

Abstract
Induction of neoangiogenesis is a hallmark feature during disease progression of hepatocellular carcinoma (HCC). Antiangiogenetic compounds represent a mainstay of therapeutic approaches; however, development of chemoresistance is observed in the majority of patients. Recent findings suggest that tumor‐initiating cells (TICs) may play a key role in acquisition of resistance, but the exact relevance for HCC in this process remains to be defined. Primary and established hepatoma cell lines were exposed to long‐term sorafenib treatment to model acquisition of resistance. Treatment effects on TICs were estimated by sphere‐forming capacity in vitro, tumorigenicity in vivo, and flow cytometry. Adaptive molecular changes were assessed by whole transcriptome analyses. Compensatory mechanisms of resistance were identified and directly evaluated. Sustained antiproliferative effect following sorafenib treatment was observed in three of six HCC cell lines and was followed by rapid regrowth, thereby mimicking responses observed in patients. Resistant cells showed induction in sphere forming in vitro and tumor‐initiating capacity in vivo as well as increased number of side population and epithelial cell adhesion molecule‐positive cells. Conversely, sensitive cell lines showed consistent reduction of TIC properties. Gene sets associated with resistance and poor prognosis, including Hippo/yes‐associated protein (YAP), were identified. Western blot and immunohistochemistry confirmed increased levels of YAP. Combined treatment of sorafenib and specific YAP inhibitor consistently revealed synergistic antioncogenic effects in resistant cell lines. Conclusion: Resistance to antiangiogenic therapy might be driven by transient expansion of TICs and activation of compensatory pro‐oncogenic signaling pathways, including YAP. Specific targeting of TICs might be an effective therapeutic strategy to overcome resistance in HCC.
Abbreviations
- 5‐FU
5‐fluorouracil
- AKT
protein kinase B
- C, Ctrl
control
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐to‐mesenchymal transition
- EpCAM
epithelial cell adhesion molecule
- GSEA
gene set enrichment analysis
- HCC
hepatocellular carcinoma
- IC50
median inhibitory concentration
- IPA
Ingenuity Pathway Analysis
- mTOR
mammalian target of rapamycin
- MYC
MYC proto‐oncogene bHLH transcription factor
- NES
normalized enrichment score
- NOD/SCID
nonobese diabetic/severe combined immunodeficient
- PI3K
phosphoinositide 3‐kinase
- pYAP
phosphorylated YAP
- SP
side population
- STAT
signal transducer and activator of transcription
- TAZ
transcriptional coactivator with PDZ‐binding motif
- TIC
tumor‐initiating cell
- YAP
yes‐associated protein 1
Hepatocellular carcinoma (HCC) ranks among the most frequent and deadliest cancers worldwide. The pronounced intratumoral and intertumoral heterogeneity in HCC represents a major challenge for molecular classification and development of effective therapeutic strategies.( 1 ) This heterogeneity is also reflected in molecular pathways that govern control of cell growth and differentiation, both critical processes in cancer progression.( 2 ) Consequently, resistance to antioncogenic therapy and tumor relapses are frequently observed.( 3 ) According to the hierarchic model of carcinogenesis, molecular diversity and acquired resistance to therapies originate in the tumor‐initiating cells (TICs), a small (<1%) subpopulation of tumor cells that share cellular properties and molecular signaling pathways with normal tissue stem cells.( 4 ) TICs have been implicated in the majority of cellular processes that confer pro‐oncogenic properties, including unlimited cell growth, invasion, generation of distant metastasis, and resistance to current therapies.( 5 ) Therefore, understanding the intrinsic properties of TICs is becoming increasingly important for translational research as the prime cellular targets for diagnostic and therapeutic strategies in HCC.( 6 )
Sorafenib affects tumor‐cell proliferation, tumor angiogenesis, and cell apoptosis by targeting numerous serine/threonine and tyrosine kinases, namely raf‐1 proto‐oncogene serine/threonine kinase (RAF1), B‐raf proto‐oncogene serine/threonine kinase (BRAF), vascular endothelial growth factor receptor (VEGFR) 1‐3, platelet‐derived growth factor receptor (PDGFR), KIT proto‐oncogene receptor tyrosine kinase (KIT), fms related receptor tyrosine kinase 3 (FLT3), fibroblast growth factor receptor 1 (FGFR1), and ret proto‐oncogene (RET).( 7 ) Sorafenib was the first systemic therapy to demonstrate a survival benefit for patients with advanced HCC.( 8 ) For more than a decade, sorafenib was the only approved drug for first‐line therapy in advanced stages. Recently, lenvatinib was demonstrated to be noninferior to sorafenib and consequently approved by the US Food and Drug Administration as an alternative first‐line therapy. More recently, immunotherapeutic approaches have shown promising clinical results.( 9 , 10 ) Concordantly, the programmed death‐ligand 1 (PD‐L1) inhibitor atezolizumab in combination with the VEGF neutralizing antibody bevacizumab have been approved for patients with unresectable or metastatic HCC who did not previously receive systemic therapy.( 10 ) However, despite an improvement in overall survival for the immunotherapeutic antiangiogenetic combination of these two drugs, only 25%‐30% of patients responded to the therapy. A group of patients with impaired liver function and increased risk of variceal bleeding did not qualify for this combination.( 10 ) Thus, sorafenib remains an important mainstay of systemic therapy and is the most widely used compound for HCC treatment worldwide. Unfortunately, despite an initial antitumorigenic response caused by sorafenib, the majority of patients rapidly develop resistance to the therapy, which is followed by the subsequent occurrence of tumor growth and/or metastatic spread.( 11 )
In order to further improve the response to sorafenib, several phase II and III clinical trials employed different combination therapies, including treatment regiments with doxorubicin, tigatuzumab, erlotinib, and hepatic arterial infusion chemotherapy( 12 , 13 , 14 , 15 ); however, all failed to show improved response and overall survival in comparison with sorafenib monotherapy.
In the present study, we designed an in vitro model based on primary and established HCC cell lines that reliably mimics the adaptive changes induced during disease progression in patients treated with sorafenib. We demonstrate that expansion of TICs during the course of HCC treatment plays a substantial role in acquired resistance to sorafenib, both in vitro and in vivo. Response of the putative TIC compartment during short longitudinal therapy accurately predicts relapse formation and subdivides sensitive from resistant cancer cells. We further define the oncogene yes‐associated protein (YAP) as the molecular target in TICs leading to formation of relapse. Finally, we show that specific targeting of TICs by a combination of the YAP inhibitor carbonic anhydrase 3 (CA3) and classic chemotherapy (i.e., sorafenib) might be an effective therapeutic strategy to overcome resistance in HCC and improve therapeutic efficacy.
Materials and Methods
Cell Lines
PLC/PRF/5 (referred to as PLC), HepG2, and Hep3B cell lines were obtained from the American Type Culture Collection (ATCC). The Huh7 cell line was obtained from Riken Cell Bank. LECHCC and HCC31 were freshly isolated, nonclonal, alpha‐fetoprotein‐negative primary HCC cell lines.( 16 ) Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 2 mM L‐glutamine, 1 unit/mL penicillin/streptomycin, and 10% fetal bovine serum (FBS).
Viability and Median Inhibitory Concentration IC50
Cell viability was measured using the WST‐1 assay according to the manufacturer's protocol (Roche). We plated 5 × 103 cells on 96‐well plates; after overnight incubation, cells were treated with increasing concentrations of sorafenib for 72 hours. Median inhibitory concentration (IC50) values were calculated by nonlinear regression using GraphPad Prism software. When IC50 concentrations were defined for each cell line, cells were treated with sorafenib and cell viability was measured at different time points (3, 7, and 14 days). Cell viability, defined as absorbance in the treatment group compared to the control group, was expressed as percentage mean change ± SD (n = 4). For combination therapy, concentrations of sorafenib were in a range from 2 to 16 µM and CA3 from 0.25 to 8 µM.
Flow Cytometry
Side Population Analysis
Side population (SP) analysis was performed as described.( 17 ) Briefly, cells were incubated at 37°C for 90 minutes with 15 μg/mL of Hoechst‐33342 (Invitrogen). A parallel sample was stained with Hoechst‐33342 in the presence of 50 μmol/L of the adenosine triphosphate binding cassette subfamily G member 2 (ABCG2) inhibitor fumitremorgin C (Sigma‐Aldrich) as a control to identify the SP cells. Cell viability was ensured by 7‐aminoactinomycin D exclusion. Analyses were performed on a BD Fortessa device.
TIC Marker Analysis
At defined time points during the course of treatment, cells were collected and fixed in 1% paraformaldehyde (PFA) for 1 hour at 4°C. Cells were further resuspended in phosphate‐buffered saline and stored at 4°C or directly stained for the analysis. Briefly, 5 × 105 fixed cells were resuspended in 100 µL staining buffer (homemade), and 1 µL of directly conjugated primary antibody was added. Cells were incubated for 15 minutes at 4°C; this was followed by removal of the staining buffer and addition of 500 µL of running buffer (self‐prepared). Cells were analyzed on a BD FACSVerse flow cytometer.
Tumorigenicity
All procedures were performed with the approval of local authorities and in accordance with the guidelines of the National Institutes of Health animal care committee. After 72 hours of sorafenib treatment (Huh7, 6.15 µM; HepG2, 12.4 µM), viability of the cells was assessed by trypan blue. Viable hepatoma cells were mixed with Matrigel (1:1) (BD Bioscience) and transplanted subcutaneously into both flanks of nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice. In total, 1 × 106 viable cells were injected, and tumor growth was monitored weekly by palpation. Three animals per group were used. Animals were euthanized when tumor size exceeded 15 mm diameter. Tumors were removed, measured, and fixed in 4% PFA or preserved at −80°C.
Soft Agar‐Based Sphere‐Formation Assay
After treatments with sorafenib (3, 7, and 14 days) or in combination with CA3, 1,000 viable single cells were resuspended in 2% agar and diluted to a final concentration of 0.25%. Cells were subsequently added on top of the precoated 48‐well plate with 1.3% agar containing DMEM with 20% FBS. During spheroid cultivation, medium was replaced every 3 days. After 14 days, spheroids were counted under a microscope. All experiments were performed in triplicate.
RNA Extraction and Reverse‐Transcription Quantitative Polymerase Chain Reaction
Total RNAs was extracted using the Qiagen RNEasy Mini‐Kit following the manufacturer’s protocol. RNA quantity and purity were estimated using a Nanodrop ND‐1000 spectrophotometer (Thermo Fisher Scientific). Two‐step reverse‐transcription quantitative polymerase chain reaction complementary DNA synthesis using SuperScript III (Invitrogen), SYBR Green Master‐Mix (Bio‐Rad), and CFX Connect System was performed.
Transcriptome Analysis
Microarray analyses were performed by the Institute of Human Genetics, Department of Genomics, Life and Brain Center, University of Bonn. Transcriptomic analyses were performed for time points of 3, 7, and 14 days for resistant cells and HCC31. Due to insufficient RNA quality or lack of viable cells, HepG2 and PLC cells were analyzed after 3 and 7 days. Following analyses and obtaining raw data, gene expression values were normalized by the quantile normalization method across all samples following subtraction of background noise in each spot by GenomeStudio (Illumina). Signal intensities with a detection P > 0.05 were treated as a missing value, and only genes with sufficient representation across the samples were included in further data analysis.( 17 ) Time course analysis was performed in Bioconductor package R version 3.12.
Gene Set Enrichment Analysis and Ingenuity Pathway Analysis
Gene set enrichment analysis (GSEA) based on obtained transcriptomic data was performed for treated and nontreated hepatoma and primary cell lines (https://www.gsea‐msigdb.org/gsea/index.jsp).( 18 ) The normalized enrichment score (NES) reflected the degree of overrepresentation for each group at the peak of the entire set. Statistical significance was calculated by the nominal P value of the enrichment score by using an empirical phenotype‐based permutation test. Canonical pathway and network analyses were performed using Ingenuity Pathway Analysis (IPA; Qiagen). The significance of each pathway was determined by the scoring system provided by the IPA tool.
Protein Isolation and Western Blot
Whole‐cell lysates (100 μg) were prepared from frozen cells using M‐PER Tissue Extraction Buffer (Thermo Fisher Scientific) containing Complete Protease Inhibitor Cocktail (Roche). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Membranes were incubated with YAP, phosphorylated YAP (pYAP), tafazzin (TAZ), and actin antibodies (Supporting Table S2). Immune complexes were detected using a LI‐COR imaging system. Quantification of expression levels was performed by densitometric analysis using ImageJ.
Immunohistochemistry
Tissue and cells were fixed in 4% formaldehyde. Tissue was further embedded in paraffin and cut in 3.5‐μm sections. Antibodies are listed in Supporting Table S2. Visualization was performed with ImmPACT DAB detection kit (VECTOR) according to the company's protocols. Stained tissues were viewed using an ECHO REB‐01_D microscope with 20× and 40× magnification objectives.
Drug Synergism
To investigate potential synergistic effects of sorafenib and YAP inhibitor (CA3), cells were seeded in a 96‐well plate at a density of 5 × 103 per well and treated with both compounds either as a single treatment or as combination. Obtained results were analyzed by R package Synergyfinder version 2.0.12.( 19 )
Statistics
Statistical analysis was performed using the Student t test and one‐way analysis of variance for multiple group comparisons. P ≤ 0.05 was considered statistically significant. Results are presented as mean ± SD.
Results
Differential Response of Cells to Sorafenib Treatment
To test the response of liver cancer cells against multityrosine kinase inhibitors, we aimed to develop a dynamic in vitro model to mimic adaptive treatment response of human patients and monitor phenotypic and molecular features (Fig. 1A). Immortalized human hepatoma (Huh7, PLC, HepG2, and Hep3B) and two primary human HCC cell lines (LECHCC and HCC31) were continuously treated with the respective IC50 concentrations of sorafenib over a period of 14 days (Fig. 1B). Subsequently, viability was assessed at days 3, 7, and 14 in comparison to untreated control cells. Interestingly, we observed two distinct types of response resembling a sensitive and a resistant phenotype (Fig. 1B). Three cell lines showed a rapid and sustained resistance to sorafenib treatment (Huh7, Hep3B, LECHCC), which was reflected in rapid regrowth after initial treatment response after 3 days (Hep3B and LECHCC) or 7 days (Huh7). In contrast, the remaining cells (HepG2, HCC31, PLC) displayed pronounced sensitivity to sorafenib, illustrated by a steady decrease of cell viability in the course of the experiment.
FIG. 1.
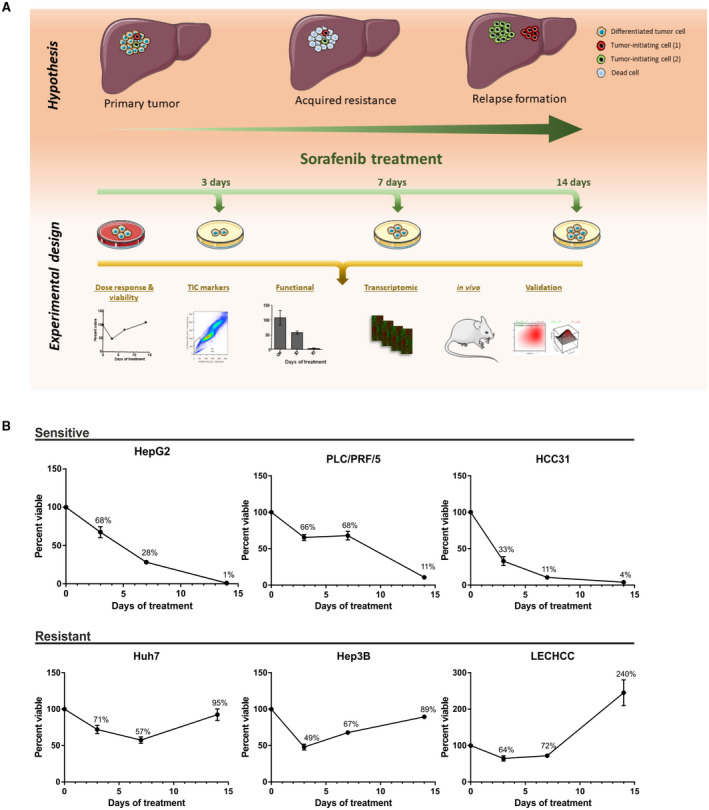
Schematic overview of the main hypothesis and experimental design and a viability analysis across cell lines. (A) Hypothesis of relapse formation during sorafenib treatment and the potential role of TICs in this process (upper panel). Workflow and analyses performed to prove the hypothesis (lower panel). (B) Viability analysis determines two types of response to antiangiogenic therapy. Different established hepatoma and primary HCC cell lines were continuously exposed to IC50 concentrations of sorafenib (PLC/PRF/5, 17.5 µM; HepG2, 12.4 µM; HCC31, 22.7 µM; Huh7, 6.1 µM; Hep3B, 7.8 µM; LECHCC, 12.6 µM). Cells were treated for a total of 14 days. Viability was assessed after 3, 7, and 14 days and compared to untreated control cells to monitor the development of drug resistance using standardized conditions and comparable drug levels. Graphs represent differential growth kinetics in response to exposure with sorafenib at the defined time points. Rapid regrowth in comparison to untreated cells was observed in half the cell lines (lower panel). Sensitive drug responses are shown in the upper panel. Data are presented as mean ± SD of three biological replicates.
In summary, these data demonstrate the existence of liver cancer cell lines that may represent a proxy for the development of HCC resistance, which is frequently observed on human hepatocarcinogenesis.
Transient Expansion of TICs During Development of Drug Resistance
To investigate if the presence of TICs in the cell lines with sorafenib resistance could explain the observed phenotype, we employed several well‐established flow cytometry‐based and functional TIC screening assays. For this, the effect of sorafenib on TICs was assessed at defined time points (3, 7, and 14 days) and compared with respective untreated controls (Fig. 2). We evaluated the relative change in frequency of putative TICs by measuring the SP, which is considered to represent a cell fraction with enhanced stemness features, and measuring relative abundance of cells expressing TIC marker epithelial cell adhesion molecule (EpCAM).( 1 , 20 ) Indeed, a significant expansion of the TIC subpopulation during the course of treatment was observed in sorafenib‐resistant cell lines (Huh7, Hep3B; Fig. 2A). For these cells, the initial expansion of TICs was subsequently followed by a gradual reduction to levels comparable to untreated control cells, indicating the transient nature of TIC expansion in the development of drug resistance. Accordingly, highest levels of TIC were observed at the time when acquisition of resistance was detected (7 and 14 days). Conversely, in sensitive cell lines, continuous and sustained antiproliferative response to the treatment was paralleled by a progressive reduction in the number of putative TICs (PLC, HCC31; Fig. 2A). These cells showed significant reduction in total number of TICs after 3 and 7 days of treatment, which ultimately led to complete abolishment of TICs in response to sorafenib after 14 days. Consistent with our findings of the SP analysis, the number of cells expressing the known TIC marker EpCAM was increased in resistant cells during the initial phase of sorafenib treatment (Fig. 2B). This expansion was further followed by reduction of EpCAM‐positive cells to a similar level as untreated control cells (Huh7). In contrast, sensitive cells did not show a comparable EpCAM induction. Instead, no EpCAM positivity was detectable in the TIC population of HCC cells that were sensitive to sorafenib (PLC).
FIG. 2.
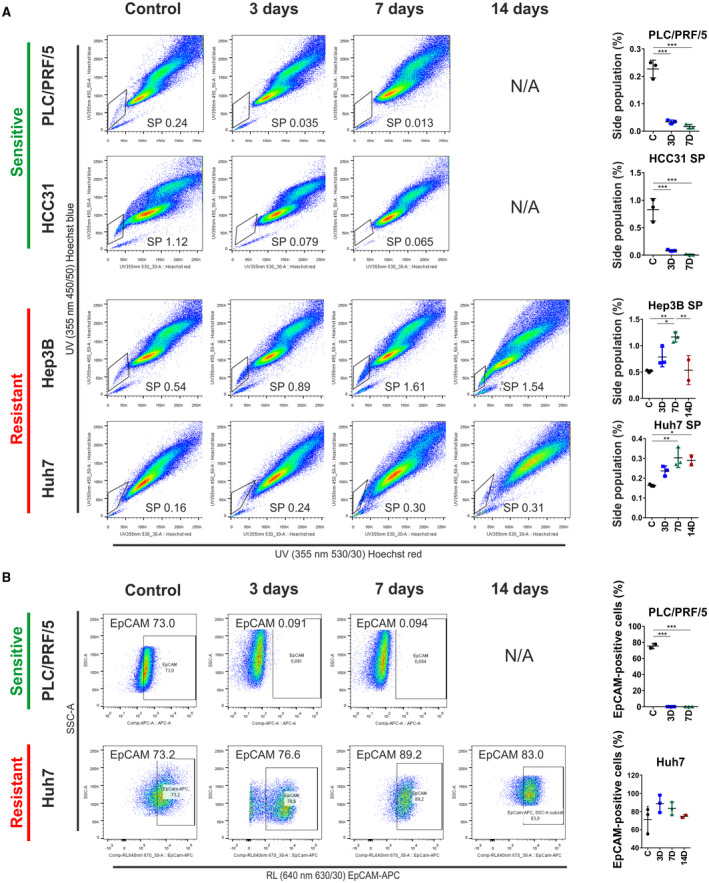
Flow cytometry analyses of putative TICs and their role in acquired drug resistance. Flow cytometry analysis of putative cancer stem cell markers as well as a dynamic SP assay in different cell lines after continuous exposure to sorafenib was performed. (A) SP analysis shows percentage of putative TICs after treatment with IC50 concentrations at different time points. Drug response (i.e., sensitive and resistant) is indicated in green and red. In the SP approach, TICs are distinguished based on their ability to actively exclude Hoechst 33342 dye; therefore, most stem‐like cells present with low staining. Cells costained with fumitremorgin C, which blocks ABCG2 drug transport, were used as the negative control. Dot plots represent SP profiles of resistant and sensitive cell lines after exposure to the drugs. Charts on the right side represent percentage of SP cells at different time points (*P< 0.05, **P < 0.01, ***P < 0.001). (B) Flow cytometry analysis of putative TICs based on surface marker staining with EpCAM before and after sorafenib treatment. Charts on the right side represent percentage of EpCAM‐positive cells at different time points (*P < 0.05, **P < 0.01, ***P < 0.001). Data are presented as mean ± SD of three biological replicates. Abbreviations: ABCG2, adenosine triphosphate binding cassette subfamily G member 2; APC, allophycocyanin; NA, not applicable; RL, red laser; SSC‐A, side scatter–area; UV, ultraviolet.
To further corroborate these findings, we performed a sphere‐formation assay, which represents a widely used approach to evaluate tumor‐initiation capacity in vitro. In accordance with the suggested transient increase of putative TICs in sorafenib‐resistant cell lines, a significant induction in sphere‐forming capacity was observed in sorafenib‐treated cells compared to nontreated controls (Fig. 3A). In contrast, the observed reduction of the SP and EpCAM‐positive cells in sensitive lines also yielded a lower capability to form spheres. Lastly, to confirm and validate our in vitro findings, we performed in vivo transplantation experiments using immunocompromised NOD/SCID mice. For this analysis, representative cell lines for sensitive (HepG2) and resistant (Huh7) cells were selected (Fig. 3B,C). As expected, Huh7 showed comparable tumor growth in both treatment and control animals. Results for HepG2 illustrated high sensitivity to sorafenib, which was reflected by significantly reduced tumor sizes and number of tumor nodules (Fig. 3B,C). Consistently, intratumoral expression of the progenitor/TIC marker cytokeratin 19 (CK19) was considerably higher in tumors obtained from Sorafenib‐pretreated Huh7 cells when compared to nontreated control cells (Supporting Fig. S1A).( 21 )
FIG. 3.
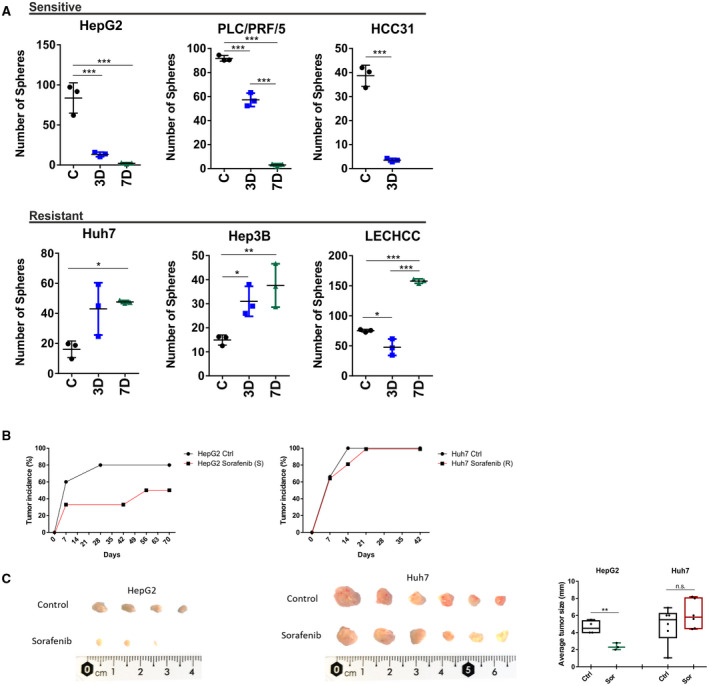
Pro‐oncogenic properties of TICs investigated by the sphere‐formation assay and in vivo tumorigenicity. (A) Graphs show results for sphere‐forming ability of sensitive and resistant cells after treatment with sorafenib. Sensitivity to the respective compounds was reflected in a significant reduction in sphere‐forming ability, whereas resistant cell lines showed a progressive expansion of cells with sphere‐forming ability reflected in significantly higher numbers of spheres after 3 and 7 days. Cells were pretreated for 3 and 7 days and then cultured under sphere‐forming conditions. A total of 1,000 cells were seeded per well, and sphere forming was monitored at the corresponding time points. All experiments were performed in triplicate and sphere‐forming capacity was estimated based on at least three independent biological replicates per cell line (*P < 0.05, **P < 0.01, ***P < 0.001). Data are presented as mean ± SD of three biological replicates. (B) Tumor incidence observed after subcutaneous injection of 1 million treated or untreated cells into NOD/SCID mice. Cells were exposed to 3 days of treatment before injection into both flanks of animals. Tumor growth was monitored weekly. Tumor incidence in the corresponding cell line is shown. (C) Tumors isolated from animals that were injected with control and treated cells. Tumors are arranged by size for side‐by‐side comparison. The graph on the right shows average size of tumor nodules for each group (*P < 0.05, **P < 0.01, ***P < 0.001). Abbreviations: n.s., not significant; R, resistant; S, sensitive; Sor sorafenib.
Overall, these results demonstrate that putative multiresistant TICs respond to chemotherapy by transient expansion of resistant cells and thereby significantly contribute to acquired drug resistance in human liver cancer. These data indicate that sustained response to antitumorigenic therapy requires inhibition of putative TICs.
Activation of Oncogenic Mechanisms and Stemness Features Drive Acquisition of Resistance to Sorafenib Therapy
To identify key molecular alterations that may lead to treatment resistance in HCC, we assessed the global pattern of transcriptome alterations associated with the observed phenotypes. We performed genome‐wide gene expression analyses in resistant and sensitive cell lines for all inspected time points (3, 7, and 14 days). Between these two groups, we identified a total of 2,096 differentially expressed genes (P < 0.01) (Supporting Table S5). These genes were highly efficient in separating sorafenib‐sensitive from sorafenib‐resistant cells, using unsupervised hierarchical cluster analysis (Fig. 4A). Subsequently, GSEA and IPA were performed to identify activated gene sets and signaling pathways characteristic for a resistant group (Fig. 4B; Supporting Fig. S2A,B). GSEA confirmed activation of signaling pathway‐associated gene signatures involved in pro‐oncogenic signaling and cell proliferation, such as MYC proto‐oncogene bHLH transcription factor (MYC), epidermal growth factor receptor (EGFR), interleukin‐6 (IL‐6)/Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3), and RAS signaling, as well as enrichment of gene sets associated with cell‐cycle regulation (mitotic spindle and E2F transcription factor 3 [E2F3] genes). Interestingly, GSEA also revealed activation of gene sets associated with cancer stemness, in particular involved in YAP activity. The majority of these genes showed higher relative expression in the resistant cells than in sensitive cells (Fig 4C). Subsequent analyses of activated canonical pathways validated our findings and disclosed regulation of the Hippo signaling pathway (Supporting Fig. S2B).
FIG. 4.
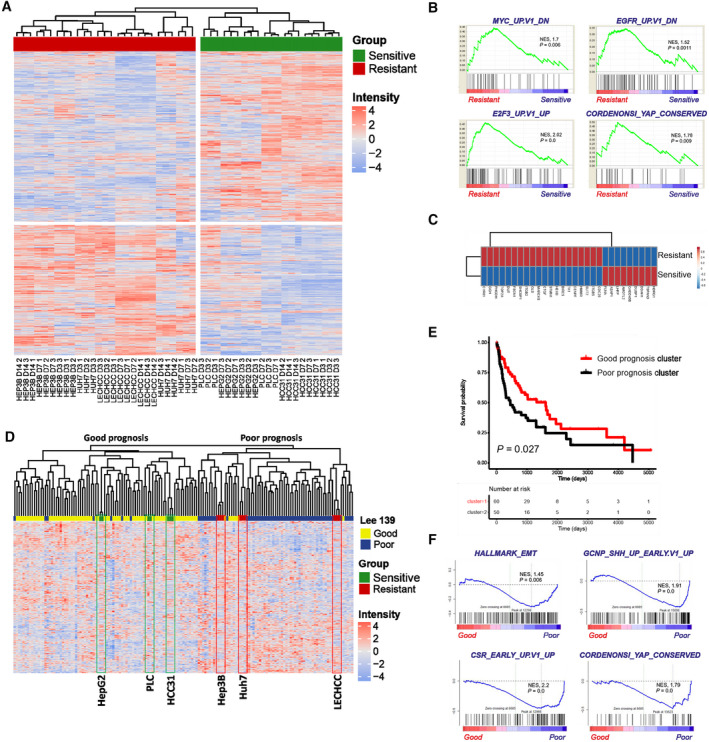
Transcriptomic analyses define molecular mechanisms responsible for acquired resistance. Time‐course analysis, survival, and GSEA based on transcriptome data of resistant and sensitive cell lines after 3, 7, and 14 days of treatment. (A) Time‐course analysis reveals clear separation of sensitive and resistant cell lines based on 2,096 differentially expressed genes between different groups. (B) GSEA for resistant cell lines in comparison to sensitive cell lines. NES reflects degree of overrepresentation for each group at the peak of the entire set. Statistical significance calculated by nominal P value of the enrichment score by using an empirical phenotype‐based permutation test. (C) Correlation analysis of Cordenonsi genes expressed in the resistant and sensitive groups. (D) Integration of resistant and sensitive cell lines based on our sorafenib resistance signature with a published data set of 139 patients with HCC.( 22 ) HepG2, PLC/PRF/5, and HCC31 cells are clustered in the good prognostic group (left). Hep3B, Huh7, and LECHCC cells are clustered in the poor prognostic group (right). (E) Kaplan‐Meier plot of overall survival of 139 individuals with HCC based on our sorafenib‐resistance signature.( 22 ) (F) GSEA for good and poor prognostic HCC groups based on the gene expression signature from Lee et al.( 22 ) NES reflects degree of overrepresentation for each group at the peak of the entire set. Statistical significance calculated by nominal P value of the enrichment score by using an empirical phenotype‐based permutation test. Abbreviations: CSR, class switch recombination; DN, down; E2F3, E2F transcription factor 3; GCNP, granule cell neuron precursor; SHH, sonic hedgehog; UP, up.
To further explore whether a sorafenib‐resistance gene signature could be used to accurately identify groups of patients with HCC with poor clinical outcome, we integrated it with published expression data from 139 patients with HCC divided into good and poor prognostic groups (Fig. 4D; Supporting Table S6).( 22 ) As expected, unbiased clustering revealed that all resistant cell lines belonged to a cluster that characterized patients with HCC with poor prognosis whereas sensitive cells clustered together with patients with liver cancer, who were characterized by better prognosis. Furthermore, we checked overall survival of the 139 patients with HCC based on our resistance signature. The findings clearly showed that patients who were characterized by the presence of the resistant gene signature had shorter survival (Fig. 4E).( 22 ) GSEA based on transcriptomic profiles of poor and good prognostic HCC subclasses confirmed enrichment of epithelial‐to‐mesenchymal transition (EMT) and TIC (sonic hedgehog and class switch recombination) but most importantly the presence of Hippo/YAP signature genes (Fig. 4F).
Our data delineate that distinct molecular alterations, such as activation of tumor‐supporting YAP, are associated with sorafenib resistance in patients with HCC with poor clinical outcome.
YAP Signaling Contributes to Sorafenib Resistance in HCC Cells
Earlier studies showed that resistance to some chemotherapies could be driven by TICs in a YAP/PDZ‐binding motif (TAZ)‐dependent manner.( 23 ) To address the relevance of YAP signaling as a potential driver of acquired drug resistance after sorafenib treatment, we first assessed protein levels of YAP, TAZ, and pYAP (also known as serine 127), which is an inhibited form of YAP. Following treatment with sorafenib, a resistant cell line (Hep3B) showed reduced levels of pYAP and slightly increased levels of TAZ, indicating an activation of YAP/TAZ and its positive effects on target genes related to TICs (Supporting Fig. S3).( 24 ) In contrast, pYAP levels in the sorafenib‐sensitive cell line PLC were only slightly decreased and remained similar to the untreated control while a prominent reduction was determined for TAZ. Notably, the presence of YAP phosphorylation has been reported to have functional consequences on the stability of the protein, leading to its degradation.( 24 ) Moreover, YAP phosphorylation in combination with TAZ reduction causes decreased activity of these transcriptional regulators.( 24 ) Thus, decreased phosphorylation and increased YAP/TAZ stability could be one of the indicators for sorafenib resistance.
To further investigate therapeutic implications of our findings, we evaluated potential synergistic effects of sorafenib and YAP inhibition (Fig. 5A). For this reason, we treated all HCC cell lines with increasing concentrations of sorafenib and CA3, an inhibitor that specifically disturbs the interaction between YAP and the transcriptional enhanced associate domain,( 25 ) as well as different combinations of both drugs. As a functional readout, we determined cellular viability and evaluated synergistic effects by using Synergyfinder.( 19 ) The results were calculated as an average synergy score and visualized as contour plots that show the range of concentrations where synergistic or antagonistic effects are most pronounced (Fig. 5A). Importantly, sorafenib‐resistant cell lines consistently showed a higher level of synergism than sorafenib‐sensitive cells; this was illustrated by a significantly higher synergy score (Fig. 5A). The presence of synergism was detectable across all tested substance concentrations (red color). The most effective drug concentrations are shown in Supporting Table S3. In contrast, sensitive cells responded only moderately to combination therapy, and two cell lines even showed antagonistic effects at specific concentrations (green color). To investigate the impact of combination therapy, we treated resistant cell lines with sorafenib at IC25 combined with increasing concentrations of CA3. As expected, combination treatment effectively reduced the number of spheres (Fig. 5B). In addition, neither sorafenib alone at different time points nor CA3 application showed direct interaction with the components of receptor tyrosine kinases (RTKs) (Supporting Fig. S4A,B).
FIG. 5.
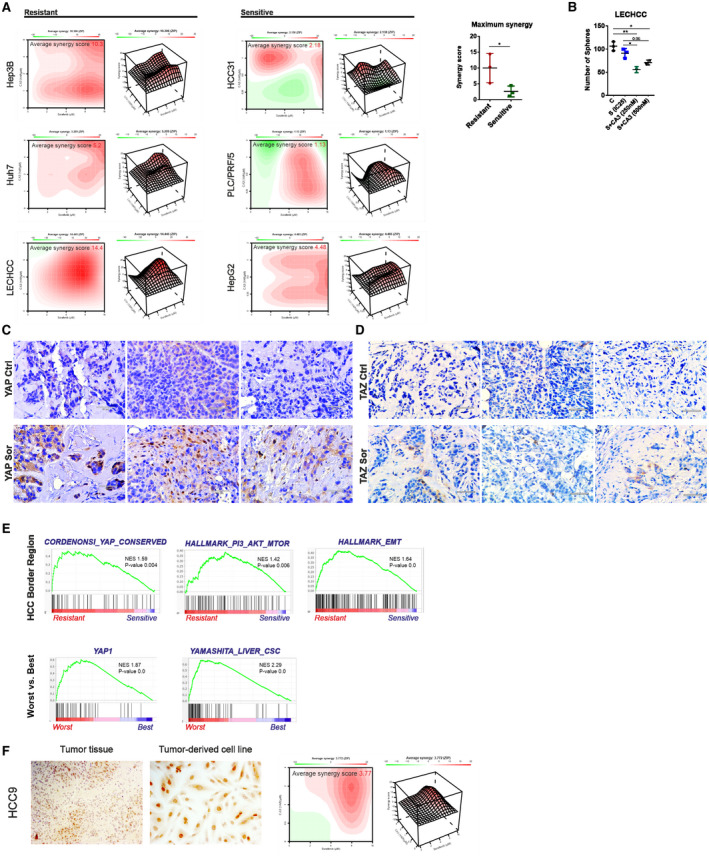
Synergistic effects of sorafenib/CA3 treatment and YAP validation in independent patient cohorts. Analysis of potential synergistic effects between sorafenib and YAP inhibitor as well as external validation of YAP signaling in two different cohorts of patients with HCC. (A) Plots indicate level of synergism between investigated drugs, where red color represents synergism and green color antagonism. Average synergy scores for each resistant cell line are shown on the top. Bars on the right represent quantification of an average synergy score between sensitive and resistant cell lines performed in three independent biological replicates per cell line (*P < 0.05). (B) Graph shows results for sphere‐forming ability of resistant cells after treatment with IC25 sorafenib (IC25, 4 µM) in combination with 250 and 500 nM of CA3. Sensitivity to the respective combination of compounds was reflected in a significant reduction in sphere‐forming ability. Cells were pretreated for 3 days and then cultured under sphere‐forming conditions. A total of 1,000 cells were seeded per well, and sphere forming was monitored and counted after 14 days. The experiments were performed in quadruplicate and sphere‐forming capacity was estimated based on two or three independent biological replicates (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Xenograft tumors, obtained after injection of resistant cells (Huh7) were stained by immunohistochemistry for YAP expression. Upper pictures represent (scale bars, 50 µm). (D) Xenograft tumors obtained after injection of resistant cells (Huh7) were stained by immunohistochemistry for TAZ expression. Upper images represent control tumors; lower images represent tumors obtained from sorafenib‐pretreated cells (scale bars, 50 µm). (E) GSEA of HCC border region in which enrichment of TIC population has previously been shown (Castven et al.( 26 )) (upper panel). Tissue samples obtained from two groups of patients who showed worst and best response to sorafenib (lower panel). (F) Primary tumor‐derived cancer cell lines can be used to predict response and level of synergism between sorafenib and YAP inhibitor. Left image shows YAP‐positive staining in HCC tissue (HCC9) (10x magnification); middle image shows YAP‐positive staining in the cell line derived from HCC9 tissue (20x magnification); right graph demonstrates that patient‐derived cell lines with YAP activation, such as HCC9, can predict response to combination treatment with sorafenib and YAP inhibitor. Abbreviations: CSC, cancer stem cell; S, Sor, sorafenib.
We therefore concluded that sorafenib‐resistant HCC cells are more susceptible to YAP‐directed perturbation approaches than sorafenib‐sensitive cells.
To further substantiate our in vitro findings and to confirm their clinical relevance, YAP activity in the context of sorafenib resistance and TICs abundances was first investigated in xenograft tumors and then in two independent cohorts of patients with HCC (Fig. 5C‐E). Immunohistochemistry showed that YAP and TAZ protein levels were markedly higher in the xenograft tumor derived from sorafenib‐pretreated Huh7 cells when compared to control cells (Fig. 5C,D). Moreover, we noticed a slight increase in pYAP staining in the control samples that was associated with reduced activity of YAP signaling (Supporting Fig. S1B). Transcriptomic validation was performed for 28 patients with HCC where tumor borders were compared with surrounding livers and tumor tissues (Fig. 5E, upper panel). In one of our previous studies, we showed that tumor border regions of patients with HCC were characterized by an enrichment of TIC features when compared to surrounding liver and tumor.( 26 ) As expected, GSEA and IPA revealed activation of gene signatures defining EMT, cellular movement, phosphoinositide 3‐kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR), and insulin‐like growth factor 1 (IGF‐1) signaling, which are processes typically associated with TIC enrichment and tumor‐invasive properties (Fig. 5E; Supporting Table S4).( 27 , 28 , 29 , 30 ) Most importantly, enrichment of YAP was noted (Fig. 5E). Our second cohort included a group of patients previously treated with sorafenib with vastly different outcomes. From this cohort, the two different subgroups were delineated as best and worst responders to therapy based on the specific survival of the patients following sorafenib treatment (Fig. 5E, lower panel). We collected tissue samples from these two cohorts and compared their transcriptomic profiles. Predictably, the worst patient group was significantly enriched for cancer stem cells as well as YAP signatures.
Finally, we tested whether primary tumor‐derived HCC cell lines could be used to predict therapeutic response and the level of synergism to combination therapy. Therefore, we first tested the expression of YAP by immunohistochemistry in tissue samples from representative patients with HCC as well as the corresponding primary cell line (Fig. 5F). Both HCC tissue and primary tumor‐derived cells showed a prominent nuclear positivity for YAP, illustrating that they represent a proxy for HCC cells in tissues with regard to YAP activation. Consistently, a positive synergistic effect of combination therapy with CA3 and sorafenib could be revealed (Fig. 5F).
Overall, elevated YAP levels in tumor cells of patients with HCC who were treated with sorafenib may contribute to sorafenib resistance after monotherapy. Simultaneous inhibition of YAP activity could increase treatment efficacy for the group of patients with HCC who develop resistance mechanisms.
Discussion
After the first identification in hematologic cancers, the existence of TICs was subsequently detected within several solid tumors, specifically in those characterized by increased rates of self‐renewal and proliferation.( 31 , 32 ) The observed complexity, which includes phenotypic, functional, and molecular heterogeneity, in several cancers, including HCC, could thus be a direct consequence of hierarchical tumor organization.( 33 ) More discoveries supported the role of TICs in adverse properties, particularly in chemoresistance, as well as their contribution to poor prognostic traits.( 34 , 35 ) In our previous studies, we showed that TIC signature and activation of proinflammatory signaling pathways possess profound prognostic implications and that specific inhibition of key signaling pathways can effectively modulate TIC properties.( 26 , 36 ) In order to delineate the role of TIC in acquired resistance to sorafenib in HCC, we performed comprehensive functional and molecular analyses and defined adaptive molecular changes during the course of treatment (Fig. 1A). We addressed the functional consequences of sorafenib administration in primary and established liver cancer cell lines as well as determined concomitant molecular adaptations that are potentially governing chemoresistance. Our study reports the identification of promising molecular targets and defines potential combination therapy for patients with HCC.
Sorafenib was the first approved systemic therapy for the treatment of advanced HCC. The Sorafenib HCC Assessment Randomized Protocol trial, a randomized controlled phase III trial for the treatment of advanced HCC, showed significant increase in median survival and the time to radiologic progression, which was nearly 3 months longer for patients treated with sorafenib than for placebo.( 37 ) However, despite exhibiting initial treatment benefit, the majority of patients still acquire resistance to sorafenib, but the exact mechanism(s) remains elusive.( 38 ) We have established an in vitro model of sorafenib resistance that allows the assessment of molecular mechanisms and functional consequences of treatment resistance as well as the evaluation of a role of TIC in these processes.
By using a dose‐adapted approach across several established and primary HCC cell lines, we treated each cell line with a corresponding IC50 concentration of sorafenib and were able to establish distinct clinical responses observed in patients with HCC treated with sorafenib. As observed in human HCC, two types of response, good response (sensitive to sorafenib) and poor response (resistant to sorafenib), were defined. Sensitive cell lines were characterized by increased cell death after an extended period of treatment. In contrast, the initial treatment benefit in resistant cell lines was followed by the rapid development of resistance and adaptations to adverse conditions, which consequently resulted in re‐establishment of cell growth and tumor relapse (Fig. 1B). These findings allowed us to address the importance of TIC‐related drug resistance in HCC cell lines. We observed a significant expansion of SP, including EpCAM+ cells, during the treatment with sorafenib in resistant cells (Fig. 2). These results showed that during initial days of treatment (3 and 7 days) the population of TICs was highly enriched, supporting the notion that TICs can survive chemotherapy. An increased number of these cells indicated the presence of more resilient phenotype(s) and was further followed by reduction to a pretreatment level when the proliferative capacity of the cells was no longer affected and relapse was fully established (Fig. 2). Acquisition of stemness features and induction of protumorigenic capacity in the resistant group was further evidenced by significant induction of spherogenicity in vitro and tumorigenicity in vivo (Fig. 3). In contrast, sensitivity to the treatment was characterized by a dramatic reduction of SP as well as the number of EpCAM+ cells (Fig. 2). A significant reduction had already occurred in the initial stages of treatment, and this impaired their proliferative potential. These results showed that successful reduction of TICs leads to reduction in proliferation and increased cell death and ultimately to a positive response to therapy in sensitive cells. The phenomenon of drug resistance and concomitant acquisition of stemness features following sorafenib treatment confirms reported studies.( 39 , 40 ) An in vivo study on a xenograft mouse model further revealed activation of stem‐like traits in the tumors that are resistant to sorafenib treatment.( 39 ) These tumors were characterized by an enhanced capacity for sphere formation in vitro, increased in vivo tumorigenicity, and a higher expression of TIC marker CK19. In addition, transcriptomic data determined IGF and FGF pathways as contributors to acquired resistance, with pronounced translational implications.( 39 ) Moreover, Cao et al.( 41 ) demonstrated the importance of leucine‐rich repeat‐containing G‐protein coupled receptor 5 (LGR5+) TICs in mouse liver cancer where these cells were able to withstand sorafenib and 5‐fluorouracil (5‐FU) and were significantly enriched following the treatment. In addition, more mechanisms of acquired resistance were proposed, and one of them includes stabilization of hypoxia‐inducible factor 1α (HIF‐1α) in a 14‐3‐3η‐dependent manner, followed by enrichment of characteristics typical for tumor progenitor cells.( 40 ) Taken together, emerging evidence clearly establishes the potential of TICs to overcome classic chemotherapy and to drive tumor progression.
After establishing the role of TICs in chemoresistance and formation of relapse, we further investigated underlying molecular mechanisms that mediate these processes. Transcriptomic profiling of our cell lines revealed clear differences in molecular profiles of resistant and sensitive groups. We further generated a specific signature of adaptive sorafenib resistance (Fig. 4A; Supporting Table S5). Resistant cells showed dysregulation in typical oncogenes and oncogenic signaling pathways, such as MYC, EGFR, KRAS proto‐oncogene guanosine triphosphatase (KRAS), and IL‐6/JAK/STAT3, as well as cell‐cycle regulation (Fig. 4B; Supporting Fig. S2A). Based on the resistance signature, corresponding cell lines reliably clustered within established good and poor prognostic subgroups of patients with HCC.( 22 ) The same poor prognostic subgroup was also characterized by activation of TIC properties and significant reduction in overall survival (Fig. 4D,E).( 22 ) An earlier study on induced sorafenib resistance proposed that HCC cells could resist some of the treatment effects by induction of EMT through PI3/AKT signaling and abnormal activation of STAT3. However, inhibition of this signaling pathway only partially restored sensitivity to sorafenib.( 38 ) In another study, EGFR activity was shown to modulate the sensitivity to sorafenib; yet, EGFR monotherapy showed only modest therapeutic benefits.( 42 ) We also observed that resistant cell lines, which tolerated extended exposure to sorafenib, had activation of EGFR signaling, confirming that this could be an important compensatory mechanism of sorafenib resistance (Fig 4B). However, considering molecular heterogeneity of HCC, resistance to sorafenib might not solely rely on increased activity of EGFR. One recent study failed to demonstrate benefits of EGFR inhibition as combination therapy with gemcitabine plus oxaliplatin (GEMOX) in HCC.( 43 ) Thus, our observation that YAP signaling might be a potential candidate for targeted therapy in sorafenib‐resistant HCC cells could be of particular relevance (Fig. 4B,C,F; Supporting Fig. S2B)
YAP and TAZ mediate important functions in various solid tumors along with HCC, and their activity is correlated with adverse tumor properties.( 44 , 45 ) Importantly, activation of YAP‐related gene sets and decreased activity of the Hippo pathway were detected in resistant cell lines (Fig. 5; Supporting Fig. S2B). These observations are in agreement with recent findings that describe the significance of YAP and Hippo pathways in solid malignancies for cell proliferation, survival, acquisition of TIC characteristics, EMT, and drug resistance.( 46 , 47 ) Recent studies also suggested that YAP could promote drug resistance (5‐FU, doxorubicin, and sorafenib) in HCC through various mechanisms. YAP up‐regulation could promote multidrug resistance through the Rac family small guanosine triphosphatase 1 (RAC1)‐reactive oxygen species‐mTOR pathway but also through interaction with the cirrhotic tumor microenvironment.( 48 , 49 ) Moreover, in a recent study, Wei et al.( 50 ) discovered that YAP promotes cell proliferation through the interaction with important cell‐cycle regulators. Importantly, 5‐FU resistance in HCC cells could be driven by TICs in a YAP/TAZ‐dependent manner.( 23 ) In concordance with these findings, a combination treatment with sorafenib and CA3, a specific YAP inhibitor, induced a considerably better response by suppressing stemness features and inducing synergistic effects exclusively present in resistant cells, indicating that combination therapy could help to overcome drug resistance (Fig. 5A,B). We could further validate these findings in xenograft tumors and human HCC (Fig. 5C‐E). Importantly, we have also noted that neither sorafenib alone nor in combination with CA3 directly interferes with the expression of the RTKs and downstream components (Supporting Fig. S4A,B), therefore supporting the hypothesis that resistance to sorafenib is compensated by activation of other molecular components, such as YAP. Finally, we showed that primary tumor‐derived cell lines can reliably predict synergistic effects of the sorafenib and CA3 combination (Fig. 5F).
The presented results provide evidence that transient expansion of TICs plays a significant role in sorafenib resistance and formation of relapse in HCC. Molecular analyses identified YAP as a potential driver of chemoresistance and a key factor regulating stemness properties in human HCC. Finally, our results show that specific targeting of YAP in combination with sorafenib could provide significant benefits to patients carrying resistant HCCs.
Supporting information
Fig S1‐S4
Table S1‐S5
Table S6
Acknowledgment
Parts of the analyses were performed at the Flow Cytometry Core Facility, Institute of Molecular Biology, Mainz. We thank all the Core Facility staff members who supported our project and helped us fluently run the experiments. D.C. thanks Monika Herr for exceptional technical support.
Supported by the German Research Foundation (MA 4443/2‐2 and SFB1292 to J.U.M.), Volkswagen Foundation (Lichtenberg program to J.U.M.), and Wilhelm‐Sander Foundation (2017.007.1 to J.U.M.).
Potential conflict of interest: Dr. Galle consults for, advises, is on the speakers’ bureau for, and received grants from Bayer and Roche; he consults for, advises, and is on the speakers’ bureau for Bristol‐Myers Squibb, MSD, Eli Lilly, Eisai, Guerbet, and Boston Scientific; he consults for and advises AstraZeneca. Dr. Marquardt advises and received grants from Leap Therapeutics; he advises Roche, Eisai, Ipsen, AstraZeneca, and MSD. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Marquardt JU, Galle PR, Teufel A. Molecular diagnosis and therapy of hepatocellular carcinoma (HCC): an emerging field for advanced technologies. J Hepatol 2012;56:267‐275. [DOI] [PubMed] [Google Scholar]
- 2. Thorgeirsson SS. Genomic decoding of hepatocellular carcinoma. Gastroenterology 2006;131:1344‐1346. [DOI] [PubMed] [Google Scholar]
- 3. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714‐726. [DOI] [PubMed] [Google Scholar]
- 4. Marquardt JU, Thorgeirsson SS. Stem cells in hepatocarcinogenesis: evidence from genomic data. Semin Liver Dis 2010;30:26‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med 2006;355:1253‐1261. [DOI] [PubMed] [Google Scholar]
- 6. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008;48:1312‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keating GM. Sorafenib: a review in hepatocellular carcinoma. Target Oncol 2017;12:243‐253. [DOI] [PubMed] [Google Scholar]
- 8. Ikeda M, Mitsunaga S, Ohno I, Hashimoto Y, Takahashi H, Watanabe K, et al. Systemic chemotherapy for advanced hepatocellular carcinoma: past, present, and future. Diseases 2015;3:360‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Personeni N, Pressiani T, Rimassa L. Lenvatinib for the treatment of unresectable hepatocellular carcinoma: evidence to date. J Hepatocell Carcinoma 2019;6:31‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T‐Y, et al.; IMbrave150 Investigators . Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 11. Mendez‐Blanco C, Fondevila F, Garcia‐Palomo A, Gonzalez‐Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia‐inducible factors. Exp Mol Med 2018;50:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu XD, Sun HC. Emerging agents and regimens for hepatocellular carcinoma. J Hematol Oncol 2019;12:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu AX, Rosmorduc O, Evans TRJ, Ross PJ, Santoro A, Carrilho FJ, et al. SEARCH: a phase III, randomized, double‐blind, placebo‐controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2015;33:559‐566. [DOI] [PubMed] [Google Scholar]
- 14. Kudo M, Ueshima K, Yokosuka O, Ogasawara S, Obi S, Izumi N, et al.; SILIUS Study Group . Sorafenib plus low‐dose cisplatin and fluorouracil hepatic arterial infusion chemotherapy versus sorafenib alone in patients with advanced hepatocellular carcinoma (SILIUS): a randomised, open label, phase 3 trial. Lancet Gastroenterol Hepatol 2018;3:424‐432. [DOI] [PubMed] [Google Scholar]
- 15. Cheng A‐L, Kang Y‐K, He AR, Lim HY, Ryoo B‐Y, Hung C‐H, et al.; Investigators’ Study Group . Safety and efficacy of tigatuzumab plus sorafenib as first‐line therapy in subjects with advanced hepatocellular carcinoma: A phase 2 randomized study. J Hepatol 2015;63:896‐904. [DOI] [PubMed] [Google Scholar]
- 16. Raggi C, Factor VM, Seo D, Holczbauer A, Gillen MC, Marquardt JU, et al. Epigenetic reprogramming modulates malignant properties of human liver cancer. Hepatology 2014;59:2251‐2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marquardt JU, Raggi C, Andersen JB, Seo D, Avital I, Geller D, et al. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology 2011;54:1031‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, et al. Methods for high‐throughput drug combination screening and synergy scoring. Methods Mol Biol 2018;1711:351‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang H, et al. EpCAM‐positive hepatocellular carcinoma cells are tumor‐initiating cells with stem/progenitor cell features. Gastroenterology 2009;136:1012‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kawai T, Yasuchika K, Ishii T, Katayama H, Yoshitoshi EY, Ogiso S, et al. Keratin 19, a cancer stem cell marker in human hepatocellular carcinoma. Clin Cancer Res 2015;21:3081‐3091. [DOI] [PubMed] [Google Scholar]
- 22. Lee J‐S, Heo J, Libbrecht L, Chu I‐S, Kaposi‐Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12:410‐416. [DOI] [PubMed] [Google Scholar]
- 23. Hayashi H, Higashi T, Yokoyama N, Kaida T, Sakamoto K, Fukushima Y, et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell‐like behaviors contributing to disease progression. Cancer Res 2015;75:4985‐4997. [DOI] [PubMed] [Google Scholar]
- 24. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 2014;94:1287‐1312. [DOI] [PubMed] [Google Scholar]
- 25. Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, et al. A novel YAP1 inhibitor targets CSC‐enriched radiation‐resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol Cancer Ther 2018;17:443‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castven D, Fischer M, Becker D, Heinrich S, Andersen JB, Strand D, et al. Adverse genomic alterations and stemness features are induced by field cancerization in the microenvironment of hepatocellular carcinomas. Oncotarget 2017;8:48688‐48700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teng CF, Jeng LB, Shyu WC. Role of insulin‐like growth factor 1 receptor signaling in stem cell stemness and therapeutic efficacy. Cell Transplant 2018;27:1313‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hart LS, Dolloff NG, Dicker DT, Koumenis C, Christensen JG, Grimberg A, et al. Human colon cancer stem cells are enriched by insulin‐like growth factor‐1 and are sensitive to figitumumab. Cell Cycle 2011;10:2331‐2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osuka S, Sampetrean O, Shimizu T, Saga I, Onishi N, Sugihara E, et al. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells 2013;31:627‐640. [DOI] [PubMed] [Google Scholar]
- 30. Du LI, Li Y‐J, Fakih M, Wiatrek RL, Duldulao M, Chen Z, et al. Role of SUMO activating enzyme in cancer stem cell maintenance and self‐renewal. Nat Commun 2016;7:12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730‐737. [DOI] [PubMed] [Google Scholar]
- 32. Ebben JD, Treisman DM, Zorniak M, Kutty RG, Clark PA, Kuo JS. The cancer stem cell paradigm: a new understanding of tumor development and treatment. Expert Opin Ther Targets 2010;14:621‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105‐111. [DOI] [PubMed] [Google Scholar]
- 34. Nunes T, Hamdan D, Leboeuf C, El Bouchtaoui M, Gapihan G, Nguyen T, et al. Targeting cancer stem cells to overcome chemoresistance. Int J Mol Sci 2018;19:4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marquardt JU, Thorgeirsson SS. Sall4 in “stemness”‐driven hepatocarcinogenesis. N Engl J Med 2013;368:2316‐2318. [DOI] [PubMed] [Google Scholar]
- 36. Marquardt JU, Gomez‐Quiroz L, Arreguin Camacho LO, Pinna F, Lee YH, Kitade M, et al. Curcumin effectively inhibits oncogenic NF‐kappaB signaling and restrains stemness features in liver cancer. J Hepatol 2015;63:661‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J‐F, et al.; SHARP Investigators Study Group . Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 38. Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin 2017;38:614‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tovar V, Cornella H, Moeini A, Vidal S, Hoshida Y, Sia D, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017;66:530‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qiu Y, Shan W, Yang Y, Jin M, Dai Y, Yang H, et al. Reversal of sorafenib resistance in hepatocellular carcinoma: epigenetically regulated disruption of 14‐3‐3eta/hypoxia‐inducible factor‐1alpha. Cell Death Discov 2019;5:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cao W, Li M, Liu J, Zhang S, Noordam L, Verstegen MMA, et al. LGR5 marks targetable tumor‐initiating cells in mouse liver cancer. Nat Commun 2020;11:1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ezzoukhry Z, Louandre C, Trécherel E, Godin C, Chauffert B, Dupont S, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer 2012;131:2961‐2969. [DOI] [PubMed] [Google Scholar]
- 43. Asnacios A, Fartoux L, Romano O, Tesmoingt C, Louafi S S, Mansoubakht T, et al. Gemcitabine plus oxaliplatin (GEMOX) combined with cetuximab in patients with progressive advanced stage hepatocellular carcinoma: results of a multicenter phase 2 study. Cancer 2008;112:2733‐2739. [DOI] [PubMed] [Google Scholar]
- 44. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell 2016;29:783‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weiler SME, Pinna F, Wolf T, Lutz T, Geldiyev A, Sticht C, et al. Induction of chromosome instability by activation of yes‐associated protein and forkhead box M1 in liver cancer. Gastroenterology 2017;152:2037‐2051.e2022. [DOI] [PubMed] [Google Scholar]
- 46. Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 2014;13:63‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zanconato F, Cordenonsi M, Piccolo S. YAP and TAZ: a signalling hub of the tumour microenvironment. Nat Rev Cancer 2019;19:454‐464. [DOI] [PubMed] [Google Scholar]
- 48. Zhou Y, Wang Y, Zhou W, Chen T, Wu Q, Chutturghoon VK, et al. YAP promotes multi‐drug resistance and inhibits autophagy‐related cell death in hepatocellular carcinoma via the RAC1‐ROS‐mTOR pathway. Cancer Cell Int 2019;19:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao J, Rong Y, Huang Y, Shi P, Wang X, Meng X, et al. Cirrhotic stiffness affects the migration of hepatocellular carcinoma cells and induces sorafenib resistance through YAP. J Cell Physiol 2019;234:2639‐2648. [DOI] [PubMed] [Google Scholar]
- 50. Wei T, Weiler SME, Tóth M, Sticht C, Lutz T, Thomann S, et al. YAP‐dependent induction of UHMK1 supports nuclear enrichment of the oncogene MYBL2 and proliferation in liver cancer cells. Oncogene 2019;38:5541‐5550. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1‐S5
Table S6