

Abstract
The electrophysiological properties of the heart include cardiac automaticity, excitation (i.e., depolarization and repolarization of action potential) of individual cardiomyocytes, and highly coordinated electrical propagation through the whole heart. An abnormality in any of these properties can cause arrhythmias. MicroRNAs (miRs) have been recognized as essential regulators of gene expression through the conventional RNA interference (RNAi) mechanism and are involved in a variety of biological events. Recent evidence has demonstrated that miRs regulate the electrophysiology of the heart through fine regulation by the conventional RNAi mechanism of the expression of ion channels, transporters, intracellular Ca2+-handling proteins, and other relevant factors. Recently, a direct interaction between miRs and ion channels has also been reported in the heart, revealing a biophysical modulation by miRs of cardiac electrophysiology. These advanced discoveries suggest that miR controls cardiac electrophysiology through two distinct mechanisms: immediate action through biophysical modulation and long-term conventional RNAi regulation. Here, we review the recent research progress and summarize the current understanding of how miR manipulates the function of ion channels to maintain the homeostasis of cardiac electrophysiology.
Keywords: microRNA, electrophysiology, arrhythmia, RNAi, biophysical modulation
Graphical Abstract
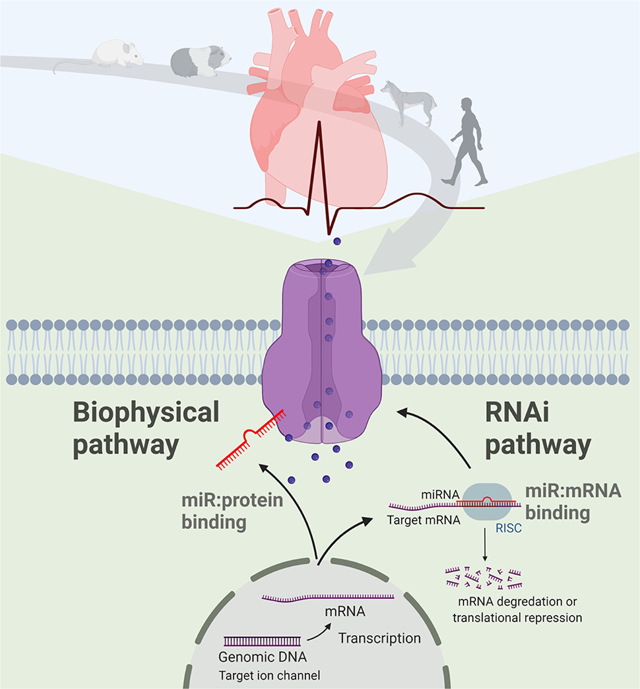
Introduction
Cardiac arrhythmia is an abnormal rate or rhythm of the heartbeat that impacts the effectiveness of blood pumping out of the heart. Arrhythmias can be classified based on heart rates such as tachyarrhythmia (above 100 beats per minute in adults), bradyarrhythmia (below 60 beats per minute), or irregular heartbeat. Arrhythmia can also be classified based on the site where they originate from such as supraventricular or ventricular arrhythmias, in which arrhythmia begins in the atria or ventricles, respectively, or such as atrial or ventricular fibrillation. Although most cases of arrhythmias are not serious, cardiac arrhythmia is a major contributor to human morbidity and mortality and resulted in more than 560,000 deaths in the United States in 2018 [1]. Atrial fibrillation (AF), the most common sustained arrhythmia, affects about 2% to 3% of the population, which is even higher among the elderly; with an aging society, the prevalence of AF is estimated to rise to 12.1 million in the United States by 2030 [2] and to 17.9 million in the European Union by 2060 [3]. Mechanistically, the generation and conduction of normal rhythmic electrical signals in the heart require the finely orchestrated activities of various molecules in cardiomyocytes, including ion channels, transporters, intracellular Ca2+-handling proteins, and other relevant factors. Abnormal impulse formation (i.e., focal activity) and conduction disturbances (i.e., reentry) are two major categories of arrhythmic mechanisms [4], which are associated with the abnormal function of ion channels. Ion channel dysregulation includes gene transcription and translation (i.e., abnormal upregulation or downregulation), protein trafficking to the sarcolemma, problematic biophysical properties, and biophysical modulation of functional channels [5].
Over the past decade, microRNA (miR), the endogenous non-coding small ribonucleic acid, has been recognized as a central player in gene expression regulation through the canonical RNA interference (RNAi) mechanism. Several review articles have summarized the critical role of miRs in cardiac development and physiology and cardiovascular diseases [6–8], the prospect of using circulating miRs and exosomal miRs as biomarkers of cardiovascular disorders [9, 10], and drug development based on targeting miRs [11, 12]. We recently discovered a novel mode of action for miRs and demonstrated that miRs control cardiac homeostasis through two different mechanisms (Figure 1) [13]. One mechanism is the well-recognized conventional RNAi mechanism of post-transcriptional regulation of gene expression, including ion channels, transporters, and transcription factors. The RNAi regulation needs a process of loading cytosolic miR into RISC and takes times (hours to days) to change the expression of target genes with a long-term effect. The other mechanism is the biophysical mechanism of direct interaction with target proteins, in which miRs act as an ion channel modulator. This biophysical action of miRs quickly (seconds to minutes) modulates the function of bound proteins, which enables miRs to rapidly respond to environmental and genetic perturbations.
Figure 1.
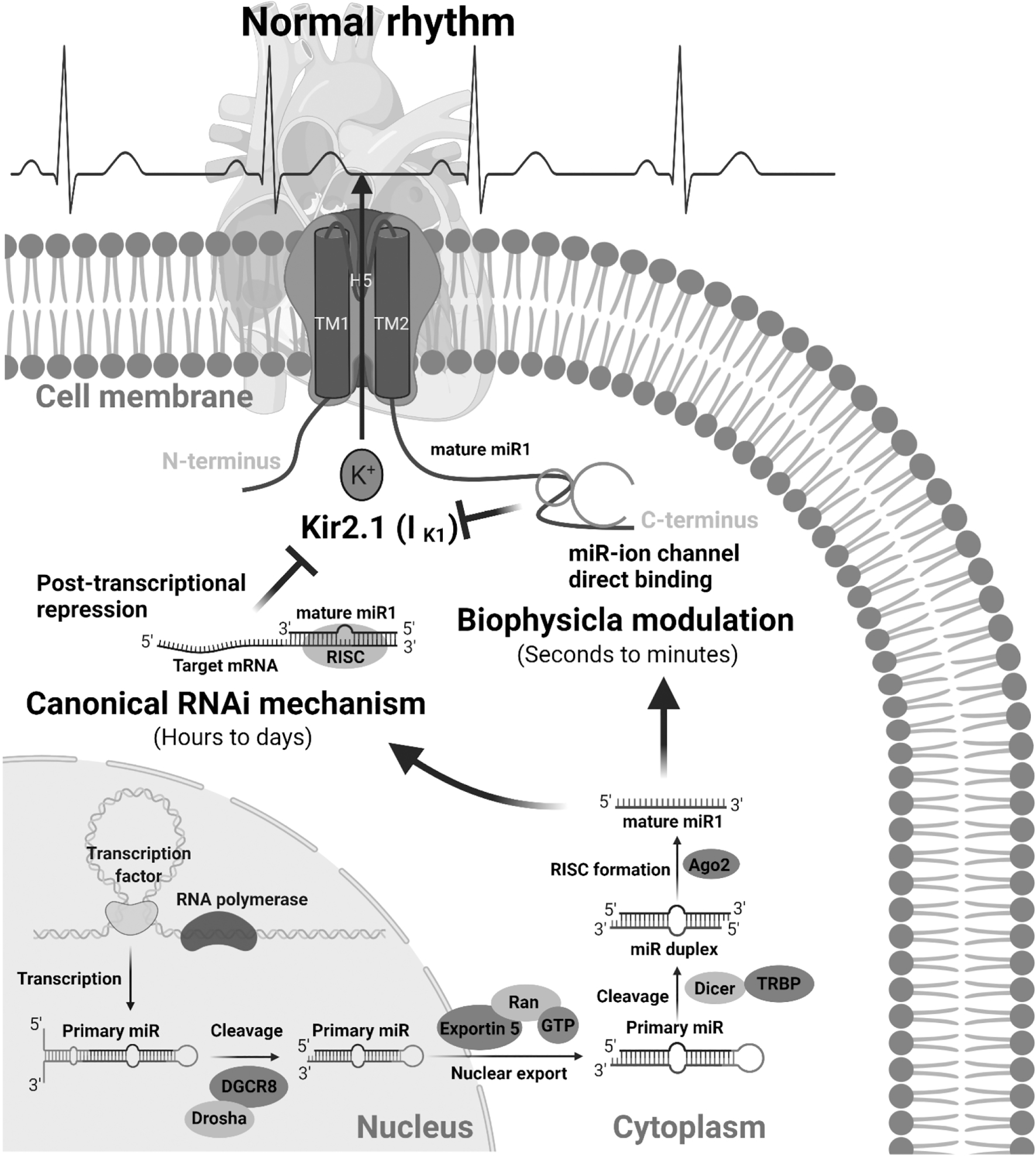
miRs regulate cardiac electrophysiology and heart rhythm through two different mechanisms: canonical posttranscriptional RNAi mechanism to regulate the expression of ion channels, which needs hours to days to take effect, and the biophysical modulation to directly and quickly (seconds to minutes) change the dynamics of ion channels.
It has been more than five years since previous review articles summarized the critical role of miRs in the regulation of cardiac conduction, excitability, and arrhythmias [14–16]. In recent years, there have been many advances associated with the conventional RNAi mechanism of miRs in regulating cardiac electrophysiology; moreover, the discovery of miRs’ biophysical function provides further insight into how miRs control cardiac electrophysiology at multilayer levels [13]. After a brief introduction of microRNA biology and cardiac electrophysiology, this review first focuses on the up-to-date RNAi discoveries of miRs controlling gene expression of ion channels/Ca2+ handling proteins and then emphasizes the new mechanism of action for miRs that biophysically modulate cardiac electrophysiology, to provide a better understanding of ion channel dysregulation in arrhythmias.
miR biogenesis and its mechanisms
miRs are ~22-nucleotide noncoding RNA that are well conserved in a wide variety of organisms, including plants, animals, bacteria, and viruses. Since the first miR was discovered in the early 1990s [17], >48,000 mature miRs have been found in 271 species, including around 2,600 mature miRs in Homo sapiens [18]. miRs are transcribed by RNA polymerases II or III and can be encoded in the intragenic (introns or exons) or intergenic regions of protein-coding genes as well as in long noncoding transcripts. miR biogenesis processing is typically classified into the well-established canonical pri-miR/pre-miR/mature miR pathway [19–21] and non-canonical pathways, such as DGCR8/Drosha-independent “miRtrons” that are spliced from introns of gene transcripts, and Dicer-independent pathway, which has been well summarized in a review article [22].
It has been broadly recognized that miRs act as fine-tuning regulators for the control of gene expression by the conventional RNAi mechanism. In brief, mature miR is loaded into an Argonaute (AGO) family protein to form the core of a miR-induced silencing complex (miRISC) and then binds to the 3’ untranslated region (UTR) of mRNA with partially complementary sequences of miR, typically resulting in mRNA degradation or translational repression [23, 24]. In addition to the 3’UTR, miRs could also bind to the 5’UTR [25, 26] or coding domain [27, 28] of the target mRNA to mediate its RNAi action, or interact with the promoter region of genomic DNA and alter the local epigenetic modification to regulate gene expression [29–31]. It has been found that miRs can even enhance gene expression at the post-transcriptional level [23, 32, 33]. In addition to interacting with mRNA or DNA, miRs have been recently shown to directly bind with and biophysically modulate the function of protein molecules, such as ion channels [13, 34–37], which implies a more complex regulatory network of miRs. Almost one-third of human miRs are expressed in cardiac tissues, and many cardiac-enriched miRs, including miR1, miR133-a/b, miR21, miR26-a/b, miR24, miR23, and miR27-a/b [38–41], are critical to the development of the heart and in cardiac remodeling of cardiovascular diseases, including ion channel (dys)regulation in cardiac electrophysiology. Therefore, miRs play a central role in regulating the homeostasis of cardiac physiology. Since each miR directly targets hundreds of genes and subsequently regulates the expression of thousands of genes with a broad impact on the functional homeostasis of cells/tissues/organs, here, we focused on discoveries where miRs directly target ion channel genes to modulate cardiac electrophysiology.
Cardiac electrical impulse and conduction
The normal cardiac impulse originates in the sinoatrial node (SAN), propagates through the atria to reach the atrioventricular node, and passes through the His-Purkinje fibers to reach the ventricle, triggering coordinated cardiac pumping actions. Cardiac electrical impulse is also referred to as action potential (AP), which typically contains five phases (Figure 2A): 1) Phase 4, or resting potential, is stable at ~−90 mV in normal working myocardial cells; 2) Phase 0 is a phase of rapid depolarization and is critical to rapid propagation of the cardiac impulse; 3) Phase 1 is a phase of rapid initial repolarization; 4) Phase 2 is a plateau phase; 5) Phase 3 is a phase of final repolarization to restore the resting membrane potential. An AP is orchestrated by multiple ion channels, transporters, and transmembrane proteins embedded across the cytoplasmic membrane. Because of regionally distinct combinations of ionic currents, cardiac cells in different regions of the heart, such as SAN pacemaker cells and atrial and ventricular myocytes, are characterized by a specific morphology of APs. The underlying molecular and ionic mechanisms of electrophysiology in different cardiac regions have been well summarized in previous publications [5, 42]. Cardiac electrophysiological properties include automaticity, excitation (i.e., depolarization and repolarization), and signal conduction within a cell (intracellular) and between cells (intercellular). An abnormality in any of these properties can induce arrhythmias in the heart.
Figure 2.
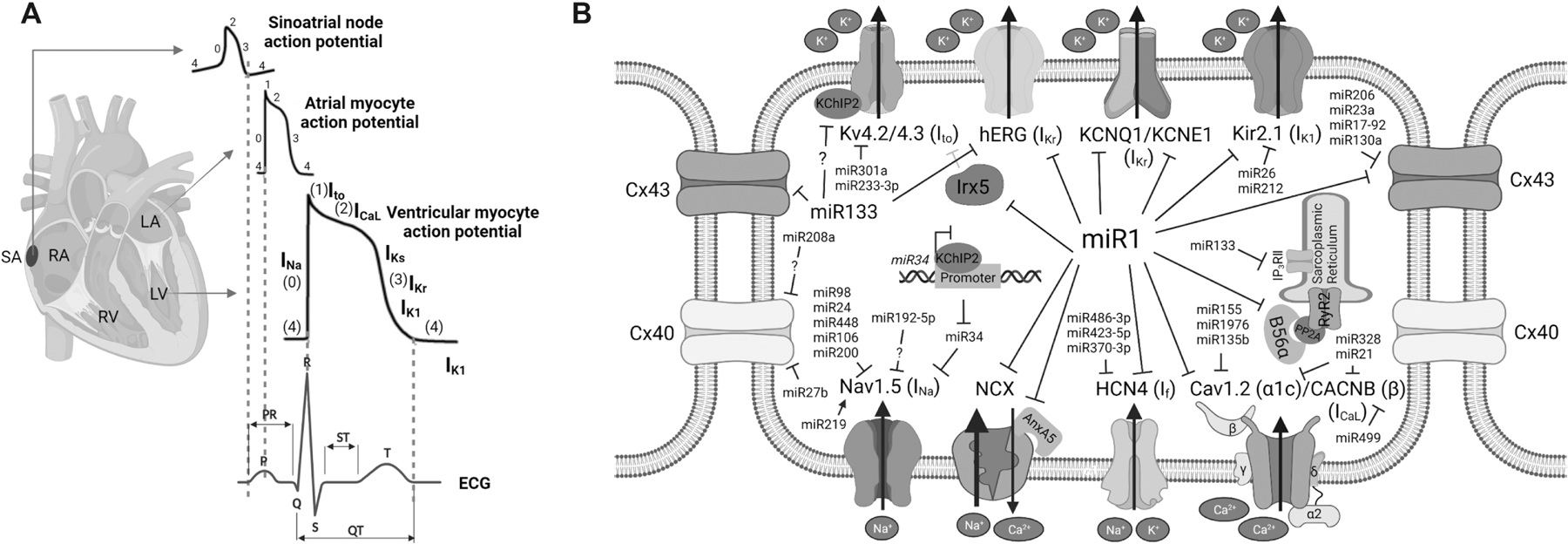
A) Schematic diagram of sinoatrial (SA) node, atrial and ventricular action potentials and surface electrocardiogram (ECG). B) Discoveries of ion channels regulation by microRNAs through RNAi mechanism.
RNAi regulation of cardiac automaticity
Automaticity of cardiac cells is the property to spontaneously generate APs. In the adult heart, the SAN, found on the top wall of the right atrium, displays the highest intrinsic automaticity, and controls the rhythm and rate of cardiac chamber contraction [43]. The SAN consists of specialized pacemaker cells, in which the spontaneously fired APs are significantly different from those in the working myocardium [44]. The membrane potential at the onset of phase 4 is more depolarized and undergoes slow diastolic depolarization to fire AP automatically. The mechanisms of SAN automaticity are described as sarcolemma voltage clock [45] and/or intracellular Ca2+ clock [46]. Pacemaker cells of the SAN highly express hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. The sarcolemma voltage clock refers to hyperpolarization-activated pacemaker current, which is also named “funny” current (If), because it is activated by hyperpolarization while most voltage-sensitive currents are activated by depolarization. At the end of the AP, If is activated and initiates the diastolic depolarization phase [45]. In contrast, the inward rectifier potassium current (IK1), encoded by Kir2.x genes, has been generally thought to play an inhibitory role in the induction of cardiac automaticity [47]. Manipulation of If and IK1 has been studied in the regulation of the automaticity of cardiomyocytes [48–51]. The Ca2+ clock refers to intracellular Ca2+ that is spontaneously released from the sarcoplasmic reticulum (SR) via the ryanodine receptor (RyR) and triggers Ca2+ extrusion by the Na+-Ca2+ exchanger (NCX) [52–54]. NCX exchanges 3 Na+ for each Ca2+ ion and generates a net inward current that is thought to contribute to pacemaking in the SAN [46].
The regulation of cardiac automaticity by miRs has been systematically investigated in the human SAN. Petkova et al utilized miR microarray analysis and found that the human SAN possesses a unique expression pattern of miRs [55]. Among 18 miRs that are significantly more abundant than in atrial muscle, luciferase assay demonstrated that miR486-3p directly targets and inhibits the expression of HCN4 and thereby reduces ex vivo rat sinus node beating rates. It was reported that downregulation of HCN4 is associated with an upregulation of miR423-5p in the SAN of athletes and rodent models; knockdown of miR423-5p could successfully reverse training-induced bradycardia in mice [56]. Intraperitoneal injection of anti-miR370-3p could restore HCN4 expression and If in the sinus node, blunting sinus bradycardia in failing mouse hearts [57]. It has been found that miR1 can reduce the If current density by repressing HCN4 translation in neonatal rat ventricular cardiomyocytes [58] and human ES cell-differentiated cardiomyocytes [41]. Kir2.1 (encoded by KCNJ2) is the main K+ channel in the heart and is responsible for setting and maintaining the cardiac resting membrane potentials (RMPs) [59]. Since Kir2.1 is one of the directly-targeted genes of miR1, abnormal expression of miR1 was found to exacerbate arrhythmogenesis, including enhanced automaticity of cardiomyocytes, via regulating the expression of Kir2.1 and IK1 [59, 60].
miR1 has been implicated in the modulation of a wide variety of Ca2+ handling proteins. Terentyev et al [61] found that overexpression of miR1 in rat myocytes enhanced the frequency of spontaneous Ca2+ sparks while reducing the sarcoplasmic reticulum Ca2+ content. In the presence of isoproterenol, miR1-overexpressed myocytes exhibited spontaneous arrhythmogenic oscillations of intracellular Ca2+. Mechanistically, miR1 directly decreases the protein level of phosphatase PP2A regulatory subunit B56α, resulting in hyperphosphorylation of RyR2 channels, which is probably conserved across species. It has been shown that miR1 targets NCX1 and annexin A5 (AnxA5), a Ca2+-binding and phospholipid-binding protein that interacts with NCX1 and regulates Ca2+ extrusion. In addition to miR1, miR133 has been shown to regulate Ca2+ release and play an anti-hypertrophic role by directly targeting cardiac Inositol 1,4,5′-triphosphate receptor II (IP3RII) calcium channel in rats [62]. Therefore, miRs control cardiac automaticity via regulation of both sarcolemma ion channels contributing to the voltage clock and intracellular Ca2+ handling proteins contributing to the Ca2+ clocks.
RNAi regulation of cardiac depolarization currents
Cardiomyocyte transmembrane potential during AP phase 4 is stable and negative (approximately −90 mV) due to the high conductance for K+ of the IK1 channel. Upon activation by electrical impulses from adjacent cells, Na+ channel activation generates a large inward Na+ current (INa), which results in the phase 0 depolarization of APs. The voltage-gated sodium channel Nav1.5 is typically considered a cardiac-specific isoform in adult cardiomyocytes and consists of a primary α-subunit and multiple secondary β-subunits. The α-subunit of Nav1.5 (encoded by SCN5A) is sufficient to generate a sodium current with characteristic features of INa in native cells, while β-subunits (β1–4) are known to regulate the trafficking and intrinsic properties of the α-subunit [63]. Daimi et al studied SCN5A expression regulated by miRs and found that miR98, miR106, and miR200 decrease the expression of SCN5A in HL-1 cells [64]. Interestingly, they observed that miR219 exceptionally enhances the expression of SCN5A, increases INa, and corrects the flecainide-induced prolongation of QRS in mice. Zhao et al found that miR192-5p, upregulated in AF patients, post-transcriptionally repressed human and rhesus SCN5A and reduced the peak current density of INa, although bioinformatic analysis didn’t reveal miR192-5p binding sites on SCN5A mRNA of other mammalian species [65]. miR448, induced by HIF-1α and NF-κB in hypoxic conditions, suppresses SCN5A expression and reduces INa, resulting in an increased risk of arrhythmia [66]. Zhang et al found that miR24 potently suppresses SCN5A expression through binding to its coding region and an arrhythmia-associated human single nucleotide-polymorphism (hSNP) of SCN5A (rs1805126) enhances this SCN5A-miR24 interaction, resulting in decreased SCN5A expression in the human and mouse heart [67].
Voltage-gated L-type calcium channels are opened in phase 0 and allow Ca2+ entry into the cells, which also contributes to phase 0 depolarization; however, ICaL current is largely responsible for the plateau of APs (phase 2) and triggering excitation-contraction (EC) coupling. ICaL channels are heterotetrameric polypeptide complexes comprising the α1c (Cav1.2, encoded by CACNA1C), β (encoded by CACNB), and α2δ (encoded by CACNA2D) subunits in ventricular myocytes. The α1 subunit harbors the ion-selective pore, voltage-sensing domain, gating machinery, and the binding sites for ICaL-modulating drugs [68–70]. The β2 and α2δ accessory subunits bind to α1 subunit and modulate the biophysical properties and proper trafficking of α1 subunit. Lu et al [71] analyzed the miR transcriptome and found that miR223, miR328, and miR664 were upregulated in left atrial samples of AF patients and AF dog models, while miR101, miR302, and miR499 were downregulated. They demonstrated that miR328 suppressed the expression of CACNA1C and CACNB1. Overexpression of miR328 in the canine atrium and mice diminished ICaL shortened atrial AP duration (APD) and increased the vulnerability to AF. Moreover, it has also been observed that miR21 was significantly upregulated in atrial myocytes of chronic AF patients. miR21 suppresses the expression of CACNA1C and CACNB2, decreases ICaL density and shortens the AP duration [72]. miR499 was also increased in atria from AF patients and regulates ICaL through CACNB2 [73]. miR155 was involved in ICaL-related electrical remodeling in AF via targeting of CACNA1C [74], which is conserved in human and mouse. CACNA1C was also reported as a targeted gene of miR1; a loss of miR1 in human myotonic dystrophy heart results in upregulation of CACNA1C [75]. Overexpression of miR133a-3p in rat atrial and ventricular cardiomyocytes resulted in increased ICaL, although mRNA expression of CACNA1C was not changed [76]. miR1976 [77] and miR135b [78] were recently reported to target CACNA1C in human sick sinus syndrome and pathological cardiac hypertrophy mouse model, respectively.
In summary, miRs are important regulators of cardiac depolarization currents (i.e. INa/ICaL) through multilayer mechanisms, including regulating the expression of ion channel proteins, controlling ion channel trafficking, and changing the biophysical properties of ion channels via accessory subunits.
RNAi regulation of cardiac repolarization currents
Cardiac repolarization is determined by a delicate balance between inward and outward ion currents and its rate is one of the most essential factors for the length of APD determination and thereby for the likelihood of developing arrhythmias. After depolarization, the inward current Na+ channel is inactivated rapidly and various outward current K+ channels are activated, and the membrane potential begins to return to the resting negative voltage state. Membrane phase 0 depolarization is immediately followed by a rapid initial repolarization phase (phase 1) due to K+ efflux through transient outward current (Ito), including fast (Ito,f) and slow (Ito,s) currents. During the phase 2 plateau, there is a balance between the inward current of ICaL and outward K+ current, including ultra-rapid (IKur) and rapid (IKr) and slow (IKs) delayed outward rectifying K+ currents, which are progressively and time-dependently sequentially activated. During the early repolarization of phase 3, L-type Ca2+ channels are inactivated, and predominant IKr and IKs currents act to repolarize the membrane potential. The final repolarization of phase 3 is mediated through the inwardly rectifying potassium current (IK1).
Ito,f is mainly generated by voltage-gated potassium (Kv) channels Kv4.2/Kv4.3, which are regulated by the auxiliary Kv channel-interacting protein (KChIP2, encoded by KCNIP2) [79]. The regulation of cardiac repolarization by miRs was noticed from transgenic miR1-deletion mouse models. In human and mouse chromosomes, there are two genes, miR1-1 and miR1-2 that are transcribed from different chromosomes, to produce the same mature miR1. While knockout of both miR1-1 and miR1-2 genes is postnatally lethal [80, 81], the deletion of miR1-2 in mice causes a developmental defect of the heart and results in sudden cardiac death with abnormal electrophysiology, such as reduced heart rate, accelerated atrioventricular conduction with shortened PR interval, and slowed ventricular conduction with prolonged QRS interval [82]. Kv4.2 (encoded by KCND2) channel for repolarizing Ito current is transcriptionally repressed by Irx5 [83], which is a gene directly targeted by miR1 conserved in mouse and human[82]. Similarly, miR301a, which is upregulated in diabetic ventricles, suppressed the expression of mouse Kv4.2 [84]; miR233-3p, which is upregulated in myocardial infarcted rat hearts, also inhibited the expression of Kv4.2, resulting in a reduction of Ito [85]. In addition, Matkovich et al found that miR133a, another cardiac and skeletal muscle-enriched miR, decreased Ito,f with a decreased expression of KCNIP2, prolonged AP duration of cardiomyocytes, and prolonged the QT interval of the mouse heart [86]. Noticeably, KChIP2 is not a direct target of miR133a, suggesting an indirect mechanism of KChIP2 regulation by miR133a.
For outwardly rectifying K+ currents, miR1 and miR133 were reported to possibly regulate IKr and IKs in canine myocytes [87]. A couple of published review articles had discussed a regulatory relationship between miR1/miR133a and ether-a-go-go related gene (ERG), KCNQ1, and KCNE1 channels; however, it remains questionable, because those cited papers of original research were retracted lately. In another study [88], arsenic trioxide was found to induce a significant upregulation of miR1 and miR133a; the authors reported that miR1 and miR133a target ERG and suppress IKr in guinea pig cardiomyocytes. In rabbit heart, right atrial tachypacing upregulated miR1 expression, which downregulated KCNE1 and KCNB2 and increased IKs [89]. The regulation of IKr and IKs by miR1 was also reported in human embryonic stem cell-derived cardiomyocytes [41].
For inwardly rectifying K+ current, miR26 and miR212 were found to promote AF vulnerability by directly targeting KCNJ2 and repressing IK1 [90, 91]. Girmatsion et al found that miR1 expression was reduced by approximately 86% in atrial tissue of AF patients, accompanied by an elevated expression of potassium channel Kir2.1 and increased IK1 [60]. Remarkably, miR1 overexpression is also arrhythmogenic. miR1 expression was found to be elevated in patients with coronary artery disease, as well as in rat hearts of myocardial infarction [92]; the exacerbated arrhythmogenesis of ischemic rat hearts was relieved by the elimination of upregulated miR1. An overexpression of miR1 in normal rat hearts suppressed the expression of Kir2.1 and gap junction protein connexin43 (Cx43, encoded by GJA1), resulting in diminished conduction and arrhythmias with prolonged QRS. Taken together with the regulation of other ion channels by miR1 (Figure 2B), these studies support a central role of miR1 for fine-tuning cardiac electrophysiology in normal and pathological conditions.
Interestingly, the crosstalk between ion channels and miRs is not a one-way regulation. It was shown that cardiac KChIP2 has a transcriptional capacity and can interact with promoter DRE elements of miR34b/miR34c to transcriptionally repress the expression of miR34b/miR34c, which subsequently target SCN5A, SCN1B and KCND3 in human and rat heart cells [93]. While KChIP2 is downregulated in heart failure, miR34b/miR34c are upregulated while the currents they target INa and Ito,f are downregulated [93]. Genetically manipulating KChIP2/miR34 axis, such as maintaining KChIP2 or inhibiting miR34, restored channel function and prevented the incidence of reentrant arrhythmias under pathologic conditions [93]. This discovery of the ion channel-miR regulatory axis suggests that cardiac repolarization currents are regulated by miRs with the more complex miR-regulatory network, which could have a broader influence on cardiac repolarization.
RNAi regulation of cardiac conduction
Cardiac conduction refers to the propagation of the electrical signal in the heart, including intracellular conduction within a cardiomyocyte and intercellular conduction between cells. The intracellular conduction is determined by the membrane depolarization of INa; the regulation of INa by miRs has been discussed previously above in this review. A proper intercellular conduction of cardiac impulse between discrete cardiac cells is critical to the whole heart function and is accomplished by gap junction channels [94]. Three types of gap junction channels are expressed in the heart; Cx43 is the main cardiac channel that is responsible for intercellular conductance in the ventricles, while connexin-40 (Cx40) and connexin-45 (Cx45) is expressed in the atria and nodal tissue respectively, as well as in the conduction system [95]. Significant changes in the expression of connexins and abnormal organization of gap junctions are commonly found in human heart diseases and are a typical feature of arrhythmogenic remodeling [96, 97]. In addition to the miR1/Cx43 study in ischemic rat heart [92], several studies have also highlighted that miR1 targets Cx43 in various cardiac diseases, such as arrhythmias [92] and viral myocarditis [98], which is covered across species. Jin et al found that miR206 inhibition alleviated ischemia-reperfusion-induced arrhythmias in a mouse model by targeting Cx43 [99]. They confirmed that miR206 directly binds to the 3’UTR of GJA1 and demonstrated that knockdown of Cx43 reversed the protective effects of miR206 inhibitor on cardiac arrhythmias. miR23a expression was upregulated after myocardial ischemia/reperfusion injury and miR23a could directly target rat GJA1 to decrease the protein expression of Cx43 [100]. GJA1 was also reported as a miR133a target in zebrafish heart [101] and a direct target of miR17-92 cluster members (i.e., miR19a/b) [102] and miR130a [103] in rodent hearts.
Callis et al compared the expression of Cx43 and Cx40 in miR208a-overexpressing transgenic mice and miR208a-knockout mice and found that miR208a is required for Cx40 expression, probably through an indirect regulation mechanism [104]. miR208-knockout mice showed decreased protein expression of Cx40, and approximately 80% of animals lacked P waves preceding QRS complexes, an indication of AF. However, Li et al observed upregulation of miR208a in human chronic AF and found a negative regulation of Cx40 by miR208a [105]. Application of a miR208a inhibitor led to a significant upregulation and miR208a mimics led to a significant downregulation of Cx40 protein in AC16 cells, while their luciferase assay showed that the Cx40-encoding gene GJA5 was not a direct target of miR208a. Takahashi et al reported that GJA5 is a direct target of miR27b and that inhibition of miR27b prevented the palmitate-induced downregulation of Cx40 in cultured cardiomyocytes and reversed the high-fat diet-induced vulnerability to atrial arrhythmia of mouse hearts [106].
In conclusion, miRs regulate all properties of cardiac electrophysiology, from cardiac automaticity to highly coordinated electrical propagation, from depolarization to repolarization of action potentials. However, the classical RNAi mechanism of miRs takes hours to days to affect gene expressions, which brings the question of whether ion channel regulation by miRs is involved in the rapid response of cardiac electrophysiology to environmental disruptions.
Biophysical modulation of ion channels by miRs
The RNAi mechanism of miR to guide target gene expression relies on the recruitment of AGO proteins, although the underlying mechanism is not entirely understood. It has been reported that the amount of intracellular miRs is 13 times higher than the amount of AGO proteins [107], and only a fraction of each miR possibly binds to AGO proteins in human cells [108, 109]. The large amount of intracellular AGO-free miRs raises the possibility for AGO-independent mechanisms. Even though these types of research are still in the early stage of the investigation, serval studies have reported that miRs play broad roles in regulating biological processes beyond being interfering RNAs [110]. Lehmann et al firstly reported that extracellular let-7 exhibits ligand-like roles by interacting with Toll-like receptors (TLRs) in neurodegeneration [34], which was evidenced by another independent group in pain-sensing [35]. Tumor-secreted miR21 and miR129a could also bind as ligands to murine TLR7 and human TLR8 in immune cells, triggering TLR-mediated premetastatic inflammatory response [36]. Han et al showed that extracellular miR711 could bind and activate transient receptor potential cation channel subfamily A member 1 (TRPA1) to mediate itch sense in neurons, which suggested that ion channels could be direct binding molecules of miRs.
This biophysical action of miR is also observed for intracellular miRs, to biophysically modulate the electrophysiology of cardiomyocytes [13]. Our group found that miR1 binds to the intracellular C-terminus of Kir2.1 on the plasma membrane, acutely suppresses IK1 within seconds/minutes, and prolongs the duration of APs of cardiomyocytes. Importantly, miR1 functions at a sub-pmol/L concentration, which is close to endogenous intracellular miR levels [111], indicating the physiological significance of endogenous miR1’s biophysical function. We showed that the biophysical modulation of ion channels by miRs requires a physical miR1:Kir2.1 binding in a sequence-dependent manner. A core sequence of 10A-15G (AAGAAG) is critical to the biophysical modulation and is outside the RNAi seed regions of miR1. Importantly, an arrhythmia-associated miR1 human single nucleotide-polymorphism (hSNP)-hSNP14A/G, in which the 14th nucleotide “A” is mutated to “G”, specifically disrupts the biophysical modulation of miR1 while maintaining normal RNAi function, validating that the biophysical modulation is independent of RNAi.
miR1-deficient hearts demonstrate pathologic phenotypes with abnormal electrophysiology, including hyperpolarized RMP, slow conduction, and high arrhythmia inducibility. An acute recovery of miR1 corrected the hyperpolarized RMP of miR1-deficient cardiomyocytes, rescued the conduction in ventricular tissues, and eliminated the high arrhythmia inducibility of miR1-deficent hearts. However, treatments with hSNP14A/G neither changed the RMP of miR1-deficient cardiomyocytes nor the arrhythmogenesis of the heart. These results demonstrated that the biophysical modulation of intracellular endogenous miRs indeed plays an important role in maintaining the homeostasis of cardiac electrophysiology and in the development of arrhythmias. Importantly, this biophysical action of miR1 is evolutionarily conserved among species, including mouse, guinea pig, canine, and human.
Previous studies have shown that SNPs within miRs are associated with cardiac disorders, such as coronary artery disease [112] and congenital heart disease [113, 114], possibly through aberrant miR processing [115–118] or disrupted miR-mRNA interaction [119] of the RNAi mechanism. The discovery of hSNPs in miR1––hSNP14A/G and hSNP15G/A, which maintain the canonical RNAi function but specifically lost the biophysical modulation, revealed a new dysregulation mechanism for hSNPs. In addition, a gain-of-function M301K mutation of Kir2.1 was reported in short-QT syndrome and AF patients and disrupts the biophysical modulation of intracellular polyamines/Mg+ on M301K/Wild-type heterotetrametric channels, leading to a larger outward current of IK1 [120, 121]. While M301 is within one of the miR1-binding residues on Kir2.1, the M301K mutation also relieves the biophysical suppression of IK1 by miR1 [13], demonstrating that biophysical modulation of miRs is involved in ion-channel dysregulation of cardiac arrhythmogenesis.
The potential for miR therapeutics in heart disease
The expression profile of miRs is dynamic in response to the metabolic or disease state of the heart. Ikeda et al comprehensively compared the profiling pattern of miRs in 67 human left ventricular samples from control and heart failure patients and found that the expression profiles of 43 miRs were differentially expressed and could be useful for clinical diagnosis [122]. Serval independent groups also identified significantly different miR expression patterns between normal and diseased human hearts [123–125]. Therefore, pathological remodeling of cardiac miRs is considered a significant signature for the diagnosis of human heart disease. Due to the role of miRs in regulating gene expression, changes in critical cardiac miRs could cause the dysregulation of various target genes and subsequently result in cardiac remodeling, heart failure, and/or arrhythmias. We focused on the regulation of ion channels and summarized the discoveries of miR’s dysregulation and cardiac electrical remodeling in various cardiovascular diseases (Table 1), showing a potential clinical application for miRs targeting and potential miR therapeutics. Indeed, some miRs have been registered in the clinicaltrials.gov database to be potentially used as biomarkers for heart disease diagnosis or as pharmacogenomic biomarkers for drug efficacy and safety assessment. However, directly targeting miRs for clinical therapeutics is still in the early stages because it is very challenging to control the functional specificity of miRs’ actions only for targeted diseases without potential side effects on the human body. Advanced discoveries of the unconventional actions of miRs in the heart, which is independent of the RNAi mechanism, could enhance the specificity of miRs’ applications.
Table 1:
Regulation of ion channels by miRs in cardiovascular diseases.
| miR | Changes in cardiac phenotypes | Species | Related diseases | Targeted ion channels | Reference |
|---|---|---|---|---|---|
| Classical RNAi mechanism | |||||
| miR423-5p | Upregulated | Human Mouse | Training-induced bradycardia | HCN4 (If) | [56] |
| miR370-3p | Upregulated | Human Mouse | Sinus bradycardia | HCN4 (If) | [57] |
| miR1 | Downregulated | Human | AF | KCNJ2 (Kir2.1, IK1) | [60] |
| Upregulated | Rabbit | Atrial tachypacing | KCNE1/KCNB2 (KV2.2, IKs) | [89] | |
| Upregulated | Human Rat | Coronary artery disease / myocardial infarction | KCNJ2 (Kir2.1, IK1) / GJA1(Cx43) | [92] | |
| Upregulated | Mouse | Viral myocarditis | GJA1 (Cx43) | [98] | |
| Upregulated | Guinea pig | Arsenic trioxide-induced QT prolongation | KCNJ2 (Kir2.1, IK1) | [88] | |
| miR448 | Upregulated | Human Mouse | Hypoxic induced arrhythmia | SCN5A (Nav1.5, INa) | [66] |
| miR328 | Upregulated | Human Canine Mouse | AF | CACNA1C (CaV1.2)/CACNB1 (ICaL) | [71] |
| miR21 | Upregulated | Human | Chronic AF | CACNA1C (CaV1.2)/CACNB2 (ICaL) | [72] |
| miR499 | Upregulated | Human Mouse | AF | CACNB2 (ICaL) | [73] |
| miR155 | Upregulated | Human Mouse | Paroxysmal AF | CACNA1C (CaV1.2, ICaL) | [74] |
| miR1976 | Upregulated | Human Rabbit Mouse | Age-related sick sinus syndrome | CACNA1C (CaV1.2, ICaL) | [77] |
| miR135b | Downregulated | Mouse | Cardiac hypertrophy | CACNA1C (CaV1.2, ICaL) | [78] |
| miR301a | Upregulated | Mouse | Diabetic ventricles | KCND2 (KV4.2, Ito) | [84] |
| miR233-3p | Upregulated | Rat | Myocardial infarction | KCND2 (KV4.2, Ito) | [85] |
| miR133-a/b | Upregulated | Guinea pig | Arsenic trioxide-induced QT prolongation | ERG (IKr) | [88] |
| miR26 | Downregulated | Human Canine Mouse | AF | KCNJ2 (Kir2.1, IK1) | [90] |
| miR34 | Upregulated | Human | Heart failure | SCN5A (Nav1.5), SCN1B (INa) and KCND3 (Kv4.3) | [93] |
| miR206 | Upregulated | Mouse | Ischemia-reperfusion-induced arrhythmias | GJA1 (Cx43) | [99] |
| miR23a | Upregulated | Rat | Myocardial ischemia / reperfusion injury | GJA1 (Cx43) | [100] |
| miR27b | Upregulated | Mouse | High-fat diet-induced atrial arrhythmia | GJA5 (Cx40) | [106] |
| Biophysical modulation | |||||
| miR1 | Deficient | Mouse | Inducibility of ventricular arrhythmia | Kir2.1 (IK1) | [13] |
| Indirect mechanism | |||||
| miR192-5p | Upregulated | Human | AF | SCN5A (Nav1.5, INa) | [65] |
| miR133a | Downregulated | Mouse | Reactive cardiac hypertrophy | KCNIP2 (KChIP2, Ito,f) | [86] |
| miR208a | upregulated | Mouse | AF | GJA5 (Cx40) | [104] |
Future directions and challenges
The interaction of long noncoding RNAs (>200nucleotides) with proteins has been reported [126], and we now found that miRs could also directly bind to and regulate protein molecules. The novel discovery of a new action for miRs to biophysically modulate ion channels significantly enhances the implications of miRs. However, several questions become important to be answered to better understand ion channel and miR biology. First, are there more ion channels biophysically modulated by miRs? The answer is most likely to be yes. In our laboratory, we have identified and been investigating two other ion channel subunits that have high affinity to and are biophysically modulated by miR1. It should be also investigated whether more miRs, such as cardiac enriched miR133a and miR499, display this type of biophysical action.
Second, what is the specific contribution of miRs’ biophysical action to the regulation of cardiac electrophysiology and function? How would these two distinct mechanisms of miRs coordinate to regulate the homeostasis of the heart? To reveal the specific physiological significance of miRs’ biophysical action, new animal models must be developed to distinguish the specific physiological contributions of biophysical vs. RNAi mechanisms.
Third, is the biophysical action of miR regulated by post-transcriptional modification of RNA? Very recently, Seok et al uncovered an epi-transcriptional modification of 8-osoguanine (o8G) that leads to mispairing of miR1 with new targeted genes, resulting in activation of cardiac hypertrophy pathways [127]. These findings suggested that epi-transcriptional modification could be a regulatory mechanism of miR, most likely including both RNAi and biophysical actions, in response to environment signal stimulus.
Lastly, in addition to ion channels on the plasma membrane, does this biophysical modulation of miRs broadly regulate the function of other intracellular proteins? As an essential regulator, intracellular miRs appear to target about 60% of mammalian genes through RNAi silencing [58]. It has been recognized that miRs are involved in most biological events, including cell proliferation, metabolism, apoptosis, cell fate determination, organogenesis, development, stress responses, and tumorigenesis [128]. However, this powerful and broad effect of the classical RNAi mechanism also results in difficulties when studying new functions of miRs, because one must provide direct evidence to separate a functional consequence of any miR new actions from its conventional RNAi mechanism. By exploiting the technical advantage of patch clamping to precisely manipulate and rapidly monitor the function of individual living cells, our study was the first to provide evidence for an intracellular biophysical action of miRs in physiological regulation beyond RNAi mechanism [13]. More expertise and efforts are required from scientists to overcome specific challenges associated with current techniques and develop new approaches that can distinguish the biophysical modulation from the conventional RNAi action. With this concept of miR’s new action, we believe that talented scientists with various expertise should be able to develop new approaches to reveal miRs’ biophysical modulations of other intracellular proteins, including ion channels and transporters. A comprehensive understanding of the dysregulation of ion channels will guide the development of novel and more effective therapeutic approaches, such as anti-arrhythmic therapies for cardiovascular disease.
Acknowledgments
We are grateful to Dr. Jill Dunham for editorial assistance. This research was supported by the National Institutes of Health (NIH)-R01HL139006 (to I.D. and J.D.F.), NIH-R01HL132520 (to I.D.), NIHR01HL096962 (to I.D.). D.Y was funded by the Kenneth M. Rosen Fellowship from the Heart Rhythm Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation, 2021. 143(8): p. e254–743. [DOI] [PubMed] [Google Scholar]
- [2].Colilla S, Crow A, Petkun W, Singer DE, Simon T, Liu X. Estimates of current and future incidence and prevalence of atrial fibrillation in the U.S. adult population. Am J Cardiol, 2013. 112(8): p. 1142–7. [DOI] [PubMed] [Google Scholar]
- [3].Krijthe BP, Kunst A, Benjamin EJ, Lip GY, Franco OH, Hofman A et al. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur Heart J, 2013. 34(35): p. 2746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Antzelevitch C, Burashnikov A. Overview of Basic Mechanisms of Cardiac Arrhythmia. Card Electrophysiol Clin, 2011. 3(1): p. 23–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Grant AO. Cardiac ion channels. Circ Arrhythm Electrophysiol, 2009. 2(2): p. 185–94. [DOI] [PubMed] [Google Scholar]
- [6].Hata A. Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol, 2013. 75: p. 69–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Barwari T, Joshi A, Mayr M. MicroRNAs in Cardiovascular Disease. J Am Coll Cardiol, 2016. 68(23): p. 2577–84. [DOI] [PubMed] [Google Scholar]
- [8].Kalayinia S, Arjmand F, Maleki M, Malakootian M, Singh CP. MicroRNAs: roles in cardiovascular development and disease. Cardiovasc Pathol, 2021. 50: p. 107296. [DOI] [PubMed] [Google Scholar]
- [9].Wang J, Chen J, Sen S. MicroRNA as Biomarkers and Diagnostics. J Cell Physiol, 2016. 231(1): p. 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kaur A, Mackin ST, Schlosser K, Wong FL, Elharram M, Delles C et al. Systematic review of microRNA biomarkers in acute coronary syndrome and stable coronary artery disease. Cardiovasc Res, 2020. 116(6): p. 1113–24. [DOI] [PubMed] [Google Scholar]
- [11].van Rooij E, Purcell AL, Levin AA. Developing microRNA therapeutics. Circ Res, 2012. 110(3): p. 496–507. [DOI] [PubMed] [Google Scholar]
- [12].Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov, 2014. 13(8): p. 622–38. [DOI] [PubMed] [Google Scholar]
- [13].Yang D, Wan X, Dennis AT, Bektik E, Wang Z, Costa M et al. MicroRNA Biophysically Modulates Cardiac Action Potential by Direct Binding to Ion Channel. Circulation, 2021. 143(16): p. 1597–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Z. The role of microRNA in cardiac excitability. J Cardiovasc Pharmacol, 2010. 56(5): p. 460–70. [DOI] [PubMed] [Google Scholar]
- [15].Kim GH. MicroRNA regulation of cardiac conduction and arrhythmias. Transl Res, 2013. 161(5): p. 381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luo X, Yang B, Nattel S. MicroRNAs and atrial fibrillation: mechanisms and translational potential. Nat Rev Cardiol, 2015. 12(2): p. 80–90. [DOI] [PubMed] [Google Scholar]
- [17].Hanieh A, David DJ. Apert’s syndrome. Childs Nerv Syst, 1993. 9(5): p. 289–91. [DOI] [PubMed] [Google Scholar]
- [18].Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res, 2019. 47(D1): p. D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Okada C, Yamashita E, Lee SJ, Shibata S, Katahira J, Nakagawa A et al. A high-resolution structure of the pre-microRNA nuclear export machinery. Science, 2009. 326(5957): p. 1275–9. [DOI] [PubMed] [Google Scholar]
- [20].Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature, 2004. 432(7014): p. 231–5. [DOI] [PubMed] [Google Scholar]
- [21].Nelson HB, Laughon A. The DNA binding specificity of the Drosophila fushi tarazu protein: a possible role for DNA bending in homeodomain recognition. New Biol, 1990. 2(2): p. 171–8. [PubMed] [Google Scholar]
- [22].O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne), 2018. 9: p. 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet, 2011. 12(2): p. 99–110. [DOI] [PubMed] [Google Scholar]
- [24].Ipsaro JJ, Joshua-Tor L. From guide to target: molecular insights into eukaryotic RNA-interference machinery. Nat Struct Mol Biol, 2015. 22(1): p. 20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5’ UTR as in the 3’ UTR. Proc Natl Acad Sci USA, 2007. 104(23): p. 9667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Orom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5’UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell, 2008. 30(4): p. 460–71. [DOI] [PubMed] [Google Scholar]
- [27].Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature, 2008. 455(7216): p. 1124–8. [DOI] [PubMed] [Google Scholar]
- [28].Friedrich M, Vaxevanis CK, Biehl K, Mueller A, Seliger B. Targeting the coding sequence: opposing roles in regulating classical and non-classical MHC class I molecules by miR-16 and miR-744. J Immunother Cancer, 2020. 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA, 2008. 105(5): p. 1608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim DH, Saetrom P, Snove OJ, Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci USA, 2008. 105(42): p. 16230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Younger ST, Corey DR. Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids Res, 2011. 39(13): p. 5682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science, 2007. 318(5858): p. 1931–4. [DOI] [PubMed] [Google Scholar]
- [33].Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell, 2014. 158(3): p. 607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J et al. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci, 2012. 15(6): p. 827–35. [DOI] [PubMed] [Google Scholar]
- [35].Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ et al. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron, 2014. 82(1): p. 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA, 2012. 109(31): p. E2110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Han Q, Liu D, Convertino M, Wang Z, Jiang C, Kim YH et al. miRNA-711 Binds and Activates TRPA1 Extracellularly to Evoke Acute and Chronic Pruritus. Neuron, 2018. 99(3): p. 449–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Luo X, Zhang H, Xiao J, Wang Z. Regulation of human cardiac ion channel genes by microRNAs: theoretical perspective and pathophysiological implications. Cell Physiol Biochem, 2010. 25(6): p. 571–86. [DOI] [PubMed] [Google Scholar]
- [39].Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics, 2007. 8: p. 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wilson KD, Hu S, Venkatasubrahmanyam S, Fu JD, Sun N, Abilez OJ et al. Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: role for miR-499. Circ Cardiovasc Genet, 2010. 3(5): p. 426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fu JD, Rushing SN, Lieu DK, Chan CW, Kong CW, Geng L et al. Distinct roles of microRNA-1 and −499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PLoS One, 2011. 6(11): p. e27417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bartos DC, Grandi E, Ripplinger CM. Ion Channels in the Heart. Compr Physiol, 2015. 5(3): p. 1423–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Van Mierop LH. Location of pacemaker in chick embryo heart at the time of initiation of heartbeat. Am J Physiol, 1967. 212(2): p. 407–15. [DOI] [PubMed] [Google Scholar]
- [44].Moorman AF, de Jong F, Denyn MM, Lamers WH. Development of the cardiac conduction system. Circ Res, 1998. 82(6): p. 629–44. [DOI] [PubMed] [Google Scholar]
- [45].Vassalle M. The pacemaker current (I(f)) does not play an important role in regulating SA node pacemaker activity. Cardiovasc Res, 1995. 30(2): p. 309–10. [PubMed] [Google Scholar]
- [46].Bogdanov KY, Maltsev VA, Vinogradova TM, Lyashkov AE, Spurgeon HA, Stern MD et al. Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ Res, 2006. 99(9): p. 979–87. [DOI] [PubMed] [Google Scholar]
- [47].Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev, 2008. 88(3): p. 919–82. [DOI] [PubMed] [Google Scholar]
- [48].Qu J, Barbuti A, Protas L, Santoro B, Cohen IS, Robinson RB. HCN2 overexpression in newborn and adult ventricular myocytes: distinct effects on gating and excitability. Circ Res, 2001. 89(1): p. E8–14. [DOI] [PubMed] [Google Scholar]
- [49].Miake J, Marban E, Nuss HB. Biological pacemaker created by gene transfer. Nature, 2002. 419(6903): p. 132–3. [DOI] [PubMed] [Google Scholar]
- [50].Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T et al. Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol, 2013. 6(1): p. 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sun Y, Timofeyev V, Dennis A, Bektik E, Wan X, Laurita KR et al. A Singular Role of IK1 Promoting the Development of Cardiac Automaticity during Cardiomyocyte Differentiation by IK1 - Induced Activation of Pacemaker Current. Stem Cell Rev Rep, 2017. 13(5): p. 631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lakatta EG, Vinogradova T, Lyashkov A, Sirenko S, Zhu W, Ruknudin A et al. The integration of spontaneous intracellular Ca2+ cycling and surface membrane ion channel activation entrains normal automaticity in cells of the heart’s pacemaker. Ann N Y Acad Sci, 2006. 1080: p. 178–206. [DOI] [PubMed] [Google Scholar]
- [53].Maltsev VA, Lakatta EG. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res, 2008. 77(2): p. 274–84. [DOI] [PubMed] [Google Scholar]
- [54].Vinogradova TM, Lakatta EG. Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J Mol Cell Cardiol, 2009. 47(4): p. 456–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Petkova M, Atkinson AJ, Yanni J, Stuart L, Aminu AJ, Ivanova AD et al. Identification of Key Small Non-Coding MicroRNAs Controlling Pacemaker Mechanisms in the Human Sinus Node. J Am Heart Assoc, 2020. 9(20): p. e16590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].D’Souza A, Pearman CM, Wang Y, Nakao S, Logantha S, Cox C et al. Targeting miR-423-5p Reverses Exercise Training-Induced HCN4 Channel Remodeling and Sinus Bradycardia. Circ Res, 2017. 121(9): p. 1058–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yanni J, D’Souza A, Wang Y, Li N, Hansen BJ, Zakharkin SO et al. Silencing miR-370-3p rescues funny current and sinus node function in heart failure. Sci Rep, 2020. 10(1): p. 11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Benzoni P, Nava L, Giannetti F, Guerini G, Gualdoni A, Bazzini C et al. Dual role of miR-1 in the development and function of sinoatrial cells. J Mol Cell Cardiol, 2021. 157: p. 104–12. [DOI] [PubMed] [Google Scholar]
- [59].Diaz RJ, Zobel C, Cho HC, Batthish M, Hinek A, Backx PH et al. Selective inhibition of inward rectifier K+ channels (Kir2.1 or Kir2.2) abolishes protection by ischemic preconditioning in rabbit ventricular cardiomyocytes. Circ Res, 2004. 95(3): p. 325–32. [DOI] [PubMed] [Google Scholar]
- [60].Girmatsion Z, Biliczki P, Bonauer A, Wimmer-Greinecker G, Scherer M, Moritz A et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm, 2009. 6(12): p. 1802–9. [DOI] [PubMed] [Google Scholar]
- [61].Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE et al. miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res, 2009. 104(4): p. 514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Drawnel FM, Wachten D, Molkentin JD, Maillet M, Aronsen JM, Swift F et al. Mutual antagonism between IP(3)RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J Cell Biol, 2012. 199(5): p. 783–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brackenbury WJ, Isom LL. Na Channel beta Subunits: Overachievers of the Ion Channel Family. Front Pharmacol, 2011. 2: p. 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Daimi H, Lozano-Velasco E, Haj KA, Chibani JB, Barana A, Amoros I et al. Regulation of SCN5A by microRNAs: miR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm, 2015. 12(6): p. 1333–42. [DOI] [PubMed] [Google Scholar]
- [65].Zhao Y, Huang Y, Li W, Wang Z, Zhan S, Zhou M et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim Biophys Acta, 2015. 1852(10 Pt A): p. 2024–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kang GJ, Xie A, Liu H, Dudley SJ. MIR448 antagomir reduces arrhythmic risk after myocardial infarction by upregulating the cardiac sodium channel. JCI Insight, 2020. 5(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhang X, Yoon JY, Morley M, McLendon JM, Mapuskar KA, Gutmann R et al. A common variant alters SCN5A-miR-24 interaction and associates with heart failure mortality. J Clin Invest, 2018. 128(3): p. 1154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Carafoli E, Santella L, Branca D, Brini M. Generation, control, and processing of cellular calcium signals. Crit Rev Biochem Mol Biol, 2001. 36(2): p. 107–260. [DOI] [PubMed] [Google Scholar]
- [69].Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol, 2000. 16: p. 521–55. [DOI] [PubMed] [Google Scholar]
- [70].Takahashi M, Catterall WA. Dihydropyridine-sensitive calcium channels in cardiac and skeletal muscle membranes: studies with antibodies against the alpha subunits. Biochemistry, 1987. 26(17): p. 5518–26. [DOI] [PubMed] [Google Scholar]
- [71].Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation, 2010. 122(23): p. 2378–87. [DOI] [PubMed] [Google Scholar]
- [72].Barana A, Matamoros M, Dolz-Gaiton P, Perez-Hernandez M, Amoros I, Nunez M et al. Chronic atrial fibrillation increases microRNA-21 in human atrial myocytes decreasing L-type calcium current. Circ Arrhythm Electrophysiol, 2014. 7(5): p. 861–8. [DOI] [PubMed] [Google Scholar]
- [73].Ling TY, Wang XL, Chai Q, Lu T, Stulak JM, Joyce LD et al. Regulation of cardiac CACNB2 by microRNA-499: Potential role in atrial fibrillation. BBA Clin, 2017. 7: p. 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wang J, Ye Q, Bai S, Chen P, Zhao Y, Ma X et al. Inhibiting microRNA-155 attenuates atrial fibrillation by targeting CACNA1C. J Mol Cell Cardiol, 2021. 155: p. 58–65. [DOI] [PubMed] [Google Scholar]
- [75].Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol, 2011. 18(7): p. 840–5. [DOI] [PubMed] [Google Scholar]
- [76].Delahunt B, Nacey JN. Renal cell carcinoma. II. Histological indicators of prognosis. Pathology, 1987. 19(3): p. 258–63. [DOI] [PubMed] [Google Scholar]
- [77].Zhang J, Wei F, Ding L, Wang L, Zhang X, Yu L et al. MicroRNA-1976 regulates degeneration of the sinoatrial node by targeting Cav1.2 and Cav1.3 ion channels. J Mol Cell Cardiol, 2019. 134: p. 74–85. [DOI] [PubMed] [Google Scholar]
- [78].Chu Q, Li A, Chen X, Qin Y, Sun X, Li Y et al. Overexpression of miR-135b attenuates pathological cardiac hypertrophy by targeting CACNA1C. Int J Cardiol, 2018. 269: p. 235–41. [DOI] [PubMed] [Google Scholar]
- [79].An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature, 2000. 403(6769): p. 553–6. [DOI] [PubMed] [Google Scholar]
- [80].Calvet JM, Sostenes C, Le Rebeller MJ. [Our experience with several types of anterior chamber implants]. Bull Soc Ophtalmol Fr, 1985. 85(6–7): p. 765–7. [PubMed] [Google Scholar]
- [81].Butler J, Attiyeh FF, Daly JM. Hepatic resection for metastases of the colon and rectum. Surg Gynecol Obstet, 1986. 162(2): p. 109–13. [PubMed] [Google Scholar]
- [82].Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell, 2007. 129(2): p. 303–17. [DOI] [PubMed] [Google Scholar]
- [83].Costantini DL, Arruda EP, Agarwal P, Kim KH, Zhu Y, Zhu W et al. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell, 2005. 123(2): p. 347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Panguluri SK, Tur J, Chapalamadugu KC, Katnik C, Cuevas J, Tipparaju SM. MicroRNA-301a mediated regulation of Kv4.2 in diabetes: identification of key modulators. PLoS One, 2013. 8(4): p. e60545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liu X, Zhang Y, Du W, Liang H, He H, Zhang L et al. MiR-223-3p as a Novel MicroRNA Regulator of Expression of Voltage-Gated K+ Channel Kv4.2 in Acute Myocardial Infarction. Cell Physiol Biochem, 2016. 39(1): p. 102–14. [DOI] [PubMed] [Google Scholar]
- [86].Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res, 2010. 106(1): p. 166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Xiao L, Xiao J, Luo X, Lin H, Wang Z, Nattel S. Feedback remodeling of cardiac potassium current expression: a novel potential mechanism for control of repolarization reserve. Circulation, 2008. 118(10): p. 983–92. [DOI] [PubMed] [Google Scholar]
- [88].Shan H, Zhang Y, Cai B, Chen X, Fan Y, Yang L et al. Upregulation of microRNA-1 and microRNA-133 contributes to arsenic-induced cardiac electrical remodeling. Int J Cardiol, 2013. 167(6): p. 2798–805. [DOI] [PubMed] [Google Scholar]
- [89].Jia X, Zheng S, Xie X, Zhang Y, Wang W, Wang Z et al. MicroRNA-1 accelerates the shortening of atrial effective refractory period by regulating KCNE1 and KCNB2 expression: an atrial tachypacing rabbit model. PLoS One, 2013. 8(12): p. e85639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest, 2013. 123(5): p. 1939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Goldoni D, Yarham JM, McGahon MK, O’Connor A, Guduric-Fuchs J, Edgar K et al. A novel dual-fluorescence strategy for functionally validating microRNA targets in 3’ untranslated regions: regulation of the inward rectifier potassium channel K(ir)2.1 by miR-212. Biochem J, 2012. 448(1): p. 103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med, 2007. 13(4): p. 486–91. [DOI] [PubMed] [Google Scholar]
- [93].Nassal DM, Wan X, Liu H, Maleski D, Ramirez-Navarro A, Moravec CS et al. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bernstein SA, Morley GE. Gap junctions and propagation of the cardiac action potential. Adv Cardiol, 2006. 42: p. 71–85. [DOI] [PubMed] [Google Scholar]
- [95].Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res, 2000. 86(12): p. 1193–7. [DOI] [PubMed] [Google Scholar]
- [96].Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation, 1997. 95(4): p. 988–96. [DOI] [PubMed] [Google Scholar]
- [97].Yao JA, Hussain W, Patel P, Peters NS, Boyden PA, Wit AL. Remodeling of gap junctional channel function in epicardial border zone of healing canine infarcts. Circ Res, 2003. 92(4): p. 437–43. [DOI] [PubMed] [Google Scholar]
- [98].Xu HF, Ding YJ, Shen YW, Xue AM, Xu HM, Luo CL et al. MicroRNA- 1 represses Cx43 expression in viral myocarditis. Mol Cell Biochem, 2012. 362(1–2): p. 141–8. [DOI] [PubMed] [Google Scholar]
- [99].Jin Y, Zhou T, Feng Q, Yang J, Cao J, Xu X et al. Inhibition of MicroRNA-206 Ameliorates Ischemia-Reperfusion Arrhythmia in a Mouse Model by Targeting Connexin43. J Cardiovasc Transl Res, 2020. 13(4): p. 584–92. [DOI] [PubMed] [Google Scholar]
- [100].Wang L, Li Q, Diao J, Lin L, Wei J. MiR-23a Is Involved in Myocardial Ischemia/Reperfusion Injury by Directly Targeting CX43 and Regulating Mitophagy. Inflammation, 2021. 44(4): p. 1581–91. [DOI] [PubMed] [Google Scholar]
- [101].Yin VP, Lepilina A, Smith A, Poss KD. Regulation of zebrafish heart regeneration by miR-133. Dev Biol, 2012. 365(2): p. 319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Danielson LS, Park DS, Rotllan N, Chamorro-Jorganes A, Guijarro MV, Fernandez-Hernando C et al. Cardiovascular dysregulation of miR-17-92 causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis. FASEB J, 2013. 27(4): p. 1460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Osbourne A, Calway T, Broman M, McSharry S, Earley J, Kim GH. Downregulation of connexin43 by microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J Mol Cell Cardiol, 2014. 74: p. 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest, 2009. 119(9): p. 2772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Li S, Jiang Z, Wen L, Feng G, Zhong G. MicroRNA-208a-3p contributes to connexin40 remolding in human chronic atrial fibrillation. Exp Ther Med, 2017. 14(6): p. 5355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Takahashi K, Sasano T, Sugiyama K, Kurokawa J, Tamura N, Soejima Y et al. High-fat diet increases vulnerability to atrial arrhythmia by conduction disturbance via miR-27b. J Mol Cell Cardiol, 2016. 90: p. 38–46. [DOI] [PubMed] [Google Scholar]
- [107].Janas MM, Wang B, Harris AS, Aguiar M, Shaffer JM, Subrahmanyam YV et al. Alternative RISC assembly: binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA, 2012. 18(11): p. 2041–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Flores O, Kennedy EM, Skalsky RL, Cullen BR. Differential RISC association of endogenous human microRNAs predicts their inhibitory potential. Nucleic Acids Res, 2014. 42(7): p. 4629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Stalder L, Heusermann W, Sokol L, Trojer D, Wirz J, Hean J et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J, 2013. 32(8): p. 1115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Dragomir MP, Knutsen E, Calin GA. SnapShot: Unconventional miRNA Functions. Cell, 2018. 174(4): p. 1038. [DOI] [PubMed] [Google Scholar]
- [111].Bissels U, Wild S, Tomiuk S, Holste A, Hafner M, Tuschl T et al. Absolute quantification of microRNAs by using a universal reference. RNA, 2009. 15(12): p. 2375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Sung JH, Kim SH, Yang WI, Kim WJ, Moon JY, Kim IJ et al. miRNA polymorphisms (miR146a, miR149, miR196a2 and miR499) are associated with the risk of coronary artery disease. Mol Med Rep, 2016. 14(3): p. 2328–42. [DOI] [PubMed] [Google Scholar]
- [113].Guo R, Feng Z, Yang Y, Xu H, Zhang J, Guo K et al. Association of a MiR-499 SNP and risk of congenital heart disease in a Chinese population. Cell Mol Biol (Noisy-le-grand), 2018. 64(10): p. 108–12. [PubMed] [Google Scholar]
- [114].Yu K, Ji Y, Wang H, Xuan QK, Li BB, Xiao JJ et al. Association of miR-196a2, miR-27a, and miR-499 polymorphisms with isolated congenital heart disease in a Chinese population. Genet Mol Res, 2016. 15(4). [DOI] [PubMed] [Google Scholar]
- [115].Gottwein E, Cai X, Cullen BR. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol, 2006. 80(11): p. 5321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hu Z, Liang J, Wang Z, Tian T, Zhou X, Chen J et al. Common genetic variants in pre-microRNAs were associated with increased risk of breast cancer in Chinese women. Hum Mutat, 2009. 30(1): p. 79–84. [DOI] [PubMed] [Google Scholar]
- [117].Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci USA, 2008. 105(20): p. 7269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum Mol Genet, 2007. 16(9): p. 1124–31. [DOI] [PubMed] [Google Scholar]
- [119].He S, Ou H, Zhao C,Zhang J, Clustering Pattern and Functional Effect of SNPs in Human miRNA Seed Regions. Int J Genomics, 2018. 2018: p. 2456076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hasegawa K, Ohno S, Ashihara T, Itoh H, Ding WG,Toyoda F et al. , A novel KCNQ1 missense mutation identified in a patient with juvenile-onset atrial fibrillation causes constitutively open IKs channels. Heart Rhythm, 2014. 11(1): p. 67–75. [DOI] [PubMed] [Google Scholar]
- [121].Hattori T, Makiyama T, Akao M, Ehara E, Ohno S,Iguchi M et al. , A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res, 2012. 93(4): p. 666–73. [DOI] [PubMed] [Google Scholar]
- [122].Ikeda S, Kong SW, Lu J, Bisping E, Zhang H,Allen PD et al. , Altered microRNA expression in human heart disease. Physiol Genomics, 2007. 31(3): p. 367–73. [DOI] [PubMed] [Google Scholar]
- [123].Cheng Y, Ji R, Yue J, Yang J, Liu X,Chen H et al. , MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol, 2007. 170(6): p. 1831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF,Newman M et al. , Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol, 2007. 42(6): p. 1137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S,van Laake LW et al. , MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation, 2007. 116(3): p. 258–67. [DOI] [PubMed] [Google Scholar]
- [126].Statello L, Guo CJ, Chen LL,Huarte M, Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol, 2021. 22(2): p. 96–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Seok H, Lee H, Lee S, Ahn SH, Lee HS,Kim GD et al. , Position-specific oxidation of miR-1 encodes cardiac hypertrophy. Nature, 2020. 584(7820): p. 279–85. [DOI] [PubMed] [Google Scholar]
- [128].Bartel DP, MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 2004. 116(2): p. 281–97. [DOI] [PubMed] [Google Scholar]