

Abstract
eIF3a (eukaryotic translation initiation factor 3a), a subunit of the eIF3 complex, has been suggested to play a regulatory role in protein synthesis and in cellular response to DNA-damaging treatments. S6K1 is an effector and a mediator of mTOR complex1 (mTORC1) in regulating protein synthesis and integrating diverse signals into control of cell growth and response to stress. Here, we show that eIF3a regulates S6K1 activity by inhibiting mTORC1 kinase via regulating Raptor synthesis. The regulation of Raptor synthesis is via eIF3a interaction with HuR (human antigen R) and binding of the eIF3a-HuR complex to the 5’-UTR of Raptor mRNA. Furthermore, mTORC1 may mediate eIF3a function in cellular response to cisplatin by regulating synthesis of NER proteins and NER activity. Taken together, we conclude that the mTOR signaling pathway may also be regulated by translational control and mediate eIF3a regulation of cancer cell response to cisplatin by regulating NER protein synthesis.
Keywords: eIF3a, protein synthesis, mTOR complex1, human antigen R, nucleotide excision repair
Introduction
Translational control is an important regulation in gene expression and occurs mainly in the initiation step involving multiple eukaryotic translation initiation factors (eIFs)1, 2. Of all eIFs, eIF3 is the largest complex consisting of 13 subunits (eIF3a-eIF3m) and plays important roles in keeping 40S ribosome from reassociation with the 60S ribosome and in recruiting a multiple-factor complex consisting of eIF1, eIF3, eIF5, and the ternary complex to form the 43S preinitiation complex2. eIF3a, the largest subunit in eIF3, is very unusual and has been suggested to have non-canonical functions in regulating translation of a subset of mRNAs in addition to participating as a subunit in eIF3 complex in global translation initiation2.
The biological significances of eIF3a in cancer disease have also been recognized, including tumorigenesis3 and prognosis4. eIF3a has been shown to cause cellular sensitivity to DNA-damaging treatments including cisplatin5, 6, doxorubicin5, 7 and ionizing radiation8, which may contribute to tumorigenesis and prognosis. These observations have been ascribed to the non-canonical function of eIF3a in suppressing synthesis of DNA repair proteins important for nucleotide excision repair (NER) of DNA crosslink adducts induced by cisplatin5, 6 and for non-homologous end joining (NHEJ) repair of double strand DNA breaks induced by doxorubicin and radiation7, 8. However, the underlying mechanism in eIF3a regulation of DNA repair protein synthesis is not yet fully understood.
The mammalian target of rapamycin (mTOR) is an evolutionarily conserved protein kinase, exists in two structurally and functionally distinct multiprotein complexes, mTOR complex1 (mTORC1) and 2 (mTORC2)9–14. mTORC1, which contains mTOR, regulatory associated protein of mTOR (Raptor), G protein β-subunit-like (GβL), proline-rich Akt substrate 40 kDa (PRAS40), and DEP domain-containing mTOR-interacting protein (DEPTOR), is a sensor of intracellular amino acid level and regulates a series of biological processes including protein synthesis and cellular response to stress such as DNA damages15–23. mTORC1 regulates protein synthesis by phosphorylating S6 kinases 1 and 2 (S6K1 and 2), and eIF4E-binding proteins 1, 2, and 3 (4EBP1, 2, and 3), while mTORC2 phosphorylates the hydrophobic motif of Akt (Akt1, 2, and 3), SGK1, and PKCα24.
S6K belongs to the AGC kinase family and regulates protein synthesis, cell survival and metabolism25, 26 with S6K1 phosphorylating multiple substrates including the cAMP-responsive activator CREM and ribosomal protein S616. Raptor, is an important signal acceptor27–32 and essential for mTOR activity10, 33. mTORC1-activating stimuli including growth factors, amino acids, and cellular energy promotes rapamycin-sensitive raptor phosphorylation, which is required for mTORC1 activation34.
In this study, we tested the hypothesis that mTORC1 mediates eIF3a regulation of cellular response to DNA damages and synthesis of NER proteins. We show that eIF3a negatively regulates Raptor synthesis, resulting in changes in mTORC1 activity and cellular response to cisplatin by binding to the Raptor mRNA 5’-UTR via interaction with the RNA-binding protein HuR. Our findings reveal a new mechanism of eIF3a regulation of mTORC1 activity by controlling Raptor synthesis in collaboration with HuR and of cellular response to cisplatin via mTORC1 regulation of NER protein synthesis.
Results
eIF3a negatively regulates mTORC1 activity.
To explore the translational regulation of mTOR signaling, we first determined mTORC1 status following eIF3a knockdown in two different cancer cell lines H1299 and A549. As shown in Fig. 1A–B, eIF3a knockdown significantly upregulated mTOR phosphorylation at Ser2448 without affecting the level of total mTOR in these cells.
Figure 1. eIF3a knockdown increases mTORC1 activity.

(A-F). Effect of eIF3a knockdown on mTORC1 and its downstream target proteins. Lysates from H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si(3a)) siRNA were subjected to immunoblot analyses of eIF3a, mTOR, and pS2448mTOR (A-B), eIF3a, 4EBP1, and pT37/464EBP1 (C-D), eIF3a, S6K1, pT389S6K1, S6, and pS240/244S6 (E-F). Actin was used as a loading control. (G-H). In-vitro mTORC1 kinase activity assay. mTOR was immunoprecipitated from H1299 cells transfected with scrambled control (Scr) or eIF3a (Si(3a)) siRNA and used for in-vitro kinase activity assay with purified recombinant S6K1 as substrates. The reaction was separated by SDS-PAGE followed by immunoblot analysis of mTOR, S6K1, and pT389S6K1. Panels B, D, F, and H show quantifications of protein intensity in immunoblots shown in panels A, C, E, and G (n=3, **p< 0.01, ***p< 0.001).
To determine if the change in mTOR phosphorylation following eIF3a knockdown alters mTORC1 activity, we analyzed the status of its downstream target proteins, 4EBP1 and S6K1. As shown in Fig. 1C–D, eIF3a knockdown significantly increased the level of pT37/464EBP with little effect on the total 4EBP1 level in both H1299 and A549 cells. Fig. 1E–F show that the level of both pT389S6K1 at and its target protein pS240/244S6 is also increased by eIF3a knockdown in both cell lines.
To ensure that the above observations were due to specific effect of eIF3a knockdown and to eliminate the possibility that the changes in mTOR activation was due to peculiar effect of scrambled control siRNAs, we examined the parental untransfected cells in comparison with scrambled control and eIF3a knockdown cells. As shown in Supplementary Fig. S1, only cells with eIF3a knockdown had upregulation in phosphorylation in pSer2448mTOR, (Fig. S1A), pT37/464EBP1 (Fig. S1B), and pT389S6K1 and pS240/244S6 (Fig. S1C). There is no difference in the level of these phosphorylated proteins between the untrasfected and scrambled siRNA-transfected cells.
To further ensure scientific rigor, we tested two additional siRNAs (#2 and #3) with different eIF3a-targeting sequences. As shown in Supplementary Fig. S2A, both siRNA#2 and #3 successfully knocked down eIF3a expression, which led to increased phosphorylation of pS2448mTOR, pT37/464EBP1, pT389S6K1, and pS240/244S6, similar to the first eIF3a siRNA. Thus, eIF3a knockdown likely increases mTORC1 activity, resulting in a cascade activation of its downstream mediators.
To confirm above findings, we performed an in-vitro mTORC1 kinase activity assay using immunoprecipitated mTORC1 from H1299 cells with eIF3a knockdown and recombinant S6K1 as a substrate. As shown in Fig. 1G–H, mTORC1 from cells with eIF3a knockdown significantly increased pT389S6K1 compared with that from cells harboring control siRNAs. Consistent with this result, mTORC1 from H1299 cells with eIF3a knockdown also significantly increased phosphorylation of recombinant 4EBP1 at Thr37/46 compared with that from cells harboring control siRNAs (Supplementary Fig. S2B–C). Thus, eIF3a knockdown promotes mTORC1 kinase activity.
eIF3a regulation of mTORC1 subunits.
Since mTORC1 consists of mTOR, Raptor, GβL, PRAS40 and DEPTOR, which affect mTORC1 activity, it is of interest to determine if eIF3a regulates the expression of other components in relationship with regulation of mTORC1 activity. For this purpose, we first knocked down eIF3a and tested the expression of Raptor, PRAS40, GβL, and DEPTOR in H1299 and A549 cells using immunoblot analysis. As shown in Fig. 2A–B, eIF3a knockdown increased Raptor, PRAS40 and GβL expression but reduced DEPTOR expression in both cell lines.
Figure 2. eIF3a regulates expression of other mTORC1 subunits.

(A-B). Effect of eIF3a knockdown on the expression of Raptor, DEPTOR, PRAS40, GβL. H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si(3a)) siRNA were subjected to immunoblot analysis of eIF3a, Raptor, DEPTOR, PRAS40, GβL and actin loading control. (C-F). Effect of eIF3a knockdown on mTORC1 subunits in the complex. Lysates from H1299 cells transfected with scrambled control (Scr) or eIF3a (Si(3a)) siRNA were subjected to co-immunoprecipitation with mTOR (C-D) or Raptor (E-F) antibody or control normal IgG (nIgG) followed by immunoblot analyses of PRAS40, GβL, and mTOR or Raptor in the precipitate. The precipitates along with input were separated by SDS-PAGE followed by immunoblot analyses of PRAS40, GβL, mTOR and Raptor. Panels B, D, and F show quantifications of protein intensity in immunoblots in panels A, C, and E (n=3, **p< 0.01, ***p<0.001).
We next immunoprecipitated mTOR and detected Raptor, PRAS40, DEPTOR, and GβL in the precipitate. As shown in Fig. 2C–D, Raptor, PRAS40, GβL were all increased in the precipitate of cells with eIF3a knockdown. However, the increase in PRAS40 and GβL is less than that of Raptor. We also immunoprecipitated Raptor for detection of these proteins. As shown in Fig. 2E–F, mTOR, PRAS40 and GβL were also increased in the precipitate of cells with eIF3a knockdown. Unfortunately, we were unable to detect DEPTOR in either mTOR or Raptor precipitates due to unknown reasons. However, it is noteworthy that Raptor was able to co-precipitate similar levels of mTOR, PRAS40, and GβL, while Raptor in mTOR precipitate has a much higher fold increase than other subunits. Thus, Raptor may be a limiting factor for mTORC1 activation under eIF3a regulation.
Raptor mediates eIF3a regulation of mTORC1 activity.
We next focused on Raptor for further study because Raptor expression was increased the most among all mTORC1 subunits following eIF3a knockdown and may be a limiting factor mediating eIF3a regulation of mTORC1. Because its phosphorylation status regulates mTORC1 with phosphorylation of Ser696, Thr706 activating and phosphorylation of Ser792 inhibiting mTORC1 activity32, 34, we first determined the phosphorylation status of these residues of Raptor following eIF3a knockdown using immunoblot analysis. As shown in Fig. 3A–B, eIF3a knockdown significantly reduced the level of pS792Raptor, but not of pS696Raptor and pT706Raptor. Similar results were also observed using different siRNAs targeting eIF3a (Supplementary Fig. S3A), suggesting that the above findings are unlikely due to off-target effects.
Figure 3. Raptor mediates eIF3a regulation of mTORC1 activity.

(A-B). Effect of eIF3a knockdown on expression and phosphorylation of Raptor. Lysates from H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si(3a)) siRNA were subjected to immunoblot analyses of total and phosphorylated Raptors at its Ser792, Thr706, Ser696, eIF3a, and actin loading control. Panel B shows quantification of eIF3a, Raptor, and pSer792, pThr706, pSer696Raptors from panel A. (C) In-vitro mTORC1 kinase activity assay. Lysate from H1299 cells were subjected to immunoprecipitation using Raptor antibody and the precipitate was used for in-vitro kinase activity assay with purified recombinant S6K1 as a substrate followed by SDS-PAGE separation and immunoblot analyses. (D). Quantification of Raptor and pT389S6K1 in panel C (n=3, **p< 0.01, ***p< 0.001). (E). Effect of Raptor and eIF3a double knockdown on mTORC1 activity. Lysate from H1299 and A549 cells transfected with scrambled control (Scr), eIF3a (Si(3a)), Raptor (Si(Rap)) siRNAs or both eIF3a and Raptor siRNAs were subjected to immunoblot analysis of eIF3a, Raptor, S6K1, pT389S6K1, S6, pS240/244S6 and actin loading control.
Because Raptor is an essential component of mTORC1 and eIF3a regulates Raptor expression and activation, we hypothesized that Raptor might mediate eIF3a regulation of mTORC1 activity. To test this hypothesis, we first performed the in-vitro kinase activity assay as described above but using Raptor-co-immunoprecipitated materials. As shown in Fig. 3C–D, mTORC1 kinase activity, as measured by phosphorylation of Thr389 in the recombinant S6K1 substrate, was dramatically increased by eIF3a knockdown. We similarly showed that the mTORC1 kinase activity was significantly increased by eIF3a knockdown using recombinant 4EBP1 as a substrate (Supplementary Fig. S3B–C).
To validate above findings, we took advantage of the stable NIH3T3 cells overexpressing eIF3a5 and tested mTORC1 kinase activity using mTOR and Raptor-immunoprecipitated materials. As shown in Supplementary Fig. S4A–D, the level of pT389S6K1 was significantly reduced in NIH3T3 cells with eIF3a overexpression compared with the vector-transfected control cells. Supplementary Fig. S4E–F show that the protein level of Raptor but not mTOR was significantly reduced in NIH3T3 cells with eIF3a overexpression compared with the control cells. These findings are consistent with the conclusion that eIF3a regulates Raptor expression and mTORC1 activity.
Finally, to determine if Raptor mediates eIF3a regulation of mTORC1, we tested if Raptor knockdown could reverse eIF3a knockdown-induced increase in mTORC1 activity by analyzing phosphorylation of endogenous S6K1 and its substrate S6 in H1299 and A549 cells. Fig. 3E shows that eIF3a knockdown increased while Raptor knockdown eliminated phosphorylation of S6K1 and its downstream target S6. However, simultaneous Raptor and eIF3a knockdown reversed eIF3a knockdown-induced phosphorylation of S6K1 and S6 in both cell lines. Thus, Raptor may mediate eIF3a regulation of mTORC1 activity.
eIF3a regulation of Raptor mRNA translation.
To determine the mechanism of eIF3a regulation of Raptor expression, we first performed a quantitative RT-PCR analysis following eIF3a knockdown in H1299 and A549 cells. As shown in Fig. 4A, eIF3a knockdown did not change the Raptor mRNA level in both cell lines. Similarly, eIF3a overexpression did not change the Raptor mRNA level in NIH3T3 cells (Supplementary Fig. S5A). Thus, eIF3a regulation of Raptor expression is unlikely at its mRNA level.
Figure 4. eIF3a regulates the 5’-UTR activity of Raptor mRNA.
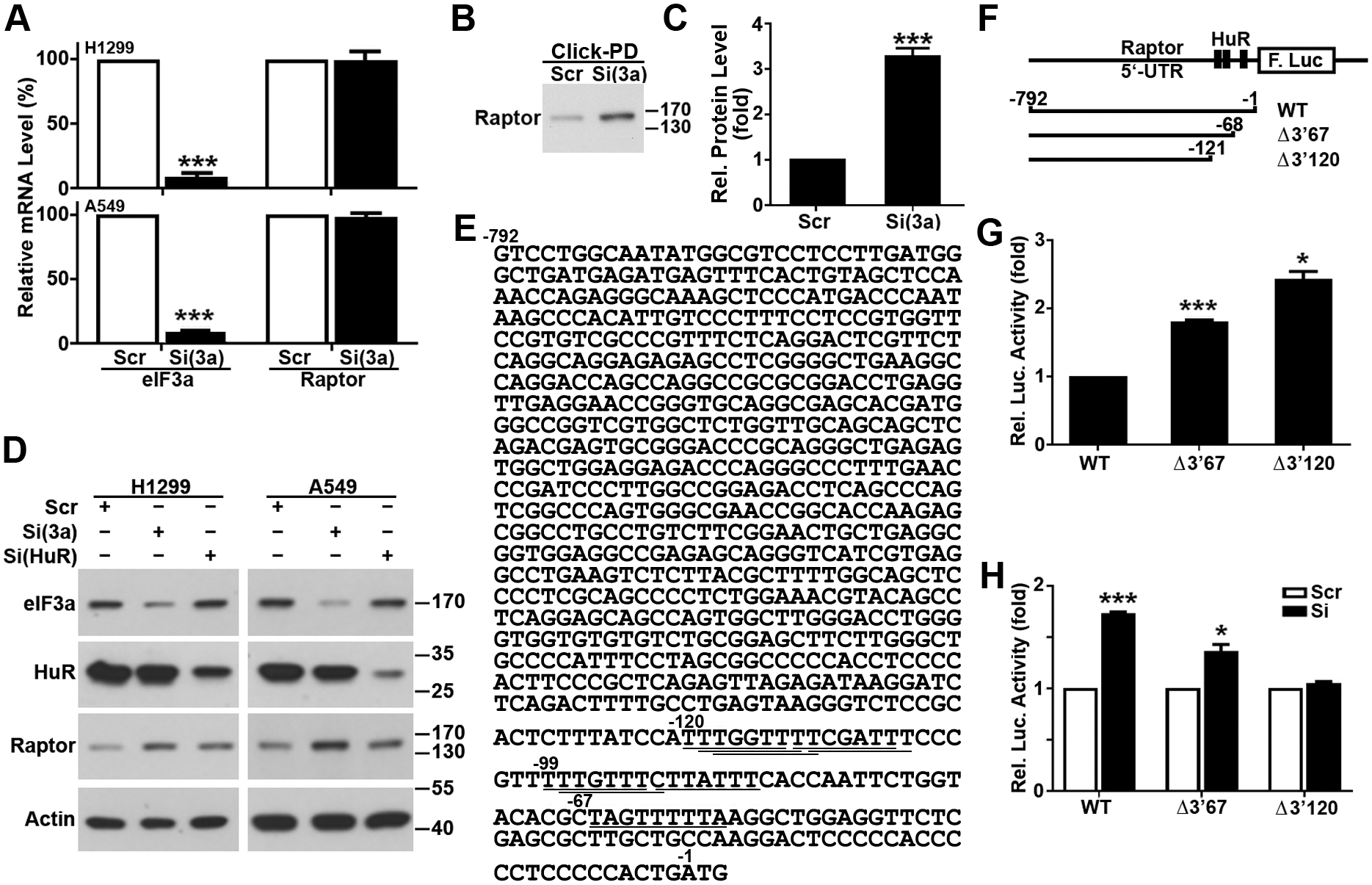
(A) Effect of eIF3a knockdown on the expression of Raptor mRNA. RNAs isolated from H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si) siRNA were used for real-time RT-PCR analysis of eIF3a and Raptor (n=3, ***p<0.001). (B-C) Effect of eIF3a knockdown on Raptor protein synthesis. H1299 cells transfected with eIF3a or scrambled control siRNAs were metabolically labeled with azidohomoalanine followed by Click-PD assay and immunoblot analysis of Raptor. Panel C shows quantifications of protein intensity in panel B (n=3, ***p< 0.001). (D) Effect of HuR knockdown on Raptor expression. Lysates from H1299 and A549 cells transfected with scrambled control (Scr), eIF3a [Si(eIF3a)], or HuR [Si(HuR)] siRNA were subjected to immunoblot analysis of eIF3a, Raptor, HuR and actin loading control. (E). The cDNA sequence encoding the 5’-UTR of Raptor mRNA. The HuR binding sites are shown by underlines. (F) Schematic diagram of reporter constructs with the wild-type and deletion mutant 5’-UTR of Raptor mRNA. (G, H) Luciferase reporter assay. Untreated (G) or eIF3a and scrambled control siRNA-transfected (H) H1299 cells were transiently co-transfected with cRNAs encoding the 5’-UTR and firefly luciferase and cRNAs encoding Renilla luciferase transfection control followed by determination of Renilla and firefly luciferase activities. The 5’-UTR activities were represented by firefly luciferase activity normalized to Renilla luciferase activity (n=3, *p<0.05, ***p<0.001).
We next determined if eIF3a regulates Raptor synthesis using pulse labeling and Click-pull down (Click-PD) assay. As shown in Fig. 4B–C, eIF3a knockdown significantly increased newly-synthesized Raptor protein. We also performed a pulse-chase experiment to determine if eIF3a knockdown changes Raptor stability. As shown in Supplementary Fig. S5B–C, eIF3a knockdown had no effect on the half-life or degradation rate of Raptor protein. Thus, eIF3a likely only regulates Raptor protein synthesis.
Recently, it was found that eIF3a regulated Chk1 synthesis via interaction with HuR that binds to the 3’-UTR of Chk1 mRNA35. To determine if HuR may also mediate eIF3a regulation of Raptor synthesis, we tested if HuR knockdown affects Raptor expression using immunoblot analysis. Similar to eIF3a knockdown, HuR knockdown increased the level of Raptor protein in both H1299 and A549 cells (Fig. 4D).
Sequence analysis of Raptor mRNA showed that the 5’-UTR of Raptor mRNA contains several overlapping putative HuR-binding sites (Fig. 4E). To investigate if these HuR-binding sites possibly function as cis-elements to mediate eIF3a regulation of Raptor mRNA translation, we engineered the 5’-UTR of Raptor mRNA into a reporter plasmid and generated two mutants with deletions of the putative HuR binding sites from the 3’-end of the 5’-UTR (Fig. 4F). Capped cRNAs were transcribed from these constructs and transfected into H1299 cells for determination of luciferase reporter synthesis. As shown in Fig. 4G, removal of 67 bases along with the 3’-end HuR-binding site (Δ3’67) significantly increased the reporter expression. More deletion to remove all putative HuR-binding sites (Δ3’120) further increased reporter expression. Thus, the HuR-binding sites in the 5’-UTR of Raptor mRNA is suppressive to the translation of the reporter gene.
We next transfected these cRNAs into H1299 cells with eIF3a knockdown followed by luciferase reporter assay. As shown in Fig. 4H, eIF3a knockdown significantly increased the reporter expression of the cRNAs with the wild-type 5’-UTR. However, this increase was much less with deletion of the 3’-end HuR binding site (Δ3’67) and was eliminated when all putative HuR-binding sites were deleted (Δ3’120). Consistently, eIF3a overexpression significantly down-regulated reporter expression with the wild-type and the Δ3’67 mutant 5’-UTR but not with the Δ3’120 mutant 5’-UTR (Supplementary Fig. S5D). Hence, eIF3a may reduce the translation of Raptor mRNAs via HuR and its binding site in the 5’-UTR of Raptor mRNA.
eIF3a and HuR binding to the 5’-UTR sequence of Raptor mRNA.
We next determined if eIF3a or HuR possibly bind to the 5’-UTR of Raptor mRNA by performing a pull-down assay of eIF3a and HuR from total cell lysate using biotin-labeled uncapped cRNA probes representing the wild-type, Δ3’67 and Δ3’120 mutant 5’-UTR sequences of Raptor mRNA. Same probes without biotin were used as negative controls. The pull-down materials were then analyzed by immunoblot for eIF3a and HuR. As shown in Fig. 5A, both HuR and eIF3a were successfully pulled down by the wild-type 5’-UTR probe. However, deletion of the HuR binding site at 3’-end (Δ3’67) dramatically reduced HuR and eIF3a binding. Further deletion to remove all HuR binding sites (Δ3’120) completely abolished HuR and eIF3a pulldown. Thus, HuR and eIF3a likely bind to the HuR-binding sites in the 5’-UTR of Raptor mRNA. Furthermore, HuR knockdown eliminated pull-down of both HuR and eIF3a by the wild-type Raptor 5’-UTR probe (Fig. 5B), suggesting that eIF3a likely binds to the Raptor 5’-UTR sequence indirectly via HuR, consistent with our previous observations on eIF3a binding via HuR to the 3’-UTR of Chk1 mRNA35.
Figure 5. HuR binds to the 5’-UTR sequence of Raptor mRNA.

Extracts from H1299 cells (A) or H1299 cells transfected with scrambled control (Scr) or HuR (Si) siRNA (B) or H1299 cells transfected with scrambled control (Scr) or eIF3a (Si) siRNA (C) were incubated with biotin-labeled cRNA probes representing the wild-type (WT) or truncated (Δ3’67, Δ3’120) 5’-UTR sequences of Raptor mRNA. The cRNA–protein complexes were then pulled down using streptavidin-conjugated beads, separated on SDS–PAGE, and subjected to immunoblot analysis of HuR and eIF3a. Probes without biotin were used as negative controls. (D) Effect of eIF3a and HuR knockdown on Raptor expression. Lysates from H1299 cells transfected with scrambled control (Scr), eIF3a [Si(eIF3a)], HuR [Si(HuR)] siRNA or both eIF3a and HuR siRNAs were subjected to immunoblot analysis of eIF3a, Raptor, HuR and actin loading control.
eIF3a regulation of HuR binding and function in controlling Raptor mRNA translation.
With above findings that eIF3a regulates Raptor synthesis via HuR binding to the 5’-UTR of Raptor mRNA, it is tempting to propose that eIF3a binding to HuR may stimulate HuR binding to Raptor mRNA and inhibiting Raptor mRNA translation. To test this hypothesis, we performed pull-down assay of HuR using wild-type 5’-UTR probe of Raptor following eIF3a knockdown. As shown in Fig. 5C, eIF3a knockdown dramatically reduced HuR binding to the wild-type 5’-UTR probe.
To validate this finding, we determined if eIF3a enhances HuR repressive function on Raptor mRNA translation by performing double knockdown. As shown in Fig. 5D, eIF3a or HuR knockdown increased Raptor expression as expected. However, simultaneous eIF3a and HuR knockdown synergistically increased the Raptor protein level. Together with the above findings using pull-down assay, we conclude that eIF3a likely promotes HuR binding to the 5’-UTR of Raptor mRNA and suppresses its translation.
eIF3a regulation of PRAS40.
Because PRAS40 negatively regulates mTORC1 activity36, 37, the finding that eIF3a knockdown increases PRAS40 expression is peculiar and inconsistent with the increased mTORC1 activity following eIF3a knockdown. However, it is also known that the phosphorylation of PRAS40 at Thr246 by Akt relieves PRAS40 inhibition of mTORC1 activity14. We, thus, determined the effect of eIF3a knockdown on pT246PRAS40. As shown in Fig. 6A–B, eIF3a knockdown increased pT246PRAS40 in both H1299 and A549 cells. Thus, PRAS40 up-regulation by eIF3a knockdown may not inhibit mTORC1 activity due to concurrent increase in Thr246 phosphorylation.
Figure 6. eIF3a regulates Akt-mediated PRAS40 phosphorylation.

(A-B). eIF3a regulation of PRAS40 phosphorylation. H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si(3a)) siRNA were subjected to immunoblot analysis of eIF3a, pThr246PRAS40 and actin loading control. Panel B shows quantifications of protein intensity in panel A (n=3, ***p<0.001). (C) Effect of eIF3a and Akt double knockdown or eIF3a knockdown and MK2206 treatment on mTORC1 activity. Lysate from H1299 cells transfected with scrambled control (Scr), eIF3a (Si(3a)), Akt (Si(Akt)) siRNA, or both eIF3a and Akt siRNAs or both eIF3a siRNA and MK2206 treatment were subjected to immunoblot analysis of eIF3a, Akt, pS473Akt, PRAS40, pT246PRAS40, S6K1, pT389S6K1 and actin loading control. (D). The cDNA sequence encoding the 5’-UTR of PRAS40 mRNA. The HuR binding sites are underlined. (E). Effect of HuR knockdown on PRAS40 expression. Lysates from H1299 and A549 cells transfected with scrambled control (Scr), eIF3a (Si(3a)), or HuR (Si(HuR)) siRNA were subjected to immunoblot analysis of eIF3a, HuR, PRAS40 and actin loading control. (F). Schematic model of eIF3a regulates mRNA translation via HuR binding to and blocking the pre-initiation complex (PIC) scanning of the 5’-UTR.
Because Akt phosphorylates PRAS40 at Thr246 37 and Akt-mediated phosphorylation of PRAS40 relieves its inhibition of mTORC1 activation38, we tested the possibility that eIF3a may regulate Akt activation that in turn mediates eIF3a regulation of PRAS40 and mTORC1. As shown in Fig. 6C, eIF3a knockdown indeed increased Akt activation (phosphorylation of Ser473) and phosphorylation of its target PRAS40 (Thr246) and mTORC1 target S6K1 (Thr389). Furthermore, Akt knockdown or treatment by Akt inhibitor MK2206 reversed eIF3a knockdown-induced phosphorylation of Akt, PRAS40 and S6K1 (Fig. 6C). Therefore, Akt phosphorylation of PRAS40 may be an important process in eIF3a regulation of mTORC1 activity.
Analysis of the 5’-UTR sequence of PRAS40 mRNA also revealed two putative overlapping HuR-binding sites (Fig. 6D), suggesting that PRAS40 mRNA translation may be regulated by eIF3a via HuR in a similar manner as the regulation of Raptor mRNA translation. Indeed, HuR knockdown increased the expression of PRAS40, similar to that following eIF3a knockdown, in both H1299 and A549 cells (Fig. 6E).
eIF3a and mTORC1 regulation of cellular response to cisplatin by increasing mTORC1 activity.
Previously, it has been shown that eIF3a upregulation contributes to increased cisplatin sensitivity in lung and nasopharyngeal carcinoma cancer cell lines5, 6. However, the underlying mechanism of eIF3a regulation of cellular response to cisplatin is not yet understood. mTOR signaling is a prototypic survival pathway and mTOR activation plays an important role in cisplatin resistance39, 40. Thus, mTORC1 may mediate eIF3a contribution to cellular sensitivity to cisplatin. To test this possibility, we used mTORC1 inhibitor everolimus. Supplementary Fig. S6 show the dose-dependent survival of H1299 and A549 cells in the presence of cisplatin and everolimus with different IC50’s. Fig. 7A and 7D show that both H1299 and A549 cells with eIF3a knockdown are more resistant to cisplatin than scrambled siRNA-transfected control cells, consistent with previous observations5, 6. Fig. 7B and 7D show that everolimus also enhanced cisplatin sensitivity in both H1299 and A549 cells. The mTORC1 inhibitor, rapamycin, similarly enhanced cisplatin sensitivity in H1299 cells (Supplementary Fig. S7A–B), confirming the role of mTORC1 in cellular response to cisplatin.
Figure 7. mTORC1 mediates eIF3a regulation of cellular response to cisplatin.

(A-C). Dose-dependent survival assay. H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si) siRNA (A), in the absence or presence of 5 nM (H1299) or 10 nM (A549) everolimus (Evr) (B), or treated with combination of both siRNA transfection and everolimus (C) were subjected to treatment with cisplatin at various concentrations for 72 h followed by methylene blue assay. (D). Relative resistance factor (RRF) derived from dose-dependent survival curves shown in panels A, B, and C. (n=5, *p<0.05, **p<0.01, ***p<0.001, # p>0.05).
Next, we performed apoptosis assay following cisplatin treatments with or without mTORC1 inhibitors. As shown in Supplementary Fig. S7C–D, cisplatin induced ~3-fold increase in apoptosis whereas 10 nM everolimus or 50 nM rapamycin alone did not induce significant cell death. However, both everolimus and rapamycin at 10 and 50 nM, respectively, significantly augmented cisplatin-induced apoptosis. These findings suggest that mTORC1 signaling contributes to cellular response to cisplatin.
Next, we conducted a rescue experiment by combining eIF3a knockdown and everolimus and determined whether inhibiting mTORC1 activity could reverse eIF3a knockdown-induced increase in cisplatin resistance in H1299 and A549 cells. As shown in Fig. 7C–D, the cisplatin resistance induced by eIF3a knockdown was successfully reversed by everolimus in both cell lines. These findings suggest that mTORC1 may mediate eIF3a regulation of cellular response to cisplatin.
eIF3a negatively regulates NER protein synthesis and activity via mTORC1.
Previously, it has been shown that eIF3a regulates the synthesis of NER proteins in cellular sensitivity to cisplatin5, 6. Indeed, eIF3a knockdown increased the level of NER proteins, XPA, XPC, RPA32, and activation of mTORC1 target S6K1 and S6 in both H1299 and A549 cells (Fig. 8A–B). Quantitative RT-PCR as well as pulse and pulse-chase labeling in combination with Click-PD assay following eIF3a knockdown showed that eIF3a knockdown had no effect on mRNA levels or protein degradation rate (Supplementary Fig. S8A–B), but dramatically increased the newly synthesized XPA, XPC, and RPA32 (supplementary Fig. S8C–E). Thus, eIF3a likely regulates NER protein synthesis.
Figure 8. mTORC1 mediates eIF3a regulation of NER gene expression.

(A-B). Effect of eIF3a knockdown (A-B) or everolimus treatment (C-D) on NER protein level. Lysate from H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si) siRNA (A-B) or treated with 10 nM (H1299) or 20 nM (A549) everolimus for 24 hours (C-D) were subjected to immunoblot analysis of eIF3a, XPA, XPC, RPA32, S6K1, pT389S6K1, S6, pS240/244S6 and actin loading control. Panels B and D show quantifications of protein intensity in panels A and C (n=3, *p<0.05, **p<0.01, ***p<0.001). (E). mTORC1 inhibitor, everolimus, ablates eIF3a knockdown-induced NER protein up-regulation. Lysates from H1299 and A549 cells transfected with scrambled control (Scr) or eIF3a (Si) siRNA and treated without or with 10 nM (H1299) or 20 nM (A549) everolimus for 24 hours were subjected to immunoblot analyses of eIF3a, XPA, XPC, RPA32, S6K1, pT389S6K1, S6, pS240/244S6, Chk1, Chk2 and actin loading control. (F) Schematic model of eIF3a regulation of cellular response to cisplatin via controlling Raptor synthesis and mTORC1 activity and NER protein synthesis.
The above findings suggest that mTORC1 may mediate eIF3a regulation of NER protein synthesis. To test this possibility, we examined if mTORC1 might regulate NER gene expression by testing the effect of mTORC1 inhibitor everolimus on NER protein level. As shown in Fig. 8C–D, everolimus treatment significantly reduced the level of NER proteins and activation of the mTORC1 targets S6K1 and S6.
Next, we determined if everolimus treatment could reverse eIF3a knockdown-induced up-regulation of XPA, XPC, and RPA32. As shown in Fig. 8E, the everolimus treatment successfully reversed eIF3a knockdown-induced up-regulation of NER protein levels and activation of mTORC1 target proteins S6K1 and S6. Interestingly, eIF3a-induced changes in Chk1 and Chk2 protein level could not be reversed by everolimus treatment, suggesting that eIF3a may regulate protein synthesis using different mechanisms and eIF3a regulation of NER protein synthesis may be via mTORC1 whereas that of Chk1 and Chk2 is not.
Finally, we examined the effect of eIF3a knockdown on NER activity using host-cell reactivation (HCR) assay as previously described41, 42. A plasmid containing luciferase reporter was UV-irradiated to generate DNA adducts, which were confirmed using PCR analysis (Supplementary Fig. S9A). The reporter plasmids with DNA adducts were then transfected into H1299 cells with eIF3a knockdown and NIH3T3 cells with eIF3a overexpression followed by cellular repair of the damaged reporter constructs before analysis of luciferase activity as an indicator of NER activity. As shown in Supplementary Fig. S9B–C, the NER activity is significantly increased in H1299 cells with eIF3a knockdown and significantly reduced in NIH3T3 cells with eIF3a overexpression compared with their respective control cells. Everolimus treatment alone significantly reduced NER activity (Supplementary Fig. S9D). Interestingly, everolimus treatment reversed eIF3a knockdown-induced increase in NER activity (Supplementary Fig. S9E). Together, these findings suggest that mTORC1 may mediate eIF3a regulation of NER protein synthesis and NER activity, which in turn contributes to cellular response to cisplatin.
Discussion
In this study, we show that eIF3a regulates synthesis of Raptor via HuR binding to the 5’-UTR of Raptor mRNA and inhibiting its translation. The reduced Raptor expression leads to reduction in mTORC1 activity and cellular sensitivity to cisplatin by reducing NER protein synthesis. eIF3a may also similarly regulate PRAS40 synthesis via HuR binding to the 5’-UTR of its mRNA and its phosphorylation via Akt.
The finding that eIF3a regulates synthesis of Raptor and activity of mTORC1 is remarkable since mTORC1 is known to regulate S6K1 and 4EBP1 that in turn regulate global mRNA translation/protein synthesis. Indeed, we found that NER protein synthesis, NER activity and cellular response to cisplatin are regulated by mTORC1. These findings suggest that global translational control of gene expression by mTORC1 is also regulated translationally by eIF3a. The finding that eIF3a regulates both Raptor and PRAS40 is very intriguing since one inhibits while the other activates mTORC1. It is possible that these two mTORC1 subunits mediate eIF3a regulation of mTORC1 with fine tuning function involving Akt. It is noteworthy that eIF3a also regulates GβL and DEPTOR expression, which may contribute to eIF3a regulation of mTORC1 activity. Unfortunately, immunoprecipitation of mTORC1 complex using GβL or DEPTOR antibody for mTORC1 kinase activity assay was unsuccessful despite extensive attempts using different antibodies. Future studies are clearly needed to investigate how eIF3a regulates synthesis of these mTORC1 subunits and if they contribute to eIF3a regulation of mTORC1.
The phosphorylation of Ser792 but not that of Ser696 and Thr706 in Raptor was reduced by eIF3a knockdown despite of the increased Raptor expression. The reduced Ser792 phosphorylation is consistent with mTORC1 activity increase following eIF3a knockdown. However, it remains unknown how eIF3a regulates Raptor phosphorylation. It is also unclear how eIF3a regulates mTOR phosphorylation. It is possible that eIF3a regulates the upstream kinases or phosphatase that affect the phosphorylation status of these proteins. Future studies are warranted to investigate the mechanism of eIF3a regulation of mTOR and Raptor phosphorylation.
While we showed that Akt may mediate eIF3a regulation of PRAS40 phosphorylation, it is unclear how eIF3a regulates Akt activation. Although mTORC2 is known to phosphorylate Akt at Ser473 43, we did not find any effect of eIF3a knockdown on the expression of Rictor (unpublished observation), a key component of mTORC244. Although this finding does not rule out the possible involvement of mTORC2 in mediating eIF3a regulation of Akt, it has been shown previously that eIF3a suppresses the synthesis of DNA-PKcs7, 8, which is known to activate and phosphorylate Akt45, 46. Thus, it is tempting to speculate that DNA-PKcs may mediate eIF3a regulation of Akt phosphorylation and activation.
It is noteworthy that the finding that eIF3a, as a known subunit of eIF3, suppresses translation of Raptor mRNA contradicts with the canonical function of the eIF3 complex in stimulating mRNA translation. While it may be possible that eIF3a knockdown disrupts47 and its overexpression enhances eIF3 complex formation, which would cause reduced47 or increased global mRNA translation, respectively, this possibility is inconsistent with our findings that eIF3a knockdown increases and its overexpression suppresses Raptor synthesis. Furthermore, it has also been shown that eIF3a knockdown did not disrupt the integrity of the eIF3(a:b:i:g) subcomplex47 despite of the fact that eIF3a may play an important role in supporting formation of this subcomplex via its spectrin domain48. Furthermore, eIF3 preparations rich in eIF3a did not differ from preparations that essentially lacked eIF3a in preinitiation complex formation49, suggesting that eIF3a may not be essential for eIF3 function in global translation initiation. Thus, eIF3a may have a non-canonical function in suppressing synthesis of proteins such as Raptor.
Indeed, eIF3a has previously been shown to inhibit translation of a subset of mRNAs encoding proteins such as p2750 while activating translation of other mRNAs encoding proteins such as Chk135. Although how eIF3a exerts this putative non-canonical function is unclear, it is known that eIF3a expression is upregulated in many cancers2, 3 and it may exist in higher abundance than other subunits such as eIF3b and g in H1299 and A549 cells. The excess eIF3a may be responsible for the additional non-canonical functions in regulating protein synthesis. Indeed, eIF3b, eIF3g and eIF3i knockdowns have all been shown to disrupt the integrity of eIF351, consistent with their lower abundance relative to eIF3a. It is noteworthy that eIF3a knockdown reduced little in one study52 while drastically reduced global protein synthesis in another study47. The reason for this difference is unknown. However, it might be due to the different level of eIF3a knockdown with the former study removing only the excess eIF3a while the latter may have essentially eliminated eIF3a, resulting in disrupted integrity of the eIF3 complex.
Together with a previous finding that eIF3a may bind to HuR and enhance translation of Chk1 mRNA via HuR binding to its 3’-UTR35, our finding here that eIF3a inhibits Raptor synthesis via HuR binding to the Raptor mRNA 5’-UTR suggests that the HuR-binding site in the 5’- or 3’-UTR may dictate the inhibition or activation function of eIF3a in mRNA translation. Consistently, the 3’-UTR of DEPTOR mRNA has 37 putative HuR binding sites and DEPTOR is the only mTORC1 subunit that is positively regulated by eIF3a. While the eIF3a-HuR complex binding to the 3’-UTR may facilitate circularization and translation of the mRNA35, eIF3a binding to HuR may stimulate HuR binding to the 5’-UTR of mRNAs and the binding of the complex to the 5’-UTR may inhibit pre-initiation complex scanning, leading to inhibition of translation initiation (Fig. 6F).
In addition to the 5’-UTR of Raptor mRNA that contains HuR-binding site and is regulated by eIF3a-HuR, the 5’-UTR of PRAS40 mRNA has two overlapping putative HuR-binding sites and its expression is also regulated by eIF3a and HuR in the same manner as Raptor. However, both the 5’- and 3’-UTRs of GβL have no putative HuR binding sites and eIF3a may regulate GβL expression indirectly via other mechanisms such as mTORC1 signaling. Further studies are needed to test these possibilities.
Previously, it has been reported that eIF3a negatively regulates synthesis of DNA repair proteins and cellular response to DNA-damaging treatments including anticancer drug cisplatin, doxorubicin and radiation5, 6, 8. It has also been reported previously that mTOR activation contributes to cellular resistance to cisplatin39, 40, doxorubicin19, 20, and radiation21, 22. Our findings here suggest that mTORC1-induced cisplatin resistance may be due to its regulation of synthesis of NER proteins, which are responsible for repair of DNA damages induced by cisplatin53–55. We also showed that inhibiting mTORC1 activity was able to reverse eIF3a knockdown-induced resistance to these DNA-damaging treatments.
Based on the findings here, we conclude that eIF3a, working with HuR, inhibits Raptor synthesis, leading to reduction in mTORC1 activity and in synthesis of NER proteins and NER activity, and increases cellular sensitivity to cisplatin (Fig. 8F). Similar mechanism and pathway may be used in cellular resistance to doxorubicin and ionizing radiation where synthesis of proteins important for homologous recombination and NHEJ repair of double strand DNA breaks may be regulated by eIF3a and HuR via Raptor. It is, thus, tempting to speculate that inhibitors targeting mTORC1 pathway may help alleviate resistance to DNA-damaging agents in cancer treatments. Finally, considering that mTORC1 signaling regulates many cellular processes56, eIF3a may contribute to the regulation of these processes via mTORC1. It, however, needs to be mindful that eIF3a may also regulate synthesis of other proteins that contribute to the cellular outcome in addition to via mTOCR1 signaling.
Materials and Methods
Materials.
ECL and His-tagged human 4EBP1 recombinant protein were from GE Healthcare (Chicago, IL, USA) and Sino Biological (Wayne, PA, USA), respectively. Cisplatin, everolimus, Cell Death Detection ELISAPLUS kit and the β-actin antibody were obtained from Sigma-Aldrich (St Louis, MO, USA). MK-2206·2HCl and rapamycin were from Selleckchem (Houston, TX, USA). Antibodies against mTOR (#2983), pS2448mTOR (#2971), Raptor (#2280), pS792Raptor (#2083), GβL (#3274), PRAS40 (#2691), pT246PRAS40 (#2997), Chk1 (#2360), S6K1 (#9202), pT389S6K (#9234), S6 (#2317), pS240/244S6 (#2215), 4EBP1 (#9644), pT37/46-4EBP1 () (#2855), Akt (#9272) and pS473Akt (#4051) were from Cell Signaling Technology (Beverly, MA, USA). Antibodies against DEPTOR (ABS222), pT706Raptor (09–230, pT696Raptor (ABS544) were acquired from Millipore (Billerica, MA, USA). Recombinant human S6K1 protein and antibodies for RPA32 (ab76420) and Chk2 (ab109413) were from Abcam (Waltham, MA, USA). Antibodies against HuR (sc-5261), XPA (sc-28353), and XPC (sc-74410), the Protein G PLUS-Agarose, and siRNAs against eIF3a and Raptor were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The eIF3a siRNA #2 and #3 of different sequences were purchased from OriGene Technologies (Rockville, MD, USA). Scrambled siRNA was purchased from Ambion (Austin, TX, USA). All other chemicals were from either Fisher Scientific (Chicago, IL, USA) or Sigma-Aldrich.
Cell lines and transfection.
H1299 and A549 cells (ATCC, Manassas, VA, USA) were maintained in RPMI1640 and DMEM from Corning (Corning, NY, USA) containing 10% fetal bovine serum (Gibco, Waltham, MA, USA), respectively. NIH3T3 cells with stable eIF3a overexpression from a previous study5 were cultured in DMEM with 10% donor bovine serum (Gibco). For RNA interference, cells were transfected with siRNAs using Lipofectamine RNAiMAX Regent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Complimentary RNA (cRNA) transfection was performed as described previously57. Briefly, 2×105 cells/well were seeded into 6-well plates on the day before and transfected with siRNAs. At 24 hours after transfection, 1×105 cells/well were seeded into a 12-well plate, incubated for 24 hours, followed by transfection with capped cRNA using Lipofectamine 3000 (Invitrogen) according to manufacturer’s instructions. At 8 hours after transfection, cells were harvested for luciferase activity assay.
Construct engineering.
The cDNA encoding the full length and truncated 5’-UTR sequences of human Raptor were generated using RT-PCR from isolated RNAs of H1299 cells with a common forward primer 5’-AATTAAGCTTGTCCTGGCAATATGGCGTCCTCCT-3’ and three individual reverse primers 5’-CATGCCATGGCAGTGGGGGAGGGGGTGGGGGAGT-3’ (for wild type), 5’- CATGCCATGGCGTGTACCAGAATTGGTG-3’ (for Δ3’67) and 5’- CATGCCATGGTGGATAAAGAGTGCGGAGACCCT-3’ (for Δ3’120). The underlined sequences representing Hind III or Nco I restriction site for subcloning. The PCR products were cloned into pGEM-T Easy vector, released by digestion with Hind Ш and Nco І, and then subcloned into reporter plasmid pSP64-Fluc-poly A containing firefly luciferase genes at the same restriction sites. All constructs were confirmed by sequencing.
In-vitro transcription.
In-vitro transcription was performed as described previously58. Briefly, DNA templates were linearized using EcoR І, and used as a template for in-vitro transcription using SP6 RNA polymerases in the presence of m7G(5’)ppp(5’)G RNA (New England Biolabs, Ipswich, MA, USA) to generate capped cRNAs for transfection and luciferase reporter assay. Biotin-11-UTP (Invitrogen) was used during transcription to generate biotinylated uncapped cRNA probes for RNA pull-down assay. The in-vitro cRNA transcripts were purified using Invitrogen PureLink™ RNA Mini Kit for reporter or RNA pulldown assay.
Cell lysis, immunoprecipitation, and immunoblotting.
Cells were washed twice with ice-cold PBS, lysed in ice-cold lysis buffer (40 mM HEPES, pH7.4, 0.3% CHAPS, 2 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1×Cocktail EDTA-free protease inhibitors), subjected to centrifugation at 13,500 rpm for 15 min at 4°C to remove cell debris and insoluble materials. Protein concentration of each lysate was determined using Bradford assay59.
Immunoprecipitation and immunoblot were performed as previously described35. Briefly, lysates were incubated with primary antibodies for 2 hours at 4°C, mixed with protein G PLUS-Agarose slurry and incubated for additional 2 hours with constant agitation. The immunoprecipitants were washed three times with wash buffer (40 mM HEPES, pH7.4, 0.3% CHAPS, 2 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 1 mM Na3VO4, 150 mM NaCl) before separation on SDS-PAGE and immunoblot analysis along with total lysate as input control. For immunoblot, proteins separated on SDS–PAGE were transferred to PVDF membranes and probed with specific primary antibodies and HRP-conjugated secondary antibodies. The signal was detected using enhanced chemiluminescence and captured on x-ray films.
mTORC1 kinase activity assay.
The mTORC1 kinase activity assay was performed as previously described14. Briefly, mTORC1 was immunoprecipitated from 700 μg cell lysate as described above, followed by buffer replacement with washes for three times in 25 mM HEPES, pH 7.4, 20 mM KCl. The precipitated mTORC1 was then mixed with 150 ng recombinant 4EBP1 in 30 μl mTORC1 kinase assay buffer (25 mM HEPES, pH 7.4, 50 mM KCl, 10 mM MgCl2, 250 mM ATP) followed by incubation for 30 min at 30°C. The reaction was stopped by addition of sample loading buffer and boiling before separation on SDS-PAGE and immunoblot analysis.
Pull-down assay.
RNA pull-down assay was performed as previously described5, 35, 60, biotin-labeled cRNA probes were incubated with H1299 cell extracts at room temperature for 1 hr. The cRNA-protein complexes were then isolated using Streptavidin MagneSphere® Paramagnetic Particles and washed for three times with 1× binding buffer (20 mM Hepes, 100 mM KCl, 10% Glycerol, 1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 1 mM PMSF). The pull-down materials were then separated on SDS-PAGE and analyzed using immunoblot probed by HuR and eIF3a antibodies.
Real-time RT–PCR.
Total RNAs were extracted using Invitrogen PureLink™ RNA Mini Kit and 1 μg RNA was used for reverse transcription using Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit according to manufacturer’s protocol. The primers used were 5’-TGATGAGGACAGAGGACCAAGAC-3’ (forward) and 5’-TCAGCATTACGCCAGGATGA-3’ (reverse) for eIF3a35, 5’-AGCTGGAGGATGAAGGATCGGATG-3’ (forward) and 5’-AGGGTCCACACCAACATTCAGG-3’ (reverse) for Raptor61 and 5’-TGGCACCCAGCACAATGAA-3’ (forward) and 5’-CTAAGTCATAGTCCGCCTAGAAGCA-3’ (reverse) for β-actin58. The reaction was performed in an Applied Biosystems 7500 PCR using SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA, USA) according to manufacturer’s instructions. The threshold cycle (Ct) value of each product was determined and normalized against that of the internal control β-actin.
Luciferase reporter assay.
Luciferase reporter assay was performed as previously described57. Briefly, cells transfected with reporter cRNA transcripts were lysed in a passive lysis buffer provided in the dual luciferase reporter assay kit. Both Renilla and firefly luciferase were then measured using the dual luciferase reporter assay kit according to manufacturer’s instructions.
Methylene blue survival assay.
Methylene blue survival assay was performed as previously described62. Briefly, H1299 and A549 cells in 6-well plates were transfected with eIF3a or scrambled control siRNA. At 24 hours after transfection, 2000 cells/well were seeded into 96-well plate and treated with cisplatin in the absence or presence of everolimus at various concentrations for 72 hours, followed by removal of medium, fixation with methanol, and staining with methylene blue. The live cell-retained dyes were released using 100% ethanol:0.1 M HCl (1:1) followed by determination of OD650nm. The data were analyzed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA) to obtain IC50 for calculating relative resistance factor (RRF) using the formula RRF=IC50 (test)/IC50 (control).
Host-cell reactivation (HCR) assay NER activity.
HCR assay was performed as previously described5, 6. Briefly, 50 μg/ml pGL3 plasmid expressing firefly luciferase in 300 μl TE buffer was UV-irradiated at different doses by a Stratalinker UV Crosslinker (Stratagene, La Jolla, CA, USA) and UV-induced DNA lesions was verified by PCR with primers 5’-GCCTCTGAGCTATTCCAGAAGTAG-3’ (forward) and 5’-ACTGCATTCTAGTTGTGGTTTGTC-3’ (reverse). The verified reporter pGL3 with UV damages or un-irradiated pGL3 control plasmids (0.1 μg) were then transfected into cells harboring scrambled or eIF3a siRNAs at 5×104 cells/well in a 24-well plate using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The pRL-TK plasmid expressing Renilla luciferase was co-transfected as a control for transfection efficiencies. Forty hours after transfection, cells were harvested for luciferase activity assayed using the Dual-Luciferase assay system (Promega, Madison, WI, USA).
Apoptosis assay.
Apoptosis assay was determined as previously described63. Briefly, H1299 cells were plated in a 12-well plate at a density of 1×105 cells/well and cultured for 24 hours, treated with cisplatin, everolimus, rapamycin or combination of cisplatin with everolimus or rapamycin for 48 hours, followed by harvesting cells for apoptosis analysis using the Cell Death Detection ELISA kit (Roche, Indianapolis, IN, USA) per manufacturer’s instructions.
Pulse and pulse-chase labeling and CLICK-PD.
Pulse and pulse-chase labeling were performed using non-radioactive chase assay. Cells were rinsed with PBS and starved of methionine for 1 hour in methionine-free medium and then metabolically labeled for 3 hours in the presence of azidohomoalanine followed by harvest or chase for different times in complete medium without azidohomoalanine before harvest. The cells are lysed with TNN buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Nonidet P-40, 50 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 1 mM DTT) and equal amount of lysates of control and eIF3a knockdown cells was mixed with and incubated for 3 hours at room temperature in 0.1 mM biotin-PEG-4-alkyne, 0.04 mM Tris[(1-benzyl-1,2,3-triazol-4-yl) methyl] amine, 1 mM tris(2-carboxyethyl) phosphine, 1 mM CuSO4. Nascent proteins-labeled with biotin by CLICK were isolated by pull down using streptavidin bead and separated by SDS-PAGE for immunoblot analysis.
Statistical Analysis.
All statistical analyses were performed using GraphPad Prism. Since minimum three independent experiments/biological replicates are required for statistical considerations, all experiments were performed at least three times with data presented as mean±standard deviation. No data were excluded, and no blinding or randomization were performed. One-way ANOVA followed by Dunnett’s test was used to compare more than two groups with similar variances, while two-tailed Student t tests was done to compare two groups.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH grant R01 CA211904.
Abbreviations used:
- cRNA
complimentary RNA
- DEPTOR
DEP domain-containing mTOR-interacting protein
- eIF
eukaryotic translation initiation factor
- mTOR
mammalian target of rapamycin
- HuR
human antigen R
- GβL
G protein β-subunit-like
- mTORC
mTOR complex
- NER
nucleotide excision repair
- PRAS40
proline-rich Akt substrate 40 kDa
- Raptor
regulatory-associated protein of mTOR
- UTR
untranslated region
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1.Clemens MJ, Bommer UA. Translational control: the cancer connection. Int J Biochem Cell Biol 1999; 31: 1–23. [DOI] [PubMed] [Google Scholar]
- 2.Dong Z, Zhang JT. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit Rev Oncol Hematol 2006; 59: 169–180. [DOI] [PubMed] [Google Scholar]
- 3.Yin JY, Dong Z, Liu ZQ, Zhang JT. Translational control gone awry: a new mechanism of tumorigenesis and novel targets of cancer treatments. Biosci Rep 2011; 31: 1–15. [DOI] [PubMed] [Google Scholar]
- 4.Yin JY, Zhang JT, Zhang W, Zhou HH, Liu ZQ. eIF3a: A new anticancer drug target in the eIF family. Cancer Lett 2018; 412: 81–87. [DOI] [PubMed] [Google Scholar]
- 5.Yin JY, Shen J, Dong ZZ, Huang Q, Zhong MZ, Feng DY et al. Effect of eIF3a on response of lung cancer patients to platinum-based chemotherapy by regulating DNA repair. Clin Cancer Res 2011; 17: 4600–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu RY, Dong Z, Liu J, Yin JY, Zhou L, Wu X et al. Role of eIF3a in regulating cisplatin sensitivity and in translational control of nucleotide excision repair of nasopharyngeal carcinoma. Oncogene 2011; 30: 4814–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Liu JY, Dong ZZ, Zou T, Wang Z, Shen Y et al. The effect of eIF3a on anthracycline-based chemotherapy resistance by regulating DSB DNA repair. Biochem Pharmacol 2021; 190: 114616. [DOI] [PubMed] [Google Scholar]
- 8.Tumia R, Wang CJ, Dong T, Ma S, Beebe J, Chen J et al. eIF3a Regulation of NHEJ Repair Protein Synthesis and Cellular Response to Ionizing Radiation. Frontiers in cell and developmental biology 2020; 8: 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal 2009; 21: 827–835. [DOI] [PubMed] [Google Scholar]
- 10.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110: 177–189. [DOI] [PubMed] [Google Scholar]
- 11.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 2003; 11: 895–904. [DOI] [PubMed] [Google Scholar]
- 12.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002; 10: 457–468. [DOI] [PubMed] [Google Scholar]
- 13.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009; 137: 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007; 25: 903–915. [DOI] [PubMed] [Google Scholar]
- 15.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004; 23: 3151–3171. [DOI] [PubMed] [Google Scholar]
- 16.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006; 124: 471–484. [DOI] [PubMed] [Google Scholar]
- 17.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102: 8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O’Reilly T et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell 2005; 120: 747–759. [DOI] [PubMed] [Google Scholar]
- 19.Babichev Y, Kabaroff L, Datti A, Uehling D, Isaac M, Al-Awar R et al. PI3K/AKT/mTOR inhibition in combination with doxorubicin is an effective therapy for leiomyosarcoma. J Transl Med 2016; 14: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res 2002; 62: 6141–6145. [PubMed] [Google Scholar]
- 21.Wei F, Liu Y, Guo Y, Xiang A, Wang G, Xue X et al. miR-99b-targeted mTOR induction contributes to irradiation resistance in pancreatic cancer. Mol Cancer 2013; 12: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nassim R, Mansure JJ, Chevalier S, Cury F, Kassouf W. Combining mTOR inhibition with radiation improves antitumor activity in bladder cancer cells in vitro and in vivo: a novel strategy for treatment. PLoS One 2013; 8: e65257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan HX, Guan KL. Structural insights of mTOR complex 1. Cell Res 2016; 26: 267–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C et al. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J Clin Invest 2011; 121: 2821–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su B, Jacinto E. Mammalian TOR signaling to the AGC kinases. Crit Rev Biochem Mol Biol 2011; 46: 527–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 2012; 441: 1–21. [DOI] [PubMed] [Google Scholar]
- 27.Carriere A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Current biology : CB 2008; 18: 1269–1277. [DOI] [PubMed] [Google Scholar]
- 28.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell 2008; 30: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gwinn DM, Asara JM, Shaw RJ. Raptor is phosphorylated by cdc2 during mitosis. PloS one 2010; 5: e9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramirez-Valle F, Badura ML, Braunstein S, Narasimhan M, Schneider RJ. Mitotic raptor promotes mTORC1 activity, G(2)/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Molecular and cellular biology 2010; 30: 3151–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P et al. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). The Journal of biological chemistry 2011; 286: 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwak D, Choi S, Jeong H, Jang JH, Lee Y, Jeon H et al. Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal Kinase (JNK)-mediated Raptor protein phosphorylation. J Biol Chem 2012; 287: 18398–18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 110: 163–175. [DOI] [PubMed] [Google Scholar]
- 34.Foster KG, Acosta-Jaquez HA, Romeo Y, Ekim B, Soliman GA, Carriere A et al. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J Biol Chem 2010; 285: 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong Z, Liu J, Zhang JT. Translational regulation of Chk1 expression by eIF3a via interaction with the RNA-binding protein HuR. Biochem J 2020; 477: 1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12: 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ et al. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 2003; 278: 10189–10194. [DOI] [PubMed] [Google Scholar]
- 38.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9: 316–323. [DOI] [PubMed] [Google Scholar]
- 39.Peng DJ, Wang J, Zhou JY, Wu GS. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem Biophys Res Commun 2010; 394: 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK et al. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004; 23: 5853–5857. [DOI] [PubMed] [Google Scholar]
- 41.Qiao Y, Spitz MR, Guo Z, Hadeyati M, Grossman L, Kraemer KH et al. Rapid assessment of repair of ultraviolet DNA damage with a modified host-cell reactivation assay using a luciferase reporter gene and correlation with polymorphisms of DNA repair genes in normal human lymphocytes. Mutat Res 2002; 509: 165–174. [DOI] [PubMed] [Google Scholar]
- 42.Ahn B, Kang D, Kim H, Wei Q. Repair of mitomycin C cross-linked DNA in mammalian cells measured by a host cell reactivation assay. Mol Cells 2004; 18: 249–255. [PubMed] [Google Scholar]
- 43.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- 44.Kocalis HE, Hagan SL, George L, Turney MK, Siuta MA, Laryea GN et al. Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis. Mol Metab 2014; 3: 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stronach EA, Chen M, Maginn EN, Agarwal R, Mills GB, Wasan H et al. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011; 13: 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30: 203–213. [DOI] [PubMed] [Google Scholar]
- 47.Wagner S, Herrmannova A, Malik R, Peclinovska L, Valasek LS. Functional and biochemical characterization of human eukaryotic translation initiation factor 3 in living cells. Mol Cell Biol 2014; 34: 3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong Z, Qi J, Peng H, Liu J, Zhang JT. Spectrin Domain of Eukaryotic Initiation Factor 3a Is the Docking Site for Formation of the a:b:i:g Subcomplex. J Biol Chem 2013; 288: 27951–27959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaudhuri J, Chakrabarti A, Maitra U. Biochemical characterization of mammalian translation initiation factor 3 (eIF3). Molecular cloning reveals that p110 subunit is the mammalian homologue of Saccharomyces cerevisiae protein Prt1. J Biol Chem 1997; 272: 30975–30983. [DOI] [PubMed] [Google Scholar]
- 50.Dong Z, Zhang JT. EIF3 p170, a Mediator of Mimosine Effect on Protein Synthesis and Cell Cycle Progression. Mol Biol Cell 2003; 14: 3942–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner S, Herrmannova A, Sikrova D, Valasek LS. Human eIF3b and eIF3a serve as the nucleation core for the assembly of eIF3 into two interconnected modules: the yeast-like core and the octamer. Nucleic Acids Res 2016; 44: 10772–10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. Oncogene 2004; 23: 3790–3801. [DOI] [PubMed] [Google Scholar]
- 53.Nouspikel T DNA repair in mammalian cells : Nucleotide excision repair: variations on versatility. Cell Mol Life Sci 2009; 66: 994–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neher TM, Rechkunova NI, Lavrik OI, Turchi JJ. Photo-cross-linking of XPC-Rad23B to cisplatin-damaged DNA reveals contacts with both strands of the DNA duplex and spans the DNA adduct. Biochemistry 2010; 49: 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neher TM, Shuck SC, Liu JY, Zhang JT, Turchi JJ. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem Biol 2010; 5: 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017; 168: 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong Z, Liu Y, Zhang JT. Regulation of ribonucleotide reductase M2 expression by the upstream AUGs. Nucleic Acids Res 2005; 33: 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yin JY, Dong ZZ, Liu RY, Chen J, Liu ZQ, Zhang JT. Translational regulation of RPA2 via internal ribosomal entry site and by eIF3a. Carcinogenesis 2013; 34: 1224–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 60.Qi J, Dong Z, Liu J, Zhang JT. EIF3i promotes colon oncogenesis by regulating COX-2 protein synthesis and beta-catenin activation. Oncogene 2014; 33: 4156–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vandamme T, Beyens M, de Beeck KO, Dogan F, van Koetsveld PM, Pauwels P et al. Long-term acquired everolimus resistance in pancreatic neuroendocrine tumours can be overcome with novel PI3K-AKT-mTOR inhibitors. Br J Cancer 2016; 114: 650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peery R, Kyei-Baffour K, Dong Z, Liu J, de Andrade Horn P, Dai M et al. Synthesis and Identification of a Novel Lead Targeting Survivin Dimerization for Proteasome-Dependent Degradation. J Med Chem 2020; 63: 7243–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang W, Dong Z, Chen Y, Wang F, Wang CJ, Peng H et al. Small-molecule inhibitors targeting the DNA-binding domain of STAT3 suppress tumor growth, metastasis and STAT3 target gene expression in vivo. Oncogene 2016; 35: 783–792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.