 The compound containing six nitrogen atoms matched to BGC6, which had only a low similarity to the known biosynthetic gene cluster of anabaenopeptin.76,77 Analysis of the BGC6 and its product identified it to produce aeruginosin 865 (18), which had no reported BGC. The product of BGC5, containing seven nitrogen atoms, was identified as the novel anabaenopeptin UIC827 (19). The compound containing three nitrogen atoms matched to BGC7, which had no similarity to known BGCs. The structure was elucidated by NMR, HRESIMS, and MS/MS and identified as a novel compound, nostopyrrolidonamide (20). The structures of compounds 17-20 are shown in Figure 6. The Orjala group was unable to detect the products of BCGs 8 and 9 under the growth conditions used.
Figure 6.
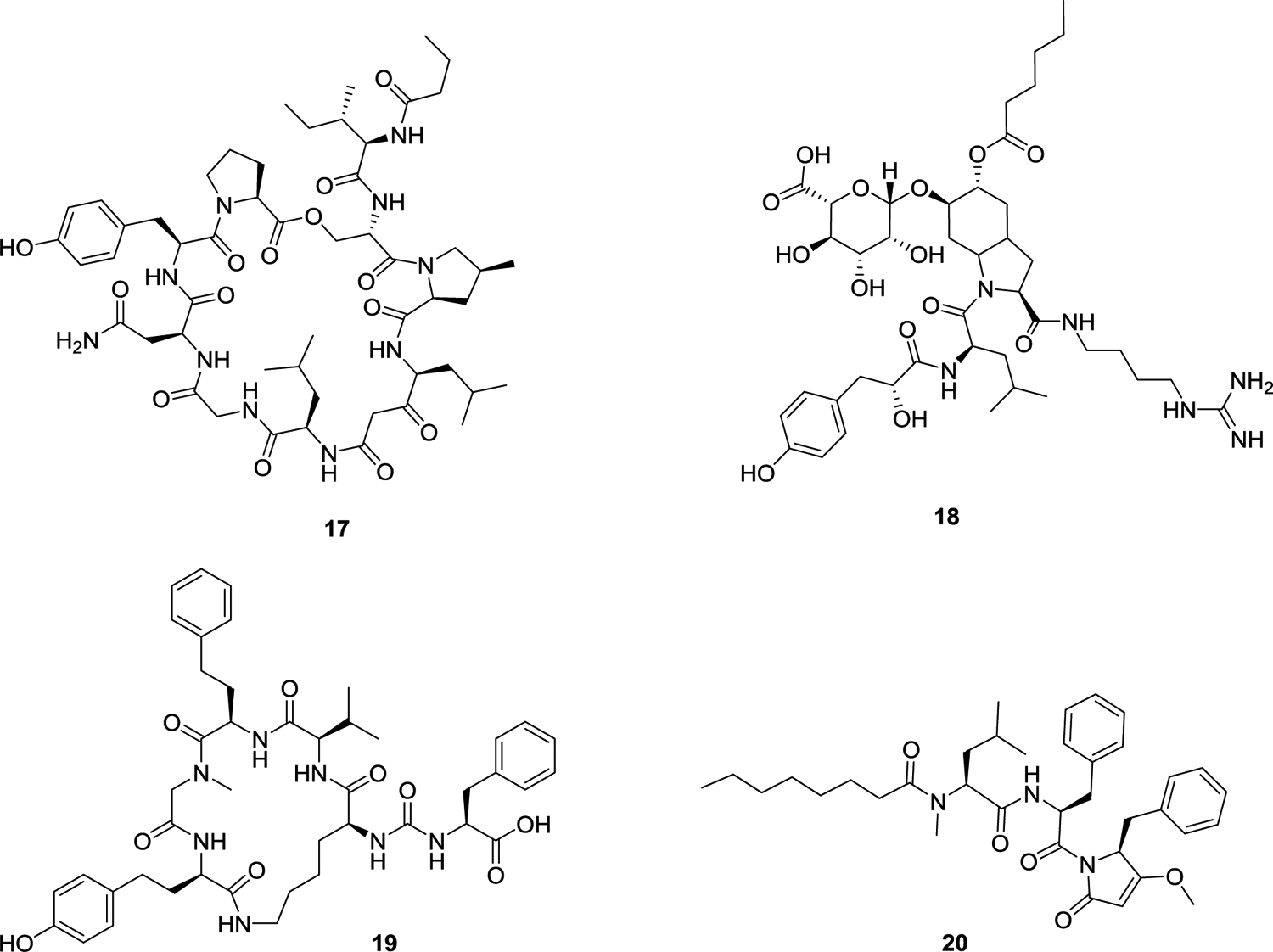
Cyanobacterial structures obtained as a result of genomic mining experiments.
Methodology using stable isotope labeling combined with comparative metabolomics has allowed the Orjala laboratory to match four of six nitrogen-containing BGCs to their respective compounds. This indicates that this approach can be useful to match cyanobacterial BGCs with their respective compounds in cell extracts and help facilitate genome-mining efforts in cultured cyanobacteria for future use.
Metabolites from Cultures of Filamentous Fungi.
The Oberlies group at UNCG has continued to study cytotoxic fungal metabolites from the Mycosynthetix library of filamentous fungi provided by Dr. Cedric Pearce. This consists of ca. 53,000 cultures that have been isolated from substrates collected from hundreds of sites, chosen to maximize the variety of ecosystems sampled and materials studied, as determined by field workers, typically botanists or mycologists. For this particular program project, a strategy designed to sample as great a variety of organisms as possible was developed; criteria included location of sample collection as well as the type of substrate investigated, and, since a historical collection is being investigated, previous data regarding biologically active metabolites produced by the fungus in question were also taken into consideration. This library has been studied since the 1980s for leads against a variety of targets (e.g., a novel herbicide is presently being evaluated for further development).78,79 However, over the last 15 years, a major focus has been on potential anticancer leads and our team has identified >625 fungal metabolites with cytotoxic activities against a variety of cancer endpoints, including about 150 compounds that were new to the literature.80–82
Fungi have been grown initially on a small scale (i.e., with a 250 mL Erlenmeyer flask) using solid substrates, particularly rice medium; this has been validated over years of in-house experience to be a consistent, inexpensive, all-around growth and production medium for the first stages of evaluation. Organic extracts which are generated via extracting with 1:1 chloroform-methanol followed by straightforward partitioning vs. H2O and hexane, are tested against human cancer cell lines to detect cytotoxic activity, which is defined as typically less than 30% survival of cancer cells when testing an extract at a concentration of 20 μg/mL. The “hit” rate, as a result of testing hundreds of fungal cultures per year, has averaged around 5% over the 15-year life of the project. However, over the past two years, where the group has both expanded the variety of locations from where the fungi were acquired and been more selective of what is considered a “hit” (i.e., using dereplication to a greater extent than previously), this rate has hovered closer to 2%. The Oberlies team has found that this recent “hit” rate, while lower, is more robust, allowing them to focus efforts on the most promising samples (i.e., those likely to lead to new chemical entities and/or potent biological activities). The active samples have been prioritized via a dereplication strategy that has evolved and expanded over the years. Essentially, it is an LC-MS based system that searches for matches in retention time, HRMS data, and MS-MS data vs. the growing database of isolated metabolites,83 and this includes the use of mass defect filtering in search of closely related analogues.84 A recent development incorporates the use of NMR data as an orthogonal prioritization procedure using the MADByTE platform.85,86 Importantly, one of the goals of the work is not only to eliminate samples where the cytotoxic activity can be ascribed to “nuisance” compounds (e.g., aflatoxins), but also to catalogue the variety of metabolites produced by each culture. There are times when this prioritization procedure can be used to select a suite of strains that biosynthesize the same compounds, an aspect that is beneficial when working on scale-up isolation studies, as needed to supply materials for in vivo evaluation of the best leads and/or to use as a starting point for semi-synthetic studies.87,88
For this review, work will be discussed on three different fungal metabolite groups (the verticillins, the resorcylic acid lactones, and the perylenequinones), with a focus on efforts to scale-up the production of these compounds and/or to generate analogues via either precursor-directed biosynthesis or via collaboration through supporting semi-synthetic medicinal chemistry studies (also see the Core 2 section of this review below).
Scaled-up Production of Verticillins and Generation of Fluorinated Analogues.
Verticillins, which are epipolythiodioxopiperazine (ETP) alkaloids, were first reported about 50 years ago,89,90 and display potent cytotoxicity with nanomolar IC50 values against a variety of cell lines.91 The goal in this digest is not to discuss their biological activity in detail; interested readers are referred to recent studies by Liu and colleagues92–94 and Burdette and associates,95 which, as discussed in the “Biological Testing and Mechanism of Action Core” section below, have shown these compounds to block histone methyltransferases. In addition, semi-synthetic studies on the verticillins are discussed in the Core 2 section of the work later in this review.
To scale up the production of these molecules, a variety of fungal cultures were sampled that were identified as biosynthesizing the verticillins via dereplication. Indeed, this is seen as a distinct advantage of working with a library of >53,000 fungal strains, where fungi from various locations can be examined to see which ones are the best for producing a particular molecule of interest. Over the past decade, more than 12 strains have been identified, isolated from a variety of sources, that all biosynthesize verticillin derivatives to a greater or lesser capacity.87 The majority of these strains were Clonostachys spp. (Ascomycota, Hypocreales, Bionectriaceae) and one, strain MSX71844, was identified as Purpureocillium lavendulum (Ascomycota, Hypocreales, Ophiocordycipitaceae). Taxonomic analysis of these96 showed that many strains had phylogenetic affinities to C. rogersoniana, the fungus from which the biosynthetic gene cluster for verticillin biosynthesis was recently reported.97
Traditional industrial approaches to medium optimization studies to enhance the production of fungal metabolites, especially in western countries, have often focused on submerged liquid cultures. However, there is evidence in the literature that solid-state fermentations of fungi can be superior, at least on a laboratory scale.98,99 Our team has spent a considerable effort in optimizing the droplet probe method for the chemical analysis of fungal cultures in situ, as reviewed recently.100 With this tool, the productivity of such solid-state fermentations can be monitored via coupling to UPLC-PDA-HRMS-MS/MS,101 essentially facilitating the use of the aforementioned dereplication/prioritization protocols83,84 on samples drawn directly from a Petri dish, where the fungi can be grown readily on many different media. Using this approach, it was determined that each of the strains that produces various verticillins has a unique biosynthetic profile, likely reflecting subtle differences in their non-ribosomal peptide synthesis biosynthetic pathways. The strains producing the highest titers were MSX59553 and MSX79542 (both of which were identified as C. rogersoniana), and, from extensive optimization studies, it was determined that both rice and oatmeal media gave good yields of the desired products, with the latter supporting faster growth and the highest titer of verticillin A (23) (Figure 7).87 From these studies, an approach has been developed to optimize the production of the verticillins, staging scale-up cultures for growth and processing approximately every two weeks. While over 500 mg of verticillin A (23) were generated in 2021, it is recognized that liquid tank cultures may be needed for possible future preclinical work.
Figure 7.
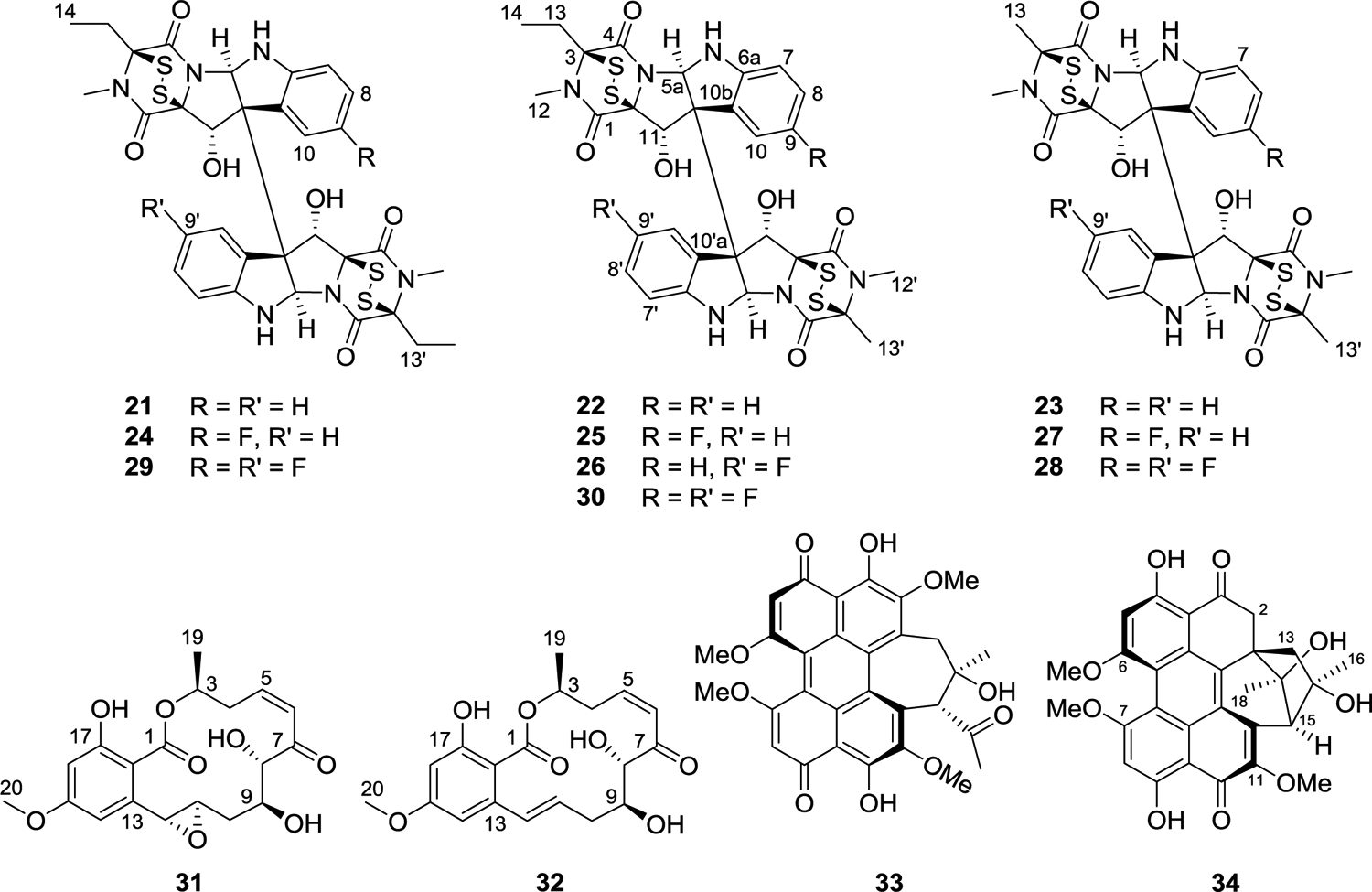
Structures of verticillins (21-30), resorcylic acid lactones (31 and 32), and perylenequinones (33 and 34) obtained from filamentous fungi.
Developing a patent position on isolated natural products is a distinct challenge,102 especially for compounds first characterized structurally decades ago. As such, efforts have been made to generate analogues of natural products, using both a semi-synthetic approach (outlined in the Core 2 medicinal chemistry section below103) and a precursor-directed biosynthetic approach. In particular, the introduction of a fluorine atom is a common strategy for drug development, as ~25% of all FDA-approved drugs include at least one fluorine atom,104–106 and fluorination at key positions in a molecule often enhances pharmacokinetic properties, particularly those associated with selected metabolically modified derivatives of a lead compound.107 Using precursor-directed biosynthesis, a technique that has been exploited throughout the history of fungal products,108 by incubating verticillin-producing fungi with fluorinated Trp, seven fluorinated analogues of verticillins were generated, essentially via the incorporation of either one or two fluorinated-Trp building blocks into a variety of verticillin backbones [i.e., verticillin H (21), verticillin A (23), or Sch52901 (22) (Figure 7)]. This is an additional example where the droplet probe method100 was advantageous for profiling a suite of growth conditions on Petri dishes. The growth conditions that produced compounds where incorporation of fluorine was evident (i.e., mass shifts of 17.99 Da for monofluorination or 35.98 Da for difluorination) were then transferred from the Petri dish to replicates of solid phase cultures grown on 10 g of oatmeal in 250 mL Erlenmeyer flasks, similar to commonly used procedures, but with the addition of 500 ppm of fluorinated Trp to the growth medium, and the desired products were isolated and characterized. The following new verticillins were produced: 9-F-verticillin H (24), 9-F-Sch 52901 (25), 9′-F-Sch 52901 (26), 9-F-verticillin A (27), 9,9′-diF-verticillin A (28), 9,9′-diF-verticilin H (29), and 9,9′-diF-Sch 52901 (30) (Figure 7). All of these compounds showed nanomolar activity against a panel of cancer cell lines, with 9-F-verticillin H (24) being approximately twice as active as the non-fluorinated natural product.
Z-Enone Resorcylic Acid Lactones.
During the course of this program project, the work in Project 3 has led to the discovery of a number of fungi that produce resorcylic acid lactones with a Z-enone moiety.109,110 This type of fungal metabolites often displays potent activity in cancer cell growth inhibition assays.111–113 In addition, these compounds are of interest due to their irreversible inhibition of oncogenic protein kinases through the formation of stable Michael addition products with the ATP-binding pocket cysteine residues,114,115 and examples of leads include hypothemycin (31) and (5Z)-7-oxozeanol (32) (Figure 7), with the latter being first reported by George Ellestad’s group at Lederle Laboratories in 1978.116
One of the goals of the work on fungi is to generate sufficient materials for both pharmacological evaluation and medicinal chemistry studies, in order to explore ways to improve the properties of potential lead compounds. An early focus was to optimize the production and isolation of the resorcylic acid lactones. For example, an initial study that generated semisynthetic analogues of the resorcylic acid lactones suggested some promise for enhancing activity against transforming growth factor-β-activated kinase 1 (TAK1),117 particularly when the Z-enone moiety was preserved across positions 5 to 7. A larger supply of these compounds was necessary as starting materials for further SAR studies via semisynthesis, and thus, upon evaluating extracts from 536 fungi from the Mycosynthetix library using our dereplication procedures,83,84 three promising isolates were identified.88 These strains were evaluated taxonomically,96 where one strain (MSX45109) was identified as Setophoma terrestris, and the other two (i.e., MSX63935 and MSX78495) were identified as Setophoma spp. (Ascomycota, Pleosporales, Phaeosphaeriaceae). These fungi were isolated from leaf litter collected at a mangrove swamp, an agricultural farm, and a semi-humid gallery forest, respectively. From a preliminary media study, it was shown that rice medium led to increased production of (5Z)-7-oxozeanol (32) by strain MSX63935, and oatmeal medium supported enhanced production of hypothemycin (31) by strain MSX78495. There are only slight differences in these two molecules (i.e., a double bond in the former vs. an epoxide in the latter across the 10 and 11 positions), yet their biosynthesis via these organisms was remarkably different. Additionally, when isolating such molecules from scaled cultures, the Oberlies group was able to circumvent the use of HPLC by precipitating these compounds from supersaturated solutions via centrifugation, leading to the rapid and efficient isolation of these molecules on a multi-gram scale; these materials are now serving as starting materials for the generation of more than 30 semi-synthetic analogues (manuscript in preparation). An additional benefit of scaling the isolation of fungal metabolites is the uncovering of minor constituents, such as 10 other resorcylic acid lactones, two radicinin analogues, and six benzopyranones, with two of the latter being new chlorinated derivatives.88
Photoactivated Perlyenequinones.
Hypocrellins (i.e., perylenequinones) were isolated initially from Hypocrella bambusae, Shiraia bambusicola and other Shiraia-like fungi.118–121 These molecules are interesting structurally, as the high level of conjugation imparts deep red and yellow colors. Unfortunately, their nomenclature, like that of many natural products, is somewhat confusing, and a recent study by the Oberlies group on these compounds has attempted to ameliorate this situation (see Figure S1 in Ref.122). Nevertheless, when first working on the bioactivity-directed fraction of a Shiraia-like species (i.e., strain MSX60519) that biosynthesized perylenequinones (Figure 7), only a few mg of a new compound, ent-shraiachrome A (33), and the known hypocrellin were isolated, and a larger supply of both was needed for further pharmacological studies (vide infra). As such, a medium optimization study was implemented, and due to the light-absorbing properties of the perylenequinones, it was decided to explore a three-by-three matrix of growth conditions, where both media types were varied (i.e., rice, oatmeal and “Cheerios”), in the presence or absence of light (i.e., growth in the dark vs. with natural light vs. white LED light).122
While the Oberlies group has studied fungal medium optimization for a number of years,79,87,88 this was the first time that varied growth conditions along two axes was observed, leading to interesting results, where it was possible to tune the production of three different types of molecules. For instance, the hypocrellins were produced in the highest yield (i.e., readily on the order of hundreds of milligrams) when growing the fungus on rice medium and either putting it through a 12-h light/dark cycle with natural light or by growing it under 24 h exposure to white LED light. Alternatively, a series of closely related molecules, the hypomycins, whereby some of the conjugation observed in the hypocrellins has been lost due to the disruption of aromaticity in ring A, were generated preferentially by the growth of the same fungal strain using oatmeal media under 24-h exposure to white LED light. This resulted in the biosynthesis of the two known compounds, hypomycins A and C, and the new compound, hypomycin E (34); this is the first report of both hypocrellins and hypomycins being isolated from the same fungus. Importantly, an addendum that adjusted how the hypomycins were drawn was also published,122 where the stereodescriptors at the C-17 position can be misconstrued if the tertiary hydroxy moiety is drawn differently. In addition, a recent study that examined the redox behavior of these perylenequinones demonstrated that hypomycins likely originate from the anaerobic reduction of hypocrellins, and this served to further refine the absolute configurations of hypomycin C and E (34).123 Finally, a follow-up manuscript was also published, noting that the biosynthesis of thielavins from this fungus could also be tuned using this approach, where their production was highest when using rice medium and a 12-h light/dark cycle with natural light.88 Interested readers may note the use of a LR-HSQMBC NMR experiment124 to refine the structure elucidation of thielavins.
Synthesis and Structural Optimization of Lead Compounds, and Preliminary Pharmacokinetics-related Studies.
Several of the natural products isolated as a part of this program project work represent potential lead compounds for the development of cancer chemotherapeutic agents. Accordingly, compounds identified through these collaborative laboratory studies are prioritized based on their structural novelty, potency in cell-based assays, and, for some natural products, their mechanism of action. In many cases, however, even the most promising of the compounds isolated may be limited in abundance or by their inherent drug-like properties. To address these limitations, medicinal chemistry and preliminary pharmacokinetics work in Core 2 of the program project, headed by Dr. James R. Fuchs at the College of Pharmacy, OSU, can be initiated to synthesize sufficient quantities of compounds for subsequent biological evaluation and to optimize the pharmacological and pharmacokinetic properties of these molecules through systematic structural modification and exploration of structure-activity relationships. In this sub-section of the review, to exemplify this process, highlighted are three recent examples of our medicinal chemistry applications, which are focused on lead compounds isolated from a tropical plant, a cyanobacterial species, and a filamentous fungus, respectively.
Several members of the phyllanthusmin class of natural products, including phyllanthusmins C and D (35 and 36, Figure 8), were isolated from the plant Phyllanthus poilanei.125 Based on the potent inhibitory activities of these arylnapthalene lignan lactone compounds against HT-29 colorectal cancer cells (e.g., for 36, IC50 = 170 nM), our program project team became interested in exploring the role of the various functional group motifs present in these molecules, in order to optimize the physicochemical properties of this class and to investigate their potential mechanism of action. Thus, synthetic analogues have been generated using a facile synthetic scheme and it was shown that their potency is dependent not only upon the diphyllin core of these molecules, but also the functionalization of the glycone unit present.126 These studies examined the introduction and substitution of various sugar moieties, ultimately leading to the preparation of PHY-34 (37),126 in which the arabinose ring found in both 35 and 36 was replaced with a functionalized galactose ring. PHY-34 is one of the most potent analogues of this series and has been shown to have in vivo activity in a murine xenograft model using OVCAR8 cells. Solubility, maximal tolerated dose (MTD) testing, and bioavailability studies on PHY-34 were carried out in Core 2 at Ohio State University by Drs. Mitch Phelps and Chris Coss.127 As mentioned in more detail in the next section of this review on biological testing, PHY-34 is a highly potent inhibitor of the ATP6V0A2 subunit of vacuolar-ATPase, a proton pump linked to cancer cell proliferation, metastasis, and the process of autophagy. In addition, it has been shown also that PHY-34 interacts with the cellular apoptosis susceptibility (CAS/CSE1L) protein, a protein involved with the nuclear export of α-importins.128
Figure 8.
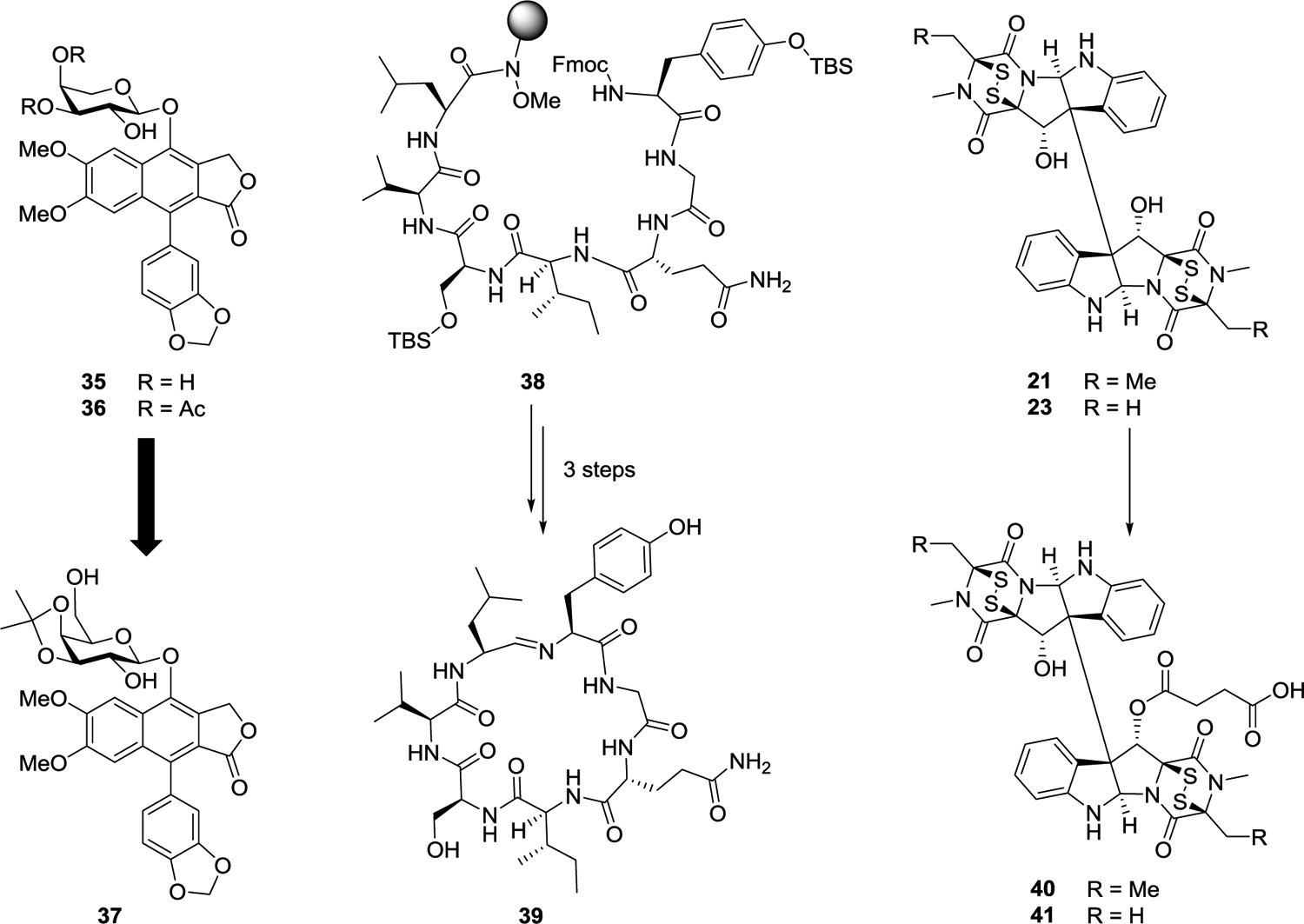
Representative examples of molecules selected for medicinal chemistry studies, including phyllanthusmins C and D (35 and 36), PHY-34 (37), a precursor and scytonemide A (38-39), and two verticillins (21 and 23) and their succinate derivatives (40 and 41).
The natural product scytonemide A (39) was reported by the Orjala laboratory at UIC (Project 2) in 2010.129 This cyclic heptapeptide was isolated from the freshwater cyanobacterium Scytonema hofmannii (UTEX 1834) and found to act as an inhibitor of 20S proteasome chymotrypsin catalytic activity (IC50 96 nM). Further evaluation of this compound in our research program was limited by the relatively low quantities that could be isolated from the slow-growing cyanobacterial species of origin. For this reason, a total synthesis of scytonemide A was initiated at OSU. The primary challenge for the synthesis of this compound proved to be the unusual imine linkage present in the macrocycle. To address this issue, the solid-phase peptide synthesis of the acyclic precursor 38 was carried out using the Weinreb AM resin, which was expected to furnish a C-terminal aldehyde upon reduction.130 After deprotection of the Fmoc group from the N-terminal tyrosine residue of 38, reduction of the Weinreb amide functionality facilitated direct cyclization of this aldehyde with the tyrosine amine to form the cyclic imine. At that stage, all that remained to complete the synthesis of scytonemide A (39) was a mild deprotection under non-acidic conditions. Optimization of normal-phase column chromatography conditions has subsequently facilitated the generation of the compound on a significantly higher scale than was possible through direct isolation.130
The epipolythiodioxopiperazine (ETP) alkaloid, verticillin H (21), was included in the most recent comprehensive review of the technical progress of our program project work.14 This complex dimeric compound was isolated from the fungal strain MSX64546, and represents a new analogue of the structurally similar verticillin A (23), a compound for which the antiproliferative effects have previously been studied in the laboratories of both our collaborators and in our own.91–95 Similar to scytonemide A (39), however, biological studies on the verticillin derivatives have been limited due to the availability of only small quantities of the purified natural products. This limitation was overcome through screening and optimization of growth conditions with multiple fungal strains shown to produce verticillin A and its analogues,87 as discussed earlier in the present review. The increased supplies thus generated of both verticillin A and verticillin H have made semi-synthetic modification of these compounds feasible (Figure 8). Using a strategy that combined both medicinal chemistry and natural products isolation, a series of ten acylated or sulfonylated derivatives was generated primarily through functionalization of the C-11 alcohol.103 Interestingly, although the compounds are dimeric, under all of the conditions examined only one of the alcohols was found to react. Both verticillin H (40) and verticillin A (41) hemisuccinates were found to show improved potency in a variety of cancer cell lines relative to the parent compounds and were also predicted to show increased water solubility.103
Biological Testing and Mechanism of Action Core.
In addition to primary screening of extracts against a small panel of cancer cell line, in vivo biological testing and mechanism of action studies have continued to be undertaken on selected pure compounds at our biological core faculty housed at UIC (Core 1), headed by Dr. Joanna E. Burdette, as described earlier.14 Currently, human cancer cell lines utilized in the initial screening procedure include HT-29 colon, MDA-MD-231 breast, MDA-MD-435 melanoma, and OVCAR-3 ovarian.31,35 In the following paragraphs, specific information is provided on follow-up biological and mechanistic aspects of an example each from a tropical plant [(+)-strebloside (4) (Figure 1)], a fungal metabolite [verticillin A (23) (Figure 7)], and a synthetic derivative [PHY-34 (37) (Figure 8)].
(+)-Strebloside (4).
(+)-Strebloside was isolated from Streblus asper in Project 1 and is a known cardiac glycoside.31,35 Cardiac glycosides are used in the treatment of heart failure, and because so many patients take this class of medication, epidemiological data have been used to reveal that they lower the incidence for certain cancers. For example, lower incidences of breast cancer and leukemia have been reported in patient populations taking cardiac glycosides.131 Inhibition of Na+/K+-ATPase is known to alter cardiac cells, but these pumps also impact cellular signaling that has implications in cancer therapy such as mutant p53 synthesis, ERK activation, and EGFR signaling. Initial studies using computer-aided molecular docking suggested that (+)-strebloside bound to Na+/K+-ATPase in a similar binding mode to other cardiac glycosides, such as digitoxin. ATPase activity assays confirmed that (+)-strebloside was roughly equal to digitoxin in its ability to inhibit the enzyme. The compound was able to significantly reduce cell viability of multiple human ovarian cancer cell lines in vitro, but was not effective against murine cell lines, which is expected as the mouse and human isoform expressions differ. Cell cycle analysis revealed that (+)-strebloside caused a G2 arrest, and the amount of p21 was significantly increased based on western blots. (+)-Strebloside was able to trigger apoptosis based on cleaved PARP and cleaved caspase-3. Two anti-apoptotic proteins, BCL2 and Mcl-1, were both reduced in response to (+)-strebloside treatment in OVCAR3 cells. Since p53 mutation occurs in almost all high grade serous tumors, and cardiac glycosides have been reported to reduce the synthesis of mutant p53, OVCAR3 cells were treated with (+)-strebloside and p53 expression was monitored. Also, pERK activation downstream of the Na+/K+-ATPase was confirmed as a mechanism for p53 degradation. Finally, (+)-strebloside was confirmed to block NFκB and did not interact with the hERG channel. Even though chemical modifications of cardiac glycosides may be able to overcome their side effects associated with established drugs, our data found that (+)-strebloside is unlikely to provide superior safety in cancer therapy due to its similar binding site and the intracellular transduction when compared to digitoxin.35
Verticillin A (23).
As mentioned earlier, the verticillins are epipolythiodioxopiperazine (ETP) alkaloids and are typically isolated from fungi.91 Of all the verticillin compounds isolated, verticillin A (23) (Figure 7) has been the most extensively studied for its potential anticancer activity. It has been evaluated in a variety of tumor types and was demonstrated to be a histone methyltransferase inhibitor that triggered apoptosis and sensitized pancreatic tumors and colon cancer to 5-fluorouracil.92 In our program project work, we focused on its role in ovarian cancer.95 Verticillin A was cytotoxic based on 2D foci assays and was able to decrease the viability of 3D spheroids of OVCAR8. It was confirmed that histone marks were altered in response to verticillin A, which is consistent with blocking histone methyltransferases. Verticillin A was equally toxic against ovarian cancer cell lines and non-tumorigenic fallopian tube epithelial cells. Therefore, when testing in vivo, verticillin A was encapsulated into an expansile nanoparticle to improve efficacy by allowing the drug to be released specifically in acidic microenvironments, such as the OVCAR8 ovarian xenograft. RNA sequencing of OVCAR8 cells treated with verticillin A uncovered that apoptosis and oxidative stress were the two major pathways modified. Interestingly, WNT signaling and cadherins were two major pathways that were repressed. Verticillin A did cause a significant increase in reactive oxygen species formation as illustrated in a cell-based DCFDA reporter assay. The formation of reactive oxygen species resulted in DNA damage in a COMET assay. The free radical scavenging agent, N-acetylcysteine, was able to reverse verticillin A-mediated reactive oxygen species, DNA damage, and cell death, although a detailed mechanism for these observations was not determined.95 Overall, the current goal is to increase the solubility of verticillin A (23) through synthetic medicinal chemistry approaches outlined earlier in the review.
The Synthetic Phyllanthusmin Derivative, PHY-34.
Following the isolation of the promising plant-derived natural products phyllanthusmins C (35) and D (36) (Figure 8),125 medicinal chemistry efforts in the P01 project have generated more than 75 synthetic analogues of the “PHY” series.126 To date, our most promising lead is PHY-34 (37) (Figure 8) and has nM potency when evaluated against numerous cancer cell lines.127 Initial studies revealed that PHY-34 was able to induce apoptosis in a dose- dependent manner. The structure of the compound, due to its diphyllin ring system, was initially thought to resemble etoposide, but several experiments conducted by Dr. Jack Yalowich at OSU demonstrated that the PHY derivatives do not bind topoisomerase and are able to induce cell death in etoposide-resistant cell models.126 However, diphyllin moieties are also associated with autophagy inhibition.132 Using cell-based reporter assays, it was discovered that PHY-34 acts as a late-stage autophagy inhibitor. When autophagy activators were combined with PHY-34 they were able to block apoptosis indicating that the inhibition of autophagy was necessary for the cytotoxic effect of the compound. In both hollow fiber assays and in OVCAR8 xenografts, PHY-34 was highly effective and reduced tumor burden significantly. No overt signs of toxicity were noted. Pharmacokinetics illustrated that the compound was bioavailable orally, subcutaneously, and intraperitoneally after administration.127
In order to find a cellular target, PHY-34 (37) was immobilized to beads using photoaffinity chemistry and lysates from OVCAR3 and OVCAR8 were incubated with the beads. Proteomics indicated that one of the proteins that interacted with PHY-34 was called CAS1 (XPO2/CSE1L).128 In turn, cas1 is part of the nuclear/cytoplasmic transport family and knockdown of this gene in cancer cells induces apoptosis. The expression of cas1 is correlated with stage and grade of high grade serous ovarian cancer based on RNA expression data from tumor databases, and a tissue microarray confirmed the protein was abundant in ovarian cancers.128 However, knockdown of cas1 did not eliminate the efficacy of PHY-34 and surface plasmon resonance suggested that the binding was in the micromolar range, when the toxicity was in the nanomolar range. A similar structure was published by Novartis in which it was shown that their compound interacted with the V0A2 subunit of the V-ATPase,133 which acidifies the lysosome and when inhibited blocks autophagy. Thus, the interaction with V0A2 explained how PHY-34 could induce late-stage autophagy inhibition. Indeed, mutants of the V0A2 subunit were resistant to PHY-34 and this helped to map not only the likely cellular target but the amino acids required on the subunit. Overall, PHY-34 has proven to be a highly potent compound with an unusual mode of action.128 There are currently no clinically approved autophagy inhibitors except hydroxychloroquine. PHY-34 would be unique based on its mechanism of action by blocking the V0A2 subunit and therefore reducing lysosomal acidification, as demonstrated using LysoTracker and acridine orange, which are both less fluorescent when the lysosome is no longer acidic. V0A2 is overexpressed in high grade serous ovarian cancer and is associated with resistance to cisplatin and related drug and metastasis.134 Therefore, PHY-34 may provide a promising chemical probe to understand the importance of V0A2 activity in ovarian cancer and should be tested for its ability to impact platinum-resistant cells.
CONCLUSIONS
The technical progress made in this program project over the last five or six years has been possible from the concerted chemical and biological work on diverse organism groups (tropical plants, U.S. lichens and their mycobionts, aquatic and terrestrial cyanobacteria, and filamentous fungi), directed towards the discovery of variety of new potential anticancer agents. Research progress in our program project has benefited not only from the frequent interactions between the components in our multidisciplinary team, but also from the valuable input of several external collaborators. Efforts have been made to investigate more intensively various lead compounds through their scale-up production and via preliminary structure-activity-relationship studies and more detailed mechanistic investigations. In addition, a large number of collaborative scientific publications (ca. 200 research and review articles) have resulted from this program work since its beginning in 2007, and over 30 Ph.D. degrees have been awarded across several different disciplines to graduate students from OSU, UIC, and UNCG.
ACKNOWLEDGMENTS
Recently, our program project group has lost two outstanding and inspirational colleagues who constantly encouraged our research initiatives. Dr. Mansukh C. Wani (Research Triangle Institute, Research Triangle Park; 1925–2020) was a senior investigator associated with Project 3 at UNCG, and he regularly attended our group meetings. Dr. G. Robert Pettit (Arizona State University) served as a member of our External Advisory Committee, and advised specifically on enhancing chemical synthesis procedures in an expert manner. The laboratory work covered in this review was supported by grants P01 CA125066 and the supplement 3P01CA125066-10S1, funded by the National Cancer Institute, NIH, Bethesda, MD, USA. We are grateful to our taxonomic collaborators in Indonesia, Vietnam and Laos for their kind cooperation concerning the plant collections. Dr. Richard W. Spjut, World Botanical Associates, Bakersfield, CA is acknowledged for the collection of taxonomically identified lichens from coastal areas of the United States, and Dr. Chad Rappleye, Department of Microbiology, Ohio State University, for the identification of fungi associated with the collected lichens. In addition, we wish to thank many current and former faculty and staff colleagues, postdoctoral associations, and graduate and undergraduate students who have participated in this multidisciplinary project, and whose names are included in the biography below.
Footnotes
DEDICATION
Dedicated to Dr. William H. Gerwick, University of California at San Diego, for his pioneering work on bioactive natural products.
B.R.S. is an inventor on patents and patent applications involving small-molecule drug discovery and ferroptosis, and co-founded and serves as a consultant to Inzen Therapeutics, Nevrox Limited, Exarta Therapeutics, and ProJenX, Inc., and serves as a consultant to Weatherwax Biotechnologies Corporation, and Akin Gump Strauss Hauer & Feld LLP. The other authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL; Miller KD; Fuchs HE; Jemal A CA Cancer J. Clin 2021, 71, 7–33. [DOI] [PubMed] [Google Scholar]
- (2).Sung H; Ferlay J; Siegel RL; Laversanne M; Soejomataram I; Jemal A; Bray F CA Cancer J. Clin 2021, 71, 209–249. [DOI] [PubMed] [Google Scholar]
- (3).Newman DJ; Cragg GM J. Nat. Prod 2020, 83, 770–803. [DOI] [PubMed] [Google Scholar]
- (4).Kasamon YL; Ko CW; Subramaniam S; Ma L; Yang Y; Nie L; Shord S; Przepiorka D; Farrell AT; McKee AE; Pazdur R Oncologist 2018, 23, 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Singh S; Jaigirdar FM; Cheng J; Hamed SS; Li Y; Liu J; Zhao H; Goheer A; Helms WS; Wang X; Agarwal R; Pragani R; Korsah J, Tang S; Leighton J; Rahman A; Beaver JA; Padzur R; Theoret MR; Singh H Clin. Cancer Res 2021, 27, 2378–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schofield MM; Jain S; Porat D; Dick GJ; Sherman DH Environ. Microbiol 2015, 17, 3964–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Agarwal G; Blanco Carcache PJ; Addo EM; Kinghorn AD Biotechnol. Adv 2020, 38, 107337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Newman DJ; Cragg GM Planta Med. 2020, 86, 891–905. [DOI] [PubMed] [Google Scholar]
- (9).Daley S-K; Cordell GA J. Nat. Prod 2021, 84, 871–897. [DOI] [PubMed] [Google Scholar]
- (10).Newman DJ J. Nat. Prod 2021, 84, 917–931. [DOI] [PubMed] [Google Scholar]
- (11).Singh SB J. Nat. Prod 2022, 85, 10.1021/acs.jnatprod.1c01135. [DOI] [PubMed] [Google Scholar]
- (12).Nakada T; Sugihara K; Jokoh T; Abe Y; Agatsuma T Chem. Pharm. Bull 2019, 67, 173–185. [DOI] [PubMed] [Google Scholar]
- (13).Kinghorn AD; Carcache-Blanco EJ; Chai H-B; Orjala J; Farnsworth NR; Soejarto DD; Oberlies NH; Wani MC; Kroll DJ; Pearce CJ; Swanson SM; Kramer RA; Rose WC; Fairchild CR; Vite GD; Emanuel S; Jarjoura D; Cope FO Pure Appl. Chem 2009, 81, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kinghorn AD; Carcache de Blanco EJ; Lucas DM; Rakotondraibe HL; Orjala J; Soejarto DD; Oberlies NH; Pearce CJ; Wani MC; Stockwell BR; Burdette JE; Swanson SM; Fuchs JR; Phelps MA; Xu L-H; Zhang X; Shen YY Anticancer Res. 2016, 36, 5623–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Henkin JM; Ren Y; Soejarto DD; Kinghorn AD In Progress in the Chemistry of Organic Natural Products; Kinghorn AD; Falk H; Gibbons S; Kobayashi J; Asakawa Y; Liu J-K; Springer International Publishing AG, Cham, Switzerland, 2018; vol. 107, pp 1–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ren Y, Carcache de Blanco EJ; Fuchs JR; Soejarto DD; Burdette JE; Swanson SM; Kinghorn AD J. Nat. Prod 2019, 82, 657–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pan L; Woodard JL; Lucas DM; Fuchs JR; Kinghorn AD Nat. Prod. Rep 2014, 31, 924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Greger H Phytochem. Rev 2021, 10.1007/s11101-021-09761-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cencic R; Carrier M; Galicia-Vázquez G; Bordeleau ME; Sukarieh R; Bourdeau A; Brem B; Teodoro JG; Greger H; Tremblay ML; Porco JA Jr.; Pelletier J PLoS One 2009, 4, e5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).An FL; Wang XB; Wang H; Li ZR; Yang MH;, Luo J; Kong LY Sci. Rep 2016, 6, 20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Othman N; Pan L; Mejin M; Voong J; Chai H; Pannell CM; Kinghorn AD; Yeo TC J. Nat. Prod 2016, 79, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Agarwal G; Kurina S; Anaya-Eugenio GD; Ninh TN; Burdette JE; Carcache de Blanco EJ; Soejarto DD; Rakotondraibe HL; Kinghorn AD J. Nat. Prod 2019, 82, 2870–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Agarwal G; Chang L-S; Soejarto DD; Kinghorn AD Planta Med. 2021, 87, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pan L; Muñoz Acuña U; Li J; Jena N; Ninh TN; Pannell CM; Chai H-B; Fuchs JR; Carcache de Blanco EJ; Soejarto DD; Kinghorn AD J. Nat. Prod 2013, 76, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dumontet V; Thoison O; Omobuwajo OR; Martin M-T; Perromat G; Chiaroni A; Riche C; Païs M; Sévenet T; Hadi AHA Tetrahedron 1996, 52, 6931–6942. [Google Scholar]
- (26).Chang L-S; Oblinger JL; Burns SS; Huang J; Anderson LW; Hollingshead MG; Shen R; Pan L; Agarwal G; Ren Y; Roberts R; O’Keefe BR; Kinghorn AD; Collins JM Mol. Cancer Ther 2020, 19, 731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Oblinger JL; Burns SS; Akhmametyeva EM; Huang J; Pan L; Ren Y; Shen R; Miles-Markley B; Moberly AC; Kinghorn AD; Welling DB; Chang L-S Neuro-Oncol. 2016, 18, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Oblinger JL; Burns SS; Huang J; Pan L; Ren Y; Shen R; Kinghorn AD; Welling DB; Chang L-S Exp. Neurol 2018, 299, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chang L-S; Oblinger JL; Zhang X; Anksapuram H; Ferrer M; Roberts RD; Kinghorn AD Connective Tissue Oncology Society, 26th Annual Meeting, Vancouver, November 10–13, 2021, Abstract P010. [Google Scholar]
- (30).King ML; Chiang CC; Ling HC; Fujita E; Ochiai M; McPhail ATJ Chem. Soc., Chem. Commun 1982, 1150–1151. [Google Scholar]
- (31).Ren Y, Chen W-L; Lantvit DD; Sass EJ; Shriwas P; Ninh TN; Chai H-B; Zhang X; Soejarto DD; Chen X; Lucas DM; Swanson SM; Burdette JE; Kinghorn AD J. Nat. Prod 2017, 80, 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Khare VK; Bhatnagar SS; Schindler O; Reichstein T Helv. Chim. Acta 1962, 45, 1515–1534. [Google Scholar]
- (33).Fiebig M; Duh C-Y; Pezzuto JM; Kinghorn AD; Farnsworth NR J. Nat. Prod 1985, 48, 981–985. [DOI] [PubMed] [Google Scholar]
- (34).Mi Q; Pezzuto JM; Farnsworth NR; Wani MC; Kinghorn AD; Swanson SM J. Nat. Prod 2009, 72, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen W-L, Ren Y; Ren J; Erxleben C; Johnson ME; Gentile S; Kinghorn AD; Swanson SM; Burdette JE J. Nat. Prod 2017, 80, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ren Y; Tan Q; Heath K; Wu S; Wilson JR; Ren J; Shriwas P; Yuan C; Ninh TN; Chai H-B; Chen X; Soejarto DD; Johnson ME; Cheng X; Burdette JB; Kinghorn AD Bioorg. Med. Chem 2020, 28, 115301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ren Y; Wu S; Chen S; Burdette JE; Cheng X; Kinghorn AD Molecules 2021, 26, 5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Anaya-Eugenio GD; Mekuria Addo E; Ezzone N; Henkin JM; Ninh TN; Ren Y; Soejarto DD; Kinghorn AD; Carcache de Blanco EJ J. Nat. Prod 2019, 82, 1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bueno Pérez L; Still PC; Naman CB; Ren Y; Pan L; Chai H-B; Carcache de Blanco EJ; Ninh TN; Thanh BV; Swanson SM; Soejarto DD; Kinghorn AD Phytochem. Rev 2014, 13, 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Henkin JM; Sydara K; Xayvue M; Souliya O; Kinghorn AD; Burdette JE; Chen W-L; Elkington B; Soejarto DD J. Med. Plants Res 2017, 11, 621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Shukla V; Joshi GP; Rawat MSM Phytochem. Rev 2010, 9, 287–307. [Google Scholar]
- (42).Tan CY; Wang F; Anaya-Eugenio GD; Gallucci JC; Gouchenour KD; Rappleye CA; Spjut RW; Carcache de Blanco EJ; Kinghorn AD; Rakotondraibe HL J. Nat. Prod 2019, 82, 2529–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang Y; Tan CY; Spjut RW; Kinghorn AD; Rakotondraibe HL Phytochemistry 2020, 180, 112521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Anaya-Eugenio GD; Tan CY; Rakotondraibe HL; Carcache de Blanco EJ Biomed. Pharmacother 2020, 127, 110124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Grobbelaar JU In Handbook of Microalgal Culture: Applied Phycology and Biotechnology; Richmond A, Hu Q, Eds.; Wiley-Blackwell: Oxford, UK, 2013; pp 123–133. [Google Scholar]
- (46).Welker M; Von Doöhren H FEMS Microbiol. Rev 2006, 30, 530–563. [DOI] [PubMed] [Google Scholar]
- (47).Tan LT Phytochemistry 2007, 68, 954–979. [DOI] [PubMed] [Google Scholar]
- (48).Chlipala GE; Mo S; Orjala J Curr. Drug Targets 2011, 12, 1654–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Singh RK; Tiwari SP; Rai AK; Mohapatra TM J. Antibiot 2011, 64, 401–412. [DOI] [PubMed] [Google Scholar]
- (50).Salvador-Reyes LA; Luesch H Nat. Prod. Rep 2015, 32, 478–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dittmann E; Gugger M; Sivonen K; Fewer DP Trends Microbiol. 2015, 23, 642–652. [DOI] [PubMed] [Google Scholar]
- (52).Kleigrewe K; Gerwick L; Sherman DH; Gerwick WH Nat. Prod. Rep 2016, 33, 348–364. [DOI] [PubMed] [Google Scholar]
- (53).Shah S; Akhter N; Auckloo B; Khan I; Lu Y; Wang K; Wu B; Guo Y-W Mar. Drugs 2017, 15, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Orjala J; Oberlies N; Pearce C; Swanson S; Kinghorn AD In Bioactive Compounds from Natural Sources, Second Edition; Tringali C, Ed.; Taylor & Francis, London, 2011; pp 37–64. [Google Scholar]
- (55).Chlipala GE; Krunic A; Lantvit D; Shen Q; Porter K; Swanson SM; Orjala JJ Nat. Prod 2010, 73, 1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kang H-S; Santarsiero BD; Kim HJ; Krunic A; Shen Q; Swanson SM; Chai H-B; Kinghorn AD; Orjala J Phytochemistry 2012, 79, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Luo S; Kang H-S; Krunic A; Chlipala GE; Cai G; Chen W-L; Franzblau SG; Swanson SM; Orjala J Tetrahedron Lett. 2014, 55, 686–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).May DS; Chen W-L; Lantvit DD; Zhang X; Krunic A; Burdette JE; Eustaquio A; Orjala J J. Nat. Prod 2017, 80, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).May DS; Kang HS; Santarsiero BD; Krunic A; Shen Q, Burdette JE,; Swanson SM; Orjala J J. Nat. Prod 2018, 81, 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Chen JL; Moore RE; Patterson GML J. Org. Chem 1991, 56, 4360–4364. [Google Scholar]
- (61).Moore BS; Chen J; Patterson GML; Moore RE Tetrahedron 1992, 48, 3001–3006. [Google Scholar]
- (62).Bui HTN; Jansen R; Pham HTL; Mundt SJ Nat. Prod 2007, 70, 499–503. [DOI] [PubMed] [Google Scholar]
- (63).Preisitsch M; Harmrolfs K; Pham HT; Heiden SE; ssel A; Wiesner C; Pretsch A; Swiatecka-Hagenbruch M; Niedermeyer TH; ller R; Mundt S J. Antibiot 2015, 68, 165–172. [DOI] [PubMed] [Google Scholar]
- (64).Preisitsch M; Niedermeyer THJ; Heiden SE; Neidhardt I; Kumpfmüller J; Wurster M; Harmrolfs K; Wiesner C; Enke H; Müller R; Mundt S J. Nat. Prod 2016, 79, 106–115. [DOI] [PubMed] [Google Scholar]
- (65).Weber T; Blin K; Duddela S; Krug D; Kim HU; Bruccoleri R; Lee SY; Fischbach MA; Müller R; Wohlleben W; Breitling R; Takano E; Medema MH. Nucleic Acids Res. 2015, 43, 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Nakamura H, Hamer HA, Sirasani G; Balskus EP J. Am. Chem. Soc 2012, 134, 18518–18521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Nakamura H, Wang JX; Balskus EP Chem. Sci 2015, 6, 3816–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Preisitsch M; Heiden SE; Beerbaum M; Niedermeyer T; Schneefeld M; Herrmann J; Kumpfmüller J; Thürmer A; Neidhardt I; Wiesner C; Daniel R; Müller R; Bange F-C; Schmieder P; Schweder T; Mundt S Mar. Drugs 2016, 14, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Nakamura H; Schultz EE; Balskus EP Nat. Chem. Biol 2017, 13, 916–921. [DOI] [PubMed] [Google Scholar]
- (70).Calteau A; Fewer DP; Latifi A; Coursin T; Laurent T; Jokela J; Kerfeld CA; Sivonen K; Piel J; Gugger M BMC Genomics 2014, 15, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Dittmann E; Gugger M; Sivonen K; Fewer DP Trends Microbiol. 2015, 23, 642–652. [DOI] [PubMed] [Google Scholar]
- (72).Shih PM; Wu D; Latifi A; Axen SD; Fewer DP; Talla E; Calteau A;, Cai F; Tandeau de Marsac N; Rippka R; Herdman M; Sivonen K; Coursin T; Laurent T; Goodwin L; Nolan M; Davenport KW; Han CS; Rubin EM;, Eisen JA; Woyke T; Gugger M; Kerfeld CA Proc. Natl. Acad. Sci. U.S.A 2013, 110, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kampa A; Gagunashvili AN; Gulder TAM; Morinaka BI; Daolio C; Godejohann M; Miao VPW; Piel J; Andresson OS Proc. Natl. Acad. Sci. U.S.A 2013, 110, E3129–E3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).May DS; Crnkovic CM; Krunic A; Wilson TA; Fuchs JR; Orjala J ACS Chem Biol. 2020, 15, 758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Hoffmann D; Hevel JM; Moore RE; Moore BS Gene 2003, 311, 171–180. [DOI] [PubMed] [Google Scholar]
- (76).Ishida K; Welker M; Christiansen G; Cadel-Six S; Bouchier C; Dittmann E; Hertweck C; Tandeau de Marsac N Appl. Environ. Microbiol 2009, 75, 2017–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Ishida K; Christiansen G; Yoshida WY; Kurmayer R; Welker M; Valls N; Bonjoch J; Hertweck C; Borner T; Hemscheidt T; Dittmann E Chem. Biol 2007, 14, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Gerwick BC; Brewster WK; Deboer GJ; Fields SC; Graupner PR; Hahn DR; Pearce CJ; Schmitzer PR; Webster JD J. Chem. Ecol 2013, 39, 253–261. [DOI] [PubMed] [Google Scholar]
- (79).Sica VP; Figueroa M; Raja HA; El-Elimat T; Darveaux BA; Pearce CJ; Oberlies NH J. Ind. Microbiol. Biotechnol 2016, 43, 1149–1157. [DOI] [PubMed] [Google Scholar]
- (80).El-Elimat T; Zhang X; Jarjoura D; Moy FJ; Orjala J; Kinghorn AD; Pearce CJ; Oberlies NH ACS Med. Chem. Lett 2012, 3, 645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Gonzalez-Medina M; Prieto-Martinez FD; Naveja JJ; Mendez-Lucio O; El-Elimat T; Pearce CJ; Oberlies NH; Figueroa M; Medina-Franco JL Future Med. Chem 2016, 8, 1399–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Gonzalez-Medina M; Owen JR; El-Elimat T; Pearce CJ; Oberlies NH; Figueroa M; Medina-Franco JL Front. Pharmacol 2017, 8, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).El-Elimat T; Figueroa M; Ehrmann BM; Cech NB; Pearce CJ; Oberlies NH J. Nat. Prod 2013, 76, 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Paguigan ND; El-Elimat T; Kao D; Raja HA; Pearce CJ; Oberlies NH J. Antibiot 2017, 70, 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Egan JM; van Santen JA; Liu DY; Linington RG J. Nat. Prod 2021, 84, 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Flores-Bocanegra L; Al Subeh ZY, Egan JM; El-Elimat T; Rafa HA; Burdette JE; Pearce CJ; Linington RG; Oberlies NH J. Nat. Prod 2022, 85, 10.1021/acs.jnatprod.1c00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Amrine CSM; Raja HA; Darveaux BA; Pearce CJ; Oberlies NH J. Ind. Microbiol. Biotechnol 2018, 45, 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Al Subeh ZY; Raja HA; Obike JC; Pearce CJ; Croatt MP; Oberlies NH J. Antibiot 2021, 74, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Katagiri K; Sato K; Hayakawa S; Matsushima T; Minato HJ Antibiot. 1970, 23, 420–422. [DOI] [PubMed] [Google Scholar]
- (90).Minato H; Matsumoto M; Katayama T J. Chem. Soc., Chem. Commun 1971, 44–45. [Google Scholar]
- (91).Figueroa M; Graf TN; Ayers S; Adcock AF; Kroll DJ; Yang J; Swanson SM; Munoz-Acuna U; Carcache de Blanco EJ; Agarwal R; Wani MC; Darveaux BA; Pearce CJ; Oberlies NH J. Antibiot 2012, 65, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Paschall AV; Yang D; Lu C; Choi JH; Li X; Liu F; Figueroa M; Oberlies NH; Pearce C; Bollag WB; Nayak-Kapoor A; Liu K J. Immunol 2015, 195, 1868–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Lu C; Paschall AV; Shi H; Savage N; Waller JL; Sabbatini ME; Oberlies NH; Pearce C; Liu K J. Natl. Cancer Inst 2017, 109, djw283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Lu C; Yang D; Sabbatini ME; Colby AH; Grinstaff MW; Oberlies NH; Pearce C; Liu K BMC Cancer 2018, 18, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Salvi A; Amrine CSM; Austin JR; Kilpatrick K; Russo A; Lantvit D; Calderon-Gierszal E; Mattes Z; Pearce CJ; Grinstaff MW; Colby AH; Oberlies NH; Burdette JE Mol. Cancer Ther 2020, 19, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Raja HA; Miller AN; Pearce CJ; Oberlies NH J. Nat. Prod 2017, 80, 756–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Wang Y; Hu P; Pan Y; Zhu Y; Liu X; Che Y; Liu G Fungal Genet. Biol 2017, 103, 25–33. [DOI] [PubMed] [Google Scholar]
- (98).Hölker U; Höfer M; Lenz J Appl. Microbiol. Biotechnol 2004, 64, 175–186. [DOI] [PubMed] [Google Scholar]
- (99).Hölker U; Lenz J Curr. Opin. Microbiol 2005, 8, 301–306. [DOI] [PubMed] [Google Scholar]
- (100).Oberlies NH; Knowles SL; Amrine CSM; Kao D; Kertesz V; Raja HA Nat. Prod. Rep 2019, 36, 944–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Sica VP; Raja HA; El-Elimat T; Kertesz V; Van Berkel GJ; Pearce CJ; Oberlies NH J. Nat. Prod 2015, 78, 1926–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Harrison C Nat. Biotechnol 2014, 32, 403–404. [DOI] [PubMed] [Google Scholar]
- (103).Amrine CSM; Huntsman AC; Doyle MG; Burdette JE; Pearce CJ; Fuchs JR; Oberlies NH ACS Med. Chem. Lett 2021, 12, 625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Müller K; Faeh C; Diederich F Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- (105).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Acena JL; Soloshonok VA; Izawa K; Liu H Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- (106).Wang J; Sanchez-Rosello M; Acena JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- (107).Meanwell NA J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- (108).Walker MC; Chang MCY Chem. Soc. Rev 2014, 43, 6527–6536. [DOI] [PubMed] [Google Scholar]
- (109).Ayers S; Graf TN; Adcock AF; Kroll DJ; Matthew S; Carcache de Blanco EJ; Shen Q; Swanson SM; Wani MC; Pearce CJ; Oberlies NH J. Nat. Prod 2011, 74, 1126–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).El-Elimat T; Raja HA; Day CS; Chen WL; Swanson SM; Oberlies NH J. Nat. Prod 2014, 77, 2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Muñoz Acuña U; Wittwer J; Ayers S; Pearce CJ; Oberlies NH; Carcache de Blanco EJ Anticancer Res. 2012, 32, 2415–2421. [PMC free article] [PubMed] [Google Scholar]
- (112).Prabhu KS; Siveen KS; Kuttikrishnan S; Iskandarani AN; Khan AQ; Merhi M; Omri HE; Dermime S; El-Elimat T; Oberlies NH; Alali FQ; Uddin S Front. Pharmacol 2018, 9, art. No. 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Prabhu KS; Siveen KS; Kuttikrishnan S; Jochebeth A; Ali TA; Elareer NR; Iskandarani A; Quaiyoom Khan A; Merhi M; Dermime S; El-Elimat T; Oberlies NH; Alali FQ; Steinhoff M; Uddin S Biomolecules 2019, 9, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Ninomiya-Tsuji J; Kajino T; Ono K; Ohtomo T; Matsumoto M; Shiina M; Mihara M; Tsuchiya M; Matsumoto KJ Biol. Chem 2003, 278, 18485–18490. [DOI] [PubMed] [Google Scholar]
- (115).Wu JQ; Powell F; Larsen NA; Lai ZW; Byth KF; Read J; Gu RF; Roth M; Toader D; Saeh JC; Chen HW ACS Chem. Biol 2013, 8, 643–650. [DOI] [PubMed] [Google Scholar]
- (116).Ellestad GA; Lovell FM; Perkinson NA; Hargreaves RT; McGahren WJ J. Org. Chem 1978, 43, 2339–2343. [Google Scholar]
- (117).Fakhouri L; El-Elimat T; Hurst DP; Reggio PH; Pearce CJ; Oberlies NH; Croatt MP Bioorg. Med. Chem 2015, 23, 6993–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Chen W-S; Chen Y-T; Wan X-Y, W.; Friedrichs E; Puff H; Breitmaier E Liebigs Ann. Chem 1981, 1981, 1880–1885. [Google Scholar]
- (119).Wu H; Lao X-F; Wang Q-W; Lu R-R; Shen C; Zhang F; Liu M; Jia L J. Nat. Prod 1989, 52, 948–951. [Google Scholar]
- (120).Morakotkarn D; Kawasaki H; Seki T; Okane I; Tanaka K Mycoscience 2008, 49, 258–265. [Google Scholar]
- (121).Dai DQ; Wijayawardene NN; Tang LZ; Liu C; Han LH; Chu HL; Wang HB; Liao CF; Yang EF; Xu RF; Li YM; Hyde KD; Bhat DJ; Cannon PF MycoKeys 2019, 58, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Al Subeh ZY; Raja HA; Monro S; Flores-Bocanegra L; El-Elimat T; Pearce CJ; McFarland SA; Oberlies NH J. Nat. Prod 2020, 83, 2490–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Al Subeh ZY; Waldbusser AL; Raja HA; Pearce CJ; Ho KL; Hall MJ; Probert MR; Oberlies NH; Hematian S J Org Chem. 2022, 87, 10.1021/acs.joc.1c0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Williamson RT; Buevich AV; Martin GE; Parella TJ Org. Chem 2014, 79, 3887–3894. [DOI] [PubMed] [Google Scholar]
- (125).Ren Y, Lantvit DD, Deng Y; Kanagasabai R; Gallucci JC; Ninh TN; Chai H-B; Soejarto DD; Fuchs JR; Yalowich JC; Yu J; Swanson SM; Kinghorn AD J. Nat. Prod 2014, 77, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Woodard JL; Huntsman AC; Patel PA; Chai H-B; Kanagasabai R; Karmahapatra S; Young AN; Ren Y; Cole MS; Herrera D; Yalowich JC; Kinghorn AD; Burdette JE; Fuchs JR Bioorg. Med. Chem 2018, 26, 2354–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Young AN; Herrera D; Huntsman AC; Korkmaz MA; Lantvit DD; Mazumder S; Kolli S; Coss CC; King S; Wang H; Swanson SM; Kinghorn AD; Zhang X; Phelps MA; Aldrich LN; Fuchs JR; Burdette JE Mol. Cancer Ther 2018, 17, 2123–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Salvi A; Young AN; Huntsman AC; Pergande M; Korkmaz MA; Rathnayake RA; Mize BK; Kinghorn AD; Zhang X; Ratia K; Schirle M; Thomas JR; Brittain SM; Shelton C; Aldrich LN; Cologna SM; Fuchs JR; Burdette JE Cell Death Dis. 2022, 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Krunic A; Vallet A; Mo S; Lantvit DD; Swanson SM; Orjala JJ Nat. Prod 2010, 73, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Wilson TA; Tokarski II RJ; Sullivan P; Demoret RM; Orjala J; Rakotondraibe LH; Fuchs JR J. Nat. Prod 2018, 82, 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Haux J; Klepp O; Spigset O; Tretli S BMC Cancer 2001, 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Lu Y; Zhang R; Liu S; Zhao Y; Gao J; Zhu L Eur. J. Pharmacol 2016, 771, 130–138. [DOI] [PubMed] [Google Scholar]
- (133).Wang AC; Pham HT; Lipps JM; Brittain SM; Harrington E; Wang Y; King FJ; Russ C; Pan X; Hoepner D; Tallarico J; Feng Y; Jain RK; Schirle M; Thomas JR ACS Chem. Biol 2019, 14, 20–26. [DOI] [PubMed] [Google Scholar]
- (134).Kulshrestha A; Katara GK; Ibrahim SA; Riehl V; Sahoo M; Dolan J; Meinke KW; Pins MR; Beaman KD J. Oncol 2019, 2019, 2343876. [DOI] [PMC free article] [PubMed] [Google Scholar]