

Abstract
CD38 enzymatic activity regulates NAD+ and cADPR levels in mammalian tissues, and therefore has a prominent role in cellular metabolism and calcium homeostasis. Consequently, it is reasonable to hypothesize about its involvement in cardiovascular physiology as well as in heart related pathological conditions.
Aim:
To investigate the role of CD38 in cardiovascular performance, and its involvement in cardiac electrophysiology and calcium-handling.
Methods and results:
When submitted to a treadmill exhaustion test, a way of evaluating cardiovascular performance, adult male CD38KO mice showed better exercise capacity. This benefit was also obtained in genetically modified mice with catalytically inactive (CI) CD38 and in WT mice treated with antibody 68 (Ab68) which blocks CD38 activity. Hearts from these 3 groups (CD38KO, CD38CI and Ab68) showed increased NAD+ levels. When CD38KO mice were treated with FK866 which inhibits NAD+ biosynthesis, exercise capacity as well as NAD+ in heart tissue decreased to WT levels.
Electrocardiograms of conscious unrestrained CD38KO and CD38CI mice showed lower basal heart rates and higher heart rate variability than WT mice. Although inactivation of CD38 in mice resulted in increased SERCA2a expression in the heart, the frequency of spontaneous calcium release from the sarcoplasmic reticulum under stressful conditions (high extracellular calcium concentration) was lower in CD38KO ventricular myocytes. When mice were challenged with caffeine-epinephrine, CD38KO mice had a lower incidence of bidirectional ventricular tachycardia when compared to WT ones.
Conclusion:
CD38 inhibition improves exercise performance by regulating NAD+ homeostasis. CD38 is involved in cardiovascular function since its genetic ablation decreases basal heart rate, increases heart rate variability and alters calcium handling in a way that protects mice from developing catecholamine induced ventricular arrhythmias.
Keywords: CD38, NAD+, heart, exercise capacity, calcium, action potential, arrhythmia
Graphical Abstract
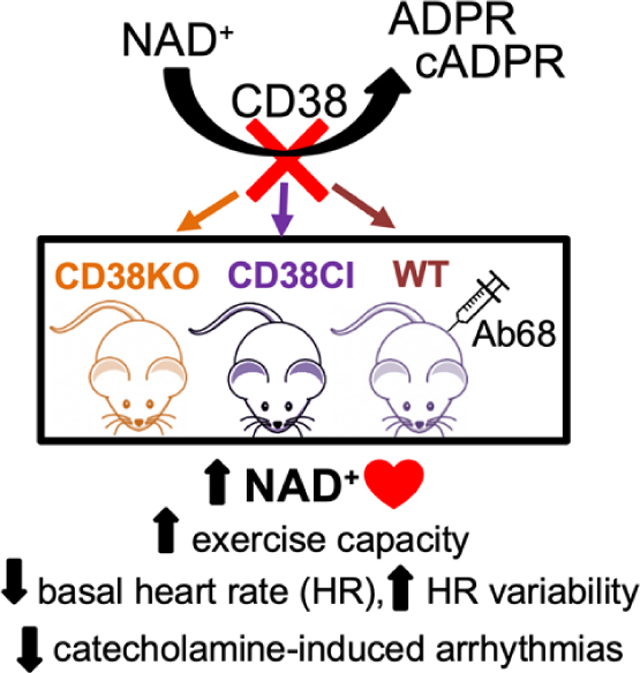
1. INTRODUCTION
CD38 is the main NAD+ consuming enzyme in most mammalian tissues [1]. Levels of CD38 can increase in different scenarios such as infections, and autoimmune or metabolic diseases, enhancing the damage in these pathologic situations by depleting tissue NAD+ levels. NAD+ is a dinucleotide involved in oxidation-reduction reactions and is substrate to a large number of enzymes related to a wide range of processes in cellular homeostasis [2][3].
In metabolic reactions, NAD+ has the potential of accepting H+ groups to form the reduced cofactor NADH. NADH is then utilized by the mitochondria to drive the oxidative phosphorylation process needed to produce ATP, the main source of energy for tissues [4]. Under physiological conditions, oxidative phosphorylation is the main process involved in the production of ATP in heart tissue (approximately 95%), and fatty acids are the primary source used as substrate [5]. The ratio between NAD+ and NADH (NAD+/NADH) is one of the most crucial features that maintains cellular homeostasis. A decrease of this ratio can impair normal cellular metabolism and lead to accumulation of reactive oxygen species [6].
NAD+ is also the substrate to a numerous set of fundamental enzymes involved in cellular homeostasis. Sirtuins (SIRTs) for instance, are NAD+ dependent deacylases that play important roles in metabolism, DNA repair, cell cycle, tumorigenesis, and cardiac function, among others [7]. These functions can therefore be influenced by the NAD+ pool which can either enhance or suppress SIRTs activity. As the main NAD+-degrading enzyme, increases in CD38 expression and activity can lead to NAD+ depletion and cellular dysfunction. It has been previously demonstrated that CD38 expression and activity rise with aging, leading to a decrease in NAD+ levels, which increases susceptibility to stress [8].
CD38 is also the main ADPR cyclase in the heart, and its catalytic product, cADPR, has been considered by several authors as a key molecule in calcium handling [9]. Despite the specific mechanism is not completely understood, it has been proposed that cADPR acts sensitizing calcium induced calcium release (CICR) probably by enhancing the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA2a) pump function [10][11][12] and also by activating the ryanodine receptors subtype 2 (RyR2) [13]. As a consequence, the calcium transient amplitude increases, as well as the frequency of calcium sparks and waves. The synthesis of cADPR as a second messenger is stimulated by beta adrenergic regulation [14][15][16]. This effect contributes to improve the contractile force when it is required, but in some pathological circumstances it could lead to different types of arrhythmias secondary to calcium overload [10][17]. One deeply known mechanism of cardiac arrhythmia is triggered activity, caused by dysregulation in calcium handling that results in delayed after-depolarizations (DADs) [18]. This mechanism has a key role in genetic disorders like Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT), and also in arrhythmias associated with heart disease. For example, the life threatening ventricular tachycardia that develops in patients with heart failure, which is one of the main causes of in-hospital admissions in the world [19][20].
Even though CD38 is thought to be involved in numerous pathological conditions related to the heart, through regulation of NAD+ levels and therefore cellular metabolism, as well as regulating the availability of cADPR [16][17][21], the mechanistic role of CD38 in cardiovascular physiology is not completely understood. It has been reported that CD38 plays a key role in the signaling pathway that leads to pathological cardiac hypertrophy in numerous models of heart failure, mainly by depleting cardiac NAD+ levels and consequently impairing SIRTs function [22]. CD38 was shown to be involved in ischemic heart disease, by NAD+ and NADP depletion leading to endothelial dysfunction [23][24][25]. It has also been proposed that CD38 expression is related to contraction-relaxation processes by regulating key proteins in the sarcoplasmic reticulum (SR), and therefore, could be implicated in the development of heart failure [26]. Because of this evidence, it is reasonable to propose CD38 as a potential therapeutic target in cardiovascular diseases [27].
Here we show that chronically blocking CD38, either genetically or pharmacologically, results in an improvement in exercise capacity that depends on NAD+ boosting. In addition to the increase in physical performance, CD38 genetic ablation or activity suppression, led to a decrease in basal heart rate and an increase in heart rate variability. We also found that despite the overexpression of SERCA2a in CD38KO hearts, the frequency of spontaneous calcium release from the cardiomyocytes sarcoplasmic reticulum under stressful conditions is decreased. Moreover, CD38KO mice were protected from developing ventricular arrhythmias induced by overstimulation of the beta-adrenergic pathway. Our results add new evidence to support the consideration of CD38 as a therapeutic target in cardiovascular related conditions.
2. RESULTS AND DISCUSSION
2.1. CD38 suppression improves exercise tolerance by boosting NAD+ levels
2.1.a. Genetic deletion of CD38 increases exercise capacity in vivo and this is dependent on the suppression of CD38 catalytic activity
We evaluated cardiovascular performance by exercise capacity in CD38KO and CD38CI mice. The animals were subjected to an uphill treadmill exhaustion test. Both CD38KO and CD38CI mice showed increased exercise capacity seen as nearly double the distance, as well as the work performed. There was also a 20% increase in time to exhaustion and 15% increase in maximal running speed (Figure 1a).
Figure 1. Genetic deletion of CD38 increases exercise capacity in vivo and induces NAD+ boosting in heart tissue, and these effects are dependent on the suppression of CD38 catalytic activity.
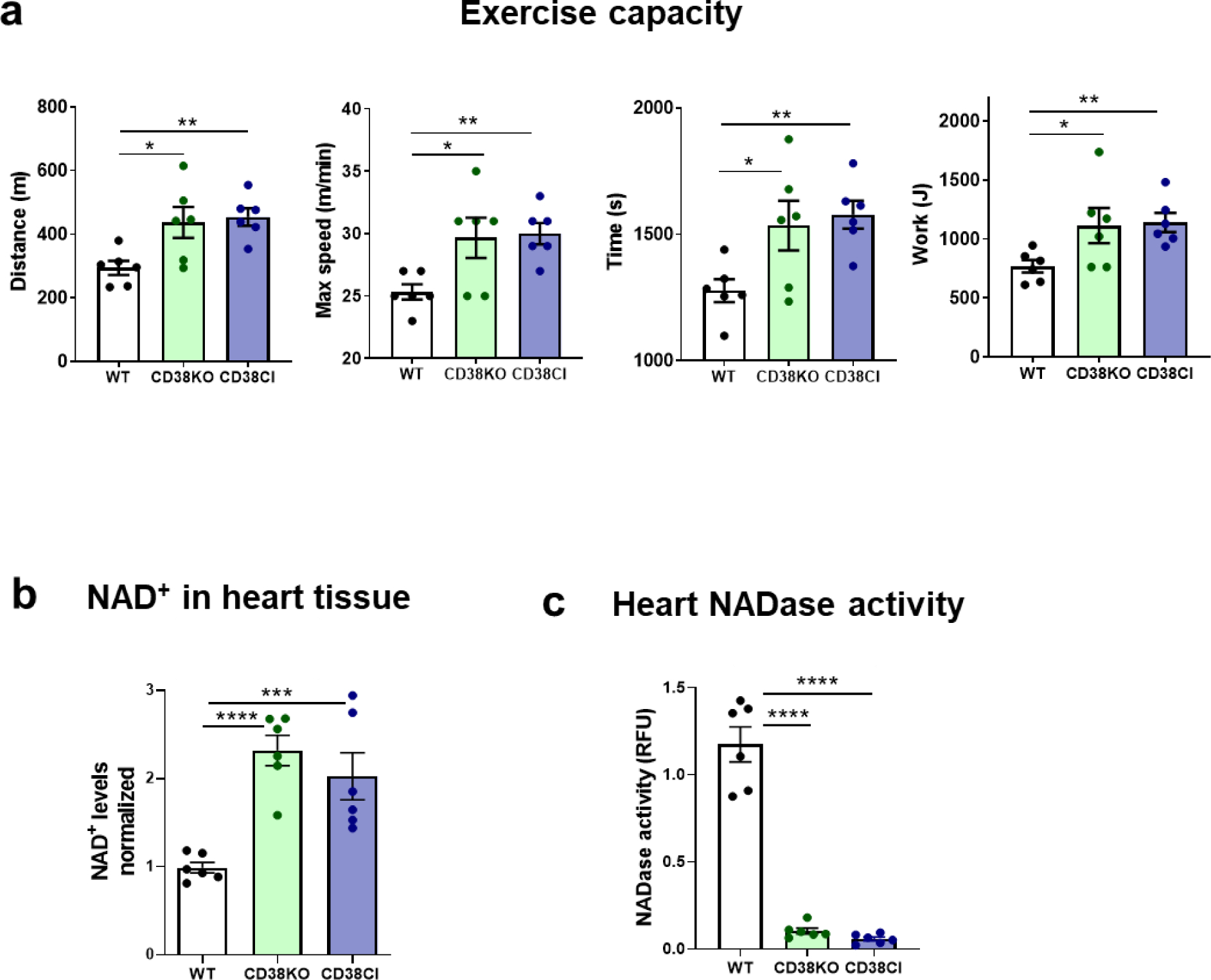
(a) WT, CD38KO and CD38CI (catalytically inactive) adult male mice (6–8-month-old) were submitted to uphill treadmill exhaustion test. The graphs show distance, maximal speed, time, and work until mice got exhausted. Both genetically modified groups present better exercise capacity than the WT group. (b) NAD+ levels in the hearts normalized for WT mice. NAD+ levels are increased in the hearts from mice without CD38 expression or activity (CD38KO and CD38CI respectively). (c) NADase activity in hearts. When CD38 is not expressed or active, NADase activity in hearts almost disappear showing that CD38 is the main NADase in hearts from these mice. Data are mean ± SEM (N=6 mice/group), analyzed by unpaired two-sided t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
These results show that CD38KO mice have improved exercise capacity and that this is not simply due to the absence of CD38, but more importantly due to the suppression of CD38 enzymatic activity, supported by the results obtained in CD38CI mice. To further confirm this hypothesis, we measured NAD+ levels in the hearts and found that they were increased in both genetically modified mice compared to WT hearts (Figure 1b). Accordingly, NADase activity measured in the hearts showed almost undetectable levels for CD38KO and CD38CI hearts (Figure 1c).
2.1.b. Pharmacologically blocking CD38 ecto-enzymatic activity mimics the benefits obtained by the genetically modified models in vivo
To better characterize the role of CD38 catalytic activity in cardiovascular performance we carried out experiments using a specific heavy chain antibody that binds and inhibits the ecto-enzymatic hydrolase activity of mouse CD38 (Ab68). We treated WT mice with either vehicle or Ab68 at a dose of 5 mg/kg, every 4 days for 2 weeks. Ab68 treated mice experienced an improvement in exercise capacity seen as a statistically significant increase in the distance elapsed, maximal running speed, time spent running and work performed during treadmill exhaustion test (Figure 2a).
Figure 2. Pharmacologically blocking CD38 ecto-enzymatic activity mimics the benefits obtained by the genetically modified models in vivo.
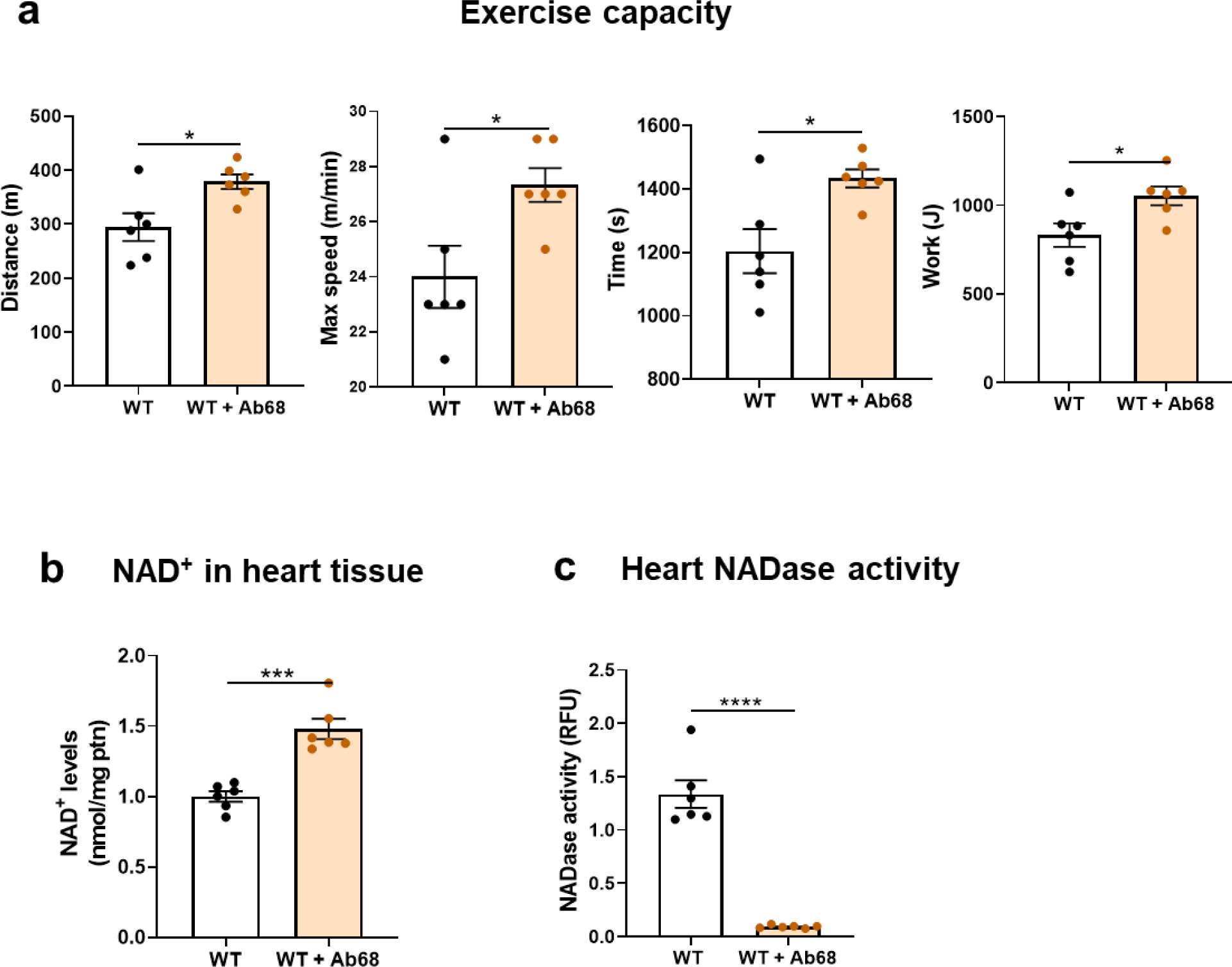
WT adult male mice (6-month-old) were treated with either vehicle or 5 mg/kg of Ab68 intraperitoneally every 4 days for 2 weeks. (a) Uphill treadmill exhaustion test. The graphs show distance, maximal speed, time, and work until mice got exhausted. Ab68-treated WT mice show increased exercise capacity when compared to control mice. (b) NAD+ levels in the hearts of Ab68-treated WT mice are significantly increased compared to controls. (c) NADase activity in heart tissue. NADase activity in the hearts was effectively blocked by treatment with Ab68. Data are mean ± SEM (N=6 mice/group), analyzed by unpaired two-sided t-test, *P<0.05, ***P<0.001, ****P<0.0001.
Again, when analyzing the hearts of these animals, NAD+ levels were significantly increased in the Ab68-treated group compared to those receiving vehicle, and this was coherent with a dramatic decrease in NADase activity in the Ab68-treated group (Figures 2b–c).
2.1.c. Pharmacologically blocking NAD+ biosynthesis pathway prevents the improvement in exercise performance obtained by genetically deleting CD38
To explore the possibility that the exercise capacity improvement observed in the previous experiments was dependent on NAD+ boosting, we blocked the NAD+ salvage biosynthesis pathway in CD38KO mice using FK866. This drug inhibits NAMPT, the rate limiting enzyme involved in the synthesis of NMN, an NAD+ precursor [28]. It has been previously shown that inhibition of NAMPT causes NAD+ depletion in an NMN-dependent manner and abrogates the NAD+ boosting derived from Ab68 treatment [29].
WT mice received only vehicle, while CD38KO mice were treated with either vehicle or FK866. Similar to what we have been demonstrating so far, CD38KO control mice presented an improvement in their physical performance. However, when CD38KO mice were treated with FK866 they showed lower exercise capacity compared to CD38KO mice treated with vehicle, suggesting an important role for NAD+ boosting in this scenario (Figure 3a).
Figure 3. Benefits in exercise performance obtained by genetically deleting CD38 are dependent on boosting NAD+ levels.
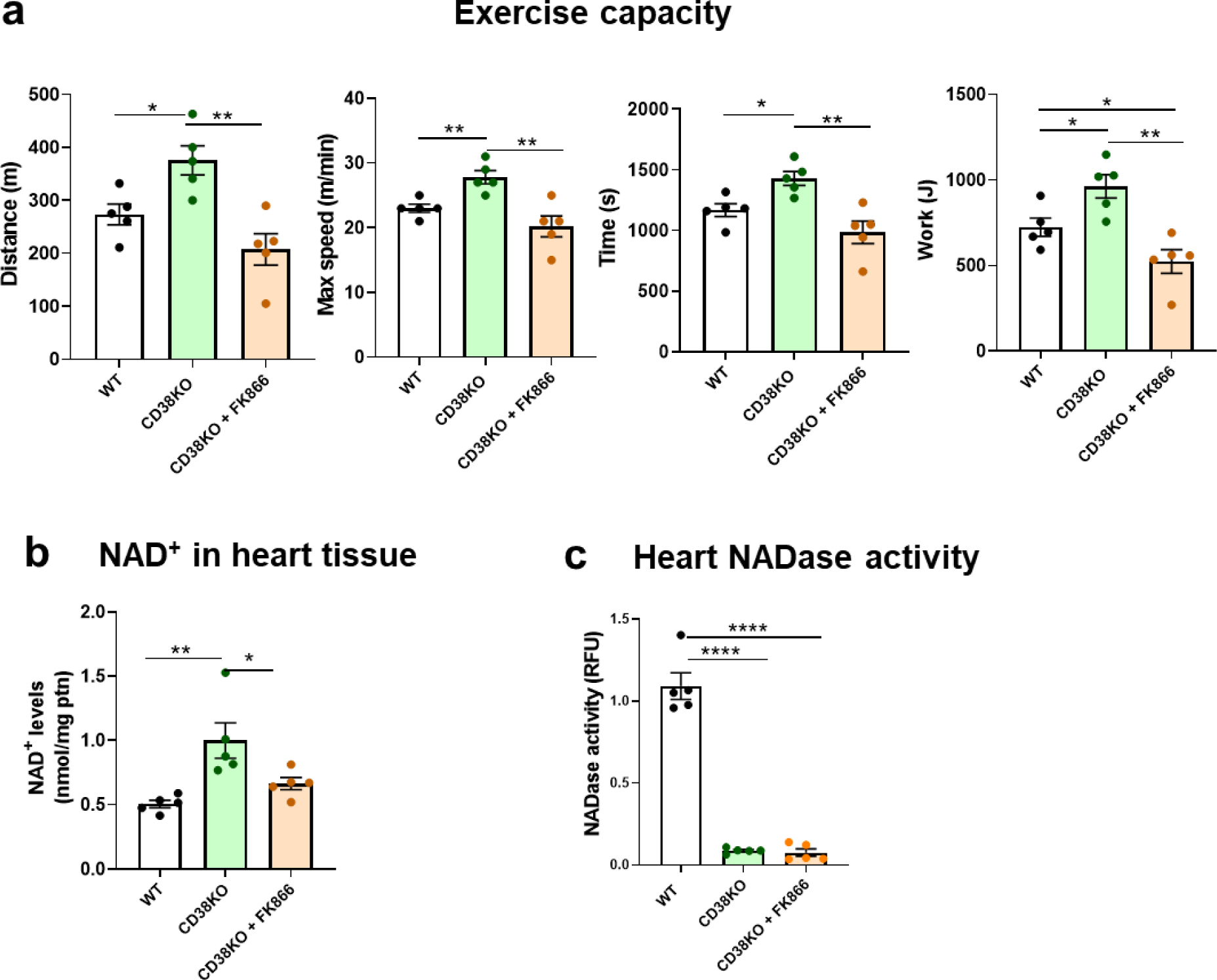
Adult male mice (6-month-old) were treated with either vehicle (WT and CD38KO mice) or 25 mg/kg of FK866, a NAMPT inhibitor, (only CD38KO mice), intraperitoneally for a week. (a) Uphill treadmill exhaustion test. The graphs show distance, maximal speed, time, and work until mice got exhausted. Treatment with FK866 prevented the improvement of exercise capacity seen in CD38KO mice. (b) NAD+ levels in the hearts. Inhibition of NAMPT reduced NAD+ levels in CD38KO mice. (c) NADase activity in heart tissue. NADase activity in the hearts was almost insignificant in both CD38KO control and treated groups when compared to WT group. Data are mean ± SEM (N=6 mice/group), analyzed by unpaired two-sided t-test, *P<0.05, **P<0.01, ****P<0.0001.
As predicted, treatment with FK866 completely abrogated the NAD+ boosting we observed in CD38KO mice (Figure 3b) although NADase activity was almost nonexistent in both control (vehicle) and treated (FK866) CD38KO groups (Figure 3c).
These results suggest the boosting in NAD+ levels as an explanation for the improvement in exercise performance. The fact that the beneficial effect of CD38 suppression was lost when CD38KO mice were pharmacologically prevented from increasing NAD+ levels, although the enzymatic activity of CD38 was absent, clearly support an NAD+-dependent mechanism in the exercise capacity amelioration (Figures 3a–c).
Altogether, NAD+ levels were increased in hearts from all these mice, i.e., CD38KO, CD38CI and Ab68-treated WT (Figures 1b, 2b and 3b). Accordingly, NADase activity in the hearts tended to zero in these models of CD38 activity suppression (Figures 1c, 2c and 3c). Therefore, our results demonstrate that in the heart of adult male mice, CD38 is the main NADase and so, the main NAD+ homeostasis regulator. In these three models, the cardiac tissue was analyzed as a whole, which means that all its forming cells were included. Therefore, we cannot conclude which type of cell(s), among those usually present in the cardiac tissue, contribute(s) to this effect. Some possible effectors rather than cardiomyocytes are endothelial cells and immune cells such as resident macrophages [30].
In the present study we only measured NAD+ levels because it is known that CD38 degrades the oxidized (NAD+), not the reduced (NADH) form of NAD. The NAD+/NADH ratio has been shown to be increased in some CD38KO tissues, since absence of CD38 causes an increase mostly in oxidized NAD+ and not NADH [8]. Furthermore, the oxidized form is the one used as substrate for non-oxidative enzymes such as sirtuins.
The beneficial effects of NAD+ boosting in exercise performance have been previously described, both in aging and disease models. Tarragó et al. [31] have reported that mice treated with 78c, a potent and specific thiazoloquin(az)olin(on)e CD38 inhibitor, show amelioration of age-related decline in exercise performance when compared to control mice.
Chiang et al [32] explored the association between CD38 and exercise tolerance in a model of western diet-induced obesity. Under this metabolic stress, CD38KO mice are protected against obesity-induced exercise impairment. Before the high-fat-high-sucrose diet started, though not statistically significant, CD38KO mice show a trend toward a better exercise performance.
Conversely, Park et al. [12] claimed that CD38KO mice have a decreased exercise capacity when compared to WT ones. However, to support this, they only present the decay in the number of mice that remain running during the intensity overload test. That number depends on the definition of exhaustion which is completely different in their protocol compared to ours and those from the above cited groups. Here we present all the parameters measured in the test and not only for CD38KO mice but also for CD38CI and Ab68-treated WT mice and we show that it is possible to improve exercise performance by modulating CD38-NAD+ axis.
It is possible that the skeletal muscle has a role as an effector in the improvement of exercise performance as well. It has been previously shown that pharmacological blockage of CD38 ameliorates age-related skeletal muscle dysfunction, reversing several morphological features of aging and increasing NAD+ levels in this tissue [28]. On the other hand, Park et al [12] found impaired positive inotropic response to adrenergic stimulation in isolated skeletal muscle. Further studies exploring the possible alterations in the skeletal muscle when inhibiting CD38 may help to clarify its contribution for the results shown in the present study.
Given the multifactorial nature of exercise, we believe that the beneficial effect seen on our results may be a consequence of several mutually non-excluding mechanisms that contribute energetically, metabolically, and/or structurally to a better exercise capacity. Heart is a key organ in the machinery of aerobic performance and exercise stress is a well-known and reliable tool to evaluate cardiac function, not only in animal models but also in the clinical practice. Here we used this valuable tool to test if boosting NAD+ levels in heart tissue through CD38 modulation could result in an increase in physical performance. We show that both CD38KO and CD38CI mice perform better and that pharmacological modulation of CD38 through Ab68 reproduced these results. This benefit was lost when CD38KO mice were treated with FK866 and NAD+ biosynthesis inhibited. These results highlight that NAD+ levels modulation can indeed alter physical performance in mice and CD38 is a potential pharmacological target for the improvement of cardiac and aerobic function.
2.2. Evaluation of CD38 involvement in cardiac electrophysiology and calcium handling
2.2.a. Genetic deletion of CD38 decreases basal heart rate and increases heart rate variability in vivo and these effects are dependent on CD38 catalytic activity suppression
In order to better understand the role of CD38 in cardiovascular physiology, we performed ECG recordings in adult male mice by telemetry. We measured heart rate (HR) and heart rate variability (HRV) of conscious unrestrained WT, CD38KO and CD38CI mice (Figure 4a). CD38KO mice exhibited lower HR and higher HRV than WT mice as shown in Figure 4b. These differences were also observed in CD38CI mice, demonstrating that they are dependent on CD38 enzymatic activity (Figure 4b).
Figure 4. Genetic deletion of CD38 decreases heart rate and increases heart rate variability in vivo, and these effects are dependent on CD38 catalytic activity suppression.
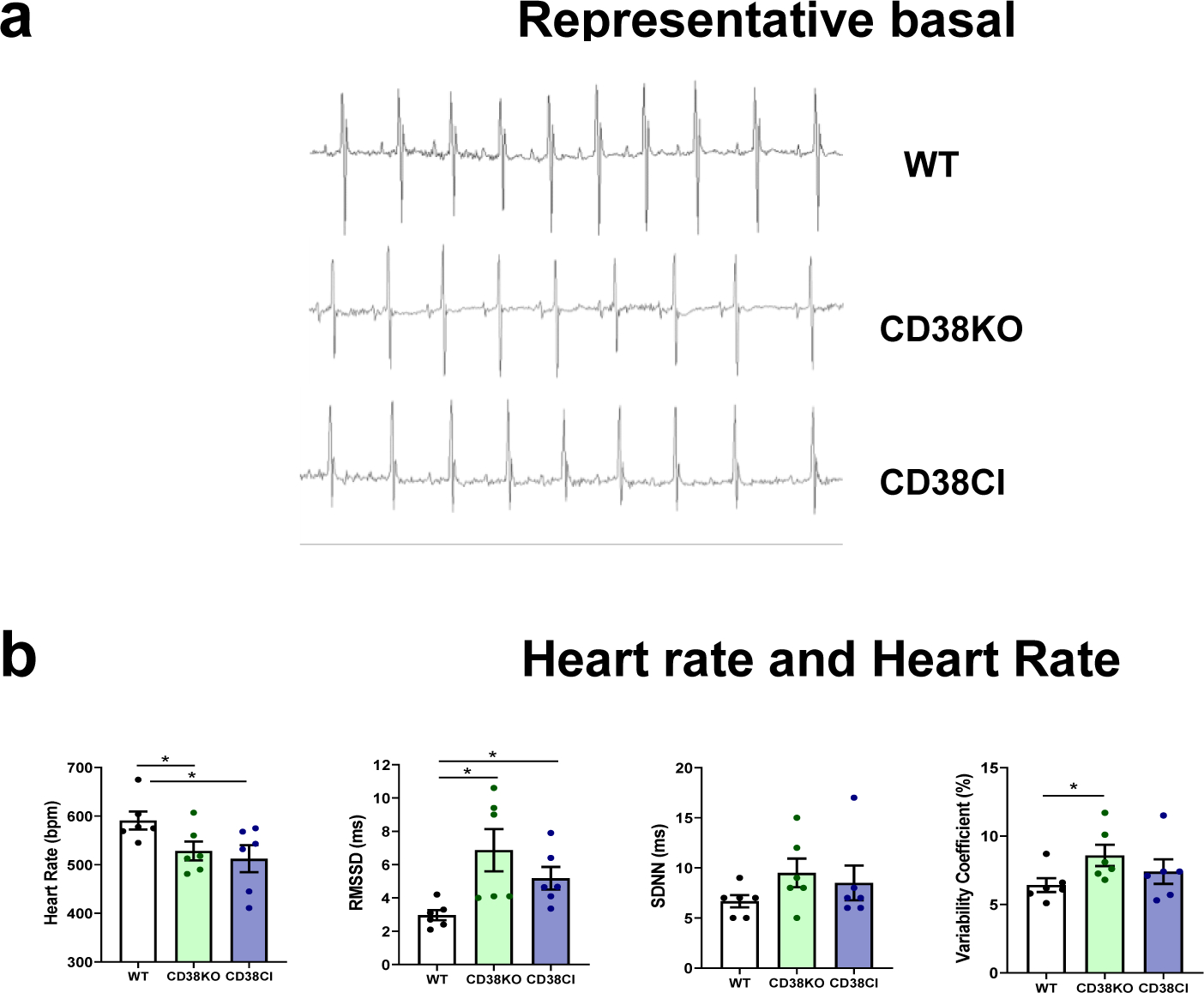
WT, CD38KO and CD38CI (catalytically inactive) male adult mice (6–8-month-old) were submitted to telemetry implant surgery and their ECG was recorded in the 3rd and 4th day after surgery. (a) Representative 1 second ECG tracings from WT, CD38KO, and CD38CI mice. (b) The graphs show heart rate (HR) and three indexes of HR variability (HRV): root mean square of successive differences (RMSSD), standard deviation of normal-normal RR intervals (SDNN), and variability coefficient (VC). HR is lower and HRV is higher in CD38KO and CD38CI groups compared to WT group. Data are mean ± SEM (N=6 mice/group), analyzed by unpaired two-sided t-test, *P<0.05.
Low HR, within the physiological range, can be considered a desirable feature for heart performance [33]. A previous study also showed lower HR values in CD38KO conscious and anesthetized mice but assessed by a less precise method than the one we used here (tail-cuff blood pressure monitoring) [34]. We used telemetry to record cardiac electrical activity and analyzed not only HR but also HRV. Beat to beat variability; i.e., RMSSD, was increased in CD38KO and CD38CI mice. This is a prominent feature and can be considered a sign of healthy modulation of HR by the autonomic nervous system [35][36], especially considering that, this kind of HRV index evaluates parasympathetic modulation of HR [37]. Increased modulation does not necessarily mean increased tone. Indeed, acetylcholine, norepinephrine and epinephrine concentrations in plasma and right atria from WT and CD38KO mice showed no differences (see Supplementary data and Figure S1). Lower HR and higher beat to beat HRV in CD38KO and CD38CI mice might be explained by a lower response to sympathetic modulation. CD38 activation has been implicated in adrenergic signaling pathway in cardiomyocytes [9] [16]. For example, Lin et al described that in the presence of isoproterenol, a beta-adrenergic agonist, calcium transient amplitudes were smaller in cardiomyocytes isolated from CD38KO mice than WT ones [9]. We evaluated the adrenergic responsiveness using a different approach, by comparing action potential modifications when paced isolated whole hearts from WT and CD38KO mice were challenged with isoproterenol. Lack of CD38 determined a different response to isoproterenol (see Supplementary data and Figure S2). Lower HR cannot be explained by a slower pacemaker in CD38KO hearts since the intrinsic HR, evaluated before pacing started, was not different when isolated hearts from WT and CD38KO mice were allowed to beat spontaneously (data not shown). The same spontaneous HR in isolated hearts from WT and CD38KO mice has also been reported by Boslett et al [38].
2.2.b. CD38 affects cardiomyocytes’ calcium handling by the sarcoplasmic reticulum
We aimed to characterize how calcium homeostasis might be affected when CD38 is absent. We recorded basal calcium transients in ventricular cardiomyocytes at room temperature and found higher amplitude and faster time decay constant in CD38KO cells (Figure 5a). The higher amplitude could be determined by higher calcium content in the SR of CD38KO cardiomyoctyes. In order to evaluate the SR calcium content, we challenged the cells with a rapid caffeine pulse. Under these conditions, the calcium transient amplitude remained higher in CD38KO cardiomyocytes, confirming a higher SR calcium content in these cells (Figure 5b, left graph). We also measured the decay time constant. This value can be explained by the action of both SERCA2a and NCX at basal conditions and only of NCX when caffeine is applied. Figure 5a–b shows that decay time constant was faster at basal conditions but not after the caffeine pulse, confirming that SERCA2a was more active in CD38KO cardiomyocytes under these conditions. Accordingly, we found an upregulation of SERCA2a levels in heart tissue from CD38KO mice (Figure 5c). The better performance of SERCA2a in CD38KO cardiomyocytes might seem paradoxical at first glance, if we consider the role of cADPR described in the Introduction section. However, CD38 and cADPR regulation operates at physiological temperature [39], and therefore at room temperature, WT cardiomyocytes might lose their effect. Under these conditions, the overexpression of SERCA2a found in CD38KO cardiomyocytes can be seen functionally. Takahashi et al. [26], also showed that male CD38KO mice overexpressed SERCA2a in heart tissue, presumably as a compensatory mechanism. This would prevent CD38KO calcium transients to differentiate from WT transients at physiological temperature. Those authors did report that CD38KO papillary muscle had no differences in calcium transient dynamics at 36°C, but when submitted to 27°C at which cADPR does not work, it showed faster calcium transient time decay constant and higher amplitude than WT muscle. In the present study we show that this compensatory mechanism is dependent on CD38 catalytic activity, as we found that CD38CI hearts overexpressed SERCA2a as well (Figure 5c). However, the exact mechanism by which this compensation occurs is not completely understood. We believe that the overexpression of SERCA2a in these mice could also be attributed to the fact that deacetylation of key proteins by SIRTs are increased, as a possible consequence of the increased NAD+ levels in both genetically modified models (Figure 1b). This could potentially lead to less SERCA2a acetylation in CD38KO and CD38CI mice, which in part would upregulate the expression of the protein [40]. Taken together, the consequence of inhibiting/ablating CD38 would be an overexpression of the calcium-reuptake protein in the SR. We did not evaluate SERCA2a expression in WT hearts after 2-week-treatment with Ab68 but we expect to find higher levels as well.
Figure 5. CD38 affects cardiomyocytes’ calcium handling by the sarcoplasmic reticulum.
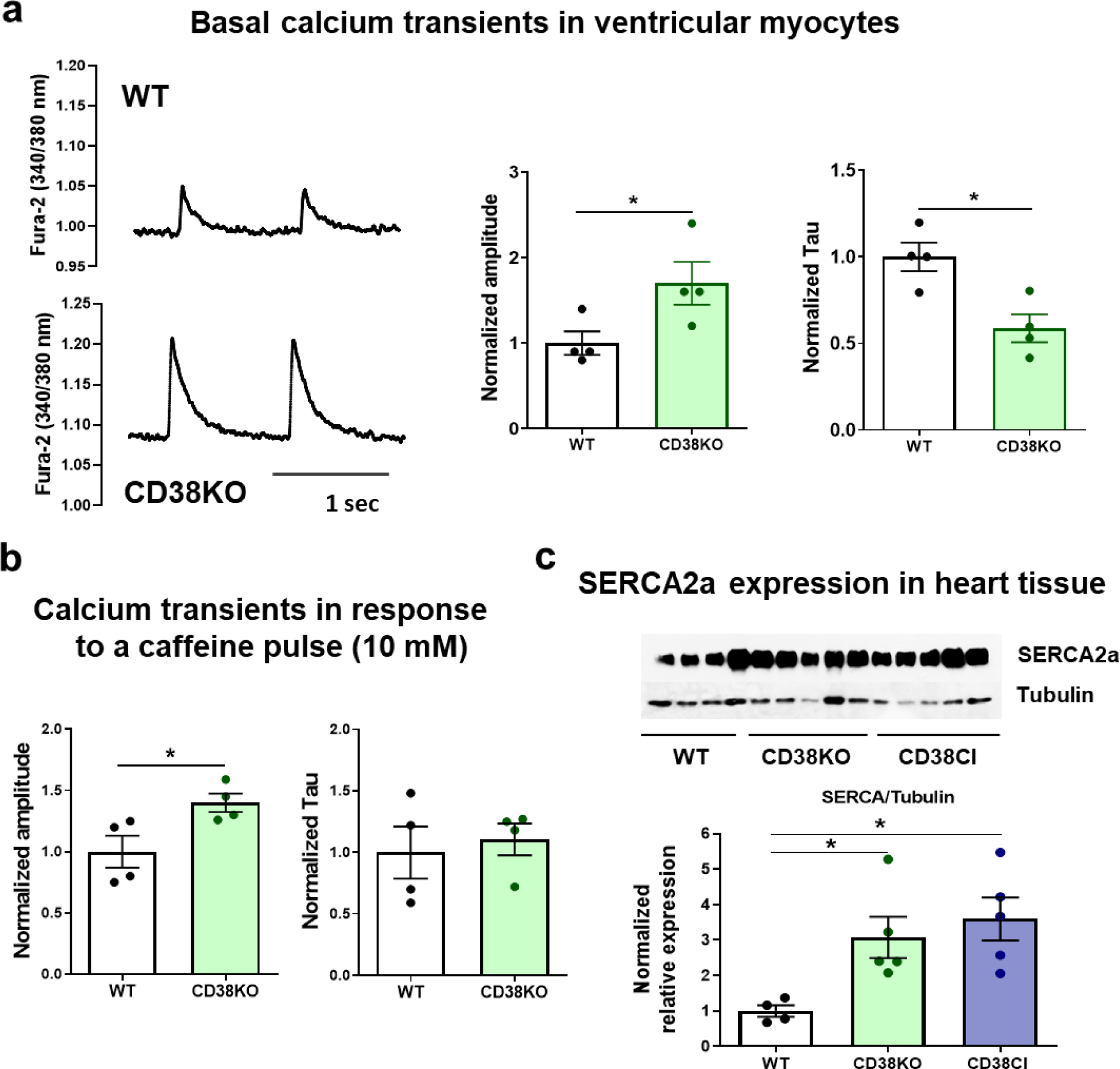
Hearts from WT and CD38KO adult male mice (6–8-month-old) were isolated in order to either obtain cardiomyocytes or lysates for immunoblotting. (a) Representative traces of calcium transients from WT and CD38KO ventricular cardiomyocytes electrically stimulated at 1 Hz at room temperature and its corresponding quantification. Isolated cardiomyocytes from CD38KO mice show increased calcium transient amplitude and faster time decay constant compared to WT cells. Each point in the graphs represents the average value for all the cells recorded for each mice (N=4 mice/group, n=22 and 30 for WT and CD38KO groups respectively). (b) Quantification of calcium transients in response to a caffeine pulse (10 mM). Isolated cardiomyocytes from CD38KO mice show increased calcium transient amplitude in response to caffeine compared to WT cells but the time decay constants are not different between both groups. Each point in the graphs represents the average value for all the cells recorded for each mice (N=4 mice/group, n=8 and 9 for WT and CD38KO groups respectively). (c) Representative immunoblottings of whole cell lysates from hearts of WT, CD38KO and CD38CI (catalytically inactive) mice and its corresponding quantification (N= 4, 5 and 5, for WT, CD38KO and CD38CI groups). SERCA2a expression in the heart was upregulated in both, CD38KO and CD38CI groups compared to WT mice. Data are analyzed by unpaired two-sided t-test, *P<0.05.
The results obtained in calcium experiments, i.e., higher calcium transient amplitude and higher SR calcium content allows to demonstrate that the overexpression of SERCA2a has a functional correlate, which, at physiological temperature might prevent the lack of cADPR from diminishing contractility. We evaluated cardiac function in vivo by echocardiography. The parameters analyzed at basal conditions with the mice mildly sedated do not differentiate CD38KO from WT mice (see Supplementary data and Figure S3). If echocardiography could be done under stressful conditions, like during exercise, it might allow the detection of potential differences when CD38KO mice can take advantage of their metabolic supremacy. Boslett et al. compared developed pressure by isolated hearts from WT and CD38KO mice and did not find differences either [38].
2.2.c. Absence of CD38 diminishes susceptibility to arrhythmias
The altered response to beta adrenergic stimulation (see Supplementary figure 2) and the upregulation of SERCA2a observed in CD38KO hearts (Figure 5c), led us to evaluate the propensity to arrhythmias under stressful conditions. First, we analyzed the spontaneous calcium release from the SR when cardiomyocytes were challenged with high extracellular calcium concentration (6 mM). CD38KO cardiomyocytes show less frequency of calcium sparks under these conditions (Figure 6a) which can evidence less susceptibility to calcium arrhythmias. Considering that these experiments were done at the same temperature than those of calcium transients (Figure 5a–b), this finding implies that a lower frequency of spontaneous events is found despite a higher SR calcium content. In this context, we can hypothesize that RyR2s are less active in CD38KO cardiomyocytes. A possible explanation is that under these stressful conditions, the metabolic advantage of the CD38KO cardiomyocytes prevents their RyR2s to become more active as a consequence of reactive oxygen species excessively emitted by the mitochondria [41].
Figure 6. Lack of CD38 reduces the frequency of spontaneous calcium release from the sarcoplasmic reticulum in vitro and diminishes susceptibility to catecholamine induced arrhythmias in vivo.
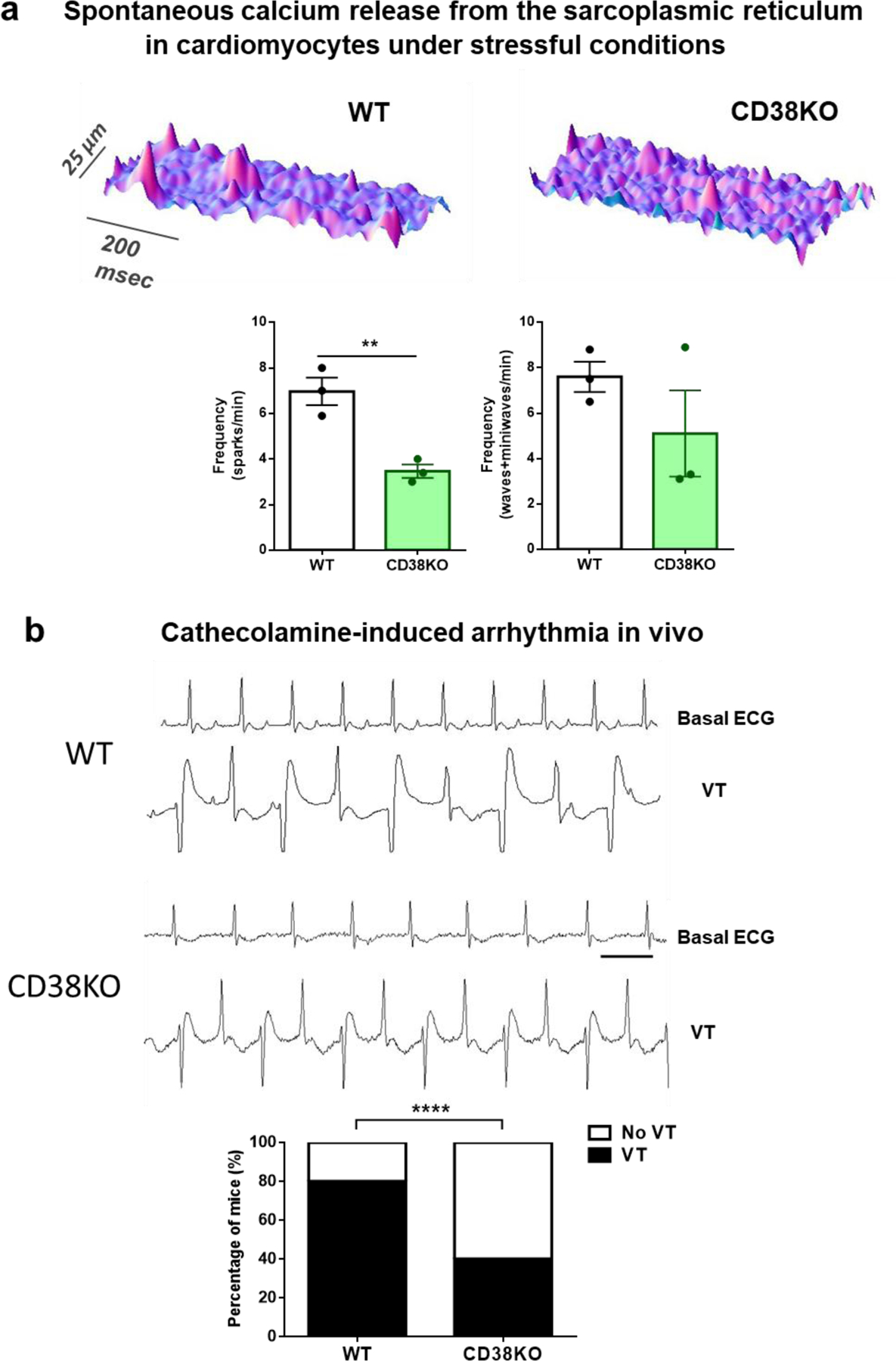
(a) Typical space-time plot of spontaneous sarcoplasmic reticulum calcium releases in representative WT and CD38KO cardiomyocytes under stressful conditions ([Ca2+]e = 6 mM). The graphs show frequency of calcium sparks (left) and frequency of both waves and miniwaves (right) in WT and CD38KO cardiomyocytes. Lack of CD38 reduces the frequency of spontaneous calcium release from the sarcoplasmic reticulum. Each point in the graphs represents the average value for all the cells recorded for each mouse (N=3 mice/group, n=8 and 15 cell for WT and CD38KO groups respectively). Data are mean ± SEM analyzed by unpaired two-sided t-test, **P<0.01. (b) Representative 1 second ECG traces obtained from a WT and a CD38KO mice before (basal) and after the injection of caffeine-epinephrine i.p. (120–2 mg/kg respectively) to induce arrhythmia. As shown in the graph, bidirectional ventricular tachycardia (VT) incidence was reduced in CD38KO group (N= 5 mice per group, aged 9-month-old). The bar represents 100 ms. Data are analyzed by Fisher’s exact test. ****P<0.0001.
We then, wanted to test whether this protection was obtained in vivo. We hypothesized that CD38KO mice would be protected against ventricular arrhythmias induced by over-drive of the sympathetic system, as seen in disorders such as CPVT and heart failure. To test this, we injected caffeine-epinephrine to WT and CD38KO mice [42][43], and registered the ECG by telemetry. We evaluated the incidence of bidirectional ventricular tachycardia (Figure 6b). A smaller proportion of CD38KO mice developed this kind of arrhythmia in response to the caffeine-epinephrine challenge (Figure 6b).
The protection found in CD38KO mice may be mediated by altered adrenergic response and/or a better calcium handling under this pathological-like condition.
The incidence of ventricular arrhythmia in the WT mice shown in Figure 6b seems higher compared to the reports in the bibliography but it was systematically obtained every time we repeated the experiment [44]. Possible explanations for this difference might be the time allowed for recovery after the surgery for telemetry transmitters implantation, the age, and/or the way arrhythmia is evaluated (in our case, both sustained and non-sustained bidirectional ventricular tachycardia was considered).
3. CONCLUSIONS
Our main finding is that inhibition of CD38 activity by different strategies causes an improvement in exercise capacity by boosting NAD+ levels. Besides, we found that as a consequence of this inhibition, cardiovascular physiology presents several modifications: lower basal heart rate and higher heart rate variability, altered response to beta-adrenergic modulation and overexpression of SERCA2a. This inhibition also diminishes the propensity to Ca2+ triggered arrhythmias in vitro and protects mice against catecholamine-induced ventricular arrhythmias. Therefore, CD38 could be considered as a therapeutic target in cardiovascular diseases. Further investigation is needed to better understand the molecular mechanisms behind all the observed effects but considering our promising results, Ab68 could be proposed as a possible pharmacological approach in cardiovascular diseases.
LIMITATIONS
In the present study we only used male mice. In order to generalize our conclusions, the same experiments should be performed in female mice not only because the deficit of CD38 has been shown to be differentially compensated in heart by gender [25] but also due to other differences like susceptibility to ventricular arrhythmias, among others [45].
4. MATERIALS AND METHODS
4.1. Animals
All protocols requiring animal manipulation in the USA were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC), and the studies were carried out in adherence to the NIH Guide for the Care and Use of Laboratory Animals (IACUC protocol #: A00003888). Male mice were used for these experiments. They were housed in standard cages at room temperature and constant humidity, with a 12-hour light-dark cycle, and maintained on a normal chow diet (PicoLab 5053 Rodent Diet 20; Lab Diets) ad libitum.
The CD38KO mice have been described previously [46]. The CD38 catalytically-inactive (CI) “knock-in” mice were generated at TransViragen (Chapel Hill, NC) using recombineering technology, as described previously [31]. All controls used for comparisons with CD38KO and CD38CI were WT mice on a C57BL/6J background, as recommended by the Jackson Laboratory. No littermates were used in this study.
Experiments performed at La Plata, Argentina were in accordance with the Guide for Care and Use of Laboratory Animals (NIH) and approved by the Ethics committee of the Faculty of Medicine, La Plata, Argentina (CICUAL). The same applies for experiments done at Facultad de Medicina, Universidad de la República, Montevideo, Uruguay. In this case, protocols were approved by the CEUA (protocol numbers 70153–000388-17 and 70153–000673-18). The animals used for these experiments were WT and CD38KO mice bred at the Pasteur Institute, Montevideo, Uruguay. WT and whole-body CD38KO mice were in a C57BL6/J pure background. CD38KO mice were backcrossed into C57BL/6J for more than 10 generations in order to ensure genetic purity.
All animals were adults, age is specified for each experiment.
4.2. Drug Treatments
CD38 ecto-enzymatic activity blocker - Ab68:
The antibody UniAb clone ID 337468 (Ab68) (TeneoBio) was used in vivo. Six-month-old WT mice were treated with PBS (vehicle) or Ab68. Mice were given intraperitoneal injections (i.p., 5 mg/kg/dose) at days 1, 5, 9 and 14. On day 16, after a treadmill exhaustion test, mice were euthanized, and tissues were harvested for assays.
NAMPT inhibitor:
Six-month-old CD38KO mice received FK866 (25 mg/kg/dose, i.p.) once daily for a week. Control groups (WT and CD38KO mice) received equivalent injections of vehicle for FK866 (1% Hydroxypropyl-β-cyclodextrin, 12% Propylene glycol) once daily also for a week. On day 8, after a treadmill exhaustion test, mice were euthanized and tissues were harvested for assays.
This drug (FK866) and all other reagents were purchased from Sigma-Aldrich unless specified.
4.3. In vivo studies
4.3.a. Exercise capacity: Treadmill exhaustion test
Exercise capacity was assessed by measuring running time, distance, maximal running speed, and work using a motorized treadmill (Columbus Instruments) in the different experiments performed as described. The mice were acclimated to the treadmill for 3 consecutive days, at an initial speed of 5 m/min and a grade of 5%, with a 2 m/min increase in speed every 2 minutes, for 5 minutes. After a day of rest, animals ran on the treadmill at an initial speed of 5 m/min and 5% grade for 2 minutes, after which the speed was increased by 2 m/min every subsequent 2 minutes until the mice were exhausted. Exhaustion was defined as the inability of the mouse to remain on the treadmill despite an electrical shock stimulus for more than 10 seconds.
Running time was recorded, running distance was given by the software (Columbus Instruments), and work was calculated (the product of body weight [kg], gravity [9.81 m/s2], vertical speed [m/s × angle], and time [s]) [47].
4.3.b. ECG recording by telemetry
4.3.b.1. Heart rate and HRV evaluation in conscious unrestrained mice
We recorded the cardiac electrical activity (1-lead ECG) in WT, CD38KO and CD38CI mice (N=6 in each group). A radiofrequency transmitter (EA-F20, Data Sciences International DSI, St. Paul, MN) with 2 wire leads was implanted subcutaneously in the mice under anesthesia with isoflurane (2.5% at 0.5 lpm O2). The electrodes were sutured to the chest muscles (lead II-like configuration) and the skin was closed with metal clips. Mice were allowed to recover for 48 h.
The ECGs of the conscious freely moving mice were recorded for 30 min in the morning (8–11 am) the subsequent 2 days.
The sampling frequency was 1000 Hz. Automatic detection of R waves and measurement of RR intervals were performed offline. The detection was visually corrected and the RR time series automatically filtered before calculation of the mean heart rate (HR) and the statistical HRV indices: SDNN (the standard deviation of all normal RR intervals) and RMSSD (square root of the mean squared successive differences of RR intervals). SDNN is an index of global variability, while RMSSD evaluates short-term variability on a beat-to-beat basis (parasympathetic influences). The variability coefficient (VC) was also calculated as SDNN (ms)/mean RR interval (ms) × 100 (relative standard deviation).
Values for each mouse were obtained as the average of HR and HRV indexes for the 2 ECG recordings.
4.3.b.2. Caffeine-epinephrine-induced ventricular arrhythmia.
We implanted telemetry transmitters in 5 WT and 5 CD38KO male mice (9-month-old) as described in 4.3.b.1. After 15 minutes of basal ECG recording, mice were injected intraperitoneally with a solution containing 120 mg/kg caffeine and 2 mg/kg epinephrine (ICU-VITA Laboratories, Uruguay). ECG recording continued for 30 minutes. Mice were then euthanized by cervical dislocation. ECG recordings were analyzed off- line by a blinded researcher looking for bidirectional ventricular tachycardia episodes.
4.4. Intracellular Ca2+ measurements in isolated cardiomyocytes
4.4.a. Basal calcium transients
Ventricular cardiomyocytes from 6–7-month-old WT and CD38KO mice were isolated via enzymatic digestion as described previously [48]. Briefly, mice hearts were perfused as indicated by the Langendorff technique via the aorta for 5 minutes with oxygenated Krebs-Henseleit buffer (containing the following in mM: NaCl, 146.2; KCl, 4.7; CaCl2 1.35; MgSO4, 1.05; NaH2PO4, 0.35; glucose, 10.0; and HEPES, 10.0; pH 7.40 at 37°C). The perfusion was then switched to Ca2+ free solution for 5 minutes. Afterwards, an enzymatic solution containing collagenase 0.7 mg/ml, pronase 0.1 mg/ml, and 1% albumin, was used and the perfusion was maintained for another 10–15 minutes. The remaining tissue was minced and shaken in enzymatic solution for another 20–30 minutes. Cardiomyocytes were collected and made Ca2+ tolerant over 15 minutes. The cells were then re-suspended in 2 mM Ca2+ Tyrode solution and maintained at room temperature.
Isolated cardiomyocytes were then loaded with Fura-2/AM (10 μM), and superfused with 2 mM Ca2+ Tyrode solution at pH 7.4, room temperature, and electrically stimulated with a biphasic pulse at 1 Hz. Cells included were only the ones with clear striation pattern, quiescent in the absence of electrical stimulation and with a clear contraction response to stimulation. Intracellular Ca2+ transients were recorded online using a dual excitation epifluorescence system (IonOptix), as described previously [48].
4.4.b. Calcium transient in response to a caffeine pulse
After recording calcium transients as described in 4.4.a., electrical stimulation was stopped and a 10 mM caffeine pulse was given to cardiomyocytes by changing the perfusion solution and increasing the perfusion flow. Fluorescence data were analyzed off-line with the ION WIZARD software.
4.4.c. Detection of spontaneous Ca2+ release by confocal microscopy
Cardiomyocytes were loaded with 10 μM Fluo-3 AM (Molecular Probes), dissolved in 20% F-127 pluronic in dimethylsulfoxide (DMSO). After 10 minutes of loading at room temperature, cells were washed and seeded on a perfusion chamber. The chamber was mounted on a Zeiss 410 inverted confocal microscope (LSMTech, Pennsylvania, USA) equipped with a 63x, 1.4 NA oil immersion objective. Cells were imaged in linescan mode along their long axis, exciting with the 488 nm line of an argon laser and collecting emission at 500–550 nm [49]. Fluo-3 fluorescence was recorded in linescan mode, with a 512 pixels line placed along the long axis of the cell. Each recording consisted of 512 consecutive linescans (4.3 msec per line), which were stacked to create space-time profiles which were analyzed with Sparkmaster pluging for Image J [50].
In order to analyze the spontaneous calcium release from the SR, the chamber solution was changed to a high calcium concentration of 6 mM. Extracellular calcium concentration was increased in steps to prevent cellular damage (2, 3, 4, 5 and 6 mM).
4.5. Measurement of NAD+ levels in the heart by Cycling Assay
Detection of NAD+ was performed as described before using a cycling assay [51]. Between 15 to 20 mg of tissue was homogenized in 10% trichloroacetic acid (TCA). Samples were centrifuged at 12,000 rpm for 2 min at 4°C. The supernatants were collected, and the pellets were resuspended in 0.2 M NaOH for protein quantification. TCA was removed with organic solvents (3 volumes 1,1,2-trichloro-1,2,2-trifluroethane: 1 volume trioctylamine) in a ratio of 2 volumes of organic solvent to 1 volume of sample. The resulting top aqueous layer containing NAD+ was recovered and 1M Tris pH 8.0 was added for the pH correction. Samples were then diluted in 100 mM sodium phosphate buffer pH 8 in a volume of 100 μL/ well and added to white 96 well plates. For the cycling assay, 100 μL of reaction mix (0.76% ethanol, 4 μM FMN, 27.2 U/ mL alcohol dehydrogenase (ADH), 1.8 U/ mL Diaphorase, and 8 μM resazurin) was added to each well. 96-well plates were read in a fluorescence plate reader (Molecular Devices, SpectraMax® Gemini™ 1579 XPS) in an excitation wavelength of 544 nm and an emission wavelength of 590 nm.
4.6. NADase activity in heart tissue
NADase activity in heart tissue was measured as described previously [52]. Briefly, we used 50 μM nicotinamide 1,N6- ethenoadenine dinucleotide (ε-NAD) as a substrate in 0.25 M sucrose and 40 mM Tris HCl (pH 7.4). Fluorescence was measured at an excitation wavelength of 300 nm and an emission wavelength of 410 nm.
4.7. Western Blot Analysis
Mouse heart tissue was lysed in RIPA buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) supplemented with 5 mM NaF, 50 mM 2-glycerophosphate, and a protease inhibitor cocktail (Roche Diagnostics). After 20 minutes of lysis, protein lysates were cleared by centrifugation at 12,000 rpm at 4°C for 10 minutes. The resulting supernatants were quantified for protein using a BioRad protein assay. The proteins were fractionated by SDS-PAGE, electro-transferred to Immobilon-P membranes, blotted with the indicated primary antibodies followed by appropriate horseradish peroxidase-conjugated secondary antibodies, and detected using SuperSignal West Pico or Femto Chemiluminescence Substrate (Thermo Fisher Scientific). Membranes were stripped and probed with tubulin antibody to control for equal gel loading and transfer. Films were scanned and densitometry was performed using ImageJ software.
4.8. Statistical Analysis
Data are shown as mean ± SEM, analyzed by unpaired 2-sided t-test. In order to compare proportions, Fisher’s exact test was used. Analyses were performed using Microsoft Excel 2010 and GraphPad Prism 6.
Supplementary Material
HIGHLIGHTS.
CD38 inhibition in male mice increases exercise capacity and NAD+ in the heart.
Improvement of exercise capacity is dependent on NAD+ boosting.
CD38 suppression lowers heart rate (HR) and increases HR variability in vivo.
CD38 knockout mice are less susceptible to catecholamine-driven arrhythmia.
ACKNOWLEDGEMENTS
The authors thank Ariel Escobar, Alicia Mattiazzi, Edith Moraes, Cecilia Vilaseca, Gina Warner and Katie Thompson for their help during the research.
FUNDING
This work was partially supported by CAP (Comisión Académica de Posgrado), CSIC (Comisión Sectorial de Investigación Científica), PEDECIBA, Universidad de la República; and FOCEM - Fondo para la Convergencia Estructural del Mercosur (COF 03/11). The work in E.N.C.’s laboratory is supported in part by grants from the Helen Diller Family Foundation, Glenn Foundation for Medical Research via the Paul F. Glenn Laboratories for the Biology of Aging at the Mayo Clinic (to E.N.C.); and NIH National Institute on Aging (NIA) grants AG-26094, AG58812, and CA233790 (to E.N.C.). The funding sources played no role in any of the stages of this study.
Abbreviations:
- cADPR
cyclic adenosine diphosphate ribose
- ATP
adenosine triphosphate
- CD38
cluster of differentiation 38
- CD38KO
CD38 knock out
- CD38CI
CD38 catalytically inactive
- CICR
calcium induced calcium release
- FK866
inhibitor of NAMPT
- HR
heart rate; HRV, heart rate variability
- NAD+
nicotinamide adenine dinucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- NMN
nicotinamide mononucleotide
- RMSSD
square root of the mean squared successive differences of RR intervals
- SERCA2a
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a
- SDNN
the standard deviation of all normal RR intervals
- SIRT
Sirtuin
- VC
variability coefficient
Footnotes
DISCLOSURES
ENC holds a patent (CD38 inhibitors and metabolism, Elysium health); is a consultant for Calico, Mitobridge, Cytokinetics, and TeneoBio (also owns stocks). His research was in compliance with Mayo Clinic conflict of interest policies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem Biophys Res Commun. 2006;345:1386–92. doi: 10.1016/j.bbrc.2006.05.042 [DOI] [PubMed] [Google Scholar]
- [2].Ball EG. The development of our current concepts of biological oxidations. Mol Cell Biochem. 1974. Nov;5(1–2):35–46. doi: 10.1007/BF01874170 [DOI] [PubMed] [Google Scholar]
- [3].Strømland Ø, Niere M, Nikiforov AA, Van Linden MR, Heiland I, Ziegler M. Keeping the balance in NAD metabolism. Biochem Soc Trans. 2019. Feb;47(1):119–30. doi: 10.1042/BST20180417 [DOI] [PubMed] [Google Scholar]
- [4].Racker E From Pasteur to Mitchell: a hundred years of bioenergetics. Fed Proc. 1980. Feb;39(2):210–5. [PubMed] [Google Scholar]
- [5].Weiss RG, Maslov M. Normal myocardial metabolism: Fueling cardiac contraction. Adv Stud Med. 2004. Jun 1;4:S457–63. [Google Scholar]
- [6].Krejčí A Metabolic sensors and their interplay with cell signalling and transcription. Biochem Soc Trans. 2012. Mar 21;40(2):311–23. doi: 10.1042/BST20110767 [DOI] [PubMed] [Google Scholar]
- [7].Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500 [DOI] [PubMed] [Google Scholar]
- [8].Camacho-Pereira J, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23(6):1127–39. doi: 10.1016/j.cet.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lin WK, Bolton EL, Cortopassi WA, Wang Y, O’Brien F, Maciejewska M, et al. Synthesis of the Ca2+-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in -adrenoceptor signaling. J Biol Chem. 2017;292(32):13243–57. doi: 10.1074/jbc.M117.789347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cui Y, Galione A, Terrar DA. Effects of photoreleased cADP-ribose on calcium transients and calcium sparks in myocytes isolated from guinea-pig and rat ventricle. Biochem J. 1999;342:269–73. [PMC free article] [PubMed] [Google Scholar]
- [11].Lukyanenko V, Györke I, Wiesner TF, Györke S. Potentiation of Ca2+ release by cADP-ribose in the heart is mediated by enhanced SR Ca2+ uptake into the sarcoplasmic reticulum. Cir Res. 2001;(89):614–22. doi: 10.1161/hh1901.098066 [DOI] [PubMed] [Google Scholar]
- [12].Park D-R, Nam T-S, Kim Y-W, Lee S-H and Kim U-H. CD38-cADPR-SERCA Signaling Axis Determines Skeletal Muscle Contractile Force in Response to β-Adrenergic Stimulation. Cell Physiol Biochem. 2018, 46(5):2017–30. doi: 10.1159/000489441 [DOI] [PubMed] [Google Scholar]
- [13].Venturi E, Pitt S, Galfré E, & Sitsapesan R (2012). From Eggs to Hearts: What Is the Link between Cyclic ADP-Ribose and Ryanodine Receptors? Cardiovascular therapeutics, 2012; 30(2), 109–116. doi: 10.1111/j.1755-5922.2010.00236.x [DOI] [PubMed] [Google Scholar]
- [14].Higashida H, Hashii M, Yokoyama S, Hoshi N, Asai K, Kato T. Cyclic ADP-ribose as a potential second messenger for neuronal Ca2+ signaling. J Neurochem. 2001;76(2):321–31. doi: 10.1046/.j1471-4159.2001.00082.x [DOI] [PubMed] [Google Scholar]
- [15].Albrieux M, Lee HC, Villaz M. Calcium signaling by cyclic ADP-ribose, NAADP, and inositol trisphosphate are involved in distinct functions in ascidian oocytes. J Biol Chem. 1998;273(23):14566–74. doi: 10.1074/jbc.273.23.14566 [DOI] [PubMed] [Google Scholar]
- [16].Gul R, Park D, Shawl AI, Im S, et al. Nicotinic Acid Adenine Dinucleotide Phosphate ( NAADP ) and Cyclic ADP-Ribose ( cADPR ) Mediate Ca2+ Signaling in Cardiac Hypertrophy Induced by β -Adrenergic Stimulation. PLoS One. 2016;11(3):e0149125. doi: 10.1371/journal.pone.0149125. eCollection 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kannt A, Sicka K, Kroll K, et al. Selective inhibitors of cardiac ADPR cyclase as novel anti-arrhythmic compounds. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(7):717–27. doi: 10.1007/s00210-012-0750-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gaztañaga L, Marchlinski FE, Betensky BP. Mechanisms of Cardiac Arrhythmias. Rev Esp Cardiol. 2012;65(2):174–85. doi: 10.1016/j.recesp.2011.09.018 [DOI] [PubMed] [Google Scholar]
- [19].Lieve KV, van der Werf C, Wilde AA. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ J. 2016;80(6):1285–91. doi: 10.1253/circj.CJ-16-0326 [DOI] [PubMed] [Google Scholar]
- [20].Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology ( ESC ) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–200. doi: 10.1093/eurjeartj/ehw128 [DOI] [PubMed] [Google Scholar]
- [21].Gul R, Park J-H, Kim S-Y, Jang KY, Chae J-K, Ko J-K, et al. Inhibition of ADP-ribosyl cyclase attenuates angiotensin II-induced cardiac hypertrophy. Cardiovasc Res. 2009;81(3):582–91. doi: 10.1093/cvr/cvn232 [DOI] [PubMed] [Google Scholar]
- [22].Guan X-H, Hong X, Zhao N, Liu X-H, Xiao Y-F, Chen T-T et al. CD38 promotes angiotensin II-induced cardiac hypertrophy. J Cell Mol Med. 2017;21(8):1492–502. doi: 10.1111/jcmm.13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guan X-H, Liu X-H, Hong X, Zhao N, Xiao Y-F, Wang L-F, et al. CD38 Deficiency Protects the Heart from Ischemia / Reperfusion Injury through Activating SIRT1 / FOXOs-Mediated Antioxidative Stress Pathway. Oxid Med Cell Longev. 2016;2016:7410257. doi: 10.1155/2016/7410257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reyes LA, Boslett J, Varadharaj S, De Pascali F, Hemann C et al. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc Natl Acad Sci. 2015;112(37):11648–53. doi: 10.1073/pnas.1505556512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boslett J, Reddy N, Alzarie YA, Zweier JL. Inhibition of CD38 with the Thiazoloquin (az)olin(on)e 78c Protects the Heart against Postischemic Injury. J Pharmacol Exp Ther. 2019;369(1):55–64. doi: 10.1124/jpet.118.254557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Takahashi J, Kagaya Y, Kato I, Ohta J, Isoyama S, Miura M, et al. Deficit of CD38/cyclic ADP-ribose is differentially compensated in hearts by gender. Biochem Biophys Res Commun. 2003;312(2):434–40. doi: 10.1016/j.bbrc.2003.10.143 [DOI] [PubMed] [Google Scholar]
- [27].Zuo W, Liu N, Zeng Y, Liu Y, Li B, Wu K, et al. CD38 : A Potential Therapeutic Target in Cardiovascular Disease. Cardiovasc Drugs Ther. 2021;35(4):815–28. doi: 10.1007/s10557-020-07007-8 [DOI] [PubMed] [Google Scholar]
- [28].Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11(9):535–46. doi: 10.1038/natrendo.2015.117 [DOI] [PubMed] [Google Scholar]
- [29].Chini CCS, Peclat TR, Warner GM, Kashyap S, Machado Espindola-Netto J, de Oliveira GC, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat Metab. 2020;2(11):1284–304. doi: 10.1038/s42255-020-00298-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boslett J, Hemann C, Christofi FL, Zweier JL. Characterization of CD38 in the major cell types of the heart: endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am J Physiol Cell Physiol. 2018;314(3):C297–309. doi: 10.1152/ajpcell.00139.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tarragó MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018;27(5):1081–95.e10. doi: 10.1016/j.cmet.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chiang SH, Harrington WW, Luo G, Milliken NO, Ulrich JC, Chen J, Billin AN, et al. Genetic ablation of CD38 protects against western diet-induced exercise intolerance and metabolic inflexibility. PLoS One, 2015;10(8), e0134927 doi: 10.1371/journal.pone.0134927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reimers AK, Knapp G, Reimers C-D. Effects of Exercise on the Resting Heart Rate : A Systematic Review and Meta-Analysis of Interventional Studies. J Clin Med. 2018;7(12):503. doi: 10.3390/jcm7120503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thai TL, Arendshorst WJ. Mice lacking the ADP ribosyl cyclase CD38 exhibit attenuated renal vasoconstriction to angiotensin II, endothelin-1, and norepinephrine. Am J Physiol Physiol Renal Phsysiol. 2009;297(1):F169–76. doi: 10.1152/ajprenal.00079.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pomeranz B, Macaulay RJ, Caudill MA, Kutz I, Adam D, Gordon D, et al. Assessment of autonomic function in humans by heart rate spectral analysis. Am J Physiol. 1985;248:H151–3. doi: 10.1152/ajpheart.1985.248.1.H151 [DOI] [PubMed] [Google Scholar]
- [36].Migliaro ER, Contreras P, Bech S, Etxagibel A, Castro M, Ricca R, et al. Relative influence of age, resting heart rate and sedentary life style in short-term analysis of heart rate variability. Braz J Med Biol Res. 2001;34(4):493–500. doi: [DOI] [PubMed] [Google Scholar]
- [37].Contreras P, Migliaro ER, Suhr B. Right atrium cholinergic deficit in septic rats. Auton Neurosci 2014;180:17–23. doi: 10.1016/j.autneu.2013.10.002 [DOI] [PubMed] [Google Scholar]
- [38].Boslett J, Helal M, Chini E, & Zweier JL Genetic deletion of CD38 confers post-ischemic myocardial protection through preserved pyridine nucleotides. Journal of molecular and cellular cardiology, 2018; 118, 81–94. doi: 10.1016/j.yjmcc.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Iino S, Cui Y, Galione A and Terrar DA. Actions of cADP-ribose and its antagonists on contraction in guinea pig isolated ventricular myocytes. Circ Res. 1997;81(5):879–84. doi: 10.1161/01.res.81.5.879 [DOI] [PubMed] [Google Scholar]
- [40].Gorski PA, Jang SP, Jeong D, Lee A, Lee P, Oh JG, et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca(2+)-ATPase in Heart Failure. Circ Res. 2019;124(9):e63–80. doi: 10.1161/CIRCRESAHA.118.313865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, Terentyev D, et al. , Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic research in cardiology, 2020; 115, 1–20. doi: 10.1007/s00395-020-0797-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].van Oort RJ, McCauley MD, Dixit SS, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122(25):2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101(10):1039–48. doi: 10.1161/CIRCRESAHA.107.148064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jelinek M, Wallach C, Ehmke H, & Schwoerer AP Genetic background dominates the susceptibility to ventricular arrhythmias in a murine model of β-adrenergic stimulation. Scientific reports, 2018; 8(1), 1–10. doi: 10.1038/s41598-018-20792-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ehdaie A, Cingolani E, Shehata M, Wang X, Curtis AB, & Chugh SS Sex differences in cardiac arrhythmias: clinical and research implications. Circulation: Arrhythmia and Electrophysiology, 2018; 11(3), e005680.. doi: 10.1161/CIRCEP.117.005680 [DOI] [PubMed] [Google Scholar]
- [46].Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med. 2001;7(11):1209–16. doi: 10.1038/nm1101-1209 [DOI] [PubMed] [Google Scholar]
- [47].LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64(9):940–8. doi: 10.1093/gerona/glp068 [DOI] [PubMed] [Google Scholar]
- [48].Palomeque J, Velez Rueda O, Sapia L, Valverde CA, Salas M, Petroff MV, et al. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105(12):1204–12. doi: 10.1161/CIRCRESAHA.109.204172 [DOI] [PubMed] [Google Scholar]
- [49].Gonano LA, Sepúlveda M, Morell M, Toteff T, Racioppi MF, Lascano E, Petroff MV, et al. Non-β-blocking carvedilol analog, VK-II-86, prevents ouabain-induced cardiotoxicity. Circulation Journal, 2018; CJ-18. doi: 10.1253/circj.CJ-18-0247 [DOI] [PubMed] [Google Scholar]
- [50].Picht E, Zima AV, Blatter LA, & Bers DM SparkMaster: automated calcium spark analysis with ImageJ. American Journal of Physiology-Cell Physiology, 2007; 293(3), C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- [51].Kanamori KS, de Oliveira GC, Auxiliadora-Martins M, Schoon RA, Reid JM, Chini EN. Two Different Methods of Quantification of Oxidized Nicotinamide Adenine Dinucleotide (NAD(+)) and Reduced Nicotinamide Adenine Dinucleotide (NADH) Intracellular Levels: Enzymatic Coupled Cycling Assay and Ultra-performance Liquid Chromatography (UPLC)-Mass Spectrometry. Bio Protocol. 2018;8(14):e2937. doi: 10.21769/BioProtoc.2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].de Oliveira GC De, Kanamori KS, Auxiliadora-Martins M, Chini CCS, Chini EN.Measuring CD38 Hydrolase and Cyclase Activities: 1,N6-Ethenonicotinamide Adenine Dinucleotide (ε-NAD) and Nicotinamide Guanine Dinucleotide (NGD) Fluorescence-based Methods. Bio Protoc. 2018;8(14):e2938.doi: 10.21769/BioProtoc.2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.