

Abstract
In COVID-19 infection, the SARS-CoV-2 spike protein S1 interacts to the ACE2 receptor of human host, instigating the viral infection. To examine the competitive inhibitor efficacy of broad spectrum alpha helical AMPs extracted from frog skin, a comparative study of intermolecular interactions between viral S1 and AMPs was performed relative to S1-ACE2p interactions. The ACE2 binding region with S1 was extracted as ACE2p from the complex for ease of computation. Surprisingly, the Spike-Dermaseptin-S9 complex had more intermolecular interactions than the other peptide complexes and importantly, the S1-ACE2p complex. We observed how atomic displacements in docked complexes impacted structural integrity of a receptor-binding domain in S1 through conformational sampling analysis. Notably, this geometry-based sampling approach confers the robust interactions that endure in S1-Dermaseptin-S9 complex, demonstrating its conformational transition. Additionally, QM calculations revealed that the global hardness to resist chemical perturbations was found more in Dermaseptin-S9 compared to ACE2p. Moreover, the conventional MD through PCA and the torsional angle analyses indicated that Dermaseptin-S9 altered the conformations of S1 considerably. Our analysis further revealed the high structural stability of S1-Dermaseptin-S9 complex and particularly, the trajectory analysis of the secondary structural elements established the alpha helical conformations to be retained in S1-Dermaseptin-S9 complex, as substantiated by SMD results. In conclusion, the functional dynamics proved to be significant for viral Spike S1 and Dermaseptin-S9 peptide when compared to ACE2p complex. Hence, Dermaseptin-S9 peptide inhibitor could be a strong candidate for therapeutic scaffold to prevent infection of SARS-CoV-2.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00894-022-05117-8.
Keywords: COVID-19, SARS-CoV-2, S1 spike protein, Antimicrobial peptide, Brevinin, Dermaseptin, Magainin, Ocellatin, Conformational sampling, Semi-empirical calculation, Conventional molecular dynamics, Steered molecular dynamics
Introduction
Owing to current global pandemic COVID-19, the entire world is facing serious consequences, since the day of coronavirus outbreak in Chinese wet markets. The causative agent of COVID-19 is found to be novel β-coronavirus SARS-CoV-2, which is a pivotal member of the sarbecovirus family that would be species-specific to humans and other related mammals [1–4]. Spike protein (S) of SARS-CoV-2 is composed of S1 and S2 subunits. S1 retains a receptor-binding domain (RBD) made up of five β-stranded sheets (β1, β2, β3, β4, and β7) from 334th to 528th positions that are arranged antiparallel to a loop area between β1 and β2 strands [3, 5, 6]. In particular, this loop region in S1-RBD consisting of 56 amino acids from 446th to 502nd residue positions forms the most unique region that specifically binds to the peptidase domain of the angiotensin-converting enzyme 2 (ACE2) receptor [7].
Distinctly, ACE2 is a zinc-containing metalloenzyme protein found on the surface of lungs, kidneys, and a number of human cells [8]. ACE2 is the functional host cell receptor for the virus to enter into the host cell and begin infection [9]. Henceforth, S1 (RDB) prevails as an appealing therapeutic target for SARS-CoV-2, for which the chemical inhibitors [10–13] and the peptide-based drugs [14–16] are developed in the line-up of therapies for COVID-19. However, there are series of complications in existing COVID-19 therapies [17]; henceforth, the antimicrobial peptides (AMPs) might be considered as an alternative therapy for SARS CoV-2 infection.
AMPs are typically cationic and amphipathic molecules that have evolved over millions of years, providing almost all multicellular animals with the first line of defense [18]. In particular, anurans (frogs and toads) exposed to both aquatic and terrestrial environments have evolved to secrete AMPs in their skin to merely survive against many pathogens [19]. According to the Antimicrobial Peptide Database-3 (APD3), out of 3000 AMPs reported, over 189 AMPs have demonstrated antiviral activity. Specifically, the potential alpha-helical AMPs obtained from frog skin retain a good antiviral activity [20, 21]. In the protein-peptide interaction analyses of AMPs focusing on S1 Middle East Respiratory Syndrome (MERS) infection, the frog skin AMPs such as Magainin, Brevinin, and Dermaseptin have been documented to be the best docked peptides with high binding affinities [22]. Moreover, we previously reported the antimicrobial properties of broad spectrum ocellatin peptides [23], which prompts us to include it in this investigation and to assess its antiviral activity. Based on it, we intended to evaluate the efficacy of potential frog skin alpha-helical AMPs with its closest analogues, such as Brevinin-1BYa (1BYa), Brevinin-1BYc (1BYc), Dermaseptin-S4 (DS4), Dermaseptin-S9 (DS9), Magainin-1 (M1), Magainin-2 (M2), Ocellatin-1 (O1), and Ocellatin-F1 (OF1) that might inhibit spike protein S1 (RDB) of SARS-CoV-2 preserving the low hemolytic activity and high antimicrobial activity.
Brevinin (1BYa and 1BYc) peptides obtained from the skin secretion of Californian foothill-yellow legged frog Rana boylii [24] have been reported to have a remarkable antiviral potency against human immunodeficiency virus-1 (HIV-1), herpes simplex virus (HSV), and Ebola virus [25, 26]. Also, Dermaseptin (DS4 and DS9) peptides which were extracted from the South American tree frogs’ dermal secretions [27, 28] hold an antiviral action for HIV-1, HSV-1, HIV-2, HSV-2, human papilloma virus (HPV), rabies virus, and SARS-CoV-2 [16, 29–33]. Magainin peptides (M1 and M2) isolated from the skin of African clawed frogs, Xenopus laevis [34, 35] showed an antiviral activity against HSV-1, HSV-2, HIV-1, and SARS-CoV-2 [36–39]. Ocellatin O1 and Ocellatin OF1 peptides were isolated from the skin secretion of Brazilian pepper frogs such as Leptodactylus ocellatus and Leptodactylus labyrinthicus respectively [24] wherein, both the ocellatin peptides have shown to possess a wide variety of antibacterial, antiviral, and antifungal activities in experimental studies [23, 40–42]. Synergic antiviral effects between OF1 and alkaloid bufotenine have shown to limit BHK-21 cell lines advanced to rabies viral infection [43]. The present study identifies a potential therapeutic scaffold among these 8 alpha helical peptides that could serve as a potential competitive ACE2 inhibitor to impede S1 (RDB) from binding to ACE2 and thus regulates the SARS-CoV-2 entry.
Materials and methods
Data set
First, the sequences of frog skin alpha-helical AMPs with its closest analogues sequences, viz., 1BYa (P84111), 1BYc (P84113), DS4 (P80280), DS9 (Q1EN15), M1 (P11006), M2 (P11006), O1 (P83951), and OF1 (C0HKF0) constituting the range of 23–27 residue length were retrieved from the UniProt database [44]. Besides, the three-dimensional (3D) structures of 1BYa (6G4U), DS4 (2DD6), M2 (2MAG), and OF1 (5UA8) available in the Protein Data Bank (PDB) were retrieved [45]. With these available 3D structural templates, the tertiary structures of their respective analogues, viz., 1BYc, DS9, M1, and O1 were modeled, through the PEP-FOLD3 server. This program follows a de novo method for estimating peptide structural orientations based on their amino acid sequences, in which the conformations of consecutive residues are calculated with the help of a hidden Markov model (HMM). Using the YASARA package, all models were energy minimized [46]. A YASARA2 force field with a 10.5 cutoff was used to perform steepest descent energy minimization on peptides, and the peptides were optimized geometrically through explicit solvent. Using the PROCHECK tool, the stereo chemical quality of energy-minimized models was confirmed by a Ramachandran plot [47]. Furthermore, the receptor 3D structure of viral S1 spike glycoprotein (ID: 6M0J: E) with a resolution of 2.45 Å was obtained from the PDB.
Protein-peptide docking and interaction analysis
To determine binding interactions of SARS-CoV-2 spike glycoprotein (S1) with all eight peptides, viz., S1-1BYa, S1-1BYc, S1-DS4, S1-DS9, S1-M1, S1-M2, S1-O1, and S1-OF1, the site-specific docking was performed using the HADDOCK (High Ambiguity Driven protein–protein DOCKing) program [48]. The HADDOCK program implements docking based on the data-driven approach that supports to a wide range of experimental data, and the best binding solution of complexes was categorized based on desolvation, Vander Waals, restraint violation, and electrostatic energies with buried surface area [48]. Furthermore, total number of intermolecular non-covalent interactions among the docked complex, such as hydrogen bonds, hydrophobic contacts, cation-π interactions, and aromatic-aromatic, were predicted, using Protein Interaction Calculator (PIC) [49]. Furthermore, the hydrogen bond interactions were visualized via the PyMol visualization tool [50].
Conformational sampling
Subsequently, the conformational ensembles of four preferred protein-peptide complexes were generated, using the tCONCOORD program [51, 52]. Wherein, tCONCOORD depicts position constraints of complexes, using Gromacs index files [53–55]. Using the Vega ZZ environment, geometrical observable measurements such as root mean square deviation (RMSD) and polar surface area (PSA) of peptide trajectories were predicted and visualized with the Xmgrace tool [56, 57]. The conformational free energies of peptide conformers were estimated, using the distance-scaled finite ideal-gas reference (DFIRE) program, based on free-energy score and knowledge-based potential [58]. Furthermore, the secondary structural profiles of conformers were computed via a Define Secondary Protein Structure (DSSP) algorithm of GROMACS package [59] and their corresponding radar map representation was made through Microsoft Excel.
Semi-empirical QM/MM calculations
AMPAC-11 package was used to perform semi-empirical QM/MM calculations of peptide structures in which the structures were optimized using Austin Model 1 (AM1) parameters [60]. Wherein, the frontier molecular orbital energies, including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), were estimated for peptides in the HOMO-5 to LUMO + 4 range applying Koopmans’ theorem [61].
Conventional MD calculations
Conventional molecular dynamics (cMD) based on Newtonian equations of motion was performed in GROMACS for S1-ACE2p and S1-DS9 complexes. In order to perform conventional MD simulations, the CHARMM36 force field was used [62]. The cubic box was generated, followed by the solvation of TIP3P water molecules. Furthermore, by embedding protein with appropriate ions, the system was neutralized. However, proceeding on to the actual dynamics, the system’s energy was reduced via steepest descent minimization, followed by appropriate NPT and NVT equilibrations. Finally, using an NVIDIA DGX 1 GPU accelerator, the production MD simulations of S1-ACE2p and S1-DS9 protein complexes were conducted for 100 ns. Furthermore, the torsional angle analyses and the principal component analysis (PCA) were performed for the generated S1-ACE2p and S1-DS9 complex trajectories. Furthermore, the contact atoms among S1-ACE2p and S1-DS9 docked complexes with respect to their key binding residues were computed through a Ligand Protein Complex-Contacts of Structural Units (LPC-CSU) program [63].
Steered MD calculations
The steered molecular dynamics (sMD) of the S1-ACE2p and S1-DS9 complexes were performed, using YASARA [64]. The dynamics were carried out in a solvent box containing water molecules using the AMBER03 force field at a constant temperature of 298 K. While executing the SMD, a particle-mesh Ewald long-range interaction and periodic boundary condition were used and the physiological pH of 7.0 was retained. The peptides from the S1 spike protein were pulled in the preferred direction using a steering potential, which kept the S1 center mass constant. An extrinsic steering force with a constant pulling acceleration of 1500 pm/picoseconds2 was used to extract the peptides from S1, and forces were calculated for every 10 ps.
Statistical analysis
A nonparametric statistical method of Wilcoxon matched pair testing was done using StatPlus software version-7 (AnalystSoft, www.analystsoft.com/en) for the conformational sampling data of RMSD and PSA. In evaluating the statistical significance of complexes, this specific test is deemed similar to Student’s t-test for matched pairs and the probability value (P value) less than 0.05 was determined to be statistically significant [65].
Results and discussion
Binding effect of protein-peptide interactions
First, the structural orientations of antiviral peptides were modeled, using proficient structure prediction platforms. Wherein, the modeled structures exhibited the alpha helical content, confirmed by visualization of their 3D structures (Supplementary Fig. 1). The modeled 3D structures were also corroborated by Ramachandran plot analysis, where all the modeled peptides were found to be sterically stable and structurally robust. Subsequently, the site-specific docking of S1 spike protein with alpha helical peptides was performed and the representations of those eight docked complexes such as S1-1BYa, S1-1BYc, S1-DS4, S1-DS9, S1-M1, S1-M2, S1-O1, and S1-OF1 were observed (Fig. 1). Consequently, the interaction between peptides and RBD of S1were quantified and assessed with the help of molecular docking (Table 1).
Fig. 1.

a) S1 docked peptide complexes. b) S1-ACE2p peptide complex derived from S1-ACE2 experimental complex structure (6M0J). The peptides-ACE2p, 1BYa, 1BYc, DS4, DS9, M1, M2, O1, and OF1 are represented as cartoon illustrated with various colors, viz., black, orange, brown, pink, red, blue, cyan, light green, and dark green, respectively. Similarly, S1 is represented as surface model
Table 1.
Binding score estimation among S1-peptide docked complexes obtained through HADDOCK tool
| S. no | Peptide complexes | HADDOCK score |
|---|---|---|
| 1 | S1-1BYa | − 64.0 |
| 2 | S1-1BYc | − 42.0 |
| 3 | S1-DS4 | − 35.5 |
| 4 | S1-DS9 | − 82.9 |
| 5 | S1-M1 | − 46.2 |
| 6 | S1-M2 | − 69.4 |
| 7 | S1-O1 | − 49.2 |
| 8 | S1-OF1 | − 49.0 |
Furthermore, the inhibitory efficacy of eight docked complexes was compared to that of the S1-ACE2 complex (ID: 6M0J) by quantifying intermolecular interactions among all docked complexes (Table 3). Regarding the inter-molecular interactions, about 43 interactions were quantified between S1 and ACE2, which serves as a reference to evaluate the corresponding interactions between antiviral peptides and S1 receptor. Similar levels of intermolecular interactions between the antiviral peptides and S1 receptor could insinuate that they could produce similar interactions that were observed between S1 and ACE2. Therefore, the peptides from each category of frog antiviral peptides which produce similar levels of interactions with S1, just as ACE2 does, were utilized for subsequent analysis. About 51 intermolecular interactions were found at the interface of S1 receptor and DS9 peptide while, about 41 and 32 inter-molecular interactions were found at the juncture of peptide-receptor for S1-M1 and S1-OF1 complexes respectively (Table 2); their corresponding interface representations in terms of hydrogen bonds and hydrophobic interactions are observed (Fig. 2).
Table 3.
Conformational and thermodynamic stability parameters of S1-1BYc, S1-DS9, S1-M1, and S1-OF1 docked complexes
| S. no | Parameters | RMSD (Å) | RMSF (Å) | Conformational free energy (kcal/mol) |
|---|---|---|---|---|
| 1 | S1-ACE2p | 2.55 | 2.26 | − 439.47 |
| 2 | S1-1BYc | 4.38 | 2.59 | − 425.45 |
| 3 | S1-DS9 | 2.37 | 2.38 | − 435.28 |
| 4 | S1-M1 | 2.70 | 2.60 | − 403.31 |
| 5 | S1-OF1 | 3.18 | 2.94 | − 413.16 |
Table 2.
Comparative analyses of intermolecular interactions of S1 docked complexes of Brevinin, Dermaseptin, Magainin, and Ocellatin in relative to S1-ACE2 and S1-ACE2p complexes
| S. no | Inter-molecular interactions | S1-1BYa | S1-1BYc | S1-DS4 | S1-DS9 | S1-M1 | S1-M2 | S1-O1 | S1-OF1 | S1-ACE2 | S1-ACE2p |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Hydrogen bonds | 15 | 17 | 12 | 30 | 25 | 20 | 10 | 26 | 38 | 37 |
| 2 | Hydrophobic interactions | 11 | 19 | 13 | 12 | 7 | 7 | 12 | 4 | 4 | 1 |
| 3 | Protein–protein ionic interactions | 2 | 0 | 2 | 1 | 3 | 3 | 2 | 0 | 0 | 3 |
| 4 | Aromatic-aromatic interactions | 1 | 1 | 1 | 6 | 2 | 2 | 0 | 0 | 1 | 0 |
| 5 | Cation-π interactions | 2 | 3 | 2 | 2 | 4 | 3 | 2 | 2 | 0 | 0 |
| Total number | 31 | 40 | 30 | 51 | 41 | 35 | 26 | 32 | 43 | 41 | |
Fig. 2.

Hydrogen bond interactions of a) S1-ACE2p, b) S1-1BYc, c) S1-DS9, d) S1-M1, and e) S1-OF1 complexes. The colors of cartoon representations: black, brown, red, blue, and green represent ACE2p, 1BYc, DS9, M1, and OF1 peptides, respectively
To ease the intense burden levied upon computational resources, we have chosen ACE2p (S1 interacting peptide derived from ACE2) instead of the entire ACE2 protein. Besides, visualizing the peptide protein complex interaction revealed that ACE2p interaction with S1 is almost on par with ACE2 complete protein interactions with S1 (Table 3). Hence, it would be a prudent choice to choose ACE2p over ACE2. Hydrogen bonds are deemed to be one of the crucial factors in ascertaining intermolecular interactions between protein and peptides [66]. Notably, the interacting regions of S1 with the peptides are similar to that of ACE2 binding site. The binding site predominantly comprised of loop region with 56 amino acids spanning between β1 and β2 strands of RBD. Strikingly, all 4 peptides interact with loop region of S1-RBD, such as 1BYc peptide interacts with amino acids of S1in the range of 448–498, while peptide DS9 forms hydrogen bond with S1 residues ranging 479–500. In the case of S1-M1 and S1-OF1 complexes, similar results were found (Fig. 2). Based on these findings, it was apparent that peptide DS9 manifests considerable interaction with S1 compared to other peptides (Fig. 2 and Table 2).
Loop region of S1 makes for a preferable drug target, since there binds the ACE2p. In recent past, several studies endorsed the targeting of loop region in S1 to competitively inhibit the binding of ACE2p, thereby proficiently arresting SARS-CoV-2 entry and averting infection [6, 7, 67, 68]. The fact that both the ACE2p and peptide DS9 associates with S1 in the similar binding vicinity coupled with an observation that the interaction of DS9 (Table 2 and Fig. 2) with S1 is a bit more adhesive than the interaction of ACE2p with S1, indicating a strong plausibility of DS9 to be ACE2p’s competitive inhibitor for binding with S1, magnificently. Although DS9 was found to be more effective in interacting with S1, the efficacy of other peptides were not negated, up till now. Accordingly, the top four peptides, viz., 1BYc, DS9, M1, and OF1, which exhibited considerable interactions with S1, were utilized for subsequent evaluation.
Exploring the effects of conformational ensembles
Most proteins perform many functions based on conformational changes in their structure without losing stability under varying circumstances [69]. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were computed to illustrate the structural stability parameters of peptide complex ensembles (Table 3). In which, S1-DS9 showed the least RMSD and RMSF values, when compared to other peptide complexes. Moreover, the RMSD measures the mean distance moved by atoms from their average positions. It is also a traditional metric for assessing conformational stability. Higher the RMSD, lower the conformational stability [70]. Our findings indicated that, among four peptide complexes, DS9 showed the least RMSD, even lower than S1-ACE2p complex which serves as positive control. This shows that the complex formed as a result of DS9’s interaction with S1 is notably stabilized. The RMSF is a distance measure between clusters of atoms in reference to a coordinate set with a well-defined average position [70]. Table 3 shows that S1-DS9 had the least fluctuation, with a mean value of 2.38 Å, indicating that it has a stronger structural stability than other peptides (Supplementary Fig. 2), further confirmed by its replicates (Supplementary Fig. 3).
Besides, its residual flexibility is the closest to positive control, compared to other peptide complexes. To further investigate the stability of S1-AMPs complexes, the conformational free energy of their trajectories was computed (Fig. 3). The data plotted in the form of kernel density estimation (KDE), illustrated that among four peptide complexes, S1-DS9 showed a least energy. The lower the free energy, the higher the thermodynamic stability [71]. Therefore, S1-DS9 is deemed to be the most stable complex. Also, it was found that S1-DS9 complex energy was quite closer to the positive control, S1-ACE2p complex energy compared to others (Table 3). Furthermore, the secondary structural profile predicted for peptide conformations from trajectory analysis showed a considerable variation in dynamic pattern of alpha helix represented as radar chart (Fig. 4). When interacting with a lipid membrane, an alpha helical peptide regulates its helicity by the distribution of intramolecular hydrogen bonds; conversely, when interacting with receptor residues, it tends to non-covalently interact with the receptor residues, resulting in perpetuation of peptide helicity.
Fig. 3.

Kernel density estimation plot of conformational free energies in protein-peptide complexes
Fig. 4.

Radar chart of secondary structural conformers of S1 spike protein and peptide complexes a) S1-1BYc, b) S1-DS9, c) S1-M1, and d) S1-OF1 where the radial values correspond to the number of residues
In comparison to DS9, other three docked complexes improperly interacted with S1 receptor during protein-peptide simulations, resulting in turn or random coil. However, a notable conservation of secondary structural feature was observed in DS9, that its overall alpha helical content of a peptide retained throughout the simulation, which influenced its interaction with S1 receptor. This further corroborated aforementioned findings which endorses DS9 to be a suitable candidate against S1.
Rationalizing the impact of DS9’s competitive binding efficacy
Based upon the aforementioned results from docking and dynamics, it was apparent that DS9 could be a viable candidate to effectively bind with S1 and potentially produce the intended effect in mitigating the virulence. We further desired to substantiate the notion that DS9 binding is on par with ACE2p binding to S1 via quantum and molecular mechanical computations. Semi-empirical QM/MM global reactivity descriptors such as HOMO (highest occupied molecular orbital) denote the ability to donate electrons, whereas LUMO (lowest unoccupied molecular orbital) denotes the ability to accept electrons [72], insinuating the chemical reactivity of the compound upon interacting with S1. The lower the energy gap, the higher the reactivity [73, 74]. Findings indicate that reactivity of DS9 with S1 is almost on par with ACE2p’s reactivity with the same. Moreover, conferring to Koopmans’ theorem, HOMO–LUMO electron density energy gap (Fig. 5) determines the global hardness of DS9 and ACE2p molecules, which is known to be an estimation of their resistance to charge transfer during small perturbations [75]. Accordingly, DS9 has a greater value (0.532 eV) than ACE2p (0.454 eV), indicating that AMP has better resistive stability in chemical reactions.
Fig. 5.

HOMOL-LUMO energy gap computed via a) ACE2p and b) DS9 peptides after docking with S1
Numerous proteins, in response to conformational changes in their 3D structure, dictate distinct cellular activity in varying cellular environments [69]. However, these conformational changes can be effectively investigated via conventional molecular dynamics (cMD) simulations with atomic precision [76]. Firstly, the torsional angle transition was investigated using cMd trajectory analysis, to substantiate the relevance of the side chains of key residues Y449, N487, and T500 highlighted in docking analyses of protein-peptide complexes (Fig. 6), which is essential for regulating the interaction between S1 and ACE2. Secondly, the comparative analysis was performed by overlapping the backbone angles (ϕ; ψ) of all three interacting residues Y449, N487, and T500 in S1 [5], since torsional angle among amino acids is essential for maintaining the structural integrity of a protein system [77]. Comparing the change in torsional backbone angle of S1 upon interacting with ACE2p and DS9 peptide could effectively validate the ability of DS9 to alter the structural integrity of S1’s pathogenic conformation that can be observed from S1-ACE2p’s interaction. Accordingly, the backbone angles (ϕ; ψ) of Y449 were located in range of (− 50°, − 100°; 0°, 50°). Similarly, the backbone angles (ϕ; ψ) of N487 and T500 were distributed in range of (50°, 100°; − 20°, 40°) and (120°, − 50°; − 50°, 50°) respectively (Fig. 6). The preceding result implicated that the angles ϕ and ψ of these three residues near the S1 binding site contribute significantly to the conformational modulation of DS9, which makes it a promising AMP analeptic. Table 4 reveals the range of nearest distance between atoms among S1-ACE2p and S1-DS9 docked complexes with respect to key binding residues, viz., Y449, Y487, and T500 computed through LPC-CSU program [63, 78]. The nearest distance between atoms of two residues in S1-ACE2p is wider (range of 2.7 to 5.4 Å) when compared to a narrow distance of S1-DS9 comparatively (range of 2.8 to 3.5 Å). This strongly suggested that DS9 has more favorable contact than ACE2p [79, 80]. Besides, the hydrophobic interaction and aromatic-aromatic interactions around key residues of DS9 and ACE2p were comparatively studied. Moreover, the destabilizing interactions (hydrophobic-hydrophilic contact) are lower in DS9 when compared to ACE2p around key residues [81]. This suggested that DS9 could possess more stable contacts towards spike protein. Therefore from Table 4, it is evident that DS9 could have more favorable binding towards spike protein when compared to ACE2p.
Fig. 6.

Torsional energy calculation of residues of a) Y449, b) N487 and c) T500 of S1-ACE2p and S1-DS9 peptide complexes, in which running averages were depicted in thin lines
Table 4.
Estimation of specific contacts among S1-ACE2p and S1-DS9 docked complexes obtained through LPC-CSU program
| S. no | S1-peptide | Specific contacts | |||||
|---|---|---|---|---|---|---|---|
| S1 residues | Peptide residues | NDA (Å) | HB | AAC | HHC | DC | |
| S1-ACE2p | Y449 | D38 | 2.7 | + | - | - | - |
| Y449 | Q42 | 2.8 | + | - | - | - | |
| Y487 | Q24 | 2.7 | + | - | - | + | |
| Y449 | F28 | 5.4 | - | - | - | + | |
| T500 | Y41 | 2.7 | + | - | + | + | |
| T500 | L45 | 4.5 | - | - | + | + | |
| S1-DS9 | Y449 | W7 | 3.5 | - | + | - | - |
| Y449 | V10 | 3.1 | - | - | + | - | |
| Y449 | L11 | 3.1 | - | - | + | - | |
| Y449 | I14 | 3.7 | - | - | + | + | |
| T500 | R3 | 2.8 | + | - | - | + | |
NDA, nearest distance between atoms of two residues
CSA, contact surface area between two residues
HB, hydrogen bond
AAC, aromatic-aromatic contact
HHC, hydrophobic-hydrophobic contact
DC, destabilizing contact ± indicates presence/absence of specific contacts
Furthermore, the Gibbs free energy landscape (ΔG) for ACE2p and DS9 was generated using the first two major principal components (PC1, PC2) as reaction coordinates from molecular dynamics trajectories. The 2D surface projections of the global energy minimum elucidated the cluster of stable states that occur in both S1-ACE2p and S1-DS9 complexes (Fig. 7). The Eigen vectors of collective motions in DS9 were clearly confined to a basin lesser than that of ACE2p. As a result, these differences in conformational structures between S1-DS9 and S1-ACE2p indicated that DS9 might regulate the conformational orientations of S1 in comparison with ACE2p’s influence over the same. Finally, steered molecular dynamics (sMD), a converse analysis for docking, was carried out on S1-ACE2p and DS9 complexes wherein the ligand is pulled away from the protein to evaluate its protein ligand interaction (Fig. 8). Specifically, the duration for dissociation is proportional to the interaction between protein and ligand; the longer the time taken to dissociate the ligand/peptide from the protein, the higher is the binding affinity between them [64]. Accordingly, it took 35.57 ps to completely dissociate ACE2p from S1, while 37.90 ps has elapsed, to completely separate DS9 from S1. In addition, free energy versus displacement result based upon Jarynski’s theory [82, 83] has been illustrated graphically (Supplementary Fig. 4). This correlates well with docking studies and SMD analysis. Besides, it reveals more favorable binding of DS9’s interaction with S1 when compared to ACE2p.
Fig. 7.
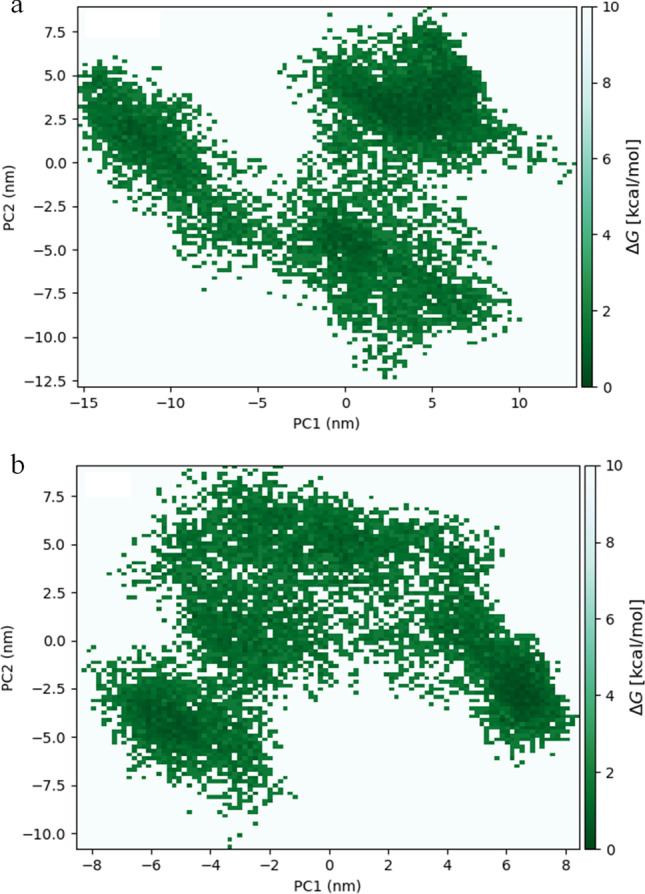
Free energy landscape of a) S1-ACE2p and b) S1-DS9 peptide complexes
Fig. 8.
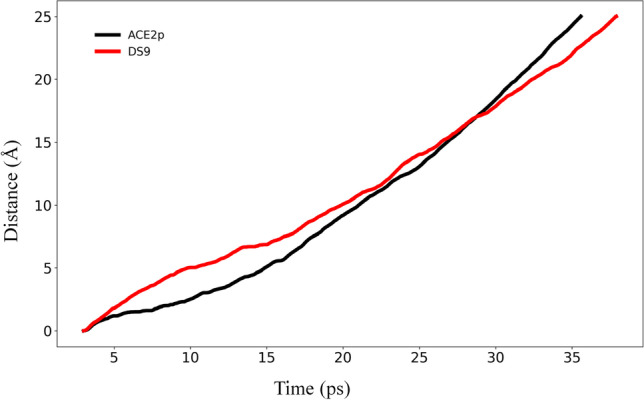
Steered molecular dynamics data illustrating the preferred pull of peptides from S1 spike protein while upholding the S1 center mass constant
Conclusions
Spike viral protein S1-DS9 peptide complex demonstrated the strongest intermolecular interactions and higher thermodynamic stability, thereby endorsing the potential competitive inhibitor role of DS9 in binding to S1. The complications with current COVID-19 therapy have urged the scientific community to find new treatments to tackle SARS-CoV-2. As an outcome of this study, we anticipate that identifying DS9 as a potential therapeutic peptide scaffold will lead to improvements in the design of peptidomimetics for the treatment of COVID-19 viral infection. As a future perspective, the incredible antiviral synergistic impact between DS9 peptide and drugs in preclinical trials might provide a creative lead to the development of promising antiviral drugs.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank VIT for providing infrastructure for carrying out this research work. In particular, we thank VIT-Centre for Technical Support for providing NVIDIA DGX 1 computational resources to carry out molecular simulations.
Author contribution
P. Chandra Sekar optimized the entire study and wrote the manuscript. E. Srinivasan standardized the computational method used in this study. G. Chandrasekhar made significant contributions to MD simulations and semi-empirical QM/MM calculations. D. Meshach Paul discussed the results and helped to draft the manuscript. G. Sanjay performed the conformational sampling and data analysis. S. Surya and NS. Arunraj Kumar collected the initial data set and performed docking and interaction analyses. R. Rajasekaran designed and meticulously evaluated the entire study and contributed significant intellectual material. The final manuscript was read and approved by all authors.
Data availability
Not applicable.
Code availability
Not applicable.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hu B, Guo H, Zhou P, Shi Z-L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol [Internet]. 2020 Oct 6 [cited 2021 Feb 12]; Available from: http://www.nature.com/articles/s41579-020-00459-7 [DOI] [PMC free article] [PubMed]
- 2.Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res. 2020;24:91–98. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo Y-R, Cao Q-D, Hong Z-S, Tan Y-Y, Chen S-D, Jin H-J, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Military Med Res. 2020;7(1):11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Liu M, Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc Natl Acad Sci USA. 2020;117(25):13967–13974. doi: 10.1073/pnas.2008209117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 8.Samavati L, Uhal BD. ACE2, Much more than just a receptor for SARS-COV-2. Front Cell Infect Microbiol. 2020;5(10):317. doi: 10.3389/fcimb.2020.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24(1):422. doi: 10.1186/s13054-020-03120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdelmageed MI, Abdelmoneim AH, Mustafa MI, Elfadol NM, Murshed NS, Shantier SW, et al. Design of a multiepitope-based peptide vaccine against the E protein of human COVID-19: an immunoinformatics approach. Biomed Res Int. 2020;11(2020):1–12. doi: 10.1155/2020/2683286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hattori S, Higashi-Kuwata N, Hayashi H, Allu SR, Raghavaiah J, Bulut H, et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun. 2021;12(1):668. doi: 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sivaraman H, Er SY, Choong YK, Gavor E, Sivaraman J. Structural basis of SARS-CoV-2– and SARS-CoV–receptor binding and small-molecule blockers as potential therapeutics. Annu Rev Pharmacol Toxicol. 2021;61(1):465–493. doi: 10.1146/annurev-pharmtox-061220-093932. [DOI] [PubMed] [Google Scholar]
- 13.Xiaojie S, Yu L, Lei Y, Guang Y, Min Q. Neutralizing antibodies targeting SARS-CoV-2 spike protein. Stem Cell Research. 2021;50:102125. doi: 10.1016/j.scr.2020.102125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baig MS, Alagumuthu M, Rajpoot S, Saqib U. Identification of a potential peptide inhibitor of SARS-CoV-2 targeting its entry into the host cells. Drugs R D. 2020;20(3):161–169. doi: 10.1007/s40268-020-00312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han Y, Král P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano. 2020;14(4):5143–5147. doi: 10.1021/acsnano.0c02857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekar PC, Rajasekaran R. Could Dermaseptin analogue be a competitive inhibitor for ACE2 towards binding with viral spike protein causing COVID19?: computational investigation. Int J Pept Res Ther [Internet]. 2021 Jan 16 [cited 2021 Feb 12]; Available from: http://link.springer.com/10.1007/s10989-020-10149-w [DOI] [PMC free article] [PubMed]
- 17.Monpara JD, Sodha SJ, Gupta PK. COVID-19 associated complications and potential therapeutic targets. Eur J Pharmacol. 2020;886:173548. doi: 10.1016/j.ejphar.2020.173548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Gallo RL. Antimicrobial peptides. Curr Biol. 2016;26(1):R14–R19. doi: 10.1016/j.cub.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Xu X, Lai R. The chemistry and biological activities of peptides from amphibian skin secretions. Chem Rev. 2015;115(4):1760–1846. doi: 10.1021/cr4006704. [DOI] [PubMed] [Google Scholar]
- 20.Elnagdy S, AlKhazindar M. The potential of antimicrobial peptides as an antiviral therapy against COVID-19. ACS Pharmacol Transl Sci. 2020;3(4):780–782. doi: 10.1021/acsptsci.0c00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44(D1):D1087–D1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mustafa S, Balkhy H, Gabere M. Peptide-protein interaction studies of antimicrobial peptides targeting Middle East respiratory syndrome coronavirus spike protein: an in silico approach. Adv Bioinform. 2019;1(2019):1–16. doi: 10.1155/2019/6815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sekar PC, Paul DM, Srinivasan E, Rajasekaran R. Unravelling the molecular effect of ocellatin-1, F1, K1 and S1, the frog-skin antimicrobial peptides to enhance its therapeutics—quantum and molecular mechanical approaches. J Mol Model. 2021;27(1):10. doi: 10.1007/s00894-020-04652-6. [DOI] [PubMed] [Google Scholar]
- 24.Conlon JM. A proposed nomenclature for antimicrobial peptides from frogs of the genus Leptodactylus. Peptides. 2008;29(9):1631–1632. doi: 10.1016/j.peptides.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 25.Savelyeva A, Ghavami S, Davoodpour P, Asoodeh A, Los MJ. An overview of Brevinin superfamily: structure, function and clinical perspectives. Adv Exp Med Biol. 2014;818:197–212. doi: 10.1007/978-1-4471-6458-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zohrab F, Askarian S, Jalili A, Kazemi OR. Biological properties, current applications and potential therapeautic applications of Brevinin peptide superfamily. Int J Pept Res Ther. 2019;25(1):39–48. doi: 10.1007/s10989-018-9723-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartels EJH, Dekker D, Amiche M. Dermaseptins, multifunctional antimicrobial peptides: a review of their pharmacology, effectivity, mechanism of action, and possible future directions. Front Pharmacol. 2019;26(10):1421. doi: 10.3389/fphar.2019.01421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mor A, Hani K, Nicolas P. The vertebrate peptide antibiotics dermaseptins have overlapping structural features but target specific microorganisms. J Biol Chem. 1994;269(50):31635–31641. doi: 10.1016/S0021-9258(18)31742-3. [DOI] [PubMed] [Google Scholar]
- 29.Belaid A, Aouni M, Khelifa R, Trabelsi A, Jemmali M, Hani K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J Med Virol. 2002;66(2):229–234. doi: 10.1002/jmv.2134. [DOI] [PubMed] [Google Scholar]
- 30.Bergaoui I, Zairi A, Tangy F, Aouni M, Selmi B, Hani K. In vitro antiviral activity of Dermaseptin S4 and derivatives from amphibian skin against herpes simplex virus type 2. J Med Virol. 2013;85(2):272–281. doi: 10.1002/jmv.23450. [DOI] [PubMed] [Google Scholar]
- 31.Lequin O, Ladram A, Chabbert L, Bruston F, Convert O, Vanhoye D, et al. Dermaseptin S9, an α-helical antimicrobial peptide with a hydrophobic core and cationic termini†. Biochemistry. 2006;45(2):468–480. doi: 10.1021/bi051711i. [DOI] [PubMed] [Google Scholar]
- 32.Lorin C, Saidi H, Belaid A, Zairi A, Baleux F, Hocini H, et al. The antimicrobial peptide dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology. 2005;334(2):264–275. doi: 10.1016/j.virol.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 33.Mechlia MB, Belaid A, Castel G, Jallet C, Mansfield KL, Fooks AR, et al. Dermaseptins as potential antirabies compounds. Vaccine. 2019;37(33):4694–4700. doi: 10.1016/j.vaccine.2018.01.066. [DOI] [PubMed] [Google Scholar]
- 34.Zerweck J, Strandberg E, Kukharenko O, Reichert J, Bürck J, Wadhwani P, et al. Molecular mechanism of synergy between the antimicrobial peptides PGLa and magainin 2. Sci Rep. 2017;7(1):13153. doi: 10.1038/s41598-017-12599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zasloff M (1987) Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA 84(15):5449–53. 10.1073/pnas.84.15.5449 [DOI] [PMC free article] [PubMed]
- 36.Aboudy Y, Mendelson E, Shalit I, Bessalle R, Fridkin M. Activity of two synthetic amphiphilic peptides and magainin-2 against herpes simplex virus types 1 and 2. Int J Pept Protein Res. 2009;43(6):573–582. doi: 10.1111/j.1399-3011.1994.tb00559.x. [DOI] [PubMed] [Google Scholar]
- 37.Balasubramaniam B, Prateek, Ranjan S, Saraf M, Kar P, Singh SP, Thakur VK, Singh A, Gupta RK (2020) Antibacterial and Antiviral Functional Materials: Chemistry and Biological Activity toward Tackling COVID-19-like Pandemics. ACS Pharmacol Transl Sci 4(1):8–54. 10.1021/acsptsci.0c00174 [DOI] [PMC free article] [PubMed]
- 38.Jacob L, Zasloff M. Potential therapeutic applications of magainins and other antimicrobial agents of animal origin. In: Marsh J, Goode JA, editors. Novartis Foundation Symposia [Internet]. Chichester, UK: John Wiley & Sons, Ltd.; 2007 [cited 2021 Feb 13]. p. 197–223. Available from: http://doi.wiley.com/10.1002/9780470514658.ch12 [DOI] [PubMed]
- 39.Zairi A, Tangy F, Bouassida K, Hani K. Dermaseptins and magainins: antimicrobial peptides from frogs’ skin—new sources for a promising spermicides microbicides—a mini review. J Biomed Biotechnol. 2009;2009:1–8. doi: 10.1155/2009/452567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomes KAGG, dos Santos DM, Santos VM, Piló-Veloso D, Mundim HM, Rodrigues LV, et al. NMR structures in different membrane environments of three ocellatin peptides isolated from Leptodactylus labyrinthicus. Peptides. 2018;103:72–83. doi: 10.1016/j.peptides.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 41.Gusmão KAG, dos Santos DM, Santos VM, Cortés ME, Reis PVM, Santos VL, et al. Ocellatin peptides from the skin secretion of the South American frog Leptodactylus labyrinthicus (Leptodactylidae): characterization, antimicrobial activities and membrane interactions. J Venom Anim Toxins Incl Trop Dis. 2017;23(1):4. doi: 10.1186/s40409-017-0094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nascimento ACC, Zanotta LC, Kyaw CM, Schwartz ENF, Schwartz CA, Sebben A, et al. Ocellatins: new antimicrobial peptides from the skin secretion of the South American frog Leptodactylus ocellatus (Anura: Leptodactylidae) Protein J. 2004;23(8):501–508. doi: 10.1007/s10930-004-7877-z. [DOI] [PubMed] [Google Scholar]
- 43.Bessa LJ, Eaton P, Dematei A, Plácido A, Vale N, Gomes P, et al. Synergistic and antibiofilm properties of ocellatin peptides against multidrug-resistant Pseudomonas aeruginosa. Future Microbiol. 2018;13(2):151–163. doi: 10.2217/fmb-2017-0175. [DOI] [PubMed] [Google Scholar]
- 44.Consortium TU UniProt: a hub for protein information. Nucl Acids Res. 2015;43(D1):D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucl Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandrasekhar G, Rajasekaran R. Investigating the pernicious effects of heparan sulfate in serum amyloid A1 protein aggregation: a structural bioinformatics approach. J Biomol Struct Dyn. 2020;14:1–15. doi: 10.1080/07391102.2020.1833756. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 48.Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: a protein−protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125(7):1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 49.Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic Acids Res. 2007;35(Web Server issue):W473–6. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 51.Seeliger D, De Groot BL. tCONCOORD-GUI: visually supported conformational sampling of bioactive molecules. J Comput Chem. 2009;30(7):1160–1166. doi: 10.1002/jcc.21127. [DOI] [PubMed] [Google Scholar]
- 52.Shruti SR, Rajasekaran R. Identification of protegrin-1 as a stable and nontoxic scaffold among protegrin family – a computational approach. J Biomol Struct Dyn. 2019;37(9):2430–2439. doi: 10.1080/07391102.2018.1491418. [DOI] [PubMed] [Google Scholar]
- 53.Meshach Paul D, Rajasekaran R. Exploration of structural and functional variations owing to point mutations in α-NAGA. Interdiscip Sci Comput Life Sci. 2018;10(1):81–92. doi: 10.1007/s12539-016-0173-8. [DOI] [PubMed] [Google Scholar]
- 54.Srinivasan E, Rajasekaran R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val) RSC Adv. 2016;6(104):102744–102753. doi: 10.1039/C6RA21927F. [DOI] [Google Scholar]
- 55.Srinivasan E, Rajasekaran R. Rational design of linear tripeptides against the aggregation of human mutant SOD1 protein causing amyotrophic lateral sclerosis. J Neurol Sci. 2019;405:116425. doi: 10.1016/j.jns.2019.116425. [DOI] [PubMed] [Google Scholar]
- 56.Pavithrra G, Rajasekaran R. Identification of effective dimeric gramicidin-D peptide as antimicrobial therapeutics over drug resistance: in-silico approach. Interdiscip Sci Comput Life Sci. 2019;11(4):575–583. doi: 10.1007/s12539-018-0304-5. [DOI] [PubMed] [Google Scholar]
- 57.Pedretti A, Villa L, Vistoli G. VEGA: a versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J Mol Graph Model. 2002;21(1):47–49. doi: 10.1016/S1093-3263(02)00123-7. [DOI] [PubMed] [Google Scholar]
- 58.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2009;11(11):2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Srinivasan E, Ravikumar S, Venkataramanan S, Purohit R, Rajasekaran R. Molecular mechanics and quantum chemical calculations unveil the combating effect of baicalein on human islet amyloid polypeptide aggregates. Mol Simul. 2019;45(18):1538–1548. doi: 10.1080/08927022.2019.1660778. [DOI] [Google Scholar]
- 60.Katritzky AR, Ignatchenko ES, Barcock RA, Lobanov VS, Mati Karelson. Prediction of gas chromatographic retention times and response factors using a general qualitative structure-property relationships treatment. Anal Chem. 1994;66(11):1799–807. doi: 10.1021/ac00083a005. [DOI] [Google Scholar]
- 61.Frau J, Flores-Holguín N, Glossman-Mitnik D. Chemical reactivity theory and empirical bioactivity scores as computational peptidology alternative tools for the study of two anticancer peptides of marine origin. Molecules. 2019;24(6):1115. doi: 10.3390/molecules24061115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M (1999) Automated analysis of interatomic contacts in proteins. Bioinformatics 15(4):327–32. 10.1093/bioinformatics/15.4.327 [DOI] [PubMed]
- 64.Isralewitz B, Gao M, Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr Opin Struct Biol. 2001;11(2):224–230. doi: 10.1016/S0959-440X(00)00194-9. [DOI] [PubMed] [Google Scholar]
- 65.Greenland S, Senn SJ, Rothman KJ, Carlin JB, Poole C, Goodman SN, et al. Statistical tests, P values, confidence intervals, and power: a guide to misinterpretations. Eur J Epidemiol. 2016;31(4):337–350. doi: 10.1007/s10654-016-0149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen D, Oezguen N, Urvil P, Ferguson C, Dann SM, Savidge TC. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci Adv. 2016;2(3):e1501240. doi: 10.1126/sciadv.1501240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(4):894–904.e9. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu Y, Wang F, Shen C, Peng W, Li D, Zhao C, et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. 2020;368(6496):1274–1278. doi: 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang L, Song G, Jernigan RL. Protein elastic network models and the ranges of cooperativity. Proc Natl Acad Sci. 2009;106(30):12347–12352. doi: 10.1073/pnas.0902159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martínez L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS One [Internet]. 2015 Mar 27 [cited 2016 Feb 29];10(3). Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4376797/ [DOI] [PMC free article] [PubMed]
- 71.Meshach Paul D, Chadah T, Senthilkumar B, Sethumadhavan R, Rajasekaran R. Structural distortions due to missense mutations in human formylglycine-generating enzyme leading to multiple sulfatase deficiency. J Biomol Struct Dyn. 2018;36(13):3575–3585. doi: 10.1080/07391102.2017.1394220. [DOI] [PubMed] [Google Scholar]
- 72.Talmaciu MM, Bodoki E, Oprean R. Global chemical reactivity parameters for several chiral beta-blockers from the Density Functional Theory viewpoint. Clujul Med. 2016;89(4):513–518. doi: 10.15386/cjmed-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srinivasan E, Rajasekaran R. Molecular binding response of naringin and naringenin to H46R mutant SOD1 protein in combating protein aggregation using density functional theory and discrete molecular dynamics. Prog Biophys Mol Biol. 2019;145:40–51. doi: 10.1016/j.pbiomolbio.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Chandrasekhar G, Rajasekaran R. In silico therapeutic peptide design against pathogenic domain swapped human cystatin C dimer. Int J Pept Res Ther. 2021;27(2):1555–1575. doi: 10.1007/s10989-021-10191-2. [DOI] [Google Scholar]
- 75.Pearson RG. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci U S A. 1986;83(22):8440–8441. doi: 10.1073/pnas.83.22.8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J, Peng C, Yu Y, Chen Z, Xu Z, Cai T, et al. Exploring conformational change of adenylate kinase by replica exchange molecular dynamic simulation. Biophys J . 2020;118(5):1009–1018. doi: 10.1016/j.bpj.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taddese B, Garnier A, Abdi H, Henrion D, Chabbert M. Deciphering collaborative sidechain motions in proteins during molecular dynamics simulations. Sci Rep. 2020;10(1):15901. doi: 10.1038/s41598-020-72766-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chwastyk M, Bernaola AP, Cieplak M (2015) Statistical radii associated with amino acids to determine the contact map: fixing the structure of a type I cohesin domain in the Clostridium thermocellum cellulosome. Phys Biol 12(4):046002. 10.1088/1478-3975/12/4/046002 [DOI] [PubMed]
- 79.Ninković DB, Filipović JPB, Hall MB, Brothers EN, Zarić SD (2020) What is special about aromatic–aromatic interactions? Significant attraction at large horizontal displacement. ACS Cent Sci 6 [DOI] [PMC free article] [PubMed]
- 80.Chandra Sekar, P., Chandrasekhar, G. & Rajasekaran, R (2021) Hydrophobic Residues Confer the Helicity and Membrane Permeability of Ocellatin-1 Antimicrobial Peptide Scaffold Towards Therapeutics. Int J Pept Res Ther 27:2459–2470. 10.1007/s10989-021-10265-1, https://link.springer.com/10.1007/s10989-021-10265-1
- 81.Sumi T, Imamura H. Water-mediated interactions destabilize proteins 12 [DOI] [PMC free article] [PubMed]
- 82.Macchiagodena M, Pagliai M, Andreini C, Rosato A, Procacci P (2020) Upgraded AMBER Force Field for Zinc-Binding Residues and Ligands for Predicting Structural Properties and Binding Affinities in Zinc-Proteins. ACS Omega 5(25):15301–15310. 10.1021/acsomega.0c01337 [DOI] [PMC free article] [PubMed]
- 83.West DK, Olmsted PD, Paci E (2006) Free energy for protein folding from nonequilibrium simulations using the Jarzynski equality. J Chem Phys 125(20):204910. 10.1063/1.2393232 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.
Not applicable.