

Abstract
Purpose of review
The application of spatial transcriptomics technologies to the interrogation of kidney tissue is a burgeoning effort. These technologies share a common purpose in mapping both the expression of individual molecules and entire transcriptomic signatures of kidney cell types and structures. Such information is often superimposed upon a histologic image. The resulting datasets are readily merged with other imaging and transcriptomic techniques to establish a spatially anchored atlas of the kidney. This review provides an overview of the various spatial transcriptomic technologies and recent studies in kidney disease. Potential applications gleaned from the interrogation of other organ systems, but relative to the kidney, are also discussed.
Recent findings
Spatial transcriptomic technologies have enabled localization of whole transcriptome mRNA expression, correlation of mRNA to histology, measurement of in situ changes in expression across time, and even subcellular localization of transcripts within the kidney. These innovations continue to aid in the development of human cellular atlases of the kidney, the reclassification of disease, and the identification of important therapeutic targets.
Summary
Spatial localization of gene expression will complement our current understanding of disease derived from single cell RNA sequencing, histopathology, protein immunofluorescence, and electron microscopy. While spatial technologies continue to evolve rapidly, their importance in the localization of disease signatures is already apparent. Further efforts are required to integrate whole transcriptome and subcellular expression signatures into the individualized assessment of human kidney disease.
Keywords: Kidney biopsy, histopathology, Visium, SlideSeq
Introduction
Spatial anchoring is essential to define the relationship between cells and structures within a tissue. Modern spatial transcriptomics platforms enable spatial localization of whole transcriptome mRNA expression, often overlaid upon histological information from the same tissue section. Gene expression profiles are then mapped back to their original location, enabling a direct link between gene expression of neighboring cells or histologic structures. By integrating cell type definitions derived from single cell or nuclear RNA sequencing (sc/snRNAseq) to improve cell type specificity, novel data outputs can be acquired which complement traditional histopathologic assessment of the kidney. These outputs include cell neighborhood definitions, receptor ligand interactions of epithelial and immune cells, and localization of relevant downstream pathways activated in disease pathology. Other highly sensitive spatial technologies may enable high resolution subcellular localization of mRNA within the kidney’s cells. Together, these outputs facilitate the ongoing development of transcriptomic atlases of the kidney.
In this discussion, the various spatial transcriptomic technologies are first introduced, followed by an evaluation of recent examples in which spatial interrogation has helped to advance our understanding of kidney disease.
Spatial transcriptomic interrogation technologies
Spatial transcriptomic technologies can be broadly grouped into those that characterize mRNA localization in tissue at the regional, cellular, or subcellular resolution. These technologies seek to link mRNA expression with a histological address for each cell type. Recent advancements have improved strategies to extract information efficiently. Six technology types worthy of highlighting include:
i). Microdissection:
Microdissection of the kidney can be accomplished through manual dissection (1) of the nephron or slide-based laser capture of labelled structures and cells (2). Traditional slide based technologies have isolated glomeruli from the tubulointerstitium, but this technique can be adapted to acquire specific cell types from all nephron subsegments using a rapid antibody-based immunofluorescence stain protocol (3). After tissue isolation, mRNA is extracted, cDNA is synthesized, and sequencing is performed in a manner analogous to bulk tissue processing for whole transcriptome measurement. The acquired tissue can be subjected to multiple interrogation techniques including proteomics (4) or bisulfite sequencing to measure methylation of cytosine residues (5). While microdissection is a form of spatial anchoring, it suffers from a number of drawbacks. First, once the tissue is dissected, the underlying histology or immunofluorescence information is lost. Second, to acquire sufficient tissue to measure mRNA expression, multiple glomeruli or renal tubular cells must be pooled, yielding an average signature with significantly less specificity than sc/snRNAseq technologies.
ii). Fluorescence in situ hybridization (FISH):
Multiplexed FISH provides targeted expression of a set of genes at subcellular resolution with high sensitivity, even for lowly expressed transcripts. In separate rounds, probes bind to mRNA, and subsequent reporter probes bind to the encoding probes. Quantitation of fluorescence in situ hybridization is obtained by counting fluorescent signals. These techniques have been used to define erythropoietin-producing cells in the murine kidney (6) and characterize co-expression of protein and mRNA (mIFISH) in human transplant kidney biopsy specimens (7). Multiplexed FISH technologies such as MERFISH (8, 9) and seqFISH+ (10) can now reach a repertoire of 10,000 co-expressed targets within a tissue. The broad application of such nearly whole transcriptome approaches is presently limited by cost, challenges in probe design, and labor-intensive experimental processes.
iii). in situ sequencing (ISS):
ISS technologies also use fluorescence output, but in contrast to FISH, the mRNA is read and converted to cDNA nucleotide by nucleotide and then sequenced with rolling circle amplification within the tissue. Examples include fluorescent in situ sequencing (FISSEQ) (11, 12) and spatially-resolved transcript amplicon readout mapping (STARmap) (13, 14). These technologies yield subcellular resolution for over 1000 multiplexed targets; however, the sensitivity tends to be lower as compared to FISH. Greater multiplexing further reduces sensitivity and increases sequencing time. Limited data is available in the application to kidney disease at the time of this review.
iv). in situ capturing (ISC):
ISC methods, sometimes referred to as solid phase-based capture technologies, utilize tissue adhered to a slide that contains equidistant capture probes, each barcoded to allow back-mapping of the mRNA signature to its original spatial location. ISC-based technologies like Slide-seq (15) and 10x Visium (16) can reduce experimental complexity, eliminate probe design requirements, and provide whole transcriptome expression. Spatial relationships are rebuilt with barcoded localization and expression quantification. Slide-seq has near single-cell resolution because the slide area is covered by 10-micron beads. The 10x Visium platform has a larger capture area, but expression is mapped over a hematoxylin and eosin stained histological image in the same section for a streamlined experimental design. Each capture zone contains five thousand spots/probes of 55 microns diameter that overlie multiple cells. Thus, slide-seq lends itself to neighborhood analyses of juxtaposed cells, while the Visium platform requires strategies for spot deconvolution for component cells. In our experience, Visium detects approximately 2,500 unique genes per 55 μm spot and 20,000 unique genes per sample. Slide-seq’s 10 μm beads detect fewer genes per spot, but when scaled to area, the 2 techniques detect comparable numbers of genes. The overall cost for one Visium sample, including reagents, array slide, tissue preparation, cDNA synthesis, and sequencing is approximately $2,000. At present. Slide-seq’s commercial availability is limited at the time of writing.
v). Full Transcriptome Spatial RNA Analysis:
The GeoMx Human Whole Transcriptome platform (offered by NanoString) is a slide based technology for FFPE or frozen samples. Tissue is adhered to a slide and visualized on the GeoMx instrument, which contains both an ultraviolet laser and microscope. Regions of interest (ROI) are selected from the slide which then undergoes ultraviolet light dissociation and the resulting RNA from each ROI is captured in a 96-well plate (17). Each well contains barcodes allowing the RNA signature to be remapped to the ROI after cDNA synthesis and sequencing. As compared to each Visium assay, the approximate cost per sample is less and total genes detected per sample is similar (~18,000). However, the GeoMx assay generally yields fewer ROIs than Visium spots and requires supervision to select each region.
vi). in silico reconstruction:
Using the expression signature of genes derived from sc/snRNAseq technologies, spatial localization can be inferred for each cell type. The technique essentially builds a virtual tissue atlas by applying computational methods. This approach, which has been applied to kidney, includes both localization of transcripts and the relative expression of contributing cells (18).
Application of spatial transcriptomics to experimental model systems of the kidney
As described above, multiple technologies are available to localize the comprehensive mRNA expression profile of the kidney. The application of in situ capturing methods to the kidney has received considerable attention in the last year. The exploration of kidney disease model systems with spatial transcriptomic ISC technologies complements recent sc/snRNAseq endeavors, by localizing cell types and injury signatures in situ. Janosevic et al. investigated a murine endotoxemia model at seven time points, defining the timeline of sepsis progression with single-cell RNA sequencing (19). Using pseudotime and velocity field analysis, the authors identify time-dependent phenotypical changes in epithelial and macrophages populations, as well as changes in receptor-ligand interactions, and the activity of genes associated with recovery in later time points. Using spatial transcriptomics, they localize the novel proximal tubule S3-Type 2 (identified in scRNAseq) to the outer medulla of the murine kidney (20).
A septic murine model was also investigated by Melo Ferreira et al. where spatial transcriptomics localized the septic injury in the murine kidney and, using a single cell reference dataset, described differences in the immune cell population when compared to a sham kidney (21). Since each capture spot of the 10X Visium platform covers multiple cells, the signature of immune cell infiltration may be disguised by the dominant transcriptomic signature of the more abundant epithelial cells. In order to uncover the subtle signature of infiltrating immune cells, the authors suppressed epithelial cell signatures from a reference scRNAseq dataset, before mapping it to the spatial transcriptomic sample. The authors applied the same methodology to an Ischemia-Reperfusion injury (IRI) model, identifying a subpopulation of proximal tubules involved in neutrophil chemotaxis localized in the outer medulla of the IRI murine model. The immune cell localization was validated for each model with codetection by indexing (CODEX) immunofluorescence.
Subsequently, the localization of sex specific characteristics of the IRI murine model were identified (22). Specifically, the authors found female mice were relatively protected from IRI with a comparable degree of injury in a 34-minute IRI model as compared to a 22-minute model in their male counterparts. Kidneys were harvested at multiple time points to define the progression of injury. The authors utilized SPOTlight and Giotto to deconvolute the cell distribution in their spatial transcriptomics samples, describing differentially expressed genes and cell population changes along the course of IRI.
Using a combination of scRNAseq and spatial transcriptomics in a murine embryonic model, Sanchez-Ferras et al. described in great detail the development of the Nephric Duct (23). Using scRNAseq, the authors defined four unique cell populations and their contribution to the development of the Nephric Duct. They describe the contribution of Gata3 and Tfap2a/2b genes as regulators of morphogenesis. In this work, spatial transcriptomics was used to validate the spatial segregation of the four Nephric duct progenitor populations.
Building a human kidney atlas
ICS-based spatial transcriptomic technologies may prove an important tool in the construction of a human kidney atlas in health and disease. The molecular interrogation of human kidney biopsy specimens has garnered considerable attention from consortia (24–26). Many spatial transcriptomic technologies are slide based and can be performed in the existing workflow of kidney biopsy processing of paraffin-embedded or Optimal Cutting Temperature (OCT) frozen blocks. Conceivably, a CLIA-validated version of a future spatial transcriptomic assay could one day supplement the existing assays a nephropathologist uses to interpret a biopsy specimen. An early application of 10X Visium spatial transcriptomics, interrogation of a healthy human nephrectomy revealed the ability to map most cell types derived from the Kidney Precision Medicine Project sc/snRNAseq atlas (21, 27). A strong alignment between the mapped spatial transcriptomic defined cell types was observed with the underlying histology, showing better than 90% alignment in all cell types.
Another ICS-based technology, Slide-seqV2, was similarly used to explore spatial relationships in the human kidney (28). The technology was applied to cortical and medullary sections of renal nephrectomies to validate appropriate cell type localization. The authors then apply Slide-seqV2 in a diabetic murine model, detecting the expansion of the juxta-glomerular apparatus and an increase in podocytes injury markers as compared to a sham rodent (28). In a UMOD-C125R knockin kidney disease model, the authors found evidence that thick ascending limb, fibroblast, and macrophage cells contribute to disease-specific neighborhoods in the medulla of the injured mice, identifying injury-related pathways in those regions that could be missed in single-cell technologies where spatial anchoring is missing.
Both of these ICS-based spatial transcriptomic technologies were adapted by Lake et al. to create a comprehensive human atlas of the kidney in health and disease (29). With over 80 kidney samples, the authors apply single-cell and single-nuclei sequencing, combined with single-nucleus chromatin accessibility and mRNA expression sequencing (SNARE-seq) (30), to characterize and uncover over 100 cell populations. Several of these populations were subtypes of common cells which were in putative injury cell states referred to as adaptative and degenerative cell types. Visium and SlideSEQ were combined to localize these novel cell populations and to establish niches of populations associated with altered cell states. 3D tissue cytometry further defined those niches and better characterized the immune cell components of the neighborhoods. With the combination of SNARE-seq and sc/snRNAseq, the authors then define trajectories for the altered cell states, which were associated with disease progression.
The link between immune cell infiltration and altered epithelial expression was also assayed using the GeoMx platform (17). In this study, the authors identified differentially expressed genes in five regions of interest in a kidney allograft with T-cell mediated rejection, and compared expression to two healthy control biopsies, demonstrating feasibility of the technique in kidney tissue.
Of considerable importance is the link between the human kidney and the mouse in order to better understand the translatable elements of common disease models. To this end, Raghubar et al. describe the species differences between mammalian kidney in mice and humans (31). The study converts human genes to murine orthologs and describes the observed differentially expressed genes. This analysis was performed in aggregate pseudo-bulk samples, without a direct comparison of individual renal structures or cell types. In the human sample, glomerular spots were defined by the expression of common marker genes and validated with immunofluorescence. The authors describe receptor-ligand pairs co-expressed within and between glomerular spots, describing over 100 potential receptor-ligand interactions. Finally, spatial transcriptomics was explored as a tool to localize disease-related single nucleotide polymorphisms.
Deconvolution
Due to the larger diameter of barcoded capture beads in certain in situ capture technologies, the cell type classification may not achieve single cell resolution currently available sc/snRNAseq datasets. Unsupervised classification of the beads often results in broad cell types, while underrepresented cell type subpopulations might not be captured with significant frequency to be correctly defined. To overcome this shortcoming, a common strategy applied to spatial transcriptomics is the use of a reference single-cell or single-nuclei dataset to inform the classification of the spatial spots or beads (32). An example is provided in Figure 1. Studies using Slide-seqV2(28, 29) have successfully applied Robust Cell Type Decomposition (RCTD) (33) and Seurat (34, 35) for deconvolution. RCTD deconvolutes by first calculating the mean expression of each cell type in the reference dataset. It then fits a statistical model where the expression of each gene is a Poisson distribution of a linear combination of the mean expression profiles. Seurat, on the other hand, projects both datasets to a common dimensionally reduced space and finds pairs of elements from both datasets sharing nearest neighbors. These pairs are called anchors and are then used to transfer scores associated with each cell type label from the reference sc/snRNAseq dataset to each bead.
Figure 1: Cell type mapping in spatial transcriptomics.
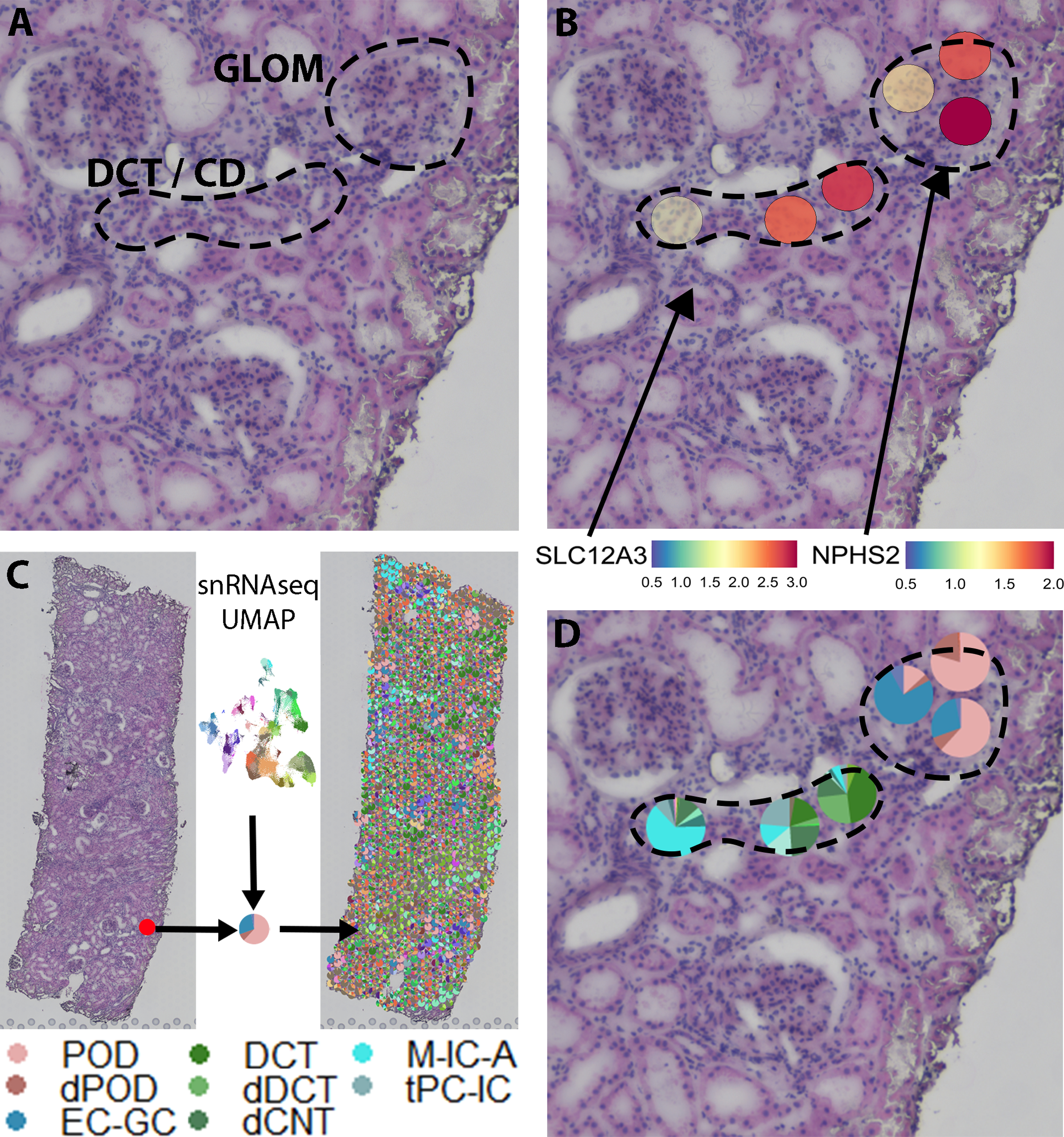
A) H+E image with a glomerulus and a transition from the distal tubule (DCT) into a collecting duct (CD). B) Podocin (NPHS2) is expressed in the glomerulus. The thiazide sensitive sodium chloride cotransporter (SLC12A3) expression decreases from right to left as the DCT transitions to CD. C) Schematic indicating that transfer scores are calculated for >100 snRNAseq clusters based on all genes expressed and then mapped onto the spatial transcriptomics sample in Seurat version 3, deconvoluting the proportion of expression in each spot corresponding to a snRNAseq cluster. D) Glomerular spots contain signature from podocytes (POD) and glomerular endothelial cells (EC-GC). Damaged POD (dPOD = cell state) signature is also seen. From right to left, a gradual transition is seen from DCT and connecting tubule (CNT) signatures to CD intercalated (MIC-A) and principal cell (tPC-IC) signatures.
Both technologies can be readily applied to Visium, but due to its spot size, multiple cell types are expected in each spot. Therefore, multiple deconvolution strategies have been designed. To uncover the transcriptomic signal of immune cells underlying the more abundant epithelial cell types, Seurat V3 can be used to map a subset of the reference dataset with immune cells and fibroblasts (21). Such an approach allowed the authors to localize immune cells, confirmed with immunofluorescence. In a later publication, the methodology is expanded to deconvolute each spot, where the contribution of each cell type in the reference is proportional to the transfer score obtained from Seurat (29). This approach helped to uncover niches of immune cells surrounding injured epithelial tissues. This cell type deconvolution strategy showed results consistent with the associated histological image and was cross-validated with the results obtained in their previous work.
Designed to deconvolute Visium spatial transcriptomics, SPOTlight (36) has been applied to kidney samples (20, 30). This method defines cell type topics, with the distribution of gene expression defining that cell type, in the reference dataset. Weights are then used to build each individual spot signature from the cell type topics. These weights can be interpreted as the proportion of each cell type captured by the spot. Other deconvolution methods have been proposed, such as Giotto (37), deconvoluting spatial transcriptomics (DSTG)(38), Tangram (14), or stereoscope (16), but applications in the kidney have yet to be examined.
Applications from other organ systems relevant to the kidney
To overcome the limitations of current spatial technologies, Chen et al. united in situ sequencing and in situ capture technologies to study brains of Alzheimer’s disease murine models (39). The authors combined immunofluorescence in sequential sections to the in situ capture technology. This strategy allowed identification of groups of genes related to amyloid plaque accumulation. When applied to the kidney, this approach could allow investigation of the relation between the whole transcriptome in the context of protein expression localization. Applications could include a link between extracellular matrix protein expression to neighboring cellular transcriptomic response in order to better define in situ pro-fibrotic processes or better characterize any immune cell infiltration which contributes to this process.
Using human embryonic hearts in four different developmental stages, Asp et al. describe the formation of multiple spatial and genetic patterns, uncovering new localized cell types (40). The Asp approach has particular relevance to the kidney given its very rich spatial structure and the unique dynamics of nephron morphogenesis. The approach could expand upon the Nephric duct developmental progression discussed above (23).
Another application of spatial transcriptomics described the structure of human white adipose tissue (41). The authors measure the propensity of cell types in close approximation to each other. The characterized white adipose tissue was found to be more organized than previously understood. The structure of the nephron is well described, but this method is complementary to the one presented by Lake et al., and could be applied to define the localization of cell states and injured cell types (29).
Lessons learned from the tumor microenvironment
A key advantage of spatial transcriptomics is the ability to define regions based on changes in expression signatures, even when the histology does not coincide with overt signs of disease. The tumor microenvironment is known to be heterogeneous with complex variables defining tumorigenesis, metastasis, and drug resistance (42). To better understand this, spatial transcriptomics has been applied to the tumor microenvironment to extract information about its architecture and tumor margins. Based on early studies, the tumor microenvironment and its near margins are composed of specialized tumor and muscle cells (43) and the margin is a transcriptionally distinct region from the tumor itself or tissue more distant from tumor margins. Analogously, spatial transcriptomics could be applied to seemingly normal histologic regions of human kidney biopsy specimens which border a patchy distribution of acute tubular necrosis or tubulointerstitial fibrosis and atrophy, in order to better understand the adaptive mechanisms of the renal penumbra adjacent to injury.
Conclusion
In the last two years, the application of spatial transcriptomic technologies to the kidney has exploded, with important insights gleaned from disease models and the creation of a spatially anchored human kidney atlas. The outputs of these technologies may one day complement the interpretation of kidney biopsy specimens by nephropathologists. Nonetheless, spatial transcriptomic techniques are constantly evolving and our understanding of cell-cell neighborhoods and localization of cell states is still a nascent endeavor. The sheer quantity of recent high impact studies reviewed here portends a vibrant landscape lies beyond the bleeding edge. Let’s go chart that landscape.
Keypoints.
Multiple spatial transcriptomic technologies have been developed to localize mRNA expression signatures. The utility and application of these technologies is constantly evolving.
Spatial transcriptomics has been applied to the kidney to better understand the pathogenesis underlying disease models and to create of a spatially anchored human kidney atlas.
Insights from other organ systems and the tumor microenvironment provide strategies which can be applied to define the distribution of injury cell states in the kidney.
Acknowledgments
Financial support and sponsorship
None.
Footnotes
Conflicts of Interest
There are no conflicts of interest.
References:
- 1.Lee JW, Chou CL, Knepper MA. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes. J Am Soc Nephrol. 2015;26(11):2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barwinska D, El-Achkar TM, Melo Ferreira R, Syed F, Cheng YH, Winfree S, et al. Molecular characterization of the human kidney interstitium in health and disease. Sci Adv. 2021;7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiten Patel JT, Emilio Poggio, Bebiak Jack, Alpers Charles, Grewenow Stephanie, Toto Robert, Eadon Michael. Molecular Signatures of Diabetic Kidney Disease Hiding in a Patient with Hypertension-Related Kidney Disease. Clinical Journal of the American Society of Nephrology. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satoskar AA, Shapiro JP, Jones M, Bott C, Parikh SV, Brodsky SV, et al. Differentiating Staphylococcus infection-associated glomerulonephritis and primary IgA nephropathy: a mass spectrometry-based exploratory study. Sci Rep. 2020;10(1):17179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park J, Guan Y, Sheng X, Gluck C, Seasock MJ, Hakimi AA, et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight. 2019;4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sciesielski LK, Felten M, Michalick L, Kirschner KM, Lattanzi G, Jacobi CLJ, et al. The circadian clock regulates rhythmic erythropoietin expression in the murine kidney. Kidney Int. 2021;100(5):1071–80. [DOI] [PubMed] [Google Scholar]
- 7.Junger H, Dobi D, Chen A, Lee L, Vasquez JJ, Tang Q, et al. Novel In Situ Hybridization and Multiplex Immunofluorescence Technology Combined With Whole-slide Digital Image Analysis in Kidney Transplantation. J Histochem Cytochem. 2020;68(7):445–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G, Moffitt JR, Zhuang X. Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci Rep. 2018;8(1):4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11(4):360–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568(7751):235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen HQ, Chattoraj S, Castillo D, Nguyen SC, Nir G, Lioutas A, et al. 3D mapping and accelerated super-resolution imaging of the human genome using in situ sequencing. Nat Methods. 2020;17(8):822–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc. 2015;10(3):442–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018;361(6400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biancalani T, Scalia G, Buffoni L, Avasthi R, Lu Z, Sanger A, et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat Methods. 2021;18(11):1352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363(6434):1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersson A, Bergenstrahle J, Asp M, Bergenstrahle L, Jurek A, Fernandez Navarro J, et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun Biol. 2020;3(1):565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salem F, Perin L, Sedrakyan S, Angeletti A, Ghiggeri GM, Coccia MC, et al. The spatially resolved transcriptional profile of acute T cell-mediated rejection in a kidney allograft. Kidney Int. 2022. Jan;101(1):131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nitzan M, Karaiskos N, Friedman N, Rajewsky N. Gene expression cartography. Nature. 2019;576(7785):132–7. [DOI] [PubMed] [Google Scholar]
- 19. Janosevic D, Myslinski J, McCarthy TW, Zollman A, Syed F, Xuei X, et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. Elife. 2021;10. *This is the first published example of in situ capturing-based spatial transcriptomics in the kidney.
- 20.Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361(6409):1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Melo Ferreira R, Sabo AR, Winfree S, Collins KS, Janosevic D, Gulbronson CJ, et al. Integration of spatial and single-cell transcriptomics localizes epithelial cell-immune cross-talk in kidney injury. JCI Insight. 2021;6(12). *This study deconvolutes cell type contributions to 10X Visium spots in the human kidney and examines the immune cell signature distribution in two models of murine injury.
- 22. Dixon E, Wu H, Muto Y, Wilson P, Humphreys B. Spatially Resolved Transcriptomic Analysis of Acute Kidney Injury in a Female Murine Model. J Am Soc Nephrol. 2021. *This study examines the relative time course of renal injury between female and male mice in an ischemia-reperfusion injury model.
- 23.Sanchez-Ferras O, Pacis A, Sotiropoulou M, Zhang Y, Wang YC, Bourgey M, et al. A coordinated progression of progenitor cell states initiates urinary tract development. Nature communications. 2021;12(1):2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Achkar TM, Eadon MT, Menon R, Lake BB, Sigdel TK, Alexandrov T, et al. A multimodal and integrated approach to interrogate human kidney biopsies with rigor and reproducibility: guidelines from the Kidney Precision Medicine Project. Physiological genomics. 2021;53(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PX, Barisoni L, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int. 2013;83(4):749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Townsend RR, Guarnieri P, Argyropoulos C, Blady S, Boustany-Kari CM, Devalaraja-Narashimha K, et al. Rationale and design of the Transformative Research in Diabetic Nephropathy (TRIDENT) Study. Kidney Int. 2020;97(1):10–3. [DOI] [PubMed] [Google Scholar]
- 27.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nature communications. 2019;10(1):2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jamie L Marshall TN, Wang Qingbow S., Silvana Bazua-Valenti, Haiqi Chen, Murray Evan, Subramanian Ayshwarya, Vernon Katherine A., Liguori Katie, Keller Keith, Stickels Robert R., Breanna McBean, Heneghan Rowan M., Weins Astrid, Macosko Evan Z., Chen Fei, Greka Anna. High Resolution Slide-seqV2 Spatial Transcriptomics Enables Discovery of Disease-Specific Cell Neighborhoods and Pathways. BioRxiv. 2021. *This is the first example of Slide-seqV2 utilization in the kidney.
- 29. Blue B Lake RM, Seth Winfree, Qiwen Hu, Ferreira Ricardo Melo, Kian Kalhor, Barwinska Daria, Otto Edgar A., Ferkowicz Michael, Diep Dinh, Plongthongkum Nongluk, Knoten Amanda, Urata Sarah, Naik Abhijit S., Eddy Sean, Zhang Bo, Wu Yan, Salamon Diane, Williams James C., Wang Xin, Balderrama Karol S., Hoover Paul, Murray Evan, Vijayan Anitha, Chen Fei, Waikar Sushrut S., Rosas Sylvia, Wilson Francis P., Palevsky Paul M., Kiryluk Krzysztof, Sedor John R., Toto Robert D., Parikh Chirag, Kim Eric H., Macosko Evan Z., Kharchenko Peter V., Gaut Joseph P., Hodgin Jeffrey B., Eadon Michael T., Dagher Pierre C., El-Achkar Tarek M., Kun Zhang, Matthias Kretzler, Sanjay Jain, for the KPMP consortium. An atlas of healthy and injured cell states and niches in the human kidney. BioRxiv. 2021. **This manuscript from the Kidney Precision Medicine Project includes a comprehensive and spatially anchored atlas of the kidney in health and disease.
- 30.Chen S, Lake BB, Zhang K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol. 2019;37(12):1452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arti M Raghubar DTP, Tan Xiao, Grice Laura F., Crawford Joanna, Lam Pui Yeng, Andersen Stacey B., Yoon Sohye, Ng Monica S.Y., Teoh Siok Min, Holland Samuel E., Anne Stewart, Leo Francis, Combes Alexander N., Kassianos Andrew J., Helen Healy, Quan Nguyen, Mallett Andrew J.. Spatially resolved transcriptome profiles of mammalian kidneys illustrate the molecular complexity of functional nephron segments, cell-to-cell interactions and genetic variants. BioRxiv. 2020. *This study compares spatial transcriptomic signatures in human and murine kidney.
- 32.Ricardo Melo Ferreira BJF, Eadon Michael T.. Deconvolution Tactics and Normalization in Renal Spatial Transcriptomics. Frontiers in Physiology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cable DM, Murray E, Zou LS, Goeva A, Macosko EZ, Chen F, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Li D, Xu X, Ziska LH, Zhu J, Liu G, et al. The potential role of sucrose transport gene expression in the photosynthetic and yield response of rice cultivars to future CO2 concentration. Physiol Plant. 2020;168(1):218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elosua-Bayes M, Nieto P, Mereu E, Gut I, Heyn H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 2021;49(9):e50. **This study relays a commonly used deconvolution method which can be applied to the kidney.
- 37.Dries R, Zhu Q, Dong R, Eng CL, Li H, Liu K, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 2021;22(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Q, Su J. DSTG: deconvoluting spatial transcriptomics data through graph-based artificial intelligence. Brief Bioinform. 2021;22(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell. 2020;182(4):976–91 e19. [DOI] [PubMed] [Google Scholar]
- 40.Asp M, Giacomello S, Larsson L, Wu C, Furth D, Qian X, et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell. 2019;179(7):1647–60 e19. [DOI] [PubMed] [Google Scholar]
- 41.Backdahl J, Franzen L, Massier L, Li Q, Jalkanen J, Gao H, et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. 2021;33(9):1869–82 e6. [DOI] [PubMed] [Google Scholar]
- 42.Wang N, Li X, Wang R, Ding Z. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnology journal. 2021;16(9):e2100041. [DOI] [PubMed] [Google Scholar]
- 43.Hunter MV, Moncada R, Weiss JM, Yanai I, White RM. Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface. Nature communications. 2021;12(1):6278. [DOI] [PMC free article] [PubMed] [Google Scholar]