

Abstract
3D organoid models have recently seen a boom in popularity, as they can better recapitulate the in vivo complexity of multicellular organs compared to other in vitro culture systems. However, organoids are difficult to image because of the limited penetration depth of high-resolution microscopes and depth-dependent light attenuation, which can limit the understanding of signal transduction pathways and characterization of intimate cell-extracellular matrix (ECM) interactions. To overcome these challenges, we developed an expansion microscopy method termed PhASE-ExM, enabling optical clearance and super-resolution imaging of organoids and their ECM in 3D. PhASE-ExM uses hydrogels prepared via photo-initiated polymerization, which is advantageous as it decouples monomer diffusion into thick organoid cultures from the hydrogel fabrication. Apart from compatibility with organoids cultured Matrigel, PhASE-ExM enables 3.25x expansion and super-resolution imaging of organoids cultured in synthetic poly(ethylene glycol) (PEG) hydrogels crosslinked via allyl-sulfide groups (PEG-AlS) through simultaneous photopolymerization and radical-mediated chain transfer reactions that complete in < 70 seconds. Further, PEG-AlS hydrogels can be in situ softened to promote organoid crypt formation, providing a super-resolution imaging platform both for pre- and post-differentiated organoids. Overall, PhASE-ExM is a useful tool to decipher organoid behavior by enabling sub-micron scale, 3D visualization of proteins and signal transduction pathways.
Keywords: expansion microscopy, intestinal organoids, synthetic hydrogels, photochemistry, extracellular matrix
Graphical Abstract
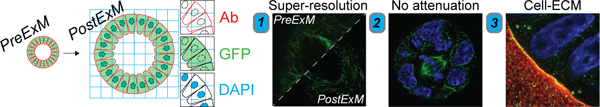
In their manuscript, Blatchley et al. devise a novel method for expansion microscopy and apply it to visualize organoids cultured in Matrigel and synthetic hydrogels. They overcome several existing limitations associated with imaging of organoids, enabling super-resolution image acquisition, eliminating signal attenuation for whole organoid imaging, and facilitating capture and quantification of cell-matrix interactions of organoids.
Organoids have emerged as the current state-of-the-art, multicellular in vitro model for nearly all mammalian tissues and organs.[1, 2] As organoid culture methods have been refined, their utility has become unmatched in developmental biology and disease modelling, owing to their biomimetic cellular composition and 3D architecture, as well as the capacity to match tissue and organ-level functionality on the benchtop. More recently, these culture methods have employed user-defined synthetic hydrogels that can precisely present physical and chemical cues to direct organoid growth and differentiation, as well as to understand the underlying mechanisms governing these morphogenetic events.[3–9] These hydrogels can be further designed to have temporally tunable mechanical properties to recapitulate extracellular matrix (ECM) dynamics throughout development and disease, thereby enabling user dependent control over organoid growth and symmetry breaking events.[10] Among others, photodegradable hydrogels, such as those that exploit cleavable o-nitrobenzyl and allyl-sulfide chemistries are particularly attractive, as they enable complete temporal and spatial control over organoid behavior.[10–13]
While hydrogels are primarily used to culture organoids, they complicate high-resolution imaging as a result of light scattering and absorption, as well as the limited penetration depth (< 100 µm) of super-resolution microscopes.[14] Traditionally, organoids have been imaged by embedding and sectioning, or by removal from their culture microenvironment and whole mounting, but the intricacies of the 3D architecture, as well as the role of ECM, are lost with these methods.[15, 16] To circumvent these limitations, we introduce Photo-transfer by Allyl Sulfide Exchange Expansion Microscopy (PhASE-ExM), enabling optical clearance and 3.25x physical expansion, and thus deep, super-resolution imaging of organoids cultured in synthetic poly (ethylene glycol) (PEG)-based hydrogels containing photodegradable allyl sulfide (AlS) crosslinks. PhASE-ExM stems from the seamless combination of two previous technologies: (1) a synthetic hydrogel amenable to organoid growth using a thiyl radical mediated AlS cleavage through a reversible addition-fragmentation chain transfer reaction (AFTC)[5, 6] and (2) photo-expansion microscopy (PhotoExM), which relies on swellable hydrogels prepared with thiol-acrylate mixed-mode photopolymerization (Figure 1a).[17] In PhASE-ExM, upon light irradiation, thiyl radicals (6 wt%, 8-arm, 10 kDa PEG-SH) are first generated via dissociation of a photoinitiator, such as lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP - 0.2 wt%). These thiyl radicals simultaneously react with sodium acrylate (16 wt%), acrylamide (3 wt%) and PEG-diacrylamide (0.875 wt%) monomers to generate a swellable hydrogel.[17] In parallel, if the concentration of thiyl radicals are much higher than AlS groups ([-SH]/[AlS] = 15.6 in this work, assuming 100% LAP initiator dissociation efficiency at t = 0), upon AFTC reaction, most of the thiols in the original AlS crosslinks (~ 94%) are replaced with the thiols permeated into the AlS crosslinked hydrogels. Since these thiols are also used to polymerize the PhotoExM formulation, the material of the AlS crosslinked hydrogel is incorporated into the PhotoExM formulation during its polymerization, resulting in a cytocompatible hydrogel platform that can be expanded without requiring an additional degradation step, herein referred to as PhASE-ExM. The onset of AlS hydrogel incorporation into PhotoExM photopolymerization is almost instantaneous (t = 1.5 s) upon mild irradiation conditions (Io = 4.5 mW/cm2, λ = 365 nm), according to in situ rheological measurements (Figure 1b). Resulting PhASE-ExM hydrogels reach > 95% conversion within 70 s, which is extremely rapid compared to previous expansion microscopy (ExM) techniques (> 90 min.),[14, 18–21] enabling the opportunity for high throughput fabrication, if desired. Upon dissociation of the pendant groups of sodium acrylate monomers via repetitive H2O washes, which promote chain repulsion, PhASE-ExM hydrogels expand ~ 3.25 ± 0.04 x of their original size (Figure 1c), resulting in a theoretical lateral imaging resolution of < 120 nm when numerical aperture (N.A.) 1.0–1.2 water immersion objectives are used with confocal microscopy imaging (λ = 488 – 594 nm). The decreased expansion factor of the PhASE-ExM method compared to PhotoExM hydrogels (4.6x)[17] was due to the ~30% shrinkage of AlS crosslinked hydrogels upon permeation with the PhotoExM solution (Figure 1d). Overall, PhASE-ExM represents a novel combination of recent advances in biomaterials design and super-resolution microscopy techniques, with potential for utility in imaging organoids cultured in tailorable microenvironments.
Figure 1. Photo-transfer by allyl sulfide exchange expansion microscopy (PhASE-ExM).
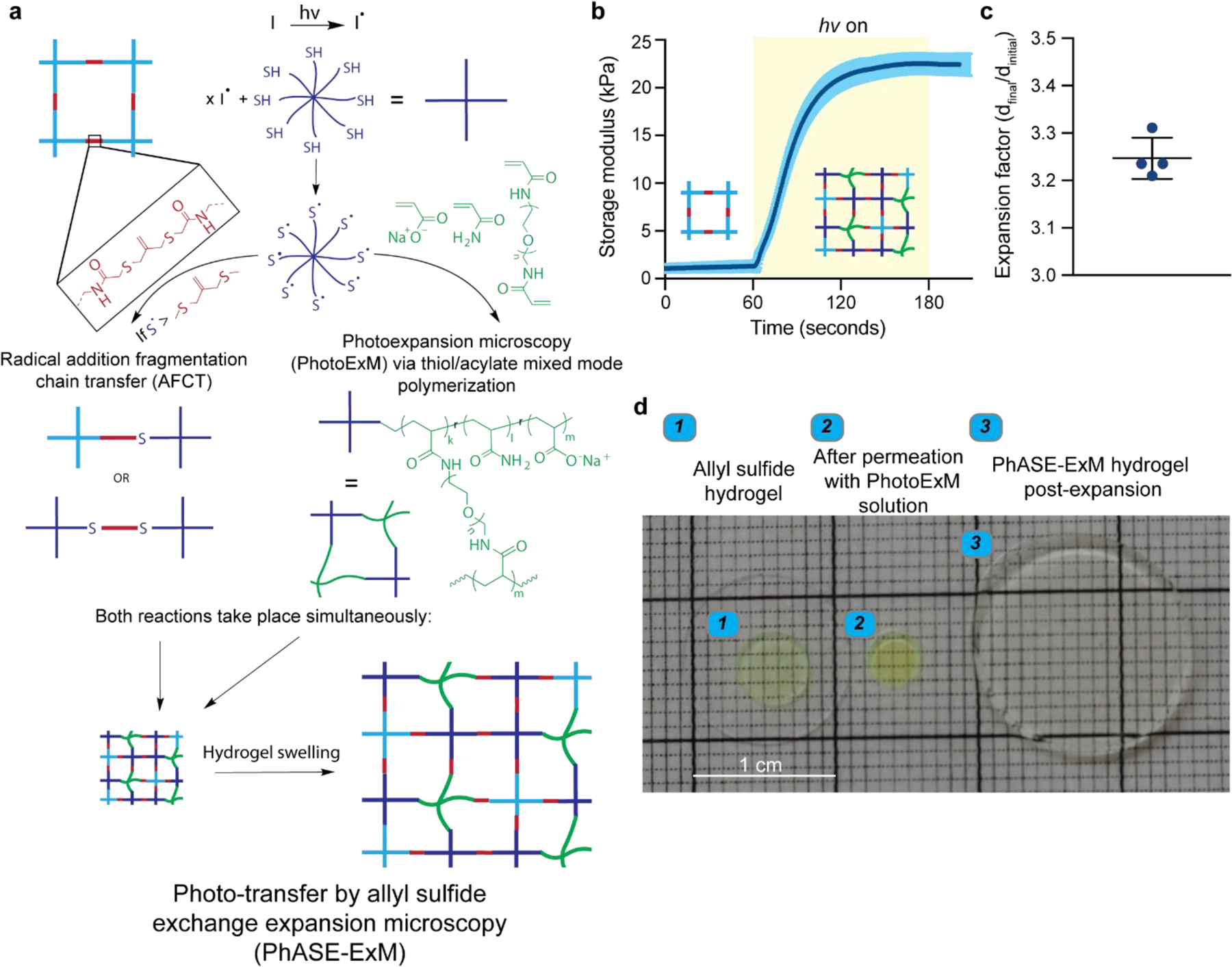
(a) Schematic illustration of photo-transfer by allyl sulfide exchange expansion microscopy (PhASE-ExM): A cytocompatible hydrogel prepared using allyl-sulfide crosslinking groups (top left) is permeated with the photoexpansion microscopy (PhotoExM) formulation, which contains an 8-arm, 10 kDa PEG-SH as the chain transfer agent, sodium acrylate as the polyelectrolyte monomer, PEG-diacrylamide as the crosslinker, acrylamide, and LAP as the photoinitiator. Upon light irradiation, two processes simultaneously occur. While photopolymerization of PhotoExM formulation yields a swellable polyelectrolyte network (middle right), radical addition fragmentation chain transfer (AFCT) reaction between the allyl-sulfide and thiyl radicals incorporates the chains of the cytocompatible hydrogel to the polymerizing PhotoExM network (middle left), which can be homogeneously expanded with repetitive H2O washes upon termination of light irradiation (bottom), enabling expansion microscopy of organoids cultured in synthetic hydrogels without requiring another degradation step for the hydrogel. (b) Gel evolution of PhASE-ExM hydrogels as measured by in situ photo-rheology: The first 60 s shows the storage modulus (G’) of the allyl-sulfide crosslinked hydrogels permeated with the PhotoExM formulation, and upon light irradiation (Io = 4.5 mW/cm2, λ = 365 nm), PhASE-ExM hydrogels reach > 95% conversion within 70 s. n = 3 independent measurements, dark blue line = mean, blue shaded area = s.d. Yellow shaded area shows the duration of light irradiation. (c) Overall size expansion factor of the PhASE-ExM hydrogels. n = 4 hydrogels, graph presented as mean +/− s.d. (d) Representative images of allyl sulfide crosslinked hydrogels on a glass coverslip (left), which shrinks ~ 30% by size following permeation with the PhotoExM solution (middle) and post-expansion (right). The allyl sulfide crosslinked hydrogels were functionalized with AlexaFluor 488 to determine its retention post-expansion.
As a first proof-of-principle to overcome limitations in existing imaging modalities, and to illustrate the broad utility of our PhotoExM method for physical expansion and optical clearance for deep, super-resolution imaging of organoids grown in 3D scaffolds, we cultured intestinal organoids in Matrigel, which is the current gold standard growth platform for nearly all types of organoids.[9, 22] Following organoid growth, samples were fixed and immunostained using established methods (Figure 2a). A tethering group, Acryloyl X (AcX) was then added to enable binding to the expansion hydrogel, and thus retain the spatial localization of the fluorophores. Next, the premixed PhotoExM hydrogel solution (Table S1) was permeated into the sample 2 times for 30 min each. Compared to other ExM methods, PhotoExM is particularly advantageous for thick organoid cultures, as photoinitiated polymerization decouples monomer diffusion into the sample from the hydrogel formation, which can otherwise cause depth dependent variations in the hydrogel crosslinking density and lead to heterogeneous expansion. As the thiol/acrylate photopolymerization used to fabricate the PhotoExM hydrogels proceeds rapidly upon photoinitiation in an oxygen insensitive manner,[23] exposure to light (λ = 365 nm, Io = 4.5 mW/cm2) for 70 s at ambient conditions links the sample to the expansion hydrogel. Next, a 16–24 h treatment with Proteinase K (16 U/mL) containing digestion buffer at 37°C was used to completely degrade the organoid, as well as the Matrigel scaffold, resulting in an optically clear sample. Finally, repetitive H2O washes resulted in a linear expansion of ~4.3x (~80x volumetric expansion) (Figure 2b), enabling super-resolution imaging of immunolabeled proteins (e.g., E-cadherin), DNA (DAPI), as well as Lgr5+ intestinal stem cells (eGFP) when reporter organoid lines were used (Figure S1a).
Figure 2. PhotoExM of intestinal organoids cultured in Matrigel.
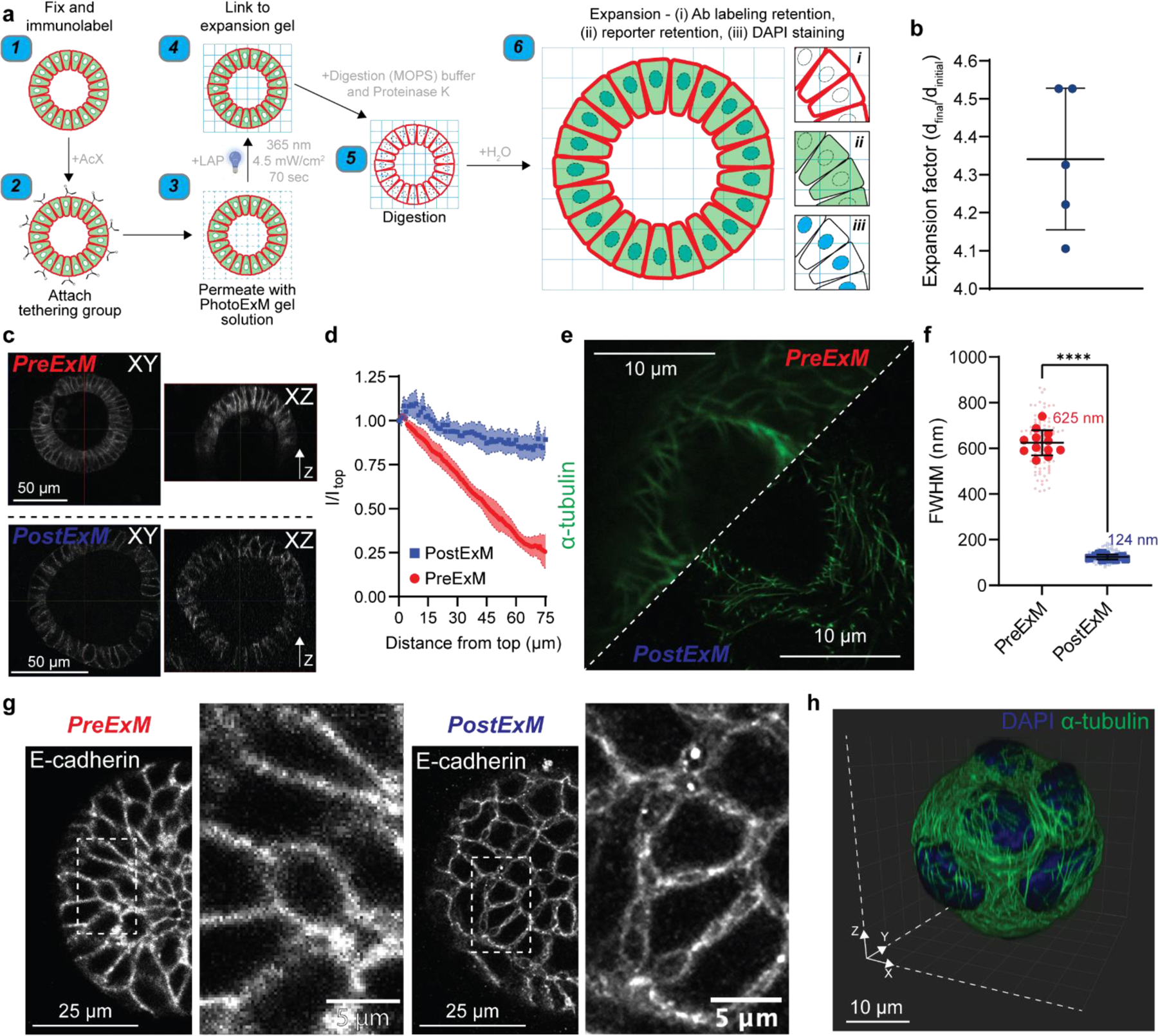
(a) Schematic for PhotoExM of intestinal organoids. Organoids are first fixed and immunolabeled using standard protocols, then the tethering group, Acryloyl X (AcX), is added. Next, the PhotoExM solution is permeated into the sample. Upon 70 s light irradiation (Io = 4.5 mW/cm2, λ = 365 nm) and the addition of a photoinitiator, LAP, the sample is linked to the expansion hydrogel. The addition of Proteinase K (16 U/mL) in the digestion buffer degrades and digests, as well as optically clears the organoids. Finally, immersion in H2O promotes dissociation of the electrolyte (sodium acrylate) monomer, resulting in physical expansion. Spatial information is retained, and immunolabeled whole organoids can be imaged in 3D. Additionally, retention of DNA allows for PostExM staining with DAPI, and retention of eGFP reporters allows for imaging of reporter organoid lines. (b) Expansion factor of organoids expanded in Matrigel. n = 5 hydrogels, graph presented as mean +/− s.d. (c) PreExM and PostExM images of E-cadherin labeled organoids. XY (single plane cross section) and XZ (orthogonal projection). (d) Fluorescent intensity of E-cadherin immunolabeling for PreExM and PostExM images across Z-stacks. Intensity normalized to the intensity at depth z=0 (Itop). The line represents the mean, with shaded regions representing 95% CI. (e) α-tubulin imaging PreExM and PostExM of organoids cultured for 2 days, starting from single cell suspensions. (f) Resolution of PreExM and PostExM samples quantified by the diameter of microtubules. n = 90–100 line scans per condition (smaller data points) across 12 individual organoids (larger data points). Graph presented as mean +/− s.d. **** p < 0.0001. (g) Single z-stack PreExM and PostExM imaging of E-cadherin. (h) 3D reconstruction in IMARIS of a whole PostExM organoid immunolabeled with α-tubulin and stained with DAPI. Organoids were cultured for 2 days, starting from a single cell encapsulation.
To determine the extent to which PhotoExM can be used to improve imaging of organoids cultured in 3D scaffolds, we imaged entire organoids (up to a z-depth of at least 75 µm) and assessed signal attenuation, which is a severe limitation of in situ imaging of whole organoids using confocal microscopy (Figure 2c; PreExM). Physical sectioning (e.g., cryosectioning) can eliminate issues with attenuation, but these methods inherently eliminate 3D whole organoid imaging. Optical clearing methods have also been established to circumvent issues with attenuation, but these methods often remove or harvest the organoid from its surrounding microenvironment, eliminating the capacity to image interactions between organoids and their extracellular matrix (ECM).[15] Using PhotoExM, attenuation can largely be eliminated, with little loss in fluorescence signal intensity of immunolabeled proteins up to a depth of 75 μm (Figure 2c; PostExM, Figure 2d).
An additional limitation of confocal microscopy is the maximum lateral resolution, which is restricted to ~300 nm by the diffraction of light, depending on the wavelength of light used and the N.A. of the objective. To assess quantitatively the resolution of PreExM and PostExM images, organoids were labeled with α-tubulin (Figure 2e). The full-width half maxima (FWHM) of Gaussian approximations of line scans perpendicular to individual microtubules were used to determine the PreExM (~625 nm) and PostExM (~124 nm) resolution. (Figure 2f). Interestingly, the resolution improvement from PhotoExM scaled larger than the expansion factor. It is likely that light scattering in PreExM imaging, where the samples are not optically cleared, contributes to the lower observed resolution compared to the theoretical resolution (298 nm). With complete optical clearance in PostExM imaging, the effective resolution was improved, especially at lower z-depths. The improved resolution was further illustrated by visualizing immunolabeled proteins (e.g., junctional E-cadherin) in single stack images before (PreExM) and after expansion (PostExM) (Figure 2g). Prior to expansion, E-cadherin appeared relatively uniform between cells, but after expansion, additional details regarding the organization and localization of E-cadherin are readily visualized. In general, a combination of optical clearance and improved resolution facilitates imaging of whole, 3D organoids, highlighting its broad utility for analyzing advanced in vitro organ and tissue models cultured in 3D scaffolds (Figure 2h, Figure S1b, c; Video S1).
Currently, in situ imaging of organoids is accomplished by embedding and sectioning, which eliminates the ability to capture 3D images, or by whole-gel imaging with minimal manipulation, which results in signal attenuation (Figure 2c, d). As PhotoExM facilitates imaging of whole organoids by optical clearance while retaining the spatial localization of extracellular proteins, it allows super-resolution imaging of cell-matrix interactions. The importance of cell-matrix interactions and ECM dynamics have been well established in embryological intestinal development,[24–28] but have not been studied in depth with regard to their specific role in shaping and guiding organoid growth and morphogenesis. Organoid studies, rather, have broadly highlighted the need for cell-ECM interactions for colony survival and growth, as well as the need for certain proteins, specifically laminin, to facilitate intestinal crypt formation.[3] In part, the lack of adequate imaging modalities to study these complex interactions has limited the ability to examine their role. As such, we first used PhotoExM to investigate cell-ECM interactions before and after crypt formation in Matrigel. To provide a more homogeneous population for analysis, we began our cultures from single intestinal stem cells (ISCs) (Figure 3a). ISCs were cultured in media favoring stem cell maintenance for 4 days (ENRCV),[29] which resulted in the formation of relatively homogeneous, spherical colonies (Figure 3a, b). This population comprised our “pre-differentiation” condition, prior to crypt formation. After 4 days, we altered the media composition to favor differentiation and crypt budding.[29] This population led to reproducible, but heterogeneous populations of organoids containing crypts; referred to as our “post-differentiation” condition (Figure 3a, c).
Figure 3. PhotoExM of cell-ECM interactions of organoids cultured in Matrigel.
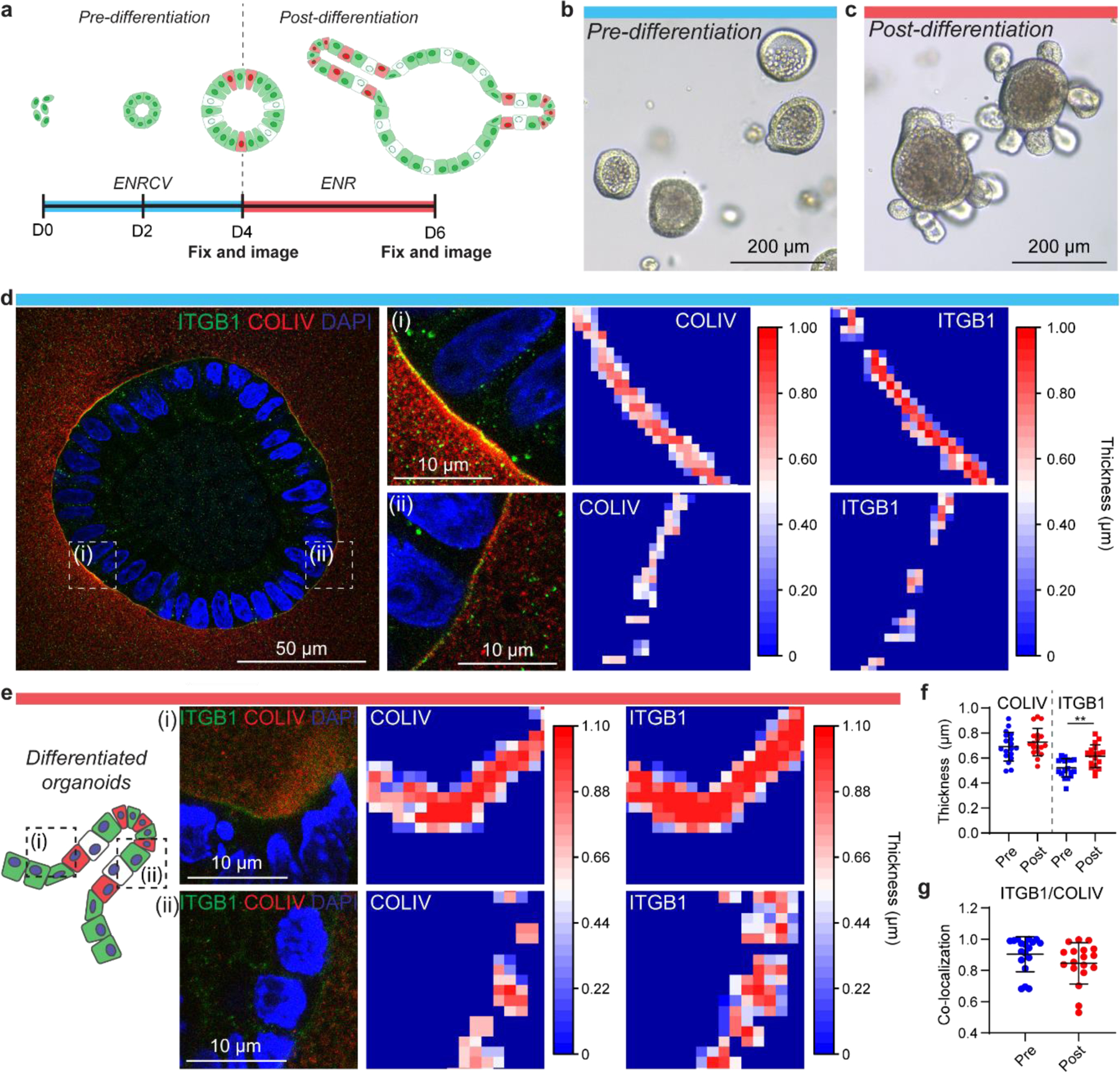
(a) Timeline of organoid growth. Organoids are generated starting from a population of single ISCs. For the first 4 days, cells were cultured in ENRCV media, favoring maintenance of stemness (“pre-differentiation”). On day 4, the media composition was changed to ENR to promote cell differentiation and crypt formation (“post-differentiation”). (b) Pre-differentiation, spherical organoids at day 4 in culture. (c) Post-differentiation organoids, containing crypts at day 6 in culture. (d) PhotoExM imaging of pre-differentiation organoids. Insets (i) and (ii) shows regions of high and low COLIV/ITGB1 thickness, respectively. (e) PhotoExM of post-differentiation organoids. The hinge region adjacent to an intestinal crypt (i) shows thicker COLIV/ITGB1 compared to the crypt region (ii). (f) Global quantification of the mean thickness of COLIV and ITGB1 in pre- and post-differentiated organoids. n = 18 organoids per condition. Graph presented as mean +/− s.d. ** p < 0.01. (g) Co-localization of ITGB1 and COLIV. Reported values are Mander’s coeffecient for the overlap of ITGB1 and COLIV. n = 18 organoids per condition. Graph presented as mean +/− s.d.
The importance of laminin in intestinal development and regeneration has been shown in model organisms in vivo, as well as in organoids in vitro.[3, 24–28] Additionally, we mined available RNA-sequencing data from intestinal organoids,[30] and saw an increase in laminin mRNA expression at the onset of crypt formation (Figure S2a), so we anticipated it would be an ideal protein to image as related to cell-ECM interactions in intestinal organoids. However, because of the high concentration of laminin in Matrigel (~60%[31]), technical limitations related to poor antibody penetration restricted our ability to analyze the role of laminin (Figure S2d). Instead, we investigated the localization and dynamics of collagen IV (COLIV), another key component of the intestinal basement membrane,[24] that, based on mRNA expression, also increased as a function of culture time (Figure S2b). To understand how cells within organoids were interacting with their local ECM, we labeled integrin β1 (ITGB1), which can bind many ECM proteins, including COLIV.[32] In pre-differentiation organoids, we identified cell-ECM interactions between ITGB1 and COLIV, which were spatially heterogeneous within the organoid on a cell-by-cell basis (Figure 3d, Figure S2e). Some regions exhibited areas of thick COLIV adjacent to the organoid, which overlapped with increased fluorescent intensity of ITGB1. Regions of thinner COLIV often corresponded to lower intensity of ITGB1, suggestive of distinct regional differences in COLIV/ITGB1 interactions. Post-differentiation, ITGB1 often overlapped with regions of locally thick COLIV at regions adjacent to crypts (“hinge regions”), while COLIV was thinner at the crypt base regions (Figure 3e). Globally, the thickness of COLIV surrounding organoids increased slightly in post-differentiation versus pre-differentiation orgnoids, although the difference was not statistically significant before and after differentiation, while the thickness of ITGB1 increased, suggesting a dynamic role of ITGB1 throughout organoid crypt formation (Figure 3f). The COLIV and ITGB1 signals were highly co-localized before and after differentiation, indicative of interactions between the two proteins during both organoid growth and crpyt formation (Figure 3g).
Based on our RNA-sequencing mining, fibronectin (FN) is also upregulated during crypt formation (Figure S2c). Since FN also interacts with ITGB1,[33] we co-stained for ITGB1 to better understand cell-FN interactions. However, we did not observe FN on the periphery of the organoids either pre- or post-differentiation (Figure S2f, g). Since only trace amounts of FN are present in Matrigel,[31] this result suggests that intestinal organoids in Matrigel did not secrete FN during growth and up to 2 days post-differentiation, even though its mRNA expression was found to increase post-differentiation, according to data curated from existing datasets (Figure S2c).[30] Overall, cells comprising organoids interact with COLIV via ITGB1 heterogeneously on a cell-by-cell basis. Heterogeneities in signaling between neighboring cells are key in regulating symmetry breaking events, where a symmetrical, spherical organoid changes its composition and shape to permit crypt budding.[30] While strictly correlative with this dataset, these heterogeneities warrant further investigation towards their potential role as upstream regulators of organoid morphogenesis. These results again highlight the utility of PhotoExM for in situ imaging of cell-ECM interactions of organoids, which can be used to discern changes in these interactions with previously unattainable resolution.
While Matrigel is used with near ubiquity in the organoid field, batch-to-batch variability, the complex and ill-defined ECM composition, and the inability to tailor its properties combine to limit its utility for mechanistic studies related to cell-matrix signaling and ECM dynamics. The proteinaceous content of Matrigel also severely limits the capacity to effectively immunolabel certain ECM proteins to study ECM dynamics, as well as the localization and accumulation of cell secreted ECM proteins. Synthetic hydrogels have been developed to overcome many of these limitations. In particular, PEG-based hydrogels provide a blank slate with which to engineer properties to mimic those of the ISC niche.[3–9] Initial matrix stiffness and integrin binding through ECM mimetic peptide sequences provide baseline cues for organoid growth, while the capacity for matrix remodeling is required for crypt formation.[3, 4]
To control organoid growth and differentiation in synthetic hydrogels, we prepared PEG-AlS hydrogels via a strain promoted-azide alkyne cycloaddition (SPAAC) reaction between an 8-arm, 20 kDa PEG-dibenzocyclooctyne (PEG-DBCO), and a bis-azide functionalized allyl sulfide (Figure 4a, Figure S4a).[5, 6] A fibronectin mimetic sequence, N3-GRGDS (0.8 mM), and laminin (0.2 mg/mL) were further incorporated into these hydrogels to promote cellular attachment and faciliate crypt formation, respectively. In these PEG-AlS hydrogels, an optimal initial stiffness (G’ ~1400 Pa) allowed for organoid colony growth (Figure 4b, Fig S4b, d), and differentiation was initiated by tuning photosoftening. Specifically, 365 nm light irradiation (4.5 mW/cm2) for 30 s in the presence of glutathione (GSH, 15 mM) and LAP (1 mM) reduced the matrix stiffness to ~50% of the initial modulus, generating an environment permissive to crypt formation (Figure S4c, d). Organoids cultured before and after differentiation were then expanded using PhASE-ExM to analyze cell-ECM interactions and ECM accumulation and localization before and after crypt formation, independent from the additional matrix cues provided by Matrigel (Figure 4b).
Figure 4. PhASE-ExM of organoids in PEG-AlS hydrogels.
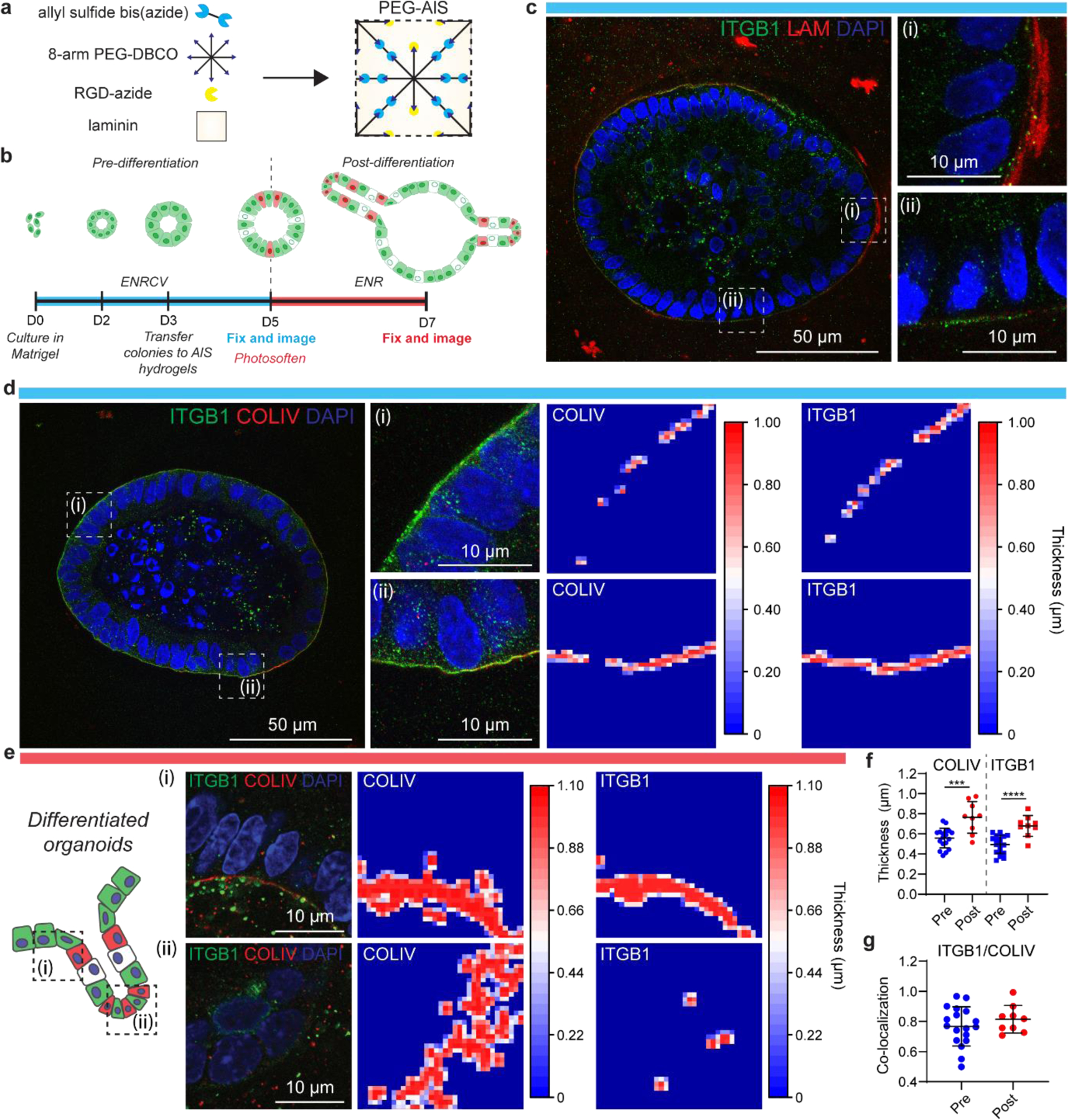
(a) PEG-AlS are synthesized by reaction of an 8-arm, 20 kDa PEG-dibenzocyclooctyne (PEG-DBCO), a bis-azide functionalized allyl sulfide, an azide functionalized RGD, and laminin. (b) Timeline of organoid growth. Organoids were generated starting from a population of single ISCs, and cultured in Matrigel for 3 days in ENRCV media, favoring stemness. At day 3, organoids were transferred to PEG-AlS hydrogels and cultured for 2 days. These organoids were the “pre-differentiation” population. On day 5, hydrogels were photosoftened and the media composition was changed to ENR to promote cell differentiation and crypt formation (“post-differentiation”). (c) Pre-differentiation PhASE-ExM imaging of LAM and ITGB1. (d) Pre-differentiation PhASE-ExM imaging and quantification of COLIV and ITGB1. (e) Post-differentiation PhASE-ExM imaging and quantification of COLIV and ITGB1. The hinge region adjacent to an intestinal crypt (i) shows thicker COLIV/ITGB1 compared to the crypt region (ii), where the COLIV is discontinuous and there is minimal ITGB1. (f) Global quantification of mean thickness of COLIV and ITGB1 in pre- and post-differentiated organoids. n = 9–18 organoids per condition. Graph presented as mean +/− s.d. *** p<0.001, **** p<0.0001. (g) Co-localization of ITGB1 and COLIV. Reported values are Mander’s coeffecient for the overlap of ITGB1 and COLIV. n = 9–18 organoids per condition. Graph presented as mean +/− s.d.
After optimizing the PhotoExM permeation and digestion conditions for organoids using Matrigel cultures, we used these same conditions to expand and image organoids cultured in PEG-AlS hydrogels with PhASE-ExM (Figure 1). Here, we assessed cell-ECM interactions by analyzing ITGB1 and ECM proteins laminin (LAM) , COLIV, and FN. While we added LAM to our PEG-AlS hydrogels, the concentration was much lower than in Matrigel, so imaging of LAM was possible, further motivating the use of our synthetic hydrogels to study organoid-ECM interactions. Interestingly, we observed heterogeneous staining for LAM around the organoid, similar to the heterogeneities present with interactions between COLIV and ITGB1 (Figure 4c).
Because PEG-AlS hydrogels have a defined composition, immunostaining can reveal localization of cell-secreted ECM proteins that are not included in the hydrogel formulation, such as COLIV and FN. We observed cell-secreted COLIV, where the accumulation varied by organoid and within each organoid (Figure 4d). Additionally, COLIV accumulated at hinge regions of differentiated organoids alongside regions of thick ITGB1, with discontinuous COLIV and minimal ITGB1 at crypt base regions (Figure 4e). Quantifications of global trends in COLIV and ITGB1 showed significant increases in thickness post-differentiation, suggesting that cells within organoids continue to secrete COLIV, and ITGB1 interactions play a role in organoid growth over the experimental time course (Figure 4f). Additionally, COLIV and ITGB1 are highly co-localized in pre- and post-differentiated organoids (Figure 4g). Finally, similar to organoids cultured in Matrigel, little FN was observed in pre- or post-differentiated organoids cultured in PEG-AlS hydrogels at the time points analyzed. (Figure S5).
Taken together, PhASE-ExM provides a novel method for optically clear, superresolution imaging of intestinal organoids cultured in Matrigel and synthetic PEG-based hydrogels. The methods presented here impart multi-factorial advancements in organoid imaging by overcoming signal attenuation, acheiving sub-micron scale resolution, and facilitating in situ imaging of cell-matrix interactions. Specifically, we used this method to gain an understanding as to how cell-matrix interactions and cell-deposited ECM proteins are localized prior to and after the initiation of crypt formation (i.e., differentiation). Heterogeneities in cell-matrix interactions are present prior to crypt formation, and differential ECM binding occurs as dependent on the location within an organoid once crypts have formed, where more ECM and a higher intensity integrin signal is present at hinge regions adjacent to crypts, compared to crypt base regions. Using PEG-AlS hydrogels, we identifed the presence of cell-deposited ECM proteins, which again vary by organoid and by location within the organoid. We anticipate that these studies will provide the basis for future investigation related to the role of cell-matrix interactions and dynamics in regulating crypt bud initiation and symmetry breaking, as well as crypt maintenance. However, further developments in antibodies enabling robust immunolabeling in 3D hydrogels is imperative; as many antibodies which provide strong signal in standard 2D cultures fail when utilized in 3D cultures. Finally, PhASE-ExM holds promise as a technique with broad utility for many types of organoids cultured in a broad array of biomaterials, to gain an appreciation for cell signaling at the sub-micron scale, which is often difficult to study with exisiting imaging techniques.
Experimental Section/Methods
Synthesis of 8-armed PEG40K-DBCO
8-armed PEG40K-DBCO was synthesized as previously described.[34]
Synthesis of allyl sulfide bis(propionamide PEG3-azide)
Allyl sulfide bis(propionamide PEG3-azide) was synthesized as previously described.[6]
Crypt Isolation and Organoid Culture
Murine small intestinal crypts were isolated from mice as previously described.[3] Crypts were maintained as organoids by encapsulation in reduced growth factor Matrigel (Corning). Organoids were expanded as colonies and cultured in ‘stem’ growth media (ENRCV[29], composed of Advanced DMEM-F12, GlutaMAX, HEPES buffer, penicillin-streptomycin, and supplemented with N2 and B27 supplements, R-Spondin conditioned media (5% v/v) (R), epidermal growth factor (50 ng/ml) (E), Noggin (100 ng/ml) (N), CHIR99021 (3 µM) (C), valproic acid (1 mM) (V). The media was changed every 2 days, and organoids were released and collected from Matrigel, sheared and re-suspended in Matrigel every 3–4 days. For all experiments, organoids were released and collected from Matrigel, then were dissociated into single cell suspensions using dissociation media comprised of 1 mL TrypLE, DNase I (~10 mg), n-acetylcysteine (1 µM), and Y-27632 (10 µM) for 8 minutes at 37°C, then added to blocking media (1 ml FBS, 8 mL base media) and passed through a 40 µm cell strainer. Cells were counted and encapsulated at a concentration of 1 million cells/ml. For the first two days following single cell dissociation, ‘stem’ growth media was supplemented with n-acetylcysteine (1 µM) and thiazovivin (2.5 µM). After 4 days in culture, ‘differentiation’ media was used (ENR).
Growth and differentiation of organoids cultured in PEG-AlS hydrogels
Starting from single cell suspensions, ISCs were cultured in Matrigel for 3 days in ENRCV. After 3 days, organoid colonies were transferred to PEG-AlS hydrogels. Hydrogel precursor solutions were prepared by diluting 8-arm PEG-DBCO in Advanced DMEM-F12 media supplemented with GlutaMAX, HEPES, and penicillin-streptomycin to 5 wt%. Azide functionalized RGD (0.8 mM) was added to the hydrogel precursor solutions, which were then kept on ice until the addition of cells (20–30 min). Organoids were released from Matrigel using cold media. The collected organoids colonies were then centrifuged (900 RPM, 4 minutes) and the pellet was resuspended in the hydrogel precursor solutions on ice. Laminin (0.2 mg/mL) was then added. Finally, gelation was initiated by the addition of the bis-azide functionalized allyl sulfide crossolinker. Immediately after the allyl sulfide crosslinker was added, the solutions were vortexed for 5 seconds and added as 10 µL drops to coverslips to gel. The gels were incubated at 37°C for 15–20 min, then cultured in media with the appropriate supplements as described above (ENRCV), along with 2.5 µM of thiazovinin (Stemgent) and 1 µM n-acetylcysteine. After two days in culture, organoids were either fixed and imaged or subjected to photosoftening Photosoftening was achieved by incubating organoids in PEG-AlS hydrogels in 15 mM GSH and 1 mM LAP in FluoroBrite media containing N2 and B27 supplements for 20–30 min at 37°C. The media was aspirated and samples were exposed to 365 nm UV light (4.5 mW/cm2) for 30 s. After photosoftening, ENR media was added. Organoids were cultured in ENR for 2 additional days, then fixed and imaged.
Rheology measurements
For the determination of the shear moduli (G’), PhASE-ExM formulations (Table S2) were in situ polymerized on a TA Instruments Discovery HR-3 rheometer equipped with a UV light guide accessory and coupled with the Omnicure 1000 light source (λ = 365 nm, Io = 4.5 mW/cm2). The storage modulus (G’) of the hydrogels was measured using an oscillatory strain of 5% and a frequency of 1.0 Hz.
For the determination of the swollen shear moduli (G’) of PEG-AlS, stock solutions of hydrogel precursors were prepared as 20 wt% PEG-8DBCO in phosphate buffered saline (PBS) and 50 mM allyl sulfide crosslinkers in DMSO. Hydrogels containing allyl sulfide bis(azide) crosslinks were formed by strain promoted azide-alkyne cycloaddition by mixing stock solutions with PBS to a final concentration of 5 wt% PEG-8DBCO and 5 mM allyl sulfide crosslinker in PBS. After mixing, the precursor solutions were vortexed for ~10 s and placed as 20 µL drops between two glass, Rain-X® coated slides separated with 0.5 mm rubber spacers. The glass slides were clamped together with paper clips and allowed to polymerize for >15 min. The samples were then swollen in PBS for >15 min, then transferred to a TA instruments’ DHR-3 rheometer with a 8 mm parallel plate geometry. The storage modulus (G’) of the hydrogels was measured using an oscillatory strain of 5% and a frequency of 1.0 Hz.
Characterization of SPAAC hydrogel degradation
PEG-AlS hydrogels were prepared as above. The fully formed gels were then placed in 500 µL baths containing a monothiol (GSH, 15 mM) in MOPS buffer contaning LAP (1 mM), and a monofunctional PEG3-azide (0.92 mM) for 30 minutes to reach a uniform concentration profile. Oscillatory rheology was performed on the gels using a TA Instruments Discovery HR-3 with an 8 mm parallel plate geometry and equipped with a UV light guide accessory, coupled with an Omnicure 1000 light source. The storage and loss moduli were recorded using a strain of 5% and a frequency of 1.0 Hz, while the sample was irradiated with 365 nm light at 5 mW/cm2.
Immunolabeling and tethering of organoid cultures
For immunolabeling of organoids with α-tubulin, samples were first extracted at room temperature for 2 min using a cytoskeletal stabilization buffer (PEM).[18] For all other samples, when organoids were cultured in Matrigel, samples were fixed in 3.2% PFA, 0.1% GA, 1X PBS for 20 mins at room temperature, then washed with PBS. After the PBS wash, samples were incubated for 5 min. at room temperature in 10 mM sodium borohydride, then washed with PBS (3x) at room temperature. For organoids cultured in PEG-AlS hydrogels, samples were fixed in 4% PFA for 30 mins at room temperature, then washed with PBS (3x) at room temperature. Permeabilization was done in 0.1% Triton-X-100 for 1 h at room temperature, followed by blocking in 2% goat serum (GS) for 1 h at room temperature for all samples in both Matrigel and PEG-AlS. Primary antibodies were incubated overnight at 4°C on a rocker. The following day, samples were washed with PBS (at least 3 times, 1 h in between), then incubated in secondary antibody overnight at 4°C on a rocker. Following immunolabeling, samples were treated with 0.1 mg/mL AcX in PBS for overnight at room temperature, and subsequently washed 3x with PBS (10 min each).
Fabrication and characterization of PhotoExM/PhASE-ExM hydrogels:
Sodium acrylate (NaAc) (33 wt%), PEGdiAcM Mn = 600 g/mol (25 wt%), AcM (40 wt%), 8-arm, 10 kDa PEG-SH (25 wt%) and LAP (2 wt%) in PBS containing additional 2 M NaCl stock solutions were premixed to obtain PhotoExM/Phase-ExM formulations shown in Table S2. The excess liquid on PEG-AlS hydrogels and Matrigel were removed, and hydrogels were permeated 2x with 100 µL of PhotoExM/PhASE-ExM formulation for 30 min at room temperature. After 2 permeations, PEG-AlS hydrogels or Matrigel were transferred into a 5.5 cm petri dish, residual liquid was wicked away, and a Rain-X® coated 12 mm glass coverslip was placed on top of the samples. The samples were irradiated with λ = 365 nm, Io = 4.5 mW/cm2 light for 70 s. Next, samples were digested with proteinase K (16 U/mL) containing digestion buffer overnight at 37°C. Samples were expanded once with a 20 min DI H2O wash, stained with DAPI (1:500) for 1 h in DI H2O and washed 2 more times with DI H2O (20 min each) to achieve final expansion. Hydrogel expansion factors were determined by measuring the diameter of the pre- and post-expansion hydrogels.
Imaging and analysis
Samples were imaged either using a Zeiss LSM710 equipped with a N.A. 1.0 water immersion (WI) objective (pinhole = 1.0 AU, Nyquist sampling) or Nikon A1R equipped with a N.A 0.95 WI objective (pinhole = 1.2 AU, Nyquist sampling). For the determination of the attenuation, entire organoids immunolabeled with E-cadherin were imaged with z-stack thicknesses of 1.2 µm for pre- and 5 µm for post-expansion organoids, respectively. Next, the post-expansion images were downscaled 4x to enhance the intensity of the features, and the E-cadherin channel was thresholded (Otsu), masked, and the average intensity of the masked regions were quantified at each z-stackin ImageJ. The average intensity values were normalized to the top z-stack of the organoid. The attenuation data was compiled from N = 20 organoids cultured in 2 independent hydrogels. The resolution was determined using organoids immunolabeled with α-tubulin. Briefly, line intensity profiles in single z-stacks were drawn perpendicular to the microtubule direction in ImageJ, curve-fitted using Gaussian approximation and the full width at half maxima (FWHM) was reported as the resolution. 18 organoids cultured in 2 independent hydrogels were analyzed. For the determination of organoid-matrix interactions, only a single stack from the middle of the organoid was imaged. N=9–18 organoids cultured in at least 2 independent hydrogels were imaged and quantified. A custom Matlab script was written to analyze cell-ECM interactions and ECM dynamics. In brief, a Gaussian filter was first applied, then the image was binarized and inverted. The perimeter of the remaining objects was determined and the ROI was isolated. The ROI was then dilated to create a mask, which was overlaid on the unbinarized image. Within the masked region, a histogram of pixel densities was calculated and plotted. These values were reported as the thickness of the ECM and ITGB1 channels. Co-localization values were reported as the Mander’s coefficient for the overlap of ITGB1 and COLIV using the JACoP plugin in ImageJ.
Supplementary Material
Acknowledgements
This work was supported in part by grants from the National Institute of Health (NIH) (R01 DK120921) and the National Science Foundation (NSF RECODE 2033723) to K.S.A. and P.J.D. F.M.Y. acknowledges the Department of Education Graduate Assistance in Areas of National Need (DoEd GAANN) fellowship and the NIH pre-doctoral fellowship (F31 DK126427). We thank the BioFrontiers Advanced Light Microscopy Core and its director Dr. Joseph Dragavon for technical advice and the use of their facilities and microscopes.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Michael R. Blatchley, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA, The BioFrontiers Institute. University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA.
Kemal Arda Günay, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA, The BioFrontiers Institute. University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA.
F. Max Yavitt, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA, The BioFrontiers Institute. University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA.
Elijah M. Hawat, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA
Peter J. Dempsey, Section of Developmental Biology, Department of Pediatrics, University of Colorado School of Medicine, 1775 Aurora Ct, Aurora, CO, 80045, USA
Kristi S. Anseth, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA, The BioFrontiers Institute. University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO, 80303 USA.
References
- 1.Hofer M and Lutolf MP, Engineering organoids. Nature Reviews Materials, 2021. 6(5): p. 402–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clevers H, Modeling Development and Disease with Organoids. Cell, 2016. 165(7): p. 1586–1597. [DOI] [PubMed] [Google Scholar]
- 3.Gjorevski N, et al. , Designer matrices for intestinal stem cell and organoid culture. Nature, 2016. 539(7630): p. 560–564. [DOI] [PubMed] [Google Scholar]
- 4.Cruz-Acuña R, et al. , Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nature Cell Biology, 2017. 19(11): p. 1326–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hushka EA, et al. , Relaxation of Extracellular Matrix Forces Directs Crypt Formation and Architecture in Intestinal Organoids. Advanced Healthcare Materials, 2020. 9(8): p. 1901214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yavitt FM, et al. , The Effect of Thiol Structure on Allyl Sulfide Photodegradable Hydrogels and their Application as a Degradable Scaffold for Organoid Passaging. Advanced Materials, 2020. 32(30): p. 1905366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez-Gordillo V, et al. , Fully synthetic matrices for in vitro culture of primary human intestinal enteroids and endometrial organoids. Biomaterials, 2020. 254: p. 120125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rezakhani S, Gjorevski N, and Lutolf MP, Low-Defect Thiol-Michael Addition Hydrogels as Matrigel Substitutes for Epithelial Organoid Derivation. Advanced Functional Materials, 2020. 30(48): p. 2000761. [Google Scholar]
- 9.Kratochvil MJ, et al. , Engineered materials for organoid systems. Nature Reviews Materials, 2019. 4(9): p. 606–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gjorevski N, et al. , Tissue geometry drives deterministic organoid patterning. Science, accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kloxin April M, et al. , Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science, 2009. 324(5923): p. 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown TE, Marozas IA, and Anseth KS, Amplified Photodegradation of Cell-Laden Hydrogels via an Addition–Fragmentation Chain Transfer Reaction. Advanced Materials, 2017. 29(11): p. 1605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKinnon DD, et al. , Design and Characterization of a Synthetically Accessible, Photodegradable Hydrogel for User-Directed Formation of Neural Networks. Biomacromolecules, 2014. 15(7): p. 2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen F, Tillberg Paul W, and Boyden Edward S, Expansion microscopy. Science, 2015. 347(6221): p. 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekkers JF, et al. , High-resolution 3D imaging of fixed and cleared organoids. Nature Protocols, 2019. 14(6): p. 1756–1771. [DOI] [PubMed] [Google Scholar]
- 16.Sato T, et al. , Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 2009. 459(7244): p. 262–265. [DOI] [PubMed] [Google Scholar]
- 17.Günay KA, et al. , Photo-expansion microscopy enables super-resolution imaging of cells embedded in 3D hydrogels in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chozinski TJ, et al. , Expansion microscopy with conventional antibodies and fluorescent proteins. Nature Methods, 2016. 13(6): p. 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tillberg PW, et al. , Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nature Biotechnology, 2016. 34(9): p. 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, et al. , Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nature Biotechnology, 2017. 35(8): p. 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truckenbrodt S, et al. , X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO reports, 2018. 19(9): p. e45836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aisenbrey EA and Murphy WL, Synthetic alternatives to Matrigel. Nature Reviews Materials, 2020. 5(7): p. 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Brien AK, Cramer NB, and Bowman CN, Oxygen inhibition in thiol–acrylate photopolymerizations. Journal of Polymer Science Part A: Polymer Chemistry, 2006. 44(6): p. 2007–2014. [Google Scholar]
- 24.Meran L, Baulies A, and Li VSW, Intestinal Stem Cell Niche: The Extracellular Matrix and Cellular Components. Stem Cells International, 2017. 2017: p. 7970385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scarpa S, et al. , Expression and synthesis of fibronectin and laminin by an intestinal epithelial cell line. Tissue and Cell, 1988. 20(3): p. 305–312. [DOI] [PubMed] [Google Scholar]
- 26.Simo P, et al. , Changes in the expression of laminin during intestinal development. Development, 1991. 112(2): p. 477–87. [DOI] [PubMed] [Google Scholar]
- 27.Simon-Assmann P, et al. , Differential expression of laminin isoforms and alpha 6-beta 4 integrin subunits in the developing human and mouse intestine. Dev Dyn, 1994. 201(1): p. 71–85. [DOI] [PubMed] [Google Scholar]
- 28.Teller IC, et al. , Laminins in the developing and adult human small intestine: relation with the functional absorptive unit. Dev Dyn, 2007. 236(7): p. 1980–90. [DOI] [PubMed] [Google Scholar]
- 29.Yin X, et al. , Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nature Methods, 2014. 11(1): p. 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serra D, et al. , Self-organization and symmetry breaking in intestinal organoid development. Nature, 2019. 569(7754): p. 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corning Matrigel matrix. Frequently asked questions Corning Incorporated Life Sciences; 2021. [Google Scholar]
- 32.Vandenberg P, et al. , Characterization of a type IV collagen major cell binding site with affinity to the alpha 1 beta 1 and the alpha 2 beta 1 integrins. Journal of Cell Biology, 1991. 113(6): p. 1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan JL, Trevithick JE, and Hynes RO, Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regulation, 1991. 2(11): p. 951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grim JC, et al. , A Reversible and Repeatable Thiol–Ene Bioconjugation for Dynamic Patterning of Signaling Proteins in Hydrogels. ACS Central Science, 2018. 4(7): p. 909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.