

Abstract
The perturbations in bile acids (BAs) in alcohol‐associated hepatitis (AH) and its relationship to disease severity is not well defined. The aims of this study were to define (1) the effects of heavy alcohol consumption on BAs and related microbiome, (2) the additional changes with AH, and (3) the relationship of these changes to disease severity. In this multicenter study, plasma and fecal BAs and related microbiome were interrogated in healthy individuals, heavy drinking controls (HDCs) without overt liver disease, and AH. Compared to healthy controls, HDCs had increased glycine‐conjugated 7α and 27α primary BAs and increased secondary BA glycocholenic sulfate (multiple‐comparison adjusted P < 0.05 for all). Plasma‐conjugated cholic and chenodeoxycholic acid increased in AH along with the secondary BAs ursodeoxycholic and lithocholic acid (P < 0.001 for all), whereas deoxycholic acid decreased; however fecal concentrations of both deoxycholic acid and lithocholic acid were decreased. Glycocholenic acid further increased significantly from HDCs to AH. HDCs and AH had distinct plasma and fecal BA profiles (area under the curve, 0.99 and 0.93, respectively). Plasma taurochenodeoxycholic acid and tauroursodeoxycholic acid were directly related to disease severity, whereas fecal ursodeoxycholic acid was inversely related. The fecal abundance of multiple taxa involved in formation of secondary BAs, especially deoxycholic acid (Clostridium cluster XIVa) was decreased in AH. Multiple genera containing taxa expressing 3α, 3β, 7α, and 7β epimerases were decreased with concordant changes in fecal BAs that required these functions for formation. Conclusion: There are distinct changes in BA‐transforming microbiota and corresponding BAs in AH that are related to disease severity.
Abbreviations
- 7‐HOCA
7‐alpha‐hydroxy‐3‐oxo‐4‐cholestenoic acid
- AH
alcohol‐associated hepatitis
- AUC
area under the curve
- BA
bile acid
- BMI
body mass index
- C4
7α‐hydroxy‐4‐cholesten‐3‐one
- HC
healthy control
- HDC
heavy drinking control
- MAH
moderate alcohol‐associated hepatitis
- mca
multiple‐comparison adjusted
- MELD
Model for End‐Stage Liver Disease
- SAH
severe alcohol‐associated hepatitis
- VCU
Virginia Commonwealth University
Alcohol‐associated hepatitis, part of the spectrum of alcohol‐induced liver disease, has a high mortality rate.( 1 ) The incidence of alcohol‐associated hepatitis is increasing, and it is a leading cause for liver‐related hospitalizations and deaths.( 2 , 3 ) The specific factors driving the onset and severity of alcohol‐associated hepatitis are incompletely understood.( 4 , 5 ) The high prevalence, variable course, and limited therapies for this condition underscore the need for better understanding of the mechanisms driving the disease and identification of potential therapeutic targets.
A hallmark of alcohol‐associated hepatitis is cholestasis and elevated circulating bile acids.( 6 ) Bile acids have a multitude of biological effects impacting metabolism, inflammation, and even oncogenesis. These are mediated both by their physicochemical properties and through specific receptors, e.g., the farnesoid X receptor, Takeda G protein receptor 5, and sphingosine 1 phosphate receptor 2.( 7 ) Different bile acids have differential biological properties due to varying agonist activity at these receptors.( 8 ) Hydrophobic bile acids with less hydroxyl groups are thought to be more toxic as they have a greater propensity to traverse the cell membrane.( 9 ) Bile acids can also undergo gut microbial‐dependent epimerization at the 3‐, 7‐, and 12‐carbon hydroxyl groups, modifying their hydrophobicity and consequently their cellular toxicity.( 10 ) The potential effects of various forms of these bile acids have spurred efforts to better understand the differential changes in bile acids in alcohol‐associated hepatitis and other liver diseases. An area of particular interest is the role of the intestinal microbiome in converting primary bile acids to secondary bile acids.( 11 ) The microbiome itself is sensitive to bile acids with some microbiota dying in the presence of bile acids while others thrive and overgrow when specific bile acids are increased in their immediate environment.( 12 , 13 ) We have previously described changes in both the fecal and circulating microbiome in alcohol‐associated hepatitis, but the changes in bile acids and the bile acid biome remain to be elucidated.( 14 , 15 )
Two recent studies have evaluated the bile acid profile in alcohol‐associated hepatitis.( 16 , 17 ) One study only evaluated 13 patients with alcohol‐associated hepatitis and observed an increase in primary bile acids despite decreased de novo synthesis but did not address the differential changes in bile acid profiles and their linkage to the intestinal microbiome.( 16 ) In another study, changes in bile acid profile and microbial taxa were noted.( 17 ) However, this study included a mix of patients with alcohol‐associated hepatitis and alcohol‐induced cirrhosis and was further limited by a lack of controls with heavy alcohol consumption but without overt liver disease, preventing assessment of changes specifically due to alcohol‐associated hepatitis. Thus, there is a need for more data to obtain clarity on the changes in primary and secondary bile acids in alcohol‐associated hepatitis and on its relationship to the bile acid biome.
The goal of the current study was to characterize the plasma and fecal bile acids in patients with alcohol‐associated hepatitis, to identify distinct profiles from heavy alcohol consumption alone versus alcohol‐associated hepatitis, and to correlate bile acid profiles to changes in the fecal microbial ecology. We aimed to further distinguish changes in bile acid profiles between patients with moderate and severe alcoholic‐associated hepatitis (MAH, SAH, respectively). The specific objectives were to define (1) the effect of heavy alcohol consumption on the bile acid profiles and their relationship to the fecal microbiome, (2) changes in bile acids and related microbial taxa and function in alcohol‐associated hepatitis, and (3) the relationship of changes in bile acid profiles with disease severity and outcomes.
Patients and Methods
The study included three clinical sites (Virginia Commonwealth University [VCU], Indiana University, and Mayo Clinic) involved in the Translation of Rehabilitation Engineering Advances and Technology (TREAT) consortium for the study of alcohol‐associated hepatitis. Whereas patients with alcohol‐associated hepatitis and controls with heavy alcohol consumption but without overt liver disease were enrolled at all sites, healthy controls were only enrolled at VCU. All participants provided informed consent, and the study was approved by the institutional review board at each center.
Patient Population
Alcohol‐associated hepatitis was defined by the sudden onset of jaundice with hepatomegaly, using the ratio of aspartate aminotransferase to alanine aminotransferase >1 in an individual with a history of heavy alcohol consumption (>5 units daily) within 6 weeks of diagnosis.( 18 ) Patients with other comorbid liver diseases, active gastrointestinal bleeding, sepsis, as well as those receiving antibiotics or ursodeoxycholic acid at the time of diagnosis were excluded from this analysis. Patients prescribed lactulose or rifaximin for hepatic encephalopathy were also excluded. Alcohol‐associated hepatitis was considered to be MAH or SAH based on the Model for End‐Stage Liver Disease (MELD) score ≤20 versus those with higher levels.( 19 )
Heavy drinking controls were defined by a history of heavy alcohol consumption (>5 units daily) without overt evidence of liver disease (normal liver enzymes, normal liver function, and absence of jaundice or hepatomegaly). Healthy controls were participants without an alcohol use disorder (Alcohol Use Disorders Identification Test score <7) or liver disease. These individuals were asymptomatic, had a normal physical examination, had normal liver enzymes and functions, and an absence of sonographic evidence of liver disease or a controlled attenuation parameter score <250 dB/second and liver stiffness measurement <6 kPa on FibroScan.( 15 )
Plasma Sample Collection and Metabolite Identification
Plasma and serum were collected under fasted conditions and spun down within 60 minutes in all cases and stored at −70°C until they were withdrawn for this study. Samples were transported to Metabolon for analyses on dry ice. All samples were thawed only once at the time of analysis for this study. Sample processing and metabolite identification are described in the Supporting Materials.
Stool Sample Collection, Fecal Metabolite, and Microbial Taxa Identification
A standardized approach to fresh stool collection was established, and a standard operating procedure was put in place. This was based on a study of the stool microbiome.( 20 ) All clinical personnel involved in stool collection were formally trained and also provided written resources and a video on YouTube as additional resources. Approximately 500 mg of feces was divided and transferred into and empty tube and another containing 10 mL of RNA later. This was then shaken thoroughly, taking care to avoid spillage. The tubes were placed in Ziplock bags packed with ice for transportation to the laboratory. Stool samples were transported on dry ice from the clinical research unit at VCU to the principal investigator’s laboratory where they were stored at −70°C until they were analyzed. Fecal microbial composition was evaluated by 16S pyrosequencing and metabolomic analyses using liquid chromatography–mass spectrometry, as described.( 14 ) Samples for bile acid analysis were transported to Metabolon for analyses on dry ice. All samples were thawed only once at the time of analysis for this study. Fecal and plasma samples for bile acids were processed by the same laboratory using the same methods. A more detailed description of these methods is provided in the Supporting Materials.
Statistical Analysis
Statistical data analysis was performed using R software. Individual metabolite compound measurements in stool and plasma were first normalized using median scaling to set the median equal to 1( 21 ); missing values were then imputed with the minimum, and final intensity data were log transformed to reduce the skewness. To assure the stability of reported results toward the median‐scaling normalization procedure, sensitivity analyses were performed by reproducing all analyses using absolute concentrations. To detect statistically significant differences between each group of interest (healthy controls and heavy drinking controls; heavy drinking controls and alcohol‐associated hepatitis; and MAH and SAH), Welsh’s unpaired two‐sample t test was performed using the R function t_test() in package rstatix. Multiple‐testing correction was performed using Benjamini‐Hochberg correction implemented in R function adjust_pvalue(). Total primary, secondary, conjugated, and unconjugated bile acid values were obtained by summing measures for corresponding compounds. Correlation between bile acids and microbial genera was calculated using the cor() function in R. Microbial genera discriminating between AH and heavy drinking control groups was identified according to described methods.( 14 ) Predictive models were built using random forest models by function randomForest() in the R package randomForest. The optimal number of bile acids used at each split of the random forest (parameter mtry) was determined using 5‐fold cross‐validation criteria by the R function train() in package caret. Mean decrease Gini was used to rank variable importance in random forest models. Overall predictive ability of the random forest model was accessed using the area under the receiving operating characteristic curve through the R function roc() in the package pROC. Association between continuous MELD score and individual metabolites was accessed using univariate regression models (function lm() in R) with MELD score as an outcome and each metabolite as a single predictor. Association between individual bile acid compounds in stool and plasma with demographic covariates (age, sex, and body mass index [BMI]) was performed using the multivariate linear regression model with bile acids as a response and demographic covariates as predictors.
Linking Bile Acid Changes to Fecal Microbiome
The microbial taxa expressing critical enzymes involved in fecal microbial bile acid transformations were first identified from existing databases using an assembly of gut organisms through reconstruction and analysis resource.( 22 , 23 ) The species were then identified to genus from the National Center for Biotechnology Information taxonomy browser. The differentially abundant taxa in alcohol‐associated hepatitis study population were mapped to this list of bile acid‐metabolizing microbiota to define the relationships between bile acid‐metabolizing microbiota and the changes in bile acid composition in alcohol‐associated hepatitis.
Results
Study Cohort
In total, 59 subjects had available plasma (20 healthy controls, 12 heavy drinking controls, 11 MAH, 16 SAH), while 49 subjects had available stool (20 heavy drinking controls, 8 MAH, 21 SAH). The distribution of patients with stool and plasma samples was similar across study groups for all demographic characteristics except white blood count. The healthy controls and heavy drinking controls were younger than those with MAH and SAH. Race, sex, and BMI distributions were not significantly different across study groups. The groups of heavy drinking controls and patients with MAH and SAH were comparable in terms of average alcohol consumption. The patients with alcohol‐associated hepatitis had lower hemoglobin, albumin, and platelets. White blood count for the patients with alcohol‐associated hepatitis who donated stool samples was significantly higher, while for the patients with alcohol‐associated hepatitis who donated plasma samples, white blood count was higher but not significantly different. The patients with alcohol‐associated hepatitis had significantly higher international normalized ratio, MELD, Child‐Pugh, and discriminant function scores. A detailed breakdown of patient demographics is provided in Table 1.
TABLE 1.
Characteristics of the Study Cohort* for Patients With Stool and Plasma Samples
| Stool | Plasma | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HDC | MAH | SAH | P Value | HC | HDC | MAH | SAH | P Value | |
| Number of participants | 20 | 8 | 21 | 20 | 12 | 11 | 16 | ||
| Age (years), mean (SD) | 46.39 (13.63) | 54.33 (7.09) | 43.21 (11.36) | 0.089 | 39.85 (11.32) | 36.50 (13.45) | 51.19 (9.57) | 48.85 (6.76) | 0.008 |
| Sex (%) | 0.148 | 0.767 | |||||||
| M | 12 (60.0) | 7 (87.5) | 10 (47.6) | 10 (50) | 8 (66.7) | 7 (63.6) | 10 (62.5) | ||
| F | 8 (40.0) | 1 (12.5) | 11 (52.4) | 10 (50) | 4 (33.3) | 4 (36.4) | 6 (37.5) | ||
| Race (%) | 0.421 | 0.187 | |||||||
| White | 18 (90.0) | 7 (87.5) | 21 (100) | 19 (95.0) | 9 (75.0) | 8 (72.7) | 15 (93.8) | ||
| Black or African American | 1 (5) | 1 (12.5) | 0 (0) | 0 (0) | 2 (16.7) | 3 (27.3) | 1 (6.2) | ||
| Asian | 0 (0) | 0 (0) | 0 (0) | 1 (5) | 0 (0) | 0 (0) | 0 (0) | ||
| More than one race | 1 (5) | 0 (0) | 0 (0) | 0 (0) | 1 (8.3) | 0 (0) | 0 (0) | ||
| BMI, mean (SD) | 29.56 (7.51) | 26.92 (2.60) | 28.35 (6.09) | 0.673 | 26.81 (6.46) | 25.08 (6.73) | 25.02 (3.82) | 27.67 (5.95) | 0.594 |
| Average number of drinks per day, mean (SD) | 17.32 (13.42) | 16.40 (13.71) | 12.44 (9.13) | 0.434 | — | 11.45 (8.30) | 6.90 (3.95) | 7.35 (4.21) | 0.110 |
| Hemoglobin (g/dL), mean (SD) | 13.21 (1.43) | 9.84 (1.82) | 10.36 (1.90) | <0.001 | 13.97 (1.61) | 11.86 (2.59) | 10.00 (2.34) | 9.78 (2.39) | <0.001 |
| AST ( U/L), mean (SD) | 25.00 (8.27) | 158.20 (91.16) | 114.50 (44.97) | <0.001 | 83.33 (171.40) | 28.27 (11.91) | 141.70 (97.87) | 109.56 (29.47) | 0.036 |
| ALT (U/L), mean (SD) | 25.11 (12.36) | 63.20 (42.86) | 40.40 (21.07) | 0.002 | 36.89 (34.44) | 21.18 (9.87) | 44.30 (23.37) | 49.31 (55.93) | 0.308 |
| Bilirubin (mg/dL), mean (SD) | 0.44 (0.27) | 7.06 (3.76) | 19.57 (11.36) | <0.001 | 0.62 (0.49) | 0.55 (0.47) | 4.88 (2.60) | 17.16 (9.64) | <0.001 |
| Albumin (g/dL), mean (SD) | 4.06 (0.52) | 2.66 (0.15) | 2.82 (0.52) | <0.001 | 4.36 (0.24) | 3.55 (0.63) | 2.74 (0.51) | 2.78 (0.61) | <0.001 |
| WBC (×109 cells/L), mean (SD) | 6.75 (2.69) | 13.74 (8.70) | 16.56 (12.83) | 0.008 | 7.18 (1.66) | 9.00 (3.34) | 9.89 (6.58) | 10.05 (5.12) | 0.451 |
| Platelets (×109/L), mean (SD) | 265.89 (52.83) | 207.60 (175.51) | 155.00 (88.64) | 0.002 | 258.33 (89.63) | 287.36 (146.47) | 145.90 (83.98) | 154.94 (88.92) | 0.003 |
| Creatinine (mg/dL), mean (SD) | 0.89 (0.35) | 0.67 (0.31) | 1.09 (0.71) | 0.257 | 0.89 (0.16) | 0.83 (0.41) | 0.74 (0.20) | 0.93 (0.54) | 0.652 |
| INR, mean (SD) | 1.00 (0.15) | 1.37 (0.20) | 2.07 (0.40) | <0.001 | — | 1.13 (0.23) | 1.50 (0.41) | 2.04 (0.45) | <0.001 |
| MELD score, mean (SD) | 7.37 (1.74) | 14.62 (3.85) | 26.81 (5.32) | <0.001 | — | 8.50 (3.50) | 15.73 (4.22) | 25.38 (3.93) | <0.001 |
| Child‐Pugh score, mean (SD) | 5.21 (0.43) | 9.00 (0.71) | 10.45 (1.64) | <0.001 | — | 5.70 (0.67) | 8.70 (1.70) | 10.31 (1.74) | <0.001 |
| DF score, mean (SD) | 12.54 (8.79) | 35.95 (7.00) | 81.33 (31.75) | <0.001 | — | ‐2.60 (11.66) | 22.04 (20.72) | 59.31 (21.56) | <0.001 |
6 patients had both stool and plasma samples.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; DF, discriminant function; F, female; INR, international normalized ratio; M, male; WBC, white blood count.
Alcohol‐Associated Hepatitis Is Associated With Significant Changes in Total Primary and Secondary Bile Acids
The total primary plasma bile acid levels were not significantly different in heavy drinking controls compared to healthy controls (Fig. 1A). The total primary bile acid levels in plasma were, however, significantly elevated in both MAH and SAH compared to either healthy controls or heavy drinking controls (multiple‐comparison adjusted [mca] P < 0.001 for all comparisons; Table 2 and Fig. 1A). The total secondary bile acid levels in plasma were not different across groups (Fig. 1A). The increase in primary bile acids in plasma in alcohol‐associated hepatitis groups was reflected in an increase in the relative proportion of primary bile acids and a decrease in secondary bile acids (Fig. 1B). This was accompanied by an increase in 7‐alpha‐hydroxy‐3‐oxo‐4‐cholestenoic acid (7‐HOCA) in both MAH and SAH groups (Fig. 1C) compared to healthy controls and heavy drinking controls, indicating the possibility of increased bile acid synthesis in alcohol‐associated hepatitis even in the face of cholestasis and increased bile acid levels.
FIG. 1.
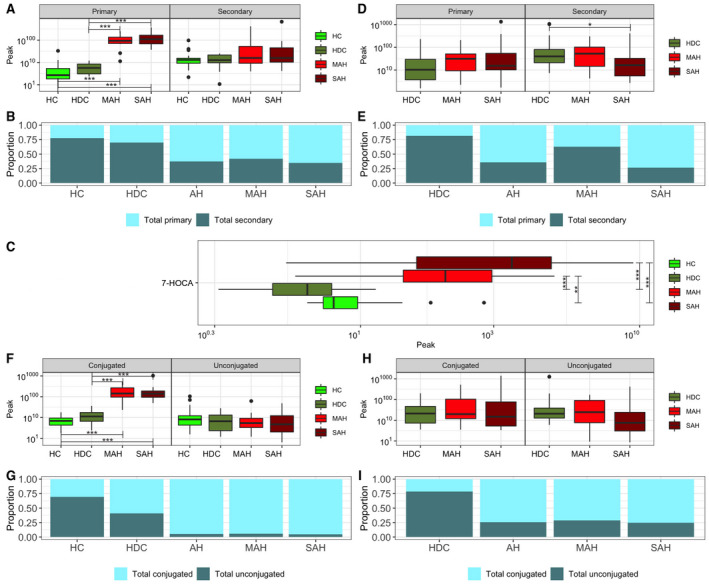
Plasma and stool bile acid profile comparison in each disease group. (A) Boxplots of total primary and secondary plasma bile acids; the y‐axis labels are displayed on the original scale even though the data are log10 transformed. (B) Proportion of total primary and secondary plasma bile acids. (C) Boxplot of 7‐HOCA in plasma; the x‐axis labels are displayed on the original scale even though the data are log10 transformed. (D) Boxplots of total primary and secondary stool bile acids; the y‐axis labels are displayed on the original scale even though the data are log10 transformed. (E ) Proportion of total primary and secondary stool bile acids. (F) Boxplots of total conjugated and unconjugated plasma bile acids; the y‐axis labels are displayed on the original scale even though the data are log10 transformed. (G) Proportion of total conjugated and unconjugated plasma bile acids. (H) Boxplots of total conjugated and unconjugated stool bile acids; the y‐axis labels are displayed on the original scale even though the data are log10 transformed. (I) Proportion of total conjugated and unconjugated stool bile acids. Horizontal segment endpoints in each plot represent the significant results for comparison between groups used in the two sample t test (e.g., between HDC and SAH); *P ≤ 0.1; **P ≤ 0.05; ***P ≤ 0.001.
TABLE 2.
Total Primary and Secondary Bile acids comparisons with Significance Values (Adjusted for Multiple Comparisons) for the Total Primary and Secondary Bile Acids Pairwise Group Comparisons (Arrows Indicate Direction of Change)
| Comparison | Plasma | Stool | ||||||
|---|---|---|---|---|---|---|---|---|
| Total Primary | Total Secondary | Total Primary | Total Secondary | |||||
| Estimate | P Value | Estimate | P Value | Estimate | P Value | Estimate | P Value | |
| HDC–HC | ↑ 0.216 | 0.3259 | ↓ −0.064 | 0.984 | — | — | — | — |
| MAH–HC | ↑ 1.413 | <0.001 | ↑ 0.299 | 0.366 | — | — | — | — |
| SAH–HC | ↑ 1.523 | <0.001 | ↑ 0.241 | 0.456 | — | — | — | — |
| MAH–HDC | ↑ 1.197 | <0.001 | ↑ 0.305 | 0.291 | ↑ 0.316 | 0.520 | ↓ −0.101 | 0.906 |
| SAH–HDC | ↑ 1.305 | <0.001 | ↑ 0.305 | 0.364 | ↑ 0.270 | 0.430 | ↓ −0.502 | 0.018 |
| SAH–MAH | ↑ 0.108 | 0.853 | ↓ −0.058 | 0.990 | ↑ 0.047 | 0.985 | ↓ −0.402 | 0.215 |
In stool, in SAH compared to heavy drinking controls, a modest nonsignificant increase in primary bile acids was noted while secondary bile acid levels decreased significantly (mca P = 0.018; Fig. 1D). These contributed to a decrease in the proportion of secondary bile acids among all measured bile acids in this group (Fig. 1E).
The increase in total plasma bile acids in both MAH and SAH was due to an increase in conjugated bile acids (mca P < 0.001 for each comparison; Fig. 1F,G). However, the fecal concentrations of conjugated bile acids were not significantly different in those with alcohol‐associated hepatitis compared to either healthy controls or heavy drinking controls (Fig. 1H). There was a borderline significant decrease in fecal concentrations of unconjugated bile acids in SAH (mca P = 0.069 vs. heavy drinking controls; Fig. 1H). However, the proportion of unconjugated bile acids in stool was decreased but similar between MAH and SAH (Fig. 1I).
Changes in Individual Bile Acids in Plasma Due to Heavy Alcohol Consumption
To characterize the effect of alcohol consumption on the plasma bile acids, the levels of plasma bile acids of individuals with heavy alcohol consumption who had not developed alcohol‐associated hepatitis were compared to healthy controls. The glycine‐conjugated 7α and 27α primary bile acids were increased in plasma of heavy drinking controls compared to healthy controls (Fig. 2). Specifically, the abundance of glycochenodeoxycholate (mca P = 0.005), glycochenodeoxycholate sulfate (mca P = 0.022), and glycocholate (mca P < 0.001) was increased in heavy drinking controls compared to healthy controls. The levels of deoxycholate were decreased, while those of taurochenodeoxycholate, tauroursodeoxycholate, and sulfated glycocholenate sulfate* were increased significantly based on unadjusted P values in heavy drinking controls, but these were not significant following correction for multiple comparisons.
FIG. 2.
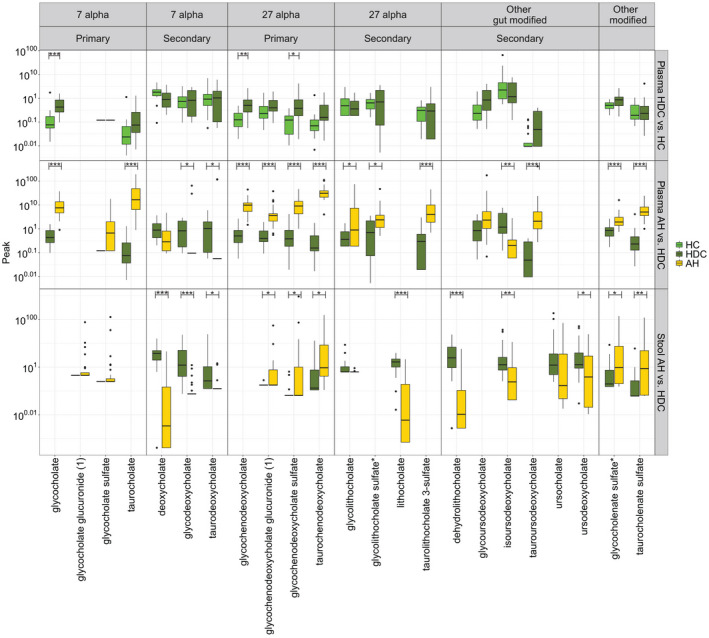
Significant differences in stool and plasma bile acids in comparisons of HDCs versus HCs and AH versus HDCs; the y‐axis labels are displayed on the original scale even though the data are log10 transformed. Horizontal segment endpoints in each plot represent the significant results for comparison between individual metabolites in the two sample t test; *P ≤ 0.1; **P ≤ 0.05; ***P ≤ 0.001.
Changes in Individual Circulating Bile Acids With Development of Alcohol‐Associated Hepatitis
In order to identify changes in bile acids specifically with alcohol‐associated hepatitis and not just alcohol exposure, the bile acid profiles in the combined alcohol‐associated hepatitis groups were compared to those in heavy drinking controls. Multiple primary and secondary bile acids were significantly different in plasma of patients with alcohol‐associated hepatitis (Fig. 2). Conjugated primary 7α bile acids (glycocholate, taurocholate) and 27α bile acids (taucochenodoxycholate, glycochenodeoxycholate) were significantly increased in alcohol‐associated hepatitis (mca P < 0.001 for all). There was also a highly significant increase in glucuronidated glycochenodeoxycholate and sulfated glycochenodeoxycholate bile acids in plasma (mca P < 0.001 for both).
Although the 7α primary bile acids were increased, their secondary gut‐derived metabolites deoxycholate (unadjusted P = 0.032; mca P = 0.090) and its taurine‐conjugated form taurodeoxycholate (mca P = 0.034) and its glycine‐conjugated form glycodeoxycholate (mca P = 0.034) were all decreased in alcohol‐associated hepatitis. However, glycocholenate sulfate* (mca P = 0.001) and taurocholenate sulfate (mca P < 0.001) were significantly increased. In contrast to the decrease in deoxycholate, glycine‐conjugated secondary bile acid glycolithocholate was increased (mca P = 0.048) as were the sulfated forms of glycolithocholate (mca P = 0.043) and taurolithocholate (mca P < 0.001). Among other gut‐modified secondary bile acids, tauroursodeoxycholate (mca P < 0.001) was increased and isoursodeoxycholate (mca P = 0.005) was decreased in patients with alcohol‐associated hepatitis.
Changes in Individual Fecal Bile Acids With Development of Alcohol‐Associated Hepatitis
Multiple primary and secondary bile acids were significantly different in stool of patients with alcohol‐associated hepatitis compared to heavy drinking controls (Fig. 2). Most fecal bile acids were decreased, and only conjugated primary bile acids remained increased. Further, all the bile acids that were decreased in stool of patients with alcohol‐associated hepatitis were gut derived. Specifically, among secondary bile acids derived from cholic acid, deoxycholate (mca P < 0.001), glycodeoxycholate (mca P < 0.001), and taurodeoxycholate (mca P = 0.042) were decreased while glycocholenate sulfate* (mca P = 0.029) and taurocholenate sulfate (mca P = 0.006) were increased in patients with alcohol‐associated hepatitis. The 27α primary bile acids glycochenodeoxycholate‐glucuronide (mca P = 0.029), glycochenodeoxycholate sulfate (mca P = 0.042), and taurochenodeoxycholate (mca P = 0.028) were increased in alcohol‐associated hepatitis. The related secondary bile acids lithocholate (mca P < 0.001), dehyrdolithocholate (mca P < 0.001), isoursodeoxycholate (mca P = 0.002), and ursodeoxycholate (mca P = 0.029) were significantly decreased in alcohol‐associated hepatitis. As noted in plasma, all of the measurable sulfated bile acids in stool were increased in alcohol‐associated hepatitis compared to heavy drinking controls.
Sensitivity analysis based on individual stool and plasma bile acid absolute concentrations revealed identical results (Supporting Figs. S4 and S5).
Relationship of Changes in Bile Acids With Varying Severity of Alcohol‐Associated Hepatitis
To interrogate the differences in plasma and stool bile acids with increasing severity of alcohol‐associated hepatitis, we compared the patients with MAH and SAH. While no statistically significant changes were detected in MAH versus SAH, we observed that most bile acids that had statistically significant differences in either (1) healthy controls to heavy drinking controls or (2) heavy drinking controls to alcohol‐associated hepatitis comparisons were decreased in SAH compared to MAH (Fig. 3A). We further investigated the association between continuous MELD score as an indicator of disease severity and the concentration of individual microbial metabolites in univariate regression models in patients with alcohol‐associated hepatitis. Among all plasma bile acids, taurochenodeoxycholate (P = 0.022, R 2 = 0.192) and tauroursodeoxycholate (P = 0.030, R 2 = 0.174) had a significant positive association with an increase in MELD score (Fig. 3B,C; Supporting Table S1). Among all stool bile acids, ursodeoxycholate sulfate (1) had a significant negative association with an increase in MELD score (P = 0.049, R 2 = 0.136) (Fig. 3D). Glycoursodeoxycholate (P = 0.073, R 2 = 0.114) and glychodeoxycholate (P = 0.102, R 2 = 0.096) had a borderline significant negative association with an increase in MELD score (Supporting Table S2). These results show that these bile acids are associated with disease severity as measured by the MELD score in the population of patients with alcohol‐associated hepatitis; however, the low R 2 values limit the clinical potential of these findings.
FIG. 3.
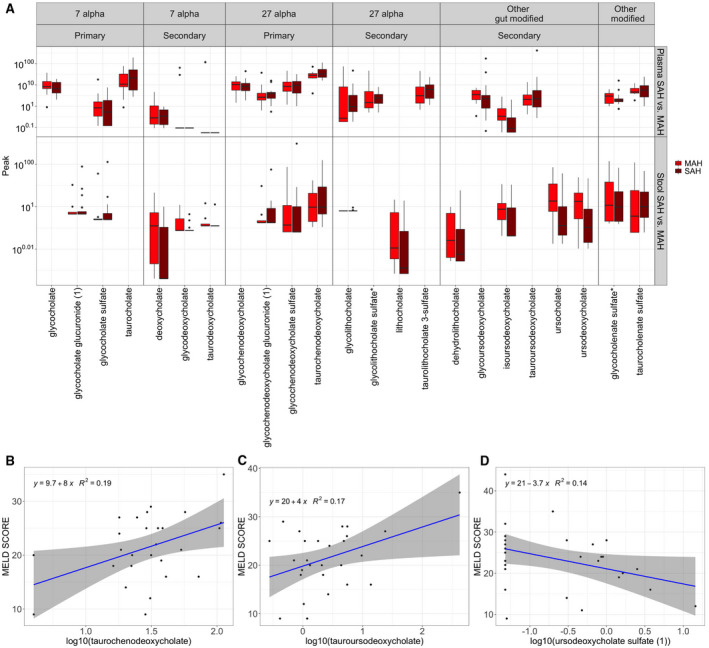
Alternations in bile acids due to severity of AH. (A) Boxplots of the plasma and stool bile acids that have significant differences across all three pairwise comparisons (HDC vs. HC, AH vs. HDC, and SAH vs. MAH); the y‐axis labels are displayed on the original scale even though the data are log10 transformed. (B) Association between MELD score and taurodeoxycholate in plasma. (C) Association between MELD score and tauroursodeoxycholate in plasma. (D) Association between MELD score and ursoodeoxycholate in stool.
Plasma and Fecal Bile Acid Signatures of Alcohol‐Associated Hepatitis
Plasma and stool bile acids discriminated between alcohol‐associated hepatitis and heavy drinking controls in random forest models (plasma area under the curve [AUC], 0.991; stool AUC, 0.936) (Fig. 4A,B). Increase in bile acids is expected in hepatic disorders, which limits the diagnostic value of these models. However, these results indicate the high quality of the random forest models fit and allow identifying a combination of key bile acids that accurately discriminate patients with alcohol‐associated hepatitis from heavy drinking controls. Taurochenodeoxycholate, glycocholate, taurocholate, tauroursodeoxycholate, and taurolithocholate 3‐sulfate were the top five plasma bile acids associated with alcohol‐associated hepatitis (Fig. 4C). Deoxycholate, glychodeoxycholate, lithocholate, dehyrdolithocholate, and isoursodeoxycholate were the top five stool bile acids diagnostic of alcohol‐associated hepatitis (Fig. 4D). The bile acid profile was not, however, able to discriminate between MAH and SAH (plasma AUC, 0.574; stool AUC, 0.446) (Supporting Fig. S1).
FIG. 4.
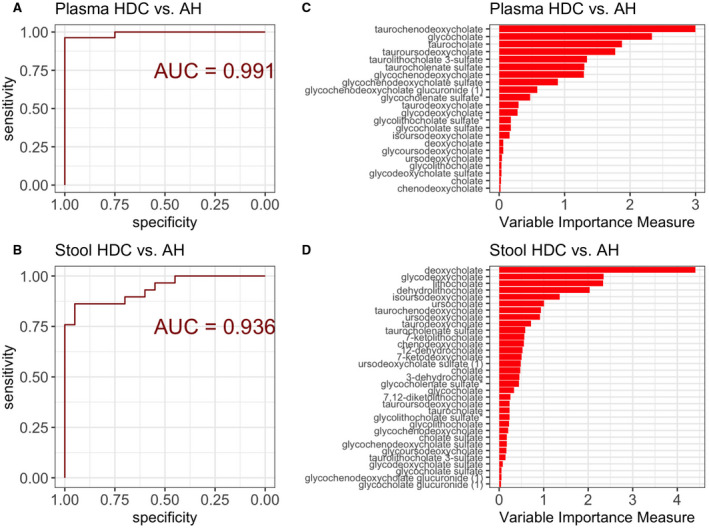
Plasma and stool metabolites predictive model of alcoholic hepatitis. (A) ROC curve for the predictive model of AH using plasma metabolites. Larger AUROC curve value corresponds to stronger predictive ability of the model to discriminate patients with HDC from AH. (B) ROC curve for the predictive model of AH using stool metabolites. Larger AUROC value corresponds to stronger predictive ability of the model to discriminate patients with HDC from AH. (C) Plasma metabolites variable importance plot in the classification model of HDC versus AH; longer bars correspond to higher importance of that metabolite. (D) Stool metabolites variable importance plot in the classification model of HDC versus AH; longer bars correspond to higher importance of that metabolite. Abbreviation: AUROC, area under the receiver operating characteristic curve.
Relationship of Changes in Fecal Bile Acids and the Microbiome
Bile acids can affect microbiota by either increasing the growth of taxa that thrive in the presence of bile acids or decreasing the abundance of taxa sensitive to the antibiotic effects of bile acids. These would be expected to be reflected in the composition and correlations of specific taxa and specific bile acids.
Within the heavy drinking controls, a positive correlation between multiple bile acids and the bile acid‐metabolizing genus Ruminococcus 2 (Lachnospiraceae family), Peptostreptococcus (Peptostreptococeae family), and Anaerofilium (Ruminococcaceae family) was noted, while a negative correlation between Blautia (Lachnospiraceae family) and dehydrolithocholate was also seen. Several members of Lachnospiraceae (e.g., genus Anaerostipes) and Coriobacteriaceae (e.g., genus Slackia) were related to ursodeoxycholate levels (Fig. 5A). Importantly, no relationships between taxa related to the family Enterobacteriaceae and bile acids were observed.
FIG. 5.
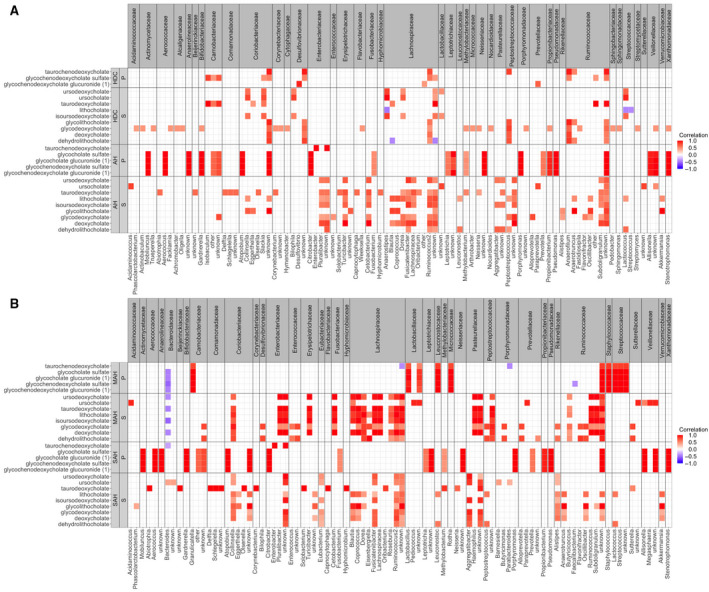
Correlation heatmaps between stool metabolites that are significant compared between HDC versus AH with stool microbiome. Family level classifications are boxed in gray on the x axis, while genus level is unboxed on the x axis. (A) Correlation heatmaps for patient groups with HDC and AH. (B) Correlation heatmaps for patient groups with MAH and SAH. Only correlations >0.4 are shown. Abbreviations: P, primary bile acids; S, secondary bile acids.
In contrast, numerous Enterobacteria, such as Citrobacter, Enterobacter, and Puralibacter, were positively correlated with several fecal bile acids in alcohol‐associated hepatitis. Of these, Citrobacter was most strongly correlated with glycine conjugates of both cholic and chenodeoxycholic acid. The levels of these specific bile acids in stool were also strongly correlated with families Actinomycetaceae, Bifidobacteriaceae, Camobacteriaceae, Prevotellaceae, Pseudomonaceae, Propionibacteriaceae, and Veillonellaceae. Other novel relationships seen principally in alcohol‐associated hepatitis included a positive correlation between members of the Erysipelotrichaceae (e.g., Turicibacter) and Fusobacteriaceae and Veillonellaceae on one hand and several fecal bile acids on the other.
Changes in Relationships Between Fecal Bile Acids and the Microbiome in MAH Versus SAH
Mostly positive correlations between microbiota and fecal bile acids were noted in both MAH and SAH (Fig. 5B). A striking exception was Bacteroidaceae (genus Bacteroides), which was inversely related to bile acid levels in both MAH and SAH. This was accompanied by an inversion of the Firmicutes to Bacteroides ratio in alcohol‐associated hepatitis compared to heavy drinking controls.( 14 ) Strong positive correlations between multiple genera in Lachnospiraceae and multiple 7α and 27α bile acids were seen in MAH and weakened or disappeared in SAH. The linkages of microbiota to glycocholate sulfate, glycochenodeoxycholate sulfate, and glucuronides noted in alcohol‐associated hepatitis above were found to be present mainly in the SAH group, with the exception of Streptoccaceae and Staphylococcaceae where such a relationship was only noted for MAH.
Functional Correlates of Changes in the Bile Acid Biome Are Linked to Altered Bile Acid Profile in Alcohol‐Associated Hepatitis
The intestinal microbiome also affects the bile acid profile by conversion of primary bile acids to secondary bile acids. To interrogate how changes in the microbiome related to changes in bile acids with heavy alcohol consumption and in alcohol‐associated hepatitis, we first screened the microbial taxa that had a differentially altered abundance in alcohol‐associated hepatitis( 14 ) to identify those that were involved in bile acid metabolism (Table 3). All bile acid‐metabolizing taxa that were differentially abundant were decreased in alcohol‐associated hepatitis compared to heavy drinking controls, with the exception of Fusobacteriaceae Fusobacterium, which was increased.
TABLE 3.
Microbial Taxa Significant in AH Compared to HDCs That are Known to Produce Bile acid‐Metabolizing Enzymes Relevant to Significant Changes in Bile Acids
| Family* (Taxonomy Identifier † ) | Lachnospiraceae | Lachnospiraceae | Eggerthellaceae | Erysipelotrichaceae | Fusobacteriaceae | |
|---|---|---|---|---|---|---|
| Genus* (taxonomy identifier † ) | Lachnoclostridium (Clostridium XIVa) | Lachnoclostridium (Clostridium XIVa) | Gordonibacter | Holdemania | Fusobacterium | |
| Species* | hylemonae; scindens | symbiosum | pamelaeae | filiformis | varium | |
| Direction of change in taxa | ||||||
| Direction of change in bile acids | ↓ in AH | ↓in AH | ↓in AH | ↓in AH | ↑ in AH | |
| Deoxycholate | ↓ in AH | 7‐deOH | ||||
| Glycodeoxycholate | ↓ in AH | 7‐deOH | ||||
| Lithocholate | ↓ in AH | 7‐deOH | ||||
| Dehydrolithocholate | ↓ in AH | 7‐deOH | 3alpha/3beta‐HSDH | 3alpha/3beta‐HSDH | 3alpha/3beta‐HSDH | |
| Isoursodeoxycholate | ↓ in AH | 7alpha/7beta‐HSDH | 3alpha/3beta‐HSDH; 7alpha/7beta‐HSDH | 3alpha/3beta‐HSDH | 3alpha/3beta‐HSDH; 7alpha/7beta‐HSDH | 7alpha/7beta‐HSDH |
| Ursocholate | ↓ in AH | 7alpha/7beta‐HSDH | 7alpha/7beta‐HSDH | 7alpha/7beta‐HSDH | ||
| Ursodeoxycholate | ↓ in AH | 7alpha/7beta‐HSDH | 7alpha/7beta‐HSDH | 7alpha/7beta‐HSDH | ||
| Tautolithocholate‐3‐sulfate | ↑ in AH | 7‐deOH | 7‐deOH | |||
| Taurodeoxycholate | ↓ in AH | 7‐deOH | ||||
Fecal levels of the secondary bile acids deoxycholate and lithocholate were decreased in alcohol‐associated hepatitis compared to heavy drinking controls. Concordant to this reduction, there was a reduction in Clostridium cluster XIVa, which contains the species Clostridium hylemonae and C. scindens, which are known to be critically related to the 7α dehydroxylation of the primary bile acids cholic acid to deoxycholate and chenodeoxycholate to lithocholate.
Bile acids formed by epimerization at the 7‐hydroxy group (ursodeoxycholate, ursocholate, isoursodeoxycholic acid) were decreased in stool in alcohol‐associated hepatitis and inversely related to the MELD score, as noted above. Ursodeoxycholate and ursocholate requires epimerization of the 7‐hydroxy group by 7α and 7β epimerases.( 24 ) There was a reduction in genera containing taxa involved in this epimerization of the 7‐hydroxy group in alcohol‐associated hepatitis; these included Clostridium cluster XIVa (C. hylemonae, C. sciendens, C. symbiosum species) and Erysipelotrichaceae Holdemania species (Holdemania filiformis species). The abundance of Fusobacteriaceae Fusobacterium (Fusobacterium varium species), which is also involved in 7‐hydroxy epimerization, was higher in alcohol‐associated hepatitis compared to heavy drinking controls.
The formation of isoursodeoxycholic acid involves further epimerization of the 3‐hydroxy group of ursodeoxycholic acid by 3α and 3β hydroxysteroid dehydrogenases. There was also a reduction in taxa containing these enzymes and involved in its formation, e.g., Clostridium cluster XIVa (C. symbiosum species), Coriobacteriaceae.unknown_gordonibacter (Gordonibacter pamelaeae species) and Erysipelotrichaceae Holdemania (Holdemania filiformis species) in alcohol‐associated hepatitis compared to heavy drinking controls.
Discussion
The current study identified a novel and distinct plasma and fecal bile acid signature associated with heavy alcohol consumption and the development of clinically overt alcohol‐associated hepatitis. It further provides novel data on bile acid metabolism and changes in secondary and tertiary bile acid derivatives in alcohol‐associated hepatitis and insights on the functional changes in the intestinal microbiome associated with altered bile acid metabolism in alcohol‐associated hepatitis.
A key finding is the increase in plasma primary bile acids derived from both the 7α and 27α pathways along with an increase in 7‐HOCA in alcohol‐associated hepatitis but not in heavy drinking controls. A previous study had also demonstrated an increase in primary bile acids in circulation but with markedly suppressed 7α‐hydroxy‐4‐cholesten‐3‐one (C4).( 16 ) The basis for this could be the small sample size of the prior study (n = 9‐15 per group) and the inclusion of patients with cirrhosis. In the current study, 7‐HOCA measurements were available in all patients, and the increase aligns with the observed increase in plasma primary bile acids. These indicate that, despite evidence of cholestasis, there was a possible increase in hepatic primary bile acid synthesis. However, C4 levels were not available in our study. C4 is involved in the rate‐limiting step in bile acid synthesis and would have provided greater evidence that hepatic synthesis of primary bile acids was truly increased.
A novel finding was the differential changes in secondary bile acids derived from the 7α versus the 27α pathways. Deoxycholate, derived from cholic acid and its conjugates, decreased in both stool and in plasma, indicative of decreased formation by the intestinal microbiome. On the other hand, lithocholate, derived from chenodeoxycholate and its conjugates, decreased in stool, but the level of its sulfated conjugates was significantly increased in plasma. One possible explanation is that the uptake of lithocholate in alcohol‐associated hepatitis is increased and the liver attempts to detoxify this toxic bile acid by conjugation. The increased systemic exposure of lithocholate conjugates, particularly in SAH, is likely to be biologically relevant given the well‐known cellular toxicity due to this molecule.( 25 )
Another novel finding is a decrease in the minor secondary bile acid ursodeoxycholate and related molecules (ursocholate and isoursodeoxycholate) in stool and an increase in tauroursodeoxycholate in plasma. The latter may reflect passive uptake of ursodeoxycholate from the intestine and its conjugation in the liver. The decrease in isoursodeoxycholate, which requires additional 3α and β epimerization of ursodeoxycholate, in alcohol‐associated hepatitis provides further evidence of specific changes in the bile acid biome in alcohol‐associated hepatitis. The biological properties of these molecules are not well established, and the current study provides a rationale for future studies to elucidate these properties.
The current study further provides evidence of functional linkages between the observed specific changes in the bile acid profile. Changes in the microbiome responsible for such changes provide novel insights on how bile acids are altered in alcohol‐associated hepatitis. Using bioinformatic analyses regressing the differential expression of specific taxa in the current study to databases containing the known function of specific taxa, a short list of taxa was identified. The specific bile acid‐metabolizing genes in these taxa are already known, allowing linkage of specific taxa to the specifically altered bile acids in alcohol‐associated hepatitis. The observed changes in Clostridium cluster XIVa is particularly of interest because it contains C. hylemonae and C. scindens, which play a major role in bile acid biotransformation.( 22 ) The current study represents a key first step to set the stage for phage‐based approaches to modify 7α‐dehydroxylase function in the gut to interrogate its ability to modify the bile acid profile and course of disease. In addition, it provides preliminary data for selection of specific taxa for inclusion in probiotics to leverage targeted microbiome supplementation to prevent development of alcoholic‐associated hepatitis among heavy drinkers.
Fusobacteria were the only bile acid‐metabolizing taxa that were increased in alcohol‐associated hepatitis compared to heavy drinking controls. They contain 7α and β epimerases needed for formation of ursocholate, ursodeoxycholate, and isoursodeoxycholate( 24 ); however, the levels of these generally cytoprotective bile acids are decreased and inversely related to the MELD score. These findings have to be taken with caution as R 2 of the inverse correlation with the MELD score was low. This may indicate a specific functional defect in the microbiome that may be clinically relevant and supports use of targeted approaches to enhance microbial 7α and β epimerase activity for the prevention and treatment of alcohol‐associated hepatitis.
Bile acids also have a major role in determining the composition of the intestinal microbiome because of their ability to both promote and inhibit the growth of various taxa.( 12 ) While much of the literature has focused on deoxycholate,( 26 ) the current study demonstrates strong positive correlations between numerous taxa and glycine‐conjugated primary bile acids in stool. These taxa are all metabolically active, and their changes in response to altered fecal concentrations in individual patients can potentially contribute to the heterogeneity in metaboinflammatory and systemic responses in alcohol‐associated hepatitis.
One such metabolite is glycocholenate sulfate, which requires monosulfation of glycocholate at the 3‐C position.( 27 ) Glycocholenate sulfate was significantly increased in alcohol‐associated hepatitis; this molecule has previously been associated with atrial fibrillation, although its mechanistic basis remains unknown.( 28 ) This warrants further study as there is a well‐known increase in atrial fibrillation in individuals with severe binge drinking.( 29 )
As with most studies, this study also has limitations. The sample size is relatively small. To account for that, we used a conservative approach to test for significance with correction for multiple testing to avoid false discovery. Also, the study cohort did not have liver histology because liver biopsies are rarely performed in routine practice in this population. This precluded our ability to be certain about the proportion when superimposed on cirrhosis. The study was also not designed to be able to differentiate the effects of varying types of alcohol and patterns of alcohol consumption on the bile acid profile.
This study was performed based on the median scaling‐normalized bile acids data rather than absolute concentrations. Sensitivity analyses presented in the Supporting Materials revealed that reproducing results based on absolute concentrations did not affect the study conclusions. Another limitation of this study is that limited patient sample size did not allow further stratifying the patient groups by race, sex, and BMI to control for these significant factors in understanding bile acid metabolism. Future studies are needed to evaluate the interaction between patient demographics and bile acid changes in alcohol‐associated hepatitis. Finally, this study is cross‐sectional, and future longitudinal studies are needed to accurately assess changes in bile acids in the development or regression of alcohol‐associated hepatitis.
Despite these limitations, the current study provides a novel signature of both circulating and fecal bile acids associated with alcohol‐associated hepatitis. It provides several novel insights on differential changes in secondary bile acid metabolism in alcohol‐associated hepatitis and their relationship to disease severity. It provides important insights on the specific microbial metabolic changes underlying these changes and will serve as a foundation for specific targeted approaches to modulate the microbiome to both better understand the role of specific microbiota and bile acids in alcohol‐associated hepatitis and to leverage this for therapeutic gain.
Supporting information
Supplementary Material
Acknowledgment
The TREAT consortium was funded by the National Institute of Alcohol Abuse and Alcoholism (NIAAA). The investigators were responsible for the design, data collection, and final analysis of this ancillary study of the TREAT consortium and take full responsibility for the manuscript. The NIAAA did not participate in the conduct of the studies.
Supported by the National Institutes of Health (NIH) (grants UO1 AA021891‐01 and T32 DK07150‐40 to A.J.S.), NIH through Virginia Commonwealth University (VCU) BERD Core (Clinical and Translational Science Award [CTSA] UL1TR002649 to E.S.), National Center for Advancing Translational Sciences through VCU Institutional Career Development Core (CTSA 5KL2TR002648 to E.S.), National Medical Research Council Singapore (Research Training Fellowship MOH‐000193 to M.D.M.), and National Institute on Alcohol Abuse and Alcoholism (UO1 grant to A.J.S.).
The NIH institutes did not have a direct role in the design and conduct of the studies.
The authors will submit the data set to the NIH data warehouse. A copy will be kept at the data coordinating center of the TREAT consortium. De‐identified data will be made available on request. R code used to produce all results and figures is publicly available on github at https://github.com/katiasmirn/STM_RCode.
Potential conflict of interest: Dr. Mirshahi owns stock in Sanyal Biotechnology. Dr. Sanyal is President of Sanyal Biotechnology and has stock options in Tiziana, Durect, Indalo, and Inversago. He has served as a consultant to Medimmune, Astra Zeneca, Nitto Denko, Nimbus, Salix, Tobira, Takeda, Terns, Conatus, Lilly, Poxel, Blade, Surrozen, Birdrock, Siemens, Madrigal, Novartis, Pfizer, Hemoshear, Novo Nordisk, Gilead, Exhalenz, Bristol Myers Squibb, Glympse, and Genfit. He has been an unpaid consultant to Intercept, Zafgen, Prosciento, Iquvia, NGM Bio, Echosens, Immuron, Syntlogic, Zafgen, Zydus, and Nordic Bioscience. His institution has received grant support from Gilead, Salix, Tobira, Intercept, Merck, Astra Zeneca, Zydus, and Novartis. Dr. Chalasani has ongoing paid consulting activities (or had in the preceding 12 months) with Abbvie, Madrigal, Foresite labs, Altimmune, Zydus, Galectin, and Boehringer‐Ingelheim. Dr. Chalasani receives research grant support from Exact Sciences and DSM where his institution receives the funding. Dr. Chalasani has equity ownership in RestUp, Inc. The other authors have nothing to report.
Contributor Information
Mark D. Muthiah, Email: mdcmdm@nus.edu.sg.
Ekaterina Smirnova, Email: ekaterina.smirnova@vcuhealth.org.
References
Author names in bold designate shared co‐first authorship.
- 1. Nguyen TA, DeShazo JP, Thacker LR, Puri P, Sanyal AJ. The worsening profile of alcoholic hepatitis in the United States. Alcohol Clin Exp Res 2016;40:1295‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Julien J, Ayer T, Bethea ED, Tapper EB, Chhatwal J. Projected prevalence and mortality associated with alcohol‐related liver disease in the USA, 2019‐40: a modelling study. Lancet Public Health 2020;5:e316‐e323. [DOI] [PubMed] [Google Scholar]
- 3. Centers for Disease Control and Prevention . Alcohol and public health: alcohol‐related disease impact (ARDI). https://nccd.cdc.gov/DPH_ARDI/default/default.aspx. Updated September 3, 2020. Accessed March 2021.
- 4. Lourens S, Sunjaya DB, Singal A, Liangpunsakul S, Puri P, Sanyal A, et al.; TREAT Consortium . Acute alcoholic hepatitis: natural history and predictors of mortality using a multicenter prospective study. Mayo Clin Proc Innov Qual Outcomes 2017;1:37‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sanyal AJ, Gao B, Szabo G. Gaps in knowledge and research priorities for alcoholic hepatitis. Gastroenterology 2015;149:4‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trinchet JC, Gerhardt MF, Balkau B, Munz C, Poupon RE. Serum bile acids and cholestasis in alcoholic hepatitis. Relationship with usual liver tests and histological features. J Hepatol 1994;21:235‐240. [DOI] [PubMed] [Google Scholar]
- 7. Chiang JY. Bile acid metabolism and signaling. Compr Physiol 2013;3:1191‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahmad TR, Haeusler RA. Bile acids in glucose metabolism and insulin signalling ‐ mechanisms and research needs. Nat Rev Endocrinol 2019;15:701‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanafi NI, Mohamed AS, Sheikh Abdul Kadir SH, Othman MHD. Overview of bile acids signaling and perspective on the signal of ursodeoxycholic acid, the most hydrophilic bile acid, in the heart. Biomolecules 2018;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doden HL, Ridlon JM. Microbial hydroxysteroid dehydrogenases: from alpha to omega. Microorganisms 2021;9:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016;7:22‐39. Erratum in: Gut Microbes 2016;7:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev 2005;29:625‐651. [DOI] [PubMed] [Google Scholar]
- 13. Urdaneta V, Casadesús J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med (Lausanne) 2017;4:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smirnova E, Puri P, Muthiah MD, Daitya K, Brown R, Chalasani N, et al. Fecal microbiome distinguishes alcohol consumption from alcoholic hepatitis but does not discriminate disease severity. Hepatology 2020;72:271‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puri P, Liangpunsakul S, Christensen JE, Shah VH, Kamath PS, Gores GJ, et al.; TREAT Consortium . The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology 2018;67:1284‐1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura‐Cots M, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ciocan D, Voican CS, Wrzosek L, Hugot C, Rainteau D, Humbert L, et al. Bile acid homeostasis and intestinal dysbiosis in alcoholic hepatitis. Aliment Pharmacol Ther 2018;48:961‐974. [DOI] [PubMed] [Google Scholar]
- 18. Crabb DW, Bataller R, Chalasani NP, Kamath PS, Lucey M, Mathurin P, et al.; NIAAA Alcoholic Hepatitis Consortia . Standard definitions and common data elements for clinical trials in patients with alcoholic hepatitis: recommendation from the NIAAA Alcoholic Hepatitis Consortia. Gastroenterology 2016;150:785‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dunn W, Jamil LH, Brown LS, Wiesner RH, Kim WR, Menon KVN, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005;41:353‐358. [DOI] [PubMed] [Google Scholar]
- 20. Gillevet P, Sikaroodi M, Keshavarzian A, Mutlu EA. Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem Biodivers 2010;7:1065‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wulff JE, Mitchell MW. A comparison of various normalization methods for LC/MS metabolomics data. Adv Bioscience Biotechnol 2018;9:339. [Google Scholar]
- 22. Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019;7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, et al. Generation of genome‐scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol 2017;35:81‐89. [DOI] [PubMed] [Google Scholar]
- 24. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006;47:241‐259. [DOI] [PubMed] [Google Scholar]
- 25. Woolbright BL, Li F, Xie Y, Farhood A, Fickert P, Trauner M, et al. Lithocholic acid feeding results in direct hepato‐toxicity independent of neutrophil function in mice. Toxicol Lett 2014;228:56‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol 2014;30:332‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alnouti Y. Bile acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci 2009;108:225‐246. [DOI] [PubMed] [Google Scholar]
- 28. Alonso A, Yu B, Sun YV, Chen LY, Loehr LR, O'Neal WT, et al. Serum metabolomics and incidence of atrial fibrillation (from the Atherosclerosis Risk in Communities Study). Am J Cardiol 2019;123:1955‐1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Larsson SC, Drca N, Wolk A. Alcohol consumption and risk of atrial fibrillation: a prospective study and dose‐response meta‐analysis. J Am Coll Cardiol 2014;64:281‐289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material