

Summary
Myotonic dystrophy type 1 (DM1) and type 2 (DM2) are common forms of adult onset muscular dystrophy. Pathogenesis in both diseases is largely driven by production of toxic-expanded repeat RNAs that sequester MBNL RNA-binding proteins, causing mis-splicing. Given this shared pathogenesis, we hypothesized that diamidines, small molecules that rescue mis-splicing in DM1 models, could also rescue mis-splicing in DM2 models. While several DM1 cell models exist, few are available for DM2 limiting research and therapeutic development. Here, we characterize DM1 and DM2 patient-derived fibroblasts for use in small molecule screens and therapeutic studies. We identify mis-splicing events unique to DM2 fibroblasts and common events shared with DM1 fibroblasts. We show that diamidines can partially rescue molecular phenotypes in both DM1 and DM2 fibroblasts. This study demonstrates the potential of fibroblasts as models for DM1 and DM2, which will help meet an important need for well-characterized DM2 cell models.
Subject areas: Biochemistry, Small molecule, Molecular physiology
Graphical abstract
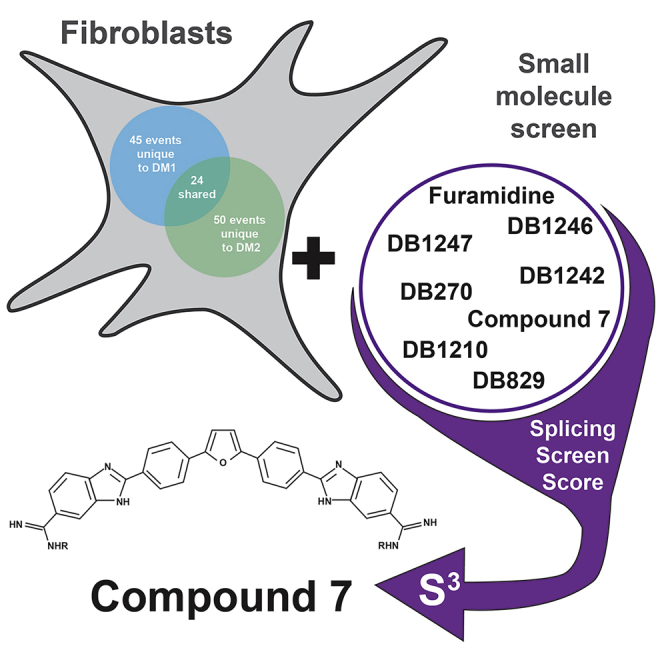
Highlights
-
•
Myotonic dystrophy 2 fibroblasts show mis-splicing and other disease features
-
•
DM1 and DM2 fibroblasts have both common and unique mis-splicing events
-
•
Select diamidines rescued mis-splicing in DM1 and DM2 fibroblasts
-
•
DM fibroblasts are a viable avenue to model disease and test therapeutic approaches
Biochemistry; Small molecule; Molecular physiology
Introduction
Myotonic dystrophy type 1 (DM1) and type 2 (DM2) are common forms of adult onset muscular dystrophy, characterized by autosomal dominant, progressive myopathy with multisystemic involvement (Harper, 2001). DM1 is caused by an expansion of a CTG repeat tract in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene, whereas DM2 is caused by an expanded CCTG repeat tract in intron 1 of the CCHC-type zinc finger nucleic acid-binding protein (CNBP) gene (Harley et al., 1992; Fu et al., 1992; Brook et al., 1992; Liquori et al., 2001; Mahadevan et al., 1992). Both DM1 and DM2 share many pathological features, including myotonia, muscle weakness and wasting, cardiac dysfunction, cataracts, insulin resistance, and cognitive impairment (Day et al., 2003; Meola, 2013). These common clinical pathologies are likely due to a similar shared RNA gain-of-function pathogenic mechanism resulting from the underlying repeat expansion. Upon transcription, the repeat expansion tracts give rise to expanded (C)CUG repeat RNA that form ribonuclear foci which sequester and disrupt normal functions of the muscleblind-like (MBNL) family of RNA-binding proteins (Taneja et al., 1995; Liquori et al., 2001; Fardaei et al., 2002; Day et al., 2003; Miller et al., 2000; Mankodi et al., 2001). The sequestration of MBNL proteins cause changes in alternative splicing, translation, polyadenylation, microRNA processing, and mRNA localization and stability (Du et al., 2010; Masuda et al., 2012; Wang et al., 2012, 2016; Batra et al., 2014). Abnormal regulation of alternative splicing is a molecular hallmark of myotonic dystrophy and can be linked directly to multiple disease features, such as myotonia, cardiac defects, and insulin resistance (Dixon et al., 2015; Mankodi et al., 2002; Savkur et al., 2001).
Although DM1 and DM2 share overlapping pathogenic mechanisms, they possess distinct molecular and clinical features. DM2 has a later onset than DM1, has no congenital form like DM1, and displays more proximal muscle wasting, whereas distal muscles are primarily affected in patients with DM1 (Day and Ranum, 2005). These differences in disease symptoms and presentation are mirrored by differences at the molecular level. For example, in DM1, but not DM2, CUG-BP Elav-like member 1 (CELF1) is upregulated via stabilization by PKC-mediated hyperphosphorylation (Kuyumcu-Martinez et al., 2007; Salisbury et al., 2009; Pelletier et al., 2009). This upregulation of CELF1 may in turn exacerbate mis-splicing caused by MBNL sequestration, as it can antagonistically regulate MBNL-dependent exons (Brinegar and Cooper, 2016). Similarly, RNA-binding Fox (RBFOX) proteins have been shown to bind and colocalize with CCUG repeat RNA foci, but not CUG repeat RNA foci; however, it is not yet clear whether the activity of RBFOX proteins is impacted in patients with DM2 (Sellier et al., 2018). CNBP is more highly expressed than DMPK in most tissues (Sznajder and Swanson, 2019), and the intronic DM2 expansions are generally larger (up to 11,000 CCTG repeats) than the 3′ UTR DM1 expansions (over 5000 CTG repeats) (Otero et al., 2021; Liquori et al., 2001). Additionally, retention of the surrounding intron caused by the expanded repeat has been shown to occur only in DM2 (Sznajder et al., 2018). Despite this potential for higher toxic RNA load in DM2, the degree of mis-splicing appears similar to or less severe than in DM1 (Meola, 2013). Though there are differences in the molecular and clinical features, the shared molecular mechanisms of MBNL sequestration and alterative splicing misregulation in DM1 and DM2 have been the target of numerous therapeutic approaches.
Our lab has previously shown that a class of small molecules called diamidines can partially alleviate the molecular phenotypes of DM1 in cell and mouse models (Jenquin et al., 2018, 2019; Coonrod et al., 2013; Siboni et al., 2015; Warf et al., 2009). Specifically, furamidine was shown to work through multiple mechanisms to rescue DM1 mis-splicing and pathology, including inhibition of the MBNL1-CUG RNA interaction, upregulation of MBNL1 and 2 proteins, and also potentially affecting the transcription and/or stability of CUG RNA (Jenquin et al., 2018). With DM1 and DM2 having similar disease pathology, we hypothesized that furamidine, and other diamidines, could rescue mis-splicing in DM2 models.
Unfortunately, unlike DM1, there are very few well characterized DM2 model systems, making studies and therapeutic development for DM2 challenging. While drosophila models of DM2 recapitulate some aspects of the disease (Cerro-Herreros et al., 2017; Souidi et al., 2018), many of the mis-spliced events observed in patients are not conserved in these model systems. Furthermore, there is no CCTG repeat expansion mouse model that recapitulates the molecular features observed in humans. While MBNL knockout mouse and zebrafish models are useful for studying the effects of MBNL depletion (Kanadia et al., 2003; Lee et al., 2013), they have limited application for testing therapeutics that target the DNA or RNA repeat expansions. Some patient-derived DM2 primary fibroblast and myoblast cell lines are available; however, they have not been well characterized or show limited mis-splicing defects (Wagner-Griffin et al., 2021; Benhamou et al., 2020a, 2020b; Arandel et al., 2017; Sznajder et al., 2018). Therefore, there is a significant need for well characterized cell models that exhibit molecular changes associated with DM2, including mis-splicing, to model the disease and test potential therapeutics.
Here, we characterized multiple primary patient-derived DM2 fibroblast lines displaying key molecular disease features, including MBNL-dependent mis-splicing changes, ribonuclear foci that colocalize with RBFOX, and CNBP intron 1 retention. We assessed mis-splicing globally by RNA-seq and compared the resulting data to mis-splicing in DM1 fibroblast lines to identify mis-splicing events unique to DM2 or common to both DM1 and DM2. Using these model systems, we showed that the small molecule furamidine was able to rescue a subset of mis-spliced events and reduce RNA foci. Additional diamidine analogs were found to rescue some mis-spliced events in both DM1 and DM2 fibroblasts, indicating that diamidines have therapeutic potential across both diseases. The characterization of these cell lines, and demonstration of their responsiveness to small molecule intervention, provides the community another viable avenue through which to model both DM1 and DM2 and test therapeutic approaches.
Results
Patient-derived DM1 & DM2 fibroblast lines display molecular markers of disease
Primary patient (two DM1 and four DM2) and non-disease (two control) fibroblast cell lines (Experimental model and subject details section in STAR Methods) were derived from skin punches collected under a University of Florida-approved IRB protocol. Using multiple assays, we performed a detailed characterization of these DM2 fibroblast cell lines. First, we assessed the CNBP CCTG repeat lengths using the Bionano Genomics Saphyr genome imaging system (Otero et al., 2021). This approach applies sequence-specific labels across the genome, images the DNA in nanochannels, and calculates the distance in nucleotides between labels. At least, 40 DNA molecules were measured across the CNBP locus in each sample, mapped to a reference genome, and the lengths of all non-zero repeat reads were averaged per sample. Repeat lengths ranged from ∼0 ± 100 CCTG units to ∼5300 ± 100 CCTG (Figures 1A–1D). The mean allele expansion length in all DM2 lines exceeded 2000 CCTG units, with DM2-C showing the shortest (∼2200 CCTG; Figure 1C) and DM2-D showing the longest (∼3550 CCTG; Figure 1D) mean allele length (marked by red boxed number and arrow in Figures 1A–1D). The DM2-D line is a homozygous line in which both alleles possess a CCTG repeat expansion, consistent with depletion of short alleles as assessed by optical mapping (Figure 1D) and previously published Southern blots (Sznajder et al., 2018). Second, we assayed retention of CNBP intron 1 in these cell lines as expanded CCTGs in CNBP have been shown to cause retention of intron 1 (Sznajder et al., 2018). All DM2 lines showed increased levels of intron 1 retention (35 ± 4, 33 ± 5, 34 ± 5, and 46 ± 2% for DM2-A, B, C, and D lines, respectively, Figure 1E) mirroring previous observations (Sznajder et al., 2018). Control (CNTRL-B) and DM1 (DM1-B) fibroblasts showed only 13 ± 4% and 9 ± 4% intron retention, respectively. These findings are consistent with the presence of the repeat expansion and the retention of CNBP intron 1 associated with DM2.
Figure 1.

Patient-derived DM1 and DM2 fibroblast lines display molecular markers of disease.
Observed Bionano reads (A–D) are shown for all DM2 fibroblast lines. Histograms of the estimated CCTG repeat lengths (green bars) are shown as read count frequency (left y axis) with the cumulative % distribution functions on the right (gray line, right y axis). The 50th, 75th, and 90th percentiles of repeat lengths are indicated with gray dotted lines. The average repeat size is displayed in the red box with arrow. Note the lack of a large peak at the shorter unaffected repeat size for the homozygous DM2-D cell line (D).
(E) Intron retention assay (E) demonstrated significant CNBP intron 1 retention in all DM2 (green) cell lines compared to control cell lines (black), but not in DM1 cells (blue). Representative gel image of PCR products using primer set amplifying intron 1 to exon3 of CNBP (product schematic shown left of gel image and molecular standard (MS) values are in base pairs to the right). Mean % intron retention ±SD is denoted below (two-tailed t-test, ∗∗p < 0.01, ∗∗∗p < 0.001).
(F) Fluorescence in situ hybridization immunofluorescence microscopy (F) in control, DM1, and DM2 fibroblasts against (C)CUG RNA using either Cy3-(CAG)8 or -(CAGG)6 probes (red). RBFOX2 immunofluorescence shown in purple and DAPI shown in blue. All images were taken using the same laser and contrast/processing settings and a representative scale bar (10μm) is included with each set of micrographs.
(G and H) Splicing analysis (G and H) of control, DM1 and DM2 fibroblasts for INSR exon 11 (G) and MBNL2 exon7 (H) alternative exon events expressed as percent spliced in (PSI). Mean % spliced in ±SD is denoted below (ANOVA two-tailed test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). All experiments were done in at least triplicate using biological replicates.
Ribonuclear foci are another characteristic feature of both DM1 and DM2 that are observed across a number of model systems and in patient-derived tissues. To assess the presence of RNA foci, we performed fluorescence in situ hybridization (FISH) using Cy3-(CAG)8 or Cy3-(CAGG)6 probes in DM1 and DM2 lines, respectively. All cell lines showed characteristic nuclear RNA foci (Figures 1F and S1). We next assessed whether RBFOX2, the predominant RBFOX paralog in fibroblasts, co-localized with RNA foci in these cell lines. Immunofluorescence (IF) microscopy revealed co-localization of RBFOX2 with CCUG foci in DM2 fibroblasts, whereas unaffected cells showed diffuse RBFOX2 staining in the nucleus as reported previously (Sellier et al., 2018) (Figures 1F and S1). Likewise, DM1 cells also showed a diffuse staining similar to the unaffected cells with some slight overlap of RBFOX2 staining with the CUG foci, consistent with previous studies (Sellier et al., 2018).
Lastly, we assessed mis-splicing, a key molecular hallmark of both DM1 and DM2. As an initial assessment, we investigated whether two known DM events, INSR exon11 and MBNL2 exon7 (Table 2), were mis-regulated in the fibroblasts. Both the DM1 and DM2 fibroblasts showed significant mis-splicing for both events, with average INSR exon11 percent spliced-in (PSI) values of 83 ± 7%, 59 ± 7%, and 62 ± 8% (Figure 1G) and average MBNL2 exon7 PSI values of 11 ± 1%, 22 ± 2%, and 18 ± 5% (Figure 1H) for control, DM1, and DM2 fibroblasts, respectively. Taken together, these studies demonstrate that the DM1 and DM2 fibroblasts display molecular features characteristic of DM.
Table 2.
Primers used for RT-PCR splicing analysis and CNBP intron 1 retention
| Target | Forward Primer | Reverse Primer |
|---|---|---|
| INSR exon11 | 5′- AGGGAGATGCTCTCGGGAAAAGTG | 5′- GTTGGCTAGAGCCTGTTGGTATTGG |
| MBNL2 exon7 | 5′- TCCTTTACCAAAGAGACAAGCAC | 5′- CTCAATGCAGATTCTTGGCATTCC |
| MYL6 exon6 | 5′- AGGAAGAAGTAGAGATGCTGGTG | 5′- ATTCACACAGGGAAAGGCAC |
| SYNE1 exon65 | 5′- GGTCCATGAAAGCAGCAATC | 5′- ACTGATTGTGTTCTGCAACG |
| FLNB exon31 | 5′- GCTTCGGTGGTGTTGATATTC | 5′- GTCACTCACTGGGACATAGG |
| MYO5A exon32 | 5′- GAACAACCGACAGCAGCAG | 5′- TTACGGACCGTCTTATCCTG |
| SH3YL1.b (exon9) | 5′- CCTCCAAAGCCATTGTCAAG | 5′- ACTGTGATTCTGTCTCCAGC |
| CPNE1 exon2 | 5′- TTTGTGAAGCGGCGAAGGAG | 5′- CGCAGAGTGGGTCAGACTTG |
| CD46 exon7 | 5′- GGTTTTTACCTCGATGGCAG | 5′- ATTTGAGACTGGAGGCTTGT |
| MPHOSPH9 exon4 | 5′- TCAGGGAAGACCAGACCATC | 5′- CTGCTGGGCAACTATCTGTTC |
| CNBP i1 3′ss (IR primers) | 5′- GAACTTTCAGTGGTTTAATGCTG | 5′- CCGCAGTTATAGCAGGCTTC |
RNA-seq identifies mis-spliced exons transcriptome-wide in DM1 & DM2 fibroblasts
Given that the initial splicing analysis showed evidence for mis-splicing in these fibroblast lines, we performed RNA-seq to measure ΔΨ, difference in PSI between control and DM cells, of alternative exons across the transcriptome. From this analysis, we identified 74 exons mis-spliced in DM2 fibroblasts versus control cells (Table S1), and 69 exons mis-spliced in DM1 fibroblasts versus control fibroblasts (Table S2) (FDR<0.1, ΔΨ ≥ 0.1, ≥25 reads per event). Of these exons, 24 were mis-spliced in both DM1 and DM2 fibroblasts relative to control cells (Figures 2A and 2B). Similar levels of mis-splicing between DM1 and DM2 were observed across the 24 overlapping events; however, slightly less severe mis-splicing was observed in the DM2 fibroblasts (Figure 2C). These 24 exons were enriched for aberrant inclusion events (79%) compared to aberrant exclusion (21%) (Figure 2B). Gene ontology analysis of all 119 mis-spliced events showed no enrichment for specific terms, but more than 85% of the events were protein coding (Table S3). We confirmed several shared mis-spliced exons (SYNE1 exon65, MPHOSPH9 exon4, MYO5A exon32, and TMEM119.a exon2) by RT-PCR (Figures 2D–2G) and observed concordance in PSI between RT-PCR and RNA-seq (Figures S2A–S2D). Similarly, PSI values for INSR exon11 via RNA-seq were similar to that observed during our initial analysis via RT-PCR (Figures S2E and 1G); however, MBNL2 exon7 was not significantly mis-spliced in the RNA-seq datasets despite showing misregulation by RT-PCR (Figure 1H). We also used RT-PCR to confirm mis-splicing of several of the 50 exons that showed dysregulation only in DM2 fibroblasts (CD46 exon4, CPNE1 exon2, MYL6 exon6, and SYH3YL.b (exon9)) and demonstrated that they were not mis-spliced in DM1 fibroblasts (Figures S3A–S3D).
Figure 2.

Global mis-splicing occurs in DM1 and DM2 fibroblasts
Splicing analysis of alternative exon events determined via RNA-seq identified both unique and overlapping events in DM1 (blue) and DM2 (green) fibroblasts.
(A) (FDR<0.1, ΔΨ ≥ 0.1, ≥25 reads across exon junction).
(B) Heatmap of change in percent spliced-in (ΔΨ) for 24 alternative exon events shared between DM1 and DM2 fibroblasts (B) shows bias toward inclusion (19 events) over exclusion (5 events).
(C) Plot of average DM1 ΔΨ (x axis) versus average DM2 ΔΨ for the 22 common alternative exon events (C) shows similar extents of mis-splicing between DM1 and DM2.
(D–G) RT-PCR splicing analysis of control, DM1, and DM2 fibroblasts for common alternative exon events in SYNE1 exon65 (D), MPHOSPH9 exon4 (E), MYO5A exon33 (F), and TMEM119.a (exon2) (G) expressed as percent spliced in (PSI). Mean % spliced in ±SD is denoted below (ANOVA two-tailed test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). All experiments were done in at least triplicate using biological replicates.
Overall, the presence of expanded repeats, intron retention, RNA foci, and mis-splicing establish these DM2 fibroblasts as viable models in which to study disease biology. Our comparison to DM1 fibroblasts demonstrates overlapping and disparate molecular phenotypes as expected based on previous studies. Given these findings, we sought to assess the response of these DM2 cell lines to compounds that we have previously shown rescue molecular phenotypes in DM1 cell and animal models.
Furamidine rescues mis-splicing and reduces ribonuclear foci in DM1 & DM2 fibroblasts
We have previously reported that diamidines can rescue molecular phenotypes in DM1 cell and animal models (Siboni et al., 2015; Jenquin et al., 2018, 2019; Coonrod et al., 2013; Warf et al., 2009), but diamidines have yet to be tested in DM2 models. To determine if patient-derived DM2 fibroblast lines also respond to diamidines, we initially treated DM1, DM2, and control fibroblasts with furamidine. We found that furamidine was able to rescue mis-splicing of INSR exon11 and did not affect splicing in non-disease control cells (Figure 3A). Interestingly, furamidine rescued mis-splicing of another shared mis-splicing event, SYNE1 exon65, in DM2, but not in DM1 fibroblasts (Figure 3B), revealing differences in responsiveness to furamidine. FLNB exon31, identified by RNA-seq as mis-spliced in every DM1 and DM2 fibroblast line except DM2-B (Figure S4), was also rescued by furamidine in all lines except DM2-B and control cells (Figure 3C). We next confirmed the MBNL1 dependence of these regulated exons via RNAi knockdown of MBNL1, as MBNL-dependent mis-splicing is considered a primary molecular marker of DM pathogenesis (Figure S5A). We initially confirmed that INSR exon11 as an MBNL1-dependent event (Figure S5B) and found that both SYNE1 exon65 and FLNB exon31 also showed changes in PSI indicative of MBNL1 dependence (Figures S5C and S5D).
Figure 3.

Furamidine rescues mis-splicing in DM1 and DM2 fibroblasts
(A–C) RT-PCR splicing analysis of common alternate exon events INSR eon11 (A), SYNE1 exon65 (B), and FLNB exon31 (C) in control (black), DM1 (blue), and DM2 (green) fibroblasts untreated (−) or treated with 2 or 4 μM furamidine (see insert in A for structure). Furamidine rescues INSR exon11 splicing across all the DM1 and DM2 fibroblast lines (A); FLNB exon31 splicing in all DM lines except DM2-B line which does not show significant mis-splicing versus control (B); and SYNE1 exon65 splicing only in DM2 lines (C).
(D) Quantification of fluorescence in situ hybridization (FISH) analysis (D) of CCUG RNA foci in DM2 fibroblasts. DM2 fibroblast untreated (−) or treated with 2 or 4 μM furamidine for a period of 4 days followed by FISH analysis using a Cy3-(CAGG)6 probe. The average number of RNA foci per nuclei within a given field was calculated and expressed as a relative value versus untreated cells.
(E) MBNL1 expression relative to GAPDH was determined via RT-qPCR (E) and displays significant increases for both DM1-B (blue) and DM2-A (green) fibroblasts when expressed as a percentage relative to untreated fibroblast.
(F) MBNL1 protein levels via western blot (F) show an increasing trend in DM1-B (blue) line and a significant increase with 2 μM furamidine treatment in DM2-A (green) fibroblasts relative to GAPDH. All experiments were done in at least triplicate using biological replicates (two-tailed t-test ∗ p<0.05, ∗∗ p<0.01, ∗∗∗ p <0.001).
Following furamidine treatment, we also assessed events that showed dysregulation only in DM2 fibroblasts (CD46 exon4 and SYH3YL.b (exon9)), but did not observe rescue of these events in the DM2-A cell line versus CNTRL-B (Figures S6A and S6B). Further, we evaluated the impact of furamidine treatment on CNBP intron 1 retention and demonstrated no significant changes in intron 1 retention in the DM2-A line (Figure S7). These data suggest that furamidine may be targeting shared, MBNL-dependent pathways between DM1 and DM2, but not MBNL1-independent DM2-specific molecular hallmarks.
Because furamidine can reduce ribonuclear foci in DM1 models (Jenquin et al., 2018, 2019), we next quantified abundance of ribonuclear foci in the DM2 fibroblast lines following furamidine treatment. At 4 μM furamidine, the average number of ribonuclear foci per nucleus was reduced by 9%–30% as compared to untreated cells in the DM2 lines (Figure 3D). Similarly, because furamidine treatment has been shown to increase MBNL1 and 2 transcript levels in DM1 models (Jenquin et al., 2018, 2019), we assessed the ability of furamidine to upregulate MBNL1 and 2 transcript levels in DM2 fibroblasts (Table 3). Furamidine increased MBNL1 (Figure 3E) and MBNL2 (Figure S8) transcript levels by ∼40% at 4 μM in both DM1-B and DM2-A fibroblast lines compared to untreated cells. The increased MBNL1 transcript levels corresponded to a trend in increased MBNL1 protein levels in the DM1-B line; however, the increases were not significant due to high variability (Figure 3F, representative blots Figure S9). MBNL1 protein levels were significantly increased by 25% in DM2-A fibroblasts at 4 μM compared to untreated cells (Figure 3F, representative blots Figure S9). Unfortunately, endogenous MBNL2 protein levels were too low to detect via western blot analysis in fibroblasts. In both the DM1 and DM2 fibroblasts, significant toxicity was only observed above concentrations corresponding to mis-splicing rescue (>8 μM, Figure S10), which is similar to that previously observed in DM1 myotubes (Jenquin et al., 2018). These data, generated with a small molecule previously shown to be a potential DM1 therapeutic candidate, support DM2 fibroblasts as a potential model system to test novel therapeutic approaches.
Table 3.
Primers used for RT-qPCR expression analysis
| Target | Forward Primer | Reverse Primer |
|---|---|---|
| MBNL1 | 5′- CGCAGTTGGAGATAAATGGACG | 5′- CACCAGGCATCATGGCATTG |
| MBNL2 | 5′- CCTGGTGCTCTTCATCCTTTAC | 5′- GTGAGAGCCTGCTGGTAGTG |
| GAPDH | 5′- AATCCCATCACCATCTTCCA | 5′- TGGACTCCACGACGTACTCA |
Screening diamidine analogs for mis-splicing rescue in DM1 and DM2 fibroblasts
To investigate the therapeutic potential of a wider range of diamidines across both DM1 and DM2 fibroblasts, we tested the responsiveness of these cells to a set of diamidine analogs with similar structural features to furamidine. The diamidine analogs used were identified from the literature and have been previously shown to have chemical properties that may be applicable to modes of rescue in DM models, such as enhanced GpC-binding affinity, rescue of molecular phenotypes in other GC-rich microsatellite expansion diseases, or the ability to cross the blood–brain barrier (Munde et al., 2007; Simone et al., 2018; Paine et al., 2010; Thuita et al., 2012; Stephens et al., 2016; Bailly et al., 2001; Hu et al., 2013). The structures of furamidine and the selected diamidine analogs, as well as more about the chemical characteristics and their breakdown into three groups, can be found in Figure S11.
To streamline the comparison process, we focused on mis-splicing rescue in the DM1-B and DM2-A lines relative to CTRL-B, as these lines showed the most robust, reproducible mis-splicing for MBNL-dependent exons and showed responsiveness to furamidine (Figures 3A–3C). Cells were treated with each diamidine analog for 4 days, and percent rescue was assessed by RT-PCR for INSR exon11, FLNB exon31, and SYNE1 exon65. Mis-splicing rescue observed at concentrations corresponding to significant decreases in cell viability below 90% were omitted for all compounds.
The response of the DM fibroblasts to treatment varied across all compounds tested. The first screening group, DB1242 and DB1210 (structures inset in Figures 4A and 4B), showed variable mis-splicing rescue. INSR showed strong rescue of ∼40% in the DM2 cell line, but no rescue in the DM1 line for either compound (Figures 4A and 4B). Interestingly, robust rescue of over 80% of FLNB with DB1242 treatment was observed in DM1, but only ∼35% in DM2 (Figure 4A). With DB1210 treatment, both cell lines showed significant rescue of FLNB of ∼35% (Figure 4B). Marginal rescue of SYNE1 (∼20%) was observed only at the highest concentration of DB1242 in DM2 cells, but no significant rescue was observed in DM1 cells (Figure 4A). Neither cell line showed rescue for SYNE1 with DB1210 treatment (Figure 4B). Both compounds showed decreases in cell viability in the 32–64 μM range: concentrations outside of the range where splicing rescue was observed (Figures S12A and S12B).
Figure 4.

Diamidine analogs show variable mis-splicing rescue in DM2 and DM1 fibroblasts
(A–H) RT-PCR splicing analysis of the common alternative exon events INSR exon 11, FLNB exon 31, and SYNE1 exon65 in DM1 (blue) and DM2 (green) fibroblasts treated for 4 days with either group one compounds, DB1242 (A) and DB1210 (B); group two compounds, DB829 (C), DB1246 (D), DB1247 (E), and DB1273 (F); and group three compounds, DB270 (G) and Compound 7 (Cmpd7) (H). All compound groups show variable mis-splicing rescue across DM1 and DM2 fibroblasts. Structures of the diamidine analogs are shown on the top right corner of each graph. All experiments were done in at least triplicate using biological replicates (two-tailed t-test ∗∗p < 0.01, ∗∗∗p < 0.001).
The second screening group, DB829, DB1246, DB1247, and DB1273 (structures inset in Figures 4C through 4F), showed more consistent rescue of INSR mis-splicing across all four compounds compared to group one. DB1247 and DB1273 demonstrated strong rescue, upward of ∼60%–100%, in both cell lines at high nM to low μM doses (Figures 4E and 4F), displaying higher efficacy over DB829 and DB1246 (Figures 4C and 4D). Analysis of FLNB showed generally greater mis-splicing rescue in DM1 than DM2 for all group two compounds (Figures 4C–4F), with DB1273 having the highest rescue of FLNB mis-splicing from ∼80%–100% in both lines (Figure 4F). Again, SYNE1 was not rescued in the DM1 cells, and only marginally rescued in DM2 cells, with all analogs in this group, with the exception of DB1247 which displayed ∼40%–50% rescue of SYNE1 in both cell lines (Figure 4E). Thus far, DB1247 was the only compound able to rescue SYNE1 mis-splicing in the DM1 line. However, dramatic decreases in cell viability were observed at levels greater than 1 μM for DB1247 and DB1273 (Figures S12E and S12F) and greater than 8 μM for DB1246 (Figure S12D), which are concentrations close to where maximum mis-splicing rescue was observed for these compounds. Cell viability was only moderately affected at 64 μM for DB829 (Figure S12C).
The two compounds in the third screening group, DB270 & Compound 7 (structures inset in Figures 4G and 4H), showed slightly different patterns of mis-splicing rescue. DB270 showed ∼40% rescue of INSR for DM2, but only ∼10% rescue for DM1 (Figure 4G). Compound 7, however, displayed a maximum percent rescue of ∼45% for both DM1 and DM2 at 1 μM (Figure 4H). In a trend similar to other groups, rescue of FLNB was better for DM1 (∼60%–70%) than DM2 (∼20%–35%) for both compounds, but Compound 7 obtained higher percent rescue of FLNB in both lines (Figures 4G and 4H) over DB270. SYNE1 showed low levels of rescue for both compounds in DM2 cells (about ∼20%). However, Compound 7 also showed ∼20% rescue in the DM1 line for SYNE1 (Figure 4H), being the only other compound besides DB1247 to show rescue of SYNE1 in the DM1 line (Figure 4E). Similar to the first group, effects on cell viability were only observed at higher μM levels (64 μM), well beyond the range where maximum rescue of splicing was observed (Figures S12G and S12H).
Taken together, these results demonstrate highly variable responses of the mis-splicing events and cell lines to the various diamidines. Overall, DB1247 and Compound 7 showed the most consistent mis-splicing rescue across all three splicing events (Figures 4E and 4H). DB1247 displayed higher mis-splicing rescue of the events over Compound 7; however, Compound 7 had significantly lower toxicity and that toxicity occurred at concentrations far outside of the range where mis-splicing rescue was observed. In order to facilitate a quantitative and rapid comparison of the compounds screened that considers the different mis-splicing events in each cell line, we developed a scoring matrix to generate a single numerical score for each diamidine analog. To generate this score, we calculated the following variables for each compound and each splicing event: (1) the maximum percent rescue or Emax, (2) the concentration required to reach 50% rescue or EC50, and (3) the concentration at which 50% cell viability was reached or IC50. A single value splicing screen score (S3) was determined by dividing the Emax by EC50 and multiplying this value by the IC50, then normalizing to the S3 of furamidine allowing for a direct comparison to the overall efficacy and toxicity of furamidine, our previous lead compound. Averaging the S3 across all three mis-splicing events (INSR, FLNB, and SYNE1) enabled a determination of the composite S3 for each compound (S3∗; Table 1). While we have chosen INSR, FLNB, and SYNE1 in this study, any events that are mis-spliced could be utilized in future screens. Based on the S3∗ for the eight compounds analyzed, Compound 7 showed the best score in both DM1 (35.48) and DM2 cell lines (4.46) (Table 1). This scoring matrix, based upon the selected mis-splicing events, provides an overview of the efficacy of the multiple diamidines screened in this study and establishes another potential tool to assess small molecules and novel therapeutic approaches for myotonic dystrophy. Overall, our use of DM1 and DM2 fibroblast cell lines in this small-scale screen of multiple diamidines with different structural profiles has identified a new lead compound, Compound 7, which has improved efficacy to toxicity profile relative to furamidine. Compound 7 is a potential candidate for future development and further investigations in additional DM1 and DM2 cell and animal model systems.
Table 1.
Variables associated with the splicing screen score (S3) for diamidine analog screen
|
INSR ex11 |
FLNB ex31 |
SYNE1 ex65 |
IC50 | S3∗ | ||||
|---|---|---|---|---|---|---|---|---|
| EC50 | Emax | EC50 | Emax | EC50 | Emax | |||
| DM1-B | ||||||||
| Furam | 3 ± 0.4 | 75 ± 7 | 3 ± 0.4 | 83 ± 4 | – | – | 18 ± 0.4 | 1.00 |
| DB1210 | – | – | 25 ± 4 | 25 ± 7 | – | – | 88 ± 3.6 | 0.09 |
| DB1242 | – | – | 2 ± 1 | 97 ± 9 | – | – | 68 ± 4.7 | 3.18 |
| DB829 | 27 ± 5 | 41 ± 15 | 2 ± 1 | 89 ± 15 | – | – | 73 ± 1.1 | 3.23 |
| DB1246 | 22 ± 4 | 22 ± 9 | 3 ± 2 | 74 ± 13 | – | – | 15 ± 0.5 | 0.42 |
| DB1247 | 0.6 ± 0.1 | 77 ± 12 | 0.5 ± 0.3 | 70 ± 4 | 1.7 ± 1.3 | 36 ± 11 | 1 ± 0.2 | 0.38 |
| DB1273 | 0.8 ± 0.3 | 45 ± 8 | 0.1 ± 0.1 | 109 ± 3 | – | – | 2 ± 0.1 | 1.64 |
| DB270 | 186 ± 26 | 13 ± 3 | 1.6 ± 1 | 57 ± 13 | – | – | 69 ± 1.3 | 2.46 |
| Cmpd7 | 0.1 ± 0.1 | 42 ± 10 | 0.4 ± 0.3 | 68 ± 5 | 20 ± 2 | 28 ± 11 | 69 ± 0.5 | 35.48 |
| DM2-A | ||||||||
| Furam | 2 ± 0.6 | 77 ± 10 | 2 ± 0.7 | 74 ± 4 | 5 ± 1 | 63 ± 12 | 19 ± 0.8 | 1.00 |
| DB1210 | 23 ± 8 | 38 ± 8 | 24 ± 6 | 34 ± 8 | – | – | 93 ± 5.1 | 0.16 |
| DB1242 | 24 ± 5 | 51 ± 6 | 71 ± 14 | 36 ± 6 | 185 ± 2 | 23 ± 16 | 75 ± 6.1 | 0.12 |
| DB829 | 10 ± 2 | 74 ± 1 | 22 ± 5 | 53 ± 4 | 129 ± 3 | 25 ± 14 | 74 ± 4.8 | 0.43 |
| DB1246 | 8 ± 3 | 57 ± 7 | 7 ± 3 | 51 ± 1 | 16 ± 2 | 28 ± 13 | 15 ± 0.9 | 0.15 |
| DB1247 | 0.9 ± 0.5 | 86 ± 11 | 0.9 ± 0.4 | 49 ± 4 | 0.9 ± 1.1 | 51 ± 7 | 2 ± 0.4 | 0.19 |
| DB1273 | 0.9 ± 0.4 | 58 ± 2 | 0.2 ± 0.3 | 77 ± 4 | 1.8 ± 1.4 | 34 ± 11 | 2 ± 0.3 | 0.43 |
| DB270 | 23 ± 4 | 43 ± 9 | 33 ± 9 | 21 ± 3 | 16 ± 2 | 25 ± 13 | 67 ± 1.1 | 0.15 |
| Cmpd7 | 0.7 ± 0.3 | 48 ± 11 | 0.8 ± 1 | 36 ± 7 | 15 ± 2 | 19 ± 14 | 68 ± 2.1 | 4.46 |
EC50 = Concentration required to reach 50% rescue; Emax = maximum percent rescue; IC50 = concentration required to reach 50% viability; S3∗ = Average of [(Emax/EC50) x IC50] normalized to score of furamidine for INSR exon11, FLNB exon31, and SYNE1 exon65 alternative exon events. DM1-B cell line values under blue column headings and DM2-B cell line values are under green column headings.Bolded entry indicate compound with the highest S3 score.
Discussion
Here, we characterized multiple patient-derived DM2 fibroblast lines confirming characteristic molecular disease features, including RNA foci, RBFOX foci co-localization, and CNBP intron 1 retention. Mis-splicing in these DM2 fibroblasts displays MBNL-dependent mis-splicing similar in severity to that observed in DM1 fibroblast lines with multiple events shared between DM1 and DM2. Additionally, we identified events unique to DM2 fibroblasts, including CPNE1 exon2, MYL6 exon6, SH3YL1.b (exon9), and CD46 exon4. We then demonstrated that DM2 fibroblasts were responsive to the small molecule furamidine, previously used to rescue molecular phenotypes in DM1 models (Siboni et al., 2015; Jenquin et al., 2018, 2019). At 4 μM treatment, furamidine showed ∼50%–95% rescue of INSR exon11, FLNB exon31, and SYNE1 exon65 mis-splicing and reduced ribonuclear foci abundance by ∼10%–30% in all DM2 fibroblast lines. Interestingly, however, DM2-specific mis-splicing events were not rescued by furamidine, potentially indicating the need for additional DM2-specific therapeutics that target additional points of regulation in the DM2 pathogenic cascade. Furthermore, CNBP intron 1 retention was not impacted by furamidine treatment; however, it is not yet clear if intron 1 retention is an appropriate readout for therapeutic efficacy. We also used these fibroblasts to test a set of diamidine analogs for their ability to rescue mis-splicing of INSR exon11, FLNB exon31, and SYNE1 exon65 events. To generate a streamlined and quantitative approach for cross comparison of compounds that rescue mis-splicing, we developed a single value splicing screen score (S3) to identify the lead candidate compound. Based upon a composite S3 for the eight compounds screened, Compound 7 showed the best score in both DM1 and DM2 cell lines. Compound 7 has an extended aromatic system with the substitution of bis-benzimidazole amidines for the amidine groups of furamidine, which can result in higher affinity for DNA sequences due to the ability to span a larger number of bases in the minor groove (Hopkins et al., 1998). In future studies, it will be interesting to determine the mechanism of action (MOA) and activity of Compound 7 across multiple model systems, including DM1 myotubes and mouse models and, when available, additional DM2 cell and animal models.
Notably, these data suggest that diamidines can modulate disease phenotypes associated with not only CUG repeats but also CCUG repeats. While determining the exact MOA for each of these compounds was outside the scope of this work, in future studies, it will be interesting to determine if any of these compounds work through multiple mechanisms like furamidine. An open question for future studies with these compounds is how treatment with these compounds could impact MBNL protein localization and potential role in the MOA. From our previous work, we know that diamidines such as pentamidine and heptamidine rescue DM1 mis-splicing primarily by affecting the transcription and/or stability of CUG RNA, while furamidine may primarily work through inhibition of the MBNL-CUG RNA interaction and upregulation of MBNL1 and 2 proteins. For instance, some compounds may have differential affinity for CUG/CTG versus CCUG/CCTG repeats and may or may not upregulate MBNL proteins. Explanations for the variability in mis-splicing rescue across the different events and cell lines by each diamidine could be that their MOAs are different and affect each system uniquely by potentially targeting one, or a combination, of these steps within the DM disease mechanism. Alternatively, the compounds may have different affinities for their targets leading to the different efficacies even with the same MOAs.
A major obstacle to understanding the disease biology of DM2 and developing therapeutic approaches has been the lack of DM2 model systems. To help address this significant need, we characterized multiple primary patient-derived DM1 and DM2 fibroblast lines, verifying multiple known molecular disease markers and identifying unique and shared mis-splicing events. We used these fibroblasts to show partial rescue of molecular features of DM1 and DM2 following treatment with furamidine and other diamidines. The identification of both shared and unique mis-splicing events for DM1 and DM2, and a biological resource to test resulting hypotheses on the cause and consequences of these events, could help to shed light on the molecular landscape of myotonic dystrophy. This same biological resource also provides a valuable platform for the pre-clinical assessment of therapeutic interventions, an especially critical resource given the lack of an animal model of DM2. Taken together, this study fills an important gap that has thus far limited our understanding of DM2 disease biology and therapeutic approaches applicable to DM2.
Limitation of the study
Variability in clinical and disease presentation is one of the hallmarks of myotonic dystrophy, making it unlikely that a single cell line or model system can accurately reflect the complex repeat expansion disease biology. This variability, in addition to the potential compound-specific mechanisms mentioned above, may also be reflected in the variable mis-splicing and rescue observed herein. Analyzing multiple molecular hallmarks and mis-splicing events across multiple cell lines provides additional data to aid in generating a better understanding of this complex disease pathogenesis. Collectively, these data establish a set of patient-derived DM2 fibroblast lines suitable for molecular studies of DM2, as well as assessment of therapeutic interventions. While fibroblasts are not the most disease-relevant cells to study neuromuscular diseases, these lines are easier to obtain, relatively robust, and easy to adapt for high-throughput therapeutic screens. Fibroblasts could also serve as candidates for reprogramming in order to generate DM myoblasts and/or iPSCs (Hirai et al., 2018; McGrath et al., 2018; Tai et al., 2018).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MBNL1 | Wolfson Centre for Inherited Neuromuscular Disease | MB1a; RRID:AB_2618248 |
| Rabbit monoclonal anti-GAPDH | Abcam | Cat# ab181602; RRID:AB_2630358 |
| Rabbit polyclonal anti-RBFOX2 | Sigma Aldrich | Cat# HPA006240; RRID:AB_1079750 |
| Deposited data | ||
| Raw and analyzed data | This paper | SRA: PRJNA795461 |
| Experimental models: Cell lines | ||
| Human: Non-disease fibroblasts | Laboratory of Laura Ranum | CNTRL-A |
| Human: Non-disease fibroblasts | Laboratory of Laura Ranum | CNTRL-B |
| Human: DM1 fibroblasts | Laboratory of Laura Ranum | DM1-A |
| Human: DM1 fibroblasts | Laboratory of Laura Ranum | DM1-B |
| Human: DM2 fibroblasts | Laboratory of Laura Ranum | DM2-A |
| Human: DM2 fibroblasts | Laboratory of Laura Ranum | DM2-B |
| Human: DM2 fibroblasts | Laboratory of Laura Ranum | DM2-C |
| Human: DM2 fibroblasts | Laboratory of Laura Ranum | DM2-D |
| Oligonucleotides | ||
| DsiRNA targeting sequence: MBNL1 | IDT | hs.Ri.MBNL1.13.1 |
| Negative Scramble DsiRNA control | IDT | Cat# 51-01-19-09 |
| FISH CTG repeat probe: Cy3-CAGCAGCAGCAGCAGCAGCAGCAG | IDT | N/A |
| FISH CCTG repeat probe: Cy3-CAGGCAGGCAGGCAGGCAGGCAGG | IDT | N/A |
| All qPCR primer sequences listed in Table 1 in Method details | IDT | N/A |
| All splicing primers sequences listed in Table 2 in Method details | IDT | N/A |
| Software and algorithms | ||
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| STAR | https://doi.org/10.1093/bioinformatics/bts635 | version 2.5.1b |
| rMATS | https://doi.org/10.1073/pnas.1419161111 | version 3.2.5 |
| GeneOntology Tools | https://doi.org/10.1038/75556 and https://doi.org/10.1093/nar/gkaa1113 | Various |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, J. Andrew Berglund (abergluund@albany.edu).
Materials availability
-
•
This study did not generate any new or unique reagents or materials.
-
•
Distribution of non-disease, DM1, and DM2 patient-derived fibroblast cell lines used in this study require a Materials Transfer Agreement (MTA) with the University of Florida Center for NeuroGenetics and inquiries should be addressed to Dr. Laura Ranum (ranum@ufl.edu).
Experimental model and subject details
Primary human cell lines
All lines were cultured in DMEM growth medium supplemented with 15% (v/v) FBS and 1% Pen-Strep at 37°C and 5% CO2. No cell aunthentification was performed. Cells were assessed for relevant disease biomarkers and phenotypes.
-
•
CNTRL-A: Female Non-disease control fibroblast line.
-
•
CNTRL-B: Male Non-disease control fibroblast line.
-
•
DM1-A: Male DM1 disease fibroblast line.
-
•
DM1-B: Female DM1 disease fibroblast line.
-
•
DM2-A: Male DM2 disease fibroblast line.
-
•
DM2-B: Male DM2 disease fibroblast line.
-
•
DM2-C: Male DM2 disease fibroblast line.
-
•
DM2-D: Female DM2 disease fibroblast line.
Method details
Culturing of fibroblasts
Primary patient and control fibroblast cell lines were derived from skin punches under a University of Florida-approved IRB protocol with informed consent from all subjects. Approximately 1 × 105 fibroblasts were plated in 12-well plates in DMEM growth medium supplemented with 15% (v/v) FBS and 1% Pen-Strep. Cells were allowed to reach >90% confluency and then treated by adding the indicated concentrations of drug. Fibroblasts were in a quiescent state at the time of harvest after 4 days of drug treatment.
RT-PCR analysis for splicing or intron retention
RNA was isolated using an AurumTM Total RNA mini kit (Bio-Rad) according to package insert with on-column DNase1 treatment. RNA concentrations were determined using a NanoDrop (Thermo) and reverse transcribed with SuperScript VI with random hexamer primers (IDT). The cDNA was then subjected to polymerase chain reaction for 32 cycles using the primer sets listed in Table 2. Resulting PCR products were run via capillary electrophoresis on Fragment Analyzer using the 1-500 bp DNF-905 kit (Agilent) for splicing or on a 1% agarose gel dyed with Gel Green for intron retention assay. Quantification for splicing was done using the integration values of the electropherogram peaks corresponding to inclusion and exclusion products from the Prosize 2.0 software (Agilent), while image J was used to determine band intensities for intron retention assay. To determine the % rescue of a given ES event, Equation 1 was used, where DM_PSI = PSI of untreated DM fibroblasts, WT_PSI = PSI of control fibroblasts, and drug_PSI = PSI of DM fibroblasts treated with indicated drug.
| (Equation 1) |
RT-qPCR for expression analysis
Quantitative real-time PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) according to the package insert. Samples were run on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) and analyzed using the Quantitative-Comparative (CT) method. The levels of MBNL1 and MBNL2 mRNA in DM fibroblasts were normalized to GAPDH mRNA and displayed graphically as relative mRNA levels setting the untreated mRNA levels to 1. Primer sets used are shown in Table 3.
Fluorescence in situ hybridization with immunofluorescence
Fibroblasts were fixed with 4% w/v PFA, permeabilized using 1% Triton X-100 in DPBS for 20 min at RT, blocked with Background Sniper (Biocare Medical), incubated with rabbit anti-RBFOX2 primary antibody at 1:500 (Sigma Aldrich) overnight at 4°C, and then incubated with 1:1000 goat anti-rabbit AlexaFluor 647 (Thermo) for 1 hr at RT. Fibroblasts samples were re-fixed, washed in DPBS, pre-hybridized for 30 min at 37°C, and then probed for 4 hrs at 50°C with a Cy3-(CAG)8 DM1 or Cy3-(CAGG)6 DM2 probe (IDT). Slides were washed with 42°C pre-warmed 40% v/v formamide in 2X SSC and mounted using ProLong Diamond Antifade mountant with DAPI (Life Technologies). Fibroblasts were imaged on a Zeiss LSM 840 confocal scanning microscope with a 40X water objective. All images were taken at the same laser intensities and settings. In DM2 fibroblasts, the foci were counted blind in at least 100 nuclei per replicate (at least 300 nuclei over three replicates) for all furamidine treatments. The number of nuclear foci for each cell was quantified using Fiji.
RNAi knockdown
Approximately 1 × 104 fibroblasts were plated in 12-well plates in DMEM growth medium supplemented with 15% (v/v) FBS and 1% Pen-Strep. The next day the media was refreshed and cells were transfected with 20nM of either a scramble control or MBNL1 siRNA (hs.Ri.MBNL1.13.1 DsiRNA from IDT) using RNAi-MAX (Thermo) according to the package insert. Cells were harvested 48 hours later and RNA was extracted using an AurumTM Total RNA mini kit (Bio-Rad) according to package insert with on-column DNase1 treatment. RNA concentrations were determined using a NanoDrop (Thermo) and reverse transcribed with SuperScript VI with random hexamer primers (IDT) for later use in RT-PCR splicing or RT-qPCR expression assays.
Western blot analysis
Protein was extracted by vortexing for 15 min at 4°C in RIPA buffer (Thermo Scientific) supplemented with complete EDTA-free Protease Inhibitor Cocktail (Roche). After centrifugation at 21,000 rcf for 15 min at 4°C, the supernatant was used to determine protein concentration with the Pierce BCA Protein Assay kit (Thermo). A total of 18 μg of protein was denatured for 5 min in 1X reducing Laemmli SDS sample buffer (Alfa Aesar) at 98°C and separated on a pre-cast 4-12% Bis-Tris gel (Bio-Rad) in 1X NuPAGE MES SDS running buffer (Invitrogen). The gel was transferred onto low fluorescence PVDF membrane (Millipore) in 1X NuPAGE transfer buffer (Invitrogen) and the membrane was blocked for 1 hr using SeaBlock (Thermo) and incubated overnight with primary antibodies [1:400 MBNL1 (MB1a, Wolfson Centre for Inherited Neuromuscular Disease), 1:5000 GAPDH (ab181602, abcam)] in SeaBlock. Blots were washed for 3 × 5minutes in 1X TBS-T and incubated at RT for 1 hr with secondary antibodies [1:7500 Goat anti-Mouse IRDye @680 (Li-Cor), 1:7500 Goat anti-Rabbit IRDye @800 (Li-Cor)] in SeaBlock, washed in 1X TBS-T for 3 × 5minutes, and imaged on a ChemiDoc MP Imaging System (Bio-Rad). Blots were analyzed using ImageJ. The relative levels of MBNL1 were calculated by normalizing lanes within the same gel to GAPDH and then by normalizing levels of MBNL1 in the untreated cells to 1.
High molecular weight DNA extraction and labeling
Cultured fibroblasts were embedded in agarose plugs and incubated with lysis buffer and Proteinase K at 50°C overnight. RNase A was added and incubated for 1 hour at 37°C. Agarose was digested with agarase and the purified DNA was subjected to drop dialysis for 4 hours and quantified by qubit BR DNA assay (Thermo). Purified DNA was labeled using the Direct Label Enzyme (DLE) method (Bionano Genomics). A total of 750ng of DNA was labeled using the DLE-1 kit following the manufacturer’s instructions and then treated with proteinase K. The DNA was stained with YOYO-1 according the DLE-1 kit instructions and homogenized by HulaMixer. The stained sample was incubated overnight at room temperature.
Genome map assembly for repeat expansion sizing
Labeled and stained DNA was loaded onto the Bionano Genomics Saphyr chip. DNA was loaded and linearized in the nanochannel array by electrophoresis. Strands were imaged and the backbone and labels detected by Bionano image detection software. Single-molecule maps were assembled into consensus maps. Consensus maps were refined and merged based on overlapping segments. The final consensus maps were aligned to the GRCh38 human reference genome. Repeat expansion in the CNBP gene were identified by distances between flanking labels in the single-molecule maps aligned to the region.
RNA-seq library preparation
RNA quality was checked via capillary electrophoresis on Fragment Analyzer using the RNA Analysis DNF-471 kit (Advanced Analytical). The NEBNext Ultra II Directional RNA Library Prep Kit for Illumina with NEBNext rRNA Depletion Kit was used to prepare RNA-seq libraries, with a total of 500 ng input RNA from each sample. The manufacturer’s protocols were followed, with the following exceptions: 40X adaptor dilutions used, all bead incubations done at room temp, used 4X lower concentrations of index primers, and ten cycles of library amplification were performed. The resulting libraries were pooled in equimolar amounts, quantified using the KAPA Library Quant Kit for Illumina, quality checked via capillary electrophoresis on Fragment Analyzer using the NGS Analysis DNF-474 kit (Advanced Analytical), and were sequenced using paired-end, 75 base pair sequencing on the Illumina NextSeq 500 massively parallel sequencer at the University of Florida Center for NeuroGenetics.
RNA-seq data analysis
For splicing, at least 50 million paired-end reads per sample were checked for quality and aligned to GRCh38.p10 human genome using STAR (version 2.5.1b). After reads were aligned, rMATS (version 3.2.5) (Shen et al., 2014) was used to analyze isoform abundances. ES events were considered significant with an FDR<0.1, p<0.01, and at least 25 reads spanning the exon junctions. Events were considered mis-spliced in the Control vs DM1/DM2 fibroblast data sets if the PSI change was ≥10% for a given ES event. Of those events, a percent rescue of ≥10% were considered ‘rescue’ with drug treatment. To determine the % rescue of a given ES event, Equation 1 was used as described in RT-PCR analysis for splicing or intron retention. Three biological replicates were sequenced for each sample. Gene ontology was performed on the gene level using the tools available at GeneOntology (Ashburner et al., 2000; Gene Ontology Consortium, 2021). The list of genes for this analysis was generated from those genes that showed exons that were significantly mis-spliced in the DM1 and DM2 RNA-seq datasets (see Table S3).
Quantification and statistical analysis
Data are expressed as mean ± standard deviation. All data shown are the summary of three or more biological replicates and statistical analyses were completed in Prism 7. For data sets where treatments were compared individually to a control, two-tailed student’s t-test was used to determine statistical significance and associated p-value. When multiple groups were compared to each other ANOVA two-tailed statistical measurements were used. Statistical values used: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Acknowledgments
Special thanks to other members of the Wang and Berglund labs and Dr. Kaalak Reddy for helpful discussions, experimental advice, and comments on the manuscript. We also thank the UF Center for NeuroGenetics, especially the Ranum and Swanson labs, as well as our colleagues at the RNA Institute for their general support and guidance. These studies were supported by funding from the MDA (516314), NIH (P50NS04843 and R01GM121862) to JAB, ETW, NIH (P01-NS058901) to LPWR, NIH supplement (P50NS04843) to JAF and a 2020 MDF postdoctoral fellowship to JRJ.
Author contributions
JRJ and JAB conceived the project and analyzed results. JRJ, JAB, JDC, and ETW wrote the manuscript. JRJ characterized and established treatment ranges in DM1 and DM2 fibroblasts, performed RT-PCR, RT-qPCR, FISH-IF, RNAi knockdown, and intron retention assay, prepared the RNA-seq libraries, and performed RNA-seq bioinformatics analyses. APO helped perform RT-PCR, RT-qPCR, and RNAi knockdown. KP, YL, and ETW developed and performed assay to determine CCTG repeat expansion size. JAF and HKS performed western and additional RT-PCR assays. JIR performed bioinformatics analyses. HM, HY, RWH, and DB synthesized & provided compounds. LPWR provided DM2 fibroblast lines.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104198.
Supplemental information
Data and code availability
-
•
RNA-seq data generated from non-disease, DM1, and DM2 fibroblasts for this study have been deposited in the Sequence Read Archive (SRA). Accession numbers are listed in the Key resources table. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
All original code will be shared by the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Arandel L., Polay Espinoza M., Matloka M., Bazinet A., De Dea Diniz D., Naouar N., Rau F., Jollet A., Edom-Vovard F., Mamchaoui K., et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model Mech. 2017;10:487–497. doi: 10.1242/dmm.027367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly C., Tardy C., Wang L., Armitage B., Hopkins K., Kumar A., Schuster G.B., Boykin D.W., Wilson W.D. Recognition of ATGA sequences by the unfused aromatic dication DB293 forming stacked dimers in the DNA minor groove. Biochemistry. 2001;40:9770–9779. doi: 10.1021/bi0108453. [DOI] [PubMed] [Google Scholar]
- Batra R., Charizanis K., Manchanda M., Mohan A., Li M., Finn D.J., Goodwin M., Zhang C., Sobczak K., Thornton C.A., Swanson M.S. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell. 2014;56:311–322. doi: 10.1016/j.molcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamou R.I., Angelbello A.J., Andrews R.J., Wang E.T., Moss W.N., Disney M.D. Structure-specific cleavage of an RNA repeat expansion with a dimeric small molecule is advantageous over sequence-specific recognition by an oligonucleotide. ACS Chem. Biol. 2020;15:485–493. doi: 10.1021/acschembio.9b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhamou R.I., Angelbello A.J., Wang E.T., Disney M.D. A toxic RNA catalyzes the cellular synthesis of its own inhibitor, shunting it to endogenous decay pathways. Cell Chem. Biol. 2020;27:223–231.e4. doi: 10.1016/j.chembiol.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinegar A.E., Cooper T.A. Roles for RNA-binding proteins in development and disease. Brain Res. 2016;1647:1–8. doi: 10.1016/j.brainres.2016.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook J.D., Mccurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T., et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Cerro-Herreros E., Chakraborty M., Pérez-Alonso M., Artero R., Llamusí B. Expanded CCUG repeat RNA expression in Drosophila heart and muscle trigger Myotonic Dystrophy type 1-like phenotypes and activate autophagocytosis genes. Sci. Rep. 2017;7:2843. doi: 10.1038/s41598-017-02829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coonrod L.A., Nakamori M., Wang W., Carrell S., Hilton C.L., Bodner M.J., Siboni R.B., Docter A.G., Haley M.M., Thornton C.A., Berglund J.A. Reducing levels of toxic RNA with small molecules. ACS Chem. Biol. 2013;8:2528–2537. doi: 10.1021/cb400431f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J.W., Ranum L.P. RNA pathogenesis of the myotonic dystrophies. Neuromuscul. Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Day J.W., Ricker K., Jacobsen J.F., Rasmussen L.J., Dick K.A., Kress W., Schneider C., Koch M.C., Beilman G.J., Harrison A.R., et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–664. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- Dixon D.M., Choi J., El-Ghazali A., Park S.Y., Roos K.P., Jordan M.C., Fishbein M.C., Comai L., Reddy S. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep. 2015;5:9042. doi: 10.1038/srep09042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H., Cline M.S., Osborne R.J., Tuttle D.L., Clark T.A., Donohue J.P., Hall M.P., Shiue L., Swanson M.S., Thornton C.A., Ares M. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei M., Rogers M.T., Thorpe H.M., Larkin K., Hamshere M.G., Harper P.S., Brook J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- Fu Y.H., Pizzuti A., Fenwick R.G., Jr., King J., Rajnarayan S., Dunne P.W., Dubel J., Nasser G.A., Ashizawa T., De Jong P., et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley H.G., Brook J.D., Rundle S.A., Crow S., Reardon W., Buckler A.J., Harper P.S., Housman D.E., Shaw D.J. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature. 1992;355:545–546. doi: 10.1038/355545a0. [DOI] [PubMed] [Google Scholar]
- Harper P.S. W.B. Saunders Company; 2001. Myotonic Dystrophy. [Google Scholar]
- Hirai H., Yang B., Garcia-Barrio M.T., Rom O., Ma P.X., Zhang J., Chen Y.E. Direct reprogramming of fibroblasts into smooth muscle-like cells with defined transcription factors-brief report. Arterioscler. Thromb. Vasc. Biol. 2018;38:2191–2197. doi: 10.1161/atvbaha.118.310870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins K.T., Wilson W.D., Bender B.C., Mccurdy D.R., Hall J.E., Tidwell R.R., Kumar A., Bajic M., Boykin D.W. Extended aromatic furan amidino derivatives as anti-Pneumocystis carinii agents. J. Med. Chem. 1998;41:3872–3878. doi: 10.1021/jm980230c. [DOI] [PubMed] [Google Scholar]
- Hu L., Patel A., Bondada L., Yang S., Wang M.Z., Munde M., Wilson W.D., Wenzler T., Brun R., Boykin D.W. Synthesis and antiprotozoal activity of dicationic 2,6-diphenylpyrazines and aza-analogues. Bioorg. Med. Chem. 2013;21:6732–6741. doi: 10.1016/j.bmc.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenquin J.R., Coonrod L.A., Silverglate Q.A., Pellitier N.A., Hale M.A., Xia G., Nakamori M., Berglund J.A. Furamidine rescues myotonic dystrophy type I associated mis-splicing through multiple mechanisms. ACS Chem. Biol. 2018;13:2708–2718. doi: 10.1021/acschembio.8b00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenquin J.R., Yang H., Huigens R.W., Iii, Nakamori M., Berglund J.A. Combination treatment of Erythromycin and furamidine provides additive and synergistic rescue of mis-splicing in myotonic dystrophy type 1 models. ACS Pharmacol. Transl. Sci. 2019;2:247–263. doi: 10.1021/acsptsci.9b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez N.M., Wang G.S., Cooper T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K., Mohan A., Warren S.A., Chamberlain C.M., Finn D., Hong H., et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013;5:1887–1900. doi: 10.1002/emmm.201303275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barceló J., O'hoy K., et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Mankodi A., Urbinati C.R., Yuan Q.P., Moxley R.T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M.S., Thornton C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C., Thornton C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- Masuda A., Andersen H.S., Doktor T.K., Okamoto T., Ito M., Andresen B.S., Ohno K. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath P.S., Diette N., Kogut I., Bilousova G. RNA-based reprogramming of human primary fibroblasts into induced pluripotent stem cells. J. Vis. Exp. 2018 doi: 10.3791/58687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol. 2013;32:154–165. [PMC free article] [PubMed] [Google Scholar]
- Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munde M., Ismail M.A., Arafa R., Peixoto P., Collar C.J., Liu Y., Hu L., David-Cordonnier M.H., Lansiaux A., Bailly C., et al. Design of DNA minor groove binding diamidines that recognize GC base pair sequences: a dimeric-hinge interaction motif. J. Am. Chem. Soc. 2007;129:13732–13743. doi: 10.1021/ja074560a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero B.A., Poukalov K., Hildebrandt R.P., Thornton C.A., Jinnai K., Fujimura H., Kimura T., Hagerman K.A., Sampson J.B., Day J.W., Wang E.T. Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep. 2021;34:108634. doi: 10.1016/j.celrep.2020.108634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paine M.F., Wang M.Z., Generaux C.N., Boykin D.W., Wilson W.D., De Koning H.P., Olson C.A., Pohlig G., Burri C., Brun R., et al. Diamidines for human African trypanosomiasis. Curr. Opin. Investig. Drugs. 2010;11:876–883. [PubMed] [Google Scholar]
- Pelletier R., Hamel F., Beaulieu D., Patry L., Haineault C., Tarnopolsky M., Schoser B., Puymirat J. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol. Dis. 2009;36:181–190. doi: 10.1016/j.nbd.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Salisbury E., Schoser B., Schneider-Gold C., Wang G.L., Huichalaf C., Jin B., Sirito M., Sarkar P., Krahe R., Timchenko N.A., Timchenko L.T. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am. J. Pathol. 2009;175:748–762. doi: 10.2353/ajpath.2009.090047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur R.S., Philips A.V., Cooper T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- Sellier C., Cerro-Herreros E., Blatter M., Freyermuth F., Gaucherot A., Ruffenach F., Sarkar P., Puymirat J., Udd B., Day J.W., et al. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 2018;9:2009. doi: 10.1038/s41467-018-04370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U S A. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siboni R.B., Bodner M.J., Khalifa M.M., Docter A.G., Choi J.Y., Nakamori M., Haley M.M., Berglund J.A. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J. Med. Chem. 2015;58:5770–5780. doi: 10.1021/acs.jmedchem.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone R., Balendra R., Moens T.G., Preza E., Wilson K.M., Heslegrave A., Woodling N.S., Niccoli T., Gilbert-Jaramillo J., Abdelkarim S., et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol. Med. 2018;10:22–31. doi: 10.15252/emmm.201707850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souidi A., Zmojdzian M., Jagla K. Dissecting pathogenetic mechanisms and therapeutic strategies in Drosophila models of myotonic dystrophy type 1. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens D.C., Kim H.M., Kumar A., Farahat A.A., Boykin D.W., Poon G.M. Pharmacologic efficacy of PU.1 inhibition by heterocyclic dications: a mechanistic analysis. Nucleic Acids Res. 2016;44:4005–4013. doi: 10.1093/nar/gkw229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajder Ł.J., Swanson M.S. Short tandem repeat expansions and RNA-mediated pathogenesis in myotonic dystrophy. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20133365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajder Ł.J., Thomas J.D., Carrell E.M., Reid T., Mcfarland K.N., Cleary J.D., Oliveira R., Nutter C.A., Bhatt K., Sobczak K., et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. U S A. 2018;115:4234–4239. doi: 10.1073/pnas.1716617115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L., Teoh H.K., Cheong S.K. Reprogramming human dermal fibroblast into induced pluripotent stem cells using nonintegrative Sendai virus for transduction. Malays. J. Pathol. 2018;40:325–329. [PubMed] [Google Scholar]
- Taneja K.L., Mccurrach M., Schalling M., Housman D., Singer R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuita J.K., Wang M.Z., Kagira J.M., Denton C.L., Paine M.F., Mdachi R.E., Murilla G.A., Ching S., Boykin D.W., Tidwell R.R., et al. Pharmacology of DB844, an orally active aza analogue of pafuramidine, in a monkey model of second stage human African trypanosomiasis. PLoS Negl. Trop. Dis. 2012;6:e1734. doi: 10.1371/journal.pntd.0001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner-Griffin S., Abe M., Benhamou R.I., Angelbello A.J., Vishnu K., Chen J.L., Childs-Disney J.L., Disney M.D. A druglike small molecule that targets r(CCUG) repeats in myotonic dystrophy type 2 facilitates degradation by RNA quality control pathways. J. Med. Chem. 2021;64:8474–8485. doi: 10.1021/acs.jmedchem.1c00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S., et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., Taliaferro J.M., Lee J.A., Sudhakaran I.P., Rossoll W., Gross C., Moss K.R., Bassell G.J. Dysregulation of mRNA localization and translation in genetic disease. J. Neurosci. 2016;36:11418–11426. doi: 10.1523/jneurosci.2352-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf M.B., Nakamori M., Matthys C.M., Thornton C.A., Berglund J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U S A. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data generated from non-disease, DM1, and DM2 fibroblasts for this study have been deposited in the Sequence Read Archive (SRA). Accession numbers are listed in the Key resources table. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
All original code will be shared by the lead contact upon request.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.