

Abstract
The host genetic background for hepatocellular carcinoma (HCC) is incompletely understood. We aimed to determine if four germline genetic polymorphisms, rs429358 in apolipoprotein E (APOE), rs2642438 in mitochondrial amidoxime reducing component 1 (MARC1), rs2792751 in glycerol‐3‐phosphate acyltransferase (GPAM), and rs187429064 in transmembrane 6 superfamily member 2 (TM6SF2), previously associated with progressive alcohol‐related and nonalcoholic fatty liver disease, are also associated with HCC. Four HCC case‐control data sets were constructed, including two mixed etiology data sets (UK Biobank and FinnGen); one hepatitis C virus (HCV) cohort (STOP‐HCV), and one alcohol‐related HCC cohort (Dresden HCC). The frequency of each variant was compared between HCC cases and cirrhosis controls (i.e., patients with cirrhosis without HCC). Population controls were also considered. Odds ratios (ORs) associations were calculated using logistic regression, adjusting for age, sex, and principal components of genetic ancestry. Fixed‐effect meta‐analysis was used to determine the pooled effect size across all data sets. Across four case‐control data sets, 2,070 HCC cases, 4,121 cirrhosis controls, and 525,779 population controls were included. The rs429358:C allele (APOE) was significantly less frequent in HCC cases versus cirrhosis controls (OR, 0.71; 95% confidence interval [CI], 0.61‐0.84; P = 2.9 × 10−5). Rs187429064:G (TM6SF2) was significantly more common in HCC cases versus cirrhosis controls and exhibited the strongest effect size (OR, 2.03; 95% CI, 1.45‐2.86; P = 3.1 × 10−6). In contrast, rs2792751:T (GPAM) was not associated with HCC (OR, 1.01; 95% CI, 0.90‐1.13; P = 0.89), whereas rs2642438:A (MARC1) narrowly missed statistical significance (OR, 0.91; 95% CI, 0.84‐1.00; P = 0.043). Conclusion: This study associates carriage of rs429358:C (APOE) with a reduced risk of HCC in patients with cirrhosis. Conversely, carriage of rs187429064:G in TM6SF2 is associated with an increased risk of HCC in patients with cirrhosis.

Abbreviations
- HU
Hounsfield unit
- APOA/B/E
apolipoprotein A/B/E
- ArLD
alcohol‐related liver disease
- AST
aspartate aminotransferase
- BMI
body mass index
- CI
confidence interval
- CVD
cardiovascular disease
- FE
finite element
- GPAM
glycerol‐3‐phosphate acyltransferase
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HSD17B13
17‐β hydroxysteroid dehydrogenase 13
- ICD‐9/10
International Classification of Diseases, Ninth/Tenth Revision
- LDLR
lipoprotein receptor
- LOR
log odds ratio
- MARC1
mitochondrial amidoxime reducing component 1
- NAFLD
nonalcoholic fatty liver disease
- OR
odds ratio
- TM6SF2
transmembrane 6 superfamily member 2
- UKB
United Kingdom Biobank
Hepatocellular carcinoma (HCC) is the third most common type of cancer death, responsible for approximately 800,000 deaths globally every year worldwide.( 1 ) Most cases of HCC develop against a background of advanced liver fibrosis and cirrhosis. Like any cancer, HCC is a product of somatic mutations acquired in pivotal driver genes but also influenced by germline (i.e., constitutional) polymorphisms modifying the susceptibility to developing HCC.( 2 ) Thus far, several such polymorphisms have been identified and robustly validated, including rs738409 in patatin‐like phospholipase domain containing 3 (PNPLA3), rs58542926 in transmembrane 6 superfamily member 2 (TM6SF2), and rs72613567 in 17‐β hydroxysteroid dehydrogenase 13 (HSD17B13).( 3 , 4 , 5 , 6 ) However, this explains only part of the host genetic background underlying HCC development. A more complete understanding of the constitutional genetic polymorphisms that predispose patients to HCC could herald several important advancements. In the short term for example, it could support risk stratification of patients with cirrhosis with respect to HCC screening decision( 7 ); in the longer term, it could guide the discovery of chemoprevention agents if any of the risk loci prove to be “druggable.”
In a recent exome association study, Jamialahmadi et al.( 8 ) identified three novel missense variants associated with hepatic fat content. These variants were rs429358 in apolipoprotein E (APOE, rs2792751 in glycerol‐3‐phosphate acyltransferase (GPAM), and rs187429064 in TM6SF2, where the latter is in complete linkage equilibrium with the better known rs58542926 locus. They also show that the rs2642438 missense variant in mitochondrial amidoxime reducing component 1 (MARC1), which we and others have recently identified as a risk factor for cirrhosis,( 9 , 10 ) is associated with liver fat content, too. In a parallel study, Bianco et al.( 11 ) indicated that higher liver fat content is causally associated with HCC occurrence. On that basis, genetic factors that alter liver fat content may also alter HCC risk; these variants therefore warrant exploration in candidate gene‐association studies for HCC. To that end, our primary objective was to explore a possible association of each of these four aforementioned variants with HCC across a variety of large data sets and etiologies.
Materials and Methods
Scientific Approach
This study uses data from the following four HCC case‐control data sets: two mixed etiology cohorts (United Kingdom Biobank [UKB] and FinnGen), one hepatitis C virus (HCV) cohort (STOP‐HCV), and one alcohol‐related liver disease (ArLD) cohort (Dresden study).
The following four candidate variants were considered for association with HCC: 1) rs429358 (APOE), 2) rs2792751 (GPAM), 3) rs2642438 (MARC1), and 4) rs187429064 (TM6SF2). Genotyping methods for these variants are described in Supporting Materials Appendix A.
In the broadest terms, our goal was to assess if the frequency of these variants was different for HCC cases versus non‐HCC controls. Two types of non‐HCC controls were considered, patients with cirrhosis without HCC and population controls without HCC. On one hand, comparing HCC cases to cirrhosis controls is essential to eliminate confounding, i.e., because variants associated with HCC tend also to be associated with progression to cirrhosis. On the other hand, population controls lend insight because HCC can also arise in patients at a precirrhosis stage (i.e., particularly nonalcoholic fatty liver disease [NAFLD]‐related HCC).( 12 , 13 ) A population perspective is also relevant to early detection case‐finding initiatives for HCC.( 14 )
Case‐Control Data Sets
UK Biobank
The UKB is a cohort of half a million middle‐aged individuals from the United Kingdom, recruited in 2006‐2010. Blood specimens donated at enrollment have been used to characterize participants in terms of genetic factors as well as being serum biomarkers (e.g., alanine aminotransferase). Participant data are also linked to UK health registries to capture medical presentations occurring both before and after enrollment.( 15 )
This study was restricted to UKB participants of White British ancestry (UKB field ID: 22006). We then excluded those with a poor quality genetic sample (defined by UKB field ID: 22027) or who were related to another participant (inferred by a kinship coefficient ≥0.1). Cases were participants with a history of HCC, defined as a hospital admission, death, or cancer registration with HCC (International Classification of Diseases, Tenth Revision [ICD‐10]: C22.0, or ICD‐9: 155.0), either before or after UKB enrollment. Liver disease etiology for the HCC cases was estimated using a hierarchical definition of a) viral hepatitis, b) autoimmune liver disease in the absence of a, c) ArLD in the absence of a‐b, d) NAFLD in the absence of a‐c, and e) other/unknown in the absence of a‐d. Risk factors for these etiologies were discerned through a combination of hospital admissions and/or information reported during the UKB enrollment interview (Supporting Table S1).
The following two control groups were considered: ontrol group 1 included UKB participants with a hospital admission for liver cirrhosis but without a history of HCC. Hospital admissions due to cirrhosis were identified using a validated set of ICD and operation/procedure codes( 16 ) (See Supporting Table S2 for further details.) Control group 2 included all UKB participants without a history of HCC. The vast majority of individuals in this group had no history of chronic liver disease. Control group 2 was broadly equivalent to a general population control group.
FinnGen
FinnGen is a public–private partnership project, combining genotyping data from Finnish biobanks with electronic health record data derived from national health registries. Genome‐wide association study (GWAS) summary statistics for more than 1,800 phenotypes/endpoints, including for primary liver cancer, have been publically released.
For this study, we used the latest R4 data released (published November 2020) pertaining to a sample size of 176,899 individuals.( 17 ) Cases were individuals with a history/diagnosis of primary liver cancer (ICD‐10: C22 and ICD‐9: 155), whereas controls were all individuals without a diagnosis of primary liver cancer. Similar to the UKB control group 2, this largely comprised individuals without any preexisting liver disease. GWAS summary statistics relating specifically to HCC were not available.
STOP‐HCV Cirrhosis Study
The STOP‐HCV cirrhosis study comprised approximately 1,200 patients with hepatitis C‐related cirrhosis. Participants were recruited from 31 specialist liver clinics in the United Kingdom between January 2015 and July 2016. Cirrhosis was defined through histologic assessment, imaging, or a validated serum biomarker consistent with liver cirrhosis (i.e., aspartate aminotransferase [AST]‐to‐platelet ratio index >2, FibroTest >0.73, or enhanced liver fibrosis score >10.48). Blood specimens collected at enrollment were used to generate host‐genotyping data through the Affymetrix UK Biobank array. Furthermore, participants from England have been linked to national hospital admission, cancer registrations, and mortality data.
The present analysis was restricted to participants from England (i.e., to ensure complete data on hospital admissions, cancer registrations, and mortality) and participants of White ethnicity. As with the UKB, cases were defined on the basis of an in‐patient hospital admission, death, or cancer registration indicating HCC (ICD‐10: C22.0; ICD‐9: 155.0) before or after study enrollment. Controls were all participants without a history of HCC.
Dresden Alcohol HCC Cohort
The Dresden HCC cohort included 2,311 patients with a history of high‐risk alcohol consumption in whom nonalcohol‐related causes of chronic liver disease had been excluded. Patients were recruited from gastroenterology and hepatology hospitals across five European countries (Austria, France, Germany, Italy, and Switzerland). For this study, cases were patients with a diagnosis of HCC determined through histologic and/or imaging (computed tomography or magnetic resonance imaging [MRI]) investigations.
As with the UKB, two control groups were considered. Control group 1 was individuals diagnosed with alcohol‐related cirrhosis but without a history of HCC. Control group 2 comprised patients without cirrhosis.
The diagnosis of alcohol‐related cirrhosis was established as described in detail.( 18 ) Briefly, the diagnosis was based on a history of prolonged sustained alcohol intake of a minimum of 40 g/day in women and 60 g/day in men, together with histologic examination of liver tissue or compatible historical, clinical, laboratory, radiologic, and endoscopic features of advanced chronic liver disease. Patients were excluded if they had any other potential cause of liver injury, specifically if they were positive for hepatitis B surface antigen, anti‐HCV, antinuclear antibodies (titer >1:80), or antimitochondrial antibodies (titer >1:40). Patients with elevated serum ferritin concentrations and a transferrin saturation >50%, a serum ceruloplasmin concentration <20 mg/dL (0.2 g/dL), or a serum alpha‐1 antitrypsin concentration <70 mg/dL (13 µmol/L) were further investigated and excluded, as appropriate. The diagnosis of HCC was based on histologic examination of tumor tissue or evidence on imaging, preferably using two modalities, of lesions that were hypervascular in the arterial phase with washout in the portal venous or delayed phases.( 19 )
Patients with alcohol misuse but no evidence of cirrhosis (control group 2) were recruited as described in detail.( 15 ) In brief, these patients had a background of alcohol consumption of at least 60 g/day for ≥10 years with or without features of alcohol dependence( 20 ); none had historical, clinical, or laboratory evidence of cirrhosis as reflected by AST‐adapted cut‐off values for liver stiffness measured by transient elastography, as described.( 21 )
All participants from the Dresden cohort were of Caucasian ancestry, and genotyping was performed using the Illumina BeadChip array (see Supporting Materials Appendix A). The study protocol was approved by the ethics committees of the participating institutions, and all patients provided written informed consent before study inclusion.
Data Analysis
Association With Liver Fat Content
We started by replicating the UKB association between each candidate variant and liver fat fraction, as reported by Jamialahmadi et al.( 8 ) This allowed us to compare each variant’s direction of association with liver fat content with the direction of association for HCC. Liver fat fraction was measured through MRI, which at the time of analysis was available for a subset of 9,893 participants (UKB Field ID: 22436). We performed log10 transformation on this variable to achieve approximate normality. Covariate adjustment was included for body mass index (BMI), age, sex, and the top five principal components of genetic ancestry. The analysis was restricted to participants in the White British ancestry subset (UKB Field ID: 22006).
Association With HCC
For each candidate variant, we computed the simple minor allele frequencies (MAFs) in cases and controls from all four cohorts. The association between each candidate variant and HCC was then quantified through multivariate logistic regression. All associations were adjusted for age, sex, and the top principal components of genetic ancestry. However, there were minor differences by data set, which are outlined in Supporting Table S3. We did not control for established HCC risk variants (i.e., rs738409 in PNPLA3, rs58542926 in TM6SF2, and rs72613567 in HSD17B13) because these were all in linkage equilibrium with the candidate variants considered (i.e., R 2 < 0.001). The exception to this was rs187429064 where we included adjustment for the rs58542926 genotype out of prudence, given that both variants lie in TM6SF2. All associations were calculated under an additive genetic model, with two‐tailed P values presented.
We then performed a fixed‐effect meta‐analysis to determine a pooled effect size across studies, using the METAL software package.( 22 ) Two pooled effect sizes were calculated. First, a pooled effect size specific to cirrhosis controls (i.e., UKB controls 1+Dresden controls 1+STOP‐HCV). Second, an overall effect size specific to population controls (i.e., UKB controls 1+FinnGen). All meta‐analyses were weighted according to the effective sample size, defined according to the formula 4/(1/number of cases + 1/number of controls). A Bonferroni‐corrected P < 0.0125 was used to judge statistical significance.
Associations were expressed either in terms of log odds ratio (LOR) or odds ratios (ORs), where the latter is simply the exponent of the LOR. In graphical figures, we present associations in terms of their LORs because these are symmetrical around the null and thus allow one to visually compare magnitude of associations for variants that affect risk in opposing directions. Various polygenic risk scores were also created, and their association with HCC was quantified (see Supporting Materials Appendix B).
Results
Association With Liver Fat Content
All four variants were strongly associated with liver fat content, with P values ranging from 2.1 × 10−6 to 3.7 × 10−10 (see Fig. 1). Two of the four variants were associated with reduced liver fat content (rs429358:C in APOE and rs2642438:A in MARC1), whereas two variants were associated with increased liver fat content (rs2792751:T in GPAM and rs187429064:G in TM6SF2). The rs187429064:G variant exhibited the strongest effect size (beta, 0.29), followed by rs429358:C (beta, −0.09), then rs279275:T (beta, 0.06), and then rs2642438:A (beta, −0.05).
FIG. 1.
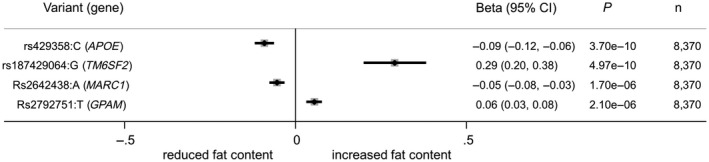
Association of candidate variants with liver fat content in the UKB study.
Case‐Control Data
In total, the four case‐control data sets included 2,070 HCC cases and 4,121 cirrhosis controls. Over half the cases were alcohol‐related HCCs from the Dresden study (n = 1,289), and 149 were hepatitis C‐related HCCs from the STOP‐HCV study. There were 366 HCC cases identified from the UKB study. Of these, we estimate that 153 (43%) were related to NAFLD, 115 (31%) related to ArLD, and 29 (9%) related to viral hepatitis. Cases were largely men (73%‐91%), with a mean age ranging from 60 to 69 years depending on the study (Table 1).
TABLE 1.
Summary of the case‐control data sets used in this study
| Data Source | Cohorts | Characteristic | Minor Allele Frequency (%) | |||||
|---|---|---|---|---|---|---|---|---|
| Number* | Mean Age, Years | Sex (% Men) | rs429358 C (APOE) | rs2792751 T (GPAM) | rs2642438 A (MARC1) | rs187429064 G (TM6SF2) | ||
| UKB | Cases: HCC | 366 | 62.1 | 77 | 10.8 | 29.1 | 25.6 | 3.6 |
| Controls 1: hospital admission for cirrhosis without HCC | 2,536 | 59.3 | 63 | 13.9 | 28.2 | 28.1 | 1.3 | |
| Controls 2: all UKB participants without HCC† | 3,49,018 | 57.5 | 47 | 15.6 | 27.4 | 29.7 | 1.1 | |
| FinnGen | Cases: primary liver cancer | 266 | 68.9 | 74 | 12.4 | 33.0 | 25.8 | 12.5 |
| Controls: all participants without primary liver cancer † | 1,76,633 | NK | NK | 18.5 | 31.9 | 28.4 | 5.1 | |
| Dresden alcohol cohort | Cases: HCC and alcohol‐related cirrhosis | 1,289 | 65.0 | 91 | 8.9 | 33.6 | 25.4 | 2.4 |
| Controls 1: alcohol‐related cirrhosis without HCC | 894 | 57.1 | 75 | 11.8 | 32.0 | 26.4 | 1.3 | |
| Controls 2: heavy drinkers with neither significant liver disease nor HCC | 128 | 60.6 | 70 | 14.6 | 32.2 | 31.1 | 0.8 | |
| STOP‐HCV | Cases: HCC and hepatitis C‐related cirrhosis | 149 | 60.3 | 73 | 9.4 | 28.9 | 24.8 | 1.7 |
| Controls: hepatitis C‐related cirrhosis without HCC | 691 | 55.8 | 77 | 13.0 | 29.9 | 29.7 | 0.9 | |
Number of cases indicated here may differ from the number used in regression analyses due to missing data for genotype and/or age, and/or sex.
Control group largely comprises individuals with no history of liver disease.
Of the cirrhosis controls, 691 were from STOP‐HCV, 2,536 from the UKB, and 894 from Dresden. Cirrhosis controls were again predominantly men (63%‐77%) but were younger than HCC cases (mean age ranging from 55 to 59 years). The UKB and FinnGen non‐liver disease control groups comprised 349,018 and 176,633 FinnGen individuals, respectively (Table 1). Of the 128 noncirrhotic controls from the Dresden cohort, 50%, 40%, and 10% were estimated to be at Metavir stage F0, F1‐2, and F3, respectively, based on AST‐adapted liver stiffness cutoffs.
In the STOP‐HCV cohort, about one fifth (19.6%) had achieved a hepatitis C sustained viral response at the time of study enrollment. This proportion was comparable for HCC cases (20.8%) and cirrhosis controls (19.4%).
Association With HCC
APOE (rs429358)
The APOE rs429358:C allele was consistently less frequent in cases versus controls across all data sets. For example, 10.8% in UKB cases versus 13.9% in non‐HCC controls (Table 1). In multivariate regression, rs429358:C was independently associated with a reduced HCC risk across all comparisons and data sets. The pooled OR for each copy of the rs429358:C allele was 0.71 (95% confidence interval [CI], 0.61‐0.84; P = 2.9 × 10−5) against cirrhosis controls and 0.66 (95% CI, 0.57‐0.78; P = 1.0 × 10−6) against population controls (see Fig. 2).
FIG. 2.
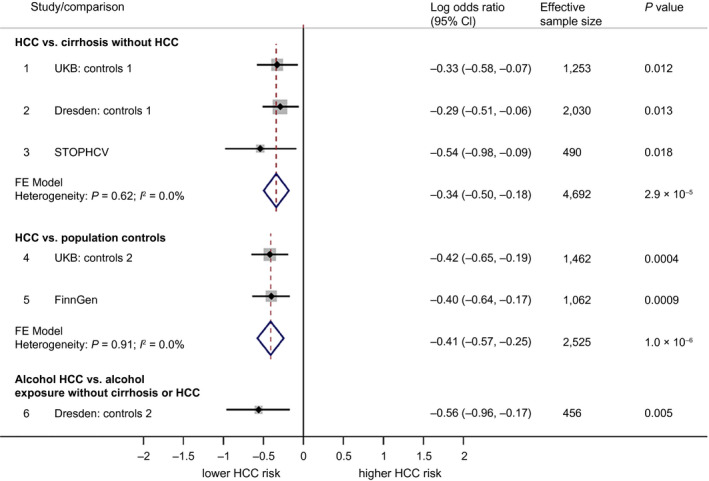
Forest plot showing association between rs429358:C (APOE) and HCC. Associations are broken down into the following three categories: 1) comparing HCC to cirrhosis controls without HCC, 2) comparing HCC to population controls (who for the most part will not have liver disease), and 3) comparing alcohol HCC to individuals with an alcohol exposure but without cirrhosis or HCC. Associations are presented in terms of the LOR. An LOR of 0 indicates that the frequency of rs429358:C is the same for cases as for controls. LORs were calculated using logistic regression under an additive genetic model. Pooled effects are based on fixed‐effect meta‐analysis, weighted by effective sample size.
GPAM (rs2792751)
The GPAM rs2792751:T allele was generally higher in HCC cases versus controls. However, the differences were modest; for example, 33.6% in Dresden HCC cases versus 32.0% in cirrhosis controls. In multivariate regression, the associations were not significant. The pooled OR for each copy of the rs2792 T allele was 1.01 (95% CI, 0.90‐1.13; P = 0.89) against cirrhosis controls and 1.04 (95% CI, 0.91‐1.17; P = 0.55) against population controls (see Fig. 3).
FIG. 3.
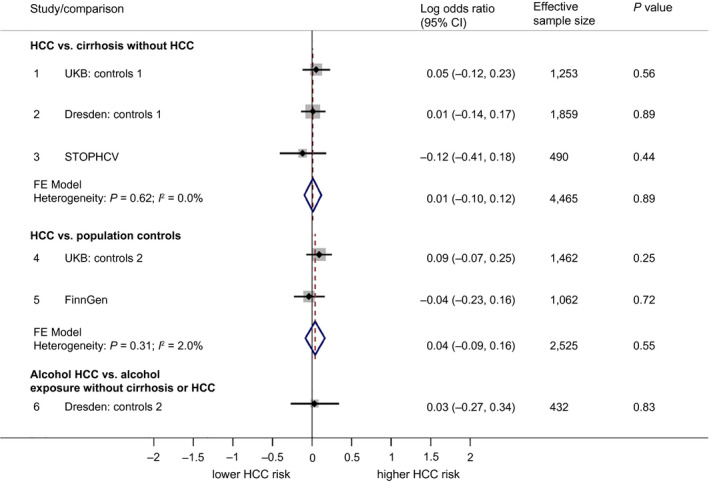
Forest plot showing association between rs2792751:T (GPAM) and HCC. Associations are broken down into the following three categories: 1) comparing HCC to cirrhosis controls without HCC, 2) comparing HCC to population controls (who for the most part will not have liver disease), and 3) comparing alcohol HCC to individuals with an alcohol exposure but without cirrhosis or HCC. Associations are presented in terms of the LOR. An LOR of 0 indicates that the frequency of rs2792751:T is the same for cases as for controls. LORs were calculated using logistic regression under an additive genetic model. Pooled effects are based on fixed‐effect meta‐analysis, weighted by effective sample size.
MARC1 (rs2642438)
The rs2642438:A variant in MARC1 was consistently less frequent in HCC cases versus controls. For example, 24.8% for HCC cases in STOP‐HCV versus 29.7% in controls. In regression analysis, the association was relatively weak. The pooled OR for each copy of the rs2642438:A allele was 0.91 (95% CI, 0.84‐1.00; P = 0.043) against cirrhosis controls and 0.83 (95% CI, 0.72‐0.95; P = 0.006) against population controls (see Fig. 4).
FIG. 4.
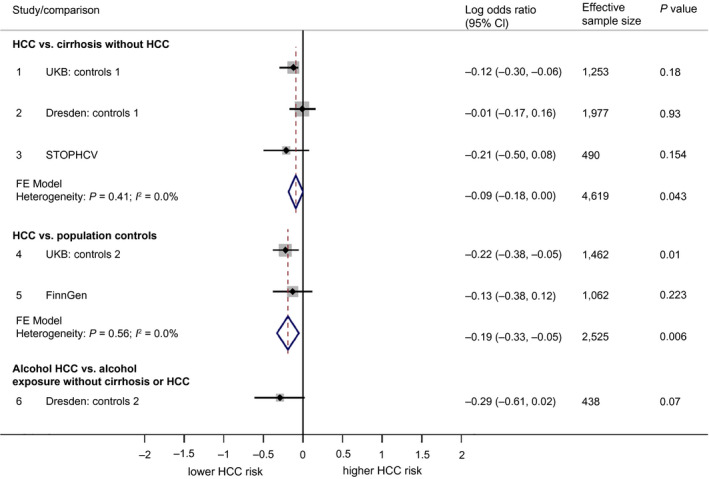
Forest plot showing association between rs2642438:A (MARC1) and HCC. Associations are broken down into the following three categories: 1) comparing HCC to cirrhosis controls without HCC, 2) comparing HCC to population controls (who for the most part will not have liver disease), and 3) comparing alcohol HCC to individuals with an alcohol exposure but without cirrhosis or HCC. Associations are presented in terms of the LOR. An LOR of 0 indicates that the frequency of rs2642438:A is the same for cases as for controls. LORs were calculated using logistic regression under an additive genetic model. Pooled effects are based on fixed‐effect meta‐analysis, weighted by effective sample size.
TM6SF2 (rs187429064)
The rs187429064:G variant in TM6SF2 was consistently higher in cases versus controls. For example, 3.6% in UKB cases versus 1.1% in all UKB controls without HCC. In regression analysis, rs187429064:G was associated with a higher HCC risk across all comparison and data sets. The pooled OR for each copy of the rs187429064 G allele was 2.03 (95% CI, 1.45‐2.86; P = 3.1 × 10−6) against cirrhosis controls and 3.86 (95% CI, 2.80‐5.31; P = 7.6 × 10−17) against population controls (see Fig. 5). Genotypic ORs were also calculated for each candidate variant and were generally consistent with allelic ORs (see Supporting Figs. S1‐S4).
FIG. 5.
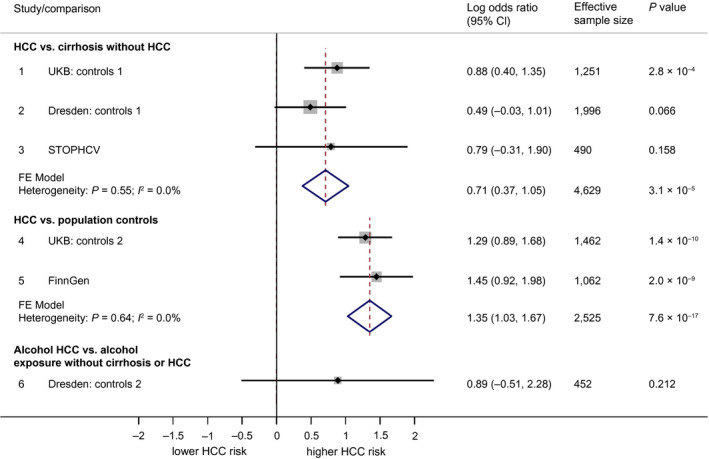
Forest plot showing association between rs187429064:G (TM6SF2) and HCC. Associations are broken down into the following three categories: 1) comparing HCC to cirrhosis controls without HCC, 2) comparing HCC to population controls (who for the most part will not have liver disease), and 3) comparing alcohol HCC to individuals with an alcohol exposure but without cirrhosis or HCC. Associations are presented in terms of the LOR. An LOR of 0 indicates that the frequency of rs187429064:G is the same for cases as for controls. LORs were calculated using logistic regression under an additive genetic model. Pooled effects are based on fixed‐effect meta‐analysis, weighted by effective sample size.
APOE Exploratory Analysis
We performed three exploratory analyses to better understand the association between rs429358 in APOE and HCC. First, we tested for interaction between rs429358:C and polymorphisms in the low‐density lipoprotein receptor (LDLR) gene. From the UKB genetic data set, 144 polymorphisms in LDLR with a MAF > 1% that did not violate the assumption of Hardy‐Weinberg equilibrium were extracted. Rs429358:C epistasis with each of these 144 polymorphisms was assessed in our UKB case‐control data set. The lead variant from this scan was rs1569372 (Supporting Figs. S5 and S6), indicating the rs429358:C association with HCC was stronger in the presence of the rs1569372 G allele (P for interaction = 0.020). However, this interaction did not replicate in the Dresden and STOP‐HCV cohorts (see Supporting Fig. S7).
Second, to generate insight into the underlying biological mechanisms, we assessed attenuation in the rs429358:C–HCC association following adjustment for selected biomarkers and/or detailed confounding factors (UKB data set only). The factors considered in this analysis were total cholesterol (Field ID: 30690), high‐density lipoprotein cholesterol (HDL‐C) (Field ID: 30760), low‐density lipoprotein cholesterol (LDL‐C) (Field ID: 30780), lipoprotein A (Field ID: 30790), tyriglycerides (Field ID: 30870), APOA (Field ID: 30630), APOB (Field ID: 30640), glycated hemoglobin A1c (Field ID: 30750), BMI (Field ID: 21001), C‐reactive protein (Field ID: 30710), statin therapy (Field ID: 20003), telomere length (Field ID: 22191), and current tobacco smoking (Field ID: 20116). All factors/biomarkers were measured at the time of UKB study recruitment. Attenuation in the rs429358–HCC association was modest/negligible for the majority of factors. The strongest attenuation occurred in relation to lipoprotein A, where the LOR attenuated to −0.19 (95% CI, −0.49 to 0.10; P = 0.20) (Supporting Figs. S8 and S9).
Third, using haplotype data from the UKB genetic data set, we characterized participants according to ε2, ε3, and ε4 APOE alleles( 23 ) and assessed the association of these alleles with HCC. Relative to ε3‐ε3, the ε4‐ε4 haplotype was associated with the greatest effect size (OR, 0.61; 95% CI, −0.23 to 0.64; P = 0.30), followed by ε3‐ε4 (OR, 0.68; 95% CI, 0.51‐0.93; P = 0.015), followed by ε2‐ε4 (OR, 0.72; 95% CI, 0.32‐1.62; P = 0.42) (Supporting Figs. S10 and S11). In this vein, we also assessed the relationship between rs7412 in APOE and case‐control status directly and did not find any significant association (Supporting Fig. S12).
Discussion
Although HCC is a leading cause of cancer mortality, the genetic factors that predispose individuals to this outcome are not fully understood. In this study, we highlight the importance of two (hitherto unrecognized) germline genetic polymorphisms with respect to HCC risk. These variants were rs429358:C in APOE, which associates with a lower risk of HCC, and rs187429064:G in TM6SF2, which associates with a higher HCC risk. Each variant exhibited a consistent trend across all data sets, and their direction of association with HCC mirrors their direction of association with hepatic fat content.
The relevance of rs2642438:A allele in MARC1 is more equivocal. Although this variant was less frequent among HCC cases in some data sets, the effect size in the pooled analysis was relatively modest and did not quite reach statistical significant when comparing against cirrhosis controls (P = 0.02). Conversely, the association between rs2792751:T in GPAM and HCC was consistently close to the null, suggesting it is not a relevant risk factor for HCC, despite its strong association with hepatic fat content.
APOE is found at the surface of lipoprotein particles and plays a pivotal role in lipid transport to and from the liver.( 23 ) Rs429358:C is a coding variant that leads to a cysteine to arginine replacement at position 112/317 of the APOE protein. This polymorphism is notorious for its adverse effect on Alzheimer’s disease (AD), yet it also modulates the risk of cardiovascular disease (CVD) and other health outcomes.( 24 , 25 ) Previous studies have shown that rs429358:C is associated with a reduced risk of cirrhosis in HCV,( 25 ) NAFLD,( 8 ) and in mixed‐etiology population cohorts.( 26 , 27 ) Our results extend this narrative by demonstrating that rs429358:C is also associated with a reduced risk of HCC in HCV cirrhosis, alcohol‐related cirrhosis, and in a mixed‐etiology cirrhosis cohort (i.e., UKB). The persistence of this association when comparing against cirrhosis controls strongly implies that APOE has a direct role in liver carcinogenesis and does not simply reduce HCC by altering progression to cirrhosis. Nevertheless, the specifics of what this direct role could be are unclear. It has been known for some time, that rs429358:C is associated with higher serum cholesterol levels.( 28 ) More recently, Qin et al.( 29 ) demonstrated that inducing higher serum cholesterol in mice (both through diet and genetic disruption of the ApoE gene) leads to enhanced HCC suppression after injection with a chemical carcinogen. The authors indicate that higher serum cholesterol may increase the cancer immunosurveillance activity of natural killer cells. Consistent with this, they also reported a correlation between the serum cholesterol and natural killer cell activity in human HCC tissue. Thus, an obvious question is whether the rs429358–HCC association is merely a corollary for differences in serum cholesterol levels according to rs429358 genotype. However, there might be additional molecular aspects involved because only modest levels of attenuation in the rs429358–HCC association were observed when adjusting for HLD‐C, LDL‐C, total cholesterol, and broader measures of dyslipidemia. We also investigated if the rs429358–HCC association varied according to polymorphisms in LDLR because interaction between loci in these genes has been observed for AD and CVD( 30 , 31 , 32 ) and because APOE is a ligand for the LDLR.( 33 ) Our results from this analysis suggest that the rs429358–HCC association may be stronger for carriers of the rs73015034:C allele in LDLR. However, further studies are needed to explore this conceivable functional link in greater detail.
We also investigated the association between the ε2, ε3, and ε4 APOE alleles and HCC, where ε2, ε3, and ε4 are determined by genotype at rs429358 and rs7412 loci. As expected, this analysis showed that the ε4 allele, defined by the presence of the rs429358:C and rs7412:C allele on the same copy of chromosome 19, was less frequent in HCC cases versus controls. This begs the question of whether it is the ε4 haplotype (i.e., combination of rs429358:C and rs7412:C on the same chromosome) or rs429358:C alone that drives the protective effect. However, because the two are effectively synonymous, i.e., the overwhelming majority of individuals with rs429358:C also carry rs7412:C on that same chromosome, it is difficult to disentangle the effect of one from the other.
This study also identifies a missense variant in TM6SF2 (rs187429064:G) as being associated with HCC among patients with cirrhosis. TM6SF2 has been widely studied in connection to rs58542926:T, another missense variant that is itself associated with HCC and also liver cirrhosis( 4 , 6 , 15 , 34 ) but interestingly protects from CVDs.( 35 ) Previous work indicates that loss of TM6SF2 function increases hepatocyte fat content by reducing APOB secretion.( 36 ) This is consistent with a study by Pelusi et al.( 37 ) showing that individuals with rare pathogenic variants in APOB are at increased risk of HCC. The present data therefore corroborate the importance of TM6SF2 in relation to HCC oncogenesis. The association we observed between the rs187429064 locus and HCC cannot be explained in terms of confounding by the rs58542926 genotype because our regression models included adjustment for rs58542926, and in any case, rs58542926 and rs187429064 genotype status are not correlated with one another in Europeans (R 2 = 0.0009). At the protein level, rs187429064:G results in a lysine to arginine substitution at position 156/377. Although the frequency of this variant is relatively rare in Europeans (allele frequency ~1%), it can vary widely from one population to another. For example, in the FinnGen cohort, rs187429064:G has an allele frequency of 5.1%, which is comparable to rs58542926:T.
Our study has a number of strengths. First, despite HCC being a relatively rare outcome, we have succeeded in assembling a large sample size with over 2,000 HCC cases. This has enabled us to generate precise effect‐size estimates for each variant. A second strength is our inclusion of both cirrhosis and population control groups, each of which complements the other. Third, we were able to draw on data from a variety of cohorts and etiologies. This affords us a level of confidence regarding the generalizability of our findings to other settings and patient groups.
A limitation of this study is that we did not have access to an equivalent case‐control data set for NAFLD, and thus we cannot say if our findings extend to this etiology specifically. Second, our base case analysis included adjustment for only age, sex, and principal components of genetic ancestry. Ideally, we would have adjusted for smoking, BMI, diabetes, and statin use, but these covariates were missing for a substantial proportion of patients in the STOP‐HCV and Dresden cohorts. However, we did perform an analysis to assess attenuation of the rs429358–HCC association following adjustment for statin use, smoking, BMI and other covariates. The level of attenuation observed was marginal, suggesting that the rs429358–HCC association cannot be explained in terms of simple confounding. Another limitation relates specifically to the FinnGen data, where the outcome event considered was primary liver cancer as opposed to HCC. However, we do not think this is likely to have exerted much bias on our results given that HCC accounts for the large majority of primary liver cancer cases. Finally, this study was restricted to individuals of Caucasian ethnicity in order to circumvent confounding by population structure. We do not know if these variants are relevant to HCC risk for individuals in other ethnic groups.
Overall, this study has helped to further elucidate the genetic background to HCC by showing that two variants, one in APOE and a second missense variant in TM6SF2, are associated with HCC across a variety of data sets and etiologies. These associations have not previously been identified or recognized hitherto. Also, despite a strong association with liver fat content, the rs2792751:T missense variant in GPAM does not appear to influence HCC risk. These findings will help fine‐tune emerging HCC risk stratification tools and allow greater insight into currently unknown molecular aspects of HCC oncogenesis.
Supporting information
Supplementary Material
Acknowledgment
This research has been conducted using the UKB Resource (application number 8764). We also acknowledge the participants and investigators of the FinnGen study. We thank participants of the STOP‐HCV cirrhosis study and STOP‐HCV Consortium members, who are as follows: Barnes E. (University of Oxford), Ball J.K. (University of Nottingham), Brainard D. (Gilead Sciences), Burgess G. (Conatus Pharmaceuticals), Cooke G. (Imperial College, London), Dillon J. (University of Dundee), Foster G.R. (Queen Mary University of London), Gore C. (Hepatitis C Trust), Guha N. (University of Nottingham), Halford R. (Hepatitis C Trust), Whitby K. (Gilead Sciences), Holmes C. (University of Oxford), Howe A. (British Columbia Centre for Excellence), Hudson E. (University of Oxford), Hutchinson S. (Glasgow Caledonian University), Irving W.L. (University of Nottingham), Khakoo S. (University of Southampton), Klenerman P. (University of Oxford), Martin N. (University of California San Diego), Massetto B. (Gilead Sciences), Mbisa T. (Public Health England), McHutchinson J. (Gilead Sciences), McKeating J. (University of Oxford), McLauchlan J. (Medical Research Council [MRC]‐University of Glasgow, Centre for Virus Research), Miners A. (London School of Hygiene and Tropical Medicine), Murray A. (OncImmune Limited), Shaw P. (Merck & Co.), Simmonds P. (University of Oxford), Spencer C. (Wellcome Trust), Thomson E. (MRC‐University of Glasgow, Centre for Virus Research), Vickerman P. (University of Bristol), Zitzmann N. (University of Oxford). We thank STOP‐HCV participating sites and Principal Investigators, who are as follows: Agarwal K. (Kings College Hospital, London), Aldersley M. (St James’s University Hospital, Leeds), Aspinall R. (Queen Alexandra Hospital, Portsmouth), Barclay S. (Glasgow Royal Infirmary), Barnes E. (John Radcliffe Hospital, Oxford), Benselin J. (University of Nottingham), Brown A. (St Mary’s Hospital, London), Ch’ng C. (Singleton Hospital, Swansea), Corless L. (Hull Royal Infirmary), Cramp M. (Derriford Hospital, Plymouth), Dillon J. (Ninewells Hospital, Dundee), English S. (Aberdeen Royal Infirmary); Forton D. (St George’s Hospital, London), Foster G. (The London Hospital), Fraser A. (Aberdeen Royal Infirmary), Gelson W. (Addenbrookes Hospital, Cambridge), Gorard D. (Wycombe Hospital), Gordon F. (Bristol Royal Infirmary), Kennedy N. (Monklands Hospital), Knowles J. (James Cook University Hospital, Middlesbrough), Leen C. (Western General Hospital, Edinburgh), McPherson S. (Freeman Hospital, Newcastle), Moreea S. (Bradford Teaching Hospitals National Health Service Foundation Trust), Mutimer D. (Queen Elizabeth Hospital, Birmingham), Prince M. (Manchester Royal Infirmary), Richardson P. (Royal Liverpool University Hospital), Rosenberg W. (Royal Free Hospital and University College Hospital), Ryder S. (Queen’s Medical Centre, Nottingham), Kara Rye (Royal Shrewsbury Hospital), Stone B. (Royal Hallamshire Hospital, Sheffield), Thursz M. (St Mary’s Hospital, London), Ustianowski A. (North Manchester General Hospital), Verma S. (Royal Sussex County Hospital, Brighton), Wiselka M. (Leicester Royal Infirmary).
Supported by the Medical Research Foundation (Viral Hepatitis Fellowship grant C0825 to H.I.), Swiss National Funds (No. 310030_169196 to F.S.), Swiss Foundation for Alcohol Research (No. 261/15 to F.S.), German Federal Ministry for Education and Research, Liver Systems Medicine Network (No. 031L0031 to J.H.), Medical Research Council (No. MR/K01532X/1 to B., Clinician Scientist Fellowship grant MR/P008348/1 to J.R.M, No. MC_UU_12014/1 to J.M.), Medical Research Foundation (No. C0365 to J.M.), Deutsche Krebshilfe (No. 70112169 to H.D.N.), European Commission European Funds for Regional Development and Regional Ministry of Economy, Science, and Digitalization (No. ZS/2018/11/95324 to A.L.), Deutsche Forschungsgemeinschaft (SFB TRR57 to P18, CRC 1382 A09 to J.T.), European Union’s Horizon 2020 Research and Innovation Programme (Galaxy, No. 668031 to J.T.; MICROB‐PREDICT, No. 825694 to J.T.; DECISION, No. 84794 to J.T.; No. 731875 to J.T.; No. 777377 to L.V.; Photonics, No. 101016726 to L.V.; and Gilead_IN‐IT‐989‐5790 to L.V.), Cellex Foundation (PREDICT to J.T.), MyFirst Grant AIRC (No. 16888 to L.V.), Ministero della Salute (No. RF‐2016‐02364358 to L.V.), Fondazione IRCCS (No. PR‐0391 to L.V., No. RC100017A to L.V.), and Cancer Research UK (No. C30358/A29725 to E.B.).
The views expressed in this article are those of the authors and not necessarily those of the National Health Service, the NIHR, or the Department of Health. The funders had no involvement in the study design; collection, analysis and interpretation of data; writing of the report; and decision to submit the article for publication.
Potential conflict of interest: Dr. Trebicka has received speaking and/or consulting fees from Gore, Bayer, Alexion, MSD, Gilead, Intercept, Norgine, Grifols, Versantis, and Martin Pharmaceutical. Dr. Valenti has received speaking fees from MSD, Gilead, AlfaSigma, and AbbVie; he has served as a consultant for Gilead, Pfizer, AstraZeneca, Novo Nordisk, Intercept, Diatech Pharmacogenetics, and Ionis Pharmaceuticals and received research grants from Gilead. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Asrani SK, Devarhbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70:151‐171. [DOI] [PubMed] [Google Scholar]
- 2. Muller M, Bird TG, Nault JC. The landscape of gene mutations in cirrhosis and hepatocellular carcinoma. J Hepatol 2020;72:990‐1002. [DOI] [PubMed] [Google Scholar]
- 3. Stickel F, Lutz P, Buch S, Nischalke HD, Silva I, Rausch V, et al. Genetic variation in HSD17B13 reduces the risk of developing cirrhosis and hepatocellular carcinoma in alcohol misusers. Hepatology 2020;72:88‐102. [DOI] [PubMed] [Google Scholar]
- 4. Stickel F, Buch S, Nischalke HD, Weiss KH, Gotthardt D, Fischer J, et al. Genetic variants in PNPLA3 and TM6SF2 predispose to the development of hepatocellular carcinoma in individuals with alcohol‐related cirrhosis. Am J Gastroenterol 2018;113:1475‐1483. Erratum in: Am J Gastroenterol 2018;113:1099. [DOI] [PubMed] [Google Scholar]
- 5. Trepo E, Romeo S, Zucman‐Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol 2016;65:399‐412. [DOI] [PubMed] [Google Scholar]
- 6. Stickel F, Moreno C, Hampe J, Morgan MY. The genetics of alcohol dependence and alcohol‐related liver disease. J Hepatol 2017;66:195‐211. [DOI] [PubMed] [Google Scholar]
- 7. Kanwal F, Singal AG. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology 2019;157:54‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jamialahmadi O, Mancina RM, Ciociola E, Tavaglione F, Luukkonen PK, Baselli G, et al. Exome‐wide association study on alanine aminotransferase identifies sequence variants in the GPAM and APOE associated with fatty liver disease. Gastroenterology 2021;160:1634‐1646.e7. [DOI] [PubMed] [Google Scholar]
- 9. Innes H, Buch S, Hutchinson S, Guha IN, Morling JR, Barnes E, et al. Genome‐wide association study for alcohol‐related cirrhosis identifies risk loci in MARC1 and HNRNPUL1. Gastroenterology 2020;159:1276‐1289.e7. [DOI] [PubMed] [Google Scholar]
- 10. Emdin CA, Haas ME, Khera AV, Aragam K, Chaffin M, Klarin D, et al.; Million Veteran Program . A missense variant in mitochondrial amidoxime reducing component 1 gene and protection against liver disease. PLoS Genet 2020;16:e1008629. Erratum in: PLoS Genet 2021;17:e1009503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bianco C, Jamialahmadi O, Pelusi S, Baselli G, Dongiovanni P, Zanoni I, et al. Non‐invasive stratification of hepatocellular carcinoma risk in non‐alcoholic fatty liver using polygenic risk scores. J Hepatol 2021;74:775‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reig M, Gambato M, Man NK, Victor D, Orci LA, Toso C. Should patients with NAFLD/NASH be surveyed for HCC? Transplantation 2019;103:39‐44. [DOI] [PubMed] [Google Scholar]
- 13. Loomba R, Lim JK, Patton H, El‐Serag HB. AGA clinical practice update on screening and surveillance for hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: expert review. Gastroenterology 2020;158:1822‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu C, Song CI, Lv J, Zhu M, Yu C, Guo YU, et al.; China Kadoorie Biobank Collaborative Group . Prediction and clinical utility of a liver cancer risk model in Chinese adults: a prospective cohort study of 0.5 million people. Int J Cancer 2021;148:2924‐2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex disease of middle and old age. PLoS Medicine 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ratib S, Fleming KM, Crooks CJ, Walker AJ, West J. Causes of death in people with liver cirrhosis in England compared with the general population: a population‐based cohort study. Am J Gastroenterol 2015;110:1149‐1158. [DOI] [PubMed] [Google Scholar]
- 17. FinnGen . Introduction. https://finngen.gitbook.io/documentation/. Updated May 2021. Accessed November, 2021.
- 18. Buch S, Stickel F, Trepo E, Way M, Herrmann A, Nischalke HD, et al. A genome‐wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol‐related cirrhosis. Nat Genet 2015;47:1443‐1448. [DOI] [PubMed] [Google Scholar]
- 19. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182‐236. Erratum in: J Hepatol 2019;70:817. [DOI] [PubMed] [Google Scholar]
- 20. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, 4th Edition. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 21. Mueller S, Englert S, Seitz HK, Badea RI, Erhardt A, Bozaari B, et al. Inflammation‐adapted liver stiffness values for improved fibrosis staging in patients with hepatitis C virus and alcoholic liver disease. Liver Int 2015;35:2514‐2521. [DOI] [PubMed] [Google Scholar]
- 22. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genome wide association scans. Bioinformatics 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frieden C, Wang H, Ho CMW. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain‐domain interactions. Proc Natl Acad Sci U S A 2017;114:6292‐6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ward H, Mitrou PN, Bowman R, Luben R, Wareham N, Khaw KT, et al. APOE genotype, lipids and coronary heart disease risk: a prospective population study. Arch Intern Med 2009;169:1424‐1429. Erratum in: Arch Intern Med 2009;169:2108. [DOI] [PubMed] [Google Scholar]
- 25. Wozniak MA, Itzhaki RF, Faragher EB, James MW, Ryder SD, Irving WL. Apolipoprotein E‐epsilon 4 protects against severe liver disease caused by hepatitis C virus. Hepatology 2002;36:456‐463. [DOI] [PubMed] [Google Scholar]
- 26. Lumsden AL, Mulugeta A, Zhou A, Hypponen E. Apolipoprotein E (APOE) genotype‐associated disease risks: a phenome‐wide, registry‐based, case‐control study utilizing the UK Biobank. EBioMedicine 2020;59:102954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emdin CA, Haas M, Ajmera V, Simon TG, Homburger J, Neben C, et al. Association of genetic variation with cirrhosis: a multi‐trait genome‐wide association and gene‐environment interaction study. Gastroenterology 2021;160:1620‐1633.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson PW, Myers RH, Larson MG, Ordovas JM, Wolf PA, Schaefer EJ. Apolipoprotein E alleles, dyslipidemia and coronary heart disease. The Framingham Offspring Study. JAMA 1994;272:1666‐1671. [PubMed] [Google Scholar]
- 29. Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, et al. High serum levels of cholesterol increase antitumour functions of nature killers cells and reduce growth of liver tumours in mice. Gastroenterology 2020;158:1713‐1727. [DOI] [PubMed] [Google Scholar]
- 30. Cheng D, Huang R, Lanham S, Cathcart HM, Howard M, Corder EH, et al. Functional interaction between APOE4 and LDL receptor isoforms in Alzheimer’s disease. J Med Genet 2005;42:129‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson LA, Olsen RHJ, Merkens LS, DeBarber A, Steiner RD, Sullivan PM, et al. Apolipoprotein E‐low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform‐dependent manner. Neurobiol Dis 2014;64:150‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vermissen J, Oosterveer DM, Hoekstra M, Out R, Berbee JFP, Blommesteijn‐Touw AC, et al. Apolipoprotein isoform E4 does not increase coronary heart disease risk in carriers of low‐density lipoprotein receptor mutations. Circ Cardiovasc Genet 2011;4:655‐660. [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto T, Choi HW, Ryan RO. Apolipoprotein E isoform‐specific binding to the low‐density lipoprotein receptor. Anal Biochem 2008;372:222‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carlsson B, Linden D, Brolen G, Liljeblad M, Bjursell M, Romeo S, et al. Review article: the emerging role of genetic in precision medicine for patients with non‐alcoholic steatohepatitis. Aliment Pharmacol Ther 2020;51:1305‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2‐rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: a meta‐analysis. Hepatology 2015;62:1742‐1756. [DOI] [PubMed] [Google Scholar]
- 36. Prill S, Caddeo A, Baselli G, Jamialahmadi O, Dongiovanni P, Rametta R, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep 2019;9:11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pelusi S, Baselli G, Pietrelli A, Dongiovanni P, Donati B, McCain MV, et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci Rep 2019;9:3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material