

Abstract
Activation of extracellular signal–regulated kinase (ERK) 1/2 promotes hepatocyte proliferation in response to growth stimuli, but whether constitutive hepatocyte ERK1/2 signaling functions in liver physiology is unknown. To examine the role of ERK1/2 in hepatic homeostasis, the effects of a knockout of Erk1 and/or Erk2 in mouse liver were examined. The livers of mice with a global Erk1 knockout or a tamoxifen‐inducible, hepatocyte‐specific Erk2 knockout were normal. In contrast, Erk1/2 double‐knockout mice developed hepatomegaly and hepatitis by serum transaminases, histology, terminal deoxynucleotide transferase‐mediated deoxyuridine triphosphate nick end‐labeling, and assays of hepatic inflammation. Liver injury was associated with biochemical evidence of cholestasis with increased serum and hepatic bile acids and led to hepatic fibrosis and mortality. RNA sequencing and polymerase chain reaction analysis of double‐knockout mouse livers revealed that the rate‐limiting bile acid synthesis gene Cyp7a1 (cholesterol 7α‐hydroxylase) was up‐regulated in concert with decreased expression of the transcriptional repressor short heterodimer partner. Elevated bile acids were the mechanism of liver injury, as bile acid reduction by SC‐435, an inhibitor of the ileal apical sodium–dependent bile acid transporter, prevented liver injury. Conclusion: Constitutive ERK1 and ERK2 signaling has a redundant but critical physiological function in the down‐regulation of hepatic bile acid synthesis to maintain normal liver homeostasis.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- Con
control
- Cyp7a1
cholesterol 7α‐hydroxylase
- EGF
epidermal growth factor
- ERK
extracellular signal‐regulated kinase
- FGF
fibroblast growth factor
- GCDCA
glycochenodeoxycholic acid
- IL
interleukin
- JNK
c‐Jun N‐terminal kinase
- KO
knockout
- MAPK
mitogen‐activated protein kinase
- mRNA
messenger RNA
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- Shp
short heterodimer partner
- Tam
tamoxifen
- TCA
taurocholic acid
- TUNEL
terminal deoxynucleotide transferase‐mediated deoxyuridine triphosphate nick end‐labeling
Activation of mitogen‐activated protein kinases (MAPKs) is central to the liver’s pathophysiological response to injury. Of the three principal MAPKs, c‐Jun N‐terminal kinase (JNK) 1/2 extracellular signal–regulated kinase (ERK) 1/2, and p38 MAPK, the functions of JNK1/2 in liver injury have been most studied and are best understood. ERK1/2 activation has been implicated in hepatocyte proliferation and resistance to injury, but evidence of ERK1/2 function has been derived largely from studies using nonspecific pharmacological inhibitors rather than genetic knockouts. In addition, little is known about the functions of constitutive ERK1/2 expression or the differential roles of the two ERK isoforms in the liver.
Activation of ERK1/2 through phosphorylation by a kinase cascade converts extracellular stimulation of cell surface receptors into changes in gene expression. Both isoforms are ubiquitously expressed and have similar regulation, subcellular expression, and substrate specificity.( 1 , 2 ) Whether ERK1 and ERK2 have unique or redundant functions is controversial.( 1 , 2 ) Global Erk1 knockouts are normal except for thymocyte abnormalities,( 3 ) whereas Erk2 loss is an embryonic lethal,( 4 ) suggesting the existence of both unique and redundant isoform functions.
Similar to nonhepatic cells, hepatocyte ERK1 and ERK2 promote proliferation and cytoprotection.( 5 , 6 , 7 ) In addition, human hepatocyte studies have implicated ERK1/2 as the downstream mediator of fibroblast growth factor (FGF) 15/19–induced inhibition of bile acid synthesis through decreased expression of the rate‐limiting enzyme cholesterol 7α‐hydroxylase (Cyp7a1).( 8 ) However, cultured hepatocytes undergo MAPK up‐regulation from the stress of perfusion and culture,( 9 ) and the findings relied on a pharmacological ERK1/2 inhibitor, which has off‐target effects on mitochondrial metabolism.( 10 ) Although this study showed no effect of a JNK inhibitor, recent investigations in transgenic mice indicate that JNK down‐regulates bile acid synthesis.( 11 ) The effects of MAPKs on bile acid homeostasis therefore remain unclear.
To delineate functions of ERK1 and ERK2 signaling in normal liver, genetic knockouts of Erk1/2 were examined. With loss of both ERK1 and hepatocyte ERK2 (but neither isoform alone), spontaneous hepatic injury, and inflammation develop, leading to fibrosis and mortality. Loss of ERK1/2 increased levels of serum and hepatic bile acids that triggered liver injury. Cyp7a1 was increased in association with decreased expression of its transcriptional repressor short heterodimer partner (Shp). These findings demonstrate that critical redundant functions of constitutive ERK1 and ERK2 signaling maintain normal liver homeostasis through the down‐regulation of bile acid synthesis.
Materials and Methods
Animals
Mice aged 9‐14 weeks were housed under 12‐hour light/dark cycles in static cages with 1/4‐inch corncob bedding, cotton nestlets, automatic water feeding, and unlimited food (PicoLab Rodent Diet 20 #5053; LabDiet, St. Louis, MO). Data were similar for the two sexes and combined in all studies. To decrease liver bile acid content, mice were fed a control diet (PicoLab Rodent Diet 20 #5001; LabDiet) alone or containing the ileal apical sodium–dependent bile acid transporter inhibitor SC‐435 (0.006% wt/wt; Shire Pharmaceuticals, Cambridge, MA),( 12 ) as previously used.( 13 ) Erk1−/− (Erk1‐KO) mice (B6.129[Cg]‐Mapk3tm1Gela /J, #019113; Jackson Laboratory, Bar Harbor, ME) were generated from crosses of Erk1+/− mice, and littermate Erk1+/+ (Erk1‐Con) mice served as controls. Hepatocyte‐specific Erk2 knockout (Erk2‐KO) mice were generated by crossing Erk2fl‐fl mice (B6.129‐Mapk1tm1Gela /J, #019112; Jackson Laboratory) with ERt‐albumin‐Cre mice with a tamoxifen‐inducible, albumin promoter–driven Cre recombinase.( 14 ) Littermate Erk2fl‐fl (Erk2‐Con) mice lacking the Cre transgene were controls for this line. Double knockout (Erk1/2‐KO) mice were generated by intercrossing the two lines with littermate mice lacking Cre as controls (Erk1/2‐Con). All mice were backcrossed to C57BL/6J mice. Cre expression was induced by a subcutaneous injection of 3 mg of tamoxifen (MilliporeSigma, St. Louis, MO) in sunflower oil. Control mice were identically injected with oil vehicle or tamoxifen. C57BL/6J male mice (#000664; Jackson Laboratory) were injected with D‐galactosamine/lipopolysaccharide, as previously described.( 15 ) Animal studies were approved by the Animal Care and Use Committee of Emory University School of Medicine and followed National Institutes of Health guidelines for animal care.
Mouse Hepatocyte Isolation and Culture
Primary mouse hepatocytes were obtained by liver perfusion and cultured as previously described.( 16 ) Cells were treated with 100 ng/mL epidermal growth factor (EGF), 100 ng/mL interleukin (IL)‐1β (R&D Systems, Minneapolis, MN), or 100 μM cholic acid (CA), taurocholic acid (TCA), chenodeoxycholic acid (CDCA), or glycochenodeoxycholic acid (GCDCA) (MilliporeSigma). Cell death was determined by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay, as previously described.( 17 )
Protein Isolation and Western Blotting
Liver total, cytosolic, and mitochondrial proteins were isolated, and western blotting was performed as previously described.( 5 , 18 , 19 ) Antibodies are listed in Supporting Table S1.
Serum Biochemistries
Serum alanine aminotransferase (ALT) was assayed by commercial kit (TECO Diagnostics, Anaheim, CA). Serum aspartate aminotransferase (AST), alkaline phosphatase, total bilirubin, and bile acids were determined by an ACE Axcel Clinical Chemistry System (Alfa Wassermann, West Caldwell, NJ).
Liver Staining
Liver sections were stained with hematoxylin and eosin (MilliporeSigma) for histology. Colorimetric terminal deoxynucleotide transferase‐mediated deoxyuridine triphosphate nick end‐labeling (TUNEL) was performed by commercial kit (Roche, Indianapolis, IN). TUNEL‐positive cells were counted under light microscopy (×400 magnification) in 10 randomly selected high‐power fields. Sections were stained with sirius red by commercial kit (Abcam, Cambridge, MA), and brightfield images taken on a Leica microscope for quantification by ImageJ software.
Quantitative Real‐Time Reverse‐Transcription Polymerase Chain Reaction
RNA extraction, reverse transcription, and quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) using the primers in Supporting Table S2 (Integrated DNA Technologies, Coralville, IA) were performed as previously described.( 20 ) Data were analyzed by the 2−ΔΔCT method for relative quantification and normalized to glyceraldehyde 3‐phosphate dehydrogenase.
Liver Collagen Content
Liver collagen content was determined by hydroxyproline quantification in 200‐300‐mg liver samples after hydrolysis in 6N HCl for 16 hours at 110°C, as previously described.( 21 )
Bile Acid Analysis
Fifty milligrams of liver tissue were homogenized in a 1:4 vol/vol water‐to‐methanol solution. After shaking at 300 rpm at 4°C for 10 minutes, samples were centrifuged at 10,000 rpm at 4°C for 5 minutes. The extract was separated, methanol added, and the samples shaken and centrifuged. This process was repeated, and the samples dried under nitrogen and reconstituted in methanol. Liquid chromatography–tandem mass spectrometry with a multiple reaction monitoring–based method was performed with a SCIEX ExionLC AD UHPLC and SCIEX QTrap 5500 Mass Spectrometer (Framingham, MA). A Thermo Scientific (Waltham, MA) Accucore C18 column with guard column was used for the analysis. Bile acids were resolved using an 8.5‐minute linear gradient consisting of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). Analysis was completed in both positive and negative mode. Data were acquired using Analyst 1.6 and processed by OS‐MQ (Sciex).
RNA Sequencing
Messenger RNA (mRNA) from flash‐frozen liver was extracted with the miRNeasy mini kit (Qiagen, Valencia, CA). mRNA were quantified using the Infinite M200 Pro (Tecan, Morrisville, NC) and quality determined with the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Novogene (Sacramento, CA) prepared the sequencing libraries with the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA). Libraries were validated with the 2100 Bioanalyzer and quantified with a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and by qRT‐PCR (Applied Biosystems, Foster City, CA). Sequencing was performed on a NovaSeq 6000 (Illumina) with a 2 × 150 paired‐end configuration. Raw sequence data were converted into fastq files and de‐multiplexed using Illumina’s bcl2fastq software. Sequences were quality‐checked using FastQC and aligned to the HG38 reference genome using STAR aligner. HTSeq‐count was used to generate raw counts per gene. Differentially expressed genes between groups were determined using DESeq2. Genes with adjusted P values, or false discovery rates, <0.05 were considered significantly differentially expressed. The data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE181803.
Statistical Analysis
Numerical data are reported as mean ± SEM from three or more independent experiments. Statistical significance was determined by one‐way analysis of variance with statistical significance defined as P < 0.05.
Results
Validation of Erk Knockout Mice
To determine the hepatic functions of ERK1/2, three transgenic mouse lines were used: Erk1‐KO mice with a global Erk1 knockout, Erk2‐KO mice with a tamoxifen‐inducible, hepatocyte‐specific Erk2 knockout, and Erk1/2‐KO mice with a double knockout. Decreases in hepatic ERK1/2 were confirmed by immunoblots of total liver protein. Mice received either oil vehicle or tamoxifen to induce ERt‐Alb‐Cre expression and knockout hepatocyte Erk2. Erk1‐KO mice have a selective decrease in p44 ERK1, which as expected is unaltered by tamoxifen (Fig. 1A). Erk2‐KO mice have normal ERK1 content and decreased levels of p42 ERK2 with tamoxifen (Fig. 1A). Tamoxifen‐injected, but not oil‐injected, Erk1/2‐KO mice have decreased levels of both isoforms (Fig. 1A).
FIG. 1.
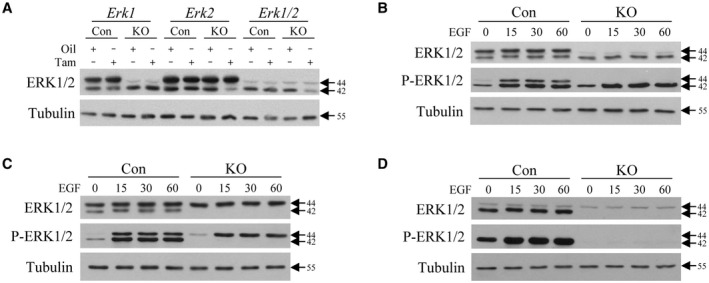
Transgenic mouse livers and hepatocytes have ERK1‐specific and/or ERK2‐specific knockouts. (A) Immunoblots of total liver protein from littermate controls and Erk1‐KO, Erk2‐KO, and Erk1/2‐KO mice probed for total ERK1/2 and tubulin. Mice were oil‐injected or tamoxifen‐injected as indicated. (B‐D) Immunoblots of total protein from primary hepatocytes isolated from tamoxifen‐injected littermate control and Erk1‐KO (B), Erk2‐KO (C), and Erk1/2‐KO (D) mice probed for total ERK1/2, phosphorylated ERK1/2 (P‐ERK1/2), and tubulin. Hepatocytes were EGF‐treated for the indicated number of minutes. Molecular weights in kilodaltons are shown by arrows. All images are representative of three independent experiments. Abbreviations: Con, control; KO, knockout; Tam, tamoxifen.
To demonstrate that hepatocyte levels of active, phosphorylated ERK are reduced in the respective knockout mice, primary hepatocytes were isolated from tamoxifen‐injected control and knockout mice and treated with the ERK1/2 activator EGF in culture. Hepatocytes from Erk1‐KO mice lacked ERK1 and P‐ERK1 at baseline and after EGF treatment, whereas P‐ERK2 induction by EGF was equivalent in control and knockout cells (Fig. 1B). Conversely, tamoxifen‐injected Erk2‐KO mice had a selective loss of ERK2 and EGF‐induced P‐ERK2 but intact P‐ERK1 activation (Fig. 1C). Erk1/2‐KO mice lacked both total and phosphorylated ERK1/2 isoforms with tamoxifen injection (Fig. 1D). Hepatocytes in the three transgenic mouse lines therefore have the appropriate, specific knockouts of total and active phosphorylated ERK1/2.
Combined KNOCKOUT of Erk1 and Erk2 Triggers Liver Injury
To determine whether baseline ERK1/2 expression functions in liver homeostasis, mice were examined 3 weeks after oil or tamoxifen injection. Body weights were equivalent in the three lines and unaffected by tamoxifen (Fig. 2A). Liver weights (Fig. 2B) and liver‐to–body weight ratios (Fig. 2C) were selectively increased in tamoxifen‐injected Erk1/2‐KO mice. The double knockout therefore led to hepatomegaly in the absence of a change in total body mass.
FIG. 2.
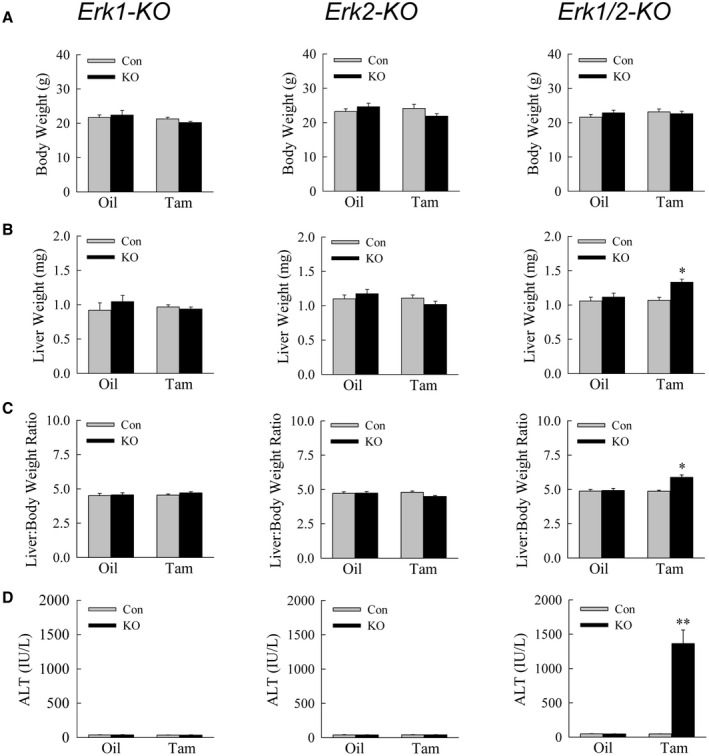
Double‐knockout mice develop hepatomegaly and liver injury. Body weights (A), liver weights (B), liver‐to–body weight ratios (C), and serum ALTs (D) in littermate controls and Erk1‐KO, Erk2‐KO, and Erk1/2‐KO (KO) mice treated with oil or tamoxifen (*P < 0.001, **P < 0.000001 compared with control oil‐injected or tamoxifen‐injected mice; n = 10‐15).
Mice were screened for liver injury by serum ALT. Erk1‐KO mice failed to develop liver injury as indicated by normal ALT levels (Fig. 2D), consistent with previous reports that these mice have a normal phenotype.( 3 ) Erk2‐KO mice similarly had normal ALTs even with tamoxifen injection (Fig. 2D). In contrast, Erk1/2‐KO mice developed markedly elevated ALT levels with tamoxifen but not oil administration (Fig. 2D).
Consistent with ALT findings, hematoxylin and eosin–stained livers from tamoxifen‐injected Erk1/2‐KO mice exhibited hepatocellular injury and inflammation (Fig. 3A). Histology was normal in Erk1‐KO and Erk2‐KO mice (Supporting Fig. S1). Injury led to hepatocyte death, as confirmed by increased TUNEL staining in tamoxifen‐injected, but not oil‐injected, Erk1/2‐KO mice (Fig. 3B,C) or Erk1‐KO and Erk2‐KO mice (Fig. 3D,E; Supporting Fig. S2). Hepatocyte death occurred in the absence of mitochondrial cytochrome c release or caspase 3/7 cleavage as detected in apoptotic liver injury from galactosamine/lipopolysaccharide, suggesting that death was from necrosis rather than apoptosis (Fig. 3F). The loss of both Erk1 and Erk2, but neither gene alone, leads to spontaneous liver injury and hepatocyte death.
FIG. 3.
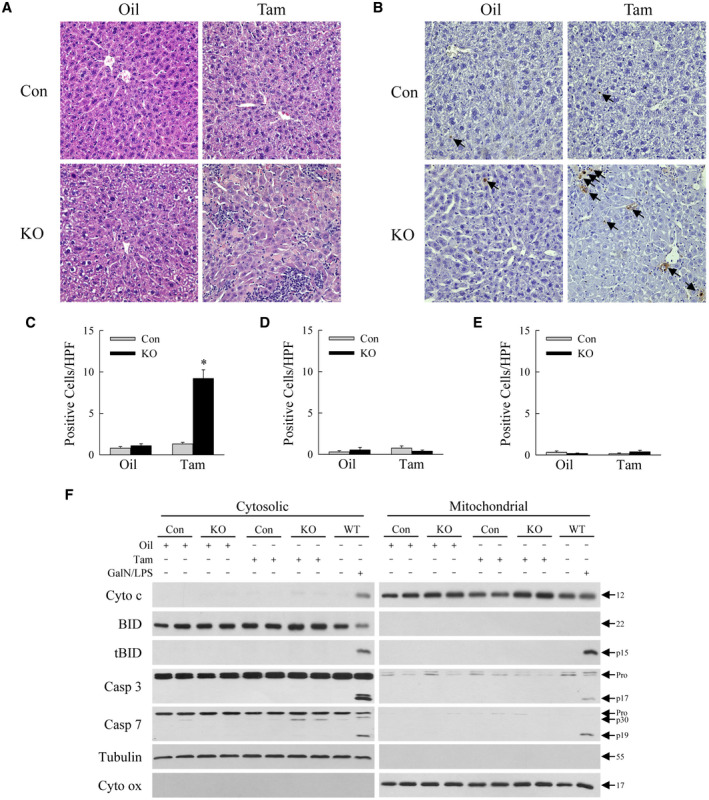
Hematoxylin and eosin and TUNEL staining confirm liver injury in Erk1/2‐KO mice. Hematoxylin and eosin (A) and TUNEL staining (B) of Erk1/2‐Con and Erk1/2‐KO mice after oil or tamoxifen treatment (×400). Arrows in (B) indicate TUNEL‐positive cells. Numbers of TUNEL‐positive cells per high‐powered field in Erk1/2‐KO (C), Erk1‐KO (D), and Erk2‐KO (E) mice (*P < 0.000001 compared with control oil‐injected or tamoxifen‐injected mice; n = 8‐10). (F) Immunoblots of liver cytosolic and mitochondrial protein from Erk1/2‐Con and Erk1/2‐KO mice after oil or tamoxifen treatment, and wild‐type mice administered galactosamine/lipopolysaccharide for 6 hours. Blots were probed for cytochrome c, BID, tBID, caspase 3, caspase 7, tubulin as a cytosolic protein loading and purity control, and cytochrome oxidase as a mitochondrial protein loading and purity control. Molecular weights in kilodaltons are indicated by the arrows. Immunoblots are representative of three independent experiments. Abbreviations: BID, BH3‐interacting domain death agonist; Casp 3, caspase 3; Casp 7, caspase 7; Cyto c, cytochrome c; Cyto ox, cytochrome oxidase; GalN/LPS, galactosamine/lipopolysaccharide; HPF, high‐powered field; KO, knockout; tBID, truncated BID; WT, wild‐type.
Liver Injury in Erk1/2‐KO Mice Has Cholestatic Features and Associated Inflammation
To further characterize the hepatic injury in Erk1/2‐KO mice, additional serum biochemical indices of liver injury were examined. Consistent with elevated ALTs, AST levels were increased in Erk1/2‐KO mice with tamoxifen treatment (Fig. 4A). Indicative of cholestatic liver injury, tamoxifen‐treated Erk1/2‐KO mice had a marked increase in alkaline phosphatase, a moderate increase in total bilirubin, and highly elevated serum bile acids (Fig. 4A).
FIG. 4.
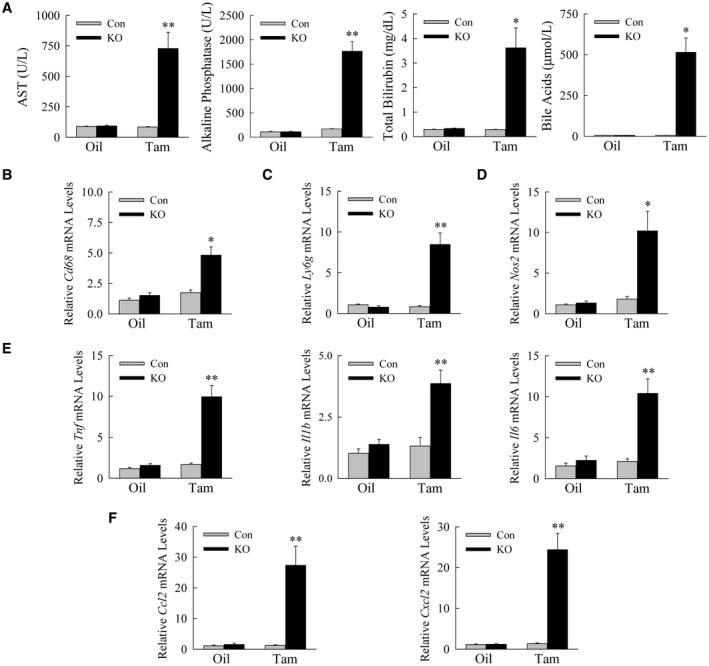
Liver injury is cholestatic and associated with inflammation. (A) Serum levels in Erk1/2‐Con and Erk1/2‐KO mice after oil or tamoxifen injection for AST, alkaline phosphatase, total bilirubin, and bile acids (*P < 0.0001, **P < 0.000001 compared with control oil‐injected or tamoxifen‐injected mice; n = 10‐15). (B‐F) Hepatic mRNA levels in the same mice for the genes Cd68 (B); Ly6g (C); Nos2 (D); Tnf, Il1b, and Il6 (E); and Ccl2 and Cxcl2 (F) (*P < 0.001, **P < 0.0001 compared with control oil‐injected or tamoxifen‐injected mice; n = 11‐13). Abbreviations: Ccl2, C‐C motif chemokine ligand 2; Cxcl2, C‐X‐C motif chemokine ligand 2; Ly6g, lymphocyte antigen 6 complex locus G6D; Nos2, nitric oxide synthase 2; Tnf, tumor necrosis factor.
Liver injury in double‐knockout mice was associated with an immune response and inflammation. Tamoxifen‐treated Erk1/2‐KO mice had increased liver infiltration with macrophages by Cd68 mRNA levels (Fig. 4B), and neutrophils as reflected in increased Ly6g (lymphocyte antigen 6 complex locus G6D) expression (Fig. 4C). Indicative of an active immune response, nitric oxide synthase 2 mRNA content was increased (Fig. 4D). Injured KO mice also had increased gene expression for the proinflammatory cytokines Tnf (tumor necrosis factor), Il1b, Il6 (Fig. 4E), and chemokines Ccl2 (C‐C motif chemokine ligand 2) and Cxcl2 (C‐X‐C motif chemokine ligand 2) (Fig. 4F), which have been reported previously to be elevated in cholestatic mouse and human liver injury.( 22 )
Liver Injury in Erk1/2‐KO Mice Leads to Hepatic Fibrosis and Mortality
ALT levels at 5 weeks after tamoxifen treatment revealed a continued absence of liver injury in the single‐knockout mice and even greater injury in tamoxifen‐treated double knockouts (Fig. 5A). In light of the sustained liver injury, Erk1/2‐KO mice were examined at 5 weeks for hepatic fibrosis. Tamoxifen‐injected Erk1/2‐KO mice had increased hepatic mRNA expression for the fibrotic genes α‐smooth muscle actin, collagen 1α1, transforming growth factor‐β1, and tissue inhibitor of metalloproteinase 1 (Fig. 5B). Fibrotic gene expression was unchanged in Erk1‐KO and Erk2‐KO mice (Supporting Fig. S3). Tamoxifen‐injected Erk1/2‐KO mice had increased hepatic fibrosis as determined by sirius red–stained liver images (Fig. 5C), quantification of the area of sirius red staining (Fig. 5D), and hepatic hydroxyproline content (Fig. 5E).
FIG. 5.
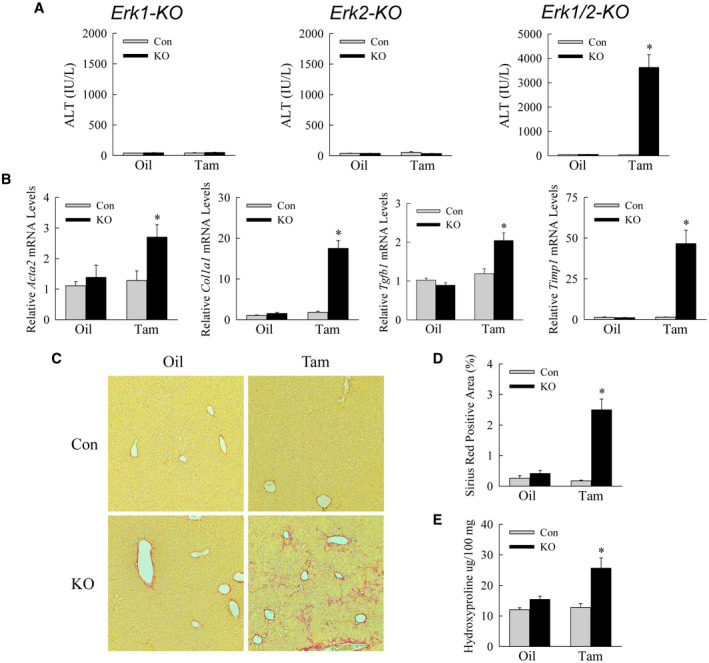
Liver injury is increased 5 weeks following tamoxifen injection. (A) Serum ALT levels in the indicated control and knockout mice at 5 weeks after oil or tamoxifen administration (*P < 0.0001 compared with control oil‐injected or tamoxifen‐injected mice; n = 10‐19). (B) Relative mRNA levels for Acta2, Col1a1, Tgfb1, and Timp1 (*P < 0.0001 compared with oil‐injected control mice; n = 13‐16). (C) Representative sirius red–stained livers. (D) Quantification of sirius red–stained liver area (*P < 0.0001 compared with control oil‐injected or tamoxifen‐injected mice; n = 6). (E) Hydroxyproline content (*P < 0.01 compared with control oil‐injected or tamoxifen‐injected mice; n = 5). Abbreviations: Acta2, α‐smooth muscle actin; Col1a1, collagen 1α1; Tgfb1, transforming growth factor‐β1; Timp1, tissue inhibitor of metalloproteinase 1.
Mouse survival was monitored for 5 weeks after oil/tamoxifen injection. No deaths occurred in tamoxifen‐injected Erk1‐KO and Erk2‐KO mice or in Erk1/2‐Con or oil‐treated Erk1/2‐KO mice, but tamoxifen‐injected double‐knockout mice had a 25% mortality (Supporting Table S3). Erk1/2‐KO mice developed chronic liver injury, leading to hepatic fibrosis and mortality.
Liver Injury in Erk1/2‐KO Mice Is Secondary to Increased Hepatic Bile Acids
Hepatic bile acid quantification by liquid chromatography–mass spectrometry revealed that tamoxifen‐treated Erk1/2‐KO mice had markedly increased levels of the unconjugated and conjugated primary bile acids CA (Fig. 6A), CDCA (Fig. 6B), and mouse‐specific α‐muricholic acid (Fig. 6C). Levels of the secondary bile acids deoxycholic acid and lithocholic acid were similarly increased (Fig. 6D,E).
FIG. 6.

Erk1/2‐KO mice have increased hepatic bile acids. Levels in oil‐injected and tamoxifen‐injected Erk1/2‐Con and Erk1/2‐KO mouse livers for the bile acids: CA, GCA, and TCA (A); CDCA, GCDCA, and TCDCA (B); α‐MCA (C); DCA and TDCA (D); and LCA and TLCA (E) (*P < 0.05, **P < 0.01, and ***P < 0.001 compared with oil‐injected control mice; n = 6). Abbreviations: DCA, deoxycholic acid; GCA, glycocholic acid; LCA, lithocholic acid; TCDCA, taurochenodeoxycolic acid; TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid; α‐MCA, α‐muricholic acid.
Elevated bile acids trigger hepatic injury and inflammation.( 23 ) Biochemical features of cholestatic liver injury, together with elevated bile acids, suggested that liver disease in Erk1/2‐KO mice resulted from increased bile acids. Alternatively, ERK promotes hepatocyte resistance to injury,( 24 ) and liver injury in Erk1/2‐KO mice could be secondary to loss of protective, or increased death‐promoting, ERK‐dependent factors. If liver injury in Erk1/2‐KO mice is secondary to altered cell death regulators, then Erk1/2‐KO hepatocytes should be sensitized to death from exogenous bile acids. Primary hepatocytes isolated from tamoxifen‐treated Erk1/2‐Con and Erk1/2‐KO mice were equally susceptible to death from primary bile acids (Fig. 7A), indicating that loss of ERK1/2 did not alter hepatocyte death pathways but rather led to hepatotoxicity by increasing bile acid levels.
FIG. 7.
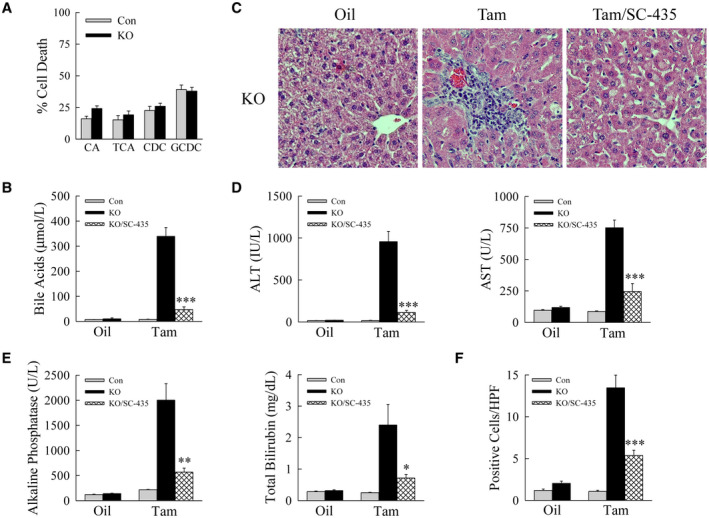
Liver injury is secondary to increased bile acids. (A) Percentage cell death by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide assay in primary hepatocytes from tamoxifen‐treated Erk1/2‐Con and Erk1/2‐KO mice treated with 100 uM CA, TCA, CDCA, or GCDCA for 24 hours (n = 11‐12). (B) Serum bile acid levels in oil‐injected or tamoxifen‐injected Erk1/2‐Con, Erk1/2‐KO, and Erk1/2‐KO mice administered SC‐435 (KO/SC‐435) (n = 10). (C) Representative hematoxylin and eosin–stained images. (D,E) Serum levels of ALT and AST (D), and alkaline phosphatase and total bilirubin (E) (n = 10). (F) Numbers of TUNEL‐positive cells per high‐powered field (n = 9) (*P < 0.05, **P < 0.001, and ***P < 0.0001 compared with tamoxifen‐injected knockout mice). Abbreviation: HPF, high‐powered field.
To prove that bile acids were the mechanism of liver injury, hepatic bile acids were reduced in Erk1/2‐KO mice by the ileal apical sodium‐dependent bile acid transporter inhibitor SC‐435.( 12 ) SC‐435 efficacy was demonstrated by the ability of this agent to lower serum bile acids by 86% (Fig. 7B). Bile acid reduction led to a marked decrease in liver injury as demonstrated by improved histology (Fig. 7C), decreased ALT, AST, alkaline phosphatase and bilirubin levels (Fig. 7D,E), and reduced TUNEL staining (Fig. 7F and Supporting Fig. S4). Liver injury in Erk1/2‐KO mice resulted from increased hepatic bile acids.
Bile Acid Synthetic Enzymes Are Up‐regulated in Double‐Knockout Mice
To delineate the mechanism of increased bile acids in the absence of ERK1/2, the hepatic ERK1/2 transcriptome was determined by RNA sequencing. Transcriptome analysis revealed that 7,276 genes were differentially expressed (3,793 increased and 3,483 decreased) in 3‐week tamoxifen‐injected Erk1/2‐KO versus Erk1/2‐Con mice. In contrast, only 77 genes were differentially expressed in tamoxifen‐injected Erk2‐KO mice with intact ERK1 signaling versus Erk1/2‐Con mice. Changes in hepatic gene expression are overwhelmingly the product of the dual knockout of ERK1 and ERK2, demonstrating that isoform functions are largely redundant in liver.
Knockout of ERK1/2 altered expression of numerous genes regulating bile acids (Fig. 8A). qRT‐PCR confirmed changes in bile acid synthetic enzymes, the most significant being a 20‐fold increase in Cyp7a1 mRNA levels (Fig. 8B). Minor two‐fold increases occurred for Cyp8b1 (sterol 12α‐hydroxylase) and Cyp27a1 (sterol 27‐hydroxylase) as well as a decrease in Cyp7b1 (oxysterol 7α‐hydroxylase) (Fig. 8B). Expression was decreased for bile acid uptake genes and increased for bile acid export genes (Supporting Fig. S5). The major effect of the loss of ERK1/2 was to increase expression of Cyp7a1, the rate‐limiting enzyme in the classic bile acid synthesis pathway.
FIG. 8.
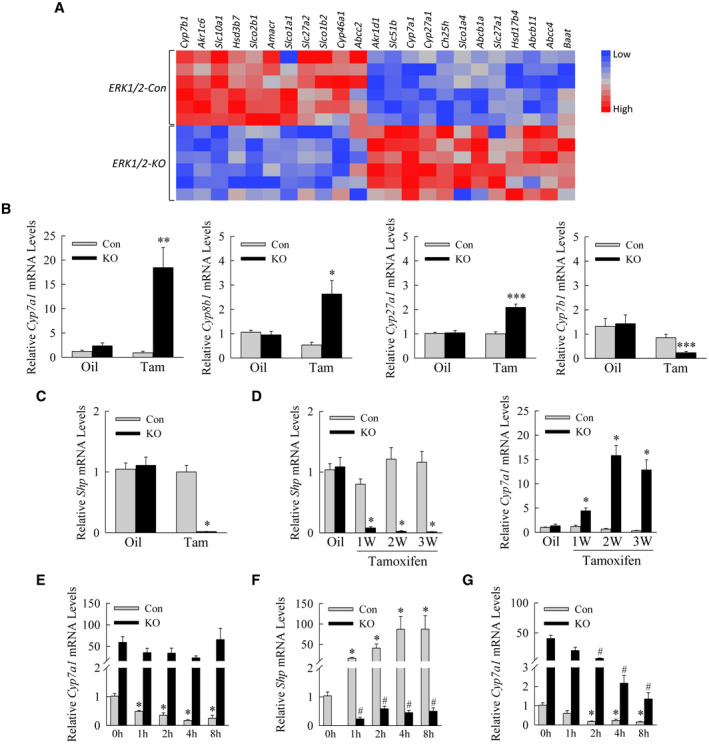
Erk1/2‐KO mouse hepatocytes have increased Cyp7a1 and decreased Shp expression. (A) Heatmap illustrates differential regulation of bile acid pathway genes in Erk1/2‐Con versus Erk1/2‐KO mouse livers. (B) Relative mRNA levels in oil‐treated or tamoxifen‐treated Erk1/2‐Con and Erk1/2‐KO mice (*P < 0.05, **P < 0.01, and ***P < 0.001 compared with oil‐injected control mice; n = 11‐15). (C) Shp mRNA levels in the same mice (*P < 0.0000001 compared with control oil‐injected or tamoxifen‐injected mice; n = 11‐15). (D) Relative liver Shp and Cyp7a1 mRNA levels over weeks after tamoxifen injection (*P < 0.0001 compared with oil‐injected control mice; n = 7‐8). (E) Cyp7a1 mRNA levels in mouse hepatocytes from Erk1/2‐Con and Erk1/2‐KO mice treated with TCA for the indicated number of hours in culture (*P < 0.0003 compared with untreated control hepatocytes; n = 6). (F) Shp mRNA levels in the same cells (*P < 0.01 compared with untreated control hepatocytes; # P < 0.01 compared with untreated knockout cells; n = 6). (G) Cyp7a1 levels in hepatocytes treated with IL‐1β for the indicated times (*P < 0.002 compared with untreated control hepatocytes; # P < 0.001 compared with untreated knockout cells; n = 4). Abbreviations: Abcb11, ATP binding cassette subfamily B member 11; Abcb1a, ATP‐binding cassette, sub‐family B (MDR/TAP), member 1A; Abcc2, ATP binding cassette subfamily C member 2; Abcc4, ATP binding cassette subfamily C member 4; Akr1c6, aldo‐keto reductase family 1, member C6; Akr1d1, aldo‐keto reductase family 1 member D1; Amacr, alpha‐methylacyl‐CoA racemase; Baat, bile acid‐CoA:amino acid N‐acyltransferase; Ch25h, cholesterol 25‐hydroxylase; Cyp46a1, cytochrome P450 family 46 subfamily A member 1; Hsd17b4, hydroxysteroid 17‐beta dehydrogenase 4; Hsd3b7, hydroxy‐delta‐5‐steroid dehydrogenase, 3 beta‐ and steroid delta‐isomerase 7; Slc10a1, solute carrier family 10 member 1; Slc27a1, solute carrier family 27 member 1; Slc27a2, solute carrier family 27 member 2; Slc51b, solute carrier family 51 subunit beta; Slco1a1, solute carrier organic anion transporter family, member 1a1; Slco1a4, solute carrier organic anion transporter family, member 1a4; Slco1b2, solute carrier organic anion transporter family, member 1b2; Slco2b1, solute carrier organic anion transporter family, member 2b1; W, weeks.
Bile Acids Fail to Induce Shp in Erk1/2‐KO Mouse Liver and Hepatocytes
Cyp7a1 is principally regulated by transcriptional repression from bile acid–induced SHP.( 25 ) Tamoxifen‐injected Erk1/2‐KO mice had a virtual total inhibition of hepatic Shp mRNA expression at 3 weeks (Fig. 8C). Levels were suppressed within 1 week of tamoxifen injection and inversely paralleled increases in Cyp7a1 over time (Fig. 8D). Decreased Shp and increased Cyp7a1 levels were confirmed in knockout primary hepatocytes (Fig. 8E,F). Cyp7a1 expression was reduced to extremely low levels in control hepatocytes by TCA, but the high level of Cyp7a1 in knockout cells was unaltered by bile acid treatment (Fig. 8E). TCA increased Shp levels markedly in control hepatocytes but minimally in knockout cells (Fig. 8F). Cyp7a1 expression was effectively inhibited in both cell types by the cytokine IL‐1β, which regulates Cyp7a1 through a JNK‐dependent effect on hepatocyte nuclear factor 4α (Fig. 8G).( 26 ) ERK1/2 signaling is required for bile acid induction of Shp in the liver, which down‐regulates Cyp7a1.
In addition to intracellular negative feedback pathways in hepatocytes, hepatic bile acid production is down‐regulated by bile acid–stimulated intestinal FGF15/19 production, which has been reported to be ERK‐dependent.( 8 ) To exclude the possibility that increased bile acids in double knockout mice were secondary to effects on intestinal FGF15, terminal ileum Fgf15 expression was examined in the mice. Fgf15 mRNA levels were markedly elevated in tamoxifen‐injected Erk1/2‐KO mice that have high bile acids (Supporting Fig. S6A). In contrast to decreased Shp expression in liver, intestinal Shp was highly induced in tamoxifen‐injected Erk1/2‐KO mice (Supporting Fig. S6B). The ileum of the hepatocyte‐specific double‐knockout mice with intact intestinal ERK2 signaling therefore exhibits normal Fgf15 induction in response to the increase in bile acids. The increased bile acids in these mice are secondary to the lack of Shp induction and failure to down‐regulate Cyp7a1 in the liver.
Discussion
The findings demonstrate that constitutive ERK1/2 signaling is critical for liver homeostasis by down‐regulating bile acid synthesis. Loss of ERK1/2 was sufficient to markedly increase hepatic and serum bile acids, confirming a central role for this MAPK in maintaining physiological levels. In the absence of ERK1/2, elevated bile acids led to liver injury and inflammation, which progressed to fibrosis and mortality. In addition to demonstrating a central role for ERK1/2 in bile acid regulation, our findings establish Erk1/2‐KO mice as a rodent model of bile acid–induced hepatic injury, inflammation, and fibrosis.
Controversy exists over whether ERK1 and ERK2 perform unique or redundant functions.( 1 , 2 ) Excessive bile acids resulted only from a combined knockout of ERK1 and ERK2, establishing bile acid synthesis as a pathway with isoform redundancy. The study also demonstrates a major overlap of isoform function in the overall regulation of hepatic gene expression, as evidenced by RNA‐sequencing findings that loss of Erk2 alone altered only 1% of the genes as a dual Erk1/2 knockout.
In vitro studies in human hepatocytes with a pharmacological ERK inhibitor demonstrated that FGF15/19 induction and activation of ERK signaling down‐regulate bile acid synthesis by decreasing gene expression of the rate‐limiting synthetic enzyme Cyp7a1.( 8 ) Our findings establish a redundant role for constitutive ERK1 and ERK2 signaling in murine bile acid synthesis in vivo with a specific genetic KO. Loss of physiological levels of ERK1/2 signaling led to Cyp7a1 overexpression, resulting in excessive hepatic bile acid synthesis and accumulation in the liver and serum. ERK1/2 inhibited bile acid synthesis independently of FGF15/19, as ERK1/2 was required to down‐regulate Cyp7a1 expression in cultured hepatocytes in the absence of intestinal FGF15/19, and Erk1/2‐KO mice have normal Fgf15 induction in the ileum. Together these findings indicate that ERK signaling is central to FGF15/19‐independent as well as FGF15/19‐dependent bile acid–mediated inhibition of hepatocyte synthesis.
Loss of ERK1/2 altered the gene expression of Shp, the transcriptional repressor of Cyp7a1. Prior studies of an ERK pharmacological inhibitor in HepG2 hepatoma cells suggested that the effect of ERK on FGF15/19‐mediated Cyp7a1 inhibition was SHP‐independent.( 8 ) However, the present findings demonstrate that ERK1/2 is the major transcriptional regulator of Shp, as mRNA levels were profoundly decreased in Erk1/2‐KO mouse livers and hepatocytes. In contrast, bile‐acid induction of Shp occurred in the ileum, which had intact ERK2, but not ERK1, signaling. Loss of ERK1/2 resulted in a greater than 20‐fold increase in Cyp7a1 expression and marked bile acid accumulation, whereas Shp knockout mice exhibit only a two‐fold increase in Cyp7a1 and a minor bile acid pool increase.( 27 ) These differences may be the result of compensatory mechanisms activated in mice with a global Shp knockout from birth, as opposed to the conditional Erk1/2‐KO mice in which hepatocyte Shp was decreased in adult mice in our study. Alternatively, ERK1/2 may regulate Cyp7a1 through additional Shp‐independent mechanisms. The absence of Shp mRNA in double‐knockout mice is likely due to the loss of an ERK1/2‐regulated transcription factor(s) required for Shp gene expression. Shp transcription has a complex regulation by multiple transcriptional factors,( 28 ) and further studies will be required to fully address the transcriptional effects of ERK1/2 signaling on Shp expression.
Bile acid homeostasis is essential to prevent excessive lipid accumulation and injury in hepatocytes.( 29 ) However, the evidence linking changes in Cyp7a1 expression and elevated bile acids to liver injury is complex and the findings contradictory. Rather than promoting liver injury, Cyp7a1 overexpression in mice protected against high‐fat diet–induced steatosis and inflammation despite increased bile acids.( 30 ) However, Cyp7a1 knockout mice with reduced bile acids were protected from hepatic injury when fed the identical diet.( 31 ) Our study clearly demonstrates that increased bile acids resulted in hepatocyte injury and death. Hepatocyte death from apoptosis secondary to the membrane permeability transition has been implicated in bile acid toxicity.( 32 ) Mitochondrial cytochrome c release and caspase cleavage did not occur in our model, consistent with the recent concept that bile acid–induced hepatocyte death is necrotic rather than apoptotic.( 33 , 34 ) Death from the loss of ERK signaling was clearly secondary to bile acid accumulation, and an ERK1/2 knockout did not alter hepatocyte death from exogenous bile acids. Loss of hepatocyte ERK1/2 led to significant fibrosis, likely secondary to liver injury and inflammation. Alternatively, increased bile acids can directly trigger fibrosis through hepatic stellate cell activation.( 35 )
Activity of another MAPK, JNK1/2, has been implicated in the down‐regulation of murine bile acid synthesis. Findings from studies in Cyp2a12 and Cyp2c70 knockout mice were interpreted to suggest a predominant role for JNK1/2 in the regulation of murine bile acid levels.( 36 ) In addition, a hepatocyte‐specific genetic deletion of JNK1/2 was demonstrated to disrupt bile acid homeostasis in mice by leading to increased serum bile acids.( 11 ) In comparison to findings in ERK1/2 knockout mice, the elevations in serum bile acids were much lower in JNK1/2 knockout mice, and only mild liver injury developed by 10 months of age.( 11 ) Loss of JNK1/2 had no effect on levels of Cyp7a1. In contrast to these two investigations of JNK1/2, our findings demonstrate that ERK1/2 is the MAPK regulating Cyp7a1, and therefore a more critical regulator of normal murine bile acid homeostasis than JNK1/2.
The study confirms a major function for ERK1/2 in the regulation of hepatic gene expression. In particular, the findings demonstrate that basal ERK1/2 activity is critical to regulate hepatocyte bile homeostasis by limiting the physiological expression of Cyp7a1. ERK1/2 signaling represents a unique potential therapeutic target for hepatic pathophysiological states of excessive bile acid accumulation.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1
Table S2
Table S3
Acknowledgment
The authors thank Pierre Chambon for the ERt‐Alb‐Cre mice; Xiao‐Ming Yin for the BID antibody; Shire Pharmaceuticals for the SC‐435; Yury Popov, Disha Badlani, and Pinzhu Huang for performing the hydroxyproline assay; Paul A. Dawson for helpful discussions; and the Emory University School of Medicine Integrated Genomics and Integrated Lipidomics Cores.
Supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK044234).
Potential conflict of interest: Nothing to report.
References
- 1. Busca R, Pouyssegur J, Lenormand P. ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front Cell Dev Biol 2016;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saba‐El‐Leil MK, Fremin C, Meloche S. Redundancy in the world of MAP kinases: all for one. Front Cell Dev Biol 2016;4:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pagès G, Guérin S, Grall D, Bonino Frédéric, Smith A, Anjuere F, et al. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999;286:1374‐1377. [DOI] [PubMed] [Google Scholar]
- 4. Yao Y, Li W, Wu J, Germann UA, Su MSS, Kuida K, et al. Extracellular signal‐regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci U S A 2003;100:12759‐12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schattenberg JM, Wang Y, Rigoli RM, Koop DR, Czaja MJ. CYP2E1 overexpression alters hepatocyte death from menadione and fatty acids by activation of ERK1/2 signaling. Hepatology 2004;39:444‐455. [DOI] [PubMed] [Google Scholar]
- 6. Frémin C, Ezan F, Boisselier P, Bessard A, Pagès G, Pouysségur J, et al. ERK2 but not ERK1 plays a key role in hepatocyte replication: an RNAi‐mediated ERK2 knockdown approach in wild‐type and ERK1 null hepatocytes. Hepatology 2007;45:1035‐1045. [DOI] [PubMed] [Google Scholar]
- 7. Frémin C, Ezan F, Guegan J‐P, Gailhouste L, Trotard M, Le Seyec J, et al. The complexity of ERK1 and ERK2 MAPKs in multiple hepatocyte fate responses. J Cell Physiol 2012;227:59‐69. [DOI] [PubMed] [Google Scholar]
- 8. Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7α‐hydroxylase gene expression. Hepatology 2009;49:297‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim SK, Woodcroft KJ, Oh SJ, Abdelmegeed MA, Novak RF. Role of mechanical and redox stress in activation of mitogen‐activated protein kinases in primary cultured rat hepatocytes. Biochem Pharmacol 2005;70:1785‐1795. [DOI] [PubMed] [Google Scholar]
- 10. Ripple MO, Kim N, Springett R. Acute mitochondrial inhibition by mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase (MEK) 1/2 inhibitors regulates proliferation. J Biol Chem 2013;288:2933‐2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Manieri E, Folgueira C, Rodríguez ME, Leiva‐Vega L, Esteban‐Lafuente L, Chen C, et al. JNK‐mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc Natl Acad Sci U S A 2020;117:16492‐16499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhat BG, Rapp SR, Beaudry JA, Napawan N, Butteiger DN, Hall KA, et al. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE−/− mice by SC‐435. J Lipid Res 2003;44:1614‐1621. [DOI] [PubMed] [Google Scholar]
- 13. Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high‐fat diet‐fed mice. Sci Transl Med 2016;8:357ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schuler M, Dierich A, Chambon P, Metzger D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis 2004;39:167‐172. [DOI] [PubMed] [Google Scholar]
- 15. Xu Y, Jones BE, Neufeld DS, Czaja MJ. Glutathione modulates rat and mouse hepatocyte sensitivity to tumor necrosis factor toxicity. Gastroenterology 1998;115:1229‐1237. [DOI] [PubMed] [Google Scholar]
- 16. Lalazar G, Ilyas G, Malik SA, Liu K, Zhao E, Amir M, et al. Autophagy confers resistance to lipopolysaccharide‐induced mouse hepatocyte injury. Am J Physiol Gastrointest Liver Physiol 2016;311:G377‐G386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao E, Amir M, Lin Y, Czaja MJ. Stathmin mediates hepatocyte resistance to death from oxidative stress by down regulating JNK. PLoS One 2014;9:e109750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor‐induced toxic liver injury results from JNK2‐dependent activation of caspase‐8 and the mitochondrial death pathway. J Biol Chem 2006;281:15258‐15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen Y, Cingolani F, Malik SA, Wen J, Liu Y, Czaja MJ. Sex‐specific regulation of interferon‐γ cytotoxicity in mouse liver by autophagy. Hepatology 2021;74:2746‐2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet‐induced murine steatohepatitis but not steatosis. Hepatology 2013;57:995‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro‐ and antifibrogenic genes. J Hepatol 2005;43:1045‐1054. [DOI] [PubMed] [Google Scholar]
- 22. Cai S‐Y, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte‐specific inflammatory response. JCI Insight 2017;2:e90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jansen PLM, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017;65:722‐738. [DOI] [PubMed] [Google Scholar]
- 24. Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol 2007;22(Suppl 1):S45‐S48. [DOI] [PubMed] [Google Scholar]
- 25. Chiang JYL, Ferrell JM. Up to date on cholesterol 7 alpha‐hydroxylase (CYP7A1) in bile acid synthesis. Liver Res 2020;4:47‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li T, Jahan A, Chiang JY. Bile acids and cytokines inhibit the human cholesterol 7 α‐hydroxylase gene via the JNK/c‐jun pathway in human liver cells. Hepatology 2006;43:1202‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell 2002;2:713‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanyal S, Kim J‐Y, Kim H‐J, Takeda J, Lee Y‐K, Moore DD, et al. Differential regulation of the orphan nuclear receptor small heterodimer partner (SHP) gene promoter by orphan nuclear receptor ERR isoforms. J Biol Chem 2002;277:1739‐1748. [DOI] [PubMed] [Google Scholar]
- 29. Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res 2017;1:3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li T, Owsley E, Matozel M, Hsu P, Novak CM, Chiang JY. Transgenic expression of cholesterol 7α‐hydroxylase in the liver prevents high‐fat diet‐induced obesity and insulin resistance in mice. Hepatology 2010;52:678‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ferrell JM, Boehme S, Li F, Chiang JY. Cholesterol 7α‐hydroxylase‐deficient mice are protected from high‐fat/high‐cholesterol diet‐induced metabolic disorders. J Lipid Res 2016;57:1144‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Botla R, Spivey JR, Aguilar H, Bronk SF, Gores GJ. Ursodeoxycholate (UDCA) inhibits the mitochondrial membrane permeability transition induced by glycochenodeoxycholate: a mechanism of UDCA cytoprotection. J Pharmacol Exp Ther 1995;272:930‐938. [PubMed] [Google Scholar]
- 33. Woolbright BL, Antoine DJ, Jenkins RE, Bajt ML, Park BK, Jaeschke H. Plasma biomarkers of liver injury and inflammation demonstrate a lack of apoptosis during obstructive cholestasis in mice. Toxicol Appl Pharmacol 2013;273:524‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woolbright BL, Dorko K, Antoine DJ, Clarke JI, Gholami P, Li F, et al. Bile acid‐induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol Appl Pharmacol 2015;283:168‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Svegliati‐Baroni G, Ridolfi F, Hannivoort R, Saccomanno S, Homan M, de Minicis S, et al. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005;128:1042‐1055. [DOI] [PubMed] [Google Scholar]
- 36. Honda A, Miyazaki T, Iwamoto J, Hirayama T, Morishita Y, Monma T, et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J Lipid Res 2020;61:54‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1
Table S2
Table S3