

Abstract
Background
Large-scale genome-wide association studies have successfully identified many genetic variants significantly associated with Alzheimer’s disease (AD), such as rs429358, rs11038106, rs723804, rs13591776, and more. The next key step is to understand the function of these SNPs and the downstream biology through which they exert the effect on the development of AD. However, this remains a challenging task due to the tissue-specific nature of transcriptomic and proteomic data and the limited availability of brain tissue.In this paper, instead of using coupled transcriptomic data, we performed an integrative analysis of existing GWAS findings and expression quantitative trait loci (eQTL) results from AD-related brain regions to estimate the transcriptomic alterations in AD brain.
Results
We used summary-based mendelian randomization method along with heterogeneity in dependent instruments method and were able to identify 32 genes with potential altered levels in temporal cortex region. Among these, 10 of them were further validated using real gene expression data collected from temporal cortex region, and 19 SNPs from NECTIN and TOMM40 genes were found associated with multiple temporal cortex imaging phenotype.
Conclusion
Significant pathways from enriched gene networks included neutrophil degranulation, Cell surface interactions at the vascular wall, and Regulation of TP53 activity which are still relatively under explored in Alzheimer’s Disease while also encouraging a necessity to bind further trans-eQTL effects into this integrative analysis.
Keywords: GWAS, Alzheimer’s Diseases, eQTL, HEIDI, SMR
Background
Alzheimer’s disease (AD) is the leading cause of brain dementia, along with which substantial failure of organs and mental issues arise. Accumulation of beta-amyloid plaques and tau tangles are two hallmarks of AD. The genetic mutations in genes such as ataxin-1 cause the misfolding of the proteins thereby starting a chain reaction of multiple neurodegenerative pathologies [1]. In the last decade, several large-scale genome-wide association studies (GWASs) have helped reveal mutations significantly associated with AD and the related traits. Yet, the functional mechanism through which these SNPs contribute to the development of AD remains largely unknown. This knowledge gap could be partly narrowed by investigating the effect of these SNPs on the downstream transcriptomic and proteomic levels. But the limited availability of gene and protein expression data in the brain tissue makes this a very challenging task.
Expression quantitative loci (eQTL) analysis aims to identify genetic variants that are significantly associated with the expression of one or more genes [2]. Recent findings show that most GWAS findings overlap with expression quantitative trait loci (eQTL), indicating the potential role of disease-related variants in gene regulation [3]. Although GWAS does not necessarily reveal the causal variants associated with the disease, with eQTL that links the genomic data to the transcriptomic data, one can isolate the location that potentially affect the downstream expression profile.
In this paper, leveraging the GWAS findings from International Genomics of Alzheimer’s Project (IGAP) and eQTL results of 3 brain regions from Brain eQTL Almanac (BRAINEAC), we applied a summary-based mendelian randomization method (SMR) to predict the associations between gene expression and AD in 3 brain regions of interest [4], including hippocampus, frontal cortex and temporal cortex as described in Fig. 1. While no significant gene-AD relationships were found from the hippocampus and frontal cortex, temporal cortex resulted in 37 SNPs from 32 unique genes that are significantly associated with AD in the transcriptomic level. Among these, 10 of them were found differentially expressed in AD brains when examined using the real gene expression data from temporal cortex tissue. For 37 significant SNPs associated with these 32 genes as in SMR, 19 of them were found to be associated with imaging phenotypes in temporal cortex, including FDG intensity, medial temporal lobe thickness, and lateral temporal lobe thickness. These results cement the theory that AD pathology variants have a higher influence on the temporal region of the brain. This is the first study that uses real brain expression data to validate the results obtained from the integrative analysis of GWAS and eQTL summary statistics in AD.
Fig. 1.

Overall pipeline of the integrative analysis of GWAS and eQTL summary statistics and the downstream validation
Results
Significant gene-AD associations
With the GWAS summary data from the IGAP and eQTL summary data from BRAINEAC, we performed both SMR and HEIDI tests to estimate the gene-AD associations in three human brain regions: frontal cortex, temporal cortex, and hippocampal regions. For the frontal cortex and hippocampal regions, we obtained 318,168 and 195,996 probes respectively that passed the genome-wide significance threshold in the SMR test. However, after FDR correction, none of them remained significant. While both frontal cortex and hippocampal regions are known to be related to AD, our results did not show any significant transcriptomic alterations in these regions, likely due to the small sample size in eQTL study. Also, some signals might be missing since BRAINEAC now only provides cis-eQTL data. In the temporal cortex, SMR test yielded 97 significant probes with FDR corrected and HEIDI test returned 2224 significant probes after correction. 37 probes corresponding to 32 unique genes were found significant in both SMR and HEIDI tests as seen in Fig. 2.
Fig. 2.

List of genes that passed both SMR and HEIDI tests in temporal cortex samples
Differential gene expression in AD brains
For all 32 significant genes identified in the temporal cortex, we further compared their expression patterns between cognitively normal controls and AD using RNA-Seq data collected from the temporal cortex region in the Mayo clinic cohort. Out of 32 genes, we identified 10 of them with significant differential expression patterns in the temporal cortex region after FDR correction. Shown in Table 1 is the summary of those differentially expressed genes (DEGs).
Table 1.
List of genes with significant differential expression levels between AD and normal brains in the temporal cortex region
| Gene | logFCa | Corrected p value | logCPMb | LRc |
|---|---|---|---|---|
| CCDC86 | − 0.2E−04 | 7.2E−05 | 11.701 | 15.750 |
| CUZD1 | − 0.3E−04 | 0.9E−05 | 9.634 | 10.850 |
| FOSB | 0.7E−05 | 4.1E−05 | 11.738 | 16.827 |
| PODXL2 | 0.3E−05 | 1.7E−05 | 15.238 | 18.474 |
| PPP1R13L | − 0.6E−04 | 6.9E−06 | 11.488 | 20.211 |
| PRPF19 | 0.4E−05 | 3.1E−09 | 15.880 | 35.106 |
| PSD2 | − 0.7E−04 | 6.9E−05 | 16.418 | 33.557 |
| PTK2B | 0.4E−05 | 3.1E−05 | 16.594 | 17.366 |
| RGS2 | 0.2E−05 | 0.1E−05 | 13.428 | 6.514 |
| SLC15A3 | − 0.7E−04 | 3.9E−09 | 12.117 | 34.668 |
aThe log of fold-change that describes the difference of expression between groups
bThe log of counts-per-million that describes the expression level of each gene
cLikelihood ratios of the genes
Pathways and networks enriched in temporal cortex
Using RectomeFA, 15 pathways in REACTOME database were found to be enriched by those 10 significant genes. Shown in Fig. 3 is a list of those pathways and the ratio between the number of significant genes and all member genes in the pathway. We further examined the interactions between those 10 genes by mapping them to the REACTOME protein interaction network. We used the ReactomeFI in Cytoscape to investigate the direct or indirect interactions between these genes. Out of 10 genes, 7 of them were found to be connected with 6 intermediate or linker genes (Fig. 4). In the subnetwork, EP300 and MAPK8 were observed as two hub genes with the highest connectivity, and PTK2B has the highest connectivity among those 10 significant genes.
Fig. 3.

Top pathways enriched by 10 significant genes
Fig. 4.

Functional interactions among 10 significant genes that passed SMR, HEIDI tests, and further showed differential expression patterns in independent cohort
Association with temporal cortex phenotypes
When examined the original GWAS and eQTL summary statistics, the significant association of those 10 genes with AD disease status was found to come from 11 unique SNPs. Using these 11 SNPs as seeds, we expanded our SNP set by including other neighboring SNPs located within the same LD block. In total, 613 SNPs were found and 518 of them were identified with genotype data from the ADNI cohort. Then, we tested their association with three AD-related brain imaging phenotypes of the temporal cortex region in PLINK. Linear regression models were used to examine the association between each pair of SNP and brain imaging phenotype. After FDR correction, our association analysis identified 18 SNPs significantly associated with FDG intensity levels, 15 SNPs associated with the thickness of the medial temporal lobe, and 15 SNPs for the lateral temporal lobe (Table 2). In total, there are 19 significant SNPs, all from chromosome 19, and 13 of them were found to be associated with all three imaging phenotypes (Fig. 5). These SNPs are all located with two LD blocks of rs73050293 and rs7669277 in NECTIN and TOMM40 respectively. 11 of the SNPs were intron variants while two other SNPs, rs11556505 and rs15758 identified as synonymous variants located in the coding sequence. Most of the SNPs in NECTIN are genic downstream transcript variants and intron variants, while one SNP rs71352238 belongs to the category of upstream transcript variant, which might be a part of the gene regulation.The intronic variants can be responsible for regulating the gene expression since there has been multiple reports of miRNAs, siRNAs, piwi-interacting RNAs (piRNAs), long noncoding RNAs (lncRNAs), and small nucleolar RNAs (snoRNAs), which do have regulatory effect in transcription , to be present in the intronic region. Variation in this region could lead to differential regulation of the gene transcription [5]. For a more detailed analysis of the intronic variants, we used the SNP Nexus online tool [6–10]. We observed that under Ensemble Regulatory Build [11], about 88 percentage of the variants feature type belonged to promoter region and was active in brain and brain related epigenomes. While the other variant feature types belong to CTCF binding sites or open chromatin sites, they were also labelled to be inactive for the above mentioned epigenomes. As for the ENCODE database results [12], it was seen that H3K36me3, H3K4me1 and, H3K4me3 histone feature and DNase open chromatin feature were the most prominent features where these variants were present. This could also imply that these variants might play a role in Alzheimer’s disease by transcriptomic regulation of the epigenetic factors.
Table 2.
Significant associations between SNPs and 3 distinct imaging phenotypes of temporal cortex region
| SNP | FDG phenotype | Lateral temporal lobe thickness | Medial temporal lobe thickness |
|---|---|---|---|
| rs34278513 | 0.004 | 0.035 | 0.445 |
| rs412776 | 0.006 | 0.036 | 0.445 |
| r73050293 | 0.069 | 0.164 | 0.028 |
| rs3865427 | 0.006 | 0.068 | 0.445 |
| rs12972156 | 1.7E−08 | 0.005 | 0.007 |
| rs12972970 | 1.7E−08 | 0.003 | 0.007 |
| rs3432646 | 1.7E−08 | 0.003 | 0.007 |
| rs283815 | 2.2E−08 | 0.009 | 0.001 |
| rs6857 | 3.2E−10 | 0.002 | 0.0003 |
| rs71352238 | 1.7E−08 | 0.002 | 0.007 |
| rs184017 | 1.8E−08 | 0.009 | 0.001 |
| rs15781 | 0.0002 | 0.078 | 0.211 |
| rs2075650 | 1.7E−08 | 0.002 | 0.007 |
| rs15781 | 2.2E−08 | 0.009 | 0.001 |
| rs34095326 | 0.0003 | 0.117 | 0.039 |
| rs34404554 | 1.7E−08 | 0.002 | 0.007 |
| rs11556505 | 1.7E−08 | 0.002 | 0.007 |
| rs157582 | 2.2E−08 | 0.009 | 0.001 |
| rs59007384 | 1.7E−08 | 0.009 | 0.001 |
Fig. 5.
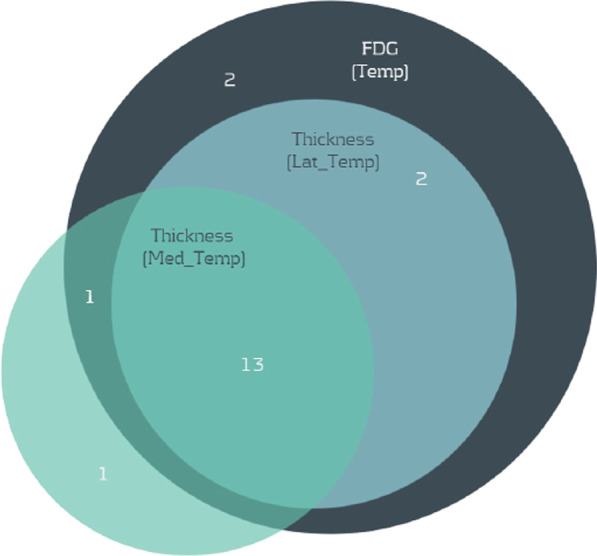
Number of significant SNPs associated with three imaging phenotypes of temporal lobe
The top three significant pathways enriched by 10 significant genes are Neutrophil degranulation, Cell surface interactions at the vascular wall, and Regulation of TP53 activity. Further probing of these pathways, neutrophils were found to migrate around AB proteins and in absence of neutrophils, improved cognitive functions, impeded microgliosis and 1–42 levels in brain homogenates [13]. Neutrophil downregulation has provided a reducing effect on levels of phosphorylated tau proteins. The secretory and the azurophil neutrophil granules have a common component, CAP37 protein, which is also seen upregulated in AD. This protein has been positively correlated with the AB-RAGE signaling pathway and also proven to activate monocytes by varying the cell adhesion pathway expression profile. Co-incidentally, we have cell surface interactions with vascular walls as one of the top significant pathways, where the process of extravasation is carried out by the vascular endothelial cells attached with neutrophils. The other important process that tangles these two pathways is NETosis (Neutrophils extracellular traps), wherein the neutrophils bind with the blood vessels and become mobile enough to target the parenchyma cells, leading to their cell death [14]. Studies have shown that NET depletion has provided positive feedback on reduced memory loss and other neuropathological features. Few studies have pointed out neutrophil levels as a potential indicator of cognitive decline [15]. Further, the proteins that belong to the Neutrophil pathway are significantly associated with Chr19, which also coincides with the location of all the significant SNPs associated with the imaging phenotype.
Regulation of TP53 is the other significant pathway along with the above two. Studies have shown that approximately 2-fold upregulation of p53 has been recorded in the superior temporal gyrus of Alzheimer’s patients. It has also been found to phosphorylate tau proteins in HEK cells [16]. Furthermore, p53 has been seen to aggregate and interact with tau oligomers in AD patients [17]. Thus, our exploration adds weight to the cause of relating AD and the cancerous pathways involving p53.
Conclusion
We performed an integrative analysis of AD GWAS and brain eQTL summary statistics to estimate the potential transcriptomic changes inside AD brains. Using real RNA-Seq gene expression data inside corresponding brain regions, identified genes with potential association with AD were further examined for altered expression patterns in AD brains. Significant gene-AD associations were found only in the temporal cortex region, but not in the frontal cortex and hippocampal regions. SNPs associated with two significant genes TOMM40 and NECTIN, as in the original eQTL summary, also showed significant association with the FDG intensity level and thickness of the temporal lobe. Further pathway and network analysis provided an enriched pathway profile, with neutrophil degranulation, Cell surface interactions at the vascular wall, and Regulation of TP53 activity as the most significant ones. Further efforts are warranted to investigate the association of neutrophils with AD. With the validation from real expression data collected from brain regions, the results of this study confirmed the potential of integrative GWAS and eQTL analysis in exploring the transcriptomic changes when lack of tissue-specific expression data. Further efforts to explore new merging methods for integrative analysis and trans-eQTL effects are warranted in the future work.
Data and methods
GWAS summary statistics
GWAS summary statistics were downloaded from the International Genomics of Alzheimer’s Project (IGAP). IGAP is a large two-stage study based upon genome-wide association studies (GWAS) on individuals of European ancestry. In stage 1, IGAP used genotyped and imputed data on 7,055,881 single nucleotide polymorphisms (SNPs) to meta-analyze four previously-published GWAS datasets consisting of 17,008 Alzheimer’s disease cases and 37,154 controls (The European Alzheimer’s disease Initiative—EADI the Alzheimer Disease Genetics Consortium—ADGC The Cohorts for Heart and Aging Research in Genomic Epidemiology consortium—CHARGE The Genetic and Environmental Risk in AD consortium—GERAD). In stage 2, 11,632 SNPs with were genotyped and tested for association in an independent set of 8572 Alzheimer’s disease cases and 11,312 controls. Finally, a meta-analysis was performed combining results from stages 1 and 2 [18].
EQTL summary data
EQTL summary statistics from the Brain eQTL Almanac (BRAINEAC) were downloaded through the UK Brain Expression Consortium (UKBEC). In total, there were 134 postmortem brains included in the study. RNA from ten brain regions were extracted and analyzed with Affymetrix Human Exon 1.0 ST and eQTLs were classified and grouped by marker type, expression type, and cis/trans type. Our study focused on three brain regions including the temporal cortex, frontal cortex, and hippocampal regions, in which the gene expression data are available in the AD brains through the AMP-AD knowledge portal.
SMR and HEIDI test
SMR (Summary-based Mendelian Randomization) software was used to integrate summary-level data from the IGAP GWAS with data from BRAINEAC eQTL studies to identify genes with potential expression levels altered in certain brain regions of AD brains [19]. Individual-level SNP genotype data from 1000 Genomes European population were used to estimate linkage disequilibrium (LD) block information. Following the default settings, SMR analysis was performed on cis-regions with a window of 2000 Kb, and LD r-squared threshold range was set between 0.9 ± 0.05. Note that the association observed in the SMR test does not necessarily indicate that gene expression and AD are affected by the same underlying causal variant. The association could be due to the top associated eQTL being in LD with two causal variants, one affecting gene expression and the other affecting AD. Compared to such linkage effect, pleiotropy effect is of more interest where gene expression and a trait (e.g., AD) share the same causal variant. Therefore, we further applied HEIDI (heterogeneity in dependent instruments) test to differentiate pleiotropy from linkage. Following the [4], we used a genome-wide significance level () for SMR test and p value threshold of 0.01 for the HEIDI test. Subsequent analyses are focused on gene-AD associations that passed both SMR and HEIDI tests.
Differential expression analysis
For all the genes that passed both SMR and HEIDI tests, we further compared their expression levels between cognitively normal controls and AD patients using RNA-Seq data from the corresponding brain tissues. In our case, significant gene-AD associations were only detected in the temporal cortex region. So we downloaded the RNA-Seq data from the temporal cortex tissue in the Mayo Clinic cohort [20]. The differential expression analysis was performed using the R package, EdgeR [21, 22], where baseline age, sex, batch, RIN, and APOE e4 status were used as covariates. EdgeR is one of the most robust packages for differential expression analysis, which uses a generalized linear model (GLM) approach based upon negative binomial distribution. Normalization of the RNA-Seq data was performed using Trimmed mean of M values (TMM method). Before analysis, genes were filtered with counts per million as very low counts provide little evidence for differential expression. p Values obtained from differential expression analysis were adjusted using the FDR method and a hard threshold was set at 0.05.
Pathway and network enrichment analysis
We further performed pathway enrichment analysis for all genes that passed the SMR, HEIDI, and differential expression tests. The ReactomePA package in R was used to perform the enrichment analysis based on the pathways in the Reactome database [23]. This package provides the best pathways for each gene-set from the input list of genes as well as the membership of genes to each pathway. Besides, we also mapped these 10 genes to the protein interaction network using ReactomeFI in Cytoscape to examine their direct or indirect functional interactions in the pathway. [24]
Genetic association analysis with brain imaging Phenotype
With the significant gene-AD associations identified from the temporal cortex, we further examined the association of these genes with the brain imaging phenotypes in the temporal cortex region. We downloaded the FDG intensity levels and mean thickness of the temporal cortex regions from the ADNI cohort. The initial phase (ADNI-1) was launched in 2003 to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment could be combined to measure the progression of MCI and early AD. ADNI-1 was extended to subsequent phases (ADNI-GO, ADNI-2, and ADNI-3) for follow-up for existing participants and additional new enrollments. More information about the data collection and preprocessing steps can be found at www.adni-info.org [25, 26]. In total, we studied 3 imaging phenotypes, including the overall FDG intensity level of the temporal lobe region and the mean thickness of medial and lateral temporal regions. To remove the potential bias introduced by confounding factors, baseline age, gender, and education years were included as covariates to adjust FDG intensity levels. Intracranial volume (ICV) was used as an additional covariate for thickness measures. The association between eQTL SNPs of those significant genes and 3 imaging phenotypes was tested using PLINK. LD information derived from 1000 Genomes using European population were used for FDR correction of the SNP p values in each phenotype.
Acknowledgements
We thank the International Genomics of Alzheimer’s Project (IGAP) for providing summary results data for these analyses. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in analysis or writing of this report. IGAP was made possible by the generous participation of the control subjects, the patients, and their families. The i–Select chips was funded by the French National Foundation on Alzheimer’s disease and related disorders. EADI was supported by the LABEX (laboratory of excellence program investment for the future) DISTALZ grant, Inserm, Institut Pasteur de Lille, Université de Lille 2 and the Lille University Hospital. GERAD was supported by the Medical Research Council (Grant n 503480), Alzheimer’s Research UK (Grant n 503176), the Wellcome Trust (Grant n 082604/2/07/Z) and German Federal Ministry of Education and Research (BMBF): Competence Network Dementia (CND) grant n 01GI0102, 01GI0711, 01GI0420. CHARGE was partly supported by the NIH/NIA grant R01 AG033193 and the NIA AG081220 and AGES contract N01–AG–12100, the NHLBI grant R01 HL105756, the Icelandic Heart Association, and the Erasmus Medical Center and Erasmus University. ADGC was supported by the NIH/NIA grants: U01 AG032984, U24 AG021886, U01 AG016976, and the Alzheimer’s Association grant ADGC–10–196728.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (A.D.N.I.) (National Institutes of Health Grant U01 AG024904) and DOD A.D.N.I. (Department of Defense award number W81XWH-12–2-0012). A.D.N.I. is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support A.D.N.I. clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. A.D.N.I. data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. ...
About this supplement
This article has been published as part of BMC Medical Genomics Volume 15 Supplement 2, 2022: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM 2021): medical genomics. The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-15-supplement-2
Abbreviations
- AD
Alzheimer’s disease
- SNP
Single nucleotide polymorphism
- GWAS
Genome wide association studies
- eQTL
Expression quantitative trait loci
- IGAP
International genomics of Alzheimer’s project
- BRAINEAC
Brain eQTL Almanac
- SMR
Summary-based Mendelian randomization
- DEG
Differentialy expressed genes
- miRNA
MicroRNA
- siRNA
Small interfering RNA
- piRNA
Piwi-interacting RNA
- lncRNA
Long non-coding RNA
- snoRNA
Small nucleolar RNA
- AB
Alpha-beta
- NET
Neutrophils extracellular trap
- HEK
Human embryonic kidney
- UKBEC
UK brain expression consortium
- AMP-AD
Accelerating medicines partnership program for Alzheimer’s disease
- GLM
Generlaized linear model
- TMM
Trimmed mean of M values
- FDR
False discovery rate
- FDG
Fludeoxyglucose
- MRI
Magnetic resonance imaging
- PET
Positron emission tomography
- ICV
Intracranial volume
Author contributions
PV: Conceptualization, methodology, visualization, formal analysis, validation, writing—original draft, writing review and editing. PG: Data curation, formal analysis. TJ: Data curation, formal analysis. DC: Data curation, formal analysis. KN: Data curation, writing—review and editing. SR: Data curation, writing—review and editing AS: Data curation, result interpretation, writing—review and editing. JY: Conceptualization, methodology, visualization, writing—original draft, writing review and editing, supervision, funding acquisition. All authors have read and approved the final manuscript.
Funding
Data collection, quality control and interpretation of results were supported by NIH Grants R01 LM013463, R21 AG066135, R01 EB022574, R01 AG019771, P30 AG010133. Design of the study and data analysis were supported by NSF CRII 1755836 and NSF CAREER 1942394. Publication costs are funded by NIH Grant R21 AG066135.
Availability of data and materials
The data that support the findings of this study are available from the AMP-AD knowledge portal and the ADNI but restrictions apply to the availability of these data. Data are however available from the AMP-AD knowledge portal and the ADNI upon reasonable application.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Rev Genet. 2010;11(4):247–58. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368(1620):20120362. doi: 10.1098/rstb.2012.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, Jansen R, de Geus EJ, Boomsma DI, Wright FA, Sullivan PF, Nikkola E, Alvarez M, Civelek M, Lusis AJ, Lehtimäki T, Raitoharju E, Kähönen M, Seppälä I, Raitakari OT, Kuusisto J, Laakso M, Price AL, Pajukanta P, Pasaniuc B. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM, Yang J. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 5.Jo BS, Choi SS. Introns: the functional benefits of introns in genomes. Genom Inform. 2015;13(4):112–118. doi: 10.5808/GI.2015.13.4.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oscanoa J, Sivapalan L, Gadaleta E, Dayem Ullah AZ, Lemoine NR, Chelala C. SNPnexus: a web server for functional annotation of human genome sequence variation (2020 update) Nucleic Acids Res. 2020;48(W1):W185–W192. doi: 10.1093/nar/gkaa420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayem Ullah AZ, Oscanoa J, Wang J, Nagano A, Lemoine NR, Chelala C. SNPnexus: assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018;46(W1):W109–W113. doi: 10.1093/nar/gky399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dayem Ullah AZ, Lemoine NR, Chelala C. A practical guide for the functional annotation of genetic variations using SNPnexus. Brief Bioinform. 2013;14(4):437–447. doi: 10.1093/bib/bbt004. [DOI] [PubMed] [Google Scholar]
- 9.Dayem Ullah AZ, Lemoine NR, Chelala C. SNPnexus: a web server for functional annotation of novel and publicly known genetic variants (2012 update) Nucleic Acids Res. 2012;40:W65–W70. doi: 10.1093/nar/gks364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chelala C, Khan A, Lemoine NR. SNPnexus: a web database for functional annotation of newly discovered and public domain single nucleotide polymorphisms. Bioinformatics. 2009;25(5):655–661. doi: 10.1093/bioinformatics/btn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The ensembl regulatory build. Genome Biol. 2015;16(1):56. doi: 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stock AJ, Kasus-Jacobi A, Pereira HA. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J Neuroinflam. 2018;15(1):240. doi: 10.1186/s12974-018-1284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pietronigro EC, Della Bianca V, Zenaro E, Constantin G. NETosis in Alzheimer’s disease. Front Immunol. 2017;8:211. doi: 10.3389/fimmu.2017.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong X, Nao J, Shi J, Zheng D. Predictive value of routine peripheral blood biomarkers in Alzheimer’s disease. Front Aging Neurosci. 2019;11:332. doi: 10.3389/fnagi.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooper C, Meimaridou E, Tavassoli M, Melino G, Lovestone S, Killick R. P53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci Lett. 2007;418(1):34–37. doi: 10.1016/j.neulet.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farmer KM, Ghag G, Puangmalai N, et al. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol Commun. 2020;8:132. doi: 10.1186/s40478-020-01012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert J-C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao T, Hu Y, Zang T, Wang Y. Integrate GWAS, eQTL, and mQTL data to identify Alzheimer’s disease-related genes. Front Genet. 2019;10:1021. doi: 10.3389/fgene.2019.01021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen M, Carrasquillo M, Funk C, et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data. 2016;3:160089. doi: 10.1038/sdata.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–97. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu G, He QY. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst. 2016;12(2):477–9. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- 24.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saykin AJ, Shen L, Yao X, Kim S, Nho K, Risacher SL, et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: progress, opportunities, and plans. Alzheimer’s Dement J Alzheimer’s Assoc. 2015;11:792–814. doi: 10.1016/j.jalz.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, et al. Recent publications from the Alzheimer’s disease neuroimaging initiative: reviewing progress toward improved AD clinical trials. Alzheimer’s Dement J Alzheimer’s Assoc. 2017;13:e1–e85. doi: 10.1016/j.jalz.2016.07.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the AMP-AD knowledge portal and the ADNI but restrictions apply to the availability of these data. Data are however available from the AMP-AD knowledge portal and the ADNI upon reasonable application.