

Abstract
Ischemic heart injury causes permanent cardiomyocyte loss and fibrosis impairing cardiac function. Tissue derived biomaterials have shown promise as an injectable treatment for the post-ischemic heart. Specifically, decellularized extracellular matrix (dECM) is a protein rich suspension that forms a therapeutic hydrogel once injected and improves the heart post-injury response in rodents and pig models. Current dECM-derived biomaterials are delivered to the heart as a liquid dECM hydrogel precursor or colloidal suspension which gels over several minutes. To increase the functionality of the dECM therapy, we developed an injectable solid dECM microparticle formulation derived from heart tissue to control sizing and extend stability in aqueous conditions. When delivered into the infarcted mouse heart, these dECM microparticles protect cardiac function, promote vessel density, and reduce left ventricular remodeling by sustained delivery of biomolecules. Longer retention, higher stiffness, and slower protein release of dECM microparticles are noted compared to liquid dECM hydrogel precursor. In addition, the dECM microparticle can be developed as a platform for macromolecule delivery. Together, the results suggest that dECM microparticles can be developed as a modular therapy for heart injury.
Keywords: decellularized extracellular matrix, microparticle, hydrogel, biomaterial, myocardial infarction, heart regeneration
Graphical Abstract
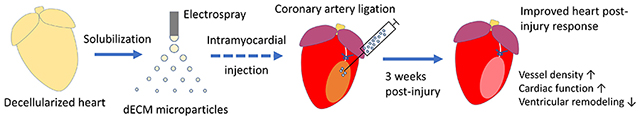
The dECM microparticle is a novel platform of dECM-derived materials for treating heart injury. Decellularized heart extracellular matrix (dECM) is solubilized and then processed (by electrospray) into microparticles. Injecting dECM microparticles into myocardial infarction by syringe improves angiogenesis, protects cardiac function, and reduces adverse ventricular remodeling.
1. Introduction
Cardiovascular disease is the leading cause of death worldwide predominated by myocardial infarction (MI). An MI results in permanent fibrosis and compromised cardiac function which has high risk for heart failure. Despite significant advances in tissue engineering, there is no regenerative therapy available for heart disease. Cell therapy and molecular reprogramming in hearts are promising approaches currently in development [1], [2]. Biomaterials provide a vehicle for cell transplantation and delivery of bioactive molecules and more recently shown evidence for directly treating the injured heart [3], [4].
Decellularized extracellular matrix (dECM) derived biomaterials have been investigated as a direct therapy for heart injury. The dECM hydrogel prepared by solubilization and gelation of adult porcine cardiac tissue improved heart recovery in post-MI pigs [5]. The robust homology of extracellular proteins allows dECM transplantation across the species [6]. The dECM derived from neonatal mice and zebrafish hearts have been shown to stimulate protection of cardiac function, cardiomyocyte proliferation, and angiogenesis in post-MI mice suggesting that bioactive cues directing cardiac repair are retained in donor dECM for transplantation [7], [8]. We reported recently that fetal porcine dECM hydrogel reduces fibroblast activation and collagen deposition, and stimulates cardiomyocyte cell cycle activity and vascularization in mice and in vitro models [9], [10]. The mechanism of dECM therapy has not been fully realized, however, dECM proteins such as agrin and transforming growth factor-beta (TGF-β) modulate cardiomyocyte proliferation and fibroblast activation [11]–[13]. We observed a rapid release of proteins from dECM hydrogel in aqueous environment supporting that the regulatory proteins of heart regeneration transfer to recipients via dECM.
An acute ischemic heart injury results in permanent loss of cardiomyocytes. The critical post-injury period that includes stabilization of the replacement scar lasts for more than 4 weeks [14]. The early cardiac post-injury response (within 72 h post-MI) with cell necrosis includes recruitment of immune cells and release of cytokines [15]–[17]. The late post-injury response can last for weeks and includes cardiomyocyte apoptosis and collagen deposition [18], [19]. The ideal degradation rate and drug release kinetics of biomaterials for tissue engineering should match the time course of tissue repair [4], [20], [21]. Cardiac dECM hydrogel is vulnerable to collagenase digestion and reported to degrade within a week [22], [23]. Thus, increasing the stability of dECM-derived biomaterials will impact the therapeutic efficacy for heart injury.
We hypothesize that solid dECM microparticles will exhibit extended stability in tissue and controlled protein release to improve the heart post-injury response. In this study, dECM microparticles were generated by electrospray, water-oil emulsion, and heat-induced gelation. The dECM microparticles were characterized for protein release kinetics, swelling capacity, stiffness, digestion resistance, and stability. Cardiac function, cardiomyocyte cell cycle activity, fibrosis, fibroblast activation, and vessel density were evaluated in post-MI hearts of mice. Similar to liquid dECM hydrogel precursor, dECM microparticles preserved cardiac function, reduced fibrosis and fibroblasts activation, and stimulated cardiomyocyte cell cycle activity. In addition, dECM microparticles protected against left ventricular remodeling and exhibited increased blood vessel density consistent with neovascularization. In situ, the dECM microparticles degraded slower than dECM hydrogel. dECM microparticles also provide a platform for surface functionalization and macromolecular loading. Together, the results suggest dECM microparticles retain the therapeutic efficacy of dECM hydrogel precursor for post-ischemic heart injury and can be employed for long-term delivery of macromolecules.
2. Results
2.1. Solid dECM microparticle generation
The dECM microparticles were generated by electrospray of solubilized dECM (Figure 1A). Solubilized dECM was generated using previously established protocols [24]. The particle size can be controlled by tuning electrospray voltage and syringe pump flow rate (Figure S1A). Detailed in Methods, dECM droplets undergo water-oil emulsion and heat-induced gelation. The microparticles used in this study are approximately 20 μm in diameter with random distribution of aggregate fibers. We observed cell attachment with dECM microparticles in 3D culture with 3T3s (Figure 1B) and capture by adherence of H9C2 cells in 2D culture (Figure 1C, D). These results suggest that cardiac cells interact directly with dECM microparticles which potentially benefits the retention of injected microparticles in tissue.
Figure 1. dECM microparticles generated by electrospray and experimental setup.
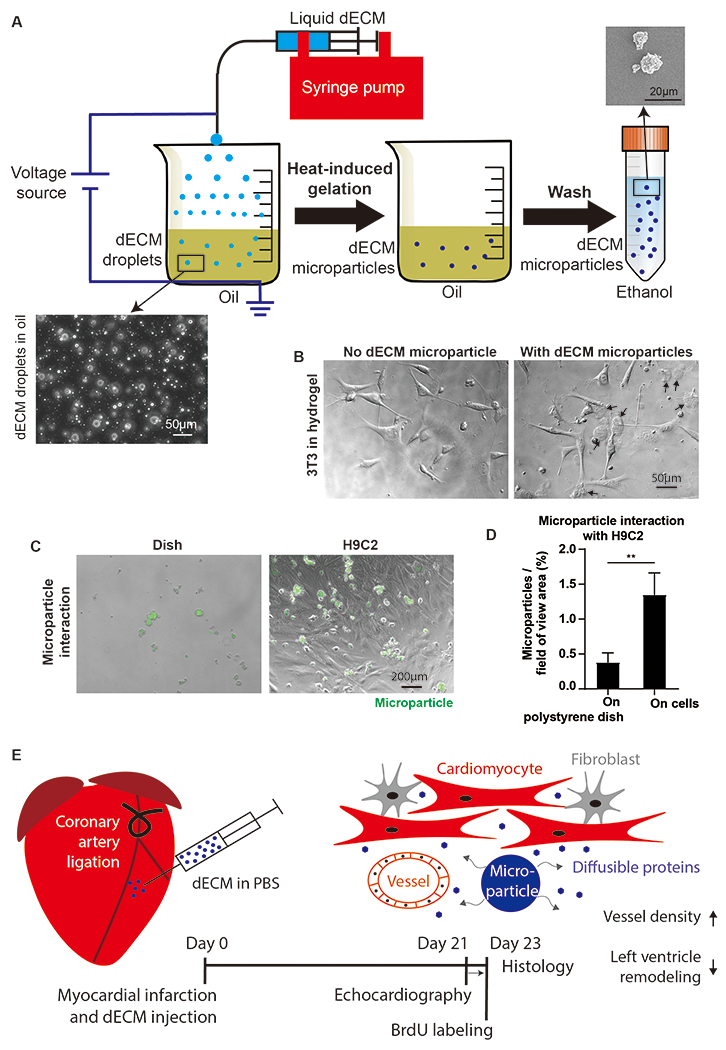
(A) Schematic of dECM microparticles fabrication. dECM droplets were generated by electrospray and stabilized in oil with surfactant. Liquid dECM droplets were gelled on hotplate and washed in acetone to remove oil and ethanol. The final solidified dECM microparticles were stable in ethanol. (B) 3T3 cells interaction with dECM microparticles. 3T3 cell protrusions were observed to make contact with dECM microparticles. (C) Microparticles captured by H9C2 cells. (D) Microparticles were labeled by WGA-Alexa Fluor 488 for quantification of fluorescent surface area after incubation on H9C2 cells and on polystyrene dish. (E) Experimental setup. dECM microparticles (resuspended in PBS) were injected into the infarct area immediately after coronary artery ligation in juvenile mice. Echocardiography was measured 21 days post-surgery. After 3 days BrdU labeling, mice hearts were harvested for histological analysis. (Panel D: n=3, t-test, **p<0.01. All data presented as mean ± SD. Black arrows in panel E indicate microparticles.)
The components of dECM were analyzed by liquid chromatography-mass spectrometry of trypsin digested dECM (Table S1). The dECM contains extracellular matrix structural proteins including collagen I, fibronectin, laminin, and fibrillin. Growth factors reported to impact cardiomyocyte proliferation and stem cell differentiation such as agrin and periostin, and angiogenic factors such as heparan sulfate proteoglycan 2 and alpha(B)-crystallin were also identified in dECM. Thus, the proteome of dECM microparticles can influence cardiac cell phenotypes by macromolecules released by passive diffusion and degradation (Figure 1E).
2.2. dECM microparticle characterization of stiffness, protein release, and swelling.
The dECM hydrogel was generated by gelation of liquid dECM hydrogel precursor at 37°C. Scanning electron microscopy (SEM) showed similar topography for dECM hydrogel and microparticles (Figure 2A, B). Atomic force microscopy analysis indicated that dECM microparticle (4.5 kPa) has a significantly higher elastic modulus than hydrogel (2.5 kPa; p < 0.001) (Figure 2C). However, the elastic moduli of both microparticles and hydrogel are within the stiffness range of the early-aged heart (<15 kPa) [25]. The dECM microparticles and hydrogels were further characterized for protein release kinetics and swelling in PBS. Morphology remained relatively constant in PBS for 14 days. Sampling the buffer, a bulk release of proteins from dECM hydrogels occurred within 15 min after immersing in PBS buffer comprising most of the 24 h solutes (Figure 2D). The dECM microparticles showed a repeatedly controlled release of proteins at a relatively constant rate over 24 h. In a 14 day study of protein release with buffer exchange every two days, approximately 70% of total released proteins from dECM hydrogels were on day 2, compared with the much lower cumulative released protein load from day 4 to 14. (Figure 2E). In contrast, the dECM microparticles released 37% of total released proteins on day 2 and then decreased to approximately 10% from days 4 to 14. The mass of total proteins released from dECM hydrogels in 14 days was 6.7 times higher than dECM microparticles.
Figure 2. dECM microparticles and hydrogels characterization.
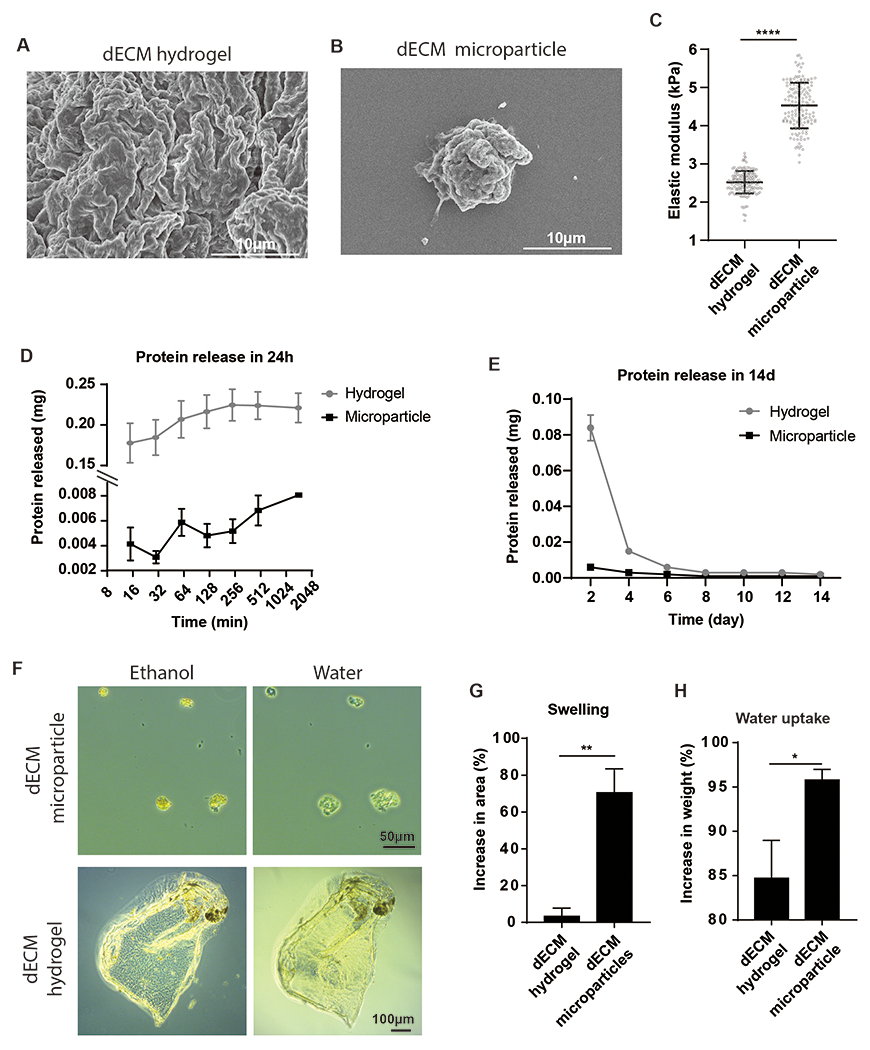
Representative SEM images of dECM hydrogels (A) and dECM microparticles (B). (C) Atomic force microscopy analysis indicates that dECM microparticles (4.5 ± 0.7 kPa) have higher elastic moduli than hydrogels (2.5 ± 0.3 kPa). (D) dECM hydrogels release soluble proteins in aqueous buffer within 24 h. The dECM microparticles exhibit a slower protein release rate. (E) In an infinite dilution experiment over 14 days, most of the diffusible proteins were released from hydrogels within 2 days, whereas dECM microparticles showed a more consistent protein release rate. (F) The dECM hydrogels and microparticles swelling and water uptake were evaluated by microscopy and analytical balance. (G) The area of dECM microparticles increases by 70% within 10 min in water, dECM hydrogels showed 3% increase in dimensions after immersing in water. (H) The weight of dECM microparticles increased by 95% after absorbing water, while that of dECM hydrogels increased by 85%. (Panel C: n = 150 to 170 measurements, unpaired t-test. Panel G, H: n=3 per treatment, unpaired t-test. *p<0.05, **p<0.01, ****p<0.0001. All data presented as mean ± SD.)
To better understand the changes in dECM microparticles and hydrogels in aqueous buffer, swelling and water uptake were examined. The dECM microparticles showed observable changes in area from dehydration to water exposure (Figure 2F), while the dECM hydrogels did not show significant changes after immersion in water. We observed that dECM microparticles swelled by 70% in water, while dECM hydrogels swelled by 3% in water (Figure 2G). Water uptake was examined by comparing dry sample weight to that after water absorption. The mass of dECM hydrogels increased by 85% after water absorption while that of dECM microparticles increased by 97% (Figure 2H). While both dECM formulations have a high water capacity, the results indicate that dECM microparticles have a slower release of diffusible proteins.
2.3. dECM microparticles collagenase digestion resistance and retention in tissue
To determine if dECM microparticles are stable in a physiological environment, collagenase digestion resistance and sample retention were evaluated. A high concentration of collagenase (0.1 mg mL−1) was used to accelerate the digestion. A larger mass of dECM microparticles was retained than hydrogels after 24 h digestion by visual inspection (Figure 3A). The dECM hydrogels achieved the maximal cumulative protein release within 2 h of digestion. In contrast, dECM microparticles released proteins over the 24 h period (Figure 3B). The retention of dECM was also examined by pre-labeling dECM with fluorescence markers. WGA-Alexa Fluor 488 labeled dECM microparticles and dECM hydrogel precursor were injected into explants from porcine hearts and examined at serial time points starting at day 1. Explants were incubated at 37°C for 2 h before adding tissue culture media to induce dECM hydrogel precursor gelation. Samples were histologically prepared for fluorescence microscopy (Figure 3C). The visible area of labeled dECM hydrogel precursor dropped rapidly in 7 days and was almost non-visible by day 14 (Figure 3D). In contrast, dECM microparticles decreased by only 50% over 14 days. The fluorescence intensity of the fluorescent labels was also quantified. The dECM microparticles and dECM hydrogel precursor showed a similar decrement of sample fluorescence intensity from day 1 to day 7 (Figure 3E). However, over the next 7 days, dECM microparticles exhibited a higher fluorescence intensity than dECM hydrogel precursor by day 14. These results suggest that dECM microparticles are more stable in natural tissue than liquid-derived dECM hydrogel.
Figure 3. Collagenase digestion and retention of dECM samples.
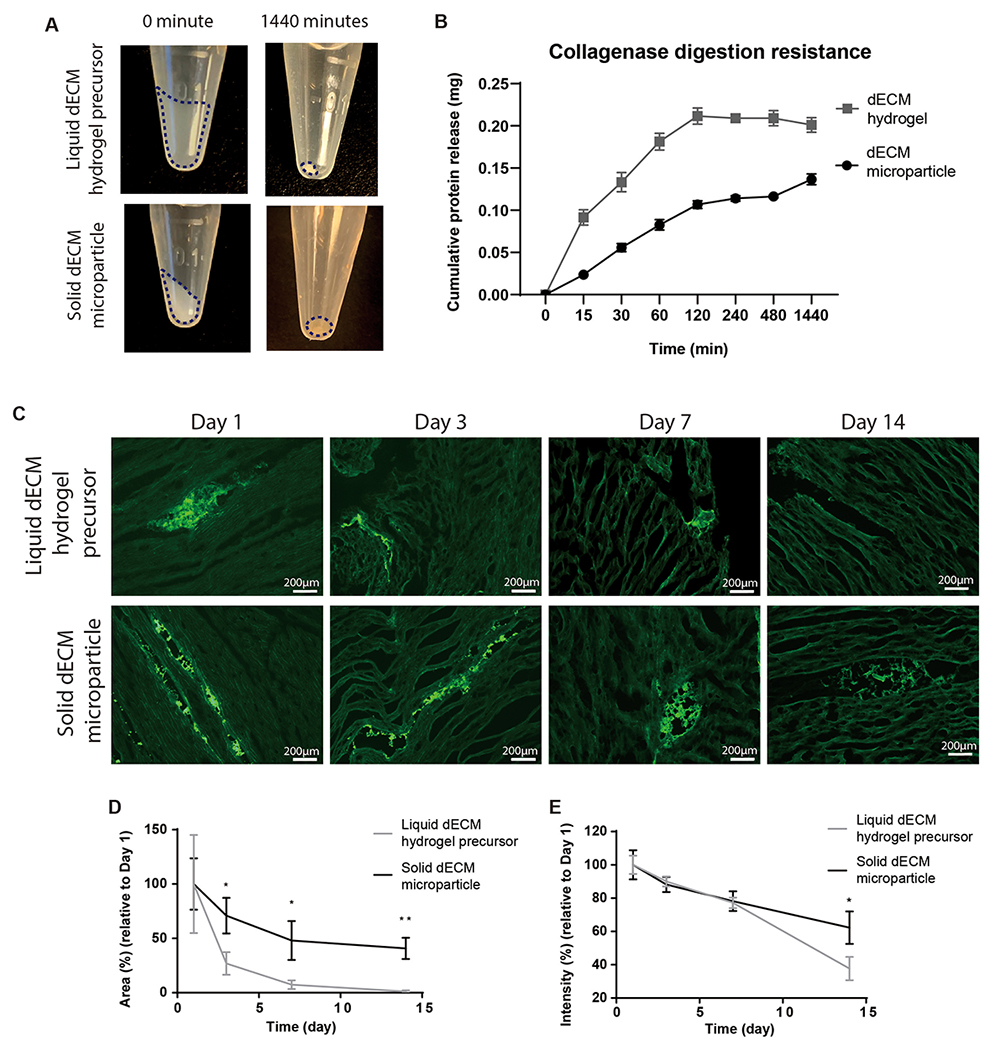
(A) dECM microparticles and hydrogels were digested by collagenase for 24 h. (B) dECM hydrogels were completed digested within 2 h, while the complete digestion of dECM microparticles took more than 24 h. (C) dECM microparticles and dECM hydrogel precursor labeled by WGA-Alexa Flour 488 were injected into porcine heart tissue explants cultured for 14 days. (D) Marginal levels of dECM hydrogel precursor can be observed on day 14. The areas of labeled dECM microparticles decreased by 50% from day 1 to day 14. (E) The average fluorescence intensity of the remaining samples decreased by 30% in dECM microparticles and 60% in dECM hydrogel precursor. (Panel B: 3 repeats per group. Panel D, E: 3 repeats per group, unpaired t-test. *p<0.05, **p<0.01. All data presented as mean ± SD. Panel A: dash line circles dECM.)
2.4. dECM microparticles increase vessel density by 3 weeks in post-MI hearts
The dECM treatments were evaluated as a therapy for the post-ischemic heart. After ligating left coronary artery in day 5 mice hearts, vessel density at week 3 post-MI was examined by fluorescent immunostaining (Figure 4A). Blood vessels were identified as α-SMA (smooth muscle) and CD31 (endothelial) double-positive structures in this study following established markers. We further analyzed blood vessel structures below 1,000 μm2 as microvasculature. The threshold reflects an inflection in the histogram data of vessel areas (Figure S2A) and corroborated dimensions from published studies for neovascularization [26], [27]. The dECM microparticles significantly increased the microvasculature density, CD31 and α-SMA positive small structures, compared to MI-control (Figure 4B) unlike the dECM hydrogel precursor. dECM microparticles treatment also showed a trend for increasing the density of all vessels (Figure 4C). The impact of dECM microparticles on vascularization was evaluated with the in vitro tube formation assay (Figure S2B). dECM microparticles increased the number of branches in human umbilical vein endothelial cells (HUVECs) network (Figure S2C, D, E). The results indicate that dECM microparticles can distinctly increase small blood vessel density in post-MI hearts.
Figure 4. dECM microparticles increase vessel density in post-MI hearts.
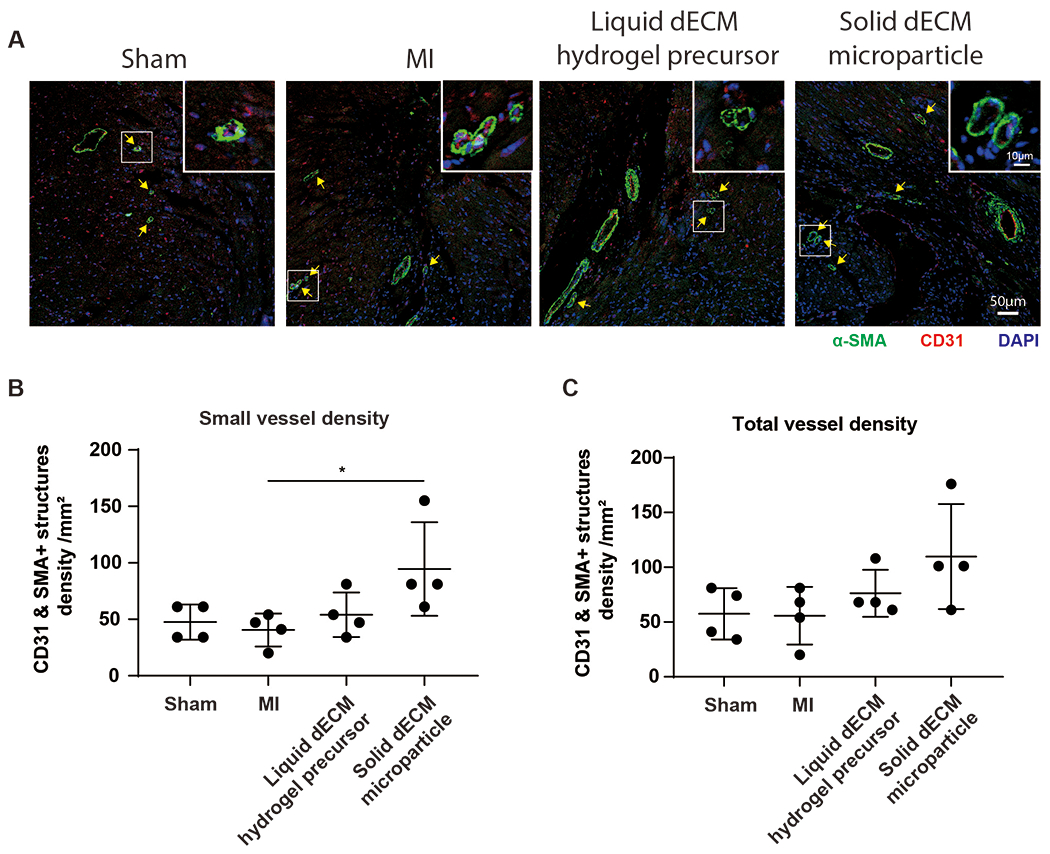
(A) Vessels were evaluated by immunostaining for α-SMA and CD31. The density and area of vessels were measured to evaluate vascularization. (B) dECM microparticles increased small vessel density compared to MI-control. (C) All vessel density. dECM microparticles show a trend towards increased vessel density. (Panel B, C: n=4 per treatment, one-way ANOVA and Tukey’s test. *p<0.05. All data presented as mean ± SD. Arrows indicate smaller blood vessels; white boxes indicate magnified areas. Single channels of a representative image in panel A are shown in Figure S6A.)
2.5. dECM microparticles preserve cardiac output 3 weeks post-MI
Cardiac function was measured by echocardiography at week 3 post-MI (Figure 5A). dECM microparticles lowered left ventricle end diastolic diameter compared to MI-control unlike the dECM hydrogel precursor (Figure 5B). Both dECM microparticles and dECM hydrogel precursor lowered end systolic diameter compared to MI-control; although dECM microparticles treated hearts was not significantly different from sham levels (Figure 5C). The ejection fraction (Figure 5D) and fractional shortening (Figure 5E) were higher with both dECM treatments relative to MI-control. The stroke volume (Figure S3A) and cardiac output (Figure S3B) were also higher in dECM microparticles and dECM hydrogel precursor treated hearts in comparison to the MI-control. Together, the results demonstrate that dECM microparticles and dECM hydrogel precursor exhibit comparable protective effects on cardiac function in post-MI hearts, while dECM microparticles show higher protection against left ventricular remodeling.
Figure 5. dECM microparticles preserve cardiac function in 3-week post-MI hearts.

(A) Cardiac function was evaluated by echocardiography. (B) dECM microparticles treatment reduced left ventricle end diastolic diameter compared to MI-control. (C) Both dECM hydrogel precursor and microparticles lowered left ventricle systolic diameter compared to MI-control. However, the end systolic diameter in dECM microparticles treated hearts was not significantly different from sham. Both dECM microparticles and dECM hydrogel precursor treated hearts showed increased (D) ejection fraction and (E) fractional shortening compared to MI-control. dECM microparticles treated group, however, had a tighter distribution than dECM hydrogel precursor group. (Panel B, C, D, E: n=4 per treatment, one-way ANOVA and Tukey’s test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. All data presented as mean ± SD.)
2.6. dECM microparticles reduce fibrosis and fibroblasts activation 3 weeks post-MI
Heart fibrosis was examined by Masson’s Trichrome staining at week 3 post-MI (Figure 6A). Since no fibrotic tissue was observed in sham hearts, they were not included in the fibrosis analysis. The wall thickness in the infarct area was higher in dECM microparticles treated hearts than MI-control (Figure 6B). Both dECM microparticles and dECM hydrogel precursor significantly lowered the fibrotic area in comparison to MI-control (Figure 6C). Because fibroblasts differentiate to α-SMA+ myofibroblasts and increase collagen deposition in the infarct area, we then evaluated fibroblast activation in the infarct and border zones. Cardiac fibroblasts are heterogeneous cell populations with no comprehensive cell marker. In this experiment, Pdgfr-α (Figure 6D) and Vimentin (Figure S4A) were used to identify fibroblasts. In Pdgfr-α positive cells, only dECM microparticles significantly reduced activated α-SMA+ fibroblasts density and total activated fibroblasts relative to MI-control, while the liquid dECM effects were only significant with the latter (Figure 6E, F). In Vimentin positive cells, only dECM microparticles treatment lowered fibroblast activation to sham levels (Figure S4C). The results demonstrate that dECM microparticles have high potency for lowering fibroblast activation and ventricular wall remodeling in post-MI hearts.
Figure 6. dECM microparticles reduce fibrosis and fibroblast activation in post-MI hearts.

(A) Fibrosis in heart was examined by Masson’s Trichrome staining. (B) Left ventricle wall thickness in the infarct area was higher in dECM microparticles treated group than MI-control. (C) dECM microparticles and dECM hydrogel precursor lowered fibrotic tissue area compared to MI-control. (D) Fibroblast activation was examined by immunostaining for Pdgfr-α and α-SMA. (E) The density of activated fibroblasts was lowered by dECM microparticles treatment compared to MI-control. (F) dECM microparticles and dECM hydrogel precursor reduced fibroblast activation. (Panel B, C, E, F: n=4 per treatment, one-way ANOVA and Tukey’s test. *p<0.05, **p<0.01, ****p<0.0001. All data presented as mean ± SD. Arrows indicate activated fibroblasts; white boxes indicate magnified areas. Single channels of a representative image in panel D are shown in Figure S6B. An example of the location chosen for wall thickness measurement is shown in Figure S7A.)
2.7. dECM microparticles promote cardiomyocyte cell cycle activity 3 weeks post-MI
Measuring cardiomyocyte renewal in vivo is challenging because of the extremely low cardiomyocyte proliferation rate and bi-nucleation. Thus, cardiomyocyte cell cycle activity was used as an indicator of proliferation in this study. Cardiomyocyte cell cycle activity was examined by immunostaining for Ki67 protein and BrdU incorporation on week 3 post-MI (Figure 7A). The number of Ki67+ cardiomyocytes is positively related to cardiomyocytes undergoing cytokinesis [28]. The dECM microparticles alone significantly increased Ki67+ cardiomyocytes compared to MI-control (Figure 7B). BrdU incorporation in cardiomyocytes was significantly increased by dECM microparticles and dECM hydrogel precursor compared to sham and MI-control (Figure 7C). A similar experiment was conducted using day 1 mice ventricle explants (Figure S5A). Cardiomyocyte BrdU incorporation (Figure S5B) and the frequency of PHH3+ cardiomyocytes (Figure S5C) were increased significantly by both dECM microparticles and dECM hydrogel precursor treatments relative to the untreated control. We then used the cardiomyocyte cell line, H9C2, to evaluate direct effects of dECM microparticles on cardiomyocyte cell cycle activity (Figure S5D). The dECM microparticles significantly increased H9C2 cell cycle activity compared to solubilized dECM treatment (Figure S5E). Together, the results indicate that dECM microparticles directly increase cell cycle activity in cardiomyocytes and in post-MI hearts.
Figure 7. dECM microparticles stimulate cardiomyocyte cell cycle activity in post-MI hearts.
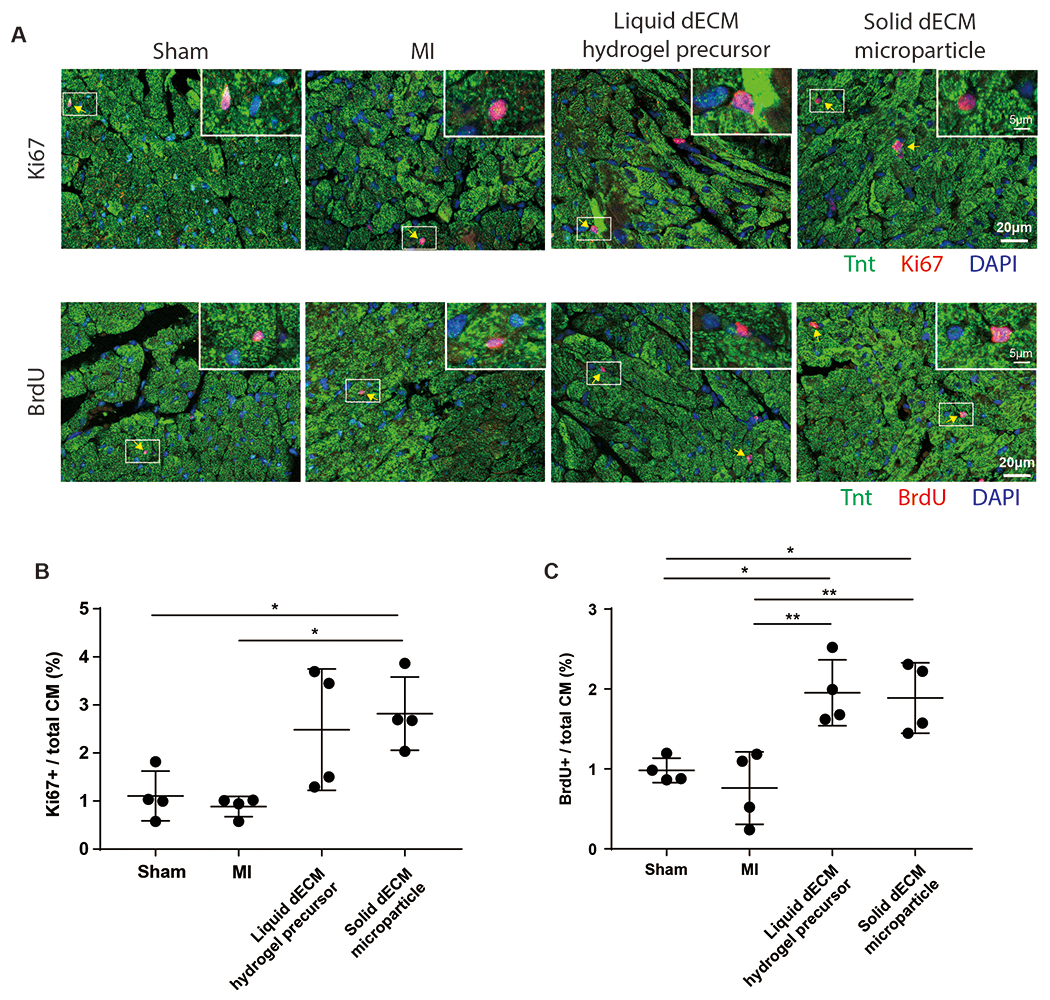
(A) Cardiomyocyte cell cycle activity was examined by immunostaining for Ki67 expression and BrdU incorporation in TnT-positive cells. (B) dECM microparticles significantly increased the percentage of Ki67+ cardiomyocytes compared to MI-control. (C) Both dECM microparticles and dECM hydrogel precursor promoted significant cardiomyocyte BrdU incorporation compared to MI-control. (Panel B, C: n=4 per treatment, one-way ANOVA and Tukey’s test. *p<0.05, **p<0.01. All data presented as mean ± SD. Arrows indicate mitotic cardiomyocytes; white boxes indicate magnified areas. Single channels of representative images in panel A are shown in Figure S6D, E.)
2.8. dECM microparticle provides a platform for drug delivery
dECM microparticles offer potential as a platform for engineered drug delivery. The dECM microparticles have an average diameter of 22.8 μm (Figure 8A). The diameter could be lowered with increased monodispersity by post-electrospray sonication to 3.4 μm (Figure 8B) and bead-homogenization to 2.3 μm (Figure 8C), respectively. For proof-of-concept of using the particle as a vehicle for macromolecule-loaded polymers, polystyrene beads (200 nm in diameter) were incorporated into dECM microparticles by mixing solubilized dECM with the fluorescently labeled nanoparticles before continuing with the pre-established fabrication protocol (Figure 8D). Nanoparticle incorporation could be titrated without any obvious impact on dECM microparticle size or aggregation. Similarly, to demonstrate direct small molecule loading, fluorescently labeled dextran (10 kDa) was incorporated into dECM microparticles at two concentrations (0.1 and 1 mg mL−1) (Figure 8E). Controlled release of dextran was observed over 7 days by fluorescence measurements of supernatant by plate reader analysis (Figure 8F). Finally, dECM microparticles were modified by chemical modification after electrospray for demonstration of altering the surface interface. NHS-AF488 was employed to determine whether NHS-ester can conjugate molecules to dECM microparticles. An increased fluorescence was observed in NHS-AF488 functionalized dECM microparticles compared to the passively adsorbed control indicating AF488 molecules enrichment (Figure 8G). FTIR analysis of NHS-AF488 functionalized dECM microparticles also indicated that NHS-AF488 was conjugated to microparticles by NHS-ester (Figure 8H). The results suggest that dECM microparticles can be modified by directly loading drugs, incorporating drug-loaded nanoparticles, or surface coating and conjugation potentially for tissue targeting and drug delivery.
Figure 8. dECM microparticles provide platform technology for drug loading and tissue targeting.
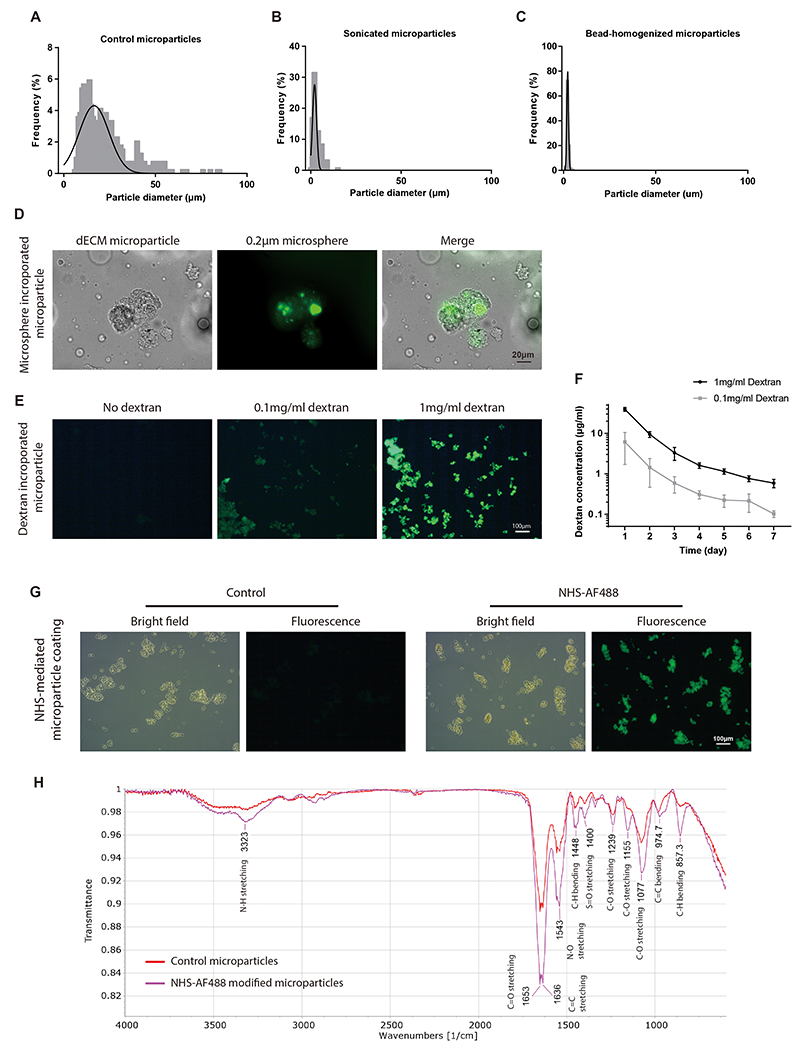
(A) Size distribution of original electrospray dECM microparticles (22.7 ± 13.7 μm). (B) Size distribution of sonicated microparticles (3.4 ± 2.4 μm). (C) Size distribution of bead-milled microparticles (2.3 ± 0.6 μm), all by microscopy. (D) Polystyrene beads were incorporated into dECM microparticles. (E) Dextran was incorporated into dECM microparticles by electrospray of pre-mixed dECM and dextran solution. (F) A controlled release of dextran was observed from dECM microparticles. (G) NHS-AF488 was used for dECM microparticle coating. A brighter fluorescent signal was observed in NHS-AF488 functionalized particles than passively adsorbed control. (H) FTIR of NHS-AF488 treated samples. Increased C-N stretching and C-H bending were observed in NHS-AF488 modified microparticles. (Panel F: n=3 repeats per condition, no statistical comparison applied. All data presented as mean ± SD.)
3. Discussion
In this study we showed evidence that dECM microparticles by electrospray achieve sustained release of diffusible bioactive molecules while being retained in tissue for at least two weeks to improve cardiac post-injury response in non-regenerative hearts. Previous studies have reported that cardiac dECM hydrogel reduces fibrosis, stimulates angiogenesis, and increases cardiomyocyte cell cycle activity in post-MI hearts [5], [8], [9], that nano- and micro-particles can be used for delivering drugs to ischemic heart [29], [30], and that extracellular matrix proteins can be processed to solid particles [31]–[33]. Nevertheless, cardiac dECM-derived microparticles and their applications in ischemic heart injury treatment have not been reported. This current study suggests that dECM microparticles increase vessel density and reduce ventricle remodeling. Our work is the first to demonstrate that dECM microparticles can be developed as a therapy for ischemic heart injury.
The dECM microparticles represent a variant of decellularized tissue-derived biomaterials. Early testing of porcine dECM hydrogel precursor treatment of heart failure patients in phase I clinical trial indicates general safety [34]. Similar to dECM hydrogel precursor, dECM microparticles are amenable to various needle gauges. In addition, dECM microparticles as a solid substrate provide a platform for further modifications such as macromolecules or drugs loading and surface functionalization or coating, as we demonstrated here. Further analysis will determine whether the dECM microparticle preparation with emulsion and solvents results in background crosslinking that distinguishes the two dECM modalities. Other groups have demonstrated that tuning hydrogel microparticle stability and protein release kinetics can be achieved by modulating particle sizes [35], [36]. Effects of particle size remain to be determined with the dECM microparticles. The dECM microparticle is primarily composed of insoluble fibrous proteins including collagen; however, the enrichment of complex biomolecules likely drives biological signaling that can drive post-ischemic signaling. The dECM investigated in this study contains extracellular matrix biomolecules reported to regulate cardiogenesis, for example agrin and periostin [37], [38]. In contrast, we did not observe putative cardiogenic growth factors such as follistatin like-1 and nidogen-1 that have been reported to stimulate cardiomyocyte proliferation [39], [40]. Potential enrichment with specific growth factors and tissue targeting may further improve the therapeutic efficacy of dECM microparticles for tissue engineering.
dECM microparticles induce increased vascularization and protection against ventricle remodeling. Otherwise, the dECM microparticles and dECM hydrogel precursor induced similar bioactivity with regards to cardiac function, cardiomyocyte cell cycle activity, fibrosis, and fibroblast activation by week-3 in post-MI hearts. Interestingly, the observed effects may be achieved by the combination of distinct factors. First, dECM microparticles release lower amounts of proteins but at a steadier rate compared to liquid-derived hydrogel which may alter signaling pathways with time. Second, the retention and stability of dECM microparticles and hydrogel precursor are different after intramyocardial injection, though it remains to be determined whether size-mediated retention facilitates this extended presence. Cardiac beating and blood flow may accelerate the spreading and dilution of dECM hydrogel precursor which result in distribution over a large area. Interestingly, nano-sized particles in dECM hydrogel precursor have been reported to infiltrate into interstitial tissue and leaky vessels which may provide its own unique advantages [41], [42]. On the other hand, dECM microparticles exhibit increased rigidity and swelling relative to dECM hydrogel precursor which suggests dECM microparticles require larger stresses to relocate. Thus, the region of dECM microparticle action mainly concentrates at the injection site for macromolecules released by diffusion. Interactions of fibroblast and cardiomyocytes with dECM microparticles were evaluated in vitro. The evidence supports cellular retention of microparticles retention by adhesion and stimulation of cardiomyocyte division. It remains to be determined whether direct cell-microparticle contact has unique benefits distinct from the released soluble factors. Interestingly, the molecular components of the final dECM products may differ. Hydrophobic molecules in the digested protein suspension may have been lost during emulsion. In contrast, hydrophilic molecules, like most growth factors, could be enriched [43]. Similarly, the slow gelation of the dECM hydrogel precursor may allow for diffusion of highly soluble factors upon injection. Further molecular characterization will elucidate differences in the composition.
There are caveats to the impact of dECM microparticles that warrant consideration. We initially observed increased surgery mortality in high-dose (> 10 μL) dECM microparticles injected groups (60% mortality vs. 20% mortality in 2 μL injected groups). A large dose delivery of microparticles or fluid volume into myocardium may damage tissue as a physical obstruction or edema increasing inflammatory responses in the heart. The diameter of the dECM microparticles can range from 5 μm to 55 μm. Pilot testing with electrospray parameters including flow rates, voltage, temperature, and emulsion formulation indicates the size range can be broadened or narrowed. We show evidence that monodispersed particles could be achieved via post-fabrication processing (e.g., sonication, homogenization) to achieve lower sizes (single-digit micron). From the literature, small particles (< 1 μm) are subject to macrophage phagocytosis and thus are potential triggers for an immunoresponse [44], [45]. Large particles (> 100 μm) have been shown to induce foreign body responses and fibrosis [46]. Although the polydispersity of dECM microparticles has been reduced to better evaluate the utility of microparticles in vivo, the inflammatory response at different time points needs to be evaluated with monodispersed particles of different sizes. In addition, optimizing the dosage for mass and volume will be important to maximizing treatment effects. Investigating post-injection dECM dynamics in vivo will provide critical information on structure-function relationships including tissue distribution based on size and extent of post-MI tissue remodeling, protein release kinetics in bulk, and defined dECM molecules (e.g., agrin) with the associated cellular signaling (e.g., Yap). Extending the duration of in vivo experiments to 4 weeks or longer will further provide detailed characterization of stability and therapeutic efficacy of dECM microparticles in late-stage heart repair. In addition, although increased vessel density was observed in dECM microparticles treated hearts, further investigation is needed for angiogenic activity. In vitro tube formation assay indicates that dECM microparticles increase tube branching. To evaluate dECM microparticles angiogenic activity, we will need to probe for established angiogenesis markers such as Ang-2 and Tie-1 and intravital stains for vessel patency.
Hydrogel particles at the micro- and nano- scales have a long history as vehicles for controlled release that can be delivered by needle and catheter [47]–[49]. The dECM-derived microparticles are a relatively new format of microspheres for biomedical application. The hydrophilicity of hydrogel microparticles makes them a promising way for delivering water-soluble biomolecules like growth factors and cells [50]. Delivering growth factors from protein-derived microparticles have been shown to increase bone and cartilage regeneration [32], [51]. Protein-derived microparticles can also be employed for delivering cells or as a scaffold for cell infiltration in tissue engineering [52], [53]. Extracellular matrix fiber proteins like collagen have also been processed to microspheres and investigated for drug delivery and tissue engineering [33], [54]–[56]. However, microparticles derived from dECM have not been investigated as a vehicle for delivering extracellular growth factors to heart-injured recipients as we show here for the first time. We also establish that the dECM microparticles provide a solid substrate for chemical modification and small molecule loading.
The juvenile mice employed in this study provide an established MI model for evaluating the post-injury response in non-regenerative hearts. Published works have demonstrated that heart regenerative capacity drops promptly after birth in mice and the incomplete recovery after heart injury is observed after day 3 in neonates [57], [58]. Similar to adult heart MI model, ischemic heart injury lowers cardiac function and results in permanent scarring in juvenile post-MI mice. The recently closed regenerative window and potential for stimulated cardiomyocyte proliferation in juvenile mice allow a useful evaluation of candidate therapies to then deploy in adults. Investigating heart repair in early-aged animals can also reveal the mechanism of complete heart regeneration in newborn animals. However, to better estimate the therapeutic efficacy of dECM microparticles for MI patients, microparticles need to be tested in adult and large mammal MI models for development towards a clinically relevant approach.
4. Conclusion
We developed a solid hydrogel microparticle derived from fetal porcine heart decellularized extracellular matrix using an electrospray emulsion approach in this study. The microparticles showed higher stability and slower macromolecule release kinetics compared to liquid forming hydrogel. dECM microparticles protected heart function and stimulated heart regeneration in post-MI mice hearts. dECM microparticles by electrospray show potential as a drug-release platform.
5. Methods
5.1. dECM isolation and solubilization
dECM harvest was based upon previous publications [59]. Porcine tissue was acquired for secondary use from animals on protocols approved by the Case Western Reserve University (CWRU) Institutional Animal Care and Use Committee (IACUC). Animals were first anesthetized by an intramuscular injection of Telazol and then euthanized by an overdose of Fatal-Plus, pentobarbital sodium (>100 mg kg−1). This method is consistent with the recommendations of the 2000 Panel on Euthanasia of the American Veterinary Medical Association. Porcine ventricles were cut to approximately 2 cm3 dices and immersed in 1 % sodium dodecyl sulfate solution (Sigma-Aldrich, St. Louis, MO, USA) to decellularize for 48 h until tissue appeared white in color. The samples were washed and soaked in a 1% solution of Triton X-100 (Sigma-Aldrich) for 4 h. The samples were then washed three times with deionized water for 12-24 h. Samples were lyophilized (SP Scientific, Gardiner, NY, USA) and kept at −80 °C for long-term storage. To generate dECM solution, lyophilized dECM was pulverized in liquid nitrogen and digested in 1 mg mL−1 pepsin (Sigma-Aldrich) in 0.01 M HCl buffer for 10 h at room temperature. Next, dECM was added at 10 mg mL−1 digestion buffer. Once completed, the pH was adjusted to 7 by adding 1 N of NaOH, and then the sample was diluted in 1:10 by volume in 10 × phosphate buffered saline (PBS; pH~7.4). Penicillin-streptomycin (P/S) (10,000 U mL−1, Cytiva, Marlborough, MA, USA) was added to reach a final concentration of 100 U mL−1. The sample was then stored at −20 °C. The dECM solution is a hydrogel precursor that aggregates into a 3D hydrogel at physiological temperature such as when administered in vivo.
5.2. Solid dECM microparticle fabrication
The solid dECM microparticles were fabricated using a custom electrospray apparatus (Spraybase, Cambridge, MA, USA) (Figure S1). Solubilized dECM solution was loaded into a 5 mL syringe (BD, Franklin Lakes, NJ, USA) and a syringe pump (New Era Pump Systems, Inc. Farmingdale, NY, USA) was set to 1.2 mL per hour flow rate. The syringe was connected to tubing which terminated in a 23-G needle. A high-power electrical source was connected to a needle and then separately the ground collection vessel via copper tape. A stir bar was placed inside the collection vessel and set to 100 RPM. The collection vessel is then filled with 7 mL olive oil with 5% surfactant (24% Tween 85 and 76% Span 85; Sigma-Aldrich). Needle distance is aligned to the beaker height. Syringe pump was then started and the voltage was set to between 8 kV to 17 kV depending on the intended sample. Once spraying is completed, the sample is transferred to another beaker with constant stirring at 600 RPM on a hot plate at 50 °C for 2 h in a desiccator. After heat-induced gelation, microparticles were washed with 3 mL acetone (Sigma-Aldrich) three times, spun at 1,700 g with each supernatant discarded, and then in 3 mL ethanol (Decon labs, Inc., King of Prussia, PA, USA) three times, with each spun at 1,700 g discarding the supernatant each time. The samples were suspended in ethanol and stored at −20 °C. The final dECM microparticles have a concentration ~10 mg mL−1.
5.3. Microparticles interaction with H9C2
Microparticles were incubated with 1:100 diluted WGA-Alexa Fluor 488 for 2h and washed in PBS 3 times before use. H9C2 cells were plated to 96 well plate at 20,000 cells per well. After overnight plating in DMEM contains 10% fetal bovine serum (FBS; ThermoFisher) and 1× penicillin-streptomycin (P/S; ThermoFisher), approximately 20 μg dECM microparticles were added to each well. Microparticles were incubated with cells or an empty well for 1h, and then gently washed in PBS 3 times. Samples were then imaged by bright field and fluorescent imaging. The area of microparticles was measured using ImageJ.
5.4. 3T3 and microparticle 3D gel culture
NIH 3T3 cell line was cultured in DMEM media with 10% FBS, 1% P/S. Matrigel (Corning) was thawed at 4 °C before use. NIH 3T3 cells were trypsinized and resuspended in culture media. NIH 3T3 (78 × 103 cells mL−1), Matrigel (final concentration 4 mg mL−1), with or without microparticles were mixed carefully on ice in a microcentrifuge tube and allowed to initiate gel setting at 37 °C for 1 min. Then 60 μL of gel suspension was added to a 96-well tissue culture plate and incubated at 37 °C for 30 min to complete gel setting. Finally, DMEM media with 10% FBS, 1% Pen-Strep was added to each dish. The gel mixtures were imaged using microscopy (Zeiss) with phase contrast after 2 days in culture.
5.5. Scanning electron microscopy
Scanning electron microscope (SEM, FEI Helios FIB, ThermoFisher) was used for imaging and surface analysis of dECM microparticles and liquid-derived hydrogels. Samples were suspended and fixed in 2.5% glutaraldehyde (Sigma-Aldrich) and 2% paraformaldehyde (PFA) (Sigma-Aldrich) in 0.1 M cacodylic acid (Sigma-Aldrich) for 1 h at room temperature. dECM microparticles and hydrogels were washed with 0.1 M cacodylic acid twice and were then serially dehydrated in ethanol (20, 50, 70, 90, 100% ethanol). After dehydration, the samples were pipetted onto glass coverslips and were left to air dry at room temperature overnight. The dried samples were then sputter-coated (Denton Vacuum, LLC., Moorestown, NJ, USA) with platinum before SEM imaging.
5.6. Atomic Force Microscopy (AFM)
A MFP-3D-Bio AFM (Asylum Research, Oxford Instruments, Santa Barbara, CA, USA) mounted on an inverted fluorescence microscope (Nikon Eclipse Ti) was used to indent the dECM hydrogels or microparticles and obtain their Young’s modulus [60]. The dECM microparticles suspended in ethanol were coated on clean glass slides and ethanol was allowed to evaporate (~2 h) prior to testing. Similarly, dECM hydrogels were placed in sterile AFM Petri dishes (Corning; 35 mm × 10 mm) and covered in PBS (37 °C). Tip-less AFM cantilevers (model Arrow TL 1, Nanoworld, nominal spring constant ~ 0.02 N m−1) were glued with a 2 ± 0.05 μm diameter polystyrene bead (Sigma) using epoxy. The spring constant was determined from a force-distance curve and using thermal calibration method in a clean culture dish containing PBS (for gel analysis) or on a clean glass slide (for microparticle analysis). Multiple force curves were obtained at random locations on the hydrogels immersed in PBS at a rate of ~ 5.1 μm s−1 and a trigger force of ~ 4 nN. The resulting force versus indentation curves were analyzed by Hertz model fit for spherical indenters using Igor Pro 6.37 software. The Young’s modulus was determined from these force curves as , where F is indentation force, EY is Young’s modulus, μ is Poisson’s ratio (~ 0.4), R is tip radius (1 μm) and δ is the indentation depth (~ 500 nm) Data were represented as mean ± standard deviation from at least n = 6 independent gels/well or multiple particles per glass slide, with at least three independent repeats of each experiment.
5.7. Protein release assay
For release kinetics, the dECM microparticles (~1 mg) and gelled dECM hydrogel precursor at 100 μL (~1 mg) were incubated in 600 μL of 1× PBS (37 °C). The dECM microparticles and hydrogel used in this experiment were from the same batch of liquid dECM hydrogel precursor. In the first 24 h, 60 μL buffer was sampled at 4 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 24 h. Fresh 1× PBS was added after sampling. After 24 h, the entire buffer was sampled at days 2, 4, 6, 8, 10, 12, and 14. Fresh 1× PBS was used to replace the old buffer after sampling. Samples were kept at −20 °C. BCA assay (ThermoFisher, Waltham, MA, USA) was used to measure the protein concentrations.
5.8. Swelling Analysis
The dECM microparticles and liquid-derived dECM hydrogels were imaged using a Zeiss fluorescence microscope (White Plains, NY, USA) in phase contrast mode. To evaluate swelling, the microparticles and hydrogels in ethanol were placed on charged glass slides (Fisher Scientific, Pittsburgh, PA, USA) and allowed to partially dry and adhere to the glass slide. A coverslip was gently placed on top, and ethanol was allowed to perfuse the area to re-solvent microparticles. After 10 min, the solution was exchanged with distilled(DI) water showing the change in size from the ethanol solvent to water after 15 min [61].
5.9. Water uptake
The dECM microparticles and liquid-derived dECM hydrogels were evaluated for water uptake following a published protocol [62]. To quantify changes in mass due to water uptake, the mass of microcentrifuge tubes was individually measured for baseline weight. The dECM microparticles and hydrogels were then added to the microcentrifuge tubes and weighed to get an initial “wet weight.” The samples were then lyophilized and weighed for the “dry weight.” Then water was added in 40 μL increments to rehydrate the samples and make sure that no excess water was added. If there was excess water, it was removed via pipette before weighing again. These masses were subtracted from each other to get changes in mass and percentage changes.
5.10. Collagenase digestion
One hundred microliters of dECM hydrogel and microparticles suspended in PBS were added to 1.5 mL centrifuge tubes. The mass of the sample in each tube is 1mg. Seven-hundred microliter of fresh collagenase (Sigma-Aldrich) digestion buffer (0.1 mg mL−1 collagenase and 0.36mM CaCl2 in 1× PBS) was added to each tube. Samples were incubated in a 37°C incubator. Seventy-two microliter of digestion buffer was sampled at time 0, 15, 30, 60, 120, 240, 480, and 1440 min, and the same volume of fresh collagenase digestion buffer was added. Protein concentrations were measured using BCA assay.
5.11. Microparticles retention and stability assay
After washing in 1× PBS three times, dECM microparticles and dECM hydrogel precursor were labeled with 100 μg mL−1 wheat germ agglutinin-Alexa fluor 488 (ThermoFisher) for 2 h at 4 °C with constant agitation. Two microliters of dECM hydrogel precursor and equivalent mass of dECM microparticles were injected through a 10 μL Hamilton syringe into a biopsy core of ~ 3 mm × 3 mm × 2 mm porcine heart cube before plating into a 48-well tissue culture dish. Samples were incubated at 37 °C for 2 h to induce dECM hydrogel precursor gelation without potential washout from buffer. Samples were then incubated in 500 μL DMEM media (ThermoFisher) for 14 days. Media was changed every 3 days. Samples were washed, cryosectioned on days 1, 3, 7, and 14, and imaged immediately. To measure the area of fluorescence-labeled dECM, images were quantified using ImageJ (NIH, USA). Histogram of pixel intensity was used to determine the threshold of non-tissue area, tissue-background, and Alexa Fluor 488-labeled signal of dECM. To measure the relative fluorescence intensity, images were measured with the defined thresholds for the averaged intensity of tissue and dECM regions of interest, and then label fluorescence intensities were normalized to the tissue.
5.12. Mice MI model and echocardiography
All animal procedures were reviewed and approved by IACUC at CWRU according to the guidelines and regulations described in the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011).
Myocardial infarction was performed on non-regenerative day 5 mice as previously published [9], [10]. Pregnant CD1 (IGS) mice were purchased from Charles River (Wilmington, MA, USA). Day 5 mice were anesthetized by hypothermia and kept on ice during the procedure. Analgesia was not available because of the age of the mice. Anesthesia was monitored by toe-pinch every 15 min if the procedures are above 15min. An incision was made on the fourth intercostal space, pericardium was removed to better visualize coronary arteries. A 10-0 Nylon suture (Arosurical instruments co., Newport Beach, CA, USA) was used to permanently ligate left coronary artery. Two microliters of dECM hydrogel precursor and dECM microparticles were injected through a 10 μL Hamilton syringe into myocardium. Two injections were made above and below the ligation site. Wound was closed by suture and skin glue (Henry Schein, Melville, NY, USA). Recovered mice were returned to the dame. Three weeks post-MI, mice were anesthetized by 5% isoflurane (Patterson veterinary, Greeley, CO, USA) vapor in air before echocardiography and maintained anesthetized using 1% to 1.5% isoflurane as previously published [63]. Body temperature was maintained at 37 °C and heart rate was kept at 400 to 500 beats per minute. M-mode and B-mode were recorded along the long axis using a Vevo 3100 (VisualSonics) equipped with a MX550D transducer. Cardiac function was analyzed using Vevo lab (VisualSonics). BrdU was injected into mice at 0.1 mg g−1 body weight for 3 days before euthanization. Animals were euthanized without anesthesia by introducing 100% CO2 to their home cage for 10min. The CO2 flow rate was about 20% of chamber volume per minute. Death was confirmed by heart removal.
5.13. Histology and immunostaining
For Masson’s Trichrome staining, heart sections were deparaffinized and stained as per protocols provided by the vendor (Electron Microscopy Sciences, Hatfield, PA, USA). For immunostaining, after deparaffinization and antigen retrieval, sections were blocked in 10% serum buffer for 1 h at room temperature and incubated in respective primary antibody staining buffer [1 × Tris-buffered saline (DOT Scientific Inc. Burton, Michigan, USA) contains 1% BSA (DOT Scientific Inc.) and 0.1% Triton X-100] overnight at 4 °C. Rabbit anti-platelet derived growth factor receptor alpha (Pdgfr-α) (Abclonal, Woburn, MA, USA), chicken anti-Vimentin (Abcam, Waltham, MA, USA), rabbit anti-phospho-histone H3 (PHH3) (Abcam), mouse anti-alpha smooth muscle actin (α-SMA) (ThermoFisher), mouse anti-cardiac troponin T (TnT) (Developmental Studies Hybridoma Bank, Iowa City, IA, USA), rabbit anti-Ki67 (ThermoFisher), rabbit anti-BrdU (ThermoFisher), and rabbit anti-CD31 (Abcam) antibodies were used. All primary antibodies except TnT antibody were 1:200 diluted in staining buffer. TnT antibody was 1:10 diluted in staining buffer. Fluorophore-conjugated goat secondary antibodies (ThermoFisher) were used for detecting primary antibodies. All secondary antibodies were 1:200 diluted in staining buffer. Nuclei were stained by DAPI (Sigma-Aldrich). A representative image of secondary antibodies-only negative control is showed in Figure S6F. Sampled were mounted in anti-fade mounting media (Vectorlab, Burlingame, CA, USA) and imaged within 2 days.
For fluorescent immunostaining, cardiomyocytes in the MI border area (within 250 μm from the edge of infarct area) were imaged. Fibroblasts in infarct and border areas (within 100 μm of collagen-rich infarct area and in the infarct area) and vessels in infarct and border areas (within 400 μm of collagen-rich infarct area and in infarct area) were imaged. Cardiomyocytes, fibroblasts, and vessels in sham group were randomly imaged in left ventricle wall. For each section, 5 images were randomly taken in the area mentioned above. Cell numbers were counted in ImageJ by thresholding using Auto Threshold (Moments, Otsu, or Percentile methods). The same auto threshold method was used to analyze the images for same cell markers. The segmented color channel of DAPI and the channel of specific cell markers were overlayed to highlight specific cells of interest. Cell number was counted using Analyze Particle function. BrdU, Ki67, α-SMA positive cells, and CD31 - α-SMA double positive structures were counted manually. For Masson’s Trichrome staining, the whole heart section was imaged and measured using imageJ Auto Thresholding. Fibrotic area was measured by Auto Thresholding and manual choosing the infarct region.
5.14. Microparticle macromolecule incorporation and conjugation
As a proof-of-concept for drug loading, dECM microparticles were fabricated after doping with latex beads and with dextran. Carboxylate-modified latex beads (ThermoFisher; 200 nm diameter; 2% w v−1 suspension) were mixed at 1:10 ratio with the dECM hydrogel precursor before electrospray. Alternatively, fluorescein isothiocyanate-dextran 10 kDa (Sigma-Aldrich) were enriched in the dECM hydrogel precursor to reach final concentrations of 0.1 mg mL−1 or 1 mg mL−1. The doped dECM mixture was then processed by the microparticle fabrication protocol by electrospray and heat-induced gelation. Doped dECM microparticles were washed in acetone, ethanol, and 1× PBS three times each, before dextran release experiments. Microsphere incorporation and dextran loading were evaluated using fluorescence microscopy.
To determine dextran release from microparticles, dextran loaded dECM microparticles (1 mg) were incubated in 400 μL PBS at 37 °C for 7 days with daily exchange of buffer. Dextran concentrations were examined by Synergy H1 spectrometry (BioTek Instruments, Winooski, VT, USA) using 490 nm excitation wavelength and 520 nm emission wavelength.
Surface functionalization was conducted for proof-of-concept of tissue targeting and surface modular motifs. NHS-AF488 (Lumiprobe, Hunt Valley, MD, USA) stock solution was added to dECM microparticles suspended in 0.1 M sodium bicarbonate buffer. Fifty micrograms of NHS-AF488 were used per milligram of dECM microparticles. dECM microparticles were incubated with NHS-AF488 at 4 °C overnight, followed by washing in PBS five times. Surface functionalized dECM microparticles were imaged using a fluorescence microscope.
5.15. Microparticles sonication and bead-homogenization
For sonication, liquid dECM droplets suspended in oil after electrospray were transferred to conical tubes and sonicated in a 0.8 L bath sonicator (Btihceuot) for 30 min using 42 kHz sonication frequency and 35 W power. Ice-cold water was used to keep the temperature at 0 °C.
For bead-homogenization, solid dECM microparticles were suspended in ethanol with polystyrene beads and metal beads for centrifugal agitation. Then washed in acetone three times (5 min each), and three more times (5 min each) in ethanol for cold storage, or switched to aqueous buffer for cell treatment.
5.16. Fourier-transform infrared spectroscopy
dECM microparticles were dried on a coverslip and transferred to a Cary 630 FTIR facility (Agilent, Santa Clara, CA, USA) for reading. Spectrum range was set to 4000 to 600 per centimeter with a resolution of 4/cm. Background calibration was conducted before reading a new sample. Averaged data of 512 scans was used for analysis. Baseline correction and peak normalization were used for processing FTIR data.
5.17. Statistical analysis
Statistical analysis was conducted using GraphPad Prism 7. A two-tailed t-test was performed to compare experiments with two groups. One-way ANOVA and Tukey’s test were used for analyzing experiments with three or more groups. The confidence interval was set to 0.95 for both t-test and one-way ANOVA. No data pairing or matching was applied. The data was assumed to exhibit Gaussian distribution. Data were represented as bar graph or dot plot with mean ± standard deviation (SD). Non-preprocessed data was used for statistical analysis except the Protein release in 14 d (Fig 2E) and dECM retention (Fig 3D, E) data. Protein release in 14 d data was normalized to the amount of total protein released in 24h. dECM retention data was normalized to the values on day 1. Number of experimental repeats is shown in figure legends. In vivo experiments were conducted on 4 individual animals for each treatment, while in vitro experiments were repeated independently 3 times. For cardiomyocytes, 300 to 550 cardiomyocytes per animal, or 1,200 to 2,200 cardiomyocytes per treatment group were analyzed. For fibroblasts, 50 to 100 cells per animal in sham group and 120 to 320 cells in MI groups, or 200 to 400 fibroblasts in sham group and 480 to 1280 fibroblasts in MI groups were analyzed.
Supplementary Material
Acknowledgements
We would like to acknowledge the use of microscopes in the Light Microscopy Imaging Facility at CWRU, made available through NIH Grant S10-OD024996. We acknowledge the support of the staffs in CWRU Case School of Engineering SCSAM center for SEM. We thank Dr. Nicholas P. Ziats and Sandra Siedlak for assistance with histology. We thank Natalie Hong, Douglas Wu, Craig Watson, and Chao Liu for providing technical support.
Funding
This work was supported by NIH R25-HL145817 (S.E.S), NIH 1 C06 RR12463-01 (CWRU), and 5T32HL134622 (A.A.). NSF Award 1937968 (K.Y.). NSF Award 1337859 (C.R.K.).
Footnotes
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The datasets generated during the current study are available from the corresponding author by reasonable request.
Reference
- [1].Sadek H and Olson EN, “Toward the Goal of Human Heart Regeneration,” Cell Stem Cell, vol. 26, no. 1. Cell Press, pp. 7–16, Jan-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hashimoto H, Olson EN, and Bassel-Duby R, “Therapeutic approaches for cardiac regeneration and repair,” Nature Reviews Cardiology, vol. 15, no. 10. Nature Publishing Group, pp. 585–600, Oct-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wu I and Elisseeff J, “Biomaterials and Tissue Engineering for Soft Tissue Reconstruction,” in Natural and Synthetic Biomedical Polymers, Elsevier, 2014, pp. 235–241. [Google Scholar]
- [4].Abdulghani S and Mitchell GR, “Biomaterials for in situ tissue regeneration: A review,” Biomolecules, vol. 9, no. 11. MDPI AG, Nov-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U, Kwan OL, Strachan GM, Wong J, Schup-Magoffin PJ, Braden RL, Bartels K, DeQuach JA, Preul M, Kinsey AM, DeMaria AN, Dib N, and Christman KL, “Safety and efficacy of an injectable extracellular matrix hydrogel for treating myocardial infarction.,” Science translational medicine, vol. 5, no. 173, p. 173ra25, Feb. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ozbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, and Adams JC, “The evolution of extracellular matrix,” Molecular biology of the cell, vol. 21, no. 24, pp. 4300–4305, Dec. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang Z, Long DW, Huang Y, Chen WCW, Kim K, and Wang Y, “Decellularized neonatal cardiac extracellular matrix prevents widespread ventricular remodeling in adult mammals after myocardial infarction,” Acta Biomaterialia, vol. 87, pp. 140–151, Mar. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen WCW, Wang Z, Missinato MA, Park DW, Long DW, Liu H-J, Zeng X, Yates NA, Kim K, and Wang Y, “Decellularized zebrafish cardiac extracellular matrix induces mammalian heart regeneration,” Science Advances, vol. 2, no. 11, pp. e1600844–e1600844, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang X, Senapati S, Akinbote A, Gnanasambandam B, Park PS-H, and Senyo SE, “Microenvironment stiffness requires decellularized cardiac extracellular matrix to promote heart regeneration in the neonatal mouse heart.,” Acta biomaterialia, vol. 113, pp. 380–392, Sep. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang X, Pierre V, Liu C, Senapati S, Park PS-H, and Senyo SE, “Exogenous extracellular matrix proteins decrease cardiac fibroblast activation in stiffening microenvironment through CAPG.,” Journal of molecular and cellular cardiology, vol. 159, pp. 105–119, Oct. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, and Tzahor E, “The extracellular matrix protein agrin promotes heart regeneration in mice,” Nature, vol. 547, no. 7662, pp. 179–184, Jul. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, and Thomas PE, “Myofibroblast Differentiation by Transforming Growth Factor-1 Is Dependent on Cell Adhesion and Integrin Signaling via Focal Adhesion Kinase*,” 2003. [DOI] [PubMed] [Google Scholar]
- [13].Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, and Steadman R, “Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts.,” The Journal of biological chemistry, vol. 288, no. 21, pp. 14824–38, May 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, and Weber KT, “Regulation of collagen degradation in the rat myocardium after infarction,” Journal of Molecular and Cellular Cardiology, vol. 27, no. 6, pp. 1281–1292, Jun. 1995. [DOI] [PubMed] [Google Scholar]
- [15].Frangogiannis NG, Smith CW, and Entman ML, “The inflammatory response in myocardial infarction,” Cardiovascular Research, vol. 53, no. 1. Oxford Academic, pp. 31–47, Jan-2002. [DOI] [PubMed] [Google Scholar]
- [16].Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, and Hausenloy DJ, “Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities,” Pharmacology and Therapeutics, vol. 186. Elsevier Inc., pp. 73–87, Jun-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu J, Wang H, and Li J, “Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: A double-edged sword,” Clinical Medicine Insights: Cardiology, vol. 10, pp. 79–84, Jun. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cahill TJ, Choudhury RP, and Riley PR, “Heart regeneration and repair after myocardial infarction: translational opportunities for novel therapeutics,” Nature Reviews Drug Discovery, vol. 16, no. 10, pp. 699–717, Jul. 2017. [DOI] [PubMed] [Google Scholar]
- [19].Sutton M. G. St. John and Sharpe N, “Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy,” Circulation, vol. 101, no. 25, pp. 2981–2988, Jun. 2000. [DOI] [PubMed] [Google Scholar]
- [20].Piraino F and Selimović Š, “A Current View of Functional Biomaterials for Wound Care, Molecular and Cellular Therapies,” BioMed Research International, vol. 2015. Hindawi Limited, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Abbott RD and Kaplan DL, “Engineering Biomaterials for Enhanced Tissue Regeneration,” Current Stem Cell Reports, vol. 2, no. 2. Springer International Publishing, pp. 140–146, Jun-2016. [Google Scholar]
- [22].Výborný K, Vallová J, Kočí Z, Kekulová K, Jiráková K, Jendelová P, Hodan J, and Kubinová Š, “Genipin and EDC crosslinking of extracellular matrix hydrogel derived from human umbilical cord for neural tissue repair,” Scientific Reports, vol. 9, no. 1, pp. 1–15, Dec. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wassenaar JW, Braden RL, Osborn KG, and Christman KL, “Modulating in vivo degradation rate of injectable extracellular matrix hydrogels,” Journal of Materials Chemistry B, vol. 4, no. 16, pp. 2794–2802, Apr. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang X, Pierre V, Senapati S, Park PS-H, and Senyo SE, “Microenvironment Stiffness Amplifies Post-ischemia Heart Regeneration in Response to Exogenous Extracellular Matrix Proteins in Neonatal Mice,” Frontiers in Cardiovascular Medicine, vol. 8, Nov. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jacot JG, Martin JC, and Hunt DL, “Mechanobiology of cardiomyocyte development.,” Journal of biomechanics, vol. 43, no. 1, pp. 93–8, Jan. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Simons M, “Angiogenesis: Where do we stand now?,” Circulation, vol. 111, no. 12. Lippincott Williams & Wilkins, pp. 1556–1566, Mar-2005. [DOI] [PubMed] [Google Scholar]
- [27].Kocijan T, Rehman M, Colliva A, Groppa E, Leban M, Vodret S, Volf N, Zucca G, Cappelletto A, Piperno GM, Zentilin L, Giacca M, Benvenuti F, Zhou B, Adams RH, and Zacchigna S, “Genetic lineage tracing reveals poor angiogenic potential of cardiac endothelial cells,” Cardiovascular Research, vol. 117, no. 1, pp. 256–270, Jan. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yahalom-Ronen Y, Rajchman D, Sarig R, Geiger B, and Tzahor E, “Reduced matrix rigidity promotes neonatal cardiomyocyte dedifferentiation, proliferation and clonal expansion,” eLife, vol. 4, Aug. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bheri S and Davis ME, “Nanoparticle-Hydrogel System for Post-myocardial Infarction Delivery of MicroRNA,” ACS Nano, vol. 13, no. 9. American Chemical Society, pp. 9702–9706, Sep-2019. [DOI] [PubMed] [Google Scholar]
- [30].Fan C, Joshi J, Li F, Xu B, Khan M, Yang J, and Zhu W, “Nanoparticle-Mediated Drug Delivery for Treatment of Ischemic Heart Disease,” Frontiers in Bioengineering and Biotechnology, vol. 8. Frontiers Media S.A., Jun-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Link PA, Ritchie AM, Cotman GM, Valentine MS, Dereski BS, and Heise RL, “Electrosprayed extracellular matrix nanoparticles induce a pro-regenerative cell response,” Journal of Tissue Engineering and Regenerative Medicine, vol. 12, no. 12, pp. 2331–2336, Dec. 2018. [DOI] [PubMed] [Google Scholar]
- [32].Kang ML, Ko JY, Kim JE, and Il Im G, “Intra-articular delivery of kartogenin-conjugated chitosan nano/microparticles for cartilage regeneration,” Biomaterials, vol. 35, no. 37, pp. 9984–9994, Dec. 2014. [DOI] [PubMed] [Google Scholar]
- [33].Yamada M, Hori A, Sugaya S, Yajima Y, Utoh R, Yamato M, and Seki M, “Cell-sized condensed collagen microparticles for preparing microengineered composite spheroids of primary hepatocytes,” Lab on a Chip, vol. 15, no. 19, pp. 3941–3951, Aug. 2015. [DOI] [PubMed] [Google Scholar]
- [34].Traverse JH, Henry TD, Dib N, Patel AN, Pepine C, Schaer GL, DeQuach JA, Kinsey AM, Chamberlin P, and Christman KL, “First-in-Man Study of a Cardiac Extracellular Matrix Hydrogel in Early and Late Myocardial Infarction Patients,” JACC: Basic to Translational Science, vol. 4, no. 6, pp. 659–669, Oct. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tsuruta LR, Lessa MM, and Carmona-Ribeiro AM, “Effect of particle size on colloid stability of bilayer-covered polystyrene microspheres,” Journal of Colloid And Interface Science, vol. 175, no. 2, pp. 470–475, Nov. 1995. [Google Scholar]
- [36].Chen W, Palazzo A, Hennink WE, and Kok RJ, “Effect of particle size on drug loading and release kinetics of gefitinib-loaded PLGA microspheres,” Molecular Pharmaceutics, vol. 14, no. 2, pp. 459–467, Feb. 2017. [DOI] [PubMed] [Google Scholar]
- [37].Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, and Tzahor E, “The extracellular matrix protein agrin promotes heart regeneration in mice,” Nature, vol. 547, no. 7662, pp. 179–184, Jul. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kühn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, and Keating MT, “Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair,” Nature Medicine, vol. 13, no. 8, pp. 962–969, Aug. 2007. [DOI] [PubMed] [Google Scholar]
- [39].Wei K, Serpooshan V, Hurtado C, Diez-Cuñado M, Zhao M, Maruyama S, Zhu W, Fajardo G, Noseda M, Nakamura K, Tian X, Liu Q, Wang A, Matsuura Y, Bushway P, Cai W, Savchenko A, Mahmoudi M, et al. , “Epicardial FSTL1 reconstitution regenerates the adult mammalian heart,” Nature, vol. 525, no. 7570, pp. 479–485, Sep. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zbinden A, Layland SL, Urbanczyk M, Carvajal Berrio DA, Marzi J, Zauner M, Hammerschmidt A, Brauchle EM, Sudrow K, Fink S, Templin M, Liebscher S, Klein G, Deb A, Duffy GP, Crooks GM, Eble JA, Mikkola HKA, et al. , “Nidogen-1 Mitigates Ischemia and Promotes Tissue Survival and Regeneration,” Advanced Science, vol. 8, no. 4, p. 2002500, Feb. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mair LO and Superfine R, “Single particle tracking reveals biphasic transport during nanorod magnetophoresis through extracellular matrix,” Soft Matter, vol. 10, no. 23, pp. 4118–4125, Jun. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Spang MT, Lazerson TS, Bhatia S, Corbitt J, Sandoval G, Luo C, Osborn KG, Cabrales P, Kwon E, Contijoch F, Reeves RR, DeMaria AN, and Christman KL, “A New Form of Decellularized Extracellular Matrix Hydrogel for Treating Ischemic Tissue via Intravascular Infusion,” bioRxiv, p. 2020.04.10.028076, Apr. 2020. [Google Scholar]
- [43].Patel ZS, Yamamoto M, Ueda H, Tabata Y, and Mikos AG, “Biodegradable gelatin microparticles as delivery systems for the controlled release of bone morphogenetic protein-2,” Acta Biomaterialia, vol. 4, no. 5, pp. 1126–1138, Sep. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Champion JA, Walker A, and Mitragotri S, “Role of particle size in phagocytosis of polymeric microspheres,” Pharmaceutical Research, vol. 25, no. 8, pp. 1815–1821, Aug. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pacheco P, White D, and Sulchek T, “Effects of Microparticle Size and Fc Density on Macrophage Phagocytosis,” PLoS ONE, vol. 8, no. 4, p. e60989, Apr. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, et al. , “Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates,” Nature Materials, vol. 14, no. 6, pp. 643–651, Jun. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Daly AC, Riley L, Segura T, and Burdick JA, “Hydrogel microparticles for biomedical applications,” Nature Reviews Materials, vol. 5, no. 1. Nature Research, pp. 20–43, Jan-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lengyel M, Kállai-Szabó N, Antal V, Laki AJ, and Antal I, “Microparticles, microspheres, and microcapsules for advanced drug delivery,” Scientia Pharmaceutica, vol. 87, no. 3. MDPI AG, p. 20, Sep-2019. [Google Scholar]
- [49].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, and Segura T, “Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks,” Nature Materials, vol. 14, no. 7, pp. 737–744, Jul. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hoare TR and Kohane DS, “Hydrogels in drug delivery: Progress and challenges,” Polymer, vol. 49, no. 8. Elsevier BV, pp. 1993–2007, Apr-2008. [Google Scholar]
- [51].Patel ZS, Young S, Tabata Y, Jansen JA, Wong MEK, and Mikos AG, “Dual delivery of an angiogenic and an osteogenic growth factor for bone regeneration in a critical size defect model,” Bone, vol. 43, no. 5, pp. 931–940, Nov. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jgamadze D, Liu L, Vogler S, Chu LY, and Pautot S, “Thermoswitching microgel carriers improve neuronal cell growth and cell release for cell transplantation,” Tissue Engineering - Part C: Methods, vol. 21, no. 1, pp. 65–76, Jan. 2015. [DOI] [PubMed] [Google Scholar]
- [53].Li F, Truong VX, Fisch P, Levinson C, Glattauer V, Zenobi-Wong M, Thissen H, Forsythe JS, and Frith JE, “Cartilage tissue formation through assembly of microgels containing mesenchymal stem cells,” Acta Biomaterialia, vol. 77, pp. 48–62, Sep. 2018. [DOI] [PubMed] [Google Scholar]
- [54].Chan OCM, So KF, and Chan BP, “Fabrication of nano-fibrous collagen microspheres for protein delivery and effects of photochemical crosslinking on release kinetics,” Journal of Controlled Release, vol. 129, no. 2, pp. 135–143, Jul. 2008. [DOI] [PubMed] [Google Scholar]
- [55].Berthold A, Cremer K, and Kreuter J, “Collagen microparticles: carriers for glucocorticosteroids,” European Journal of Pharmaceutics and Biopharmaceutics, vol. 45, no. 1, pp. 23–29, Jan. 1998. [DOI] [PubMed] [Google Scholar]
- [56].Yang C and Wang J, “Preparation and characterization of collagen microspheres for sustained release of steroidal saponins,” Materials Research, vol. 17, no. 6, pp. 1644–1650, Nov. 2014. [Google Scholar]
- [57].Notari M, Ventura-Rubio A, Bedford-Guaus SJ, Jorba I, Mulero L, Navajas D, Martí M, and Raya Á, “The local microenvironment limits the regenerative potential of the mouse neonatal heart,” Science Advances, vol. 4, no. 5, p. eaao5553, May 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, and Sadek HA, “Transient regenerative potential of the neonatal mouse heart,” Science (New York, N.Y.), vol. 331, no. 6020, pp. 1078–1080, Feb. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang X, Senapati S, Akinbote A, Gnanasambandam B, Park PS-H, and Senyo SE, “Microenvironment stiffness requires decellularized cardiac extracellular matrix to promote heart regeneration in the neonatal mouse heart,” Acta Biomaterialia, Jun. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].J J, G M, and CR K, “Three-dimensional collagenous niche and azacytidine selectively promote time-dependent cardiomyogenesis from human bone marrow-derived MSC spheroids,” Biotechnology and bioengineering, vol. 115, no. 8, pp. 2013–2026, Aug. 2018. [DOI] [PubMed] [Google Scholar]
- [61].Kesenci K and Piykin E, “Production of poly[(ethylene glycol dimethacry1ate)-co-acrylamide] based hydrogel beads by suspension copolymerization,” Macromol. Chem. Phys, vol. 199, no. 3, p. 998, Mar. 1998. [Google Scholar]
- [62].Elçin YM, “Encapsulation of urease enzyme in xanthan-alginate spheres,” Biomaterials, vol. 16, no. 15, pp. 1157–1161, Oct. 1995. [DOI] [PubMed] [Google Scholar]
- [63].Lindsey ML, Kassiri Z, Virag JAI, de C. Brás LE, and Scherrer-Crosbie M, “Guidelines for measuring cardiac physiology in mice,” 10.1152/ajpheart.00339.2017, vol. 314, no. 4, pp. H733–H752, Apr. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author by reasonable request.