

SUMMARY
Tight regulation of mRNA isoform expression is essential for neuronal development, maintenance, and function; however, the repertoire of proteins that govern isoform composition and abundance remains incomplete. Here, we show that the RNA kinase CLP1 regulates mRNA isoform expression through suppression of proximal cleavage and polyadenylation. We found that human stem-cell-derived motor neurons without CLP1 or with the disease-associated CLP1 p.R140H variant had distinct patterns of RNA-polymerase-II-associated cleavage and polyadenylation complex proteins that correlated with polyadenylation site usage. These changes resulted in imbalanced mRNA isoform expression of long genes important for neuronal function that were recapitulated in vivo. Strikingly, we observed the same pattern of reduced mRNA isoform diversity in 3′ end sequencing data from brain tissues of patients with neurodegenerative disease. Together, our results identify a previously uncharacterized role for CLP1 in mRNA 3′ end formation and reveal an mRNA misprocessing signature in neurodegeneration that may suggest a common mechanism of disease.
In brief
LaForce et al. utilize motor neuron disease models to examine CLP1 function in mRNA 3′ end formation. CLP1 suppresses proximal polyadenylation to regulate mRNA isoform balance and maintain neuronal health. The authors find that diminished isoform diversity is a signature of neurodegenerative disease.
Graphical Abstract
INTRODUCTION
Differential co-transcriptional processing is the foundation of mRNA isoform diversity. Multiple transcript isoforms can be generated from a single gene, and the ratio of expressed isoforms across all genes is a key determinant of cell-type specification (Belgard et al., 2011; Gupta et al., 2018; Kazantseva et al., 2009; Lerch et al., 2012; Shulman and Elkon, 2019). The usage of alternative transcription termination sites (TTSs) (alternative polyadenylation, APA) results in transcriptome 3′ end diversity, a major driver of tissue-specific isoform diversity (Reyes and Huber, 2018). APA generates mRNA isoforms with different 3′ untranslated region (UTR) sequences harboring regulatory information that dictate mRNA localization, stability, and translation (Davis and Shi, 2014; Guhaniyogi and Brewer, 2001; Mayr, 2016; Turner et al., 2018). APA can also alter the coding potential of mRNA isoforms to create distinct proteins that differ in activity or function (Hwang et al., 2017). Neurons express a greater diversity of mRNA isoforms as well as longer genes than most cell types (Gabel et al., 2015; He et al., 1998; King et al., 2013; Liu et al., 2011; Miura et al., 2013). Together, these determinants may contribute to selective neuronal vulnerability to RNA splicing defects (Alcott et al., 2020; Hutton et al., 1998; Lefebvre et al., 1995; Lin et al., 1998). The importance of transcriptome 3′ end diversity in neurons and how its disruption impacts neuronal function and survival is unclear.
At least 20 proteins comprise the core cleavage and polyadenylation (CPA) machinery involved in mRNA 3′ end processing in mammalian cells (Shi et al., 2009; Turner et al., 2018). The machinery was initially purified as four distinct subcomplexes—cleavage and polyadenylation specificity factor (CPSF), cleavage stimulating factor (CstF), mammalian cleavage factor I (CFIm), and mammalian cleavage factor II (CFIIm). CFIIm is the smallest subcomplex, consisting only of PCF11 and CLP1 (de Vries et al., 2000). PCF11 enhances transcription termination and 3′ end processing of closely spaced genes and attenuates expression of genes typically encoding transcriptional regulators that are enriched for intronic polyadenylation (IPA) sites (Kamieniarz-Gdula et al., 2019; Wang et al., 2019). In contrast, CLP1 function in mRNA 3′ end processing is poorly characterized. CLP1 is an RNA kinase (Weitzer et al., 2015), first identified as a component of the CPA complex (de Vries etal., 2000) and later as a component of the intron-containing tRNA-splicing complex (Paushkin et al., 2004; Weitzer and Martinez, 2007). CLP1 function in tRNA processing is well characterized (Weitzer et al., 2015), but its role in co-transcriptional mRNA 3′ end processing has proven elusive (de Vries et al., 2000; Guyon et al., 2020; Li et al., 2015; Ogorodnikov et al., 2018).
We previously identified a homozygous p.R140H variant in CLP1 as causative for the neurodegenerative disease pontocerebellar hypoplasia type 10 (PCH10) (Schaffer et al., 2014). Patients with PCH10 present with cerebellar and brainstem atrophy, progressive microcephaly, tonic-clonic seizures, and motor neuron disease (Karaca et al., 2014; Schaffer et al., 2014; Wafik et al., 2018). CLP1 p.R140H has reduced kinase activity and impaired association with PCF11 (Karaca et al., 2014; Schaffer et al., 2014, Figure S1A), which may disrupt mRNA 3′ end processing. Given the profound neurodevelopmental impact of this variant and its observed disruption of CFIIm-complex integrity, we sought to understand the role of CLP1 in the regulation of mRNA isoform diversity in neurons. Therefore, we generated mutation-specific in vitro and in vivo models to define the function of CLP1 in mRNA 3′ end processing in motor neurons. We show CLP1 suppresses proximal polyadenylation site (PAS) usage, thereby regulating expression of long genes that are important for neuronal function. Lastly, we establish this novel mRNA transcriptomic misprocessing signature as a shared feature among neurodegenerative diseases.
RESULTS
CLP1 is required for motor neuron differentiation
Patients with PCH10 have penetrant motor neuron degeneration (Karaca et al., 2014; Schaffer et al., 2014); therefore, we chose to profile the motor neuron transcriptome to gain insight into the molecular changes associated with CLP1 perturbation. We utilized in-vitro-derived human motor-neuron-enriched cell cultures to measure transcriptome 3′ end diversity (Figure 1A). We generated a compound heterozygous deletion in exon two of CLP1 in H9 human embryonic stem cells (hESCs) using CRISPR-Cas9 gene editing (Figures S1B and S1C), creating a CLP1 null (CLP1KO) cell line (Figure 1B). As a control, we used isogenic H9 hESCs that underwent mock gene editing (control, CLP1WT). We then differentiated CLP1KO and CLP1WT hESCs to spinal motor neurons using an established protocol (Markmiller et al., 2018). CLP1KO motor neuron cultures differentiated at a significantly lower density than CLP1WT controls but were otherwise morphologically indistinct (Figure 1C). Both cultures expressed the motor-neuron-specific transcription factors ISLET-1/2 and HB9 (Figure S1D, left) as well as SCIP and HOXA5 (Figure S1D, right). Medial and hypaxial motor column-like motor neuron markers FOXP1, HOXC6, LHX3, and HOXC9 were undetected (Dasen et al., 2008; Jung et al., 2010; data not shown), indicating our hESC-derived motor neurons had a phrenic-like identity (Machado et al., 2014; Philippidou et al., 2012).
Figure 1. CLP1 is not required in hESCs but impairs directed differentiation to motor neurons.
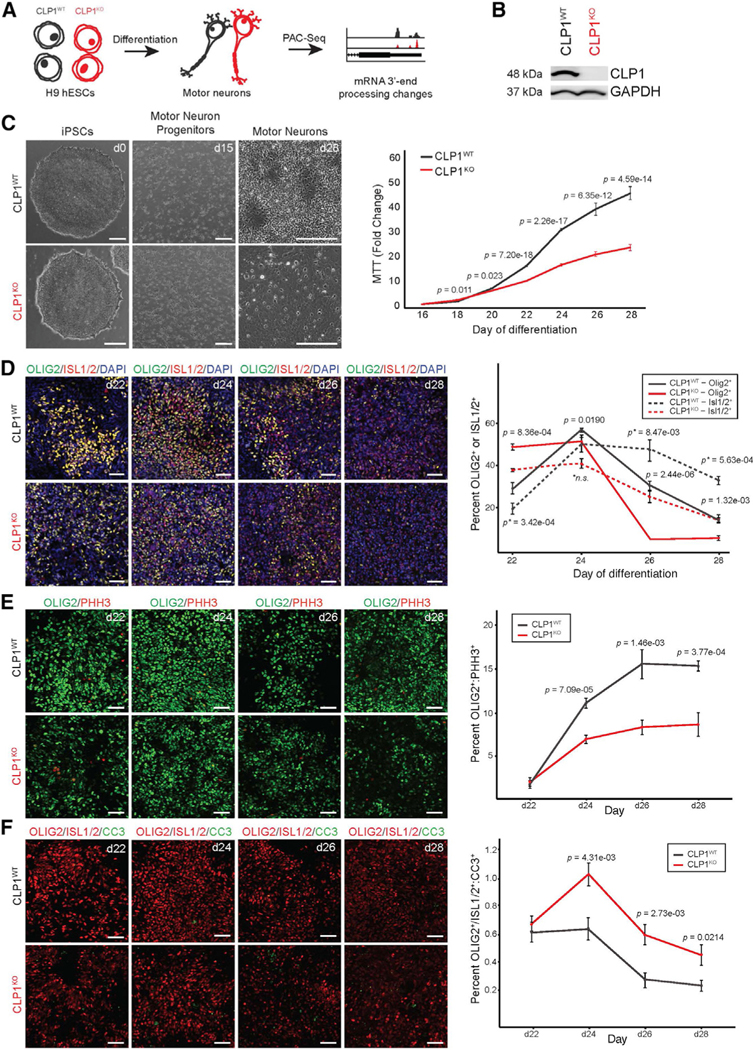
(A) Study schematic. H9 hESCs lacking CLP1 (CLP1KO) and isogenic controls (CLP1WT) were differentiated into motor neurons and profiled for changes in mRNA 3′ end processing by PAC-seq.
(B) Western blot for CLP1 in CLP1KO and isogenic CLP1WT H9 hESCs. GAPDH, loading control; n = 3 replicates.
(C) Representative brightfield images of CLP1KO and CLP1WT hESCs during motor neuron differentiation (left). Scale bars, 250 μm. MTT assay of CLP1KO and CLP1WT hESC-derived motor neurons during differentiation is depicted (right); n = 8 technical replicates. Three clonal hESC lines were used as biological replicates; two-sided Student’s t test.
(D) Immunostaining for OLIG2 and ISL1/2 in CLP1KO and CLP1WT cultures at day 22 of motor neuron differentiation. p, OLIG2+ statistics; p*, ISL1/2+ statistics; n.s., non-significant (p > 0.05); two-sided Student’s t test; n = 6, 20× fields-of-view per clone. Representative images are shown from three clonal lines per genotype. Scale bars, 50 μm.
(E) Analysis of OLIG2+ motor neuron progenitor cell proliferation by PHH3 immunostaining in CLP1WT and CLP1KO cultures during differentiation (days 22–28); two-sided Student’s t test; n = 6, 20× fields-of-view per clone. Representative images are shown from three clonal lines per genotype. Scale bars, 50 μm.
(F) Analysis of motor neuron (OLIG2+ or ISL1/2+) apoptosis by CC3 immunostaining in CLP1WT and CLP1KO cultures during differentiation; two-sided Student’s t test; n = 6, 20× fields-of-view per clone. Representative images are shown from three clonal lines per genotype. Scale bars, 50 μm. See also Figure S1.
To investigate whether the reduced cell density observed in CLP1KO motor neuron cultures was caused by an altered rate of differentiation, we performed immunostaining for markers of motor neurons (ISL1/2) and motor neuron progenitors (OLIG2) from day 22 to day 28 of differentiation (Figure 1D, left). CLP1KO motor neuron cultures had fewer progenitors and mature motor neurons throughout differentiation compared to CLP1WT controls (Figure 1D, right). An early increase in the proportion of OLIG2 and ISL1/2 co-positive cells in CLP1KO motor neuron cultures at day 22 suggested an arrest in differentiation. However, the proportion of OLIG2 and ISL1/2 co-positive cells significantly decreased from day 24 to day 28 of differentiation, likely due to the loss of cells expressing these markers (Figure S1E).
Next, we investigated whether diminished progenitor cell proliferation or elevated apoptosis was responsible for the decrease of motor neuron cell types and density observed in CLP1KO motor neuron cultures. Immunostaining for p-Histone H3 (PHH3) and OLIG2 between day 22 and day 28 of differentiation revealed that the proportion of proliferating OLIG2+ motor neuron progenitor cells was reduced in CLP1KO compared to CLP1WT motor neuron cultures after day 22 (Figure 1E). The overall proliferation rate of the CLP1WT and CLP1KO motor neuron cultures was comparable (Figure S1F), suggesting the decrease in cell density in the CLP1KO motor neuron cultures is not due to altered proportions of mitotic cell types. To assess cell death, we quantified the percent of cleaved caspase 3 (CC3)-positive motor neuron cell types (ISL1/2+ or OLIG2+) in CLP1WT and CLP1KO cultures between day 22 and day 28 of differentiation. Consistent with a neurodegeneration phenotype, CLP1KO cultures from day 24 to day 28 had increased apoptosis of OLIG2+ and ISL1/2+ cells compared to CLP1WT controls (Figure 1F).
CLP1 regulates mRNA isoform diversity by suppressing IPA
To assess changes in mRNA 3′ end formation caused by CLP1 mutation, we first constructed an atlas of all PASs utilized in human motor neurons. We used poly(A)click-sequencing (PAC-seq) to capture and sequence the 3′ ends of mRNAs in our motor neuron cultures (Routh et al., 2017). We combined data from 16 human motor neuron PAC-seq datasets to generate a motor-neuron-specific PAS database (Data S1A) that we cross-referenced to the PolyA_DB database (Wang et al., 2018). We found that 66.4% of the PASs used in motor neurons were not previously annotated in the PolyA_DB database (91,089 novel sites; 46,032 PolyA_DB-matching sites; Figure S2A). The majority of novel sites (>60,000) identified in our database fell within introns or the coding sequence (CDS) of their annotated gene (Figure S2B). Indeed, PASs in positions proximal to the 3′ UTR represent 60% of the sites that we identified; a substantial proportional increase relative to PolyA_DB (27%), despite our database containing significantly fewer samples and detecting 56% less PASs. In addition to cell-type specificity, our database may contain more proximal PASs than others because our highly stringent filtering criteria enabled us to use fewer a priori assumptions (see STAR Methods).
To assess mRNA 3′ end formation in the absence of CLP1, we analyzed PAC-seq data from CLP1KO and CLP1WT motor neuron cultures by assigning PAC-seq reads to PASs present in our motor-neuron-specific PAS database and compared usage using DEXseq (Anders et al., 2012) (Figure 2; Data S1B). We determined the positional distribution of novel and previously annotated PASs that were differentially used in CLP1KO and CLP1WT hESC-derived motor neurons. We found that the majority of differentially used PASs in the CLP1KO motor neurons were previously annotated in PolyA_DB, while novel sites comprised the majority of intronic and CDS APA events (Figure 2A). At a false discovery rate (FDR) of 0.05, our analysis found 2,191 significant APA events in the CLP1KO motor neurons, with 1,320 sites showing increased usage and 871 with decreased usage (Figure 2B). This increase in the total number of PASs used in the CLP1KO motor neurons indicates that CLP1 functions to suppress mRNA 3′ end diversity in neurons (Figure 2B).
Figure 2. CLP1 regulates mRNA isoform diversity by repressing intronic and proximal polyadenylation site usage.

(A) Novel and previously annotated PASs in CLP1KO versus CLP1WT hESC-derived motor neurons binned by intragenic position. 3′ UTR subdivisions are “F,” “S,” “M,” and “L” for first, single, middle, and last, respectively. Intergenic corresponds to ≥ 10 kb beyond the annotated end of genes.
(B) M-A plot (top) of polyadenylation sites (PASs) in CLP1KO versus CLP1WT hESC-derived motor neurons, red points = p value < 0.05. Quantification of sites with increased and decreased usage is shown (bottom); two-sided binomial test.
(C) APA events binned by intragenic position in CLP1KO versus CLP1WT hESC-derived motor neurons. 3′ UTR subdivisions are “F,” “S,” “M,” and “L” for first, single, middle, and last, respectively. p values are derived from a two-sided binomial test.
(D) Positional distribution of PASs used in CLP1KO stem-cell-derived motor neurons compared to CLP1WT controls. TSS, transcription start site; TTSs, transcription termination sites; K-S test.
(E) Genome browser tracks (top) of CLP1KO and CLP1WT hESC-derived motor neuron 3′ end sequencing reads mapped to CPSF6 at a differentially used intronic polyadenylation (IPA) site (yellow, increased usage). Representative gel images are shown (bottom) with qRT-PCR validation values below; n = 3.
(F) Genome browser tracks (top) of CLP1KO and CLP1WT hESC-derived motor neuron 3′ end sequencing reads mapped to the PCF11 gene (yellow, increased usage). Representative gel images (bottom) are shown with qRT-PCR validation values below; n = 3.
(G) Co-immunoprecipitation of RNA polymerase II (RNAPII) in CLP1KO and CLP1WT hESC-derived motor neurons. Western blot of input and affinity purified proteins for cleavage and polyadenylation subcomplexes; (PCF11 [CFIIm], CPSF100 [CPSF], CstF64 [CstF], and CFIm68 [CFIm]). β-actin, loading control; *p < 0.05; n.s., non-significant (p > 0.05); two-sided Student’s t test, n = 3.
To probe whether the pattern of APA observed in CLP1KO motor neurons indicated a specific regulatory function for CLP1 in PAS selection, we binned all APA events by their annotated intra-genic region. The most significant increase in PAS usage in the CLP1KO motor neurons occurred in introns, while the only significant decrease in usage was at the most distal 3′ UTR position (Figure 2C). Significant increases were also observed for PASs positioned within the 5′ UTR, the CDS, and the proximal region of the 3′ UTR (Figure 2C). Indeed, when we examined the normalized gene position of all PASs used in the CLP1KO motor neurons compared to CLP1WT controls, we observed a trend toward a diffuse, proximal distribution (Figure 2D).
This mRNA 3′ end processing pattern was present in several genes related to mRNA processing, including CPSF6, which encodes CFIm68 of the CPA complex. Increased usage of an IPA site in CPSF6 in CLP1KO motor neurons resulted in 3′ UTR lengthening (Figure 2E) and a ~50% decrease in protein (Figure 2G). CLP1KO motor neurons also had increased usage of an IPA site recently shown to generate a short PCF11 isoform (Kamieniarz-Gdula et al., 2019; Wang et al., 2019), required for autoregulation of full-length PCF11 expression (Figure 2F) (Kamieniarz-Gdula et al., 2019). Usage of this IPA site in CLP1KO motor neurons correlated with an ~85% reduction in PCF11 protein (Figure 2G). Furthermore, protein levels of CstF and CPSF subcomplex proteins, CstF-64 and CPSF100, respectively, were reduced by ~30%–35% (Figure 2G). This decrease in mRNA-processing proteins signaled that the integrity of the CPA complex may be compromised in the absence of CLP1. Indeed, co-immunoprecipitation of RNA polymerase II (RNAPII) to assess association of CPA subcomplexes revealed a broad loss of interaction (Figure 2G). These findings demonstrate CLP1 modulates expression of CPA co-factors and is required for their binding to RNAPII in motor neurons.
To validate our PAC-seq findings, we selected 8 PASs for qRT-PCR validation. These sites matched their predicted direction of change (Pearson r = 0.48, n.s; Figure S2D). In addition to mRNA co-transcriptional regulators, we also observed changes in genes related to neuronal mRNA transport and translation, such as CAPRIN1. We identified increased usage of an IPA site in CAPRIN1 in CLP1KO motor neurons resulting in 3′ UTR shortening (Figure S2E, top, middle) and ~50% less protein (Figure S2E, bottom). Together, these results show CLP1 regulates IPA of several genes encoding RNA binding proteins.
Consensus sequences within pre-mRNAs are recognized by several CPA subcomplexes to define the site of mRNA cleavage and polyadenylation; however, a consensus sequence for the CFIIm subcomplex has not been determined (Chan et al., 2011; Mandel et al., 2008). Positional motif enrichment analysis (Bailey and Machanick, 2012) identified a mild enhancement of the canonical AU-rich polyadenylation signal in PASs with differential usage in the CLP1KO motor neurons compared to CLP1WT controls (Figure S2C), suggesting that CFIIm does not utilize a consensus sequence.
CLP1 p.R140H iPSCs show precocious motor neuron differentiation and reduced cell density
Next, we wanted to investigate the impact of the pathogenic CLP1 p.R140H variant on mRNA 3′ end formation in motor neuron disease. Patients with homozygous CLP1 p.R140H variants present with penetrant, severe motor neuron disease characterized by progressive sensorimotor polyneuropathy and pronounced demyelination (Karaca et al., 2014; Schaffer et al., 2014; Wafik et al., 2018). To examine mRNA 3′ end diversity in motor neuron disease and define the genome-wide effect of CLP1 p.R140H on mRNA 3′ end processing, we reprogrammed family-matched fibroblasts derived from an unaffected parent (CLP1 p.R140H carrier, referred to as “unaffected”) and an affected child (homozygous for CLP1 p.R140H, referred to as “affected”) (Schaffer et al., 2014) to induced pluripotent stem cells (iPSCs). The iPSCs expressed stem cell markers, were pluripotent, and had a normal karyotype (Figures S3A–S3C). The iPSCs were then differentiated to motor neurons (Markmiller et al., 2018) to examine mRNA 3′ end formation (Figure 3A).
Figure 3. iPSC-derived motor neurons from patients with PCH10 precociously differentiate, resulting in reduced neuronal density at maturity.
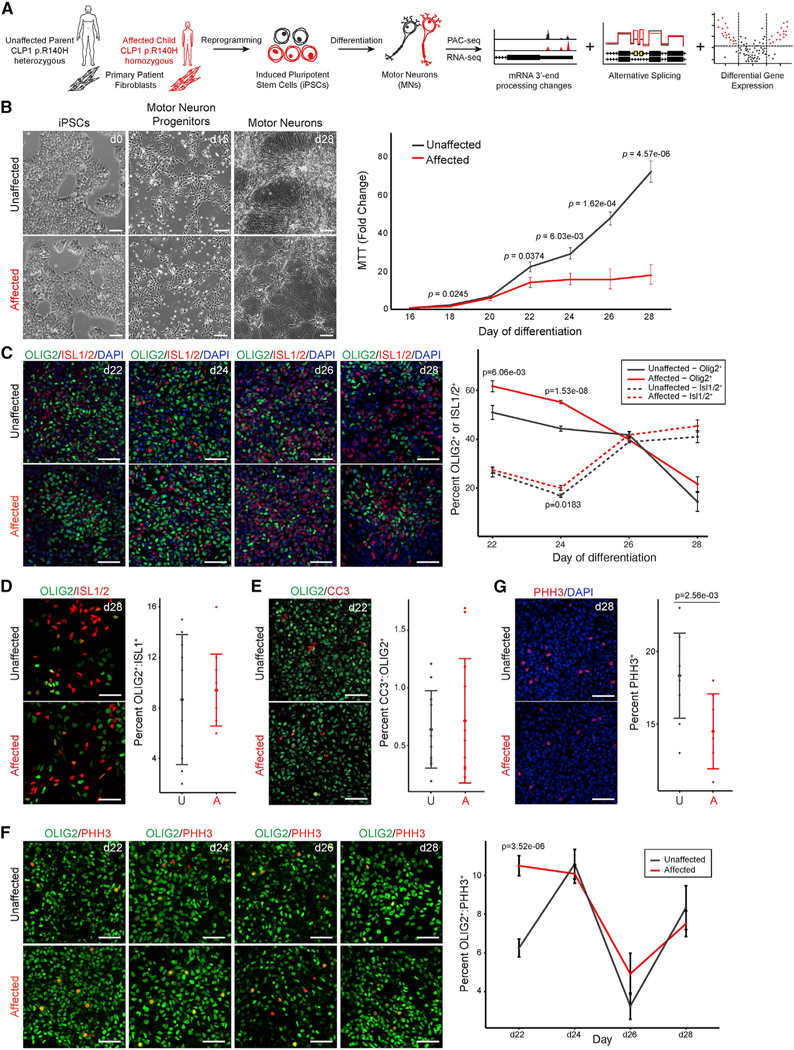
(A) Study schematic. Homozygous and heterozygous CLP1 p.R140H fibroblasts from patients with PCH10 and unaffected individuals, respectively, were re-programmed into iPSCs and differentiated into motor neurons, followed by transcriptomic analysis.
(B) Representative images of iPSCs derived from affected and unaffected patients shown during motor neuron differentiation (left). Scale bars, 100 μm. MTT assay of motor neurons derived from affected and unaffected patients during differentiation is depicted (right). Two clonal lines per individual (biological replicates); n = 8 technical replicates; two-sided Student’s t test.
(C) Immunostaining for OLIG2 and ISL1/2 in affected and unaffected motor neuron cultures during differentiation. d, day of differentiation; p, OLIG2+ statistics, p*, ISL1/2+ statistics; two-sided Student’s t test; n = 6, 20× fields-of-view per clone. Representative images are shown from two clonal lines per individual. Scale bars, 50 μm.
(D) Co-immunostaining for OLIG2 and ISL1/2 in affected (A) and unaffected (U) motor neuron cultures at day 28; two-sided Student’s t test; n = 6, 20× field-of-view per clone. Representative images are shown from two clonal lines per individual. Scale bars, 50 μm.
(E) Co-immunostaining for OLIG2 and CC3 at day 22 of differentiation in affected (A) and unaffected (U) cultures; two-sided Student’s t test; n = 6, 20× field-of-view per clone. Representative images are shown; two clonal lines per individual. Scale bars, 50 μm.
(F) OLIG2 and PHH3 co-immunostaining in affected (A) and unaffected (U) motor neuron cultures during differentiation; two-sided Student’s t test. n = 6, 20× field-of-view per clone. Representative images are shown from two clonal lines per individual. Scale bars, 50 μm.
(G) PHH3 immunostaining with DAPI in affected and unaffected cultures at day 28 of differentiation; two-sided Student’s t test. n = 6, 20× field-of-view per clone.
Representative images are shown from two clonal lines per individual. Scale bars, 50 mm.
See also Figure S3.
Unaffected and affected iPSCs were morphologically indistinguishable as they differentiated into motor neuron progenitor cells and mature neurons that co-expressed the motor-neuron-specific transcription factors, ISLET-1/2 and HB9 (Figures 3B, left and S3D). Unaffected and affected motor neurons expressed the phrenic motor neuron subtype identity markers, SCIP and HOXA5 (Figure S3E) but were largely negative for FOXP1, HOXC6, LHX3, or HOXC9 expression (data not shown). Cell density measurements during motor neuron development revealed a progressive reduction in cell numbers between day 22 and day 28 of differentiation in affected compared to unaffected motor neuron cultures (Figure 3B). Immunostaining for markers of motor neurons (ISL1/2) and motor neuron progenitors (OLIG2) across this time period (Figure 3C) indicated that affected motor neuron cultures had a significantly higher proportion of progenitors early in differentiation (day 22 through day 24) compared to unaffected controls, while the proportion of mature motor neurons was mildly elevated throughout differentiation (Figure 3C). We did not observe a significant difference in the proportion of OLIG2 and ISL1/2 co-positive cells at any time point for either genotype (Figure 3D), indicating affected motor neurons were not developmentally arrested.
Next, we investigated whether progenitor cell proliferation or apoptosis was responsible for the increased proportion of motor neuron cell types and decreased density observed during neurogenesis in affected motor neuron cultures. We quantified the number of CC3-positive motor neuron cell types (ISL1/2+ or OLIG2+) in unaffected and affected motor neuron cultures between day 22 and day 28 of differentiation and observed no differences (Figure 3E; data not shown). Immunostaining for PHH3, and OLIG2 between day 22 and day 28 of differentiation revealed that the proportion of proliferating OLIG2+ motor neuron progenitors was elevated in affected motor neuron cultures at day 22 but similar to unaffected controls between day 24 and day 28 (Figure 3F). Lastly, analysis of the overall proliferation rate showed that the unaffected cultures contained a higher proportion of mitotic cells only at the end of differentiation (day 28; Figure 3G). These findings indicate that the reduction in cell density in the affected motor neuron cultures throughout differentiation is likely due to enhanced proliferation and differentiation of motor neuron progenitor cell types into post-mitotic neurons. While the CLP1KO and affected motor neuron cultures both show a relative decrease of motor neuron density, the cellular mechanism in affected cell cultures in vitro parallels the agenesis phenotype observed in patients with PCH10 (Karaca et al., 2014; Schaffer et al., 2014), justifying the use of this model as a tool to uncover molecular changes associated with motor neuron disease.
CLP1 p.R140H and CLP1 deficiency have opposing effects on 3′ end formation
Next, we carried out PAC-seq experiments similar to those described above, using the iPSC-derived motor neuron disease model of PCH10 to measure and compare changes in mRNA 3′ end formation. We assessed the positional distribution of novel and previously annotated PASs that were differentially used between motor neurons derived from affected and unaffected iPSCs. Overall, we found that most differentially used PASs were annotated in PolyA_DB (Figure 4A). Applying the same analysis methods, we detected 906 differential APA events in the affected motor neurons (379 increased usage, 527 decreased usage; Figure 4B; Data S1C). These findings demonstrate CLP1 p.R140H alters mRNA 3′ end processing in motor neurons.
Figure 4. Motor neurons derived from PCH10 patient iPSCs display suppression of proximal polyadenylation and reduced mRNA isoform diversity.
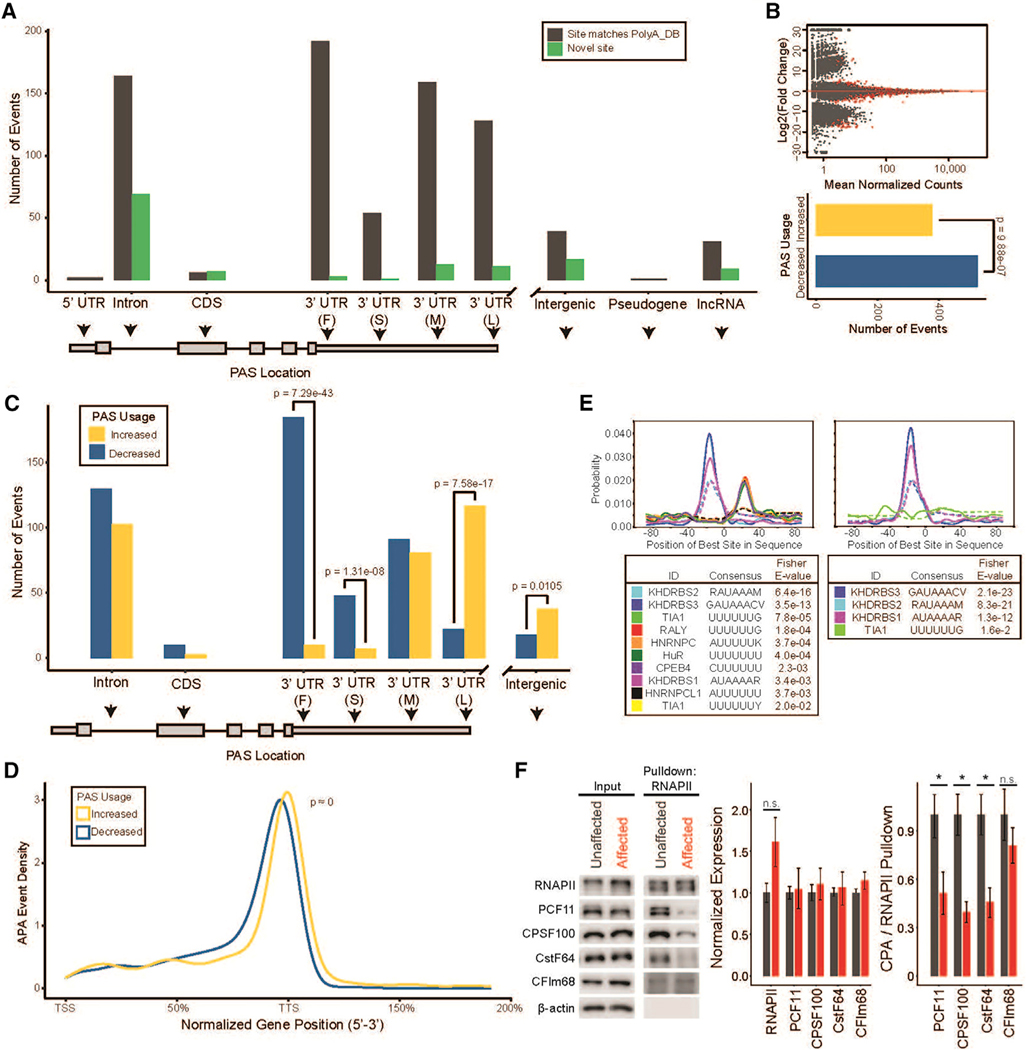
(A) Novel and previously annotated PASs in PCH10 patient versus control iPSC-derived motor neurons binned by intragenic position. 3′ UTR subdivisions are “F,” “S,” “M,” and “L” for first, single, middle, and last, respectively. Intergenic corresponds to ≥ 10 kb beyond the annotated end of genes.
(B) M-A plot (top) of PASs detected in affected motor neurons compared to unaffected controls; red points indicate an adjusted p value < 0.05. Quantification of sites with increased and decreased usage is shown (bottom); two-sided binomial test.
(C) APA events binned by intragenic position in affected and unaffected motor neurons. 3′ UTR subdivisions are “F,” “S,” “M,” and “L” for first, single, middle, and last, respectively. p values are derived from a two-sided binomial test.
(D) Positional distribution of PASs in affected and unaffected motor neurons. TTS, transcription termination site; TSS, transcription start site. K-S test.
(E) Positional motif enrichment of PASs with increased usage (left) and decreased usage (right) in affected versus unaffected iPSC-derived motor neurons. Plots are centered on the cleavage site.
(F) Co-immunoprecipitation of RNAPII in affected and unaffected iPSC-derived motor neurons for association of the cleavage and polyadenylation (CPA) subcomplexes PCF11 (CFIIm), CPSF100 (CPSF), CstF64 (CstF), and CFIm68 (CFIm). β-actin, loading control; *p < 0.05; n.s., non-significant (p > 0.05); two-sided Student’s t test; n = 3.
To detect a regulatory pattern of APA in the CLP1 p.R140H mutant motor neurons, we binned all APA events by their annotated intragenic region. Because the CLP1 p.R140H variant maintains weak interactions with its canonical binding partners (Figure S1A [Schaffer et al., 2014]), we hypothesized that we would find a similar but attenuated pattern of APA in the CLP1 p.R140H motor neurons to that observed in the CLP1KO cultures. In direct contrast, affected motor neurons had a significant reduction in the usage of proximal PASs with a corresponding increase in distal 3′ UTR and downstream, intergenic PASs (Figure 4C), cumulatively resulting in lengthened 3′ UTRs. Despite detecting fewer APA events, the proximal-to-distal shift was more pronounced in affected motor neurons than the distal-to-proximal shift seen in CLP1KO motor neurons (Figure 4D).
We selected 18 PASs to validate our findings by qRT-PCR and observed a statistically significant correlation between PAS fold changes in vitro and in silico (Pearson r = 0.61, p = 6.7e–03; Figure S4). Sequencing tracks for a subset of these validated genes are shown in Figures S4B and S4C. KLC1, encoding a member of the kinesin light-chain family, showed increased usage of an IPA site and decreased usage of the lowly expressed distal 3′ UTR, resulting in increased expression of a short isoform (Gen-Bank: NM_005552; Figure 4B). GRID1, encoding the delta-1 subunit of the glutamate receptor (GluD1), displayed decreased usage of a novel IPA site, in addition to readthrough transcription into downstream long non-coding (lnc) RNA, SALNG0079379 (Figure S4C). The biological impact of these two APA events, and the hundreds of others that we identified, remains to be determined.
Next, we examined differentially used PASs in affected motor neurons for sequence motifs that may predict dysregulation by CLP1 p.R140H. Positional motif enrichment analysis (Bailey and Machanick, 2012) revealed a U-rich sequence motif located in the +20 position to the cleavage site within PASs that show increased usage in affected motor neurons (Figure 4E, left), while less-used PASs contained a more diffuse U-rich sequence element (Figure 4E, right). Consistent with the CLP1KO motor neurons, we did not detect a difference in the sequence composition of the AU-rich polyadenylation signal at sites with differential usage in affected motor neurons (Figure 4E).
Since CLP1 is important for intron-containing tRNA splicing (Weitzer et al., 2015), we next assessed tRNA splicing and abundance in affected and unaffected motor neurons by sequencing (Pinkard et al., 2020) and northern blot analysis and found only marginal differential expression of 5 of the 48 tRNA isodecoder groups (Figures S5A–S5C; Data S1D and S1E). No overt changes in intron-containing tRNA splicing or fragment accumulation were observed in affected motor neurons (Figures S5B and S5C), possibly due to compensation by the parallel tRNA ligation pathway active in human cells (Popow et al., 2011).
Given that CLP1 p.R140H has impaired association with PCF11, a component of the CPA complex, we next tested whether the expression of CPA-complex proteins or their association with RNAPII was altered in the presence of the pathogenic CLP1 allele. Western blot analysis for PCF11, CPSF100, CstF64, and CFIm68 in affected motor neurons found no difference in protein abundance compared to unaffected controls (Figure 4F). However, co-immunoprecipitation experiments revealed a striking decrease in the association of PCF11, CPSF100, and CstF64 but not CFIm68 with RNAPII in the disease state (Figure 4F). These results show CLP1 p.R140H not only disrupts the interaction of CFIIm but significantly impairs association of CPA-subcomplex proteins with the transcriptional machinery.
Suppression of proximal polyadenylation is associated with patterned transcriptomic dysregulation of long genes
Co-transcriptional cleavage at proximal PASs leads to expression of shorter mRNA isoforms with differential stability, coding potential, and translation efficiency. To understand how alternative usage of these proximal PASs impact mRNA isoform ratios and gene expression in the context of motor neuron disease, we performed RNA sequencing on day 28 unaffected and affected motor neurons. We used established bioinformatic pipelines to detect changes in gene expression and mRNA splicing (Hartley and Mullikin, 2016; Love et al., 2014; Shen et al., 2014). At an FDR threshold of 0.05, we identified 7,172 differentially expressed genes and 4,270 cassette exon, inclusion, or skipping events, with strong overlap between genes called for APA, alternative splicing, and differential expression (p = 9.4e–154, likelihood ratio test; Figures 5A, 5B, and S6A–S6D; Data S1F–S1H).
Figure 5. PCH10-patient iPSC-derived motor neurons show broad transcriptomic dysregulation.

(A) Transcriptomic events detected by bioinformatic analysis of RNA sequencing of unaffected and affected motor neurons (bar graph). n = 2 unaffected and n = 2 affected clonal replicates. Alternative exon events are cassette exons showing differential inclusion (“Up”) or exclusion (“Down”).
(B) Venn diagram of overlapping genes with mRNA processing events or differential expression. DGE, differential gene expression; APA, alternative poly-adenylation. p = 9.4e–154, likelihood ratio test.
(C) Normalized gene position of differentially used splice acceptor sites in affected versus unaffected motor neurons; Wilcoxon rank sum test; TSS, transcription start site; TTS, transcription termination site. Whiskers extend the full range of the data.
(D) APA events with gene expression changes by intragenic position. Correlation, two-sided binomial test. 3′ UTR subdivisions are “F,” “S,” “M,” and “L” for first, single, middle, and last, respectively.
(E) Length of significantly differentially expressed genes colored by adjusted p value.
(F) Genome-wide correlation of gene length and number of annotated IPA sites from RNA sequencing of two independent iPSC-derived motor neuron clones per individual. n = 40,619 intron-containing genes; default ggplot geom_smooth trendline; Pearson r correlation test; x axis in log10 base pairs.
(G) Violin plot (top) of gene length for differentially expressed genes with or without annotated IPA sites; quantiles drawn at 25%, 50%, and 75% of data density; Welch’s t test; y axis in log10 base pairs.
(H) Genome browser tracks of affected (A1, A2) and unaffected (U2, U3) iPSC-derived motor neuron RNA-sequencing and PAC-sequencing reads mapped to CAMTA1, decreased IPA (blue), increased distal 3′ UTR polyadenylation (yellow). RPM, reads per million. A subset of representative isoforms are shown. Representative gel images with qRT-PCR validation values are shown below; n = 3. Western blot (right) for CAMTA1 in CLP1KO and CLP1WT H9 hESC-derived motor neurons. β-actin, loading control.
See also Figures S5 and S6 and Data S1.
The affected motor neurons had a strong increase in distal PAS usage; therefore, we theorized that a subset of genes would likely evade proximal CPA and show upregulation of distal exons positioned downstream of these sites. We calculated the normalized gene position of all splice acceptor sites showing differential usage in the affected motor neurons. In agreement with our observation of APA within the CDS, we found a significant 3′ end bias in upregulated splice junctions relative to those that were downregulated (Figure 5C). Taken together, our data suggest proximal CPA evasion as a potential mechanism for the mRNA processing dysregulation observed in the affected motor neurons. Next, we examined whether gene expression changes in the affected motor neurons were correlated with increased or decreased usage of PASs by intragenic regions. Decreased usage of IPA sites was highly correlated with upregulated gene expression (Figure 5D), indicating that evasion of IPA likely contributes to the dysregulation of gene expression observed in the affected motor neurons.
Given the positive correlation between gene length and number of putative IPA sites (Figure 5F), we next asked whether expression of long genes might be influenced by reduced IPA. Our differential gene expression analysis revealed a strong positive length bias in upregulated genes from affected motor neurons (Figure 5E), which was dependent upon annotated IPA sites (Figure 5G). For example, affected motor neurons had decreased usage of IPA sites that generate short or non-coding isoforms of calmodulin-binding transcription activator 1 (CAMTA1) and a corresponding increase in full-length coding transcripts and protein expression (Figure 5H). Thus, CLP1-p.R140H-dependent modulation of IPA site usage can influence the balance of mRNA isoform expression and abundance, particularly for long genes like CAMTA1.
Altered mRNA processing in motor neurons disproportionately affects genes encoding RNA-binding and synapse-associated proteins
Next, we utilized gene ontology (GO) term enrichment analysis with clustering visualization methods to categorize the differentially expressed genes, with and without APA, observed in the affected motor neurons. Upregulated genes were strongly enriched for synapse-specific proteins, ion channels, and cytoskeleton components, whereas downregulated genes function in mRNA processing and chromosome maintenance (Figures 6A, 6B, and S7A; Data S1I and S1J). We compared the expression of various cell-type-specific marker genes in affected and unaffected motor neuron cultures and found no differences (Figure S7B; Data S1K), in agreement with our immunofluorescence-based analysis (Figures 3C and S3D–S3G). These results suggest the transcriptomic dysregulation observed in affected motor neurons is likely due to the pathogenic CLP1 p.R140H variant rather than culture composition.
Figure 6. Enrichment analysis of differentially expressed genes with APA in PCH10 patient iPSC-derived motor neurons predicts neuronal dysfunction.

(A) ClueGO visualization of biological processes in differentially expressed genes with alternative polyadenylation in affected motor neurons compared to unaffected controls.
(B) Top five genes ranked by adjusted p value from the six GO term clusters. See also Figure S7 and Data S1.
The genes upregulated by CLP1 p.R140H in the affected motor neurons are consistent with a model of intronic PAS suppression enhancing expression of a subset of long genes, as genes that are important for neuronal function are disproportionately long (Gabel et al., 2015; King et al., 2013; Polymenidou et al., 2011; Takeuchi et al., 2018). We noted increased expression of GRIN2B and GRIN1 (Data S1F), both of which encode components of ionotropic glutamate NMDA receptors (Dingledine et al., 1999), in addition to a broad spectrum of ion channel genes, indicating that ion homeostasis is probably disrupted in affected motor neurons. Cumulatively, the differentially expressed genes in affected motor neurons are likely to influence neuronal activity, which may lead to hyperexcitability (Wainger et al., 2014) and the neuropathy observed in patients with PCH10 (Karaca et al., 2014).
Mouse model of PCH10 shows correlated gene expression changes with patient-derived motor neurons
Next, we generated and studied an in vivo model of PCH10. We employed CRISPR-Cas9-mediated gene editing to introduce the CLP1 p.R140H variant (Clp1R140H/R140H) into the murine genome on a B6SJLF1/J genetic background (Figures S8A, S8B, and S8D). Mice with genomic insertions and deletions disrupting Clp1 were also present (Clp1KO/+; Figures S8A, S8C, and S8E). Consistent with prior reports, we found loss of Clp1 was embryonic lethal in mice (Figure S8G), while Clp1R140H/R140H mutant mice were produced at expected Mendelian ratios (Figure S8F; Hanada et al., 2013; Morisaki et al., 2021).
We analyzed the brain and motor neurons of Clp1R140H/R140H mutant mice. Compared to littermate controls, Clp1R140H/R140H mice exhibited a 17.6% reduction in anterior isocortex cell density (Figure S8H), akin to frontal cortex atrophy. Tonic-clonic seizures were present in Clp1R140H/R140H mice by postnatal day 3′ (P30) and were often lethal (~75%). Clp1R140H/R140H mice did not show overt changes in cerebellar folia morphology (granule cell layer and molecular layer widths) or granule cell and Purkinje cell density at P30 compared to controls (Figures S8I–S8M), consistent with a subset of patients with PCH10 (Karaca et al., 2014). Diaphragm neuromuscular junction (NMJ) formation and area appeared normal in Clp1R140H/R140H mice at P0 (Figure 7A). However, by P30, NMJ size was reduced by 22.7% in Clp1R140H/R140H mice (Figures 7B–7D), without changes in colocalization of pre- and post-synaptic density markers (Figure 7E). Clp1R140H/R140H mice that survived beyond P30 demonstrated reduced stride length of 4%, 17%, and 13% at 3, 4, and 5 months old, respectively (Figures 7F and 7G), indicative of impaired motor function and progressive motor neuron degeneration. Collectively, we find shared brain phenotypes and penetrant motor neuron dysfunction in patients with PCH10 and mice.
Figure 7. CLP1 p.R140H homozygous mutant mice model motor neuron dysfunction and show correlated gene expression changes to motor neurons derived from PCH10 patient iPSCs.

(A) Immunofluorescence staining of control and homozygous Clp1 p.R140H (Clp1R140H/R140H) mutant diaphragms at P0 for presynaptic (neurofilament) and postsynaptic (α-bungarotoxin) markers; ImageJ tracing (left). Quantification; α-bungarotoxin, red, area: 1.00 ± SD 0.134 versus 0.959 ± SD 0.105; n = 3 mice per genotype. Scale bars, 1 mm.
(B) Control and Clp1R140H/R140H neuromuscular junction (NMJ) patterning is shown by α-bungarotoxin staining at P30. Scale bars, 1 mm.
(C) High-magnification images of immunofluorescence staining for presynaptic (GFP) and postsynaptic (α-bungarotoxin) markers in ChAT-GFP;Clp1R140H/R140H mice compared to control littermates at P30. Scale bars, 100 μm.
(D) Quantification of NMJ size. Boxplot represents the distribution of average NMJ area for n = 3 mice per genotype, individual points represent the average NMJ area for 6 fields-of-view from a single diaphragm. Whiskers extend the full range of the data; two-sided Student’s t test.
(E) Quantification of presynaptic (GFP) and postsynaptic (α-bungarotoxin) markers at diaphragm NMJs in control and ChAT-GFP;Clp1R140H/R140H mice at P30. Boxplot represents the distribution of colocalization coefficients for n = 3 mice per genotype, individual points represent the Pearson r (no threshold) for 6 NMJs from 4 fields-of-view from a single diaphragm. Whiskers extend the full range of the data, excluding outliers (>1.5× interquartile range); two-sided Student’s t test. (F and G) Stride length of 3-, 4-, and 5-month-old Clp1R140H/R140H mice compared to wild-type sex-matched littermates. Representative images of strides (F) from 4-month-old male mice. Scale bars, 1 cm. Data in (G) are mean values ± SEM, n = 4 mice per genotype and age; two-sided Student’s t test.
(H) Schematic of experimental design for in vivo gene expression validation. e, embryonic day; FACS, fluorescence activated cell sorting.
(I) qRT-PCR of FACS and whole embryonic spinal cords for motor neuron markers, Hb9/Mnx1 and Isl1; two-sided Student’s t test, n = 3.
(J) Scatterplot correlation of differential expression for genes assessed by qRT-PCR from PCH10-patient-derived motor neurons versus Clp1R140H/R140H mouse primary spinal motor neurons compared to controls (CLP1 p.R140H heterozygous individuals or mice), n = 3. Pearson r = 0.61.
To evaluate molecular changes in murine motor neurons, we crossed Clp1R140H/+ mutant mice to mice expressing an Hb9-driven GFP reporter (Hb9-GFP) to generate Hb9-GFP;Clp1R140H/R140H mice. We isolated GFP+ spinal motor neurons from embryonic day (e) 12.5 Hb9-GFP;Clp1+/+, Hb9-GFP;Clp1R140H/+, and Hb9-GFP;Clp1R140H/R140H dissociated spinal cords by fluorescence-activated cell sorting (FACS) and extracted RNA for qRT-PCR and PAC-seq (Figures 7H and 7I). Hb9-GFP;Clp1R140H/+ littermates were used as a control to match heterozygous parental controls for the homozygous patients. We compared differential gene expression of Khdrbs1, Pspc1, Snca, Kcnma1, Grin2b, and Kcnj6 in primary murine spinal motor neurons to PCH10-patient-derived motor neurons and found that 5 out of the 6 genes showed concordant changes (Pearson r = 0.61; Figure 7J). PAC-seq detected few APA events, none of which could be validated across litters (data not shown). Although the genotypes and tissue examined were different, recent work in CLP1R140H/KO mice support our finding that CLP1 p.R140H suppresses proximal PAS usage (Monaghan et al., 2021).
Reduced APA-mediated mRNA isoform diversity is a shared transcriptomic signature among neurodegenerative diseases
Our findings prompted us to ask whether altered mRNA 3′ end diversity might underlie neurodegeneration more broadly. To address this question, we surveyed publicly available datasets to identify pre-existing mRNA 3′ end sequencing data generated from primary tissues isolated from patients with neurodegenerative disease, and performed our APA analysis. Two datasets utilized postmortem brain tissue for mRNA 3′ end sequencing in the context of neurodegenerative disease with motor phenotypes: myotonic dystrophy (DM) (Goodwin et al., 2015) and sporadic Parkinson’s disease (sPD; Rhinn et al., 2012). The first identified subtype of DM, DM1, is caused by a CUG-repeat expansion in the DMPK gene, which sequesters MBNL RNA-binding proteins into nuclear RNA foci (Jiang et al., 2004). sPD has an unknown molecular etiology but shows molecular phenotypes of α-synuclein aggregation away from synaptic terminals and into cytoplasmic Lewy bodies.
We tabulated PAS usage and gene expression from mRNA 3′ end sequencing of patients with DM1 and control postmortem frontal cortex samples (GEO: GSE68890) using our neuronal PAS annotation set. Our analysis identified 768 APA events in DM1, with a bias toward decreased PAS usage and isoform diversity (n = 326 upregulated, n = 442 downregulated PASs; Figure 8A). DM1 patient frontal cortex samples showed decreased polyadenylation near the canonical transcript termination site and upregulation of PASs throughout the gene bodies, as opposed to upregulated proximal PAS usage detected in motor neurons of patients with PCH10 (Figure 8B). This mRNA processing pattern was also observed in frontal cortex samples from DM2 patients (Goodwin et al., 2015; Figures 8C and 8D).
Figure 8. Loss of mRNA isoform diversity is a shared transcriptomic signature among neurodegenerative diseases.

(A) APA events detected in mRNA 3′ end sequencing datasets from autopsy frontal cortex tissues of patients with myotonic dystrophy type 1 (DM1); two-sided binomial test. PAS, poly-adenylation site.
(B) The aggregate normalized positional distribution of DM1 APA events within gene bodies; K-S test.
(C) APA events detected in mRNA 3′ end sequencing from autopsy frontal cortex tissues of patients with myotonic dystrophy type 2 (DM2); two-sided binomial test.
(D) The aggregate normalized positional distribution of DM2 APA events within gene bodies; K-S test.
(E) APA events detected in mRNA 3′ end sequencing datasets from autopsy BA9 region tissues of patients with the sPD; two-sided binomial test.
(F) The positional distribution of differential APA events within gene bodies in sPD tissues.
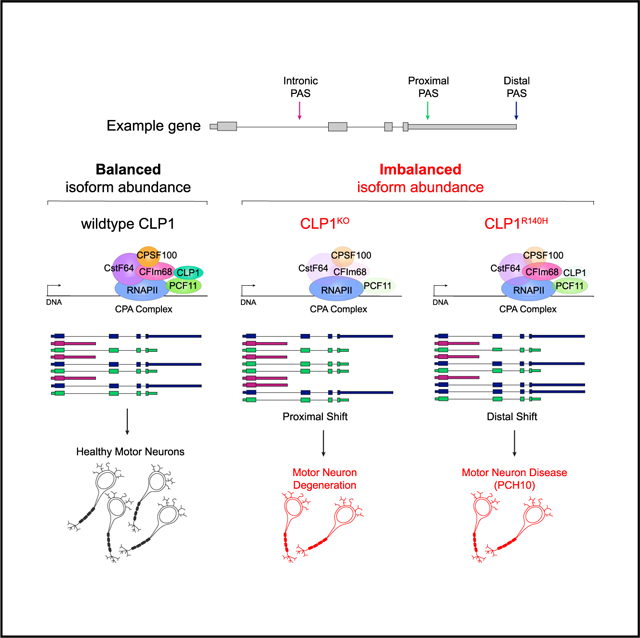
(G) Model of neurodegeneration caused by 3′ end processing bias and limited isoform diversity. An example transcript can be cleaved in several places to produce mRNA isoforms with 3′ end diversity. Healthy neurons use a balance of isoforms, whereas diseased neurons show reduced isoform diversity with a proximal or distal cleavage bias.
This analysis was also performed with publicly available mRNA 3′ end sequencing data generated from postmortem brain area 9 (BA9) tissue samples of patients with sPD and control patients (GEO: GSE40710). We detected 72 APA events with significant bias toward less sites used (n = 21 upregulated, n = 51 downregulated PASs; Figure 8E). While our analysis detected far fewer APA events in the sPD dataset than in the DM1/2 datasets, we still saw a similar trend of reduced polyadenylation at canonical transcript termination sites (Figure 8F). Collectively, we have uncovered a previously unrecognized molecular signature of mRNA misprocessing associated with neurodegenerative disease, characterized by imbalance mRNA isoform expression driven by diminished mRNA 3′ end diversity (Figure 8G).
DISCUSSION
Here, we present evidence for a regulatory role for the RNA kinase CLP1 in balancing mRNA isoform composition and abundance in human motor neurons. We found CLP1 perturbation led to distinct patterns of mRNA isoform expression stemming from altered mRNA 3′ end processing that correlated with disease. Mechanistically, we show CLP1 regulates proximal polyadenylation, especially IPA, and mRNA 3′ end diversity in motor neurons. In addition, we discover a common transcriptional signature of reduced mRNA isoform diversity driven by aberrant CPA in neurodegeneration. This signature was also present in postmortem brain tissues from patients with DM and sPD, potentially suggesting a common pathophysiological mechanism.
CLP1 and PCF11 comprise the CFIIm subcomplex of the mammalian CPA complex (de Vries et al., 2000). Our findings from CLP1KO hESC-derived motor neurons suggest CLP1 re-presses PCF11. In the absence of CLP1, we observed increased IPA site usage and reduced distal PAS usage, paralleling PCF11 overexpression (Kamieniarz-Gdula et al., 2019; Wang et al., 2019). We also observed CLP1-dependent suppression of PCF11 IPA at its autoregulatory site, suggesting CLP1 may promote long-gene expression in differentiated neuronal subtypes through direct repression of PCF11. Based on our findings from CLP1KO and isogenic control hESC-derived motor neurons, we postulate that CLP1 p.R140H is not strictly a loss-of-function variant because of the opposing 3′ end processing changes observed in iPSC-derived motor neurons from patients with PCH10. CLP1 p.R140H has drastically reduced affinity for PCF11 (Figure S1A; Schaffer et al., 2014); however, its interaction with other mRNA 3′ end processing factors has not been thoroughly investigated.
Our studies in CLP1KO motor neurons suggest that the integrity of the CPA complex may be dependent on CLP1 association. It remains unknown how and when CLP1 is co-transcriptionally recruited to the CPA complex. In CLP1KO and CLP1 p.R140H motor neurons, the presence or absence of CFIm68 bound to RNAPII correlates well with the changes in PAS usage observed with each CLP1 mutation (Li et al., 2015; Martin et al., 2012). It is possible that CLP1 p.R140H maintains association with some CPA-complex components, while PCF11 and others are lost. In corroboration, PCF11 knockdown and CLP1 p.R140H both show increased usage of distal PASs and suppression of proximal PAS usage (Li et al., 2015; Wang et al., 2019). A comparison of genes with APA events in neuroblastoma cells with PCF11 knockdown (Ogorodnikov et al., 2018) to CLP1KO hESC-derived motor neurons found substantial overlap (22%; p value = 2.7e–9, Fisher’s exact test), suggesting CFIIm may regulate a common subset of genes in neuronal subtypes. Overall, this highlights the need for additional understanding of CFIIm functionality.
While it is likely that the loss of individual factors or overall disruption of the CPA complex could lead to the mRNA 3′ end processing changes we observed, it is also possible that CLP1 p.R140H gains interactions with neuron-specific factors that enhance PCF11 function, resulting in more full-length isoforms of long, neuronal genes. It is common for missense variants in RNA-binding proteins to display gain- and loss-of-function activities. For instance, variants in TARDBP, encoding TDP-43, cause the motor neuron disease amyotrophic lateral sclerosis (ALS) (Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008). Loss of TDP-43 in the nucleus alters mRNA splicing (loss-of-function; Polymenidou et al., 2011; Tollervey et al., 2011), while toxic cytoplasmic aggregation leads to sequestration of other proteins and impaired nuclear import/export (neomorphic gain-of-function; Chou et al., 2018; Liu-Yesucevitz et al., 2010; Neumann et al., 2006). Similar to mutant TDP-43, CLP1 p.R140H is stable and mislocalized to the cytoplasm, which could enable novel protein-protein and protein-RNA interactions that may be pathogenic.
tRNA biogenesis defects have been implicated in many subtypes of PCH (van Dijk et al., 2018), including PCH10 (Karaca et al., 2014; Schaffer et al., 2014). CLP1 associates with the tRNA splicing endonuclease (TSEN) complex to participate in intron-containing tRNA splicing (Paushkin et al., 2004; Weitzer and Martinez, 2007) and this interaction is reduced by p.R140H. Defects in tRNA processing were identified in PCH10-patient-derived fibroblasts and induced neurons (Karaca et al., 2014; Schaffer et al., 2014). In contrast, we did not observe tRNA splicing defects or tRNA expression changes in affected motor neurons. While tRNA processing defects could be pathogenic for other neuronal subtypes, they do not appear to cause motor neuron dysfunction in PCH10.
Our findings demonstrate that transcriptome 3′ end diversity is important in neurons. This diversity is achieved by selective regulation of gene isoforms derived from differential PAS choice. Our CLP1KO studies identify a role for CLP1 in suppressing IPA and other proximal PASs in favor of expression of full-length transcripts. The selective regulation of PAS usage is linked to 3′ UTR position and length, which mediate protein structure and function, as well as mRNA stability and abundance, respectively. Since CLP1 is an established component of the CPA complex, it is likely that CLP1 directly regulates the generation of a subset of mRNA isoforms. Neurons generally express longer genes (King et al., 2013) as well as a greater diversity of mRNA isoforms (Gabel et al., 2015; He et al., 1998; King et al., 2013; Liu et al., 2011; Miura et al., 2013; Polymenidou et al., 2011; Takeuchi et al., 2018) that commonly encode ion channels, synapse-specific proteins and transmembrane proteins, suggesting that unbalanced isoform expression and abundance will likely affect neuronal identity or homeostasis, leading to neurodevelopmental defects and neurodegeneration (Alcott et al., 2020; Hutton et al., 1998; Lefebvre et al., 1995; Lin et al., 1998).
Our findings in human motor neurons suggest mRNA 3′ end processing defects may be major contributors to pathogenesis of neurodegenerative diseases, including those without a defined genetic etiology. The transcriptomic signature we discovered reveals IPA and usage of upstream PASs as a regulatory mechanism for controlling mRNA isoform balance and attenuating long gene expression. Whether the loss of short mRNA isoforms with potentially important biological roles or the overexpression of long mRNA isoforms is toxic in neurons remains to be determined; however, overexpression of a long SCNA isoform was recently shown to be pathogenic in cortical neurons from patients with PD (Rhinn et al., 2012). Thus, we propose a model of neurodegeneration stemming from overall loss of mRNA isoform diversity, due to biased cleavage at proximal or distal PASs (Figure 8G). Normal usage of proximal PASs attenuates long-gene expression and enhances mRNA 3′ end isoform diversity in healthy neurons, while reduced usage of proximal PASs leads to lower mRNA 3′ end isoform diversity, increased expression of long genes, including genes encoding modulators of neuronal activity that likely contribute to neurodegeneration.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ashleigh Schaffer (ashleigh.schaffer@case.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
RNA-sequencing, Poly(A) Click-sequencing, and tRNA-sequencing data used in this study will be available from the Gene Expression Omnibus through series accession number GEO: GSE183872.
All other data referenced in this publication or code used to perform meta-analysis will be shared by the lead contact upon request.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice were maintained according with the National Institutes of Health Guidelines for the Care and Use of Laboratory animals and were approved by the Case Western Reserve Institutional Animal Care and Use Committee. CRISPR genome editing was performed in the Case Western Reserve University Transgenic and Targeting Facility. B6SJLF1/J hybrid mice (Jackson Laboratory, 100012) were used for CRISPR editing of the Clp1 locus. Founder mice with the Clp1R140H/+ and Clp1KO/+ alleles were intercrossed with B6SJLF1/J mice to maintain a mixed background for disease modeling and behavioral analysis, or ChAT-GFP mice (Jackson Laboratory, 007902) for diaphragm characterization. Clp1R140H/+ mice were crossed to Hb9-GFP mice (Jackson Laboratory, 005029) for in vivo validation of molecular findings. All ex vivo analyses were performed on tissue collected from mice of both sexes at postnatal day (P) 0 or P30. No sex-dependent differences were observed.
Cell Culture
H9 human embryonic stem cells (hESCs) were co-electroporated with px330 (Addgene, #42230) containing a 20 nucleotide single guide sequence targeting exon 2 of human CLP1 and pCMV-GFP (Addgene, #11153). The GFP+ single cells were FACS isolated and seeded at a low density on Matrigel (Corning) coated plates to establish clonal lines for genotyping, as previously described (Schaffer et al., 2018). Clones containing bi-allelic indel variants in CLP1 (CLP1KO), or unedited isogenic clones (CLP1WT) were selected for downstream analysis.
Reprogramming of patient fibroblasts into induced pluripotent stem cells (iPSCs) was performed using the Epi5 Episomal iPSC Reprogramming Kit (Thermo Fisher) except Matrigel (Corning) was used as a substrate. Reprogramming vectors were prepared in house and the medium was changed to StemFlex (Thermo Fisher) after 15 days. iPSC clones were selected for expansion and plated on vitronectin-coated plates.
Motor neurons were differentiated from patient-derived iPSCs or hESCs using a previously described protocol (Markmiller et al., 2018).
METHOD DETAILS
Animal Genotyping
Litters were genotyped by allele-specific polymerase chain reaction (AS-PCR). Genomic DNA was prepared from mouse tissue samples as previously described (Truett et al., 2000). AS-PCR for each allele was assembled using a modified GoTaq DNA polymerase (Promega) protocol. Modifications include the use of four primers – common forward, common reverse, mutant reverse, and wild type forward (see Data S1L). The wild type forward primer was used at double the concentration of all other primers. Primers were designed using an established workflow (Gaudet et al., 2009). Reaction conditions were executed as recommended by the manufacturer. PCR for the green fluorescent protein (GFP) gene was assembled using the standard GoTaq (Promega) protocol and GFP-specific primers (see Data S1L).
Stride Length Analysis
The hind limb paws were painted with blue ink (Sheaffer) and mice were guided down a runway over a white paper strip toward a hut. Tracks were collected, and hind limb stride lengths were measured and analyzed for male mice only.
Immunohistochemistry
Whole-mounts of diaphragm muscles were stained as described in Philippidou et al. (2012). Briefly, diaphragm muscles from neonate and adult mice were dissected and fixed in 1% paraformaldehyde (Tsuchida et al., 1994) for 1 hr at RT followed by three washes in PBS. Diaphragms were stained overnight at 4°C with rabbit anti-neurofilament (Synaptic Systems, 1:1000), or rat anti-GFP (gift from C. Kioussi, 1:3000) antibodies in 0.5% Triton X-100/PBS and 4% goat serum, washed three times for 1 hr in 0.5% Triton X-100/PBS, and incubated overnight at 4°C with Alexa 488-conjugated goat anti-rabbit or goat anti-rat (Invitrogen, 1:1,000) and Alexa 555-conjugated anti-α-bungarotoxin (ThermoFisher, 1:1,000) in 0.5% Triton X-100/PBS and 4% goat serum. Diaphragms were washed three times for 1 hr in Triton X-100/PBS, post-fixed in 1% PFA for 10 min and rinsed in PBS before flat mounting with Fluoromount-G (Invitrogen).
Whole brains were dissected from adult mice that had been perfused with 4% PFA and fixed overnight at 4°C in 4% PFA, followed by dehydration overnight in 30% sucrose. Brain tissue was divided into hemispheres and each were embedded in OCT compound and rapidly frozen on dry ice. Serial sagittal sections of 12 mm thickness were cut with a cryostat. Standard hematoxylin and eosin (H&E) staining was performed on sagittal sections spanning the width of one hemisphere.
Immunocytochemistry
Cultured human iPSCs or stem cell-derived motor neurons were fixed for 10 min in 4% PFA, then permeabilized and blocked for 1 hr in 1% goat serum and 0.1% Triton X-100 in PBS. Primary antibodies were applied overnight at 4°C in Shandon coverplates. Primary antibodies used for stem cell characterization include mouse anti-TRA-1–81 (Millipore, 1:200), rabbit anti-Oct4 (ThermoFisher, 1:1,000), goat anti-LIN-28A (R&D Biosystems, 1:50), mouse anti-Nanog (Santa Cruz, 1:200), rabbit anti-α fetoprotein (Dako, 1:400), mouse anti-α smooth muscle Actin (Sigma, 1:400), mouse anti-Nestin (ThermoFisher, 1:200), rabbit anti-PAX6 (BioLegend, 1:2000).
Primary antibodies used for motor neuron characterization include mouse anti-Islet-1/2 (DSHB, 1:500), mouse anti-HB9/Mnx1 (DSHB, 1:500), goat anti-SCIP (Santa Cruz, 1:5000), anti-HOXA5 (Dasen et al., 2005), anti-FOXP1 (Dasen et al., 2008), anti-HOXC6 (Liu et al., 2001), anti-HOXC9 (Jung et al., 2010), anti-LHX3 (Tsuchida et al., 1994), rabbit anti-OLIG2 (Proteintech, 1:500), mouse anti-p-Histone H3 (Santa Cruz, 1:400), Alexa 488 pre-conjugated rabbit anti-Cleaved Caspase-3 (Cell Signaling, 1:50). For detection, we used Alexa fluor [488, 555, 594, 647]-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies (Invitrogen, 1:1,000).
Quantification of Neuromuscular Junction Morphology
Neuromuscular junction size was determined by outlining junctions from α-bungarotoxin staining and measuring the area within each outline in ImageJ. Colocalization of α-bungarotoxin and GFP staining was determined using the Coloc2 plugin with default parameters in ImageJ with individual neuromuscular junctions as regions of interest (ROI).
FACS Isolation of e12.5 Spinal Motor Neurons
Mouse embryos from Hb9-GFP;Clp1R140H/+ x Clp1R140H/+ or Hb9-GFP;Clp1R140H/+ x Hb9-GFP;Clp1R140H/+ timed matings were collected at embryonic day (e)12.5 and screened for GFP expression. A GFP-negative embryo was retained as a negative control for sorting. GFP-positive embryos were genotyped as described above. Spinal cords were dissected and dissociated using a Papain dissociation kit (Worthington). Briefly, spinal cords were incubated at 37°C for 30min in a solution of Papain and DNase dissolved in EBSS. Spinal cords were dissociated by gentle pipetting. Cells were spun down and resuspended in 1% FBS and DNase in PBS. The cell suspension was passed through a cell strainer (Falcon). Cells were spun down and resuspended in 1% FBS and DNase in PBS, 200 μl per embryo. GFP-positive motor neurons were isolated by fluorescence activated cell sorting (FACS) using the GFP-negative sample as the gating control. Motor neurons were sorted into PBS and spun down to remove excess PBS before adding the extraction buffer from the Picopure RNA isolation kit (Thermo Fisher) using a ~4:1 extraction buffer to cell suspension ratio.
Cell Density Measurement
An MTT cell assay was performed on human stem cells during motor neuron differentiation (day 16 – day 28) as previously described (Schaffer et al., 2014) with the following considerations. Motor neuron progenitors at day 15 of differentiation were plated at 105 cells/well and measurements were captured every two days.
Western Blot
Western blot analysis for protein expression was performed according to standard protocols. Protein lysates collected in RIPA lysis buffer (25mM Tris-HCl, pH 7.5, 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, Protease Inhibitor Cocktail (ThermoFisher) from CLP1WT, CLP1KO, CLP1 p.R140H heterozygous (unaffected, U), and CLP1 p.R140H homozygous (affected, A) cell lines were separated by SDS-PAGE and transferred to a nitrocellulose membrane using the TransBlot Turbo (Biorad). The membrane was blocked with 5% non-fat milk in PBS with 0.05% Tween-20 for 3′ minutes at room temperature. Primary antibodies against the protein of interest were applied overnight at 4°C. Primary antibodies for analyzing protein expression include rabbit anti-CLP1 (Abcam, 1:2000), rabbit anti-CLP1 (ThermoFisher, 1:1000), rabbit anti-CAPRIN1 (ThermoFisher, 1:1000), rabbit anti-CAMTA1 (ThermoFisher, 1:1000), rabbit anti-FLAG (Sigma-Aldrich, 1:500), mouse anti-β-actin (loading control; Proteintech, 1:20,000), and mouse anti-GAPDH (loading control; Proteintech, 1:20,000). The membrane was washed in PBS with 0.05% Tween-20 and HRP-conjugated goat anti-rabbit and goat anti-mouse secondary antibodies (Abcam, 1:20,000) diluted in 5% non-fat milk in PBS with 0.05% Tween-20 were incubated at room temperature for 1–2 hours. Supersignal West Dura substrate (Thermofisher) was used for detection and blots were imaged with a Li-Cor Fc instrument. Quantification of western blots was performed in ImageJ.
Co-immunoprecipitation
Co-immunoprecipitation of RNA polymerase II was performed with Dynabeads Protein G for Immunoprecipitation (ThermoFisher) according to the manufacturer protocol with the following modifications. Protein lysates were collected in NP-40 lysis buffer (50mM HEPES-KOH, pH 7.5, 150mM KCl, 2mM MgCl2, 2% NP-40, Protease Inhibitor Cocktail (ThermoFisher)) from CLP1WT, CLP1KO, CLP1 p.R140H heterozygous (unaffected, U), and CLP1 p.R140H homozygous (affected, A) cell lines. Primary antibodies for the co-immunoprecipitation experiments include mouse anti-RNAPII (Active Motif, 10μg per IP) and mouse IgG Isotype Control (ThermoFisher, 10μg per IP). Following antigen binding, bead-Antibody-Antigen complexes were washed in PBS with 0.1% Tween-20. Immunoprecipitated proteins were eluted using the denaturing elution protocol. Western blot analysis of immunoprecipitated proteins was performed as previously described. Primary antibodies for analysis include mouse anti-RNAPII (Active Motif, 1:1000), rabbit anti-PCF11 (Proteintech, 1:1000), rabbit anti-CPSF2 (Proteintech, 1:1000), rabbit anti-CPSF6 (ThermoFisher, 1:1000), rabbit anti-CstF64 (Proteintech, 1:500), and mouse anti-β-actin (loading control, Proteintech, 1:20,000). Quantification of western blots was performed in ImageJ.
Northern Blot
Northern blot analysis was performed as previously described (Schaffer et al., 2014). Probe sequences for intron-containing tRNAs are as follows: Chr14.tRNA19-TyrGTA intron probe 5′-GAT GTC CAC AAA TGT TTC TAC AGG CTA C-3′, Chr19.tRNA10-IleTAT intron probe 5′-TGC TCC GCT CGC ACT GTC A-3′, Tyr-GTA 5′exon probe 5′-CTA CAG TCC TCC GCT CTA CC-3′, Ile-TAT 5′-exon probe 5′-TAT AAG TAC CGC GCG CTA AC-3′, Leu-CAA 5′-exon probe 5′-CTT GAG TCT GGC GCC TTA GAC-3′. A U6 probe 5′-GCA GGG GCC ATG CTA ATC TTC TCT GTA TCG-3′ was used as a loading control.
Quantitative Real-time Reverse Transcriptase PCR
Total RNA was isolated from hESC- or patient-specific, iPSC-derived motor neurons using TRIzol as per manufacturer protocol. Random hexamer primed cDNA for validation of differential gene expression, alternative splicing, and APA was synthesized using the SuperScript IV VILO Master Mix with ezDNase Enzyme (ThermoFisher) as per manufacturer protocol.
Total RNA was isolated from sorted mouse embryonic spinal motor neurons using the Picopure RNA isolation kit (ThermoFisher) as per manufacturer protocol, and total RNA from whole embryonic spinal cords was isolated using TRIzol as described above. Reverse transcription and qPCR of mouse RNA samples spinal motor neuron total RNA were carried out as described above with the following modifications. RNA prepared using the Picopure kit were not treated with ezDNase during cDNA synthesis.
Quantitative PCR was performed with either PowerUP or Power SYBR™ Green PCR Master Mix (ThermoFisher) as per manufacturer protocol on a CFX384 thermal cycler (Bio-Rad).
Primers to validate differential gene expression were designed to span constitutive exon junctions. Primers for validation of splicing events were designed to flank both constitutive and alternative exons. Two reference genes for DGE and alternative splicing validation, RANBP2 and EIF2B5, were chosen from the DESeq2 results as highly expressed and with low change. APA validation primers were designed to amplify regions upstream of the 100bp bioinformatic counting window for chosen polyadenylation sites. Primer sequences are listed in Data S1L.
Digital PCR gel analysis
A subset of alternative splicing events with cassette exon inclusion/exclusion were validated by RT-PCR followed by digital gel analysis. Random hexamer primed cDNA was synthesized as described above. Primers were designed to flank the alternative exon and PCR was carried out using GoTaq® DNA Polymerase (Promega) as per manufacturer protocol for 3′ cycles. The resultant products were resolved by TapeStation (Agilent) and quantified.
RNA sequencing and Bioinformatics
Two biological replicates of CLP1WT H9 hESC-derived motor neurons, CLP1KO H9 hESC-derived motor neurons, and two clonal replicates of CLP1 p.R140H heterozygous iPSC-derived motor neurons (unaffected parent), and CLP1 p.R140H homozygous (affected child) were used for total RNA-seq analysis. Two clonal replicates of the following cell lines were used for total tRNA-seq analysis: CLP1 p.R140H heterozygous iPSC-derived motor neurons and CLP1 p.R140H homozygous. RNA was extracted from hESC- or iPSC-derived motor neurons at day 28 of differentiation with TRIzol as per manufacturer protocol.
tRNA sequencing was performed as in Pinkard et al. (2020). Briefly, 1ug of total RNA was ligated to annealed adapters under conditions as in Shigematsu et al. (2017). cDNA was synthesized according to the manufacturer’s suggested conditions using Super-Script IV Reverse Transcriptase (Thermo Fisher) and gel purified with denaturing polyacrylamide gel electrophoresis. Gel purified cDNA product was ligated using CircLigase (Lucigen) according to manufacturer’s recommendations. Libraries were amplified using Q5 DNA polymerase (New England Biosciences) for 7–8 cycles and purified on a native agarose gel following the manufacturer’s suggested parameters. Libraries were sequenced with single end reads on an Illumina NextSeq 550. The sequencing reads were aligned to predicted tRNA genes from GtRNAdb with Bowtie 2 (Langmead and Salzberg, 2012) following demultiplexing and CCA trimming, and reads per million (RPM) counted by featureCounts (Liao et al., 2014) in R. Normalized tRNA isodecoder reads were summed and differential gene expression analysis was performed with DESeq2 (Love et al., 2014) using default parameters.
For mRNA sequencing, stranded paired-end 150bp total RNA-sequencing with ribosomal depletion was performed by Novogene, Inc. using the dUTP method (Parkhomchuk et al., 2009). Reads were aligned to the Hg38 human genome build with STAR (Dobin et al., 2013) using default parameters, returning a unique mapping rate of 94–95% (31–33 million uniquely mapping reads) per sample. Differential gene expression was tested using DESeq2 (Love et al., 2014) with default parameters through Galaxy (Afgan et al., 2018) using genewise count files generated by featureCounts (Liao et al., 2014) against the Ensembl Release 95 (Frankish et al., 2019; Zerbino et al., 2018) gene annotation set. Any results on the Y chromosome were filtered out. Alternative exon events were tested with rMATS (Shen et al., 2014) using default parameters and the Ensembl Release 95 gene annotation set. Per-exon and per-splice junction events which were used to determine positional bias were discovered using JunctionSeq (Hartley and Mullikin, 2016) with default parameters against the Ensembl Release 95 gene annotation set. Relative cell type composition bioinformatic analysis was performed on the same RNA-seq samples as above. In brief, genewise abundance estimates were calculated with pseudoalignment using Salmon (Patro et al., 2017) and genewise aggregation with tximport (Soneson et al., 2016). We calculated the geometric mean TPM of cell type-specific marker genes gathered from D’Erchia et al., (2017) and compared marker expression averages between the affected and unaffected motor neuron samples.
For mRNA 3’ end sequencing analysis, two batches of PAC-seq were run. One batch was a collection of RNA from 16 human stem cell-derived motor neuron cultures from the PCH10 index patient family and H9 hESC genetic backgrounds. A smaller batch containing H9 CLP1WT and CLP1KO iPSC-derived motor neuron RNA samples, with two replicates each, was also run. PAC-Seq library preparation and sequencing was performed as previously described (Routh et al., 2017). The larger PAC-seq batch showed ample polyadenosine homopolymeric stretches within the 3’ end of its reads, whereas the smaller batch did not. As such, only samples from the larger batch were used to construct in-house annotation databases of human and mouse motor neuron polyadenylation sites. To construct these polyadenylation site annotations, each sample’s raw reads underwent pre-alignment trimming which ensured polyadenosine stretches at the 3’ end of reads remained intact. Reads with any 2-color sequencing chemistry artifacts were trimmed with Cutadapt (Martin, 2011) using settings -a G{150} -e 0.4. Any reads smaller than 40 bp after trimming were filtered out. 12 nucleotide unique molecular identifiers (UMIs) were removed from the 5’ end of each read and appended to the read header using umi_tools (Smith et al., 2017). Processed reads were aligned to the hg38 genome build with STAR (Dobin et al., 2013) using an alignment strategy which only performs softclipping on the 3’ end of reads (–alignEndsType Extend5pOfRead1). Because untemplated poly(A) tails could cover large portions of input reads, STAR was also set to be less strict with the minimum proportion of the read aligned (–outFilterMatchNminOverLread 0.3 and –outFilterScoreMinOverLread 0.3). In total, this alignment strategy produced ~75% uniquely mapped reads and optimized for alignments which ended directly at the polyadenylation site but still contained some poly(A) tail sequence within the softclipped data.
These alignments were then filtered with an in-house script to remove any entries with no softclipping before deduplication using umi_tools. A final filtering step performed with in-house scripts inspected the sequence data that was soft-clipped and removed any read alignments which did not show enough untemplated adenosines to meet the standards followed by Gencode for their polyadenylation site annotations (Derti et al., 2012). Specifically, each read’s softclipped sequence needed to contain 1) greater than 66% adenosine content and 2) at least 3 untemplated adenosines. This filtering step was made more strict for sites where the sequence just upstream of the softclip indicated that the read came from an internal priming hotspot, in which case the sum of terminal adenosines must exceed the length of the oligo d(T) primer, then show an additional 3+ untemplated adenosines beyond this range. Using these high-confidence alignments, each sample had its unique polyadenylation sites defined by taking slices of read coverage greater than a minimum read threshold, keeping the most 3’ end nucleotide of each slice region, then reducing the read threshold repeatedly, keeping the higher coverage slice region when there were overlaps. This coverage slicing process was repeated when merging each sample’s unique polyadenylation sites to output a final set of sites that showed at least 3 reads in at least 2 samples, and were at least 100 bp apart.
Providing each polyadenylation site with an assigned gene and intragenic feature followed logic that replicates how one annotates polyadenylation sites by hand. Each polyadenylation site was checked for overlap or proximity to specific intragenic features in order of likelihood, and removed from the pool for annotation when a probable match was found. These features in order were transcript ends, 3’ UTRs, terminal exons, regions up to 10 kb downstream of transcript ends, introns, CDS, 5’ UTRs, and exons, respectively. Any site which had more than one potential match in a step would be assigned to the longest transcript and its corresponding gene. Determining which intragenic feature of an assigned gene that a polyadenylation site was part of followed the rules applied to Poly-A_DB (Wang et al., 2018) with the change that we used Gencode gene and transcript annotations v37 for human (Frankish et al., 2019).
With final polyadenylation site annotations available, APA analysis was performed with DEXSeq (Anders et al., 2012). Polyadenylation site counts were acquired using countOverlaps in the GenomicRanges R package (Lawrence et al., 2013), with inputs of either fully-filtered per-sample alignments as described above, or in the case of samples in the smaller sequencing batch, unfiltered alignments were used as count inputs and counts were mathematically adjusted before use in DEXSeq to reflect the site-specific proportion of reads that passed filters in the 16 other samples. Any genes with only one annotated polyadenylation site were excluded from analysis.
Analysis of external 3’ end sequencing datasets for sporadic Parkinson’s disease (GEO: GSE40710) or Myotonic Dystrophy (GEO: GSE68890) took place in a near-identical fashion to what was performed for the smaller sequencing batch because untemplated adenosines were relatively scant within each read. Polyadenylation site counts based on unfiltered alignments were multiplied by the inverse of the empirically determined internal priming rate at each given site before use in DEXSeq.
Positional motif enrichment analysis was performed using CentriMo (Bailey and Machanick, 2012) within the MEME Suite (Bailey et al., 2015). Genomic sequence at each polyadenylation site +/− 100bp was extracted using the BSgenome.Hsapiens.UCSC.hg38 R package (Pagès, 2018). Sequences flanking non-significant polyadenylation sites for a given comparison were used as the control sequence input and sequences flanking upregulated or downregulated polyadenylation sites were used as the primary sequence input. Positional enrichment settings were such that enrichment could occur anywhere, and only strand-matching motifs could be searched. Positional motif matching was performed against the Ray 2013 Homo sapiens database (Ray et al., 2013). Differentially enriched motifs required a Fisher E-value less than 0.05 to be deemed statistically significant.
Gene ontology (GO) term enrichment visualization was performed using ClueGO, a Cytoscape plug-in developed by Bindea et al., (2009). The input for ClueGO was a list of genes with differential expression and alternative polyadenylation, grouped by fold change sign.
Clustering was performed using the GO-BiologicalProcess-EBI-UniProt-GOA-ACAP-ARAP-13.05.2021 ontology database, showing pathways with p-value ≤ 0.05. Default settings were used for all other parameters. Additional GO term enrichment analysis (see Figure S7) was performed using DAVID (Huang et al., 2009a, 2009b) with a gene list of upregulated or downregulated genes (FDR < 0.05) against a background set of all genes expressed in our dataset at or above 1 TPM. DAVID enrichment was performed using the website’s biological process, molecular function, and cellular compartment annotations (see Data S1I and S1J).
QUANTIFICATION AND STATISTICAL ANALYSIS
Error bars in figures are shown as ± 1 SEM unless otherwise stated. The number (n) and type of replicates analyzed for each experiment is described within the figure legends. Boxplots are displayed with lines at the median, 25th, and 75th percentiles, with “whiskers” extending the full range of the data. Statistical tests were performed using R (Fisher’s exact test, K-S test, Binomial test, Wilcoxon Rank Sum test, Welch’s t test, Pearson correlation test, Chi-squared test) or Microsoft Excel (Student’s t test). All tests were performed two-sided unless otherwise noted. Sample sizes were not statistically pre-determined. Experiments were not randomized. Non-normally distributed data were tested by Wilcoxon Rank Sum for means or K-S test for distribution. Logarithmic or other scaling transformations were performed as written in graph axes. Collection and analysis of data was not blinded with the exception of cell counting in immunofluorescence images. Tests for overlap or enrichment were performed using the Chi-squared likelihood ratio test (3-way Venn diagram) or Fisher’s exact test (2-way Venn diagram).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-synaptophysin | ThermoFisher | Cat#: MA5–14532; RRID: AB_10983675 |
| Mouse anti-α-Bungarotoxin | ThermoFisher | Cat#: B35451; RRID: AB_2617152 |
| Mouse anti-TRA-1–81 | Millipore | Cat#: MAB4381; RRID: AB_177638 |
| Rabbit anti-Oct4 | ThermoFisher | Cat#: PA5–27438; RRID: AB_2544914 |
| Goat-anti-LIN-28A | R&D Biosystems | Cat#: AF3757; RRID: AB_2234537 |
| Mouse anti-Nanog (1E6C4) | Santa Cruz | Cat#: sc-293121; RRID: AB_2665475 |
| Rabbit anti-α-fetoprotein | Dako | Cat#: A000829–2 |
| Mouse anti-α smooth muscle Actin | Sigma-Aldrich | Cat#: M085101–2 |
| Mouse anti-Nestin | ThermoFisher | Cat#: MA1–110; RRID: AB_2536821 |
| Rabbit anti-PAX6 | BioLegend | Cat#: 901301; RRID: AB_2565003 |
| Mouse anti-islet-1/2 | DSHB | Cat#: 39.4D5; RRID: AB_2314683 |
| Mouse anti-HB9/Mnx1 | DSHB | Cat#: 81.5C10; RRID: AB_2145209 |
| Goat anti-SCIP | Santa Cruz | Cat#: sc-11661; RRID: AB_2268536 |
| Anti-HOXA5 | Dasen et al., 2005 | N/A |
| Anti-FOXP1 | Dasen et al., 2008 | N/A |
| Anti-HOXC6 | Liu et al., 2001 | N/A |
| Anti-HOXC9 | Jung et al., 2010 | N/A |
| Anti-LHX3 | Tsuchida et al., 1994 | N/A |
| Rabbit-anti-OLIG2 | Proteintech | Cat#: 13999–1-AP; RRID: AB_2157541 |
| Mouse anti-p-Histone H3 | Santa Cruz | Cat#: sc-374669; RRID: AB_11150094 |
| Mouse anti-KI 67 | Cell Signaling | Cat#: 9449S; RRID: AB_2797703 |
| Rabbit anti-Cleaved Caspase-3 | Cell Signaling | Cat#: 9603S; RRID: AB_11179205 |
| Rabbit anti-GPIP137 (CAPRIN1) | ThermoFisher | Cat#: PA5–114941 |
| Rabbit anti-CAMTA1 | ThermoFisher | Cat#: PA5–78440; RRID: AB_2735569 |
| Rabbit anti-CLP1 | Abcam | Cat#: ab133669 |
| Rabbit anti-CLP1 | ThermoFisher | Cat#: PA5–22386; RRID: AB_11155231 |
| Rabbit anti-PCF11 | Proteintech | Cat#: 23540–1-AP; RRID: AB_2879293 |
| Mouse anti-RNA Polymerase II | Active Motif | Cat#: 91151; RRID: AB_2793789 |
| Rabbit anti-CPSF2 (CPSF100) | Proteintech | Cat#: 17739–1-AP; RRID: AB_2084368 |
| Rabbit anti-CstF64 | Proteintech | Cat#: 26825–1-AP; RRID: AB_2880649 |
| Rabbit anti-CPSF6 (CFIm68) | ThermoFisher | Cat#: PA5–41830; RRID: AB_2607690 |
| Mouse anti-β-Actin | Proteintech | Cat#: 66009–1-Ig; RRID: AB_2687938 |
| Mouse anti-GAPDH | Proteintech | Cat#: 60004–1-Ig; RRID: AB_2107436 |
| Mouse IgG Isotype Control | ThermoFisher | Cat#:10400C; RRID: AB_2532980 |
| Rabbit anti-FLAG | Sigma-Aldrich | Cat#: F7425; RRID: AB_439687 |
|
| ||
| Biological samples | ||
|
| ||
| Mouse brain tissue | Strains listed in this table | N/A |
| Mouse diaphragm tissue | Strains listed in this table | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Thiazoyl Blue Tetrazolium Bromide | Sigma-Aldrich | Cat#: M5655; CAS: 298–93-1 |
| Corning Matrigel Basement Membrane Matrix | VWR | Cat#: 47743–715 |
| Vitronectin (VTN-N) Recombinant Human Protein, Truncated | ThermoFisher | Cat#: A14700 |
| Dynabeads Protein G for Immunoprecipitation | ThermoFisher | Cat#:10004D |
| Pierce Protease Inhibitor Mini Tablets, EDTA-free | ThermoFisher | Cat#: A32955 |
|
| ||
| Deposited data | ||
|
| ||
| Analyzed RNA-sequencing data | This paper | GEO: GSE183872 |
| Analyzed Poly(A)Click-sequencing data | This paper | GEO: GSE183872 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: 1810-VI-1 (affected) Fibroblasts | Schaffer et al., 2014 | N/A |
| Human: 1810-V-8 (unaffected) Fibroblasts | Schaffer et al., 2014 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Clp1R140H/+: B6SJLF1/J-Clp1em1Aes | This paper | N/A |
| Mouse: Clp1KO/+: B6SJLF1/J-Clp1em2Aes | This Paper | N/A |
| Mouse: Hb9-GFP+: B6.Cg-Tg(Hlxb9-GFP) 1Tmj/J | Jackson Laboratory | Stock No: 005029 |
| Mouse: ChAT-GFP+: B6.Cg-Tg(RP23–268L19-EGFP)2Mik/J | Jackson Laboratory | Stock No: 007902 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for genotyping (see Data File S1L) | Eurofins Genomics | N/A |
| Primers for qPCR (see Data File S1L) | Eurofins Genomics | N/A |
| CRISPR constructs (see Data File S1L) | PNA Bio, IDT | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| px330 | Cong, et al. 2013 | Addgene Plasmid #42230 |
| pCMV-GFP | Matsuda and Cepko, 2004 | Addgene Plasmid #11153 |
| pCXLE-hOCT3/4-shp53-F | Okita et al., 2011 | Addgene Plasmid #27077 |
| pCXLE-hSK | Okita et al., 2011 | Addgene Plasmid #27078 |
| pCXLE-hUL | Okita et al., 2011 | Addgene Plasmid #27080 |
| pCXWB-EBNA1 | Okita et al., 2013 | Addgene Plasmid #37624 |
|
| ||
| Software and algorithms | ||
|
| ||
| Bowtie 2 v2.3.5.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| DESeq2 v1.26.0 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| STAR v2.7.3a | Dobin et al., 2013 | https://github.com/alexdobin/STAR/releases |
| featureCounts v2.0.1 | Liao et al., 2014 | http://bioconductor.org/packages/release/bioc/html/Rsubread.html |
| RNASeq-MATS v4.0.2 | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net/ |
| JunctionSeq v1.16.0 | Hartley and Mullikin, 2016 | https://bioconductor.org/packages/release/bioc/html/JunctionSeq.html |
| Cutadapt v2.8 | Martin, 2011 | https://pypi.org/project/cutadapt/ |
| DEXSeq v1.32.0 | Anders et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| GenomicRanges v1.34.0 | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| GenomicAlignments v1.18.1 | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| GenomicFeatures v1.34.8 | Lawrence et al., 2013 | http://bioconductor.org/packages/release/bioc/html/GenomicFeatures.html |
| BSgenome v1.50.0 | Pagès, 2018 | https://bioconductor.org/packages/release/bioc/html/BSgenome.html |
| Rsamtools v1.34.1 | Morgan et al., 2019 | https://bioconductor.org/packages/release/bioc/html/Rsamtools.html |
| rtracklayer v1.42.2 | Lawrence et al., 2009 | https://bioconductor.org/packages/release/bioc/html/rtracklayer.html |
| ggplot2 v3.3.5 | Wickham, 2016 | https://ggplot2.tidyverse.org |
| CentriMo v5.4.1 | Bailey and Machanick, 2012 | https://meme-suite.org/meme/tools/centrimo |
| GSEA v4.0.0 | Mootha et al., 2003; Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ClueGO v2.5.8 | Bindea et al., 2009 | https://apps.cytoscape.org/apps/cluego |
| DAVID v6.8 | Huang et al., 2009a, 2009b | https://david.ncifcrf.gov/home.jsp |
| RStudio | RStudio | https://rstudio.com/products/rstudio/download/ |
| Fiji | Fiji | http://fiji.sc |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij |
| Illustrator 2021 | Adobe Inc. | https://www.adobe.com/products/illustrator.html |
| Photoshop 2021 | Adobe Inc. | https://www.adobe.com/products/photoshop.html |
Highlights.
CLP1 limits mRNA isoform diversity by suppressing proximal poly(A) site choice
CLP1 knockout and pathogenic CLP1 p.R140H have opposing effects on mRNA processing
Intronic polyadenylation evasion drives overexpression of long genes in neurons
Neurodegenerative diseases show a pattern of reduced mRNA 3′ end diversity
ACKNOWLEDGMENTS
The authors would like to thank Drs. Baker and Serdar for their technical support and Drs. Licatalosi and Lou for their scientific insights and feedback. Dr. Dasen provided the HOXA5, FOXP1, HOXC9, HOXC6, and LHX3 antibodies to P.P.; the HB9 and ISL1/2 monoclonal antibodies were developed by T.M. Jessell and S. Brenner-Morton and were obtained from the Developmental Studies Hybridoma Bank. This work made use of the HPCC Resource and Case Transgenic & Targeting Core Facilities at Case Western Reserve University. This study was supported by NIH R00HD082337 and R01NS5123524, the Mt. Sinai Foundation, and CWRU Startup Funds to A.E.S.; NIH R00NS085037, NIH R01NS114510, the Mt. Sinai Foundation, Weidenthal Family Designated Professorship in Career Development, and CWRU Startup Funds to P.P.; NIH K01NS116119 and R01NS121374 to H.C.M.; NIH R03CA223893 to P.J.; NIH T32NS077888 and F31NS122207 to G.R.L.; the Chinese Scholarship Council (CSC) Fellowship to J.L.; UTMB BMB Startup Funds to E.J.W.; and NIH R01GM125086 to J.C.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2022.01.018.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46, W537–W544. 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcott CE, Yalamanchili HK, Ji P, van der Heijden ME, Saltzman A, Elrod N, Lin A, Leng M, Bhatt B, Hao S, et al. (2020). Partial loss of CFIm25 causes learning deficits and aberrant neuronal alternative polyadenylation. Elife 9, e50895. 10.7554/eLife.50895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, and Huber W. (2012). Detecting differential usage of exons from RNA-seq data. Genome Res 22, 2008–2017. 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res 43, W39–W49. 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, and Machanick P. (2012). Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40, e128. 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgard TG, Marques AC, Oliver PL, Abaan HO, Sirey TM, Hoerder-Suabedissen A, García-Moreno F, Molnár Z, Margulies EH, and Ponting CP (2011). A transcriptomic atlas of mouse neocortical layers. Neuron 71, 605–616. 10.1016/j.neuron.2011.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, and Galon J. (2009). ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Choi EA, and Shi Y. (2011). Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2, 321–335. 10.1002/wrna.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C-C., Zhang Y., Umoh ME., Vaughan SW., Lorenzini I., Liu F., Sayegh M., Donlin-Asp PG., Chen YH., Duong DM., et al. (2018). TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 21, 228–239. 10.1038/s41593-017-0047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Erchia AM, Gallo A, Manzari C, Raho S, Horner DS, Chiara M, Valletti A, Aiello I, Mastropasqua F, Ciaccia L, et al. (2017). Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 7, 10046. 10.1038/s41598-017-10488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasen JS, De Camilli A, Wang B, Tucker PW, and Jessell TM (2008). Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor. FoxP1. Cell 134, 304–316. 10.1016/j.cell.2008.06.019. [DOI] [PubMed] [Google Scholar]
- Dasen JS, Tice BC, Brenner-Morton S, and Jessell TM (2005). A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell 123, 477–491. 10.1016/j.cell.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Davis R, and Shi Y. (2014). The polyadenylation code: a unified model for the regulation of mRNA alternative polyadenylation. J. Zhejiang Univ. Sci. B 15, 429–437. 10.1631/jzus.B1400076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries H, Rüegsegger U, Hübner W, Friedlein A, Langen H, and Keller W. (2000). Human pre-mRNA cleavage factor II(m) contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J 19, 5895–5904. 10.1093/emboj/19.21.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, MacIsaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, and Babak T. (2012). A quantitative atlas of polyadenylation in five mammals. Genome Res 22, 1173–1183. 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, and Traynelis SF (1999). The Glutamate Receptor Ion Channels. Pharmacological Reviews 51, 7–62. [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet M, Fara AG, Beritognolo I, and Sabatti M. (2009). Allele-specific PCR in SNP genotyping. Methods Mol. Biol. 578, 415–424. 10.1007/978-1-60327-411-1_26. [DOI] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL III, Bigio EH, Caselli R, et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin M, Mohan A, Batra R, Lee K-Y, Charizanis K, Fernández Gómez FJF, Eddarkaoui S, Sergeant N, Buée L, Kimura T, et al. (2015). MBNL sequestration by toxic RNAs and RNA misprocessing in the Myotonic dystrophy brain. Cell Rep 12, 1159–1168. 10.1016/j.celrep.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guhaniyogi J, and Brewer G. (2001). Regulation of mRNA stability in mammalian cells. Gene 265, 11–23. 10.1016/S0378-1119(01)00350-X. [DOI] [PubMed] [Google Scholar]
- Gupta I, Collier PG, Haase B, Mahfouz A, Joglekar A, Floyd T, Koopmans F, Barres B, Smit AB, Sloan SA, et al. (2018). Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 36, 1197–1202. 10.1038/nbt.4259. [DOI] [PubMed] [Google Scholar]
- Guyon C, Jmari N, Padonou F, Li Y-C, Ucar O, Fujikado N, Coulpier F, Blanchet C, Root DE, and Giraud M. (2020). Aire-dependent genes undergo Clp1-mediated 3′UTR shortening associated with higher transcript stability in the thymus. Elife 9, e52985. 10.7554/eLife.52985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, et al. (2013). CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 495, 474–480. 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SW, and Mullikin JC (2016). Detection and visualization of differential splicing in RNA-seq data with JunctionSeq. Nucleic Acids Res 44, e127. 10.1093/nar/gkw501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Ruknudin A, Bambrick LL, Lederer WJ, and Schulze DH (1998). Isoform-specific regulation of the Na+/Ca2+ exchanger in rat astrocytes and neurons by PKA. J. Neurosci. 18, 4833–4841. 10.1523/JNEUROSCI.18-13-04833.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Hwang H-W, Saito Y, Park CY, Blachère NE, Tajima Y, Fak JJ, Zucker-Scharff I, and Darnell RB (2017). cTag-PAPERCLIP reveals alternative polyadenylation promotes cell-type specific protein diversity and shifts Araf isoforms with microglia activation. Neuron 95, 1334–1349.e5. 10.1016/j.neuron.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, and Thornton CA (2004). Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13, 3079–3088. 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Jung H, Lacombe J, Mazzoni EO, Liem KF Jr., Grinstein J, Mahony S, Mukhopadhyay D, Gifford DK, Young RA, Anderson KV, et al. (2010). Global control of motor neuron topography mediated by the repressive actions of a single hox gene. Neuron 67, 781–796. 10.1016/j.neuron.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde CV, Bouchard J-P, Lacomblez L, Pochigaeva K, Salachas F, et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kamieniarz-Gdula K, Gdula MR, Panser K, Nojima T, Monks J, Wiśniewski JR, Riepsaame J, Brockdorff N, Pauli A, and Proudfoot NJ (2019). Selective roles of vertebrate PCF11 in premature and full-length transcript termination. Mol. Cell 74, 158–172.e9. 10.1016/j.molcel.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, et al. (2014). Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157, 636–650. 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva A, Sepp M, Kazantseva J, Sadam H, Pruunsild P, Timmusk T, Neuman T, and Palm K. (2009). N-terminally truncated BAF57 isoforms contribute to the diversity of SWI/SNF complexes in neurons. J. Neurochem. 109, 807–818. 10.1111/j.1471-4159.2009.06005.x. [DOI] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. (2013). Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62. 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, and Carey V. (2009). rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842. 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comp. Biol. 9, e1003118. 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lerch JK, Kuo F, Motti D, Morris R, Bixby JL, and Lemmon VP (2012). Isoform diversity and regulation in peripheral and central neurons revealed through RNA-seq. PLoS One 7, e30417. 10.1371/journal.pone.0030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, and Tian B. (2015). Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet 11, e1005166. 10.1371/journal.pgen.1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, and Rothstein JD (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20, 589–602. 10.1016/S0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- Liu JP, Laufer E, and Jessell TM (2001). Assigning the positional identity of spinal motor neurons: rostrocaudal patterning of Hox-c expression by FGFs, Gdf11, and retinoids. Neuron 32, 997–1012. [DOI] [PubMed] [Google Scholar]
- Liu X, Bates R, Yin D-M, Shen C, Wang F, Su N, Kirov SA, Luo Y, Wang J-Z, Xiong W-C, and Mei L. (2011). Specific regulation of NRG1 isoform expression by neuronal activity. J. Neurosci. 31, 8491–8501. 10.1523/JNEUROSCI.5317-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang Y-J, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al. (2010). Tar DNA binding Protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5, e13250. 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado CB, Kanning KC, Kreis P, Stevenson D, Crossley M, Nowak M, Iacovino M, Kyba M, Chambers D, Blanc E, and Lieberam I. (2014). Reconstruction of phrenic neuron identity in embryonic stem cell-derived motor neurons. Development 141, 784–794. 10.1242/dev.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel CR, Bai Y, and Tong L. (2008). Protein factors in pre-mRNA 3′-end processing. Cell. Mol. Life Sci. 65, 1099–1122. 10.1007/s00018-007-7474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, et al. (2018). Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–604.e13. 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, and Zavolan M. (2012). Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep 1, 753–763. 10.1016/j.celrep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proceedings of the National Academy of Sciences 101, 16. 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C. (2016). Evolution and biological roles of alternative 3′UTRs. Trends Cell Biol 26, 227–237. 10.1016/j.tcb.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Shenker S, Andreu-Agullo C, Westholm JO, and Lai EC (2013). Widespread and extensive lengthening of 3′ UTRs in the mammalian brain. Genome Res 23, 812–825. 10.1101/gr.146886.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan CE, Adamson SI, Kapur M, Chuang JH, and Ackerman SL (2021). The Clp1 R140H mutation alters tRNA metabolism and mRNA 3’ processing in mouse models of pontocerebellar hypoplasia. Proc. Natl. Acad. Sci. USA 118, e2110730118. 10.1073/pnas.2110730118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Morgan M, Pagès H, Obenchain V, and Hayden N. (2019). Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import, R package version 1.34.1. http://bioconductor.org/packages/release/bioc/html/Rsamtools.html. [Google Scholar]
- Morisaki I, Shiraishi H, Fujinami H, Shimizu N, Hikida T, Arai Y, Kobayashi T, Hanada R, Penninger JM, Fujiki M, and Hanada T. (2021). Modeling a human CLP1 mutation in mouse identifies an accumulation of tyrosine pre-tRNA fragments causing pontocerebellar hypoplasia type 10. Biochem. Biophys. Res. Commun. 570, 60–66. 10.1016/j.bbrc.2021.07.036. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ogorodnikov A, Levin M, Tattikota S, Tokalov S, Hoque M, Scherzinger D, Marini F, Poetsch A, Binder H, Macher-Göppinger S, et al. (2018). Transcriptome 30end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 9, 5331. 10.1038/s41467-018-07580-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K. i., et al. (2011). A more efficient method to generate integration-free human iPS cells. Nature Methods 8, 409–412. 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Okita K, Yamakawa T, Matsumura Y, Sato Y, Amano N, Watanabe A, Goshima N, and Yamanaka S. (2013). An Efficient Nonviral Method to Generate Integration-Free Human-Induced Pluripotent Stem Cells from Cord Blood and Peripheral Blood Cells. Stem Cells 31, 458–466. 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- Pagès H. (2018). BSgenome: Software infrastructure for efficient representation of full genomes and their SNPs. R package version 1.50.0. http://bioconductor.org/packages/BSgenome/. [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, and Soldatov A. (2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37, e123. 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin SV, Patel M, Furia BS, Peltz SW, and Trotta CR (2004). Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3′ end formation. Cell 117, 311–321. 10.1016/s0092-8674(04)00342-3. [DOI] [PubMed] [Google Scholar]
- Philippidou P., Walsh CM., Aubin J., Jeannotte L., and Dasen JS. (2012). Sustained Hox5 gene activity is required for respiratory motor neuron development. Nat. Neurosci. 15, 1636–1644. 10.1038/nn.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkard O, McFarland S, Sweet T, and Coller J. (2020). Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun. 11, 4104. 10.1038/s41467-020-17879-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lührmann R, Söll D, and Martinez J. (2011). HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331, 760–764. 10.1126/science.1197847. [DOI] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177. 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, and Huber W. (2018). Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res 46, 582–592. 10.1093/nar/gkx1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, and Abeliovich A. (2012). Alternative a-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 3, 1084. 10.1038/ncomms2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh A, Ji P, Jaworski E, Xia Z, Li W, and Wagner EJ (2017). Poly(A)- ClickSeq: click-chemistry for next-generation 3-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res 45, e112. 10.1093/nar/gkx286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Breuss MW, Caglayan AO, Al-Sanaa N, Al-Abdulwahed HY, Kaymakç alan H, Yılmaz C, Zaki MS, Rosti RO, Copeland B, et al. (2018). Biallelic loss of human CTNNA2, encoding aN-catenin, leads to ARP2/3 complex overactivity and disordered cortical neuronal migration. Nature Genetics 50, 1093–1101. 10.1038/s41588-018-0166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. (2014). CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157, 651–663. 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR III, Frank J, and Manley JL (2009). Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 33, 365–376. 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu M, Honda S, Loher P, Telonis AG, Rigoutsos I, and Kirino Y. (2017). YAMAT-seq: an efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res 45, e70. 10.1093/nar/gkx005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman ED, and Elkon R. (2019). Cell-type-specific analysis of alternative polyadenylation using single-cell transcriptomics data. Nucleic Acids Res 47, 10027–10039. 10.1093/nar/gkz781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I. (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love M, and Robinson M. (2016). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000 Res. 4, 1521. 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Iida K, Tsubota T, Hosokawa M, Denawa M, Brown JB, Ninomiya K, Ito M, Kimura H, Abe T, et al. (2018). Loss of Sfpq causes long-gene transcriptopathy in the brain. Cell Rep 23, 1326–1341. 10.1016/j.celrep.2018.03.141. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Župunski V, et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, and Warman ML (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). BioTechniques 29, 52–54. 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, and Pfaff SL (1994). Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell 79, 957–970. 10.1016/0092-8674(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Turner RE, Pattison AD, and Beilharz TH (2018). Alternative polyadenylation in the regulation and dysregulation of gene expression. Semin. Cell Dev. Biol. 75, 61–69. 10.1016/j.semcdb.2017.08.056. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7, 409–416. 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk T, Baas F, Barth PG, and Poll-The BT (2018). What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J. Rare Dis. 13, 92. 10.1186/s13023-018-0826-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wafik M, Taylor J, Lester T, Gibbons RJ, and Shears DJ (2018). 2 new cases of pontocerebellar hypoplasia type 10 identified by whole exome sequencing in a Turkish family. Eur. J. Med. Genet. 61, 273–279. 10.1016/j.ejmg.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 7, 1–11. 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng D, Wei L, Ding Q, and Tian B. (2019). Regulation of intronic polyadenylation by PCF11 impacts mRNA expression of long genes. Cell Rep 26, 2766–2778.e6. 10.1016/j.celrep.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng D, Yehia G, and Tian B. (2018). A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res 28, 1427–1441. 10.1101/gr.237826.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzer S, Hanada T, Penninger JM, and Martinez J. (2015). CLP1 as a novel player in linking tRNA splicing to neurodegenerative disorders. Wiley Interdiscip. Rev. RNA 6, 47–63. 10.1002/wrna.1255. [DOI] [PubMed] [Google Scholar]
- Weitzer S, and Martinez J. (2007). The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature 447, 222–226. 10.1038/nature05777. [DOI] [PubMed] [Google Scholar]
- Wickham H. (2016). ggplot2: Elegant Graphics for Data Analysis (Springer; –Verlag: ). [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. (2018). Ensembl 2018. Nucleic Acids Res 46, D754–D761. 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing, Poly(A) Click-sequencing, and tRNA-sequencing data used in this study will be available from the Gene Expression Omnibus through series accession number GEO: GSE183872.
All other data referenced in this publication or code used to perform meta-analysis will be shared by the lead contact upon request.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.