

SUMMARY
The host immune response is a potent defense mechanism against cancer development and progression. Therefore, cancer cells must develop mechanisms to evade the immune response to survive. Based on this knowledge, a series of new therapies collectively referred to as immunotherapies have been developed and translated to the clinic for treating cancer patients. Although subsets of certain cancer types have shown strong clinical responses, including curative outcomes in some patients, for some subtypes and forms of cancers, immunotherapies have not worked as desired. Here, we provide an overview of the transcriptional mechanisms that drive response and resistance to immunotherapies. We also discuss possible interventions to enhance the outcomes of immunotherapies by targeting dysregulated transcription networks in cancer cells.
Keywords: Transcription, gene regulation, epigenetics, immunotherapy
INTRODUCTION
The ability to overcome the host immune system by thwarting or evading its action is one of the key features of cancer cells that allows cancer development and metastatic progression [1–3]. Cancer cells employ a diverse array of techniques to disarm both adaptive and innate immune responses, which include secretion of immunosuppressive cytokines [4], suppression of MHC class I expression on cancer cells [5], reduced expression of natural killer (NK) cell activating ligands, increased expression of NK cell inhibitory ligands [6], and expression of immune checkpoint proteins, such as PD-L1 on cancer cells [3]. Additionally, resistance to immunotherapies can also emerge as an unwarranted effect of immune attack on cancer cells leading to immunoediting-mediated development of acquired resistance [7].
Recent studies have shown that it is possible to overcome the immune resistance of cancer cells [8–11]. These therapeutic approaches are collectively referred to as immunotherapies. Striking responses are observed in subsets of several cancer types, including curative effects in some cancer patients [12–14]. However, immunotherapies are limited due to the unavailability of reliable and widely applicable predictive biomarkers, as well the lack of full understanding of the mechanisms that drive intrinsic or acquired resistance towards these therapies.
With new knowledge gleaned over the past several years regarding the mechanism of resistance to immunotherapies, it is likely that mechanisms of resistance to immunotherapies can be therapeutically targeted to broaden the patient population that can benefit from these drugs. Several recent studies have shown that immunotherapy-resistant tumors display distinct transcription profiles due to altered expression or function of various transcriptional regulators [15–19]. Here, we describe the transcriptional mechanisms that predict response and resistance to cancer immunotherapies and how these transcriptional drivers can be targeted to improve the outcomes of cancer immunotherapies.
Approaches for cancer immunotherapies
Cancer immunotherapies include the use of cancer vaccines, cytokine therapies, antibody-based therapies, cell therapies (CAR-T cells, CAR-NK cells, and CRISPR-engineered T cells), and immune checkpoint blockade (ICB) therapies (Figure 1). Akin to traditional vaccines against pathogens, cancer vaccines are developed to mount antigen-specific immune responses (e.g., neoantigens or tumor associated antigens in cancer) [20,21]. The idea behind cancer vaccines is to provide a boost to the immune system of the host by promoting expression of cancer-associated neoantigens leading to their presentation on antigen presenting cells. This, in turn, can result in robust T cell activation, proliferation, and T cell-mediated tumor eradication. Among the several advantages of cancer vaccines is the ability to use various platforms for vaccine generation, such as mRNA vaccines and viral vaccines [20]. Additionally, because each cancer patient and their tumor are somewhat unique, personalized cancer vaccines can be generated based on the neoantigens present in a specific cancer patient’s tumor. Finally, like traditional vaccines, cancer vaccines can generate immunological memory and thereby prevent cancer recurrence. However, generation of effective cancer vaccines is not without challenges. These challenges can manifest in many forms, such as challenges in identifying appropriate immunogenic neoantigens, differences in in silico neoantigen prediction and patient outcomes, and tumor heterogenetic and immunoediting-mediated selection of cancer cells that lack neoantigen expression.
Figure 1. Approaches for cancer immunotherapies.
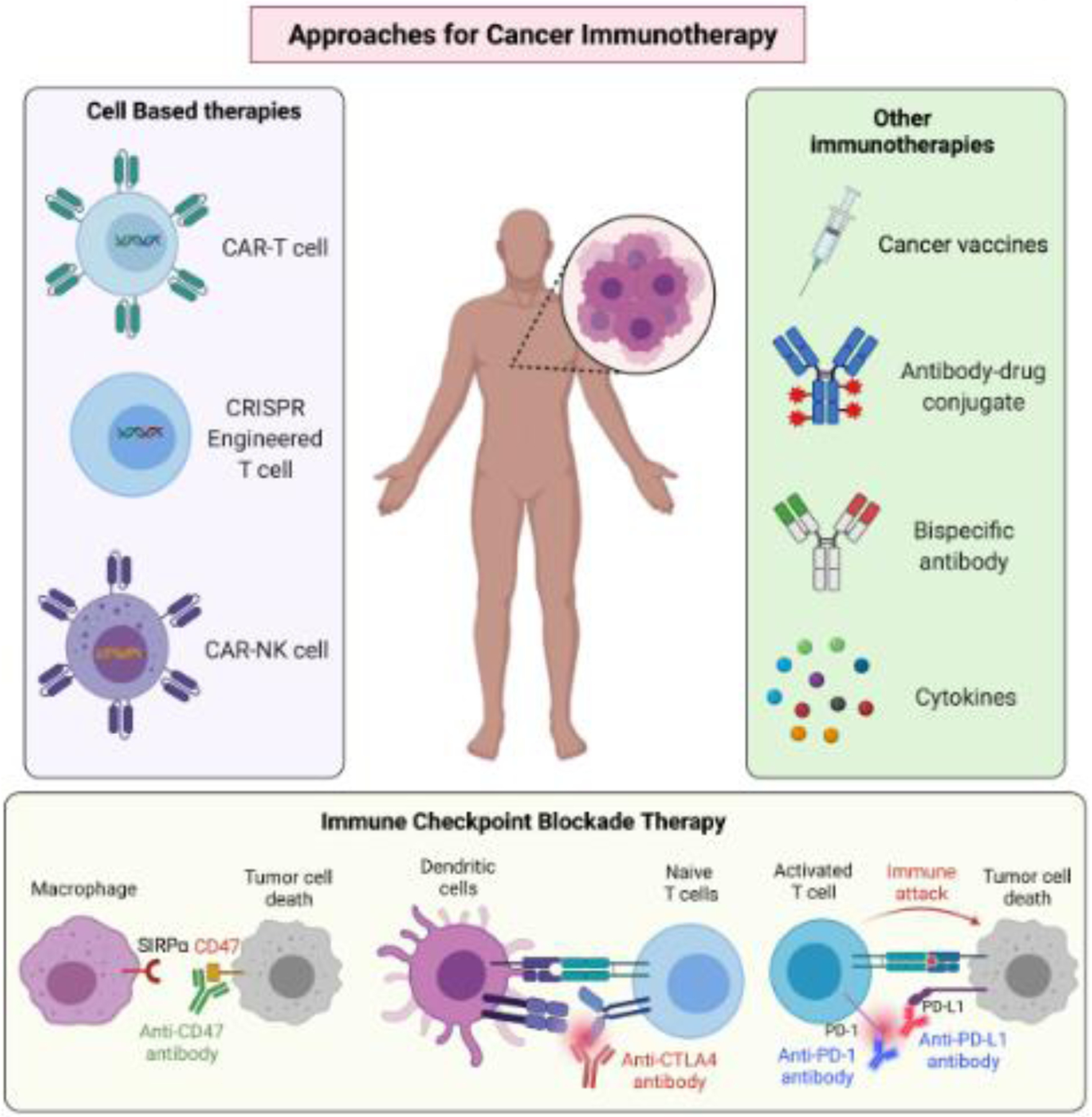
Based on the understanding of immune evasion mechanisms by cancer cells, a series of approaches to enhance immune system-mediated eradication of cancer cells have been implemented. Key immunotherapeutic approaches are depicted.
Cytokines are another immunotherapeutic approach to treat cancer. Among these, interleukin 2 (IL-2) is used to treat various cancers, such as metastatic melanoma and metastatic renal cancer [22,23]. In addition to the positive immune stimulatory effects of IL-2, there are some negative effects of using IL-2 for immunotherapy. These include the stimulation of regulatory T cells (Tregs) by IL2, which negatively impact the response to immunotherapy. However, this negative impact can be controlled by combining IL-2 with IL-21 to prevent Treg stimulation [24]. Like IL-2, other cytokines are also used to stimulate immune cells for treatment. This includes IL-15 by using IL-15 superagonists, such as ALT-803, IL-21, interferons (IFNs), and GM-CSF [23].
Antibody-based immunotherapy [25] originally used antibodies based on the tumor antigens and worked quite well in exerting anti-tumor activities. Examples of such antibodies include trastuzumab for treating HER2 breast cancer and rituximab for treating CD20+ non-hodgkin B-cell lymphoma. However, more recently, antibody-based therapies that are independent of the presence of tumor-specific antigens were designed [25] even though most antibody-based immunotherapies are dependent upon the presence of an antigen on the tumor cell surface. However, some recent findings have suggested that monoclonal antibodies can be targeted to intracellular cancer antigens [26,27].
Adoptive cell therapy is another form of cancer immunotherapy that has been clinically approved for several forms of hematological malignancies, including multiple myeloma, leukemia, and lymphoma [28,29]. Adoptive cell therapy covers various approaches, including tumor-infiltrating lymphocytes (TILs)-based therapies [30], engineered T cell receptor (TCR) therapy [31], chimeric antigen receptor T cell (CAR-T) therapy [32], and NK cell therapy [33]. Each of these therapies can be tailored to a patient’s cancer to provide the maximum cancer suppressive effect. An exciting and emerging field of cell-based immunotherapy that has gained traction is the development of CRISPR-engineered T cells. A recent study showcased the possibility of using this approach and reported the first in-human phase 1 clinical trial to test the safety and feasibility of multiplex CRISPR-Cas9 editing to engineer T cells in three patients with refractory cancer [34]. Based on the initial success, it is likely that similar trials will be conducted and that CRISPR-engineered T cells may provide a new therapeutic opportunity for cancer patients and may even be beneficial against solid tumors.
More recently, ICB therapies that target T cell checkpoints by blocking CTLA4, PD-L1, and PD-1 using antibodies have shown exceptional outcomes in clinical trials [3,10]. Because of their clinical success, anti-CTLA4, anti-PD-1, and anti-PD-L1 antibodies now represent the most used immune checkpoint therapies for treating a variety of cancer types.
Some new therapies targeting additional targets, are being tested, and have yielded promising clinical results. For example, CD47 expression on cancer cells activates the “do not eat me” signal; thus, anti-CD47 blocks the macrophage checkpoint and promotes phagocytosis [35–37]. To this end, magrolimab, a humanized monoclonal antibody targeting CD47, is being used for the treatment of acute myelogenous leukemia and non-hodgkin’s lymphoma and is showing excellent preliminary activity in the clinic [38]. Magrolimab is also being explored for solid tumors [39]. Another anti-CD47 antibody worth mentioning is lemzoparlimab that has shown a better safety and pharmacokinetic profile because it is designed to limit inherent binding to red blood cells while preserving its anti-tumor activity. Of note, based on the promising effects of antibodies that block the CD47-mediated macrophage checkpoint, bispecific antibodies, such as CD47XCD20, have been developed and are being used in the clinic for cancer treatment [40]. A subset of metastatic cancer patients showed remarkable responses to these immune-checkpoint therapies, including curative effects in some cases. However, a significant number of cancer patients do not respond or acquire resistance to these immune-checkpoint therapies. Therefore, additional studies and other combination therapeutic approaches are required to further improve the outcomes of ICB therapies.
Transcriptional Profiling to Predict Outcomes of Cancer Immunotherapies
mRNA-based transcription profiling of cancer cells has been used to establish and distinguish biologically and pathologically distinct groups [41]. Recently, similar approaches have been used to predict the outcomes of cancer immunotherapies [15,16,42–44]. An example of using mRNA expression profiling to predict ICB therapy response was documented in a study that used bilateral tumor implantation and syngeneic cell line-based breast cancer models to study the response to anti-PD-1 therapy [15]. In this approach, E07771 cells were orthotopically implanted bilaterally into the mammary fat pad of the same female mice and one of the tumors was resected early on to identify the predictors of the response and the second tumor was used to monitor the effect of anti-PD-1 therapy. A response rate of 60–70% was observed in this model allowing 30–70% of the mice to be used as non-responders to anti-PD-1 therapy to identify the determinants of immunotherapy response. Because the CD8+ T cell population in tumors was identified as an early predictor of the anti-PD-1 response, the authors performed RNA sequencing of CD8+ T cells isolated from size-matched tumors from responder and non-responder mice. This RNA-seq analysis showed that the responder gene signature was enriched for pathways related to T cell activation and the inflammatory response, whereas the gene signature of non-responders was enriched for pathways indicative of T cell exhaustion [15]. Finally, analysis of breast cancer data from the METABRIC database showed that the mouse CD8+ T cells responder gene signature was associated with better breast cancer survival and that the non-responder gene signature was associated with poorer breast cancer survival. A similar response correlation was also observed for bulk breast and melanoma samples and single cell RNA-seq of CD38+ T cells from patient samples. Collectively, this study highlighted the utility of the bilateral tumor implantation mouse model to identify clinically relevant transcriptional signatures that can predict the immunotherapy response.
A more comprehensive approach beyond the transcriptome would allow for more in-depth identification and characterization of mediators of the immunotherapy response and immunotherapy resistance. This approach was well-documented in a study in which the genomic and transcriptomic features of responses to anti-PD-1 therapy in metastatic melanoma were analyzed [16]. This study showed that a high mutational load might associate with better survival; however, it could not predict the anti-PD-1 therapy response. This was consistent with observations in clinical settings that the neoantigen load/mutational load alone was not predictive of various ICB therapies for various cancer types and that other genetic and non-genetic factors may also be at play. Interestingly, this study identified that BRCA2 mutations were specifically enriched in melanomas that were responsive to anti-PD-L1 therapy. This study also identified a transcriptional signature that was related to innate anti-PD-1 resistance (IPRES). Notably, the IPRES transcriptional signature was enriched in heightened mesenchymal transition, angiogenesis, hypoxia, and wound healing pathways. IPRES was predictive of innate resistance to anti-PD-1 therapy in melanoma and for other types of advanced cancers, such as pancreatic cancer.
Finally, because of the abundance of RNA-seq data from The Cancer Genome Atlas (TCGA) and from pre- and post-immunotherapy cancers, one can envision the development of computational tools to identify signatures that can predict the immunotherapy response. In this regard, a study developed a computational tool called Tumor Immune Dysfunction and Exclusion (TIDE) to predict the ICB response in models of two primary mechanisms of tumor immune evasion: T cell dysfunction in tumors with high infiltrations of cytotoxic T lymphocytes (CTL) and the prevention of T cell infiltration (T cell exclusion) in tumors with low CTL levels [45]. To compute T cell dysfunction scores for different datasets, authors analyzed 73 datasets with at least 50 samples from TCGA [41], PRECOG [46], and METABRIC [47] that had both tumor mRNA expression profiles and patient survival data. This study showed that in five datasets (melanoma, neuroblastoma, triple negative breast cancer, endometrial cancer, and acute myeloid leukemia), over 1% of genes showed significant interactions with CTL to affect survival. These genes included genes with previously known roles in tumor immunity, such as PD-L1, and genes with no previously documented roles in tumor immunity. Furthermore, the TIDE T cell dysfunction scores were consistent with the transcription signatures of tumor immune evasion. Similarly, TIDE was able to model the gene expression signature of T cell exclusion based on expression profiles of cancer-associated fibroblasts, myeloid-derived suppressor cells, and the M2 subtype of tumor-associated macrophages, cell types that are known to restrict T cell infiltration into tumors [48]. Overall, this study used transcription profiles of cancer and immune cells to identify gene signatures that predict either T cell dysfunction or T cell exclusion.
Collectively, the studies highlighted above document three distinct approaches: using pre-clinical mouse models, cancer patient samples, and computational approaches and integration thereof to identify transcriptional signatures and associated biological pathways that predict responses to ICB therapies. One can expect to utilize similar approaches for other types of immunotherapies and for better patient stratification. These approaches can also be used to develop new therapeutic interventions for patients with tumors that are either intrinsically resistant or become resistant post-therapy (acquired resistance). An example of this is documented in the melanoma study in which the IPRES signature showed enrichment of mesenchymal and angiogenesis signatures [16]; if these signatures can be suppressed therapeutically by using anti-angiogenic agents, it might lead to better therapeutic outcomes.
Drivers of dysregulated transcriptional programs in immunotherapy-resistant tumors
Dysregulated transcription in immunotherapy-resistant tumors can arise because of various factors, including changes in expression and activity of transcription factors, as well as DNA modification and chromatin regulatory proteins. Over the last few years, the roles of these factors in driving dysregulated transcriptional programs and causing immunotherapy resistance has been recognized [18,19,43,44] (Figure 2, Key Figure).
Figure 2, Key Figure. Transcriptional mechanisms that drive response and resistance to cancer immunotherapies.
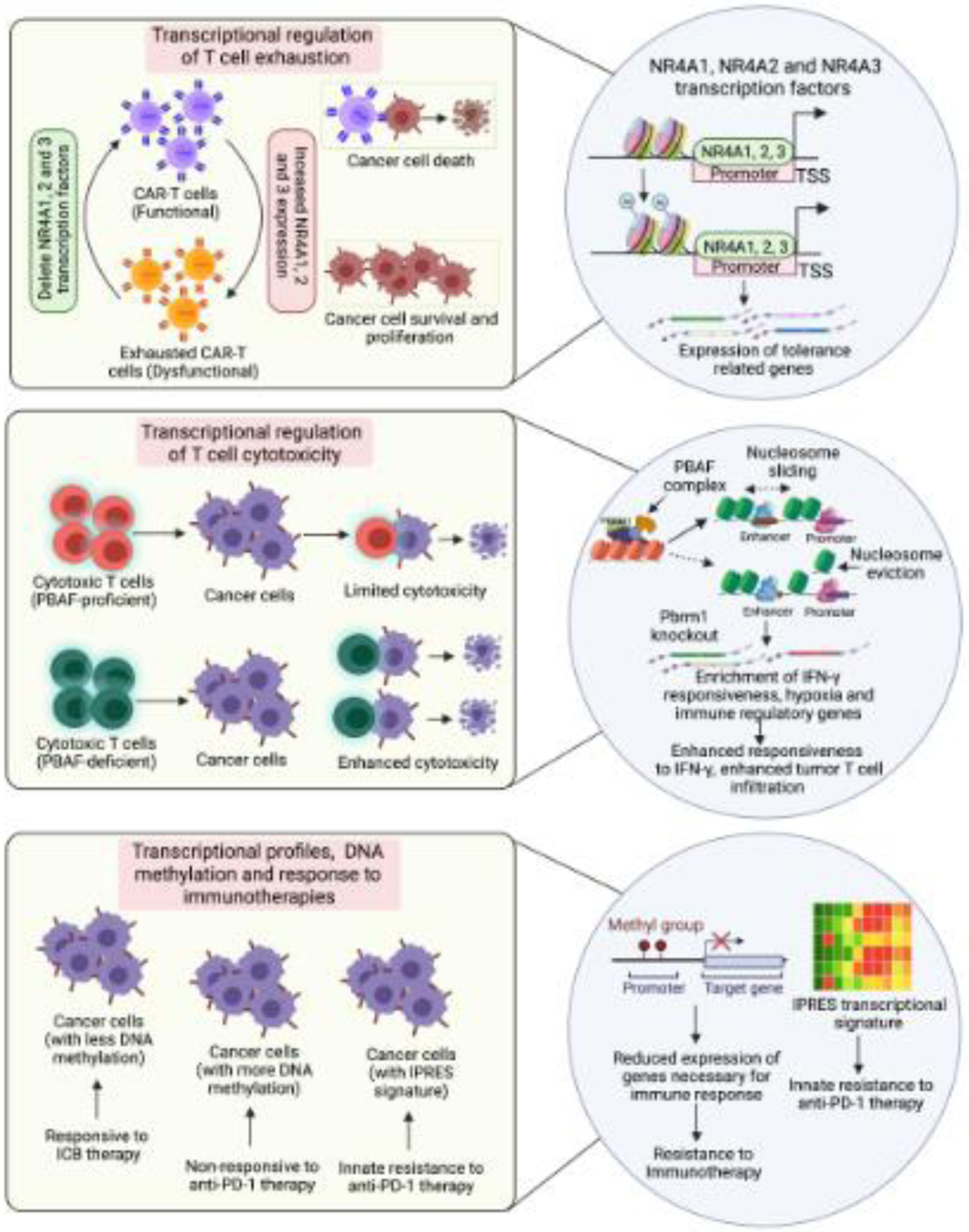
Schematic depicting the role of transcription profile changes, DNA methylation changes, and changes in transcription factor and chromatin regulators on immune cell function and the immunotherapeutic response.
The role of transcription factors in the regulation of immune function was documented in a pair of recent studies that identified NR4A transcription factors in T cell exhaustion in solid tumors [18] (Figure 2, Key Figure). The goal of these studies was to understand the mechanism behind T cell exhaustion/dysfunction. The first study used CD19 (hCD19)-reactive CAR-T cells because of their ability to clear hCD19+ solid tumors in mice. CAR-T cells are effective for treating hematological malignancies but are less effective for solid tumors due, in part, to their entry into the exhausted state [49,50]. Therefore, if the mechanism behind T cell exhaustion is understood, it can be targeted to enhance tumor eradication by T cells. The first study found that CD8+CAR+ tumor-infiltrating lymphocytes and CD8+ endogenous tumor-infiltrating lymphocytes expressing the inhibitory receptors PD-1 and TIM3 exhibited similar gene expression and chromatin accessibility profiles [18] and associated with increased expression of the nuclear receptor transcription factors NR4A1, NR4A2, and NR4A3. Upregulation of these transcription factors was also observed in CD8+ cells from human cancer patients. Furthermore, CAR-T cells lacking all three NR4A transcription factors promoted tumor regression and prolonged the survival of tumor-bearing mice further establishing their functional role in CAR-T cell exhaustion. In the second study, the authors probed chromatin and transcription regulation associated with T cell dysfunction [19]. To this end, they generated tolerant T cells using inhibitory co-stimulation [51] and identified NR4A1 as an important mediator of T cell dysfunction. This study also noted that NR4A1 binding promoted acetylation of histone 3 at lysine 27 (H3K27Ac) leading to activation of tolerance-related genes. Collectively, these studies identified the NR4A family of transcription factors as the key mediators of T cell exhaustion. They also demonstrated that targeting NR4A transcription factors can improve the outcomes of immunotherapies for solid tumors by overcoming the T cell exhaustion phenotype.
Like the relevance of transcription factor driven regulation of immunotherapy phenotypes, direct changes in DNA methylation have also been shown to predict or modulate immunotherapy responses [17,52,53]. An example of this is a study that analyzed the DNA methylome at the pathway level of 141 advanced non-small cell lung cancer (NSCLC) samples from two independent cohorts that were subjected to ICB therapy [17]. Integration of DNA methylation data with transcriptome data revealed significant overlaps between increased DNA methylation and transcriptional repression in NSCLCs that were non-responsive to ICB therapies. This study identified 15 immune-related pathways, including IFN signaling, that were enriched for both DNA methylation and transcriptional repression. Additionally, an eight gene DNA methylation-based (IRF6, CTSD, GRN, LTBR, TRIM36, EVL, CD3E, and LCP1) prediction model was generated to accurately classify patient survival outcomes and was validated using the TCGA NSCLC dataset and the IDIBELL dataset.
Chromatin regulators and chromatin states have also been shown to suppress or enhance immune cell function [52,54–56]. The role of chromatin regulators was well-highlighted in two studies that showed that proteins of the SWI/SNF complex were important in predicting the T cell response (Figure 2, Key Figure). The first study by Pan et al. used CRISPR-CAS9-based screening to identify mechanisms of resistance to cytotoxic T cell-mediated tumor eradication [57]. This study identified over 100 genes whose inactivation sensitized mouse melanoma B16F10 cells to T cell-mediated killing. These genes included several chromatin regulators, such as Pbrm1, Arid2, and Brd7. Pbrm1, Arid2, and Brd7 encode the PBAF form of the SWI/SNF complex. In many human cancers, expression of PBRM1 and ARID2 correlated inversely with T cell cytotoxicity markers in TCGA datasets. Prbm1 knockout tumors showed enhanced chromatin accessibility for IFN-gamma responsive genes and consequently higher responsiveness to IFN-gamma stimulation. This study also found that Pbrm1 knockout mouse melanoma tumors showed increased infiltration of T cells.
Similarly, another study aimed to identify genomic alterations in clear cell renal cell carcinoma (ccRCC) that correlate with anti-PD-1 monotherapy [58]. This study found loss-of-function mutations in the PBRM1 gene, which, as mentioned above, forms a subunit of the PBAF form of the SWI/SNF chromatin remodeling complex. Furthermore, gene expression profiles of PBAF-deficient ccRCC cell lines and PBRM1-deficient tumors revealed enrichment of JAK-STAT, hypoxia, and immune signaling pathway gene signatures. Collectively, these studies identified the PBAF form of the SWI/SNF chromatin remodeling complex as a major determinant of immune and immunotherapy responses against cancer cells.
Overall, the studies described above showcase how transcription factors, DNA methylation modifications, and chromatin regulators drive immune responses against cancer cells and demonstrate that their statuses can predict outcomes of various immunotherapies.
Targeting deregulated transcriptional networks and mediators thereof to improve outcomes of immunotherapeutic agents
As the awareness of transcription regulators and transcriptional networks in conferring intrinsic or acquired resistance to immunotherapies has increased, it has become clear that these factors represent therapeutic targets to improve immunotherapy outcomes (Figure 3). An example of drug combinations that regulate transcription is highlighted by the PEMDAC phase 2 study that used the histone deacetylase (HDAC) inhibitor entinostat along with the PD-1 inhibitor pembrolizumab to treat patients with metastatic uveal melanoma (Clinicaltrials.gov identifier: NCT02697630) [59]. Objective responses and/or prolonged survival were seen in patients with BAP1 wild-type tumors and in one patient with an iris melanoma that exhibited a UV signature [59]. This study concluded that dual HDAC and PD-1 inhibition can result in durable responses in a subset of patients with metastatic uveal melanoma. Although the mechanism behind the enhanced therapeutic benefit was not fully resolved, the fact that BAP1 wild-type tumors responded better than the mutant indicated that the status of BAP1, in part, was responsible for the observed effects and that BAP1 might have immune enhancing effects. Similarly, the iris melanoma correlated with a better response indicating that this subtype of uveal melanoma might be more responsive to pembrolizumab and entinostat therapy.
Figure 3. Clinical translation of transcription targeting therapies in combination with cancer immunotherapies.
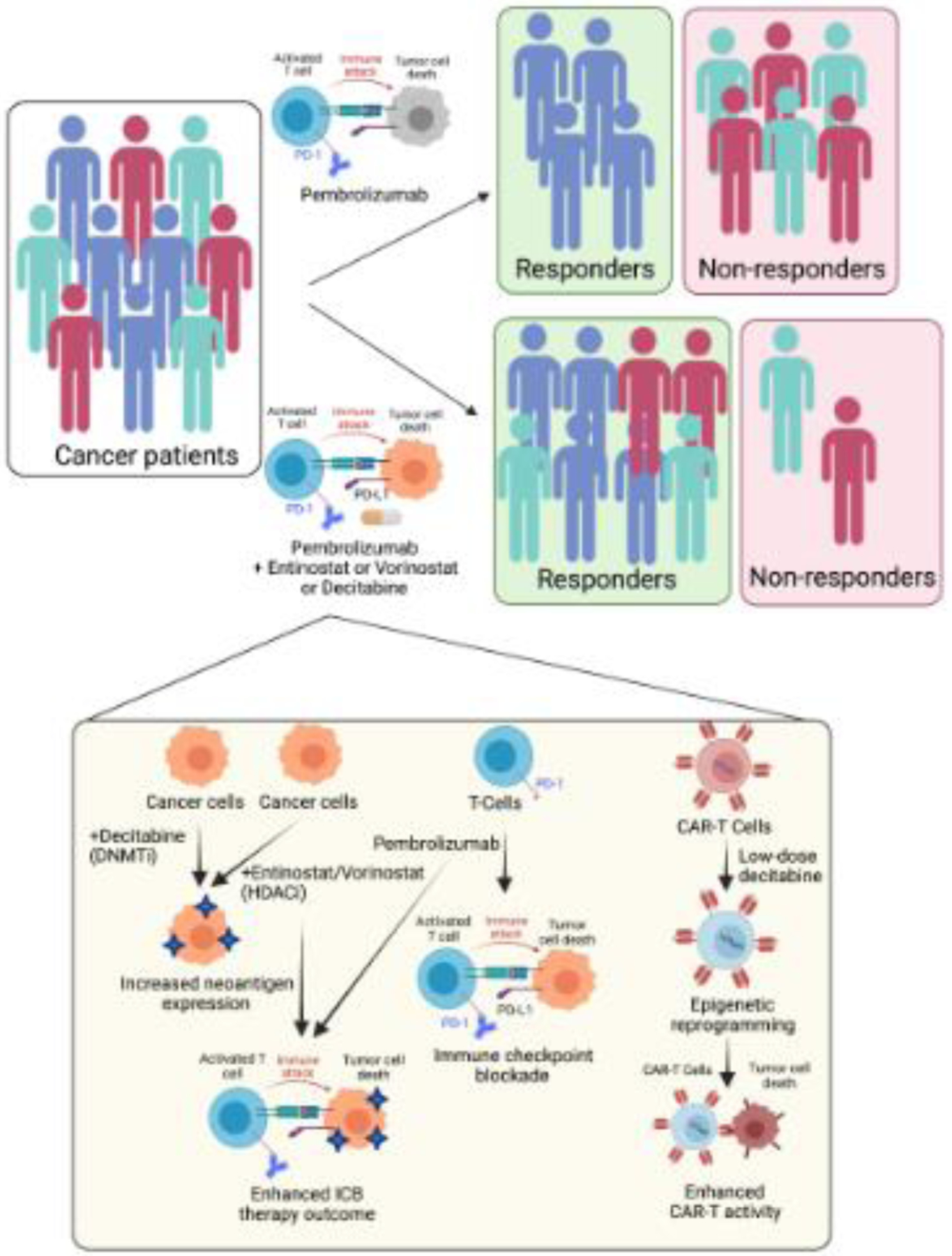
New studies have revealed the immune stimulatory effects of DNA methylation and chromatin regulatory proteins resulting in the use of the DNMT inhibitor decitabine and the HDAC inhibitors entinostat and vorinostat in combination with ICB therapies and CAR-T cells.
Furthermore, a similar phase 1/1b study of advanced/metastatic NSCLC used a combination of pembrolizumab and the HDAC inhibitor vorinostat (Clinicaltrials.gov identifier: NCT02638090). A total of 33 patients who were ICB inhibitor naïve or who progressed on the ICB inhibitor were treated with this combination. The study concluded that pembrolizumab and vorinostat combination therapy was well tolerated and showed preliminary tumor suppressive activity in ICB inhibitor naïve patients and in patients that progressed on ICB inhibitors. Another combination that was tested in a phase 1b trial was the HDAC6 inhibitor ACY-241 (citarinostat) plus nivolumab, an anti-PD-1 antibody for treating NSCLC. The results of the study suggested that this combination may be feasible in patients with advanced NSCLC. Furthermore, partial response or stable disease was observed in a subset of patients treated with ACY-241 plus nivolumab. Of note, the response was also observed in patients whose tumors progressed on ICB inhibitor treatment. The mechanism behind the enhanced pembrolizumab and vorinostat-mediated tumor suppressive effect was not determined but would be instrumental to improve treatment and to identify the correct patient populations for treatment.
Other agents, such as the DNA methylation inhibitor decitabine, have also been combined with immunotherapies to improve therapeutic outcomes in cancer patients. An example of this was a phase 2 clinical trial that used low-dose decitabine in combination with camrelizumab to treat relapsed/refractory classical hodgkin lymphoma [60]. The combination therapy with decitabine and camrelizumab was more effective in achieving a complete response than camrelizumab alone. Notably, naïve patients treated with anti-PD-1 treatment showed an impressively higher level of complete response compared to patients who previously underwent anti-PD-1 treatment. Based on previous studies, possible explanations for the superior clinical efficacy of decitabine and camrelizumab combination therapy could be due, in part, to the ability of this combination to overcome T cell exhaustion by reduced DNA methylation as well as by promoting CD4+ and CD8+ infiltration; however, this was not investigated directly in this particular study.
Notably, immunotherapy combinations with HDAC inhibitors or DNA methyltransferase (DNMT) inhibitors, such as entinostat, vorinostat, and decitabine, are not restricted to ICB therapies and are now being combined with other forms of immunotherapies, such as CAR-T cell-based immunotherapies (Clinicaltrials.gov identifier: NCT04553393). This is due, in part, to the ability of decitabine and HDAC inhibitors to increase tumor antigen and HLA expression, enhance antigen processing, promote T cell infiltration, and boost effector T cell function. Similarly, some new observations have shown that low-dose decitabine priming enhances the persistent anti-tumor potential of CAR-T cells [61].
Furthermore, similar to the HDAC and DNMT inhibitors, other drugs that target chromatin modifiers, such as EZH2 inhibitors, are also being tested in combination with immunotherapeutic agents [56]. Collectively, these studies highlight the beginning of a new phase of cancer immunotherapy in which immunotherapeutic agents are combined with transcriptional regulatory agents to enhance the outcomes of immunotherapies.
CONCLUDING REMARKS
Although impressive clinical outcomes have been observed for subsets of patients with various cancer types treated with immunotherapeutic agents, many cancer patients still either fail to respond to immunotherapies or become resistant to these agents after the initial response. Therefore, due to their improved clinical benefits, combination therapies with drugs that influence transcription (e.g., DNMT or HDAC inhibitors) are expected to become the common approach for cancer treatment. Furthermore, identifying the drivers of transcription deregulation that cause intrinsic or acquired resistance to immunotherapies will uncover new drug targets that can be combined with immunotherapies to achieve better clinical outcomes (see Outstanding Questions). Recent studies of CAR-T cells and solid tumors are of interest because CAR-T cells have displayed impressive efficacy against hematological malignancies but have displayed limited efficacy against solid tumors due to phenomena, such as T cell exhaustion. Similarly, with an increased interest in CRISPR-engineered T cells for cancer immunotherapies and with the recent success of the first in-human clinical trial, it can be envisioned that the use of CRISPR-engineered T cells will become more common for treating cancer patients. As this field progresses, it would be good to see if, in addition to known immune checkpoint proteins, other transcription regulatory factors that limit T cell function can also be targeted to enhance T cell function and to potentially limit their off-target effects, such as the ability to induce autoimmune side effects.
OUTSTANDING QUESTIONS.
What transcriptional regulators and states modulate immune cell-mediated eradication of cancer cells?
What roles do age and gender driven transcriptional changes play in determining the outcomes of immunotherapies?
How can the optimal combination of transcription regulatory drugs and immunotherapies be identified?
What can be done to prevent immunotherapy-related adverse effects in cancer patients?
Although a lot is now known regarding factors that determine the outcomes of immunotherapies, there are still several mechanisms of response and resistance to immunotherapies that are not fully understood and require further research. In this regard, some of the less understood aspects include the impact of aging and gender on the immunotherapy response. For example, although age did not clearly demarcate the benefits of immune checkpoint-based therapy, surprisingly, some studies, including a meta-analysis, have shown better clinical benefits for older patients than younger patients [62]. It would be interesting to see if age-related transcriptional changes can predict the better benefit for older patients and if this information can be used to enhance immunotherapy outcomes for younger patients. Similarly, gender (male or female) has also been associated with the response to immunotherapies [63,64]. However, this area of immunotherapy is largely underexplored; thus, further studies linking these studies to transcriptional alterations might identify new opportunities to enhance the outcomes of immunotherapies for both genders.
HIGHLIGHTS.
Cancer cells acquire a number of alterations that allow them to escape the host immune system leading to tumor initiation and progression.
New therapeutic approaches focused on restoring the sensitivity of cancer cells to immune-mediated eradication has now moved to the forefront of cancer therapeutics and are collectively referred to as immunotherapies.
Transcriptional mechanisms play a key role in regulating immune cell function and the sensitivity of cancer cells to immune-mediated eradication.
Combination therapy with DNA methylation or HDAC inhibitors and cancer immunotherapies have moved into clinical use and have shown significant benefits in patients.
ACKNOWLEDGEMENTS
We gratefully acknowledge grants from the National Institutes of Health: R01CA195077-01A1 (NW), R01CA200919-01 (NW), R01 CA218008-01A1 (NW), R03 CA230815 (RG) R03CA248913 (RG), and R01CA233481 (RG).
CONFLICT OF INTEREST STATEMENT
A.M. has received research funding from Incyte, Takeda, Forty seven Inc/Gilead, Juno Pharmaceuticals/BMS, Celgene/BMS, Innate Pharmaceuticals, Seattle Genetics, TG Therapeutics, Affimed, Merck, Kite/Gilead, Roche-Genentech, and I-MAB. A.M. has also served as a consultant, speaker, or on the advisory board of Gilead, Astra Zeneca, Pharmacyclics, Seattle Genetics, Incyte, Morphosys/Incyte, TG Therapeutics, Kyowa Kirin, Novartis, and BMS. R.G. and N.W. have no conflicts of interest to declare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hiam-Galvez KJ et al. (2021) Systemic immunity in cancer. Nat Rev Cancer 21, 345–359. 10.1038/s41568-021-00347-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez H et al. (2018) Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32, 1267–1284. 10.1101/gad.314617.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P and Allison JP (2020) Dissecting the mechanisms of immune checkpoint therapy. Nat Rev Immunol 20, 75–76. 10.1038/s41577-020-0275-8 [DOI] [PubMed] [Google Scholar]
- 4.Kim R et al. (2006) Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 66, 5527–5536. 10.1158/0008-5472.CAN-05-4128 [DOI] [PubMed] [Google Scholar]
- 5.Dersh D et al. (2021) A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol 21, 116–128. 10.1038/s41577-020-0390-6 [DOI] [PubMed] [Google Scholar]
- 6.Bugide S et al. (2018) Epigenetic Mechanisms Dictating Eradication of Cancer by Natural Killer Cells. Trends Cancer 4, 553–566. 10.1016/j.trecan.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Donnell JS et al. (2019) Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol 16, 151–167. 10.1038/s41571-018-0142-8 [DOI] [PubMed] [Google Scholar]
- 8.Waldman AD et al. (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20, 651–668. 10.1038/s41577-020-0306-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegde PS and Chen DS (2020) Top 10 Challenges in Cancer Immunotherapy. Immunity 52, 17–35. 10.1016/j.immuni.2019.12.011 [DOI] [PubMed] [Google Scholar]
- 10.Sharma P et al. (2021) The Next Decade of Immune Checkpoint Therapy. Cancer Discov 11, 838–857. 10.1158/2159-8290.CD-20-1680 [DOI] [PubMed] [Google Scholar]
- 11.Milone MC et al. (2021) Engineering enhanced CAR T-cells for improved cancer therapy. Nat Cancer 2, 780–793. 10.1038/s43018-021-00241-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robert C et al. (2015) Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 372, 2521–2532. 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 13.Larkin J et al. (2019) Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 381, 1535–1546. 10.1056/NEJMoa1910836 [DOI] [PubMed] [Google Scholar]
- 14.Herbst RS et al. (2020) Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N Engl J Med 383, 1328–1339. 10.1056/NEJMoa1917346 [DOI] [PubMed] [Google Scholar]
- 15.Chen IX et al. (2020) A bilateral tumor model identifies transcriptional programs associated with patient response to immune checkpoint blockade. Proc Natl Acad Sci U S A 117, 23684–23694. 10.1073/pnas.2002806117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hugo W et al. (2017) Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 168, 542. 10.1016/j.cell.2017.01.010 [DOI] [PubMed] [Google Scholar]
- 17.Kim JY et al. (2020) Genome-wide methylation patterns predict clinical benefit of immunotherapy in lung cancer. Clin Epigenetics 12, 119. 10.1186/s13148-020-00907-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J et al. (2019) NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. 10.1038/s41586-019-0985-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X et al. (2019) Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529. 10.1038/s41586-019-0979-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blass E and Ott PA (2021) Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 18, 215–229. 10.1038/s41571-020-00460-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shemesh CS et al. (2021) Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol Ther 29, 555–570. 10.1016/j.ymthe.2020.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchbinder EI et al. (2019) Therapy with high-dose Interleukin-2 (HD IL-2) in metastatic melanoma and renal cell carcinoma following PD1 or PDL1 inhibition. J Immunother Cancer 7, 49. 10.1186/s40425-019-0522-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chulpanova DS et al. (2020) Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front Cell Dev Biol 8, 402. 10.3389/fcell.2020.00402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battaglia A et al. (2013) Interleukin-21 (IL-21) synergizes with IL-2 to enhance T-cell receptor-induced human T-cell proliferation and counteracts IL-2/transforming growth factor-beta-induced regulatory T-cell development. Immunology 139, 109–120. 10.1111/imm.12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiner LM et al. (2012) Antibody-based immunotherapy of cancer. Cell 148, 1081–1084. 10.1016/j.cell.2012.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo K et al. (2011) Targeting intracellular oncoproteins with antibody therapy or vaccination. Sci Transl Med 3, 99ra85. 10.1126/scitranslmed.3002296 [DOI] [PubMed] [Google Scholar]
- 27.Wang Y et al. (2015) Intracellular antigens as targets for antibody based immunotherapy of malignant diseases. Mol Oncol 9, 1982–1993. 10.1016/j.molonc.2015.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayes C (2021) Cellular immunotherapies for cancer. Ir J Med Sci 190, 41–57. 10.1007/s11845-020-02264-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullard A (2021) FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov 20, 166. 10.1038/d41573-021-00031-9 [DOI] [PubMed] [Google Scholar]
- 30.Creelan BC et al. (2021) Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med 27, 1410–1418. 10.1038/s41591-021-01462-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Q et al. (2021) Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front Immunol 12, 658753. 10.3389/fimmu.2021.658753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao L and Cao YJ (2019) Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol 10, 2250. 10.3389/fimmu.2019.02250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers JA and Miller JS (2021) Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol 18, 85–100. 10.1038/s41571-020-0426-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stadtmauer EA et al. (2020) CRISPR-engineered T cells in patients with refractory cancer. Science 367. 10.1126/science.aba7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vonderheide RH (2015) CD47 blockade as another immune checkpoint therapy for cancer. Nat Med 21, 1122–1123. 10.1038/nm.3965 [DOI] [PubMed] [Google Scholar]
- 36.Liu X et al. (2015) CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med 21, 1209–1215. 10.1038/nm.3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sikic BI et al. (2019) First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J Clin Oncol 37, 946–953. 10.1200/JCO.18.02018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarr KU et al. (2021) Effect of CD47 Blockade on Vascular Inflammation. N Engl J Med 384, 382–383. 10.1056/NEJMc2029834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Upton R et al. (2021) Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2026849118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaur S et al. (2020) Preclinical and Clinical Development of Therapeutic Antibodies Targeting Functions of CD47 in the Tumor Microenvironment. Antib Ther 3, 179–192. 10.1093/abt/tbaa017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Research, N. et al. (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu M et al. (2020) Transcription factor c-Maf is a checkpoint that programs macrophages in lung cancer. J Clin Invest 130, 2081–2096. 10.1172/JCI131335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott AC et al. (2019) TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. 10.1038/s41586-019-1324-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seo H et al. (2019) TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A 116, 12410–12415. 10.1073/pnas.1905675116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang P et al. (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. 10.1038/s41591-018-0136-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gentles AJ et al. (2015) The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945. 10.1038/nm.3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curtis C et al. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. 10.1038/nature10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeNardo DG and Ruffell B (2019) Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19, 369–382. 10.1038/s41577-019-0127-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srivastava S and Riddell SR (2018) Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J Immunol 200, 459–468. 10.4049/jimmunol.1701155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marofi F et al. (2021) CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther 12, 81. 10.1186/s13287-020-02128-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nurieva R et al. (2006) T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J 25, 2623–2633. 10.1038/sj.emboj.7601146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung H et al. (2019) DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun 10, 4278. 10.1038/s41467-019-12159-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filipski K et al. (2021) DNA methylation-based prediction of response to immune checkpoint inhibition in metastatic melanoma. J Immunother Cancer 9. 10.1136/jitc-2020-002226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bugide S et al. (2018) Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U S A 115, E3509–E3518. 10.1073/pnas.1802691115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bugide S et al. (2021) EZH2 inhibits NK cell-mediated antitumor immunity by suppressing CXCL10 expression in an HDAC10-dependent manner. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2102718118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emran AA et al. (2019) Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol 40, 328–344. 10.1016/j.it.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 57.Pan D et al. (2018) A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. 10.1126/science.aao1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miao D et al. (2018) Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. 10.1126/science.aan5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ny L et al. (2021) The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun 12, 5155. 10.1038/s41467-021-25332-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nie J et al. (2019) Addition of Low-Dose Decitabine to Anti-PD-1 Antibody Camrelizumab in Relapsed/Refractory Classical Hodgkin Lymphoma. J Clin Oncol 37, 1479–1489. 10.1200/JCO.18.02151 [DOI] [PubMed] [Google Scholar]
- 61.Wang Y et al. (2021) Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun 12, 409. 10.1038/s41467-020-20696-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan X et al. (2020) Impact of Age on the Efficacy of Immune Checkpoint Inhibitor-Based Combination Therapy for Non-small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Front Oncol 10, 1671. 10.3389/fonc.2020.01671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klein SL and Morgan R (2020) The impact of sex and gender on immunotherapy outcomes. Biol Sex Differ 11, 24. 10.1186/s13293-020-00301-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye Y et al. (2020) Sex-associated molecular differences for cancer immunotherapy. Nat Commun 11, 1779. 10.1038/s41467-020-15679-x [DOI] [PMC free article] [PubMed] [Google Scholar]