

Abstract
The degeneration and death of motor neurons leads to motor neuron diseases such as Amyotrophic Lateral Sclerosis (ALS). Although the exact mechanism by which motor neuron degeneration occurs is not well understood, emerging evidence implicates the involvement of ferroptosis, an iron-dependent oxidative mode of cell death. We reported previously that treating Gpx4NIKO mice with tamoxifen to ablate the ferroptosis regulator glutathione peroxidase 4 (GPX4) in neurons produces a severe paralytic model resembling an accelerated form of ALS that appears to be caused by ferroptotic cell death of spinal motor neurons. In this study, in support of the role of ferroptosis in this model, we found that the paralytic symptoms and spinal motor neuron death of Gpx4NIKO mice was attenuated by a chemical inhibitor of ferroptosis. In addition, we observed that the paralytic symptoms of Gpx4NIKO mice were malleable and could be tapered by lowering the dose of tamoxifen, allowing for the generation of a mild paralytic model without a rapid onset of death. We further used both models to evaluate mitochondrial reactive oxygen species (mtROS) in the ferroptosis of spinal motor neurons and showed that overexpression of peroxiredoxin 3, a mitochondrial antioxidant defense enzyme, ameliorated symptoms of the mild but not the severe model of the Gpx4NIKO mice. Our results thus indicate that the Gpx4NIKO mouse is a versatile model for testing interventions that target ferroptotic death of spinal motor neurons in vivo.
Keywords: Ferroptosis, Motor Neuron Degeneration, Glutathione Peroxidase 4, Amyotrophic Lateral Sclerosis, mitochondrial reactive oxygen species, Peroxiredoxin 3
Introduction:
Developing effective treatments for motor neuron diseases such as Amyotrophic Lateral Sclerosis (ALS) has proven to be a tremendous challenge. Despite spending hundreds of millions of dollars conducting preclinical scientific research and dozens of human clinical trials over the past few decades, there are no treatments that are considered effective in curing ALS, or even in extending patient lifespan by more than a few months (Kiernan et al. 2020). One major impediment of the development of effective treatments is the lack of in vivo models of rapid motor neuron degeneration. Some of the most commonly used mouse models of ALS involve transgenic mice expressing mutated forms of the human Superoxide Dismutase 1 (SOD1) gene, which were identified in familial ALS cases (Gurney et al. 1994). Other non-SOD1 mouse models of motor neuron diseases have been developed as well (Stephenson and Amor 2017). However, almost all of these mouse models of motor neuron death and degeneration do not exhibit onset and progression of symptoms for many weeks or months, vastly increasing the amount of time and expense required to test novel therapeutic treatments.
Another issue impeding the development of treatments for ALS and motor neuron diseases is that the precise modalities of cell death involved in these diseases are not well understood. Although apoptosis is believed to be a mechanism of motor neuron death, interventions that target apoptosis has failed to arrest the progression of these diseases in human clinical trials (Gordon et al. 2007). The disappointing results of these trials suggest that apoptosis alone cannot account for the loss of neurons observed in these diseases. Ferroptosis is a mode of cell death caused by iron-mediated oxidation of phospholipids in membranes which leads to a buildup of hydroperoxides in phospholipids that ultimately result in the demise of the cells (Dixon et al. 2012; Yang and Stockwell 2016). Interestingly, characteristics of ferroptosis are associated with motor neuron diseases. For example, excessive iron accumulation (Hall et al. 1998; Barber and Shaw 2010; Do et al. 2016) and increased lipid peroxidation (Pedersen et al. 1998; Simpson et al. 2004; Perluigi et al. 2005; Devos et al. 2019) were widely reported in human ALS patients and mouse models of motor neuron diseases. The presence of ferroptotic markers in motor neuron diseases suggests that ferroptosis is a modality of cell death in motor neuron degeneration
Glutathione Peroxidase 4 (GPX4) is the master regulator of ferroptosis, a dysfunction of which can result in excessive lipid peroxidation which will ultimately initiate ferroptotic cell death (Yang et al. 2014). As previously reported, the Gpx4 Neuron Inducible Knockout (Gpx4NIKO) mouse (Chen et al. 2015) is a model which allows for the neuron-specific, inducible knockout of Gpx4 by treatment with Tamoxifen (TAM). When adult Gpx4NIKO mice were treated with TAM, they experienced a rapid degeneration and death of spinal motor neurons within days. As a result, the mice developed a paralytic syndrome with symptoms that included loss of locomotor function, loss of muscle mass and bodyweight, and died within two weeks. These symptoms resembled those observed in mouse models of motor neuron disease as well as those experienced by human motor neuron disease patients, though they occurred and progressed over a much shorter duration (Drey et al. 2013; Gordon et al. 2013). In addition, the spinal motor neuron death observed in Gpx4NIKO mice was associated with markers of ferroptotic cell death, including increased 4-HNE protein adducts and elevated neuroinflammation, suggesting ferroptosis is the main mechanism of motor neuron death in this model (Chen et al. 2015).
In the present study, we wished to further test the role of ferroptosis in the spinal motor neuron death in Gpx4NIKO mice by determining whether direct inhibition of ferroptosis via treatment with Liproxstatin-1 (Lip-1), a small molecule compound inhibitor of ferroptosis, would slow the progression of motor neuron disease in this model. In addition, we wondered if the severity of the paralytic symptoms of Gpx4NIKO mice could be modulated with different doses of TAM; something which could aid in the testing of interventions. Furthermore, we tested whether overexpression of Peroxiredoxin 3 (Prdx3), an enzyme known to reduce mitochondrial reactive oxygen species (mtROS) levels, could affect the paralysis development of Gpx4NIKO mice.
Method:
2.1. Animal cohorts, conditional ablation of Gpx4 and liproxstatin-1 treatment
The generation of Gpx4(f/f) mice was described previously (Yoo et al. 2012). The SLICK mice, which express a tamoxifen-activatable form of Cre recombinase in neurons under the direction of Thy1 promoter (Heimer-McGinn et al. 2011), were obtained from The Jackson Laboratory (Bar Harbor, Maine). Gpx4NIKO mice with floxed Gpx4 alleles and the SLICK transgene were generated by cross-breeding Gpx4(f/f) mice with SLICK mice. Gpx4NIKO mice were subsequently cross-bred with Gpx4(f/f) mice to generate mice used in this study. To investigate the role of mtROS in the ferroptosis of spinal motor neurons, we also generated Gpx4NIKO mice that overexpress Prdx3, a mitochondria-specific antioxidant defense mechanism (i.e., Gpx4NIKO/PRDX3 mice). Gpx4NIKO/PRDX3 mice were produced by successive breeding of Gpx4NIKO mice with Tg(PRDX3) transgenic mice generated in our lab (Chen et al. 2008). Gpx4NIKO mice at 2–4 months of age were enrolled in experimental cohorts.
Tamoxifen (T5648, Sigma Chemical Co., St. Louis, MO) was dissolved in corn oil at a concentration of 10 mg/ml. Tamoxifen (TAM) was administered to Gpx4NIKO mice intraperitoneally (i.p.) at a dose of 60 mg/kg of body weight for a total of five injections (once daily). Control Gpx4NIKO mice without tamoxifen received daily injection of corn oil only. Additionally, TAM was administered to mice via single injections (i.p.) at dose of 90mg/kg or 45mg/kg of bodyweight.
Liproxstatin-1 (Cat #-S7699, Selleckem.com) was dissolved in DMSO, then diluted in PBS. After first tamoxifen treatment, mice were injected i.p. daily with liproxstatin-1 at a dose of 10 mg/kg or with the vehicle (1.5% DMSO in PBS).
This study was reviewed and approved by Institutional Animal Care and Use Committees (IACUCs) of the University of Texas Health San Antonio and the Audie Murphy Memorial Veterans Hospital, South Texas Veterans Health Care System.
2.2. Locomotor function assays
Rotarod performance was measured with a Rotamex 4/8 (Columbus Instruments, Columbus, OH) using an accelerating rod protocol. The initial speed of the rod was set to 2 rpm with a linear acceleration to 40 rpm over 300 seconds. The latencies to fall were used as indicators of rotarod performance.
The hang-wire test is a motor task measuring balance and grip strength that involves placing the mice on a wire-mesh cage top and inverting it so that the hang upside down (Weydt et al. 2003; Miana-Mena et al. 2005). The duration for which the mice are able to hang upside down, known as the latency to fall, was evaluated in 2–3 trials a maximum time of 60 seconds.
2.3. Survival determination
Mice were checked twice daily for general appearance and locomotor behavior including movement initiation, walking and turning. A mouse that was immobile for a period of 20 seconds due to paralysis in four limbs would be euthanized humanly and recorded as dead.
2.4. Tissue preparation and immunofluorescence staining
After a mouse was sacrificed humanely, the spine was quickly dissected out on ice. The spine was put in a weigh-boat filled with ice-cold saline and cut into sections. Using a 5-ml syringe with an attached blunt needle (#9), the spinal cord was gently flushed out from the spine. The spinal cord was fixed in 4% paraformaldehyde at 4°C overnight and equilibrated in 30% sucrose in PBS for 1–2 days at 4°C. The spinal cord was then snap-frozen by submersion in 2-metylbutane chilled in dry ice. The spine cord sections at a thickness of 16 μm were made using a cryostat.
For immunofluorescence staining, spinal cord sections were blocked with blocking buffer (5% BSA, .3% Triton X-100 in PBS) for 60 minutes and incubated with primary antibody in PBS at 4°C overnight. The sections were then washed 3 times with PBS and incubated with the fluorophore-conjugated secondary antibody (AlexaFluor546- labeled rabbit antibody) for 2–4 hours at room temperature in PBS. After washing 3 times, slides were mounted with ProLong Gold Antifade Reagent (P36930, Invitrogen, Carlsbad, CA), observed and imaged with a confocal microscope.
2.5. Antibodies and Western blots
Antibodies used: Anti-NeuN (MAB377, Millipore, Billerica, Massachusetts); anti-ChAT, Anti-PSD95, anti-Actin, anti-GFAP (Cell Signaling Technology, Beverly, MA); anti-GAPDH (Sigma-Aldrich, St. Louis, MO); anti-Iba-1 (Wako Chemicals USA, VA); anti-4-HNE antibody from R&D Systems (Minneapolis, MN); and anti-GPX4 (Santa Cruz Biotechnology, CA).
Protein levels in tissues were determined by Western blots using respectively antibodies. The protein level of β-Actin was used to adjust for loading. The bands were visualized using the ECL Kit (RPN2132, GE Healthcare, Piscataway, NJ). The bands were quantified using NIH ImageJ software, and normalized to the loading control. The mean level of protein of interest (the ratio of protein to Actin) in controls was assigned as 1 arbitrarily, and relative data are expressed as mean ± SEM.
2.6. Real-Time quantitative PCR (RT-qPCR)
Total RNA was isolated from spinal cord tissues using Tri Reagent (Molecular Research Center, Cincinnati, OH) and reverse-transcribed using random hexamers and Multi-Scribe Reverse Transcriptase (Applied Biosystems, Foster City, CA). The mRNA levels of GFAP and Iba-1 were quantified using standard RT-qPCR protocol and were normalized to β-Actin to control for input RNA. The primers used are as follows: β-Actin (forward: 5’-ATC TGG CAC CAC ACC TTC TAC-3’; reverse: 5’-CAG GTC CAG ACG CAG GAT G-3’), GFAP (forward: 5’- GTT GGC AAC CTA TGG TTC GT −3’; reverse: 5’- CAG GAG CTG GTT GCT TTT CT −3’). Iba-1 (forward: 5’-AGC TTT TGG ACT GCT GAA GG −3’; reverse: 5’- GGC AGA TCC TCA TCA TTG CT −3’).
2.7. Detection of Cre-mediated Gpx4 ablation
A PCR-based method was used to detect Cre-mediated recombination of the floxed Gpx4 allele in genomic DNA isolated from spinal cord tissues (Chen et al, 2015). PCR reactions were performed using primers (P1, 5’-TAC TGC AAC AGC TCC GAG TTC-3’; P2, 5’-CTT CAC CAC GCA GCC GTT CT-3’) which produce a 700bp-amplicon from the recombined Gpx4 (rGpx4) allele.
2.8. Statistical Analysis
All data are expressed as mean±SEM. Significant differences in bodyweight, rotarod and hang-wire test performances were calculated using repeated measures ANOVA for day (average of 2–3 trials) and genotype/treatment. Significance was assigned using one-way ANOVA for all other measures. p<0.05 was considered significant for all analysis.
Results:
3.1. Treatment with ferroptosis inhibitor Lip-1 retarded onset of paralysis and extended survival of Gpx4NIKO mice.
We reported previously that conditional ablation of Gpx4 in Gpx4NIKO mice (Gpx4 Neuronal Inducible Knockout mouse, which has two floxed Gpx4 alleles and a tamoxifen-activatable Cre recombinase gene directed by a neuron-specific Thy1 promoter) resulted in rapid onset of paralysis, loss of muscle mass and bodyweight, and death driven by a dramatic degeneration of spinal motor neurons (Chen et al. 2015). Given the role of Gpx4 in inhibiting ferroptosis, a non-apoptotic mode of cell death that is oxidative and inflammatory, we hypothesized that the motor deficits, neuronal loss, and greatly diminished lifespan of the Gpx4NIKO mouse model are the result of ferroptotic cell death. Indeed, the degeneration of spinal motor neurons in Gpx4NIKO mice was associated with features of ferroptosis.
To further determine the importance of ferroptosis in spinal motor neuron degeneration induced by Gpx4 ablation, we treated Gpx4NIKO mice with Liproxstatin-1 (Lip-1), a small-molecule compound that can reduce lipid ROS and was shown to be a specific inhibitor of ferroptosis that does not impair other cell death modalities like apoptosis or necrosis (Friedmann-Angeli et al. 2014). After the initiation of tamoxifen (TAM) treatment, the animals were divided into two cohorts. The first cohort of Gpx4NIKO mice received Lip-1 via i.p. injection daily at a dose of 10 mg/kg, while the other cohort of mice were injected with the vehicle only. We assessed the locomotor function of these cohorts of Gpx4NIKO using a rotarod task and determined the time of paralysis onset in each cohort based on when the mice showed a significant decline of rotarod performance. As shown in Fig. 1a, Gpx4NIKO mice treated with vehicle showed a significant decline of rotarod performance on day 7 after initiating TAM treatment, indicating onset of paralysis. In comparison, Gpx4NIKO mice treated with 10 mg/kg of Lip-1 had onset of paralysis on day 10 after initiating TAM treatment, indicating that Lip-1 treatment delayed the onset of paralysis. We also measured the change in bodyweight of both cohorts of mice, as we have previously shown that Gpx4NIKO mice show a rapid and precipitous decline in bodyweight following TAM injections. As is apparent in Fig. 1b, treatment with Lip-1 prevented the Gpx4NIKO mice from dropping below the threshold of 80% of their initial bodyweight that the vehicle treated mice surpassed on average around 9 days following TAM injections. Additionally, we measured the survival times of the two cohorts of mice. Gpx4NIKO mice treated with vehicle lived for 10–12 days after initiating TAM treatment, while Gpx4NIKO mice treated with Lip-1 at a dose of 10 mg/kg lived for 13–16 days. The respective median survival times for these cohorts was 10 days for vehicle treated mice and 15 days for Lip-1 treated mice. The Kaplan-Meier survival curves for the two cohorts of mice are presented in Fig. 1c. The hazard rate of the Gpx4NIKO mice treated with Lip-1 was significantly different from that of Gpx4NIKO mice treated with Vehicle judged by the log-rank test (p < 0.05), indicating that Lip-1 extended the survival of the Gpx4NIKO mice.
Fig. 1.
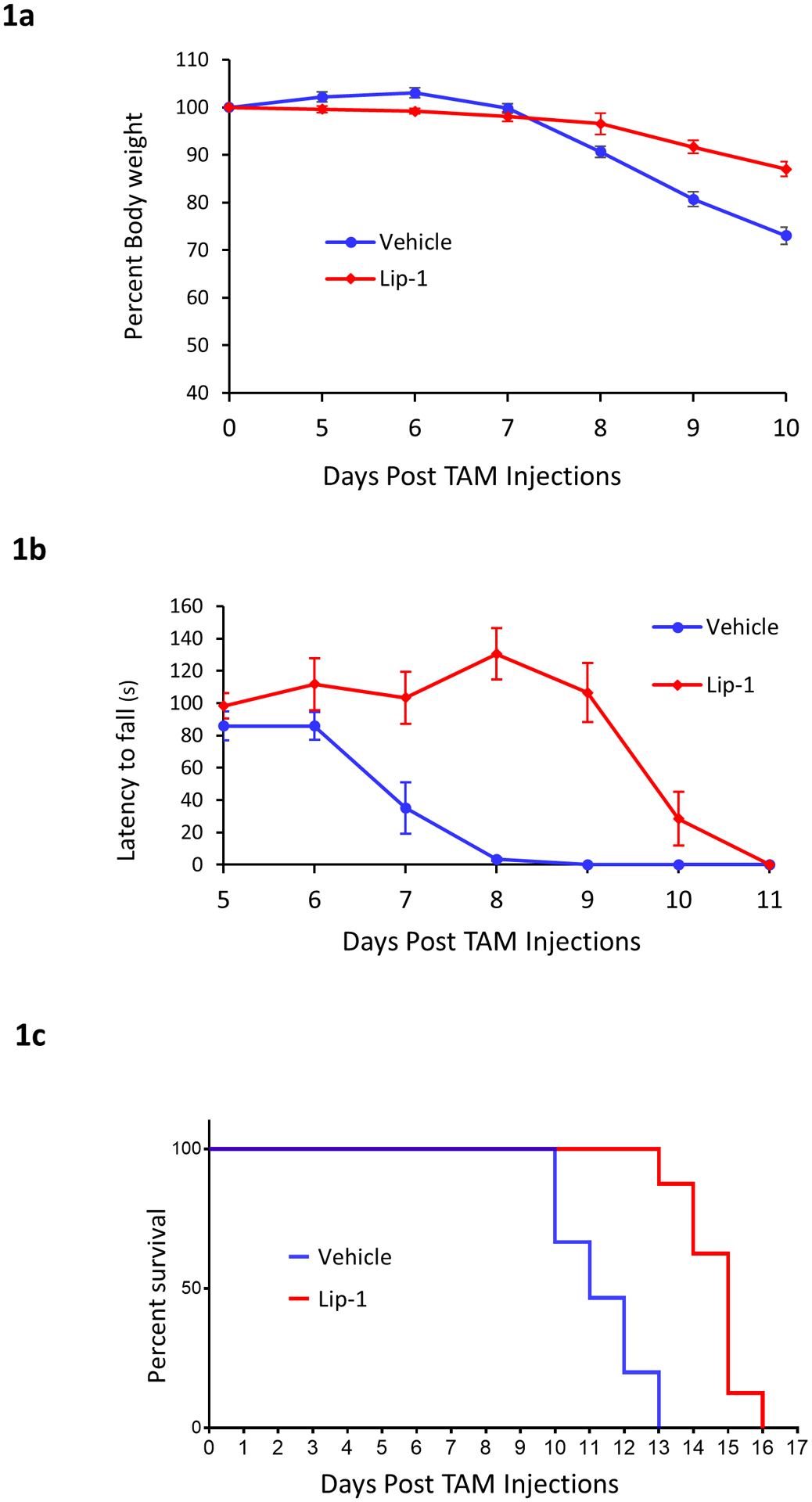
Ferroptosis inhibitor Lip-1 delayed paralysis onset and extended survival of Gpx4NIKO mice. Gpx4NIKO mice were treated with high dose (60 mg/kg i.p. injection daily for five consecutive days) of tamoxifen (TAM) to ablate Gpx4. Afterwards, one cohort of mice were given vehicle (1.5% DMSO in PBS) and served as controls (Vehicle), another cohort were treated daily with liproxstatin-1 (Lip-1) at a dose of 10 mg/kg. a. Rotarod performance of Vehicle mice and Lip-1 mice. b. Change in percent of initial bodyweight of mice treated with Vehicle or Lip-1. C. Survival curves of Gpx4NIKO mice treated with vehicle or Lip-1. n=8 for all cohorts
3.2. Lip-1 treatment ameliorated spinal motor neurodegeneration in Gpx4NIKO mice.
To assess the effect of Lip-1 on spinal motor neuron degeneration, we collected lumbar spinal cord tissues from Gpx4NIKO mice treated with vehicle or Lip-1 (10 mg/kg) at day 10 after TAM treatment, as well as from control Gpx4NIKO mice without any TAM or Lip-1 treatment. We then compared levels of neural marker proteins between control Gpx4NIKO mice and the TAM-treated Gpx4NIKO mice that were administered either Lip-1 or vehicle. Choline acetyltransferase (ChAT) is a motor neuron specific protein. As shown in Fig. 2a and 2b, vehicle treated mice had a significantly decreased level of ChAT protein compared with control mice without TAM treatment, indicating a severe degeneration of spinal motor neurons triggered by Gpx4 ablation. Consistent with the delayed paralysis development, Lip-1 treated mice had a significantly higher level of ChAT protein (Fig. 2a and 2b). We also compared levels of two other neural specific proteins: PSD95 and NeuN. Levels of both PSD95 and NeuN proteins were decreased in vehicle treated mice. Notably, Lip-1 treated mice also had higher levels of PSD95 and NeuN than vehicle treated mice. These results thus indicate that Lip-1 treatment ameliorated spinal motor neuron degeneration induced by Gpx4 ablation.
Fig. 2.
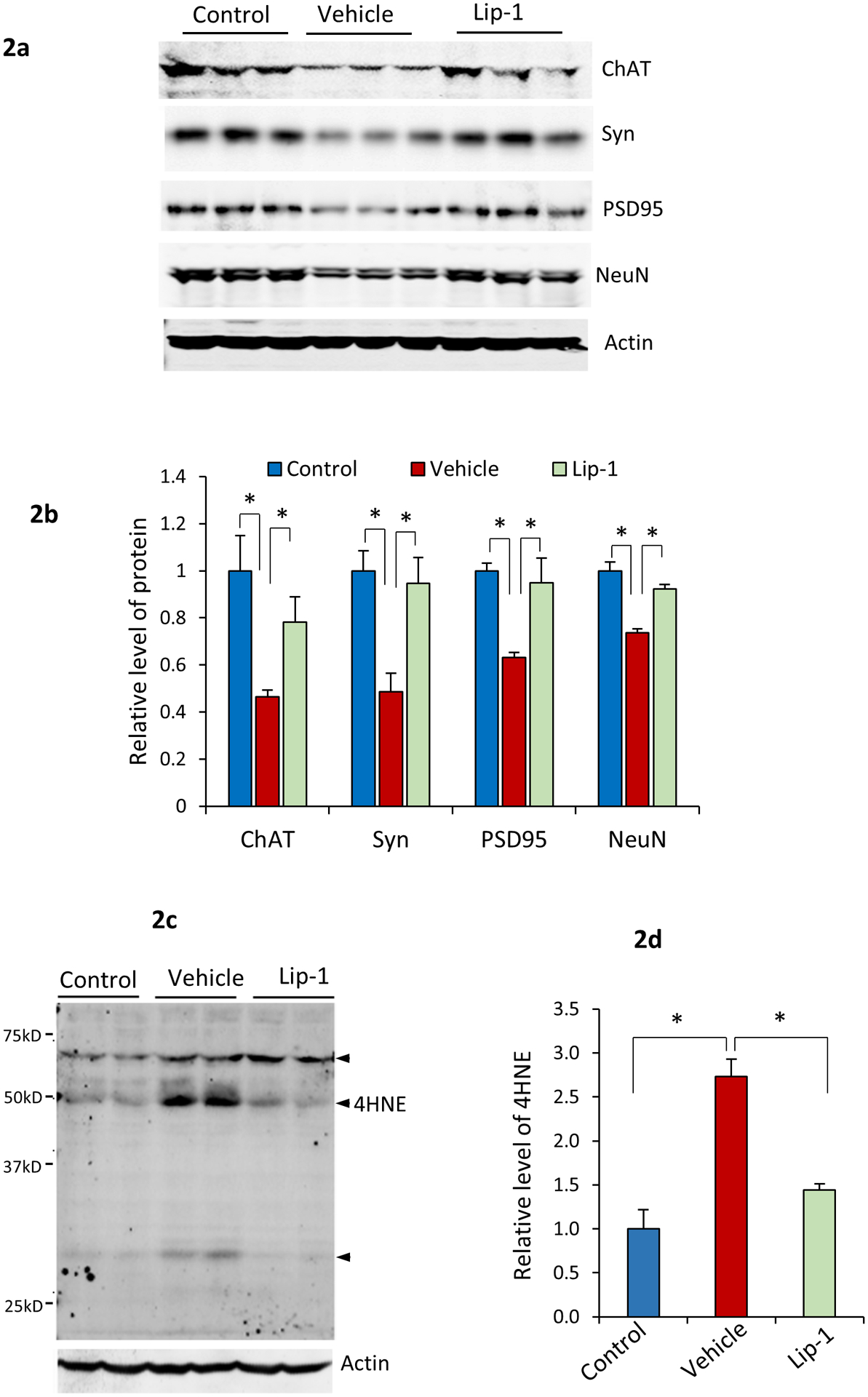
Lip-1 ameliorated spinal motor neuron degeneration in Gpx4NIKO mice. a. Western blots showing levels of ChAT, PSD95, and NeuN in spinal cord tissues from Gpx4NIKO mice treated with high dose of TAM then with vehicle (Vehicle), Gpx4NIKO mice treated with high dose of TAM then with Lip-1, and Gpx4NIKO mice without TAM treatment (Control). b. Quantified levels of ChAT, PSD95, and NeuN in spinal cord tissues from Control mice, Vehicle mice and Lip-1 mice. n=3, *: p<0.05. c. A graph of Western blots of spinal cord proteins from Control mice, Vehicle mice and Lip-1 mice probed with an anti-4-HNE antibody. Arrows indicate 4-HNE protein adducts in proteins. d. Quantified levels of 4-HNE adducts. n=4, *p<0.05.
3.3. Liproxstatin-1 treatment reduced lipid peroxidation in Gpx4NIKO mice
Lipid peroxidation is the driving force of ferroptosis (Yang et al. 2016), and we showed previously that motor neuron degeneration in TAM-treated Gpx4NIKO mice was associated with elevated levels of 4-hydroxynonenal (4-HNE) protein adducts. To determine if Lip-1 decreased lipid peroxidation in Gpx4NIKO mice, we compared 4-HNE adducts levels in spinal cord tissues from Control mice, Vehicle-treated mice, and Lip-treated mice by Western blots. As shown in Fig. 2c and 2e, consistent with our previous report, Gpx4NIKO mice treated with Vehicle had an increased level of 4-HNE protein adducts, indicating elevated lipid peroxidation. As expected, compared with Vehicle-treated mice, Lip-1-treated mice had a decreased level of 4-HNE protein adducts, indicating that Lip-1 significantly reduced lipid peroxidation in Gpx4NIKO mice.
3.4. Reduced astrogliosis and microgliosis in Gpx4NIKO mice treated with Lip-1.
Inflammation is a characteristic of ferroptosis because cells dying through ferroptosis release damage-associated molecular patterns (DAMPs) and lipid metabolites that are immunogenic (Friedmann-Angeli et al. 2014; Linkermann et al. 2014). We showed previously that spinal motor neuron degeneration was associated with elevated neuroinflammation in spinal cord of Gpx4NIKO mice. To determine the effect of Lip-1 on neuroinflammation, we first compared microgliosis and astrogliosis by immunofluorescence staining using the most common glial markers, Iba-1 for microglia and GFAP for astrocytes. The confocal microscopy images of lumbar spinal cord (ventral horn region) are shown in Fig. 3a–b. Vehicle treated mice had numerous Iba-1 positive mature microglia compared with control mice. Notably, spinal cord from Lip-1 treated mice had less Iba-1 positive microglia. Lip-1 treated mice also had a remarkable reduction of astrogliosis, as evidenced by the decreased staining intensity of GFAP. To corroborate the immunofluorescence staining results, we also compared levels of Iba-1 and GFAP proteins by Western blots (Fig. 3c). As showed in Fig. 3d, compared with vehicle treated mice, mice treated with Lip-1 had significantly decreased levels of Iba-1 and GFAP proteins. We further compared levels of Iba-1 and GFAP mRNA by quantitative real-time RT-PCR. Similar to the results of proteins, Lip-1 treatment significantly reduced Iba-1 and GFAP message levels (Fig. 3e).
Fig. 3.
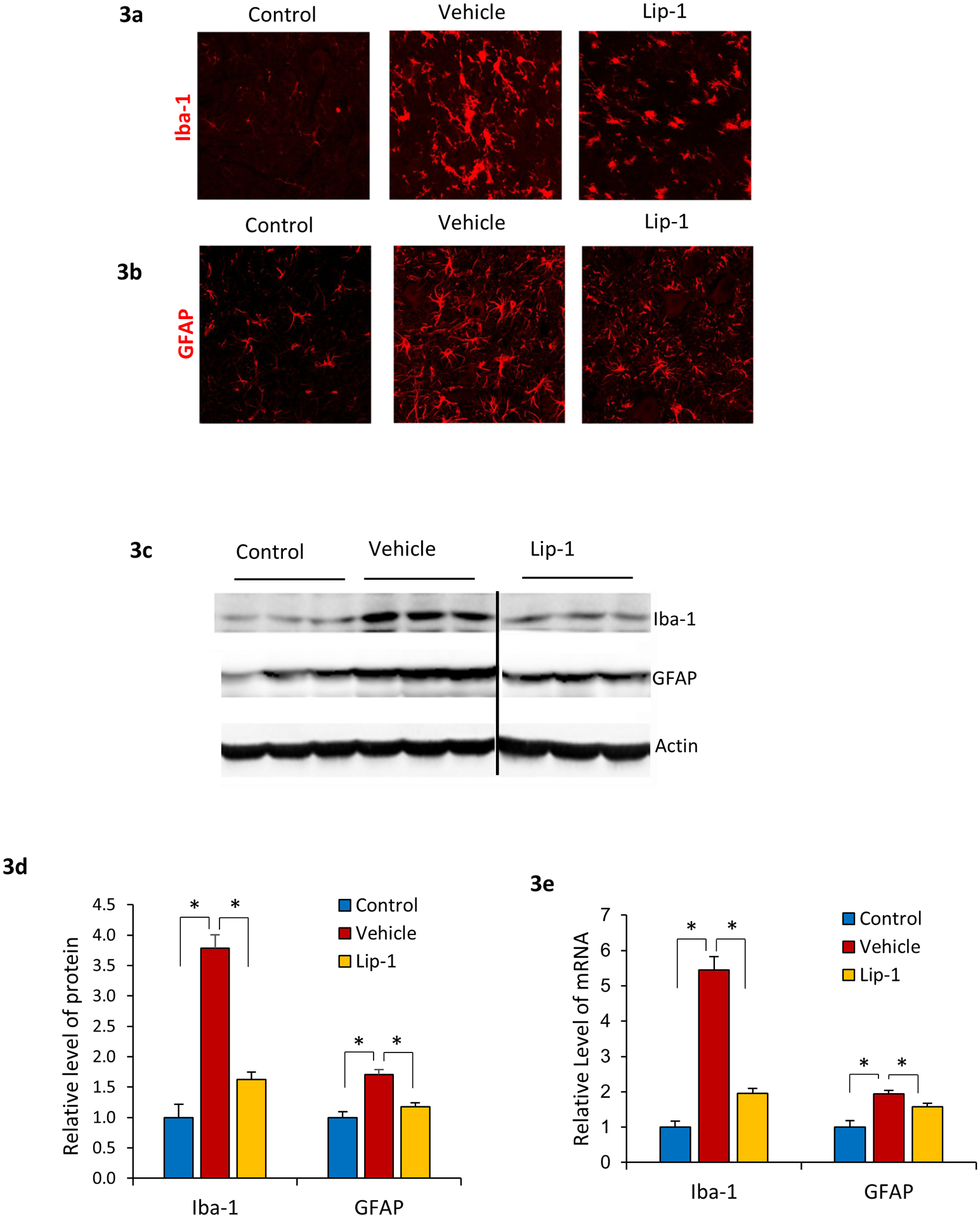
Suppressed neuroinflammation in Gpx4NIKO mice treated with Lip-1. Images of lumbar spinal cord sections from Control mice, Vehicle mice and Lip-1 mice at day 10 of treatment stained with an anti-Iba-1 antibody (a) or an anti-GFAP antibody (b). c. Western blots showing levels of GFAP and Iba-1 in spinal cord tissues from Control mice, Vehicle mice and Lip-1 mice. d. Quantified results of GFAP and Iba-1 protein levels. e. Results of GFAP and Iba-1 mRNA levels. n=3, *: p<0.05.
3.5. Severity of paralysis development in Gpx4NIKO mouse model is TAM dose-dependent.
We previously induced Gpx4 ablation in Gpx4NIKO mice using a protocol consisting of five consecutive days of 60 mg/kg TAM injections (totaling 300 mg/kg). Low doses of TAM can cause incomplete cre-loxP recombination, which leads to partial gene deletion and mild phenotypes (Reinert et al. 2012). To determine if the phenotypes of Gpx4NIKO mice are TAM dose-dependent, we treated Gpx4NIKO mice with different TAM dose regimens and assessed the motor neuron disease phenotypes of the treated mice. We found that when given a single 90 mg/kg TAM i.p. injection, mice exhibited nearly identical timeline for developing the neurodegenerative symptoms compared to five consecutive days of 60mg/kg TAM injections (i.e. onset of bodyweight loss and paralytic symptoms 7 days after starting TAM treatment, with the majority subjects dying by day 12). Therefore, we called Gpx4NIKO mice treated with either the 90 mg/kg dose of TAM administered by a single injection, as well as the 300 mg/kg cumulative dose administered over 5 days of injections, the severe paralytic model because they experience a rapid decline of locomotor function and bodyweight as well as death.
We also evaluated Gpx4NIKO mice treated with a single injection of TAM at a dose of 45 mg/kg. Fig. 4a shows that none of the subjects treated with 45mg/kg of TAM had reached the 80% of initial bodyweight threshold by the end of the 2-week observation period, whereas all of those in the high TAM dose condition (90 mg/kg or 300 mg/kg) had done so by this point. Similarly, it took several days longer for many of the 45mg/kg TAM treated mice to show a decline in behavioral assessments of motor control including the rotarod and hang-wire tests compared to the mice treated with higher TAM doses, and required a notably longer time to reach a state of total paralysis (Fig. 4b–c). Thus, Gpx4NIKO mice treated with 45 mg/kg TAM in a single injection produced similar symptoms as those observed in the higher TAM doses, but these symptoms developed more slowly and were often less pronounced. Therefore, we called Gpx4NIKO mice treated with 45 mg/kg TAM in a single dose a mild paralytic model. Thus, by varying the doses of TAM, two distinct variants of Gpx4NIKO mice can be generated: a severe paralytic model that results in rapid spinal motor neuron degeneration followed by the onset of motor impairments, paralysis, and death, as well as a mild paralytic model that has a slow progression of motor deficits and does not result in the development of complete paralysis, severe loss in bodyweight, or rapid onset of death.
Fig. 4.
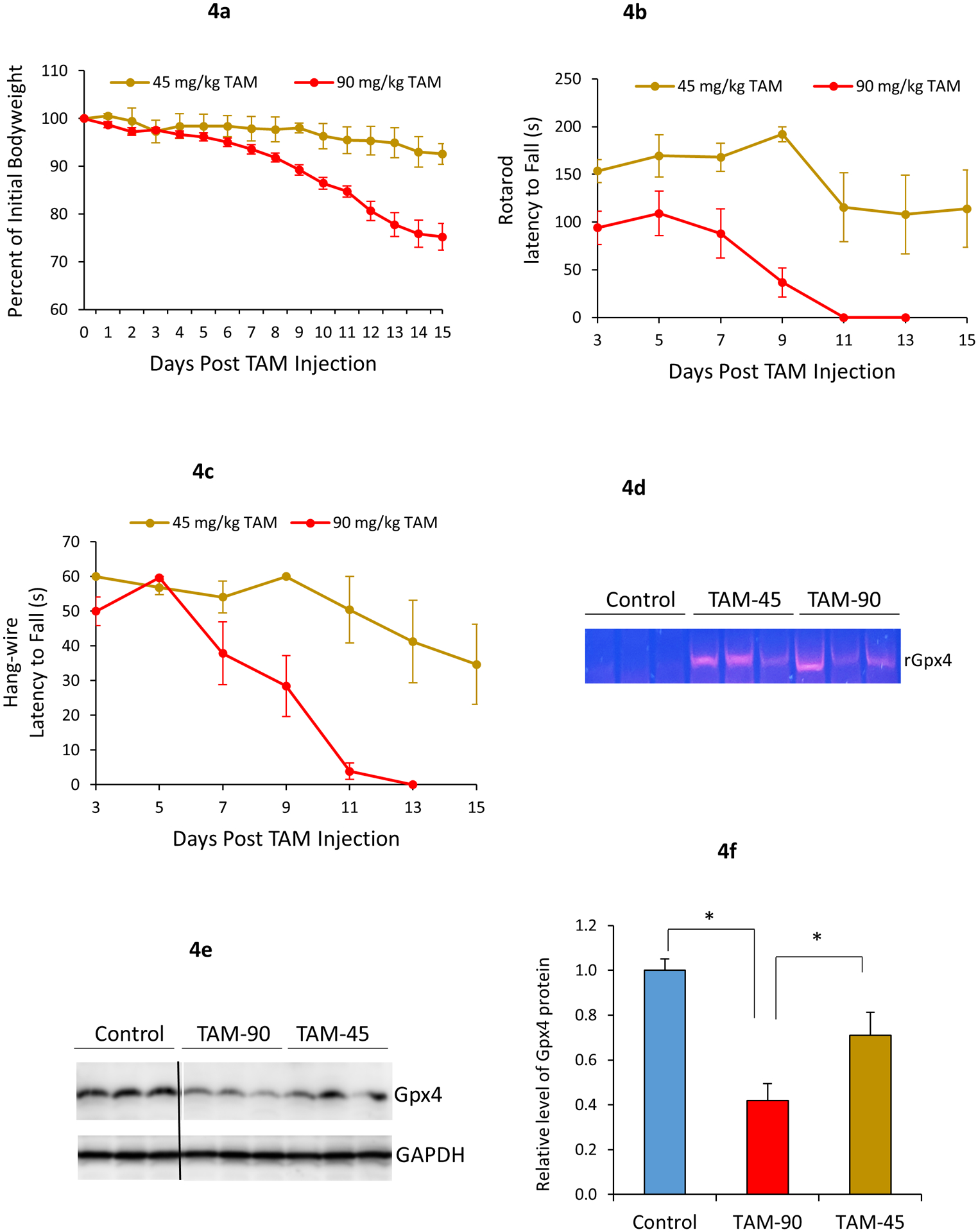
Severity of paralytic phenotypes of Gpx4NIKO mice induced by TAM is dose-dependent. Gpx4NIKO mice were treated with either a high dose (single 90 mg/kg i.p. injection) or a low dose (single 45 mg/kg i.p. injection) of TAM to ablate Gpx4. a. Change in percent of initial bodyweight of mice treated with high or low dose of TAM. b. Rotarod performance of Gpx4NIKO mice treated with high or low dose of TAM. c. Wire hang test performance of Gpx4NIKO mice treated with high or low dose of TAM. n=8 for the high dose TAM cohort and n=6 for the low dose TAM cohort. d. Gel image showing detection of rGpx4 by PCR in spinal cord tissues from Gpx4NIKO mice treated with TAM. e. Graph of western blots showing levels of Gpx4 in Control mice, Gpx4NIKO mice treated with 45 mg/kg (TAM-45) or 90 mg/kg (TAM-90) of TAM. f. Quantified results of western blots. n=3. *: p<0.05.
We further inspected the status of Gpx4 in spinal cord tissues of Gpx4NIKO mice treated with the high or low dose of TAM. To confirm Gpx4 ablation, we used a PCR-based method to detect the presence of recombined Gpx4 allele (rGpx4) which is derived from cre-mediated recombination of floxed-Gpx4 allele (Chen et al 2015). As expected, rGpx4 was readily detected in spinal cord tissues from Gpx4NIKO mice treated with both 45 mg/kg and 90 mg/kg of TAM, but not in Gpx4(f/f) control mice treated with 90 mg/kg of TAM (Fig. 4d). We next compared Gpx4 levels by Western blots. Consistently with Gpx4 ablation, Gpx4NIKO mice treated with both 45 mg/kg and 90 mg/kg of TAM had reduced Gpx4 protein compared with control Gpx4(f/f) mice treated with 90 mg/kg of TAM (Fig. 4e and Fig. 4f). However, mice treated with 45 mg/kg TAM exhibited a higher level of Gpx4 protein than mice treated 90 mg/kg TAM. Thus, the mild paralytic model induced by treatment with 45 mg/kg TAM appears to be due to partial ablation of Gpx4.
3.6. Overexpression of Prdx3 delayed onset and reduced severity of locomotor impairment in the mild paralytic Gpx4NIKO model.
While the mitochondria are the major source of ROS in many cells, mitochondrial ROS (mtROS) were not originally thought to participate in ferroptosis (Dixon et al. 2012). However, more recent studies have shown a connection between mitochondrial dysfunction and ferroptosis in certain cell types (Abdalkader et al. 2018; Battaglia et al. 2020). Peroxiredoxin 3 (Prdx3) is an antioxidant defense enzyme effective in reducing mtROS (Zhang et al. 2007). We previously generated Tg(PRDX3) mice that overexpress human Prdx3 ubiquitously, and showed that mitochondria from Tg(PRDX3) mice have significantly reduced mtROS (Chen et al. 2008). To test whether mtROS contribute to the ferroptosis of spinal motor neurons, we crossed Gpx4NIKO mice with Tg(PRDX3) mice, generating Gpx4NIKO/PRDX3 mice. To determine the effect of Prdx3 overexpression on paralysis development in Gpx4NIKO mice, we first treated Gpx4NIKO/PRDX3 mice and control Gpx4NIKO mice with a high dose (90 mg/kg) of TAM to induce severe paralysis. However, the results showed that Gpx4NIKO/PRDX3 mice did not have an altered trajectory of the bodyweight decline that is characteristic of the severe paralytic model of Gpx4NIKO mice, indicating that Prdx3 overexpression was not sufficient to impact the paralysis development of this model (Data not shown).
We theorized that it could be possible that the paralytic symptoms of Gpx4NIKO mice treated with 90 mg/kg TAM may have been too severe for Prdx3 overexpression to overcome. Therefore, we also treated Gpx4NIKO/PRDX3 mice and control Gpx4NIKO mice with the low dose (45 mg/kg) of TAM to induce the mild paralytic model. Given that survival and loss of bodyweight are not characteristic of this mild model, we instead focused on the effects of Prdx3 overexpression on the development of locomotor impairment in these mice. Fig. 5a–b show that when treated with a single 45 mg/kg TAM injection, the Gpx4NIKO/PRDX3 mice showed little to no decline in performance on the rotarod and hang-wire tests respectively over the course of the experiment compared to the notable reduction in performance observed in the control Gpx4NIKO mice. These results indicate that overexpression of Prdx3 is sufficient to reduce the severity of the motor impairments associated with the mild paralytic model of the Gpx4NIKO model, suggesting that mtROS are involved in ferroptosis of spinal motor neurons observed in Gpx4NIKO mice.
Fig. 5.
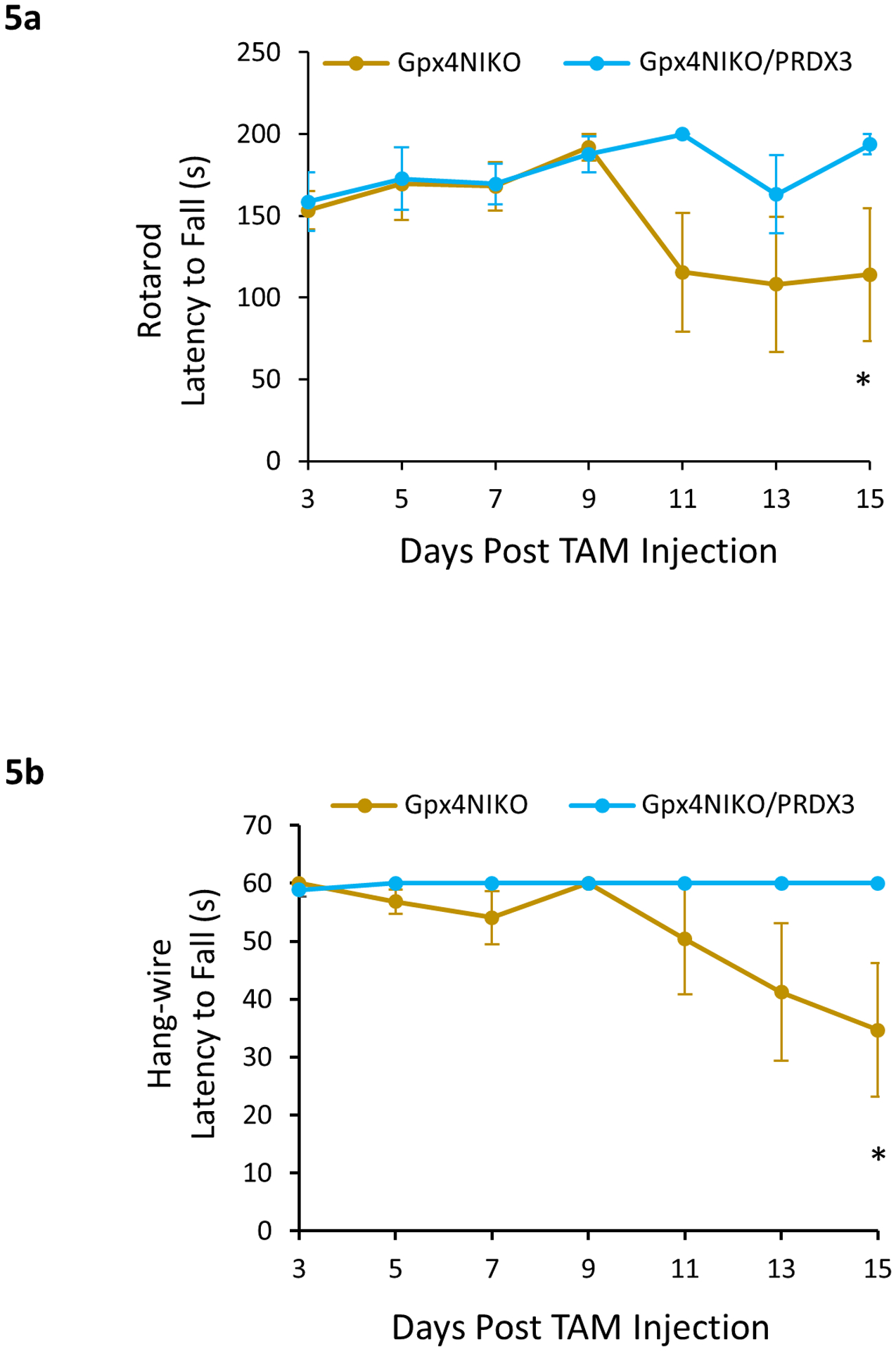
Prdx3 overexpression prevented motor impairment in the mild paralytic Gpx4NIKO model. Gpx4NIKO/PRDX3 and control Gpx4NIKO mice were treated with a low dose (single 45 mg/kg i.p. injection) of TAM to ablate Gpx4. a. Rotarod performance of Gpx4NIKO/PRDX3 and control Gpx4NIKO mice treated with the low dose of TAM. b. Hang-wire test performance of Gpx4NIKO/PRDX3 and control Gpx4NIKO mice treated with the low dose of TAM. n=6–8. *: p<0.05.
Discussion:
Previously, we have shown that the death of spinal motor neurons in Gpx4NIKO mice was associated with markers of ferroptotic cell death, suggesting that ferroptosis is the key mechanism of neurodegeneration in this model (Chen et al. 2015). Our findings from this current study show that treatment with Lip-1, a known small-molecule inhibitor of ferroptosis, significantly reduced the severity of the spinal motor neuron degeneration, lipid peroxidation, neuroinflammation, and locomotor dysfunction of Gpx4NIKO mice. These results support the notion that the pathology of the Gpx4NIKO mouse model was driven by the ferroptotic death of spinal motor neurons, and indicates that the Gpx4NIKO mouse model is suitable for testing therapeutic interventions targeting ferroptosis of neurons in neurodegenerative diseases. Despite its beneficial effects on delaying the onset of the paralytic syndrome, Lip-1 treatment did not completely rescue the Gpx4NIKO mice from paralysis development. At present, we do not know why this is the case, but it is likely that factors such as pharmacological dynamics of the compound, dose, and tissue/cell availability contributed to its inability to fully rescue the paralysis induced by Gpx4 ablation.
The Gpx4NIKO mouse model is a robust in vivo model of spinal motor neuron degeneration that develops symptoms similar to human motor neuron diseases (Chen et al. 2015). One of the primary benefits of this model over other mouse models of motor neuron disease is that the onset of the pathology occurs very rapidly and progresses in an accelerated, consistent manner that can be initiated at any specific point in the animal’s lifespan. Because of this, we believe the Gpx4NIKO model has the potential to serve as a valuable tool in the evaluation of the efficacy of therapeutic interventions designed to treat neurodegeneration compared to other mouse models of motor neuron diseases in which the paralytic phenotypes take much longer to develop (Stephenson and Amor 2017). To further increase the utility of Gpx4NIKO mice, we experimented with administering lower doses of TAM in hopes of producing less severe paralytic models in which some therapeutic treatments may exhibit a greater efficacy. We determined that a single, high-dose of TAM (90 mg/kg) was sufficient to produce a nearly identical severe paralytic model to what occurred with the 5-day series of TAM injections (300 mg/kg), but that a single, low-dose injection of TAM (45 mg/kg) actually caused a mild paralytic model with a significant delay in the onset of pathology and reduction in the severity of symptoms. This indicates the severity of the degenerative phenotype observed in the Gpx4NIKO model caused by TAM treatment is dose-dependent, and suggests that the lower TAM doses are failing to produce a complete neuronal ablation of Gpx4 in the mouse model. Thus, by varying the dosing regimens of TAM treatment, a severe or a mild paralytic model can be generated from Gpx4NIKO mice, allowing for flexibility of testing interventions in different conditions.
Prdx3 is an enzyme known to reduce mitochondrial ROS (Chen et al. 2008). We showed previously that mitochondria from Prdx3 overexpressing mice have significantly decreased ROS production. In this study, we showed that Gpx4NIKO mice overexpressing Prdx3 (i.e. Gpx4NIKO/PRDX3) showed no difference in survival and body weight loss compared to control Gpx4NIKO mice when using the severe paralytic model. However, in the mild paralytic model, Gpx4NIKO/PRDX3 showed slowed decline in motor performance compared to Gpx4NIKO controls. Although mtROS were not thought to contribute to ferroptosis originally, recent studies have linked mitochondrial dysfunction to ferroptosis in various cell types (Abdalkader et al. 2018; Battaglia et al. 2020). Because of the key role of ferroptosis in the development of paralysis of Gpx4NIKO mice, this result suggests the involvement of mtROS in the ferroptosis of spinal motor neurons.
Our results indicate that the Gpx4NIKO model develops an accelerated paralytic syndrome similar to ALS and that the degeneration and death of spinal motor neurons in Gpx4NIKO mice appears to be driven by ferroptosis. Ferroptosis has been implicated as playing a key role in ALS related neurodegeneration (Pedersen et al. 1998; Simpson et al. 2004; Perluigi et al. 2005; Devos et al. 2019, Chen et al. 2021). Our results from this study indicate that the Gpx4NIKO mouse is a versatile model that is useful for testing therapeutic interventions targeting ferroptosis-driven death of spinal motor neurons in vivo.
It is worth noting some limitations of the mild and severe paralytic models of the Gpx4NIKO mouse in modeling ferroptosis-based neurodegeneration and in testing therapeutic interventions. For instance, because ferroptosis of motor neurons in Gpx4NIKO models is driven by loss of Gpx4, interventions aiming to increase Gpx4 expression and/or Gpx4 activity may not be suitable for testing with these models. In addition, while Gpx4NIKO mice afflicted with the mild paralytic model do experience a slower onset and progression of symptoms with less severity, there is still only a short period of time between the initial TAM treatment and the plateau of symptom progression. Despite these limitations, the Gpx4NIKO models may serve as quick and cost-effective models for testing of interventions aiming to retard ferroptosis of motor neurons at early stage of therapeutic development.
Funding:
This work was supported in part by Merit Review Award I01 BX003507 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program and by an award from Owens Foundation San Antonio to Q.R. Q.R. is also supported by grant AG064078 from NIA, NIH.
Footnotes
Conflicts of interest/Competing interests: The authors declare no competing interests.
Ethics approval: Procedures for handling mice in this study were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Texas Health San Antonio and the Audie Murphy Memorial Veterans Hospital, South Texas Veterans Health Care System. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication: All authors have given their permission for publication.
Availability of data and material:
Available upon request.
References:
- Abdalkader M, Lampinen R, Kanninen KM, Malm TM, Liddell JR (2018) Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front Neurosci 12:1–9. 10.3389/fnins.2018.00466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber SC, Shaw PJ (2010) Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 48:629–641. 10.1016/j.freeradbiomed.2009.11.018 [DOI] [PubMed] [Google Scholar]
- Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F (2020) Ferroptosis and cancer: mitochondria meet the “iron maiden” cell death. Cells 9:1–26. 10.3390/cells9061505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hambright WS, Na R, Ran Q (2015) Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J Biol Chem 290:28097–28106. 10.1074/jbc.M115.680090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Na R, Gu M, Salmon AB, Liu Y, Liang H, Qi W, Van Remmen H, Richardson A, Ran Q (2008) Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell 7:866–878. 10.1111/j.1474-9726.2008.00432.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Na R, McLane KD, Thompson CS, Gao J, Wang X, Ran Q (2021) Overexpression of ferroptosis defense enzyme Gpx4 retards motor neuron disease of SOD1G93A mice. Sci Rep 11:1–13. 10.1038/s41598-021-92369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Moreau C, Kyheng M, Garçon G, Rolland AS, Blasco H, Gelé P, Lenglet TT, Veyrat-Durebex C, Corcia P, Dutheil M, Bede P, Jeromin A, Oeckl P, Otto M, Meininger V, Danel-Brunaud V, Devedjian J, Duce JA, Pradat PF (2019) A ferroptosis-based panel of prognostic biomarkers for Amyotrophic Lateral Sclerosis. Sci Rep 9:1–6. 10.1038/s41598-019-39739-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B III, B.R. Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do VB, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC (2016) Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis 94:169–178. 10.1016/j.nbd.2016.05.011 [DOI] [PubMed] [Google Scholar]
- Drey M, Grosch C, Neuwirth C, Bauer JM, Sieber CC (2013) The Motor Unit Number Index (MUNIX) in sarcopenic patients. Exp Gerontol 48:381–384. 10.1016/j.exger.2013.01.011 [DOI] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Forster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16:1180–1191. 10.1038/ncb3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PH (2013) Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis 4:295–310. https://doi.org/10.14336%2FAD.2013.0400295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, Spitalny GM, MacArthur RB, Mitsumoto H et al. (2007) Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol 6:1045–1053. 10.1016/S1474-4422(07)70270-3 [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD et al. (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264:1772–1775. 10.1126/science.8209258 [DOI] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME (1998) Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J Neurosci Res 53:66–77. [DOI] [PubMed] [Google Scholar]
- Heimer-McGinn V, Young P (2011) Efficient inducible Pan-neuronal cre-mediated recombination in SLICK-H transgenic mice. Genesis 49:942–949. 10.1002/dvg.20777 [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Vucic S, Talbot K, McDermott CJ, Hardiman O, Shefner JM, Al-Chalabi A, Huynh W, Cudkowicz M, Talman P, Van den Berg LH, Dharmadasa T, Wicks P, Reilly C, Turner MR (2021) Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol 17:104–118. 10.1038/s41582-020-00434-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De ZF, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Brasen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U.S.A. 111:16836–16841. 10.1073/pnas.1415518111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miana-Mena FJ, Muñoz MJ, Yagüe G, Mendez M, Moreno M, Ciriza J, Zaragoza P, Osta R (2005) Optimal methods to characterize the G93A mouse model of ALS. Amyotroph Lateral Scler Frontotemporal Degener 6:55–62. 10.1080/14660820510026162 [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, Kasarskis E, Mattson MP (1998) Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol 44:819–824. 10.1002/ana.410440518 [DOI] [PubMed] [Google Scholar]
- Perluigi M, Poon HF, Hensley K, Pierce WM, Klein JB, Calabrese V, De Marco C, Butterfield DA (2005) Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice--a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med 38:960–968. 10.1016/j.freeradbiomed.2004.12.021 [DOI] [PubMed] [Google Scholar]
- Reinert RB, Kantz J, Misfeldt AA, Poffenberger G, Gannon M, Brissova M, Powers AC (2012) Tamoxifen-induced Cre-loxP recombination is prolonged in pancreatic islets of adult mice. PloS One 7:e33529. 10.1371/journal.pone.0033529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH (2004) Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology 62:1758–1765. 10.1212/WNL.62.10.1758 [DOI] [PubMed] [Google Scholar]
- Stephenson J, Amor S (2017) Modelling amyotrophic lateral sclerosis in mice. Drug Discov Today Dis Models 25–26:35–44. 10.3389/fnmol.2020.00093 [DOI] [Google Scholar]
- Weydt P, Hong SY, Kliot M, Möller T (2003) Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport 14:1051–1054. 10.1097/01.wnr.0000073685.00308.89 [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156:317–331. 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26:165–176. 10.1016/j.tcb.2015.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SE, Chen L, Na R, Liu Y, Rios C, Van RH, Richardson A, Ran Q (2012) Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med 52:1820–1827. 10.1016/j.freeradbiomed.2012.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Go YM, Jones DP (2007) Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys 465:119–126. 10.1016/j.abb.2007.05.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Available upon request.