

Abstract
Protein kinase D (PRKD) family kinases are required for formation and function of osteoclasts. However, the substrates of PRKD in osteoclasts are unknown. To identify PRKD-dependent protein phosphorylation in osteoclasts, we performed a quantitative LC-MS/MS phosphoproteomics screen for proteins showing differential phosphorylation in osteoclasts after treatment with the PRKD inhibitor CRT0066101. We identified 757 phosphopeptides showing significant changes following PRKD inhibition. Among the changes, we found a group of 13 proteins showing decreased phosphorylation at PRKD consensus phosphorylation motifs. This group includes histone deacetylase 5 (HDAC5), which is a previously validated PRKD target. Considering this known interaction, work suggesting HDACs may be important regulators of osteoclasts, and studies suggesting potential functional redundancy between HDACs, we further investigated the relationship between PRKD and class IIa HDACs in osteoclasts. We confirmed that CRT0066101 inhibits phosphorylation of endogenous HDAC5 and to a lesser extent HDAC4, whereas HDAC7 phosphorylation was not affected. Osteoclast cultures from Hdac5 global knockout mice displayed impaired differentiation and reduced ability to resorb bone, while conditional knockout of Hdac4 in osteoclasts showed no phenotype in vitro or in vivo. The inhibitory effect of CRT0066101 was reduced in Hdac5 KO osteoclasts. Together these data indicate that the PRKD/HDAC5 axis contributes to osteoclast formation in vitro and suggest that this pathway may contribute to regulation of skeletal dynamics in vivo.
Keywords: osteoclast differentiation, protein kinase D, bone remodeling, histone deacetylases, protein phosphorylation
1. Introduction
Destruction of bone by osteoclasts is a hallmark of diseases such as periodontal disease, arthritis, osteoporosis and many cancers. The spectrum of clinical complications associated with this heightened bone destruction includes fragility fractures, joint destruction, pain, and hypercalcemia. Additionally, high levels of osteoclast-mediated bone resorption can contribute to failure of dental or orthopedic implants. Particularly in light of the aging population, these conditions represent a substantial and costly burden[1]. Osteoclasts are formed by fusion of mononucleated myeloid precursors into larger multinucleated cells. Following differentiation, they bind tightly to the bone surface and secrete a potent mixture of acids and proteases to demineralize the hydroxyapatite and digest the organic collagen-rich matrix[2]. Current anti-resorptive therapeutics are often efficacious but are associated with a range of adverse effects[3, 4]. Thus, deeper understanding of the molecular pathways regulating osteoclasts is an important scientific and translational goal to enable new directions in management of bone resorption.
Our previous studies discovered that protein kinase D (PRKD) is a positive regulator of osteoclast differentiation and resorptive function [5, 6]. Over the course of several days in culture, murine bone marrow macrophages stimulated with macrophage colony stimulating factor (M-CSF) and receptor activator of NF-KB ligand (RANKL) become lineage committed mononucleated pre-osteoclasts, proliferate and then undergo cell-cell fusion to generate large, mature multinucleated osteoclasts capable of forming actin rings and resorbing bone. We showed that treating such cultures with PRKD inhibitors CRT0066101, Gö6976 or CID755673 reduces their survival, cell-cell fusion into multinucleated cells, reduces resorptive activity, and disrupts specialized actin cytoskeletal structures called actin rings that are an important component of the bone resorptive machinery. These observations indicate that PRKD promotes multiple distinct aspects of osteoclastogenesis and suggest that PRKD might be a locus for novel antiresorptive therapies.
PRKD consists of three closely related serine/threonine kinases (see [7, 8]for review). Of the three Prkd genes, osteoclasts express Prkd2 and Prkd3 [6]. The PRKD kinases are activated by binding to phospholipids such as diacylglycerol, phosphorylation by protein kinase C and SRC and through autophosphorylation by PRKD itself. Analysis of the known PRKD substrates and in vitro studies using degenerate peptide arrays reveal that PRKDs preferentially phosphorylate a consensus motif of leucine at −5 and arginine at −3 relative to the phosphorylated serine/threonine residue[9, 10]. There is a modest but growing list of known direct substrates of PRKDs including histone deacetylases, SNAIL, CERT, E-cadherin, cortactin, SSH1L, HSP27, TRPV1. These substrates have connected PRKD to a wide range of cellular processes: cell proliferation and survival, actin cytoskeleton dynamics, adhesion and motility, gene expression, vesicle trafficking. Through these cellular processes PRKDs have been linked to diseases including cancer, cardiovascular disease, CNS disorders and inflammation-related conditions. PRKD inhibitors are being investigated as potential therapeutic agents in a range of human diseases.
While our work has shown that PRKD is important in osteoclasts, the molecular mechanisms responsible for its effects in these specialized cells are untested and no specific endogenous PRKD substrates have been demonstrated. In the present study, we sought to investigate PRKD’s mode of action and identify PRKD targets in osteoclasts. To achieve this, we performed a quantitative mass-spectroscopy-based phosphoproteomics screen comparing control and PRKD-inhibited osteoclasts. From this we identified 757 phosphopeptides showing significant changes in abundance, including 13 proteins that represent candidates for endogenous direct phosphorylation substrates for PRKD including Histone deacetylase 5 (HDAC5), a known substrate of PRKD in other contexts[11–13]
Histone deacetylases are divided into four subgroups based on their domain compositions and overall sequence homologies[reviewed by 14, 15]. HDAC5 is one of four highly similar proteins in class IIa, along with HDACs 4, 7 and 9. HDACs are best known as transcriptional co-repressors that participate in repressing gene expression by locally deacetylating histone core proteins to reduce transcription of that locus. The class IIa HDACS have all been shown to be phosphorylated by PRKD and other protein kinases at three to four conserved serine residues in their N-terminal regulatory domains, thereby influencing their cellular localization, interactions and activities. HDACs have been implicated in osteoclast formation, with individual HDACs reported to show distinct functional activities. Deletion of the class I deacetylase Hdac3 has shown both positive and negative effects on osteoclasts which may be due to differences in the exact method and developmental timing of gene knockout involved[16–18]. Deletion of the class IIa deacetylase Hdac7 enhances osteoclasts, suggesting that it acts negatively [18–20], while deletion of Hdac9 accelerated osteoclast differentiation, indicating a positive role[21, 22]. A study in which the remaining two class IIa HDACS, Hdac4 and Hdac5, were individually knocked down using shRNAs reported increased osteoclast formation, suggesting that they both inhibit differentiation[21]. Their in vitro and in vivo phenotypes using genetic knockout specifically in osteoclast-lineage cells have not been described, although HDAC5 has been implicated in several other cell lineages affecting bone physiology[23–25].
In the current study, we present phosphoproteomics data that identify HDAC5 as a substrate of PRKD in osteoclasts. Molecular studies confirm PRKD phosphorylation of HDAC5, while we were unable to validate other class IIa HDACs as endogenous PRKD substrates in these cells. Given our prior work with PRKD in osteoclasts, proteomics data showing PRKD phosphorylation of HDAC5, and the reports with Hdac5 and HDAC4 shRNA, we characterized osteoclast phenotypes from genetic knockout of these Hdacs. In vitro cultures from Hdac5 knockout mice showed impaired osteoclast formation, while we did not detect any phenotype from Hdac4 conditional knockout osteoclasts either in culture or in vivo. Collectively, our data suggest a pathway in which PRKD/HDAC5 contributes to regulation of osteoclast formation and functional activity.
2. Materials and methods
2.1. Osteoclast culture
Primary murine bone marrow derived osteoclasts were isolated and cultured by the protocol of Xing & Boyce[26]. Mice were euthanized at 2–4 months age by CO2 inhalation. Marrow was flushed from tibiae and femurs, treated with red blood cell lysis buffer (150 mM ammonium chloride, 10 mM potassium bicarbonate, 0.1 mM EDTA pH 7.4) and cultured overnight in 100 mm tissue culture dishes at 37° in phenol red-free αMEM supplemented with 5% FBS, 1% penicillin-streptomycin and 1% CMG14-12 cell supernatant as a source of M-CSF[27]. The following day, the nonadherent monocyte population was collected, counted and re-plated to multiwell tissue culture plates at a density of 100,000 cells/cm2. Any remaining non-adherent cells were discarded 48 hours after seeding. The adherent bone marrow macrophages were then stimulated with 20 ng/mL RANKL (R&D Systems) and 1% CMG14-12 supernatant for up to 4 days to generate mature osteoclasts. Cell growth media was changed every two days. For resorption pit staining, cells were seeded onto bone slices (Immunodiagnostic Systems) and cultured as above until day 3, at which point they were changed into media at pH 6.8 to promote survival and resorptive activity of mature osteoclasts[28]. On day 6, cells were removed with a cotton swab. Resorption pits were stained with HRP-conjugated wheat germ agglutinin [5] or hematoxylin[29] using standard methods. After staining, the bone slices were mounted in glycerol on a glass slide and imaged using reflective light microscopy on an Olympus BX51 microscope.
For cell staining and analysis, osteoclasts were fixed with 4% formaldehyde solution and washed with PBS, permeabilized with 0.3% tritonX-100 in PBS for 5 minutes then stained with DAPI to visualize nuclei and rhodamine-phalloidin to visualize actin cytoskeleton. For TRAP staining, cells were incubated with 50 mM sodium acetate buffer pH 5.0, 0.1% Triton X-100, 30 mM sodium tartrate, 100 μg/mL naphthol AS-MX, 3 μg/mL fast red violet LB at 37°C for 5–10 minutes until stained sufficiently. Color development was stopped by washing twice with PBS. Stained cells were photographed on an Olympus IX70 microscope equipped with a DP71 digital camera. The number of DAPI-stained nuclei and osteoclasts were determined using Adobe Photoshop and NIH ImageJ, with osteoclasts defined as TRAP-positive cells containing three or more nuclei. Actin cytoskeletal organization was quantified manually with the investigator blinded to treatment group.
2.2. Phosphoproteomics screen & Gene Ontology Functional Annotation analysis
Osteoclasts were cultured and differentiated as above until day 3 after addition of RANKL. They were treated with 200 nM CRT0066101 (Tocris) or an equal volume of sterile PBS vehicle for 5 hours. This dosage and timing for CRT0066101 treatment was selected based on prior dose response studies in osteoclasts[5]. Cells were scraped, collected by centrifugation, and lysed on ice for 5 minutes in 7M urea, 2M thiourea, 0.4M tris pH 7.5, 20% acetonitrile, 4mM TCEP, 5mM EDTA, 10 mM sodium fluoride, 1mM sodium orthovanadate. Lysates were subjected to barocycling (37°C, 35000psi × 20 seconds, 0psi × 10 seconds for 60 cycles). Proteins were then alkylated with iodoacetamide at room temperature for 15 minutes and digested with trypsin overnight at 37°. Samples were acidified and passed through with Oasis HLB cartridges (Waters). Phosphopeptides were enriched using titanium dioxide resin (ThermoFisher). Samples were dried under vacuum centrifuge, resuspended in 25μL of 3% acetonitrile, 0.25% formic acid and centrifuged at 10,000g × 2 minutes. An aliquot of this solution was collected and used for liquid chromatography and mass spectrometry (LC-MS/MS) analysis on an LTQ Orbitrap instrument with six replicates per group. Analysis of the MS data were performed with Proteome Discoverer 1.4 (ThermoFisher) and Scaffold (Proteome Software) software packages. Lists of the MS fragmentation spectra were generated and searched in PEAKS software (Bioinformatics Solutions, Inc) against the Mus musculus component of the UniProt database. The estimated false discovery rate was 0.5% at the peptide level and 1.1% at the protein group level. Further bioinformatics analyses were performed using R Studio. Protein Functional Annotation Clustering was performed using DAVID Bioinformatics Resources v6.8 using UniProt Keywords, Gene Ontology Biological Process and Gene Ontology Molecular Function groupings under medium stringency.
2.3. Transfection, Immunoprecipitation and Western blotting
For overexpression studies, HEK293T cells were transfected using Fugene HD (Promega) according to the manufacturer’s protocol with plasmids encoding PRKD2 (pCDNA3.1 PRKD2-FLAG, Genscript), PRKD3 (pCMV6 PRKD3-MYC-FLAG, Origene), HDAC5 (pCDNA3 HDAC5-FLAG[30]) or the corresponding empty vectors.
Osteoclasts or transfected HEK293T cells were lysed in ice-cold NP40 lysis buffer (50 mM Tris pH 7.4, 250 mM NaCl, 5 mM EDTA, 1% NP40) supplemented with HALT protease and phosphatase inhibitor cocktail (ThermoFisher). Lysates were cleared by centrifugation at 12,000 × g for 10 minutes at 4°. For immunoprecipitations, the appropriate primary antibody was added and incubated with rocking at 4° overnight. The following day, the immunoprecipitations were collected with EZview Red Protein G Affinity Gel (Sigma Aldrich) for one hour at 4°, spun down at 3000 × g and washed with lysis buffer 3 times for 5 minutes each. Proteins were separated by SDS-PAGE electrophoresis and transferred to Immobilon-P PVDF Membrane (ThermoFisher). Blots were blocked with 3% BSA in TBST for 15 minutes and incubated with primary antibodies overnight at 4°. They were washed 4 times, incubated with HRP-conjugated secondary antibodies (Advansta) in 5% nonfat milk in TBST for one hour, washed again, visualized using WesternBright Sirius chemiluminescent substrate (Advansta) and imaged on a ChemiDoc Touch imaging system (Bio-Rad).
Antibodies used for immunoprecipitations and Western blotting were: Phospho-PRKD Ser916 (Cell Signaling #2051), Phospho-HDAC (Cell Signaling #3424), Phospho-HDAC (Cell Signaling #3443), α-tubulin (Cell Signaling #2144), HDAC4 (Millipore Sigma H9536), HDAC5 (Santa Cruz sc-133225), HDAC7 (Millipore Sigma H2662), Phospho-p38 (Cell Signaling #9211), Phospho-ERK/2 (Cell Signaling #9101), Phospho-AKT (Cell Signaling #4058), Phospho-p65 NF-kB (Cell Signaling #3033), Phospho-CREB (Cell Signaling #9198), P-Cofilin (Cell Signaling #3313), P-LIMK (ECM Biosciences LP1891), β-Actin (Cell Signaling #4970), P-SRC (Cell Signaling #6943), SRC (Cell Signaling #2123), Protein Kinase D3 (Cell Signaling #5655), normal rabbit IgG (Cell Signaling #2729) and DYKDDDDK (FLAG) Tag (Cell Signaling #14793).
2.5. Mice breeding & genotyping
Wild-type mice from Jackson Laboratory and all of the mutant mice described below are in the C57Bl/6 genetic background. Hdac4 floxed mice and Hdac5 global knockout mice were created by Eric Olson (UT Southwestern Medical Center) and obtained from Hank Kronenberg (Massachusetts General Hospital). cFms-Cre mice were purchased from the Jackson Laboratory. Prkd3flox mice[31] were obtained from Sho Yamasaki (Kyushu University). Hdac4flox or Prkd3flox mice were mated to cFms-Cre to generate homozygous flox/flox; cFms-Cre conditional knockout (cKO) and flox/flox non-Cre (flox) control littermates. The presence of Hdac4flox and Hdac4 wild-type alleles was determined by PCR using the primers 5’-atctgcccaccagagtatgtg (forward) and 5’-cttgttgagaaca-aactcctgcagct (reverse). Prkd3 floxed mice were genotyped using primers 5’-gttgaaggtctggagcttgagagtctgctgtacttaac (forward) and 5’-gaatagaaaagaatcaactggcagctcaca (reverse). The Cre transgene was detected using primers 5’-ttggcagaacgaaaacgctg (forward) and 5’-tcagctacaccagagacgga (reverse). Hdac5+/− mice were interbred to give Hdac5−/− (Hdac5 KO) and Hdac5+/+ wild-type (WT) control littermates that were genotyped by PCR using primers 5’-ccttgtgcatgctgggctgg (Hdac5 forward), 5’-gtcaagtggtcgtgctgct (wild-type reverse) and 5’-gtttgaggggacgacgacag (mutant reverse).
2.5. Micro Computed Tomography (μCT) Imaging and analysis
μCT imaging of mouse femurs were performed as previously described[32]. Bones were scanned in 70% ethanol using a Nikon XT H 225 μCT instrument equipped with a 1 mm aluminum filter. The scan settings were 120 kV, 61 μA, 720 projections, 2 frames per projection, and an integration time of 708 milliseconds, with an isotropic pixel size of 7.11 μm. Scans were reconstructed using CT Pro 3D software (Nikon Metrology) and converted to bitmaps using Studio MAX 3.2 (Volume Graphics GbH). Morphometric analysis was performed using CT-Analyzer software (Bruker microCT). The region of interest for trabecular bone analysis in the distal metaphysis started 0.5 mm proximal to the growth plate and extended 1.5 mm proximally towards the diaphysis. The region for cortical bone analysis was a 0.5 mm region at the mid-diaphysis. Automated contouring was used to determine the region of interest boundaries for both trabecular and cortical bone with manual editing as needed. Global thresholding was used to segment bone from surrounding tissue for analysis.
2.6. Quantitative RT-PCR
RNA was harvested from osteoclast cultures in triplicate using Trizol (Thermo Fisher) and reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturers’ protocols. Quantitative real-time PCR was performed using the iTaq SYBR Green Supermix (Bio-Rad) on a CFX Connect Real Time PCR System. Target gene expression was normalized to Hprt1. The data shown in Figure 3 represent the mean expression from three independent experiments, each with triplicate biological replicates and giving similar results. Primer sequences used were: Hprt1 For 5’-gaggagtcctgttgatgttgccag; Hdac5 For 5’-ttgcttgggcccatgacag, Rev 5’-actgcttctcctctcggct; Nfatc1 For 5’-tcatcctgtccaacaccaaa, Rev 5’-Rev 5’-tcaccctggtgttcttcctc ; cFos For 5’-actgcttctcctctcggct ; Nfatc1 For 5’-tcatcctgtccaacaccaaa, Rev 5’-tcaccctggtgttcttcctc ; cFos For 5’-ccaagcggagacagatcaactt, Rev 5’-tccagtttttccttctctttcagcaga ; Apc5 For 5’-cgtctctgcacagattgca, Rev 5’-gagttgccacacagcatcac ; Dcstamp For 5’-gggcaccagtattttcctga, Rev 5’-tggcaggatccagtaaaagg; Atp6v0d2 For 5’-tcagatctcttcaaggctgtgctg, Rev 5’-gtgccaaatgagttcagagtgatg; Ctsk For 5’-agggaagcaagcactggatc, Rev 5’-gctggctggaatcacatctt.
Figure 3.

In vitro culture of Hdac5 KO osteoclasts (A) real-time RT-PCR (left) and Western blotting (right) against HDAC5 expression in wild-type and Hdac5 KO osteoclast cultures (B) Proliferation/ survival curves showing the number of nuclei per field in cultures of WT cells (dark bars) and Hdac5 KO (light bars) from Day -1 (the day prior to RANKL), Day 0 (the day of RANKL addition), and Days 1–3 of osteoclast differentiation. Data are graphed as mean number of nuclei per field relative to WT Day -1. (C) TRAP staining (top row), rhodamine-phalloidin (middle row) and resorption pits on bone slices visualized by hematoxylin staining (bottom row) of wild type control and Hdac5 KO osteoclast cultures. (D) Quantitation of mature osteoclasts comparing mean number of osteoclasts per field, mean number of nuclei per osteoclast, largest number of nuclei per osteoclast determined from cells double stained for TRAP and DAPI, and total resorbed area fraction from resorption assay on bone slices. (E) Western blotting for HDAC5, phospho-SRC Y416. Total SRC and ACTIN were visualized as loading controls P-SRC to total SRC ratios are indicated below the blots. (F) Real-time RT-PCR from wild-type and Hdac5 KO osteoclast cultures. Graphs show the mean + SD from three independent experiments * p<0.05, ** p<0.005.
2.7. Experimental Replication and Statistical analysis
Statistical testing was performed using Prism statistical software (Graphpad) or Excel (Microsoft) with a p<0.05 considered as statistically significant. Cell culture experiments were performed independently at least three times with at least three technical replicates per group. Within each experiment, control and knockout cells were always from the same sex. Similar results were obtained using cells from either sex, so they were grouped together for analysis. The data graphed represent pooled results from the independent trials. For cell proliferation assays, the counts within each experiment were normalized to the Control Day -1 group to correct for experimental variability in the starting number of cells between separate experiments. Statistical testing for differences between actin cytoskeletal distributions in Figure 4 was performed using the Chi-Square test.
Figure 4.

CRT0066101 treatment of wild-type and Hdac5 knockout osteoclast cultures. Cells were treated with CRT0066101 at 0, 20nM or 100 nM beginning at the time of RANKL stimulation and stained for TRAP (top rows) or rhodamine-phalloidin (red) and DAPI (blue), middle rows. Bottom rows show resorption pit staining on bone slices. Hdac5 KO or wild-type osteoclasts were cultured on bone slices for 6 days, swabbed to remove the cells and visualized with hematoxylin staining. Graphs at the right present comparisons in mean and maximum nuclei per osteoclast and total resorbed area for wild-type (dark bars) and Hdac5 KO (lighter grey bars). Quantitative data for CRT0066101-treated cells are presented as change relative to the corresponding untreated WT or KO cells. Actin morphology was manually scored for each multinucleated osteoclast and the population distribution is graphed for peripheral actin belts, smaller internal actin rings and disordered/other. * p<0.05, ** p<0.005.
2.7. Ethics Statement
Mice were kept in University of Minnesota Research Animal Resources housing and maintained according to applicable NIH and University of Minnesota guidelines. All experimental procedures received prior approval from the University of Minnesota IACUC (Protocols 2010-38523A, 2006-38192A and 1806-36053A).
3. Results
3.1. Quantitative Phosphoproteomics Screen for PRKD-dependent phosphorylation
Our previous work[5, 6] revealed that PRKDs are required for distinct cellular processes throughout the osteoclast differentiation process but did not identify the molecular mechanisms or targets responsible. In those studies, mouse bone marrow monocytes were treated with M-CSF and RANKL to stimulate differentiation into multinucleated osteoclasts over the course of 4–5 days. Cultures treated with PRKD inhibitors including CRT0066101, Gö6976 and CID755673 showed unaffected induction of TRAP-expressing committed mononucleated pre-osteoclasts. However, their ability to fuse into multinucleated mature osteoclasts, organize their actin cytoskeleton into podosome rings or resorb bone in culture were all disrupted. These data establish positive roles for PRKD in osteoclast differentiation and raise the question of what are the phosphorylation substrates of PRKD responsible for its effects on osteoclast formation?
To identify specific changes in the phosphoproteome of osteoclasts deficient for PRKD activity, we made use of a label-free quantitative LC-MS/MS based screen to compare protein phosphorylation between control cultures of primary murine bone marrow-derived osteoclasts with cultures treated with PRKD inhibitor, illustrated in Fig. 1A. Bone marrow monocytes were cultured in the presence of M-CSF and RANK Ligand (RANKL) for three days to stimulate osteoclast differentiation and then treated with the PRKD inhibitor CRT0066101 at 200 nM for 5 hours before harvesting cellular protein for analysis.
Figure 1.

Phosphoproteomics analysis of CRT0066101-responsive protein phosphorylation (A) Schematic of workflow. Osteoclast cultures on day 3 of RANKL stimulation were treated with 200 nM CRT0066101 for 5 hours, lysed and subjected to quantitative phosphoproteomics screening. (B) Concentric Venn diagram illustrating total peptides (grey), phosphopeptides (green), and significantly changed phosphopeptides (innermost circle), divided into increased abundance (blue) and decreased abundance (red) areas. (C) Volcano plot of the 15,269 phosphopeptides that were quantitatively identified comparing Log2 fold change versus adjusted p-value. Significantly increased phosphopeptides are in blue, decreased abundance in red. (D) Top 15 functional annotation clusters from the 757 significantly altered phosphopeptides, analyzed using the DAVID gene ontology tool. (E) Significantly decreased phosphopeptides mapping to PRKD consensus motifs.
CRT0066101 was chosen for PRKD inhibition because it appears to be the most specific PRKD inhibitor available. It was screened against a large panel of protein kinases[33] and used in at least 20 publications with no off-target activity to our knowledge. The decision to perform the treatment at day 3 was based on our previous findings[5, 6]. Osteoclast-forming BMM cultures treated with CRT0066101 or other PRKD inhibitors proliferate normally and form TRAP-positive pre-osteoclasts on days 1–2. On day 3 control cultures begin to fuse into multinucleated osteoclasts, but CRT0066101-treated cultures show significantly impaired cell-cell fusion, which suggests that PRKD activity is required on day 3. Moreover, since the control cultures at this timepoint contain a mixture of committed mononucleated preosteoclasts, small multinucleated osteoclasts and a few larger, more differentiated osteoclasts, we anticipated being able to potentially detect changes in proteins involved in both early and later steps of the differentiation program.
Following inhibitor treatment, we prepared cell lysates, digested them with trypsin, performed titanium dioxide phosphopeptide enrichment and characterized the resulting peptides using LC-MS/MS. From this we identified 20,820 peptides mapping to 4293 unique proteins. Of those, 15,269 were phosphopeptides (Fig. 1B). Further screening of this group for phosphopeptides showing at least 1.5-fold change in abundance (log2 fold-change greater than ±0.5850) and adjusted P-value < 0.05 in the PRKD inhibitor-treated group versus the untreated control group (Fig. 1B–C) identified 757 significantly changed phosphopeptides that mapped to 522 unique proteins (Supplemental Data 1). 322 phosphopeptides showed decreased abundance while 435 phosphopeptides exhibited increased abundance. The 522 significantly altered proteins were submitted for gene ontology analysis using the DAVID Functional Annotation Clustering tool, which identified 45 functional clusters from our proteins. The top 16 clusters based on their enrichment scores greater than 1.5 are summarized in Figure 1D, full results are in Supplemental Data 2. These top 16 enriched functional annotation clusters include several processes where PRKD proteins have been linked, including transcriptional regulation, protein transport, cell-cell adhesion, cadherins, endosomes/lysosomes, serine/threonine protein kinases, vesicle-mediated transport and actin binding. Other enriched clusters implicated terms such as mRNA processing, RNA binding, and translational initiation.
In vitro phosphorylation arrays and comparative sequence analysis of known PRKD phosphorylation substrates have led to identification of a consensus PRKD motif of [LVI]-x-R-x-x-p[ST], with a preference for leucine at the -5 position and arginine at -3 relative to the targeted serine/threonine[9, 10]. Sequence analysis of the candidate phosphopeptides showing significantly decreased abundance in the CRT0066101 group identified 14 phosphopeptides for which the decreases mapped to a PRKD consensus motif (Fig. 1E). Within our data, these proteins represent the most immediate candidates for direct substrates for phosphorylation by PRKD.
3.2. Class IIa HDACs are endogenous targets of PRKD in osteoclasts
Among the phosphoproteins with CRT0066101-responsive decreases at consensus PRKD motifs, we noted histone deacetylase 5 (HDAC5). HDAC5 and closely related class IIa histone deacetylase proteins are direct substrates of PRKD in cardiac and skeletal muscle, lymphocytes, endothelial cells, neurons, and osteoblasts[11, 13, 34–38]. Functional Annotation Clustering placed HDAC5 into annotation clusters 3 – transcriptional regulation and 9 – zinc-binding. The proteomics data indicated a 75% reduction in the abundance of a phosphopeptide containing phosphoserine 650, one of the previously known PRKD target sites (Fig. 2A). HDAC5 and the closely related class IIa HDAC proteins HDAC4, HDAC7 and HDAC9 are phosphorylated at 3 or 4 conserved serine residues in their N-terminal non-catalytic domain by protein kinases including PRKD (Fig. 2B, reviewed in [15]). Phosphorylation of these sites is thought to reduce HDAC transcriptional repressive activity by promoting export out of the nucleus and retention in the cytoplasm via interactions with 14-3-3 chaperones. Phosphorylation of HDACs by PRKD specifically in osteoclasts has not been previously reported, but since HDAC5 Ser650 is one of the residues that was previously shown to be a direct substrate of PRKD in vitro and in cardiomyocytes[39], along with ser250 and ser488 these data strongly suggest HDAC5 as an endogenous target of PRKD in osteoclasts. To further test this hypothesis, we treated Day 3 osteoclasts with CRT0066101 and performed western blotting using two different antibodies from Cell Signaling Technology against phospho-HDACs: P-HDAC #3424, which recognizes phospho-serine488 of HDAC5 and the analogous residue in other HDACs, and P-HDAC #3443, which recognizes HDAC5 phospho-serine250 and the corresponding residues (Fig. 2B). We are unaware of any commercially available antibody against P-Ser650. As shown in Fig. 2C, treating osteoclasts with CRT006101 for times between 15 minutes and 5 hours led to a strong reduction in phosphorylation of PRKD serine 916, which is a site of autophosphorylation commonly used as a measure of PRKD kinase activity. Similarly, blotting with anti-phospho-HDAC antibody #3424 detected bands at 100–150 kDa that were clearly reduced in the presence of CRT0066101. P-HDAC antibody #3443 sometimes showed reductions after CRT006601 treatment, although the effects seen with this antibody were more variable and inconsistent than those with the #3424. These data confirm that CRT0066101 decreases HDAC phosphorylation, suggesting that class IIa HDACs can be endogenous substrates for PRKD phosphorylation in osteoclasts.
Figure 2.

Protein kinase D regulates HDAC phosphorylation (A) mean abundance of HDAC5 phosphopeptide (amino acids 648–659, phosphoserine 650) in CRT0066101 and untreated osteoclast cultures. (B) Schematic of class IIa HDACs and conserved regulatory phosphoserine residues. Amino acid numbering is based on mouse HDAC proteins. Position of conserved phosphorylated residues and the sites targeted by anti-P-HDAC antibodies #3424 and 3443 are indicated above. CtBP and MEF2 interaction domains, Nuclear Localization Sequence (NLS) and Nuclear Export Sequences (NES) and the deacetylase catalytic functional domains are also illustrated (C-D) Western blotting against phosphorylated PRKD, P-HDACs and additional phospho-proteins following treatment of day 3 osteoclasts with 200 nM CRT0066101 for the indicated times. α-tubulin was blotted as a loading control (E) Osteoclasts treated with 200 nM CRT0066101 for 60 minutes were lysed and immunoprecipitated (IP) with antibodies against HDAC4, HDAC5, HDAC7 or normal rabbit IgG (control IP). Western blots were blotted with the indicated P-HDAC and total HDAC antibodies. (F) HEK293T cells were transfected with FLAG-tagged HDAC5, PRKD2 and PRKD3 proteins as indicated, lysed and immunoblotted for the FLAG and P-HDAC. (G) Western blotting against Prkd3 control and Prkd3 cKO osteoclast cultures. ** p< 0.005.
A number of PRKD substrates or PRKD-responsive pathways have been reported in the literature. To further define CRT0066101-responsive pathways in osteoclasts and further test the specificity of CRT0066101’s effects, we examined its effect on phosphorylation of a panel of potential PRKD responsive targets obtained the literature. CRT0066101 strongly reduced P-HDAC Ab #3424 as expected, but we were unable to detect consistent reproducible changes to p38 MAPK, p44/42 ERK1/2, AKT, p65 NF-κB, CREB, COFILIN, or LIMK (Fig. 2D).
Because of the high degree of sequence conservation between class IIa HDACs around the phosphorylation sites illustrated in figure 2B, the P-HDAC antibodies are cross-reactive against all the HDACs. To determine specifically which HDACs are being affected, we treated osteoclast cultures with CRT0066101, performed immunoprecipitations using antibodies specific for individual HDACs or non-specific negative control antibody and then blotted with the P-HDAC antibodies (Fig. 2E). In some experiments, phosphorylation of HDAC4 was modestly reduced by CRT0066101, although this effect was not observed consistently. The HDAC5 band detected with P-HDAC #3424 was reproducibly and strongly reduced by CRT treatment, whereas P-HDAC #3443 showed less change. No significant bands were detected in negative control antibody immunoprecipitations. Blotting against HDAC7 in cell lysates and HDAC7 IPs detected a doublet of 115–120 kDa. However, the major band detected by P-HDAC antibodies in these samples migrated at roughly 140 kDa, with weaker bands at 110–115 kDa, none of which were convincingly affected by CRT0066101. We are uncertain whether these higher weight bands reflect a shift in mobility of phosphorylated HDAC7, co-precipitation of HDAC4 or 5, or some other explanation. Attempts to show co-immunoprecipitation of HDAC7 with other HDACs from osteoclasts were not successful (data not shown). Considering these ambiguities, the data do not currently give support to PRKD phosphorylating HDAC7 in osteoclasts. In agreement from previous reports[21, 22], HDAC9 protein levels at this stage of osteoclast differentiation were quite low and we were unable to consistently detect phospho-HDAC9 (data not shown).
All three PRKD kinases have been reported able to phosphorylate HDAC5[11–13, 40]. Since both PRKD2 and PRKD3 are expressed in osteoclasts[5], we sought to verify their ability to phosphorylate HDAC5. We performed co-transfection experiments in which HEK293T cells were transfected with plasmids encoding FLAG-tagged HDAC5 alone and with FLAG-PRKD2 or FLAG-PRKD3 expression vectors, lysed and examined by western blotting. Figure 2F presents anti-FLAG blots that confirmed the expected pattern of overexpression. Phosphorylation of both HDAC5 Ser250 (P-Hdac Ab#3443) and Ser488 (P-Hdac Ab #3424) were strongly increased by co-transfection with PRKD3. When HDAC5 was overexpressed with PRKD2 we saw a clear increase in phosphorylation of serine 488 (antibody #3424), while serine 250 (antibody #3443) exhibited less of an increase. These data confirm that PRKD2 and 3 can phosphorylate HDAC5. Finally, as an additional direct test of whether HDAC5 is an endogenous substrate of PRKD in osteoclasts, we generated mice conditionally deleted for PRKD3 in the osteoclast lineage by mating floxed Prkd3 mice to the cFms-Cre line, which is expressed in osteoclasts and related myeloid lineage cells[41, 42]. As expected, western blotting (Fig. 2G) showed a very strong reduction in PRKD3 expression in osteoclasts cultured from the Prkd3 cKO mice compared to controls. HDAC5 protein levels were unaffected. Similar to what we observed with osteoclasts treated with PRKD inhibitor CRT0066101, in the Prkd3 knockout cultures there was a clear reduction in P-HDAC #3424 but little change in P-HDAC #3443. Taken together, these data support that HDAC5 is an endogenous substrate of PRKD in osteoclasts at Ser488, Ser650 and perhaps Ser250, while HDACs 4, 7 and 9 were not shown to be PRKD substrates.
3.3. Loss of Hdac5 impairs osteoclast formation in vitro
HDAC5 has been implicated in bone physiology through effects on chondrocyte and osteoblast-lineage cells[23–25], but its role directly in osteoclasts has not been closely examined. We asked whether genetic deletion of Hdac5 affected osteoclasts in vitro. To test this, we obtained global knockout mice for Hdac5 (Hdac5 KO)[43]. As expected, Hdac5 mRNA measured by qPCR was strongly reduced and HDAC5 protein was undetectable on western blots against osteoclasts cultured from bone marrow from these mice (Fig. 3A). Counting the number of nuclei present throughout differentiation showed no reproducible difference between Hdac5 KO and wild-type control cultures (Fig. 3B), suggesting that proliferation and cell survival are unaffected. However, TRAP staining of the cultures at day 4 of RANKL treatment revealed that osteoclast formation in Hdac5 KO was reduced compared to the control groups (Fig. 3C, top photos). Quantitative analysis revealed that the total number of osteoclasts (containing 3 or more nuclei) formed was not significantly different, but the average and maximum number of nuclei per osteoclast both showed statistically significant reductions by about a third (Fig. 3D). Despite this reduction in their size, formation of podosome belts or actin rings was not strongly disrupted in the Hdac5 KO cultures, either on tissue culture plastic (Fig. 3C middle photos) or on bone slices (data not shown). Moreover, as a further test of whether Hdac5 disrupted cell spreading or actin cytoskeleton, we performed western blotting against phospho-SRC (Fig. 3E). Densitometeric analysis of these blots revealed no change in levels of SRC phosphorylation at Tyr416. To test their resorptive capacity, Hdac5 KO and control cells were cultured on bone slices. Staining for bone resorption revealed that Hdac5 KO osteoclasts resorbed significantly less area than the control osteoclasts (Fig. 3C bottom photos and 3D). To further characterize the phenotype of the Hdac5 KO osteoclasts we performed qPCR on a panel of osteoclast genes including transcriptional regulators Nfatc1 and cFos, Acp5 which codes the TRAP enzyme, Atp6v0d2 and Dcstamp genes involved in osteoclast fusion, and Ctsk which is involved in resorption. Although these cultures showed the expected knockout of Hdac5 mRNA (Fig. 3A), none of the marker genes examined showed significant changes in their expression (Fig. 3F). Collectively, these observations indicate that loss of HDAC5 inhibits osteoclast differentiation and resorptive activity, thus suggesting that HDAC5 activity positively contributes to osteoclasts.
Loss of PRKD activity and Hdac5 knockout both impair osteoclasts. This observation and our phosphorylation data led us to hypothesize that if HDAC5 is a downstream effector of PRKD, then knockout of Hdac5 should reduce the ability of CRT0066101 to inhibit osteoclast differentiation. To test this, we compared the effects of 20 and 100 nM CRT0066101 on wild-type versus Hdac5 KO cells. These doses of CRT0066101 were based on our previous studies[5]. Consistent with our hypothesis, our data show that CRT0066101 is less able to inhibit osteoclasts in Hdac5 KO cultures than in wild-type. Untreated Hdac5 KO cells exhibit reductions in osteoclast differentiation and in vitro bone resorption but little difference in actin belt morphology (Fig. 3 & Fig. 4 photos). Since the baseline measurements are different between untreated WT and Hdac5 KO osteoclasts, to quantitatively assess the magnitude of CRT0066101’s effects, we normalized the data for CRT0066101-treated WT and KO cells each relative to their untreated control group. CRT0066101 gave dose-dependent inhibition of wild-type osteoclasts, which formed smaller multinucleated osteoclasts that stained only weakly for TRAP (Fig. 4, top rows), disrupted actin belt formation (Fig. 4, middle rows) and reduced resorptive activity (Fig. 4, bottom rows). CRT0066101 did give reductions in Hdac5 KO osteoclasts, but its effects were weaker. In wild-type cells, 100 nM CRT0066101 reduced the mean number of nuclei per osteoclast by 81%; in Hdac5 KO cells it gave only 72% reduction, statistically significant at p=0.033. There was a similar trend at 20 nM CRT0066101, with 63% reduction in wild-type versus 52% reduction in Hdac5 KO. Maximum nuclei/osteoclast and total resorbed area also showed this pattern where CRT0066101 gave weaker inhibition on Hdac5 knockout cells than wild-type. In contrast, there was no significant difference observed between Hdac5 genotypes in terms of CRT0066101’s ability to disrupt peripheral actin belts and smaller internal actin rings. The reduced ability of CRT0066101 to inhibit osteoclast formation and resorptive ability in the absence of HDAC5 supports our hypothesis that HDAC5 is functionally downstream of PRKD during osteoclastogenesis.
3.4. Hdac4 conditional knockout in osteoclasts has little effect in vitro or in vivo
There is close homology between the class IIa histone deacetylases, literature suggesting functional redundancy between HDAC4 and HDAC5[23, 24], and previous work reporting that shRNA knockout of HDAC4 or HDAC5 increased osteoclast differentiation in culture[21]. Although our data did not show phosphorylation of HDAC4 by PRKD in osteoclasts, based on these considerations and our results with HDAC5 knockout, we were curious to determine how knockout of HDAC4 affects osteoclasts. To test this, we generated mice conditionally deleted for Hdac4. Mice carrying a floxed allele of the Hdac4 gene were mated to the cFms-Cre transgene, which is expressed in osteoclast precursors and other cells of the monocyte/macrophage lineage, to generate homozygous Hdac4flox/flox; cFms-Cre conditional knockout (Hdac4 cKO) and Hdac4flox/flox; Cre-negative (Hdac4 flox) control littermates. Western blotting lysates from osteoclasts cultured from these mice revealed a strong loss in the HDAC4 protein (Fig. 5A). We counted the number of DAPI-stained nuclei on successive days of culture to test whether there is indication of changes to cellular survival or proliferation. No difference was observed in the number of nuclei between Hdac4 flox control and Hdac4 cKO cultures (Fig. 5B), suggesting no effect on cell proliferation, survival, or death. TRAP and rhodamine-phalloidin staining on day 4 of RANKL treatment to visualize osteoclasts showed robust formation of large multinucleated osteoclasts that could properly organize actin belts when grown on tissue culture plastic or actin rings when grown on a bone substrate (Fig. 5C and data not shown). Finally, culture on osteoassay surface (not shown) or bovine bone slices revealed that Hdac4 cKO and Hdac4 flox osteoclasts showed similar resorptive activity in vitro (Fig. 5C). As well as looking at cultured Hdac4 cKO osteoclasts in vitro, we performed in vivo analyses of the Hdac4 cKO mice (Fig. 6). Each genotype was born at the expected Mendelian ratio and showed no overt developmental phenotype. Mice were subjected to analysis of 3 months of age, with the number of mice analyzed for each group Hdac4 flox male: 11, Hdac4 cKO male: 11, Hdac4 flox female: 11, Hdac4 cKO female: 12. Measurements of body mass and length (measured from the tip of the nose to the base of the tail) indicated no difference between sex-matched littermate flox controls and cKO mice (Fig. 6A). Likewise, μCT analysis of femurs from male and female cKO and flox mice revealed no difference in cortical parameters cross-sectional thickness (Ct.Th) or cortical bone area percentage (Ct.Ar/Tt.Ar) (Fig. 6B). Trabecular bone parameters showed no significant difference in bone volume to tissue volume (BV/TV), trabecular thickness (Tb.Th) or trabecular number (TB.N). The data did show a small but statistically significant increase in trabecular spacing (Tb.Sp) seen only in the male Hdac4 cKO versus Hdac4 flox (0.120 vs 0.135 mm, P=0.023) and a small but significant increase in Tb.Th seen only in female Hdac4 cKO versus Hdac4 flox (0.0362 vs 0.0382 mm, P=0.026). Collectively these data suggest that deletion of Hdac4 in osteoclasts has no observed effect on osteoclasts in vitro and little to no impact on the intact skeleton under these assay conditions.
Figure 5.

In vitro culture of Hdac4 cKO osteoclasts (A) Western blotting against HDAC4 or β-actin as a loading control from osteoclast cultures during differentiation. The blots from Hdac4 flox and cKO cells shown are taken from the same photo of a single western blot. (B) Proliferation/ survival curves graphing number of nuclei per field in cultures of WT cells (dark bars) and Hdac4 cKO (light bars) in osteoclast cultures from Day -1 (the day prior to RANKL), Day 0 (the day of RANKL addition), and Days 1–3 of osteoclast differentiation. Data are graphed as mean number of nuclei per field relative to WT Day -1. (C) TRAP staining (left), rhodamine-phalloidin (middle column, red) and resorption pits on bone slices (right column, wheat germ agglutinin-HRP staining) of wild type control and Hdac4 cKO osteoclast cultures. (D) Quantitation of mature osteoclasts, comparing number of osteoclasts per field, number of nuclei per osteoclast, largest number of nuclei per osteoclast determined from cells double stained for TRAP and DAPI, and total resorbed area fraction from resorption assay on bone slices.
Figure 6.
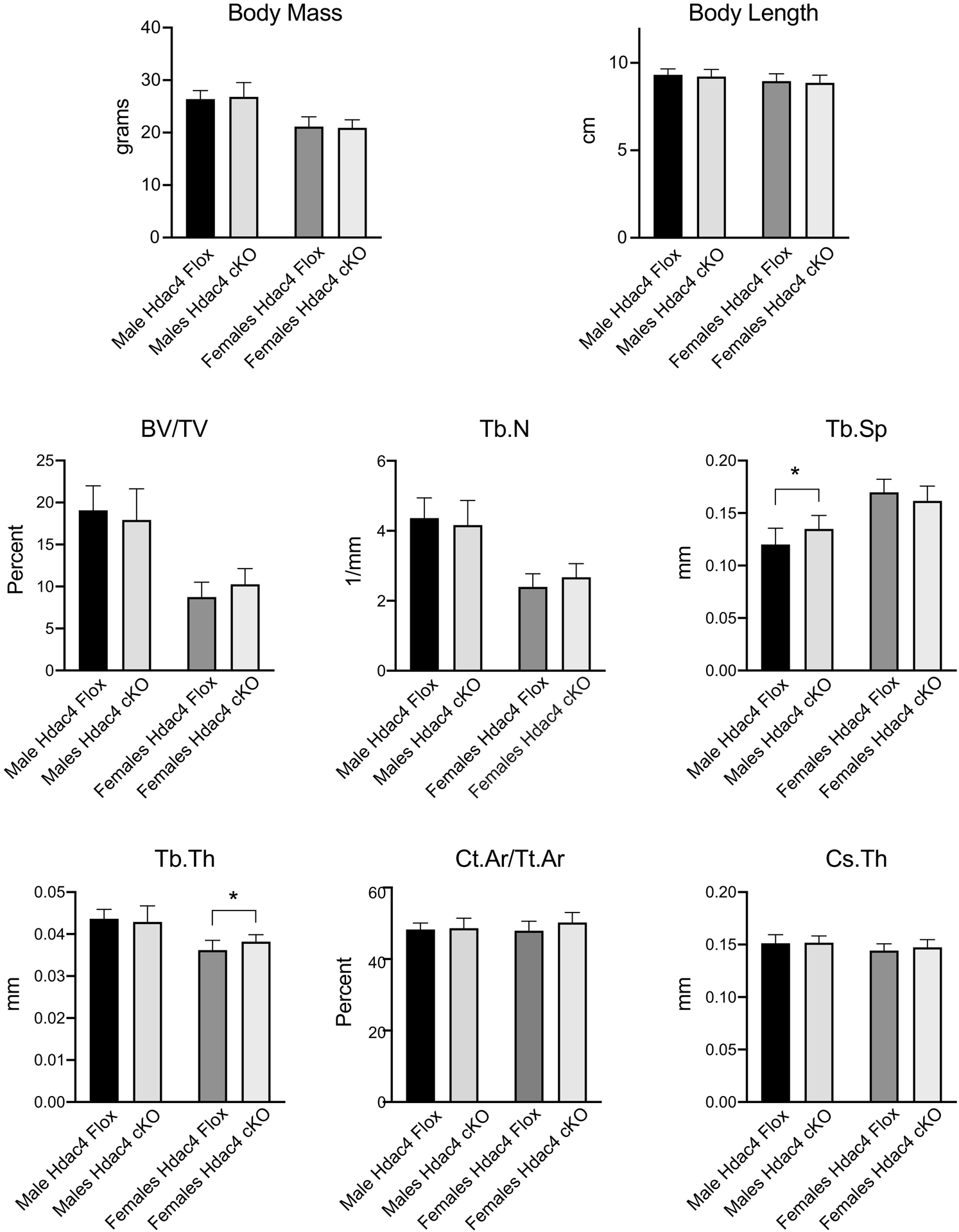
Analysis of skeletal parameters of Hdac4 cKO male and female mice at 12 weeks age. Mass and body length were measured at sacrifice. μCT analysis of trabecular bone was performed at the distal femur; cortical measures were obtained at the mid-diaphysis. Differences between Hdac4flox and Hdac4 cKO of the same sex did not reach statistical significance unless specifically indicated. * p<0.05.
4. Discussion
The overall goal of our research is to better understand novel regulators of osteoclasts. From our prior studies, protein kinase D had emerged as an important kinase to promote osteoclast formation and resorptive function. However, the cellular and molecular mechanisms through which PRKD acts in the osteoclast lineage remain poorly understood. A number of PRKD-responsive targets and pathways have been identified from other contexts. Based on those literature, we treated osteoclasts with CRT0066101 and looked for effects on a variety of potentially significant pathways. We were unable to convincingly show effects of CRT0066101 on these pathways in osteoclasts (Fig. 2D and data not shown). Consequently, we turned to an unbiased phosphoproteomics approach to search for novel targets of regulation by PRKD. This strategy enabled us to identify multiple clusters of proteins involved in a diverse range of biological processes. Some of these clusters include cellular processes where PRKD is known to be active such as cadherin-binding and cell adhesion, vesicle-mediated transport, signal transduction cascades and regulation of transcription. Rather less is known of PRKD’s involvement in mRNA processing and splicing, where significant numbers of PRKD-responsive phosphoproteins were clustered. Similar enrichment of proteins associated with mRNA processing and splicing were reported in a study that used phosphoproteomics characterization of breast cancer cells treated with CRT0066101 [44]. The significance of these findings remains an intriguing question for future study.
In addition to the functional annotation clustering analysis of our phosphoprotein data, an important analysis was to query the decreased abundance phosphopeptides to identify potential direct substrates of PRKD in osteoclasts. Known direct phosphorylation substrates of PRKD have a well-characterized consensus motif. This knowledge enabled us to identify 13 proteins where decreased phosphorylation in the CRT0066101 treated group was mapped to PRKD consensus sites. The validity of our screen was boosted by the identification of HDAC5, an experimentally validated PRKD substrate[11, 13, 15]. Consequently, we chose to further investigate the PRKD/HDAC5 pathway in osteoclasts. HDAC5 and the related class IIa HDACs are recruited to gene regulatory regions via interactions with DNA-binding transcription factors where they catalyze deacetylation of histones, leading to a repressed chromatin state. Understanding the spectrum of HDAC functions has come to be of translational importance in recent years due to substantial interest in uses of HDAC inhibitors as therapies against various cancers and in other diseases[45]. Further, work from our group and others have already implicated HDACs as regulators of osteoclasts[18–22, 46, 47]. These considerations made HDAC5 an interesting and important protein for further study.
A crucial first step was to confirm whether PRKD inhibition really reduces HDAC5 phosphorylation in osteoclasts. Using western blotting and a series of immunoprecipitation studies, we confirmed that CRT0066101 reduces phosphorylation of HDAC5 at Ser488, while exogenous HDAC5 can be phosphorylated at Ser488 by overexpressed PRKD3 or PRKD2, confirming previous results reported by Huynh et al. [11]. Phosphorylation of HDAC5 Ser250 and of HDAC4 and HDAC7 were less clearly responsive to CRT0066101, suggesting that another kinase, possibly calcium/calmodulin kinase II [48, 49] contributes more strongly to phosphorylation of those sites. As another consideration, the level of HDAC phosphorylation at each site is determined by the relative rate of phosphorylation by PRKD and other protein kinases, balanced by the rate of dephosphorylation by protein phosphatases. If the rate of HDAC dephosphorylation is slow, we might inhibit PRKD but still see little change in in P-HDAC levels.
CRT0066101 showed less ability to inhibit osteoclasts in Hdac5 KO cells (Fig. 4), which supports a functional role for HDAC5 downstream of PRKD to promote osteoclast differentiation. Although statistically significant, the differences in the strength of CRT0066101’s effects between Hdac5 KO and WT cells were modest, with CRT0066101 retaining significant repressive activity even in the absence of HDAC5. One reasonable interpretation of these data is that HDAC5 is not the only important target of PRKD during osteoclastogenesis. Additional possible PRKD phosphorylation targets in osteoclasts include the 12 other proteins identified by our proteomics screen (Fig. 1E).
In some situations, class IIa HDACs appear to be functionally redundant. For instance, Chang et al found synergistic effects of deleting Hdac5 and Hdac9 around cardiac hypertrophic responses greater than the phenotype of losing either gene individually, which suggests that the two proteins have overlapping actions and compensate for each other[43]. Similar findings have been reported for HDAC4 and HDAC5 redundancy in hypertrophic chondrocytes[23, 24]. However, it is also clear that these closely related HDACs can localize to different cellular compartments in the same cells and exert distinct functional activities, as reviewed by [50, 51]. Our Hdac knockout osteoclast cultures revealed a clear difference between Hdac4 and Hdac5 knockouts. After careful characterization we were unable to detect any reproducible effect of Hdac4 knockout on osteoclasts either in vitro or on the intact skeleton. In contrast Hdac5 KO osteoclasts showed a significant decrease in their ability to form multinucleated osteoclasts or to resorb bone. This defect seems to be centered around the differentiation or cell-cell fusion process, rather than impaired progenitor viability or growth. Unlike PRKD-inhibited osteoclasts, the Hdac5 KO multinucleated osteoclasts that did form showed were able to organize their actin cytoskeleton, forming normal looking mature actin belts and smaller actin rings, suggesting that this activity of PRKD may be independent of HDAC5. We conclude that loss of HDAC5 primarily reduces the ability of mononucleated preosteoclasts to differentiate, fuse into mature multinucleated osteoclasts, and to resorb bone, while HDAC4 is not required for osteoclastogenesis, at least in the presence of other HDACs. These conclusions are distinct from the prior study describing shRNA-mediated knockdown of Hdac4 or Hdac5 that reported accelerated in vitro osteoclast formation from either gene knockdown[21]. We speculate that this may be due to differences in the relative timing and efficiency of gene knockdown by conditional genetic deletion versus shRNA during osteoclastogenesis or due to non-specific effects of viral infection on osteoclast cultures.
We did not test the in vivo skeletal phenotype with the Hdac5 global KO mice, as several previous studies have already characterized bone phenotypes using this mouse model. Hdac5 KO mice display low bone mass that has been explained by effects on hypertrophic chondrocytes in the growth plate [23, 24]. In addition, they were found to exhibit increased sclerostin expression by osteocytes leading to low bone formation and an osteopenic phenotype in female mice[52]. Histological examination of their bones revealed a trend towards reduced osteoclast parameters (Oc.S/B.Pm, N.Oc/B.Pm, ES/BS), which is consistent with the in vitro phenotype we observed. Further complicating understanding of the Hdac5 KO skeletal phenotype, Obri et al.[25] proposed that their osteopenia stems from enhanced expression of RANKL by osteoblast lineage cells causing enhanced bone resorption. Synthesizing from these studies, we speculate that direct negative effects of Hdac5 deletion on osteoclasts in vivo might be offset by increased RANKL from the osteoblast lineage, thereby rescuing osteoclast formation and masking what might otherwise be a stronger Hdac5 KO osteoclast defect. The net balance between bone formation and bone resorption is a delicate equilibrium subject to multiple overlapping physiological inputs from a variety of cellular lineages, and the skeletal effects of HDAC5 deficiency on chondrocytes, osteocytes and osteoblasts make clear that unambiguous determination of HDAC5’s significance to osteoclast lineage cells in vivo will require a targeted knockout strategy.
What is the functional and molecular significance of PRKD/HDAC5 pathway in osteoclasts? Inhibition of PRKD impairs osteoclasts, implying that PRKD functions positively to promote osteoclast formation and function. In the model of PRKD/HDAC interactions found in the literature, PRKD antagonizes class IIa HDAC transcriptional repression activity by directing their export out of the nucleus to the cytoplasm and binding to 14-3-3 chaperones [12, 13, 35–39]. From this predicted antagonism, we expected that their knockouts would show opposite effects on osteoclasts. However, our data showed that loss of HDAC5 impaired osteoclastogenesis, which indicates HDAC5 activity also acts positively towards their differentiation. This was unexpected. Based on our experimental results and the literature about PRKD-HDAC interactions we propose a model (Fig. 7) in which HDAC5 promotes osteoclast formation by limiting expression of not-yet-known osteoclast inhibitory factors. PRKD stimulates factors promoting osteoclast development, actin cytoskeletal organization, and is among the factors contributing to HDAC5 phosphorylation. This de-represses HDAC5 targets and allows a moderate expression of inhibitory factors that act as to counterbalance the PRKD-responsive activating factors to prevent excessive or premature osteoclast formation. Hdac5 knockout removes repression of these inhibitory factors, increasing their expression and limiting osteoclastogenesis. In the case of PRKD inhibition, decreased HDAC5 phosphorylation reduces expression of the inhibitory factors, but this is offset by reduced activation of the stimulatory factors by PRKD, resulting in a net decrease in osteoclast formation. This balancing act between multiple positive and negative factors might allow for careful fine-tuning of the level of osteoclastogenesis and bone resorption in response to physiological needs. Further studies will be required to better test this model and to elucidate the cellular and molecular mechanisms around HDAC5 and protein kinase D as regulators of osteoclasts and skeletal physiology.
Figure 7.

Model of regulation of osteoclasts by PRKD and HDAC5. Under normal conditions (upper schematic), PRKD activates factors that promote osteoclast formation, regulates actin cytoskeleton and phosphorylates HDAC5 to give a moderate level of inhibitory factors, ultimately giving balanced level of osteoclastogenesis. In Hdac5 KO osteoclasts (middle schematic), loss of HDAC5 dysregulates expression of osteoclast inhibitory factors thereby reducing osteoclasts. Treatment with PRKD inhibitors (bottom panel) reduces HDAC5 phosphorylation thus reducing expression of HDAC5-responsive inhibitory factors, but loss of PRKD-responsive stimulatory factors results in diminished osteoclast formation.
Supplementary Material
Highlights.
We found 522 of PRKD-responsive phosphoproteins and 13 putative direct PRKD substrates
Histone deacetylases 5 is an endogenous substrate of PRKD in osteoclasts
Loss of HDAC5 impairs osteoclast differentiation in vitro
Loss of HDAC4 in osteoclasts did not affect cultured osteoclasts or in vivo bone
Acknowledgements
We thank LeeAnn Higgins and Todd Markowski (University of Minnesota Center for Mass Spectroscopy & Proteomics) for technical support with mass spectroscopy, Laurie Parker for advice and assistance around analysis of the phosphoproteomics data and Bonnie VanHeel for assistance with microCT imaging.
Funding:
This work was supported by a grant to EDJ and KM from the National Institute Arthritis and Musculoskeletal and Skin Diseases (NIH R01AR061352).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
CMGM: Investigation, Formal analysis; SDB: Investigation, Formal Analysis; BF: Investigation; Formal Analysis PK: Investigation, Formal analysis, JC: Investigation; KM: Conceptualization, Supervision, Writing – editing; EJ: Conceptualization, Supervision, Formal analysis, Writing- original, Writing- editing. All authors have read and approved the final manuscript.
Conflicts of Interest: none
References
- [1].Briggs AM, Cross MJ, Hoy DG, Sànchez-Riera L, Blyth FM, Woolf AD, March L, Musculoskeletal Health Conditions Represent a Global Threat to Healthy Aging: A Report for the 2015 World Health Organization World Report on Ageing and Health, The Gerontologist 56(Suppl_2) (2016) S243–S255. [DOI] [PubMed] [Google Scholar]
- [2].Boyce BF, Advances in the regulation of osteoclasts and osteoclast functions, J Dent Res 92(10) (2013) 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Brown JP, Antiresorptives: Safety Concerns-Clinical Perspective, Toxicol Pathol 45(7) (2017) 859–863. [DOI] [PubMed] [Google Scholar]
- [4].Ukon Y, Makino T, Kodama J, Tsukazaki H, Tateiwa D, Yoshikawa H, Kaito T, Molecular-Based Treatment Strategies for Osteoporosis: A Literature Review, Int J Mol Sci 20(10) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Leightner AC, Mello Guimaraes Meyers C, Evans MD, Mansky KC, Gopalakrishnan R, Jensen ED, Regulation of Osteoclast Differentiation at Multiple Stages by Protein Kinase D Family Kinases, Int J Mol Sci 21(3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mansky KC, Jensen ED, Davidova J, Yamamoto M, Gopalakrishnan R, Protein kinase D promotes in vitro osteoclast differentiation and fusion, J Biol Chem 288(14) (2013) 9826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rozengurt E, Protein kinase D signaling: multiple biological functions in health and disease, Physiology (Bethesda) 26(1) (2011) 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang X, Connelly J, Chao Y, Wang QJ, Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases, Biomolecules 11(3) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Döppler H, Storz P, Li J, Comb MJ, Toker A, A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D, J Biol Chem 280(15) (2005) 15013–9. [DOI] [PubMed] [Google Scholar]
- [10].Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC, Determination of the specific substrate sequence motifs of protein kinase C isozymes, J Biol Chem 272(2) (1997) 952–60. [DOI] [PubMed] [Google Scholar]
- [11].Huynh QK, McKinsey TA, Protein kinase D directly phosphorylates histone deacetylase 5 via a random sequential kinetic mechanism, Arch Biochem Biophys 450(2) (2006) 141–8. [DOI] [PubMed] [Google Scholar]
- [12].Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM, Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes, Mol Cell Biol 26(4) (2006) 1569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA, Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5, Mol Cell Biol 24(19) (2004) 8374–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bahl S, Seto E, Regulation of histone deacetylase activities and functions by phosphorylation and its physiological relevance, Cell Mol Life Sci 78(2) (2021) 427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Martin M, Kettmann R, Dequiedt F, Class IIa histone deacetylases: regulating the regulators, Oncogene 26(37) (2007) 5450–67. [DOI] [PubMed] [Google Scholar]
- [16].Molstad DHH, Mattson AM, Begun DL, Westendorf JJ, Bradley EW, Hdac3 regulates bone modeling by suppressing osteoclast responsiveness to RANKL, J Biol Chem 295(51) (2020) 17713–17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Molstad DHH, Zars E, Norton A, Mansky KC, Westendorf JJ, Bradley EW, Hdac3 deletion in myeloid progenitor cells enhances bone healing in females and limits osteoclast fusion via Pmepa1, Sci Rep 10(1) (2020) 21804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pham L, Kaiser B, Romsa A, Schwarz T, Gopalakrishnan R, Jensen ED, Mansky KC, HDAC3 and HDAC7 have opposite effects on osteoclast differentiation, J Biol Chem (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jin Z, Wei W, Dechow PC, Wan Y, HDAC7 inhibits osteoclastogenesis by reversing RANKL-triggered beta-catenin switch, Mol Endocrinol 27(2) (2013) 325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stemig M, Astelford K, Emery A, Cho JJ, Allen B, Huang TH, Gopalakrishnan R, Mansky KC, Jensen ED, Deletion of histone deacetylase 7 in osteoclasts decreases bone mass in mice by interactions with MITF, PLoS One 10(4) (2015) e0123843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Blixt NC, Faulkner BK, Astleford K, Lelich R, Schering J, Spencer E, Gopalakrishnan R, Jensen ED, Mansky KC, Class II and IV HDACs function as inhibitors of osteoclast differentiation, PLoS One 12(9) (2017) e0185441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jin Z, Wei W, Huynh H, Wan Y, HDAC9 Inhibits Osteoclastogenesis via Mutual Suppression of PPARgamma/RANKL Signaling, Mol Endocrinol 29(5) (2015) 730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nishimori S, Lai F, Shiraishi M, Kobayashi T, Kozhemyakina E, Yao TP, Lassar AB, Kronenberg HM, PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy, JCI Insight 4(5) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nishimori S, Wein MN, Kronenberg HM, PTHrP targets salt-inducible kinases, HDAC4 and HDAC5, to repress chondrocyte hypertrophy in the growth plate, Bone 142 (2021) 115709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Obri A, Makinistoglu MP, Zhang H, Karsenty G, HDAC4 integrates PTH and sympathetic signaling in osteoblasts, J Cell Biol 205(6) (2014) 771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xing L, Boyce BF, RANKL-based osteoclastogenic assays from murine bone marrow cells, Methods Mol Biol 1130 (2014) 307–313. [DOI] [PubMed] [Google Scholar]
- [27].Takeshita S, Kaji K, Kudo A, Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts, J Bone Miner Res 15(8) (2000) 1477–88. [DOI] [PubMed] [Google Scholar]
- [28].Orriss IR, Arnett TR, Rodent osteoclast cultures, Methods Mol Biol 816 (2012) 103–17. [DOI] [PubMed] [Google Scholar]
- [29].Tamura T, Takahashi N, Akatsu T, Sasaki T, Udagawa N, Tanaka S, Suda T, New resorption assay with mouse osteoclast-like multinucleated cells formed in vitro, J Bone Miner Res 8(8) (1993) 953–60. [DOI] [PubMed] [Google Scholar]
- [30].Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, Verdin E, A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p, J Biol Chem 274(17) (1999) 11713–20. [DOI] [PubMed] [Google Scholar]
- [31].Ishikawa E, Kosako H, Yasuda T, Ohmuraya M, Araki K, Kurosaki T, Saito T, Yamasaki S, Protein kinase D regulates positive selection of CD4(+) thymocytes through phosphorylation of SHP-1, Nat Commun 7 (2016) 12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Blixt N, Norton A, Zhang A, Aparicio C, Prasad H, Gopalakrishnan R, Jensen ED, Mansky KC, Loss of myocyte enhancer factor 2 expression in osteoclasts leads to opposing skeletal phenotypes, Bone 138 (2020) 115466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherzadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guha S, A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo, Mol Cancer Ther 9(5) (2010) 1136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Choi M, Lee SH, Wang SE, Ko SY, Song M, Choi JS, Kim YS, Duman RS, Son H, Ketamine produces antidepressant-like effects through phosphorylation-dependent nuclear export of histone deacetylase 5 (HDAC5) in rats, Proc Natl Acad Sci U S A 112(51) (2015) 15755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dequiedt F, Van Lint J, Lecomte E, Van Duppen V, Seufferlein T, Vandenheede JR, Wattiez R, Kettmann R, Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis, J Exp Med 201(5) (2005) 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jensen ED, Gopalakrishnan R, Westendorf JJ, Bone morphogenic protein 2 activates protein kinase D to regulate histone deacetylase 7 localization and repression of Runx2, J Biol Chem 284(4) (2009) 2225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Parra M, Kasler H, McKinsey TA, Olson EN, Verdin E, Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation, J Biol Chem 280(14) (2005) 13762–70. [DOI] [PubMed] [Google Scholar]
- [38].Sinnett-Smith J, Ni Y, Wang J, Ming M, Young SH, Rozengurt E, Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling, Am J Physiol Cell Physiol 306(10) (2014) C961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD, AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway, Mol Cell 32(2) (2008) 169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Just S, Berger IM, Meder B, Backs J, Keller A, Marquart S, Frese K, Patzel E, Rauch GJ, Tubingen Screen C, Katus HA, Rottbauer W, Protein kinase D2 controls cardiac valve formation in zebrafish by regulating histone deacetylase 5 activity, Circulation 124(3) (2011) 324–34. [DOI] [PubMed] [Google Scholar]
- [41].Deng L, Zhou JF, Sellers RS, Li JF, Nguyen AV, Wang Y, Orlofsky A, Liu Q, Hume DA, Pollard JW, Augenlicht L, Lin EY, A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis, Am J Pathol 176(2) (2010) 952–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA, A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse, Blood 101(3) (2003) 1155–63. [DOI] [PubMed] [Google Scholar]
- [43].Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN, Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development, Mol Cell Biol 24(19) (2004) 8467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu Y, Wang Y, Yu S, Zhou Y, Ma X, Su Q, An L, Wang F, Shi A, Zhang J, Chen L, The Role and Mechanism of CRT0066101 as an Effective Drug for Treatment of Triple-Negative Breast Cancer, Cell Physiol Biochem 52(3) (2019) 382–396. [DOI] [PubMed] [Google Scholar]
- [45].Ho TCS, Chan AHY, Ganesan A, Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight, J Med Chem 63(21) (2020) 12460–12484. [DOI] [PubMed] [Google Scholar]
- [46].Algate K, Haynes D, Fitzsimmons T, Romeo O, Wagner F, Holson E, Reid R, Fairlie D, Bartold P, Cantley M, Histone deacetylases 1 and 2 inhibition suppresses cytokine production and osteoclast bone resorption in vitro, J Cell Biochem 121(1) (2020) 244–258. [DOI] [PubMed] [Google Scholar]
- [47].Rahman MM, Kukita A, Kukita T, Shobuike T, Nakamura T, Kohashi O, Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages, Blood 101(9) (2003) 3451–9. [DOI] [PubMed] [Google Scholar]
- [48].Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, Khochbin S, Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7, J Biol Chem 276(50) (2001) 47496–507. [DOI] [PubMed] [Google Scholar]
- [49].McKinsey TA, Zhang CL, Olson EN, Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14–3-3 to histone deacetylase 5, Proc Natl Acad Sci U S A 97(26) (2000) 14400–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB, Histone deacetylases (HDACs): characterization of the classical HDAC family, Biochem J 370(Pt 3) (2003) 737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Di Giorgio E, Brancolini C, Regulation of class IIa HDAC activities: it is not only matter of subcellular localization, Epigenomics 8(2) (2016) 251–69. [DOI] [PubMed] [Google Scholar]
- [52].Wein MN, Spatz J, Nishimori S, Doench J, Root D, Babij P, Nagano K, Baron R, Brooks D, Bouxsein M, Pajevic PD, Kronenberg HM, HDAC5 controls MEF2C-driven sclerostin expression in osteocytes, J Bone Miner Res 30(3) (2015) 400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.