

Abstract
Severe traumatic brain injury (TBI) is associated with high rates of mortality and long-term disability linked to neurochemical abnormalities. Although purine-derivatives play important roles in TBI pathogenesis in preclinical models, little is known about potential changes in purine levels and their implications in human TBI. We assessed cerebrospinal fluid (CSF) levels of purines in severe TBI patients as potential biomarkers that predict mortality and long-term dysfunction. This was a cross-sectional study performed in 17 severe TBI patients (Glasgow Coma Scale < 8) and 51 controls. Two to four hours after admission to ICU, patients were submitted to ventricular drainage and CSF collection for quantification of adenine and guanine purine-derivatives by HPLC. TBI patients survival was followed up to 3 days from admission. A neurofunctional assessment was performed through the modified Rankin Scale (mRS) two years after ICU admission. Purine levels were compared between control and TBI patients, and between surviving and non-surviving patients. Relative to controls, TBI patients presented increased CSF levels of GDP, guanosine, adenosine, inosine, hypoxanthine, and xanthine. Further, GTP, GDP, IMP, and xanthine levels were different between surviving and non-surviving patients. Among the purines, guanosine was associated with improved mRS (p=0.042; r= −0.506). Remarkably, GTP displayed predictive value (AUC=0.841, p=0.024) for discriminating survival vs. non-survival patients up to three days from admission. These results support TBI-specific purine signatures, suggesting GTP as a promising biomarker of mortality, and guanosine as an indicator of long-term functional disability.
Keywords: CSF nucleotides derivatives, GTP, guanosine, IMP, biomarkers, severe traumatic brain injury
Graphical Abstract
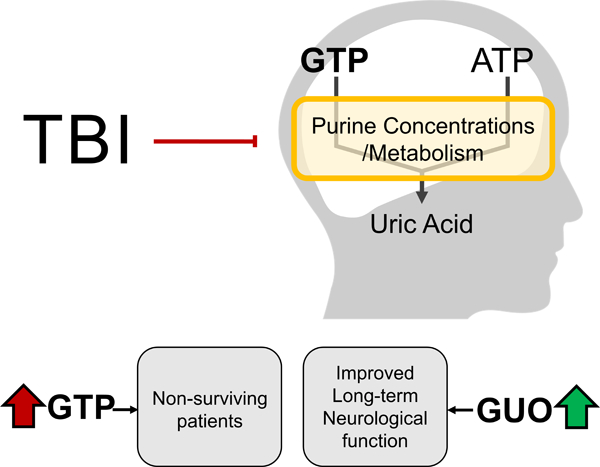
Severe Traumatic Brain Injury (TBI) is a major cause of death worldwide and is also associated with long-term neurological deficits. The availability of complementary prognostic tools to identify TBI patients at risk of death, or delayed unfavorable neurological outcomes is scarce. We identified that TBI altered the concentration of specific purines in the CSF, which represents a neurobiological signature with potential clinical and metabolic relevance. Particularly, increased GTP level, a guanine-derived purine, displayed value as a biomarker for mortality after TBI, and the nucleoside guanosine, also derived from guanine, was associated with impaired two-years neurological function.
1. Introduction
Traumatic brain injury (TBI) is considered a major health concern with a high incidence of disability worldwide, and it is the leading cause of mortality among young adults from low- and middle-income countries (Nguyen et al. 2016). In severe TBI, immediate mechanical damage to brain tissue can be life-threatening (Kuenzler et al. 2015) and can trigger a series of secondary neurochemical cascades that further damage the brain. Some of the most well-characterized of these processes include excitotoxicity, formation of proteotoxic aggregates, neuroimmune dysfunction, and changes in brain metabolism (Doran et al. 2019; Stefani et al. 2017; Johnson et al. 2013; Johnson et al. 2012; Lyons et al. 2018; Henry et al. 2020). However, while adenine- and guanine-based purines have been shown to play important roles in the pathogenesis of other brain disorders, little is known about their contribution to the post-TBI sequelae.
Beyond their central role in the formation of nucleic acids and bioenergetics, purines have a wide range of neuromodulatory effects that may be disrupted in various neurological disorders. Indeed, purinergic receptors have been explored as therapeutic targets in animal models of epilepsy, amyotrophic lateral sclerosis, Alzheimer’s, and Parkinson’s disease (Burnstock 2020; Burnstock 2017). An important feature of normal purine-mediated brain responses is the strict control of their individual concentrations, performed by intra- and extracellular enzymes (Robson et al. 2006). In particular, the Ecto-NTPDases are a family of enzymes responsible for the extracellular degradation of purines, thereby controlling binding to specific purinergic receptors and the magnitude of receptor activation (Robson et al. 2006). This includes the stepwise extracellular degradation of ATP and GTP, resulting in the formation of ADP and AMP, or GDP and GMP, respectively. Further catabolism leads to correspondent nucleosides adenosine and guanosine, which consequently generate IMP, inosine, hypoxanthine, xanthine, and uric acid (Pelligrino et al. 2010; Cunha 2016; Zeng et al. 2018). Purine-derived metabolites also cross plasma membranes through specific transporters, influencing their intra- and extracellular levels (Beal et al. 2004). Remarkably, from the extracellular triphosphate forms to their downstream metabolites, each purine derivative may exert characteristic physiological roles in the brain mediated by purinergic receptors and GTPase-activating proteins, neuroinflammatory effectors, and glutamatergic neurotransmission. Hence, changes in purine levels in the brain after TBI could play an important role in TBI-related outcomes.
While clinical studies on purines in TBI have been limited, Headrick and colleagues demonstrated that acute increases in extracellular adenosine levels were associated with impaired neuroenergetic and neurological functions in a preclinical rat TBI model (Headrick et al. 1994). In addition, inhibition of adenosinergic signaling with caffeine has been shown to decrease the mortality rate of rats submitted to severe TBI (Lusardi et al. 2012). Furthermore, intraperitoneal administration of guanosine after experimental TBI attenuated cognitive and mitochondrial dysfunction (Gerbatin et al. 2017; Dobrachinski et al. 2019). Finally, in vivo, extracellular levels of adenosine, inosine, and hypoxanthine were found to increase soon after TBI, which was mechanistically linked to energy failure mirrored by the rapid ATP catabolism to adenosine (Bell et al. 1998). Although preclinical studies have already unveiled associations between TBI and purine metabolism regarding neuroprotective properties, the potential clinical relevance of extracellular purine levels and interconversion, as prognostic biomarkers have received little attention in the TBI settings..
In this study, we assessed cerebrospinal fluid (CSF) levels of purines in severe TBI patients in order to identify specific signatures associated with mortality and long-term neurological dysfunction.
2. Subjects and Methods
2.1. Study population and clinical management
This is a single-center cross-sectional study, carried out in the Emergency Unit of the Cristo Redentor Hospital (Porto Alegre, RS, Brazil). This study included a total of 21 consecutive severe TBI patients, presenting on hospital admission both Glasgow Coma Scale (GCS) ≤ 8 and an abnormal brain CT scan (Böhmer et al. 2011; Stefani et al. 2017), and due to the exploratory nature of this study, no sample size calculation was performed, and the sample size was based in previous studies exploring TBI biomarkers (Stefani et al. 2017; Böhmer et al. 2011). Power analysis performed a posteriori through G*Power 3.1 software for our primary outcome, CSF purines as biomarkers of mortality in TBI (namely GTP), showed a power of 0.9851 (98.5%). Our secondary outcome, purines as prognostic biomarkers of long-term neurological function (namely guanosine), showed a power of 0.8411 (84.1%). TBI Patients were predominantly young (mean [SD] age, 29 [13] years; M/F ratio, 9:1), with an average Glasgow Coma Scale upon admission of 6, and Intracranial pressure of 12.89 [11.92] (mean [sd]). Inclusion criteria were an isolated severe TBI, with no previous history of comorbidities that could influence the biomarkers’ concentrations and clinical outcomes. Exclusion criteria and clinical management were previously reported (Stefani et al. 2017; Böhmer et al. 2011). CSF was collected between 2 and 4 h after hospitalization through an intraventricular catheter.
Additionally, 51 healthy subjects (ASA I status) scheduled for elective urological, gynecological, general, or vascular procedures were selected as age and sex-matched controls (Böhmer et al. 2011). Control subjects were predominantly young (mean [sd] age, 27.60 [6] years, M/F ratio, 8:1). Experienced anesthesiologists collected the CSF after successful subarachnoid puncture and before the intrathecal injection of anesthetics or analgesics. The anesthetic procedure was standardized, all patients received a combined spinal injection, at the level of L 3 ⁄L 4, of hyperbaric bupivacaine 0.5% (10 to 20 mg), associated with fentanyl 20 µg and/or morphine 100 µg, depending on the surgical procedure. The first 0.5 mL of CSF aspirated was discarded, and the subsequent 0.5 mL sample was collected and inspected visually for blood contamination.
Immediately after CSF collection, the samples from controls and patients were centrifuged at 10,000 x g for 5 min to obtain a supernatant free of cells and cellular debris. The supernatant was then transferred to sealed plastic tubes and stored at −70°C within 30 min of collection. Up to 48 hours from collection, CSF samples were thawed over ice for purines assessment.
Written informed consent for participating in this study was obtained from patients’ family members and directly from healthy individuals, according to the Declaration of Helsinki. Concomitantly, family members were questioned about the patient’s lifestyle and pre-existent diseases. This study was not pre-registered and was approved by the local institutional Ethics Committee approved this protocol (project number 0038.0.164.165–05).
2.2. Outcome measures
The primary patient outcome was defined accordingly: deterioration to brain death (non-survival n = 6) or survival (survival, n = 11), within 3 days after hospital admission. Deterioration to brain death occurred up to 3 days after admission to the ICU in non-surviving patients. The ICP, hemodynamic, and metabolic variables including mean arterial blood pressure and cerebral perfusion pressure were assessed daily and reported previously (Böhmer et al. 2011). Two years after discharge, telephone calls were placed to confirm survival, and investigate the level of long-term functional disability. TBI patients were then assessed for the modified Rankin Scale (mRS), the secondary outcome, and scored by an experienced neurologist. The mRS ranks disability following stroke and cerebral injuries, ranging from 6 (dead) to 0 (fully independent), and is considered a reliable endpoint for clinical neurological studies.
2.3. CSF Purinomics
We carried out measures in 10µL CSF aliquots at a Shimadzu Class-VP high-performance chromatography system (HPLC) in which the separation of the adenine- and guanine-based purines was achieved with a Supelcosil™ LC18 250mm, 4.6 mm diameter size (Sigma Aldrich, 2011, Catalog number: 58298), as previously described by our group and others (Schmidt et al. 2015; Oses et al. 2007; Domanski et al. 2006). Absorbance was read at 254 nm, and quantification of all purines from each subject was obtained in a single run.
The following purines and metabolites were determined: adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), adenosine (ADO), guanosine triphosphate (GTP), guanosine diphosphate (GDP), guanosine monophosphate (GMP), guanosine (GUO), inosine monophosphate (IMP), inosine (INO), hypoxanthine (HXN), xanthine (XAN), and uric acid (UA). HPLC procedure was performed simultaneously between control and TBI samples in a randomized manner. Collection of samples from control and TBI patients was performed concomitantly, patient information from each sample was stored, anonymized, and sequentially assigned an internal identification number that did not indicate grouping or outcome, randomly assigning sequential numbers to control and TBI patients. HPLC procedure was performed utilizing the sequential identification numbers by a researcher blinded to patient information. The acquired data was stored in a data bank for future comparisons.
2.4. Statistical analysis
Experimental outline, with included patients, and a step by step schematic of data analysis is presented in Figure 1. Before performing statistical analysis, obtained data was submitted to Kolmogorov-Smirnov testing for normality, and purine levels of controls and patients were screened for mathematical outliers. Subjects that presented purines value above 1.5-fold of interquartile range from the median, or 3-fold of standard deviation from mean were excluded from all analyses (Mowbray et al. 2019). A total of 4 patients were excluded from all analyses due to outlier levels of purines. Excluded patients frequently presented outlier values for more than one purine concurrently, with the purines that most diverted from average values being AMP (Patient #4: 11.85µM), Inosine (Patient #3 33.10µM and Patient #7: 23.70µM), and Uric acid (Patient #14: 314µM). A complete report of purine concentrations of excluded patients is available as supplementary table 1.” Data were analyzed using RStudio (1.2.5001), and associated packages were further indicated. Data are presented as mean ± SD, or median ± IQR and the graphical representation was made using GraphPad Prism 8. Comparison between groups was performed by two-tailed Student’s independent t-test or Mann-Whitney test. Receiver Operating Characteristic (ROC) curves were created to explore the ability of biomarkers to predict survival, indicated by the Area Under the Curve (AUC). The cutoff value was calculated by the Youden method, using the R package “OptimalCutpoints” (López-Ratón et al. 2014). Multivariate correlations between all purines and clinical data were assessed through the Spearman correlation test, with corrections by Bonferroni, using the R package “Psych” (Revelle 2018) for statistical testing, and “Corrplot” (Wei and Simko 2017) for graphical representation.
Figure 1: Flow diagram of patient inclusion and data analysis.

Following eligibility criteria, 21 TBI patients and 51controls were initially included in the study, with CSF collected and purines quantified. Homogeneity of data analysis led to exclusion of 4 subjects from all further analyses. Step by step description of data analysis shows that first, all purines were investigated regarding TBI patients relative to controls, to assess the impacts of TBI over physiological purine profiles, and associate purine levels impact over a long-term neurofunctional score. Posterior analysis investigated differential purine profiles within surviving and non-surviving TBI patients, identifying only 4 altered purines. Finally, the 4 altered purines in non-surviving patients were investigated relative to their predictive values of mortality through a receiver operating characteristic curve.
The multivariate correlation of purine derivatives was utilized to create a model, that superimposes correlations with the classical cascade of purine degradation obtained from the KEGG database (Kanehisa 2019; Kanehisa and Goto 2000).
Briefly, based on the significant purine correlations present in controls, but absent in TBI patients, we obtained a list of correlations that were impaired by TBI. Within this list of correlations impaired by TBI, we selected only the correlations between purines that both presented altered concentration in TBI patients (Highlighted in the model by arrowheads). We then graphically present these impaired correlations over the theoretical enzymatic degradation cascade of purines. The concept behind this model is to provide a topographical overview of the impact of TBI on the purine interconversion and profile.
For all tests, the statistical significance was considered when p ≤ 0.05.
3. Results
3.1. Severe TBI triggers specific changes in CSF purine metabolites
Among guanine-derived purines, GDP and guanosine were significantly increased in TBI patients, compared to controls (Figure 2A and B, and Table 1), while no difference was observed in GTP and GMP levels (Supplemental Figure 1A and B). In non-surviving TBI patients, there were increased CSF concentrations of GTP, and GDP compared with surviving patients (Figure 3A and B, respectively). Concentrations of GMP and guanosine did not differ between surviving and non-surviving patients (Table 2, supplemental figures 2A and B).
Figure 2. Purine profile is altered in the CSF of TBI patients.
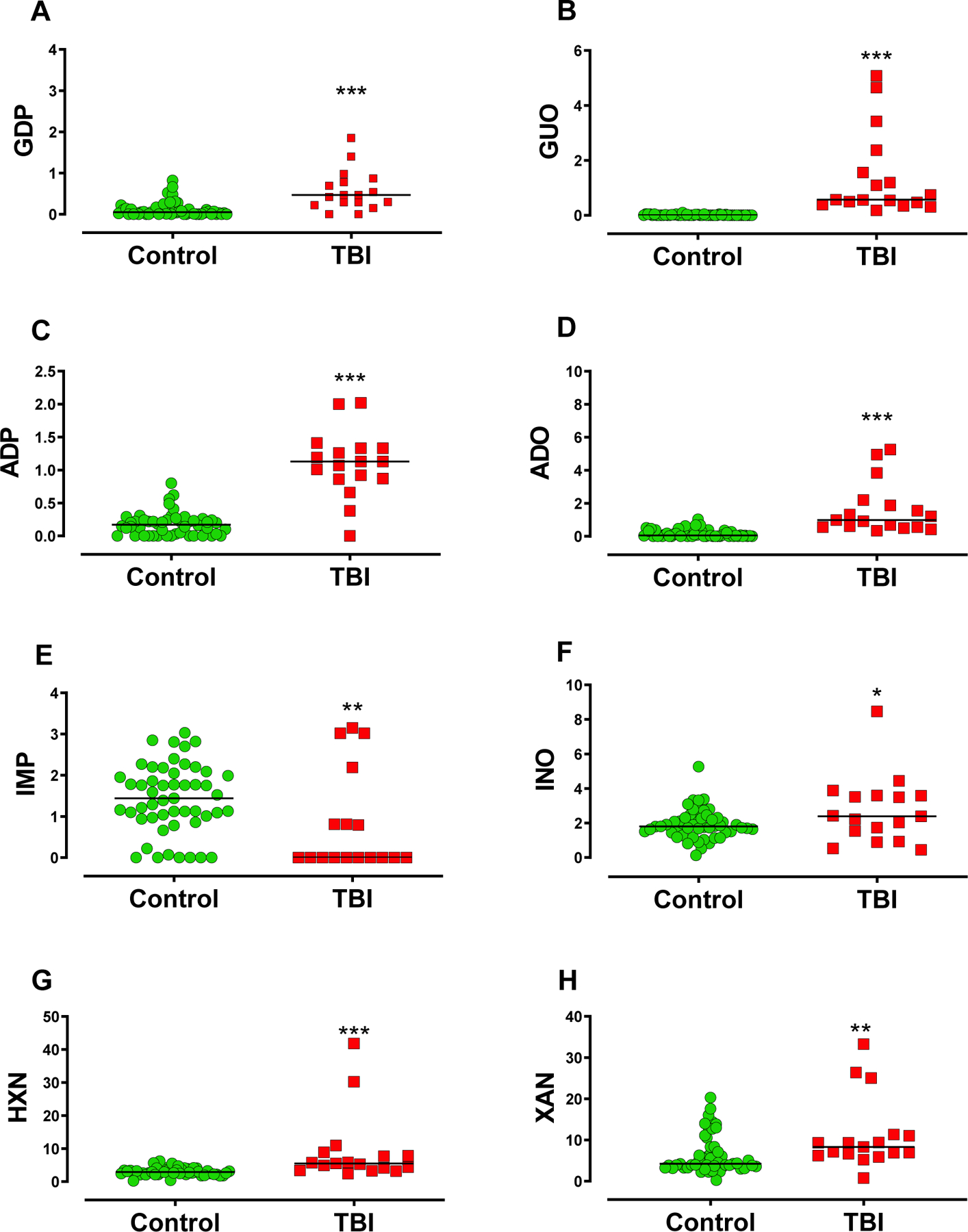
This setting displays the composite of purines with statistically significant differences between TBI patients and controls. (A) Guanine derivatives GDP, (B) and guanosine (GUO) presented increased levels in TBI patients compared to controls. (C) Adenine derivatives ADP and (D) adenosine (ADO) were increased in TBI patients compared to controls. (E) CSF IMP levels were reduced in TBI patients compared to control, (F) whilst inosine (INO), (G) hypoxanthine (HXN), and (H) xanthine (XAN) were increased. Horizontal lines indicate the median value. Statistical significance was assessed through Mann-Whitney or Student’s independent T-test, with *, **, *** indicating p ≤ 0.05, p ≤ 0.01, p ≤ 0.001, respectively (Controls n= 51 patients, TBI n = 17 patients
Table 1: Descriptive data of purine profile, comparing TBI patients and matched controls.
Descriptive statistics are presented as mean ± SD, or median and interquartile range (IQR) (Q1 – Q3). Purine levels are shown as µM concentration. Statistical values are reported for independent t-test, or Mann-Whitney U test, with the respective t-values or U-values, degrees of freedom (Df), and obtained p-values for the tests.
| Gaussian distribution | |||||
|---|---|---|---|---|---|
| Controls (mean ± sd) |
sTBI Patients (mean ± sd) |
t-value | Df | p-value | |
| Inosine | 1.930 ± 0.855 | 2.711 ± 1.933 | 2.308 | 66 | *0.0241 |
| Non-Gaussian distribution | |||||
|
Controls (Median (IQR)) |
sTBI
Patients
(Median (IQR)) |
U-value | Df | p-value | |
| GTP | 2.140 (1.60 – 2.34) |
1.580 (1.24 – 2.65) |
347 | 66 | 0.2241 |
| GDP | 0.050 (0.000 – 0.120) |
0.470 (0.255 – 0.825) |
136.5 | 66 | ***<0.0001 |
| GMP | 0.000 (0.000 – 0.000) |
0.000 (0.000 – 0.000) |
454 | 66 | 0.9408 |
| Guanosine | 0.000 (0.000 – 0.0200) |
0.570 (0.430 – 1.965) |
0 | 66 | ***<0.0001 |
| ATP | 0.000 (0.000 – 0.000) |
0.000 (0.000 – 0.040) |
353 | 66 | 0.1073 |
| ADP | 0.170 (0.000 – 0.240) |
1.130 (0.865 – 1.330) |
50.50 | 66 | ***<0.0001 |
| AMP | 1.710 (0.000 – 2.680) |
0.830 (0.520 – 2.370) |
403 | 66 | 0.6883 |
| Adenosine | 0.050 (0.00 – 0.290) |
0.990 (0.560 – 2.040) |
37 | 66 | ***<0.0001 |
| IMP | 1.440 (0.970 – 2.050) |
0.000 (0.000 – 1.500) |
252 | 66 | **<0.01 |
| Xanthine | 4.250 (3.290 – 8.340) |
8.330 (6.430 – 11.19) |
232 | 66 | **0.0037 |
| Hypoxanthine | 2.890 (2.210 – 3.660) |
5.510 (3.740 – 8.335) |
116 | 66 | ***<0.0001 |
| Uric Acid | 24.35 (18.03 – 33.76) |
22.41 (15.13 – 31.08) |
386 | 66 | 0.5091 |
Indicates p-values <0.05,
indicates p-values <0.01 and
indicates p-values < 0.001. (Controls n = 51 patients, TBI n = 17 patients.
Figure 3. Increased concentration of CSF purines is observed in non-surviving TBI patients.
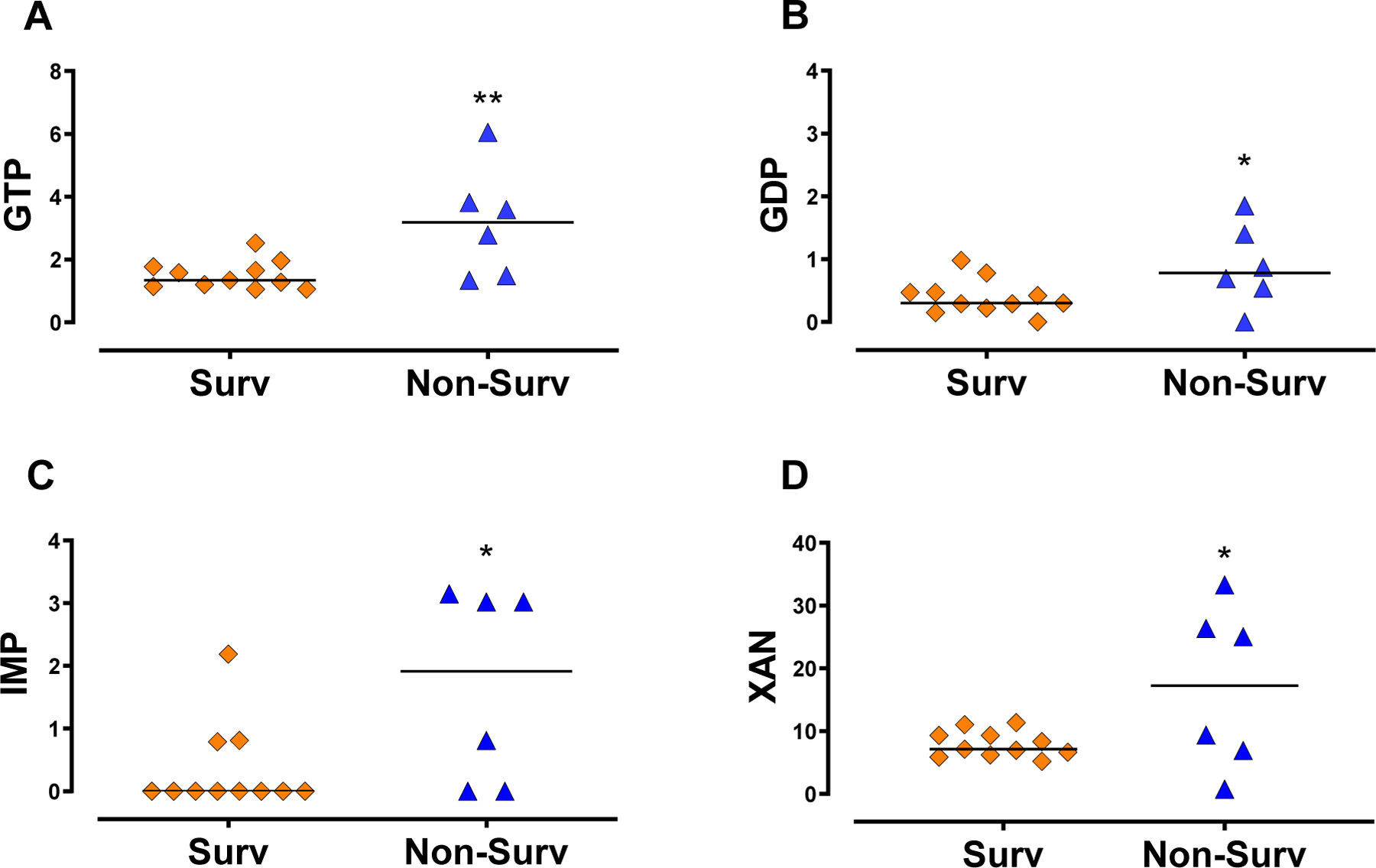
(A) GTP (B) and GDP concentrations were increased in non-surviving (Non-Surv), relative to surviving TBI patients (Surv). (C) Non-surviving patients also presented increased IMP (D) and xanthine (L) CSF levels compared to surviving patients. Horizontal lines indicate the median value. Statistical significance was assessed through Mann-Whitney or Student’s independent T-test, with *, **, *** indicating p ≤ 0.05, p ≤ 0.01, p ≤ 0.001, respectively ( Surv n =11 patients, Non-Surv n=6 patients).
Table 2: Descriptive data of purine profile, comparing surviving and non-surviving severe TBI patients.
Descriptive statistics are presented as mean ± SD, or median and interquartile range (IQR) (Q1 – Q3). Purine levels are shown as µM concentration. Statistical values are reported for independent t-test, or Mann-Whitney U test, with the respective t-values or U-values, degrees of freedom (Df) and obtained p-values for the tests.
| Gaussian distribution | |||||
|---|---|---|---|---|---|
| Surviving (Mean ± sd) |
Non-surviving (Mean ± sd) |
t-value | Df | p-value | |
| GTP | 1.505 ± 0.452 | 3.178 ± 1.74 | 3.075 | 15 | **0.0077 |
| GDP | 0.397 ± 0.28 | 0.819 ± 0.654 | 2.209 | 15 | *0.0431 |
| ADP | 1.138 ± 0.362 | 1.008 ± 0.720 | 0.5017 | 15 | 0.6232 |
| Inosine | 3.124 ± 2.200 | 1.953 ± 1.100 | 1.210 | 15 | 0.2449 |
| Xanthine | 7.943 ± 2.076 | 16.96 ± 12.97 | 2.315 | 15 | *0.0352 |
| Non-Gaussian distribution | |||||
| Surviving (Median (IQR)) |
Non-surviving
(Median (IQR)) |
U-value | Df | p-value | |
| GMP | 0.000 (0.000 – 0.000) |
0.000 (0.000 – 1.170) |
27.5 | 15 | 0.353 |
| Guanosine | 0.740 (0.540 – 1.560) |
0.485 (0.338 – 3.048) |
26 | 15 | 0.524 |
| ATP | 0.000 (0.000 – 0.040) |
0.000 (0.000 – 0.088) |
32.50 | 15 | >0.999 |
| AMP | 0.830 (0.570 – 2.220) |
0.880 (0.298 – 2.620) |
30 | 15 | 0.7885 |
| Adenosine | 1.210 (0.560 – 1.880) |
0.655 (0.545 – 5.028) |
35.20 | 15 | 0.9812 |
| IMP | 0.000 (0.000 – 0.790) |
1.915 (0.000 – 3.053) |
15.5 | 15 | *0.0438 |
| Hypoxanthine | 5.260 (3.340 – 7.660) |
6.805 (3.705 – 33.15) |
24 | 15 | 0.4043 |
| Uric Acid | 25.95 (15.58 – 27.95) |
20.01 (12.50 – 129.7) |
32 | 15 | 0.9612 |
indicates p-values <0.05,
indicates p-values <0.01 and
indicates p-values < 0.001 (Surviving n = 11 patients, Non-surviving n = 6 patients).
Adenine-based purines, ADP and adenosine, were increased in TBI patients, relative to controls (Figure 2C and D, respectively, and Table 1), whilst no difference was observed in ATP and AMP levels (Supplemental figure 1C and D). Further, ATP, ADP, AMP, and adenosine were not different between surviving and non-surviving patients (Table 2, supplemental figure 2C to F).
The downstream guanine and adenine purine metabolism converge to the formation of IMP, inosine, hypoxanthine, xanthine, and uric acid. While CSF levels of IMP were reduced in TBI patients, inosine, hypoxanthine, and xanthine were increased (Figure 2E to H, respectively, also Table 1). No significant difference was observed in uric acid CSF levels (supplemental figure 1E). Among TBI patients, IMP and xanthine levels were increased in non-surviving compared with surviving patients (Figure 3C and D). Similar levels of inosine, hypoxanthine and uric acid were found in both surviving and non-surviving patients (supplemental figures 2G, H, and I, respectively).
3.2. CSF purinomics yield prognostic signatures for neurological outcomes
Considering that four different purine metabolites presented statistical differences between surviving and non-surviving TBI patients, we investigated the accuracy of these metabolites as prognostic biomarkers for mortality due to severe TBI.
The AUROC indicates how well GTP, GDP, IMP, and xanthine can discriminate patients that will survive from patients that will die up to 3 days from the admission to ICU (Figure 4A to D). With a cut-off value of > 2.780, GTP provided 66.67% of sensitivity, and 100% of specificity (AUROC: 0.841, p = 0.0237, 95% CI, CL: 95.45% to 100%). GDP, IMP, and xanthine did not present statistically significant predictive values (p > 0.05). However, the AUROC values for IMP barely reached significance (p = 0.06).
Figure 4. Predictive accuracy of CSF purines relative to death up to 3 days from ICU admission.
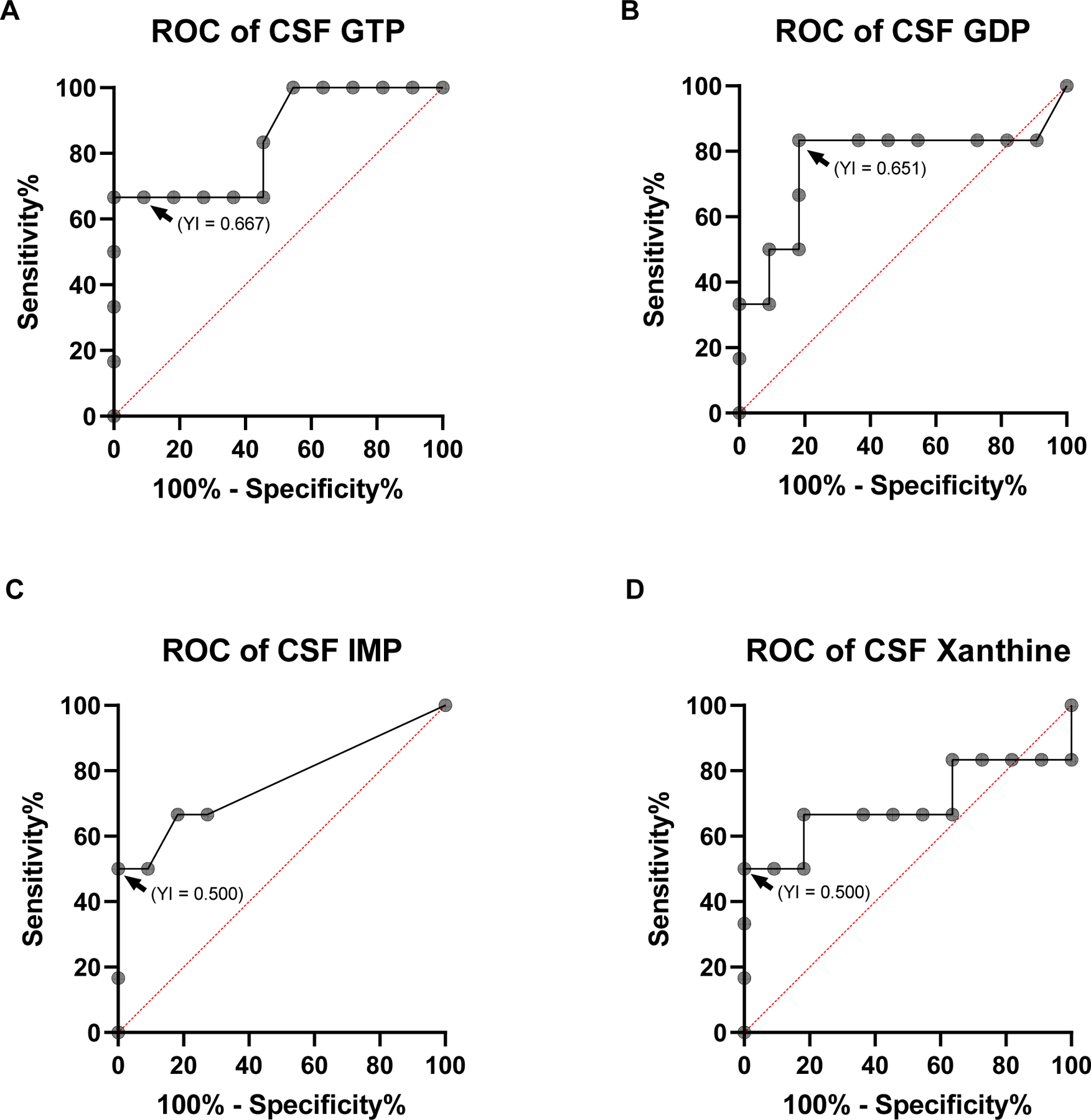
The area under the curve of the receiver operating characteristic curve (AUROC) for GTP (A), GDP (B), IMP (C), and Xanthine (D). (A) GTP presented at a cut-off value of >2.780 µM (indicated by arrowhead) a sensitivity of 66.67%, and 100% of specificity (AUROC: 0.841, p = 0.0237, 95% CI, CL (Confidence Levels): 62.26% to 99.53%, Youden Index (YI) for >2.780 µM = 0.667). CSF GDP (B), IMP (C) and xanthine (D) levels presented AUROC with lower predictive value (p > 0.05). Arrowheads indicate the optimal cut-off point indicated by the Youden criteria, followed by the respective Youden Index (YI) obtained, presented within parenthesis. (Survival n =11 patients, Non-Survival n=6 patients)
Also, we performed bivariate correlations between CSF purine levels and clinical neurological outcomes. We found that the modified Rankin Scale (mRS) did not correlate with TBI severity at admission (GCS) (r: −0.32, p = 0.207). Only guanosine correlated with mRS (r: −0.50, p = 0.042) (Figure 3 A and B, respectively), with all remaining purines and derivatives not presenting statistical correlation with long-term neurological outcomes (supplementary figure 3). Clinical parameters measured at ICU such as the medium arterial pressure, and cerebral perfusion pressure were not statistically correlated to mRS (supplementary table 2).
3.3. Severe TBI disrupts CSF purinomic networks
Correlations between purines were assessed to estimate functional purinomic networks. Briefly, a stepwise metabolism of ATP generates ADP and AMP, and downstream metabolites, meaning that correlations between these molecules represent physiological interconnections. Here we show that TBI patients displayed a distinct profile of associations from controls, thereby suggesting a rupture in the physiological purinomic networks (Figure 6A and B).
Figure 6. Multivariate correlations between purine derivatives in the CSF.
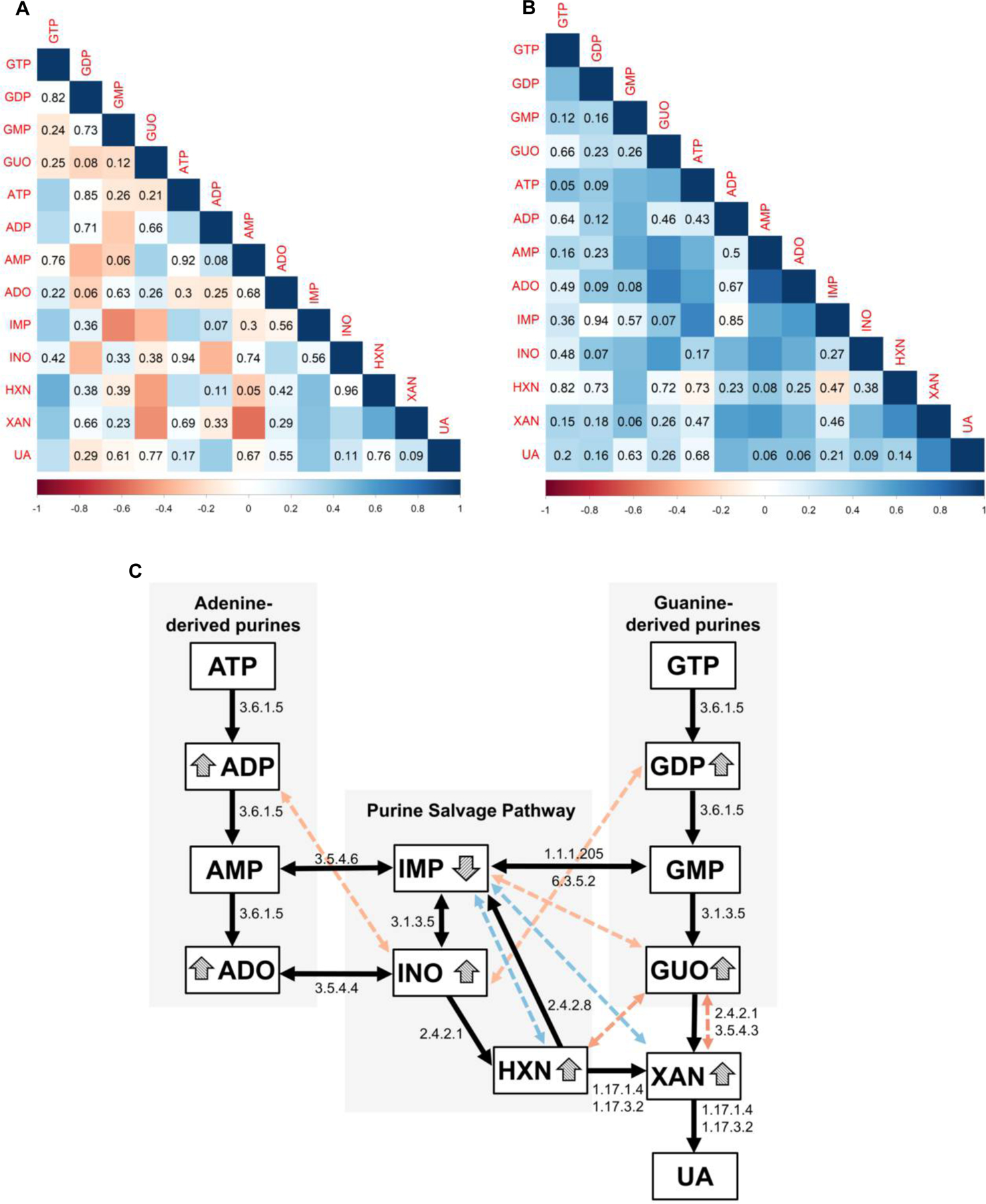
(A), and (B) report Spearman’s r values adjusted by Bonferroni, indicated by color scale ranging from −1 (red) to +1 (blue), and obtained p-values are displayed for non-statistically significant correlations in both panels. Squares with no indication of p-value represent correlations with p<0.05. (A) Spearman r correlations between purines in controls. The positive and negative correlations between adenine and guanine derivatives and their products of downstream degradation suggest an integrated physiological metabolic network. (B) Only positive correlations between purine derivatives were observed in TBI patients, with a limited association between adenine- and guanine-derived metabolites and a rupture of the proposed physiological negative correlations.
(C) Integrative model based on classical metabolic pathways of purine degradation indicated by the black flowchart, accompanied by the E.C. (Enzyme Commission) numbers of the enzymes related to the respective metabolic steps leading from GTP and ATP to uric acid (UA) formation. The colored dotted double-headed arrows indicate correlations in purine metabolism of controls, that are lost in TBI patients. The color of double-headed arrows indicates Spearman r values, following the color scale displayed in (A) and (B). The specific purine derivatives modified by TBI are highlighted by the arrowheads.(Controls n= 51 patients, TBI n = 17 patients).
Additionally, we propose a model based on the classical purine metabolism cascade, as described in the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways. The model displays in the double-headed arrows correlations between purines present in controls, but absent in TBI patients (the color scales represent Spearman r correlation values). Here, we intended to explore how TBI impairs interconversion of purines within the topographical map of purine metabolism (Classical degradation and purine salvage pathways).
In this model, we primarily showed that correlations between purines in controls subjects match the stepwise cascades of the classic purine degradation and salvage pathways. For instance, a negative correlation between ADP and GDP with INO is observed in control subjects. This finding fits the physiological degradation of ADP and GDP driving the formation of INO.
However, in TBI patients such correlations are lost, as indicated in our model by the colored dashed lines (Figure 6C). For instance, the negative correlation between guanosine (GUO) and xanthine (XAN) is not present in TBI patients, suggesting that this classical degradation step may be impaired following injury. Similarly, the absence of the positive correlation between hypoxanthine (HXN) and IMP may suggest an impairment in the salvage of HXN to IMP.
4. Discussion
This study provides the first evidence that examination of purine levels in CSF in the acute setting after TBI may serve as a promising biomarker in predicting outcomes. Such “purinomics” approach showed that CSF GTP levels assessed within a short time after ICU admission appears to be a prognostic indicator of death and survival outcomes. Also, guanosine levels were associated with the impairment in a functional neurological score (mRS) two years after ICU admission. Correlations between ATP- and GTP-derived purines, resulted in a mechanistic model that highlighted the impact of TBI on the enzymatic degradation of purines.
There is rapidly increasing interest in examining body fluids in search of biomarkers that diagnose pathological changes and predict outcomes after TBI (Atkinson et al. 2001; Zetterberg et al. 2013). The purinergic system has established components in the brain that share specific localization and functions with glutamatergic tripartite synapse, microglia, and oligodendrocytes (Burnstock 2017; Burnstock 2020). Among those components, are transporters, receptors, and Ecto-NTPDase (E-NTPDase) enzymes, which at the mechanistic level control the balance of extracellular purine levels, and consequently, their effects on specific extrasynaptic and synaptic players.
We have previously shown that after a single convulsive seizure in a rat model, CSF adenine and guanine E-NTPDase activities increased in a similar time profile to classical biomarkers of neuronal (NSE) and astrocyte (S100B) death, implying that such increased activities also reflected neural cell damage (Oses et al. 2004; Cruz Portela et al. 2002; Oses et al. 2007). Similarly, pentylenetetrazol-kindling rats showed alterations in adenine and guanine enzymatic degradation along with increased CSF GTP, GDP, ADP, and uric acid levels (Oses et al. 2007). Notably, it has been described that TBI promotes disturbances in purinergic signaling that may favor the development of epilepsy (Boison 2008; Fedele et al. 2005; Englander et al. 2003; Jackson et al. 2016). These findings suggest that an acute alteration in extracellular purine concentrations after TBI could influence neurological outcomes.
The findings of the present study demonstrate that several extracellular purines are altered after a severe TBI in patients, and may serve as brain biomarkers of both short- and long-term injury caused by mechanisms associated with glutamatergic excitotoxicity (Stefani et al. 2017). Remarkably, increased extracellular purine catabolism has the potential to generate endogenous neuroprotective anti-glutamatergic responses through adenosine and guanosine (Dunwiddie and Masino 2001; Schmidt et al. 2000), and an antioxidant defense through the uric acid. Accordingly, exogenous administration of guanosine after mild TBI in rats displayed neuroprotective properties (Courtes et al. 2020). However, considering the extravasation of active biomolecules caused by the rupture of cell membranes and axons (Johnson et al. 2013) in the first hours after TBI, it is unlikely that these endogenous neuroprotective mechanisms are ready-to-use and overcome the biochemical components of secondary damage (Stefani et al. 2017).
While clinical studies investigating purine levels in biological fluids after TBI are scarce, previously published reports in rodent models of TBI (Verrier et al. 2012; Bell et al. 1998; Marklund et al. 2006) have demonstrated acute increases in the levels of ADP, adenosine, inosine, and hypoxanthine, which are consistent with the concentration profile found in our current study (Fig 1E, F, H and I respectively). However, we did not find reports of changes in GDP, guanosine, and xanthine levels in the biological fluids of patients, or in preclinical models of TBI. Hence, this work primarily demonstrates that CSF levels GDP, guanosine, ADP, adenosine, inosine, xanthine, and hypoxanthine increased above the control levels within the first 3 hours after TBI, whereas IMP was consistently decreased. Although this composite of purinergic abnormalities likely reflects damage to the brain cells, it further requires associations with clinical and neurological endpoints to be featured as a feasible biomarker in the ICU (Atkinson et al. 2001).
Among the adenine- and guanine-derived purines, GTP, GDP, IMP, and xanthine were significantly increased in non-surviving compared to surviving patients. Despite IMP being decreased in the TBI group relative to control, comparison between non-surviving and surviving patients within the TBI group displayed differences (see Figures 2 E and 3 C). The discriminatory profile of these particular purines may encompass features with potential validity to be explored as predictive biomarkers (Cristofori et al. 2005; Laketa et al. 2015).
A previous case report investigating energy failure and oxidative damage in one severe TBI patient has shown that the CSF levels of adenosine, hypoxanthine, xanthine, and uric acid increased up to 100 h after ventricular catheter insertion. The concentrations of hypoxanthine, xanthine, and uric acid increased 4- to 6-fold 72 h after catheter insertion and before brain death, which also suggests a putative prognostic value (Cristofori et al. 2005). Albeit the time between the catheter insertion and CSF collection differs from our work, we found similar increments in xanthine levels before brain death. Also, our work further highlighted the discriminatory profile of GTP, GDP, IMP, and xanthine, expanding the opportunity to identify candidate biomarkers and clinical associations.
Regarding the sensitivity and specificity of GTP, GDP, IMP, and xanthine in determining the short-term neurological prognosis of severe TBI patients, interestingly, only GTP levels displayed predictive value relative to mortality within 3 days after TBI (sensitivity of 66.67% and specificity of 100%). A potential mechanism underlying these properties has been identified in a post-mortem examination of the brains of TBI patients. The GTP binding receptors RhoA and RhoB expression levels were acutely increased after TBI, which has been sustained for months after the head impact (Brabeck et al. 2004). These receptors participate in cerebral responses to injuries, regulating cerebrovascular tonus, astrocytic and microglial activation, axonal regeneration, and neuroplasticity (Stankiewicz and Linseman 2014). In animal models of TBI, it has been reported an increase in the downstream GTP-binding receptor pathways, such as the RhoA-ROCK cascade, which is known to exacerbate neuronal death (Sabirzhanova et al. 2013; Labandeira-Garcia et al. 2017; Dubreuil et al. 2006; Rikitake et al. 2005). Such role is apparently corroborated by the increased GTP levels observed in non-surviving patients in our study. In addition, we sought associations between purine levels and long-term neurologic disability. Our data demonstrate that only guanosine levels at admission in the ICU were inversely associated with mRS scores two years after TBI. Exogenous administration of guanosine has well-recognized effects against glutamatergic excitotoxicity and mitochondrial dysfunction, important components of secondary damage following TBI (Gerbatin et al. 2017; Dobrachinski et al. 2019; Gerbatin et al. 2019). Based on these properties, we posit that even a build-up of endogenous levels of guanosine at admission may attenuate the progression of neuronal damage and functional decline following severe TBI.
Here, we further attempted to uncover the mechanism of purine degradation after TBI using a modeling based on the multivariate correlations analysis between purines. Primarily, we showed that purine metabolism in controls is tethered to the classical enzymatic degradation steps (KEGG pathways). The correlation network, present in controls, reflects an integrated enzymatic degradation, endorsing the biological plausibility of these proposed associations. However, many of these correlations present in the controls disappear in the TBI group, suggesting an uncoupling of the enzymatic steps, leading to the accumulation of specific purine derivatives. For instance, in the TBI group, negative correlations (Figure 6C, red double-headed arrows) between inosine and ADP, guanosine and xanthine are lost, as well as the positive correlations (Figure 6C, blue double-headed arrows) between IMP and both, hypoxanthine and xanthine. The loss of negative correlations was accompanied by the accumulation of GDP, ADP, inosine, xanthine, and guanosine (indicated in the model by the arrowheads), likely suggesting the impairment of some enzymes, including the E-NTPDases.
Noteworthy, the loss of the positive correlations between IMP and both, hypoxanthine and xanthine paralleled with a decrease in IMP levels, and consequent accumulation of hypoxanthine and xanthine. A previous study demonstrated that changes in ectonucleotidase activity in the rodent cortex, hippocampi, and caudate nucleus caused reduced AMP hydrolysis, but not ATP hydrolysis 4 hours after injury, emphasizing that purine degradation enzymatic activities are actually altered by TBI (Bjelobaba et al. 2009). Also, accumulation of xanthine and hypoxanthine in both CSF and serum, and alterations in xanthine oxidase activity have been previously reported in preclinical studies of TBI, supporting the mechanistic involvement of purine metabolism (Laketa et al. 2015; Solaroglu et al. 2005; Tayag et al. 1996). Overall, our mathematical model in CSF of patients suggests that the imbalance of purine metabolism was likely mediated by altered enzymatic activity caused by TBI. These collective findings support further investigations of key effectors mediating purine degradation after TBI, as well as the role of specific enzymes on clinical outcomes.
Our study presents limitations that should be disclosed. This is a unicentric study, that enrolled primary 17 patients with severe TBI. The low number of TBI patients limits the power of the clinical associations with purine levels. Nonetheless, this cohort has been previously explored for well-established biomarkers, including NSE, GFAP, and S100B (Böhmer et al. 2011) and replicates studies with large TBI populations. Also, a a posteriori power analysis suggests that our results present appropriate power with the current study population, for our two main outcomes; a. GTP as an indicator of mortality (98.5%), and to a lesser extent b. GUO as a prognostic indicator of long-term neurofunctional status (84.1%). Also, this is a hypothesis-generating work and was not designed to determine GTP or GUO as definitive biomarkers. Therefore, considering the primary nature of this exploratory study, and the novelty of these findings, the results presented here reveal potentialities that need to be better explored in further studies. Also, the comparison between TBI and controls could be somewhat biased considering the location of the CSF collection. Whereas the TBI patients had their CSF samples collected from external ventricular drains, controls had a lumbar puncture for CSF collection, which could affect the composition and content of various purines and their metabolites (Hegen et al. 2018; Brandner et al. 2013; Podkovik et al. 2020).
5. Conclusion
In the present study, severe TBI imprinted particular purine-derived signatures in the CSF of patients. We identified GTP as a sensitive and specific predictive biomarker of mortality, and guanosine as an indicator of long-term functional disability.
Supplementary Material
Supplemental Figure 1. Composite of purines in the CSF ofTBI patients relative to controls (surviving and non-surviving). This setting displays the remaining purines that did not show a statistically significant difference between TBI patients and controls. (A) GTP, (B) GMP, (C) ATP, (D) AMP, and (E) Uric Acid. Statistical significance was assessed through Mann-Whitney or Student’s independent T-test (Controls n= 51 patients, TBI n = 17 patients).
Supplemental Figure 2. Composite of purines in the CSF of Non-surviving and surviving TBI patients. This setting displays the remaining purines that did not show a statistically significant difference in non-surviving and surviving TBI patients. (A) GMP, (B) Guanosine, (C) ATP, (D) ADP, (E) AMP, (F) Adenosine, (G) Inosine, (H) Hypoxanthine, and (I) Uric Acid. Statistical significance was assessed through Mann-Whitney or Student’s independent T-test (Surv n =11 patients, Non-Surv n=6 patients).
Supplementary Figure 3: Long-term neurological outcome is not associated with CSF levels of most purines levels. This panel displays correlation plots between the modified Rankin Scale (mRS) and (A) GTP, (B) GDP, (C) GMP, (D) ATP, (E) ADP, (F) AMP, (G) Adenosine, (H) IMP, (I) Inosine, (J) Hypoxanthine, (K) Xanthine and (L) Uric acid; where none of the purines assessed displayed significant correlations (Spearman correlation, significance considered when p<0.05) with the two-year mRS of severe TBI patients (n = 17 TBI patients).
Figure 5. Spearman bivariate correlation between CSF purine levels and clinical neurological outcomes.
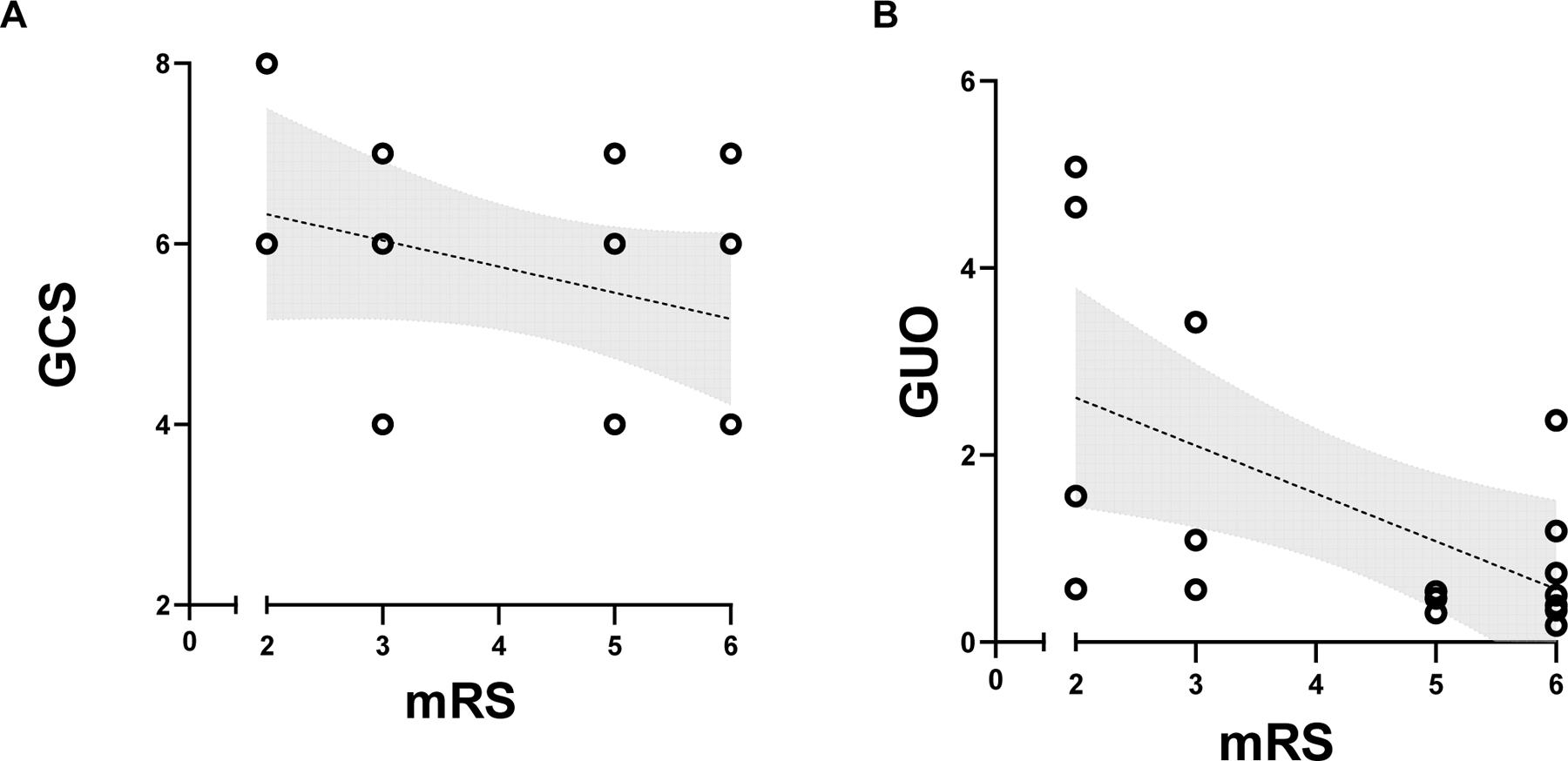
(A) Glasgow Coma Scale (GCS) at admission did not present a statistically significant correlation with two years modified Rankin Scale (mRS) (p = 0.2070, r = −0.302, n= 17 patients). (B) Guanosine (GUO) levels at admission were statistically correlated with disability 2 years after discharge (mRS) (p = 0.0428, r = −0.506, n = 17 patients) (Survival n =11 patients, Non-Survival n=6 patients). Lines indicate linear regression model, and shaded area indicates the of 95% confidence intervals.
10. Acknowledgments
The authors would like to thank the Center for Brain Injury and Repair, University of Pennsylvania (CBIR-UPENN) for the technical and scientific support, the Brazilian Agencies/Programs FAPERGS #1010267, FAPERGS/PPSUS#17/2551–0001, FAPERGS/PRONEX#16/2551–0000499-4, Programa de Internacionalização de Ciência FAPERGS/CAPES #19/25510000717–5, Program Science without Borders CNPQ #4011645/2012–6, and CNPq INNT #5465346/2014–6. Also, this research was made available with the support from National Institutes of Health grants R01NS092398, R01NS038104, R01NS094003, R01EB021293, Paul G. Allen Family Foundation, and the PA Consortium on Traumatic Brain Injury 4100077083.
Abbreviation List
- ADO
Adenosine
- ADP
Adenosine Diphosphate
- AMP
Adenosine Monophosphate
- ATP
Adenosine Triphosphate
- AUC
Area Under the Curve
- CSF
Cerebrospinal Fluid
- CT
Computed Tomography
- CL
Confidence Levels
- GUO
Guanosine
- GDP
Guanosine Diphosphate
- GMP
Guanosine Monophosphate
- GTP
Guanosine Triphosphate
- HPLC
High Performance Liquid Chromatography
- HXN
Hypoxanthine
- INO
Inosine
- IMP
Inosine Monophosphate
- ICU
Intensive Care Unit
- ICP
Intracranial Pressure
- KEGG
Kyoto Encyclopedia Of Genes And Genomes
- mRS
Modified Rankin Scale
- NSE
Neuron-Specific Enolase
- TBI
Traumatic Brain Injury
- UA
Uric Acid
- XAN
Xanthine
Footnotes
Declarations of interest: none.
12 References
- Atkinson AJ, Colburn WA, DeGruttola VG, DeMets DL, Downing GJ., Hoth DF, Oates JA, et al. (2001) Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework [Google Scholar]
- Beal PR, Yao SYM, Baldwin SA, Young JD, King AE, Cass CE (2004) The equilibrative nucleoside transporter family, SLC29. Pflügers Archiv European Journal of Physiology 447, 735–743. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Kochanek PM, Carcillo JA, Mi Z, Schiding JK, Wisniewski SR, Clark RSB, Dixon CE, Marion DW, Jackson E (1998) Interstitial Adenosine, Inosine, and Hypoxanthine Are Increased after Experimental Traumatic Brain Injury in the Rat. Journal of Neurotrauma 15, 163–170. [DOI] [PubMed] [Google Scholar]
- Bjelobaba I, Stojiljkovic M, Lavrnja I, Stojkov D, Pekovic S, Dacic S, Laketa D, Rakic L, Nedeljkovic N (2009) Regional changes in ectonucleotidase activity after cortical stab injury in rat. General physiology and biophysics 28 Spec No, 62–8. [PubMed] [Google Scholar]
- Böhmer AE, Oses JP, Schmidt AP, Perón CS, Krebs CL, Oppitz PP, D’Avila TT, Souza DO, Portela LV, Stefani MA (2011) Neuron-Specific Enolase, S100B, and Glial Fibrillary Acidic Protein Levels as Outcome Predictors in Patients With Severe Traumatic Brain Injury. Neurosurgery 68, 1624–1631. [DOI] [PubMed] [Google Scholar]
- Boison D (2008) The adenosine kinase hypothesis of epileptogenesis. Progress in Neurobiology 84, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabeck C, Beschorner R, Conrad S, Mittelbronn M, Bekure K, Meyermann R, Schluesener HJ, Schwab JM (2004) Lesional Expression of RhoA and RhoB following Traumatic Brain Injury in Humans. Journal of Neurotrauma 21, 697–706. [DOI] [PubMed] [Google Scholar]
- Brandner S, Thaler C, Lewczuk P, Lelental N, Buchfelder M, Kleindienst A (2013) Neuroprotein dynamics in the cerebrospinal fluid: intraindividual concomitant ventricular and lumbar measurements. European neurology 70, 189–194. [DOI] [PubMed] [Google Scholar]
- Burnstock G (2017) Purinergic Signalling and Neurological Diseases: An Update. CNS & Neurological Disorders – Drug Targets 16, 257–265. [DOI] [PubMed] [Google Scholar]
- Burnstock G (2020) Introduction to Purinergic Signalling in the Brain, in Advances in Experimental Medicine and Biology, pp. 1–12. [DOI] [PubMed] [Google Scholar]
- Courtes AA, Gonçalves DF, Hartmann DD, Rosa P. C. da, Cassol G, Royes LFF, Carvalho N. R. de, Soares FAA (2020) Guanosine protects against behavioural and mitochondrial bioenergetic alterations after mild traumatic brain injury. Brain Research Bulletin 163, 31–39. [DOI] [PubMed] [Google Scholar]
- Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Signoretti S, Amorini AM, Fazzina G, Lazzarino G (2005) Biochemical analysis of the cerebrospinal fluid: evidence for catastrophic energy failure and oxidative damage preceding brain death in severe head injury: a case report. Clinical Biochemistry 38, 97–100. [DOI] [PubMed] [Google Scholar]
- Cruz Portela LV, Oses JP, Silveira AL, Schmidt AP, Lara DR, Oliveira Battastini AM, Ramirez G, Vinade L, Freitas Sarkis JJ, Souza DO (2002) Guanine and adenine nucleotidase activities in rat cerebrospinal fluid. Brain research 950, 74–78. [DOI] [PubMed] [Google Scholar]
- Cunha RA (2016) How does adenosine control neuronal dysfunction and neurodegeneration? Journal of Neurochemistry 139, 1019–1055. [DOI] [PubMed] [Google Scholar]
- Dobrachinski F, Gerbatin RR, Sartori G, Golombieski RM, Antoniazzi A, Nogueira CW, Royes LF, et al. (2019) Guanosine Attenuates Behavioral Deficits After Traumatic Brain Injury by Modulation of Adenosinergic Receptors. Molecular Neurobiology 56, 3145–3158. [DOI] [PubMed] [Google Scholar]
- Domanski L, Sulikowski T, Safranow K, Pawlik A, Olszewska M, Chlubek D, Urasinska E, Ciechanowski K (2006) Effect of trimetazidine on the nucleotide profile in rat kidney with ischemia–reperfusion injury. European Journal of Pharmaceutical Sciences 27, 320–327. [DOI] [PubMed] [Google Scholar]
- Doran SJ, Ritzel RM, Glaser EP, Henry RJ, Faden AI, Loane DJ (2019) Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. Journal of Neurotrauma 36, 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Marklund N, Deschamps K, McIntosh TK, McKerracher L (2006) Activation of Rho after traumatic brain injury and seizure in rats. Experimental Neurology 198, 361–369. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA (2001) The Role and Regulation of Adenosine in the Central Nervous System. Annual Review of Neuroscience 24, 31–55. [DOI] [PubMed] [Google Scholar]
- Englander J, Bushnik T, Duong TT, Cifu DX, Zafonte R, Wright J, Hughes R, Bergman W (2003) Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Archives of Physical Medicine and Rehabilitation 84, 365–373. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rülicke T, Crestani F, Boison D (2005) Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 128, 2383–2395. [DOI] [PubMed] [Google Scholar]
- Gerbatin R. Da R., Cassol G, Dobrachinski F, Ferreira APO, Quines CB, Pace I. D. Della, Busanello GL, et al. (2017) Guanosine Protects Against Traumatic Brain Injury-Induced Functional Impairments and Neuronal Loss by Modulating Excitotoxicity, Mitochondrial Dysfunction, and Inflammation. Molecular Neurobiology 54, 7585–7596. [DOI] [PubMed] [Google Scholar]
- Gerbatin RR, Dobrachinski F, Cassol G, Soares FAA, Royes LFF (2019) A1 rather than A2A adenosine receptor as a possible target of Guanosine effects on mitochondrial dysfunction following Traumatic Brain Injury in rats. Neuroscience Letters 704, 141–144. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Bendall MR, Faden AI, Vink R (1994) Dissociation of Adenosine Levels from Bioenergetic State in Experimental Brain Trauma: Potential Role in Secondary Injury. Journal of Cerebral Blood Flow & Metabolism 14, 853–861. [DOI] [PubMed] [Google Scholar]
- Hegen H, Walde J, Auer M, Deisenhammer F (2018) Cerebrospinal fluid:serum glucose ratio in the ventricular and lumbar compartments: implications for clinical practice. European journal of neurology 25, 373–379. [DOI] [PubMed] [Google Scholar]
- Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, Szeto GL, et al. (2020) Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. The Journal of Neuroscience 40, 2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Boison D, Schwarzschild MA, Kochanek PM (2016) Purines: forgotten mediators in traumatic brain injury. Journal of Neurochemistry 137, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2012) Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain pathology (Zurich, Switzerland) 22, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Experimental Neurology 246, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M (2019) Toward understanding the origin and evolution of cellular organisms. Protein Science 28, 1947–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenzler M, Braun CT, Maeder MB (2015) Mortality and Outcome of Severe Traumatic Brain Injury in a Swiss Level One Trauma Center. Emergency Medicine: Open Access 05. [Google Scholar]
- Labandeira-Garcia JL, Rodríguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ (2017) Brain Renin-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Frontiers in Aging Neuroscience 9, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laketa D, Savić J, Bjelobaba I, Lavrnja I, Vasić V, Stojiljković M, Nedeljković N (2015) Brain Injury Alters Ectonucleotidase Activities and Adenine Nucleotide Levels in Rat Serum / Povreda Mozga Menja Ektonukleotidazne Aktivnosti I Nivo Adeninskih Nukleotida U Serumu Pacova. Journal of Medical Biochemistry 34, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Ratón M, Rodríguez-Álvarez MX, Suárez CC, Sampedro FG (2014) OptimalCutpoints : An R Package for Selecting Optimal Cutpoints in Diagnostic Tests. Journal of Statistical Software 61, 1–36. [Google Scholar]
- Lusardi TA, Lytle NK, Szybala C, Boison D (2012) Caffeine prevents acute mortality after TBI in rats without increased morbidity. Experimental Neurology 234, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DN, Vekaria H, Macheda T, Bakshi V, Powell DK, Gold BT, Lin A-L, Sullivan PG, Bachstetter AD (2018) A Mild Traumatic Brain Injury in Mice Produces Lasting Deficits in Brain Metabolism. Journal of Neurotrauma 35, 2435–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Salci K, Ronquist G, Hillered L (2006) Energy Metabolic Changes in the Early Post-injury Period Following Traumatic Brain Injury in Rats. Neurochemical Research 31, 1085–1093. [DOI] [PubMed] [Google Scholar]
- Mowbray FI, Fox-Wasylyshyn SM, El-Masri MM (2019) Univariate Outliers: A Conceptual Overview for the Nurse Researcher. The Canadian journal of nursing research 51, 31–37. [DOI] [PubMed] [Google Scholar]
- Nguyen R, Fiest KM, McChesney J, Kwon C-S, Jette N, Frolkis AD, Atta C, et al. (2016) The International Incidence of Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques 43, 774–785. [DOI] [PubMed] [Google Scholar]
- Oses JP, Leke R, Portela LV, Lara DR, Schmidt AP, Casali EA, Wofchuk S, Souza DO, Sarkis JJF (2004) Biochemical brain markers and purinergic parameters in rat CSF after seizure induced by pentylenetetrazol. Brain Research Bulletin 64, 237–242. [DOI] [PubMed] [Google Scholar]
- Oses JP, Viola GG, Paula Cognato G. de, Júnior VHC, Hansel G, Böhmer AE, Leke R, et al. (2007) Pentylenetetrazol kindling alters adenine and guanine nucleotide catabolism in rat hippocampal slices and cerebrospinal fluid. Epilepsy Research 75, 104–111. [DOI] [PubMed] [Google Scholar]
- Pelligrino DA, Xu H-L, Vetri F (2010) Caffeine and the Control of Cerebral Hemodynamics. Journal of Alzheimer’s Disease 20, S51–S62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podkovik S, Kashyap S, Wiginton J 4th, Kang C, Mo K, Goodrich M, Wolberg A, Wacker MR, Miulli DE (2020) Comparison of Ventricular and Lumbar Cerebrospinal Fluid Composition. Cureus 12, e9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelle W (2018) psych: Procedures for Psychological, Psychometric, and Personality Research Evanston, Illinois. [Google Scholar]
- Rikitake Y, Kim H-H, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK (2005) Inhibition of Rho Kinase (ROCK) Leads to Increased Cerebral Blood Flow and Stroke Protection. Stroke 36, 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson SC, Sévigny J, Zimmermann H (2006) The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signalling 2, 409–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirzhanova I, Liu C, Zhao J, Bramlett H, Dietrich WD, Hu B (2013) Changes in the GEF-H1 Pathways after Traumatic Brain Injury. Journal of Neurotrauma 30, 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AP, Böhmer AE, Hansel G, Soares FA, Oses JP, Giordani AT, Posso IP, et al. (2015) Changes in Purines Concentration in the Cerebrospinal Fluid of Pregnant Women Experiencing Pain During Active Labor. Neurochemical Research 40, 2262–2269. [DOI] [PubMed] [Google Scholar]
- Schmidt AP, Lara DR, Faria Maraschin J. De, Silveira Perla A. Da, Souza DO (2000) Guanosine and GMP prevent seizures induced by quinolinic acid in mice. Brain Research [DOI] [PubMed] [Google Scholar]
- Solaroglu I, Okutan O, Kaptanoglu E, Beskonakli E, Kilinc K (2005) Increased xanthine oxidase activity after traumatic brain injury in rats. Journal of Clinical Neuroscience 12, 273–275. [DOI] [PubMed] [Google Scholar]
- Stankiewicz TR, Linseman DA (2014) Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Frontiers in Cellular Neuroscience 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MA, Modkovski R, Hansel G, Zimmer ER, Kopczynski A, Muller AP, Strogulski NR, et al. (2017) Elevated glutamate and lactate predict brain death after severe head trauma. Annals of Clinical and Translational Neurology 4, 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayag EC, Nair SN, Wahhab S, Katsetos CD, Lighthall JW, Lehmann JC (1996) Cerebral uric acid increases following experimental traumatic brain injury in rat. Brain Research 733, 287–291. [DOI] [PubMed] [Google Scholar]
- Verrier JD, Jackson TC, Bansal R, Kochanek PM, Puccio AM, Okonkwo DO, Jackson EK (2012) The brain in vivo expresses the 2′,3′-cAMP-adenosine pathway. Journal of Neurochemistry 122, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, Simko V (2017) R package “corrplot”: Visualization of a Correlation Matrix [Google Scholar]
- Zeng X-J, Li P, Ning Y-L, Zhao Y, Peng Y, Yang N, Zhao Z-A, Chen J-F, Zhou Y-G (2018) Impaired autophagic flux is associated with the severity of trauma and the role of A2AR in brain cells after traumatic brain injury. Cell Death & Disease 9, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg H, Smith DH, Blennow K (2013) Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nature Reviews Neurology 9, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Composite of purines in the CSF ofTBI patients relative to controls (surviving and non-surviving). This setting displays the remaining purines that did not show a statistically significant difference between TBI patients and controls. (A) GTP, (B) GMP, (C) ATP, (D) AMP, and (E) Uric Acid. Statistical significance was assessed through Mann-Whitney or Student’s independent T-test (Controls n= 51 patients, TBI n = 17 patients).
Supplemental Figure 2. Composite of purines in the CSF of Non-surviving and surviving TBI patients. This setting displays the remaining purines that did not show a statistically significant difference in non-surviving and surviving TBI patients. (A) GMP, (B) Guanosine, (C) ATP, (D) ADP, (E) AMP, (F) Adenosine, (G) Inosine, (H) Hypoxanthine, and (I) Uric Acid. Statistical significance was assessed through Mann-Whitney or Student’s independent T-test (Surv n =11 patients, Non-Surv n=6 patients).
Supplementary Figure 3: Long-term neurological outcome is not associated with CSF levels of most purines levels. This panel displays correlation plots between the modified Rankin Scale (mRS) and (A) GTP, (B) GDP, (C) GMP, (D) ATP, (E) ADP, (F) AMP, (G) Adenosine, (H) IMP, (I) Inosine, (J) Hypoxanthine, (K) Xanthine and (L) Uric acid; where none of the purines assessed displayed significant correlations (Spearman correlation, significance considered when p<0.05) with the two-year mRS of severe TBI patients (n = 17 TBI patients).