

Abstract
The Zika virus (ZIKV) disease caused a public health emergency of international concern that started in February 2016. The overall number of ZIKV-related cases increased until November 2016, after which it declined sharply. While the evaluation of the potential risk and impact of future arbovirus epidemics remains challenging, intensified surveillance efforts along with a scale-up of ZIKV whole-genome sequencing provide an opportunity to understand the patterns of genetic diversity, evolution, and spread of ZIKV. However, a classification system that reflects the true extent of ZIKV genetic variation is lacking. Our objective was to characterize ZIKV genetic diversity and phylodynamics, identify genomic footprints of differentiation patterns, and propose a dynamic classification system that reflects its divergence levels. We analysed a curated dataset of 762 publicly available sequences spanning the full-length coding region of ZIKV from across its geographical span and collected between 1947 and 2021. The definition of genetic groups was based on comprehensive evolutionary dynamics analyses, which included recombination and phylogenetic analyses, within- and between-group pairwise genetic distances comparison, detection of selective pressure, and clustering analyses. Evidence for potential recombination events was detected in a few sequences. However, we argue that these events are likely due to sequencing errors as proposed in previous studies. There was evidence of strong purifying selection, widespread across the genome, as also detected for other arboviruses. A total of 50 sites showed evidence of positive selection, and for a few of these sites, there was amino acid (AA) differentiation between genetic clusters. Two main genetic clusters were defined, ZA and ZB, which correspond to the already characterized ‘African’ and ‘Asian’ genotypes, respectively. Within ZB, two subgroups, ZB.1 and ZB.2, represent the Asiatic and the American (and Oceania) lineages, respectively. ZB.1 is further subdivided into ZB.1.0 (a basal Malaysia sequence sampled in the 1960s and a recent Indian sequence), ZB.1.1 (South-Eastern Asia, Southern Asia, and Micronesia sequences), and ZB.1.2 (very similar sequences from the outbreak in Singapore). ZB.2 is subdivided into ZB.2.0 (basal American sequences and the sequences from French Polynesia, the putative origin of South America introduction), ZB.2.1 (Central America), and ZB.2.2 (Caribbean and North America). This classification system does not use geographical references and is flexible to accommodate potential future lineages. It will be a helpful tool for studies that involve analyses of ZIKV genomic variation and its association with pathogenicity and serve as a starting point for the public health surveillance and response to on-going and future epidemics and to outbreaks that lead to the emergence of new variants.
Keywords: Zika virus, arbovirus, phylogeography, molecular epidemiology, evolutionary biology
1. Introduction
Determination of viral genetic variants/subtypes/groups is paramount for epidemiological purposes and for the surveillance of endemic and emerging infectious diseases associated with human viruses, e.g. human immunodeficiency virus HIV-1 (Theys et al. 2018), hepatitis C virus (HCV) (Paraschiv et al. 2017), Zika virus (ZIKV) (Aubry et al. 2021), and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Rambaut et al. 2020). Identification of emerging mutations linked to phenotypic traits related to infectiousness, preferred routes of transmission or pathogenicity of these virus variants is also crucial for surveillance purposes that aid the implementation of public health measures in order to mitigate the impact of outbreaks.
ZIKV form the species Zika virus of the genus Flavivirus in the family Flaviviridae, which includes also other species of the arthropod-borne flaviviruses like dengue, West Nile, and yellow fever (Simmonds et al. 2017). It gathered international attention when it was declared a public health emergency of international concern by the World Health Organization (WHO) during the 2015–2016 pandemic in the Americas, Caribbean, and Africa (Roos 2016; Musso, Ko, and Baud 2019; Wilder-Smith and Osman 2020).
While the number of disease cases has declined sharply after 2016, ZIKV may still be circulating undetected. The majority of the infections are asymptomatic and diagnosis is challenging due to cross-reactivity in diagnostic testing of persons previously exposed to other flaviviruses, which are endemic in South America (e.g. Dengue) (Felix et al. 2017). Additionally, there is limited availability of viral samples in the short window period after the onset of illness (Lanciotti et al. 2008). Furthermore, for the detection of congenital Zika virus syndrome, current diagnostic testing remains suboptimal. Insufficient information on ZIKV seroprevalence, combined with uncertainty about waning immunity, hinders the evaluation of the potential of future epidemics among the more than 2 billion people who live in regions at risk for ZIKV transmission. Furthermore, the characterization of mutations in the ZIKV genome potentially associated with altered transmission patterns, increased pathogenicity, and/or congenital complications is important for the surveillance of future outbreaks.
Whole-genome sequencing analyses of the virus allows the characterization of the genetic variability of ZIKV to identify mutations associated with particular phenotypes or with signs of selection. This is important to understand the evolution of the virus and its epidemics and may provide an indication of potential targets for therapeutic/vaccine development. The full-length RNA genome sequence of ZIKV, composed of 10.8 kb, was first published by Kuno and Chang (2007) (GenBank accession number [GAN] AY632535). It is based on a strain isolated from a sentinel rhesus monkey (Macaca mulatta) and lacks a N-glycosylation motif (VNDT) in the envelope region that is present in all human ZIKV genomes (Theys et al. 2017). In 2016, a candidate WHO reference strain (GAN KX369547), isolated from serum from a French Polynesian patient in 2013, was proposed as being representative of clinically significant viruses with widespread distribution (Trösemeier et al. 2016). However, this sequence does not cover the full length of the genome and in 2017 another sequence (GAN KJ776791) was proposed as the ZIKV reference (Theys et al. 2017), along with the correct positioning of the mature proteins in the polyprotein (https://rega.kuleuven.be/cev/reference-sequences/rega-zikv—last accessed: 10 January 2022). Reference-guided and codon-corrected sequence alignments are essential to avoid frameshift errors within the coding area (Gorbalenya et al. 2010; Libin et al. 2019).
Phylogenetic analyses have been essential to understand the evolution and spread of ZIKV. Two major lineages have been identified, the ‘African’ and the ‘Asian’ lineages, named after the geographical region of their first identification (Faye et al. 2014). The ‘Asian’ lineage circulating in Southeast Asia and the Pacific (termed ‘PreAm-ZIKV’) has differentiated into the American epidemic lineage (termed ‘Am-ZIKV’) (Faria et al. 2016; Faria et al. 2017). Several studies have reported the nucleotide (NT) mutations and AA changes across the entire diversity of ZIKV (Pettersson et al. 2016; Wang et al. 2016; Zhu et al. 2016; Metsky et al. 2017; Rossi 2018; Smith et al. 2018; Collins et al. 2019). The occurrence of particular mutations in all sequences of a certain clade of the phylogeny may be important for disease control, especially when that clade is associated with increased pathogenicity. However, a verification of the causal relationship between a mutation and a phenotype has always to be done using reverse genetics since the mutation may become fixed due to chance effects rather than a fitness advantage.
Two naturally occurring AA variations have been shown to be associated with a distinct phenotype in an experimental setting. One is the serine-to-asparagine S139N substitution in the Pr peptide that occurred in the clade that diverged from Asian strains and lead to the lineage that includes French Polynesia and American sequences (Pettersson et al. 2016). This mutation has been shown to increase neurovirulence in neonatal mouse models (Yuan et al. 2017). The other is the A982V (position 188 of the NS1 protein), which confers enhanced mosquito infectivity (Liu et al. 2017) and is absent in the early Asian sequences and present in African and recent American strains (Delatorre, Mir, and Bello 2017; Liu, Shi, and Qin 2019). Contrary to initial expectations, ZIKV strains from the Asian lineage, and associated with microcephaly, have been shown to be less transmissible and less virulent (ex vivo and in vivo) than the African lineages (Simonin et al. 2017; Rossi 2018). Other hypotheses that could explain increased transmission and virulence in outbreak areas have been put forward, e.g. the dispersal to a massive susceptible host population, presence of more efficient vectors, and/or high mobility of people (Rossi 2018).
Analyses of the genetic variation within and between ZIKV lineages and across the genome may inform about the genotype of circulating virus and help to identify gene regions prone to diversification. A few studies have done detailed analyses on the genome-wide diversity of ZIKV. Faria et al. (2017) found higher genetic diversity for the ‘PreAm-ZIKV’ strains compared to the ‘Am-ZIKV’. This study reported this pattern consistently across the genome, as would be expected given the longer duration of ZIKV circulation in Asia, and suggests a founder effect at the origin of the American lineage. Metsky et al. (2017), analysed 174 ZIKV genomes from the Americas and found 1030 mutations, 202 of which were non-synonymous and evenly distributed across the genome. Collins et al. (2019) analysed two strains from each of the African, Asian, and American lineages and also found inter-lineage single-NT variants dispersed throughout the genome. This study reported the highest genomic diversity in the prM/M and NS1 gene regions for the Asian/American lineage.
Intensified surveillance efforts following the American epidemics expanded ZIKV whole-genome sequencing for public health purposes and diagnostics, providing a unique opportunity to revisit the classification and reconstruction of ZIKV evolution. The current classification of ZIKV into two genotypes, ‘African’ and ‘Asian’, is inadequate and needs updating. It does not reflect the range of genetic diversity that the virus accumulated after the pandemic. Classifications of other viruses have typically been based on phylogenetic relationships and genetic distances between clades using consensus criteria for the definition of the groups and adopting hierarchical levels of classification, e.g. HIV (Robertson et al. 2000), influenza A (WHO ⁄OIE ⁄FAO 2012), HCV (Smith et al. 2014), and dengue (Cuypers et al. 2018). However, these systems have not been able to deal with the complexity of the evolutionary changes occurring in dynamic epidemics. The on-going pandemic of SARS-CoV-2 and the huge number of sequences produced have boosted efforts to develop a dynamic nomenclature that identifies lineages contributing most to the global transmission and viral genetic diversity progression, capturing local and global patterns of diversity. Its most important advantage when compared to other viruses’ classifications is its flexibility to incorporate new diversity, which, in fast-evolving viruses, is being generated in real time (Rambaut et al. 2020). This nomenclature proposal was implemented in Pangolin software and allows rapid classification of new strains after sequencing (O’Toole et al. 2021), available at https://cov-lineages.org/resources/pangolin.html (last accessed: 20 January 2022).
In this study, we present an in-depth genome-wide diversity analysis of ZIKV from over 800 complete coding sequences available in GenBank (NCBI), including strains covering the geographical distribution of ZIKV. We propose a classification and a new naming system for ZIKV lineages, inspired by the above-mentioned SARS-CoV-2 nomenclature system and based on phylogenetic analyses, clustering techniques, within- and between-group pairwise genetic distances, and evolutionary analyses to define genetic groups and subgroups. This nomenclature proposal avoids geographical terminology, using instead alpha-numerical labels, and provides a dynamic system that can be rapidly updated as new lineages are identified.
2. Materials and methods
2.1. Dataset compilation and sequence alignment
A total of 2179 sequences of ZIKV were downloaded from the GenBank database (Benson et al. 2013) in December 2021. The filtering steps to select the sequences for analyses are detailed in Supplementary Fig. S1, and the complete list of sequences and exclusion criteria are found in Supplementary Table S1.
Step 1: Before alignment—Criteria: size and nature of the sequence
Sequences shorter than 7000 bp were excluded, after which 1377 sequences remained. From these, the ones identified as patent, recombinant, clone, synthetic construct, or mutant were eliminated, leaving 966 sequences that had been isolated from natural hosts.
Step 2: After NT alignment—Criteria: removal of identical sequences and quality control (ACTG content, presence of stop codons in coding region)
The 966 sequences were aligned against the curated ZIKV reference sequence with GenBank accession number (GAN) KJ776791 as described in Theys et al. (2017) using the codon-correct alignment tool VIRULIGN v1.0.1 (Libin et al. 2019). The codon-correct NT alignment was then cleaned to:
Remove sequences with ACTG content lower or equal to 70 per cent of the complete coding region,
Remove sequences harbouring a stop codon within the coding region, which resulted in 874 sequences,
Remove identical sequences (with 100 per cent NT identity) using a custom script (available at https://github.com/seabrasg/zika_diversity—last accessed: 23 March 2022). We kept only the most recent sequence (and the first in alphabetical order, by accession id, when several sequences were encountered from the same date) resulting in 770 sequences. With one exception, all sequences that were excluded for being identical belonged to the same country (Supplementary Table S2). The exception concerns one sequence from Mexico that was identical to a sequence from the USA. For this case, we kept the most recent sequence, i.e. the sequence originating from the USA.
Step 3: After AA alignment—Criteria: quality control of AA alignment
The AA alignment of the 770 sequences from the previous step was manually verified, guided by a quality assurance heuristic that focuses on the most unexpectedly diverse sequence fragments. By visual inspection of those mutations in the alignment, we identified the ones that were adjacent to regions with missing data, i.e. flanking regions of fully ambiguous NTs (N symbols) (Supplementary Fig. S2). If these mutations were unique to the sequence with the missing data, i.e. indicating sequencing errors in these flanking regions, we masked them by extending the fully ambiguous NTs (Ns) to those positions in the corresponding NT alignment. Sequences that had several of these issues along the alignment and originated from the same study were excluded, resulting in a final dataset of 762 sequences.
In this final alignment (762 sequences), all sequences had collection date information and only four sequences did not have information about the collection geographical location (i.e. three imported cases in China and one in Japan). The classification of ZIKV sequences as ‘African’ or ‘Asian’ genotypes was done using the Genome Detective ZIKV typing tool (Alcantara et al. 2009; Fonseca et al. 2019).
Recombination was assessed for this full curated dataset using the Phi-test (Bruen, Philippe, and Bryant 2006) available in SPLITSTREE version 4.17.0 (Huson and Bryant 2006) and using the methods RDP (Martin and Rybicki 2000), GENECONV (Padidam, Sawyer, and Fauquet 1999), BOOTSCAN (Salminen et al. 1995), Maxchi (Smith 1992), CHIMAERA (Posada and Crandall 2001), SISscan (Gibbs, Armstrong, and Gibbs 2000), PhylPro (Weiller 1998), LARD (Holmes, Worobey, and Rambaut 1999), and 3SEQ (Boni, Posada, and Feldman 2007), implemented in RDP4 (Martin et al. 2015). Sequences with potential recombinant regions were identified in RDP4 when at least four methods had a Bonferroni-corrected P-value lower than 0.05.
From the ‘Full curated dataset’ (762 sequences) we excluded the sequences with potential recombinant regions (‘Afro-Asian’ dataset—759 sequences). This dataset included the ‘African’ and ‘Asian’ genotypes and was used in the phylogenetic, genetic diversity, clustering, and selection analyses. We also did the clustering and selection analyses after excluding the African genotype (‘Asian genotype’ dataset—752 sequences). For the phylogeographical reconstruction, we excluded the sequences that did not have information about the geographical location of collection (Geo-referenced ‘Asian’ dataset—748 sequences). A subset of sequences from the early American expansion and the 5 years before that (2010–2015) (‘Early Am’ dataset—68 sequences) was also used in the selection analyses (Supplementary Fig. S1).
2.2. Phylogenetic reconstruction
The tree reconstruction for the full dataset of ZIKV was done to confirm the separation of the ‘African genotype’ and ‘Asian genotype’ clades. The maximum-likelihood (ML) method was applied using the software package IQ-TREE version 2.0.3 (Nguyen et al. 2015) with the GTR + F + G4 substitution model according to the best fit models identified by IQ-TREE ModelFinder. We evaluated the robustness of the tree with 1000 ultrafast bootstraps (UFBoot) (Hoang et al. 2018) and 1000 SH-like approximate likelihood ratio tests (aLRTs) (Guindon et al. 2010).
The obtained ML tree was annotated using the online tool iTOL version 6.3 (Letunic and Bork 2021) according to the variables: host (human, monkey, and mosquito) and geographical regions based on the United Nations M49 coding classification of geographic regions (https://unstats.un.org/unsd/methodology/m49/—last accessed: 10 January 2022)—Africa (Eastern, Middle, and Western), Asia (Eastern, South-Eastern, and Southern), North America (Caribbean and Central and North America), South America, and Oceania (Melanesia, Micronesia, and Polynesia).
2.3. Genetic diversity and distances
AA and NT mutation tables were obtained from VIRULIGN and were used for the genetic diversity analyses using custom R scripts (available at https://github.com/seabrasg/zika_diversity—last accessed: 23 March 2022). The mutation tables were filtered to set the ambiguities as missing values. The proportion of missing data was analysed per AA position and per NT position. The most frequent AA and NT at each position for the entire dataset of sequences (consensus sequence) were compared with two reference genomes, the curated reference genome GAN KJ776791 (Theys et al. 2017) and the NCBI reference Natal genome GAN KU527068, sequenced from brain tissue of an autopsied microcephalic foetus (Mlakar et al. 2016). For each position, AA and NT variation were characterized by estimating the frequency of the minor variants and the Shannon entropy diversity index, determined using the Chao–Shen estimator (Chao and Shen 2003), as implemented in the R software package entropy version 1.3.0.
Considering that the present ZIKV nomenclature recognizing the ‘African’ and ‘Asian’ genotypes does no longer reflect the actual geography of the virus circulation for the vast Asian clade, we revisited the classification of these clades that was used to propose a new nomenclature. We used the Bayesian model-based hierarchical clustering algorithm hierBAPS (Cheng et al. 2013) as implemented in the software R package rhierbaps version 1.1.3 (Tonkin-Hill et al. 2018). This method combines model-based techniques with an initial fast distance-based complete-linkage agglomerative clustering (Tonkin-Hill et al. 2019). Given an a priori upper bound for the number of genetic clusters, K, hierBAPS estimates K as part of the model fitting procedure and it finds the partition of the data (allocation of each sequence to one of K possible clusters) that maximizes the posterior probability of that allocation (Tonkin-Hill et al. 2018). Two hierarchy levels of clustering were set with a prior upper boundary of 20 clusters. Ten independent runs were performed to assess the congruence of cluster assignment. This analysis was separately applied to genome-wide NT alignments of the ‘Afro-Asian’ and the ‘Asian genotype’ datasets.
To visualize the relationships between haplotypes and co-occurrence in the different geographical areas, we obtained a TCS haplotype network (Clement, Posada, and Crandall 2000), as implemented in the software PopART (Leigh and Bryant 2015). The TCS method starts by calculating pairwise distances between sequences and relies on statistical parsimony to find the connections between sequences (Clement, Posada, and Crandall 2000). The method removes the sequences with more missing data than others and then masks the sites with gaps or characters other than NTs from the original alignment.
We verified if the clustering obtained with these methods were phylogenetically supported in the ML tree and if they were consistent with the within- and between-group genetic distances. Pairwise Kimura’s two-parameter distances were obtained with the dist.dna function in R package ape version 5.5. The analysis of molecular variance (AMOVA) was performed to test between-group differentiation, using 1000 permutations, on R package pegas version 0.14.
2.4. Phylogeographic reconstruction
The transmission routes of ZIKV ‘Asian genotype’ were explored using the fast likelihood method of PastML version 1.9.34 (Ishikawa et al. 2019) to reconstruct ancestral scenarios. This method uses a rooted tree and the states (geographical location, in this case) of each terminal node to reconstruct the ancestral node states. Once all ancestral node states have been reconstructed, a two-step compression is performed for the visualization by clustering the regions of the tree where no state change happens, as well as by merging identical subtree configurations (Ishikawa et al. 2019). In the ancestral character reconstruction (ACR) graph, each node represents the ancestral state, and the size of the node is proportional to the number of tips collapsed into that node. This represents the likely transmissions happening in the same geographical location and with the same source within that location. This analysis was performed on a rooted ML tree obtained in IQ-TREE using an African outgroup (sequence KX601166_Senegal_17 November 1984) that was subsequently removed. PastML was run with the marginal posterior probabilities approximation method. This analysis was also done, for comparison, on a time-calibrated tree inferred using the least-squares dating (LSD2) method (To et al. 2016), based on sampling dates of the sequences, as implemented in IQ-TREE.
2.5. Mutation tracking and selection analyses
We also investigated for evidence of selective pressure in the full and in the ‘Asian genotype’ datasets. We used two methods that detect selection by examining patterns of synonymous and non-synonymous substitutions on a per-site basis: (1) the mixed-effects model of evolution (MEME) that detects sites under pervasive or episodic positive/diversifying selection (Murrell et al. 2012) and (2) the Fast, Unconstrained Bayesian AppRoximation (FUBAR) method that detects sites under pervasive positive/diversifying selection and also those under negative/purifying selection (Murrell et al. 2013). Both methods were used as implemented in HyPhy version 2.2 (Kosakovsky Pond, Frost, and Muse 2005) on the Datamonkey server (Weaver et al. 2018). The MEME method uses phylogenetic models and an ML approach to describe the evolution of codons along tree branches by a continuous-time stationary Markov process. It identifies sites only when some of the lineages evolved under positive selection (Murrell et al. 2012). A P-value threshold of 0.05 was used to consider a site to be under positive selection with this method (Murrell et al. 2012). The FUBAR method uses a Bayesian approach, providing more power to detect selection compared to ML-based approaches. Yet, it assumes that the selection pressure is constant across the phylogeny for each site (Murrell et al. 2013), which is a disadvantage. FUBAR infers non-synonymous (dN) and synonymous (dS) substitution rates. In this framework, an AA position was considered to be under positive/diversifying selective pressure if the posterior probability of dN/dS being larger than 1 at that position was higher than 0.9 (Murrell et al. 2013). An AA position was considered to be under negative/purifying selection if the posterior probability of dN/dS being lower than 1 was higher than 0.9. With the aim of detecting the sites with evidence of selection during or immediately after the selective process (Martin et al. 2021), we also restricted the analysis to the period of the earlier Am-ZIKV expansion and the years just before that expansion by analysing sequences sampled from 2010 to 2015 (dataset ‘early Am’). To understand the spread of the mutations on sites with evidence of positive selection, we analysed the AA frequencies at those sites for each genetic group obtained previously.
2.6. Lineage naming system for ZIKV
The nomenclature proposal for ZIKV presented here is based on the phylogenetic lineages and genetic groups found, their genetic diversity, genetic divergence, and evolutionary dynamics. It aimed to avoid geographical terminology, using instead alpha-numerical labels, and to provide a dynamic system that reflects the progression of the epidemics.
3. Results
3.1. ZIKV full-genomes dataset
The full curated dataset consisted of 762 ZIKV aligned genomes. The alignment had 10,269 NTs, corresponding to 3423 AA positions. Only the two terminal noncoding regions (5ʹ and 3ʹ-NCR) were not included in the analyses. The ZIKV genome encodes for three structural proteins: C (122 aa), Pr/M (168 aa, including 93 of the peptide Pr), E (504 aa), and seven non-structural proteins: NS1 (352 aa), NS2A (226 aa), NS2B (130 aa), NS3 (617 aa), NS4A (150 aa, including 23 of the peptide 2K), NS4B (251 aa), and NS5 (903 aa).
Viral strains analysed here originated from 46 different countries across the world and have been sampled between 1947 and 2021 (Supplementary Table S3). Genome Detective ZIKV typing tool indicated the ‘African genotype’ for 7 sequences, the ‘Asian genotype’ for 754 sequences, and signalled one sequence as ‘Related to but not part of Asian’ (OK054351_India_28 July 2021). The distribution of the samples according to continent of collection was: Africa (1.6 per cent), Asia (15 per cent), North America (57.3 per cent), Oceania (2.1 per cent), and South America (24.0 per cent), and according to geographical region of collection: Eastern Africa (0.3 per cent), Western Africa (0.7 per cent), Middle Africa (0.7 per cent), South-Eastern Asia (13.3 per cent), Southern Asia (0.8 per cent), Eastern Asia (0.9 per cent), Melanesia (0.1 per cent), Micronesia (0.1 per cent), Polynesia (1.9 per cent), South America (24.0 per cent), Central America (28.1 per cent), Caribbean (23.0 per cent), and Northern America (6.2 per cent) (Supplementary Table S3).
While a broad range of time, spanning 74 years of viral spread was covered in the dataset, the detection and public health impact of ZIKV outbreaks in the Americas, together with the availability of advanced next-generation (real time) sequencing methods, caused a dramatic sequencing scale-up effort in samples collected in 2016 (83.1 per cent of the sequences). The human host was the most frequently sampled to characterize ZIKV infections (94.9 per cent), while there were 34 sequences from mosquito host (4.5 per cent) and only 5 sequences (0.7 per cent) from a non-human primate host.
3.2. Recombination analyses
No evidence of recombination was found with Phi-test in SPLITSTREE (P = 0.1023806). Using the methods implemented in RDP4, three sequences were identified as having small genomic regions that presented evidence for potential recombinant history with African sequences: KY241700_Singapore_27 August 2016 (region 3950–4358 bp), KY241712_Singapore_30 August 2016 (region 52–269 bp) and EU545988_Micronesia_2007-06 (region 1–52 bp) (Supplementary Table S4). We excluded these three sequences from the alignment to produce the ‘Afro-Asian’ dataset with 759 full-coding sequences that was further analysed for the phylogenetic reconstruction and genetic diversity analyses.
3.3. Phylogenetic reconstruction
The ML tree of the ‘Afro-Asian’ dataset of 759 sequences obtained in IQ-TREE confirmed the pattern of genetic differentiation of the ‘African genotype’ clade (n = 7) from the remaining samples (Fig. 1; Supplementary Fig. S3), forming the ‘Asian genotype’ clade. The sequence from the recent 2021 outbreak in India (GAN OK054351), previously identified by Genome Detective as ‘Related to but not part of Asian’, grouped with the early Asian Malaysia sequence. Sequences obtained from mosquito hosts formed clusters in African and early Asian clades, while they were dispersed in the rest of the tree.
Figure 1.
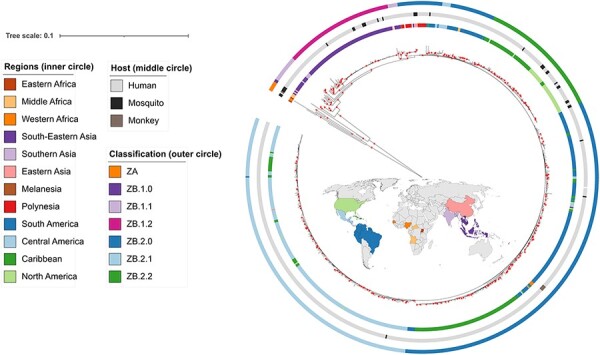
ML tree of the full dataset (759 sequences). Colour annotations are given in the circles around the terminal nodes. From inner to outer circle: geographical regions (same colours as in the map); host; proposed classification. Red circles indicate support values SH-aLRT ≥80 per cent and UFBoot ≥95 per cent. A version of this tree including the node labels is provided in Supplementary Fig. S3.
3.4. Genetic diversity and genetic distances
In the ‘Afro-Asian’ dataset of 759 sequences, the proportion of missing data per NT position was at most 15.2 per cent (minimum coverage of 84.8 per cent) (Supplementary Fig. S4A).
A total of 2663 parsimony-informative NT sites out of 10,269 total sites were found in the ZIKV alignment of the full dataset. The consensus NT genome sequence differed at 11 positions from the reference genome KJ776791, of which 3 showed a consensus NT with a prevalence above 95 per cent (Supplementary Table S5). The consensus NT genome differed at 21 positions from the NCBI reference Natal genome KU527068, of which 17 positions had a consensus NT with a prevalence above 95 per cent.
Overall, 99 per cent of the NT positions had a prevalence of the consensus NT above 95 per cent (Supplementary Table S6). The mean entropy at NT level considering the complete dataset was 0.021 (95 per cent confidence interval: 0.020–0.022; range: 0–0.69). The most variable positions were distributed across the genome and were present in every gene region (Fig. 2A; Supplementary Fig. S4B). From the 139 NT positions with frequency of alternative NT higher than 5 per cent, 18 were non-synonymous, located in proteins C (I80T, D107E), PR (N139S/K), E (V503A/E), NS1 (G894A, V988A, M1143V/T, T1145S), NS2A (I1162M), NS3 (M2074L, H2086Y/R), NS4B (I2367M/V), and NS5 (V2634M, I2842V, R3045C/S, Q3282W, T3328I/N, D3398E) (Supplementary Table S6).
Figure 2.
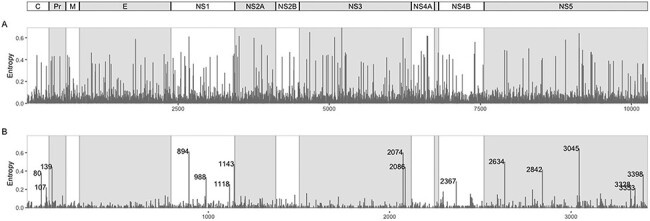
A) Entropy per nucleotide position. B) Entropy per AA position, with labelled AAs having entropy values higher than 0.2. Shaded region between NS4A and NS4B is the peptide 2K.
The consensus AA genome sequence (Supplementary Fig. S5) differed at position 2634 (position 114 in the NS5 protein) from the reference genome KJ776791, with AA V instead of M, showing a prevalence of 81.3 per cent (Supplementary Table S5). When considering the NCBI reference Natal genome KU527068, the consensus differed at four positions: position 940 (NS1_146) with AA K at 98.8 per cent, instead of E; position 1027 (NS1_233) with AA T at 98.9 per cent, instead of A; position 1143 (NS1_349) with AA M at 82.9 per cent, instead of V; position 2509 (NS4B_240) with AA T at 99.6 per cent, instead of I.
Overall, 96.9 per cent of the AA genome positions were highly conserved, showing a prevalence of the consensus AA above >99 per cent. The proteins with higher proportion of positions below 99 per cent consensus AA prevalence were PR (7.6 per cent of positions) and C (4.1 per cent of positions) (Supplementary Table S6).
The mean entropy at AA level considering the complete dataset was 0.0097 (95 per cent confidence interval: 0.0085–0.011; range: 0–0.642). The positions with higher diversity (higher frequency of AA changes and higher entropy levels) were located in proteins C, Pr, NS1, NS3, NS4B, and NS5 (Fig. 2B; Supplementary Fig. S4C).
3.5. ZIKV nomenclature
The hierarchical clustering method implemented in hierBAPS identified four clusters for the full dataset in the first level of analysis, one composed of the African sequences and the others subdividing the ‘Asian genotype’ clade into three groups (Supplementary Table S3). The second level of analysis on this dataset subdivided the African group into three groups: Western Africa (Guinea and Senegal), Eastern Africa (Uganda), and Middle Africa (Central African Republic). However, when analysing the ML tree, not all these groups had good phylogenetic support. Due to the small number of samples in this analysis (n = 7), we restricted the naming proposal to one group (new name: ZA).
Conducting the hierBAPS analysis for the ‘Asian genotype’ dataset only (752 sequences), 6 clusters were obtained in the first level of analysis, while 12 nested subclusters were found in the second level of analysis. The TCS network obtained in POPArt also allowed identifying several clustering patterns. When visualizing the clusters from both methods and relating them to geographical sampling location (Fig. 3; Supplementary Fig. S6), we were able to propose a subdivision into groups that were afterwards verified by analysing the phylogenetic tree (Fig. 4) and the between- and within-group genetic distances and AMOVA results (see below). This approach helped in the following step of the nomenclature proposal of the lineages. A basal group in the tree that was also identified in hierBAPS was composed of the sequence from Malaysia (South-Eastern Asia) sampled in 1966 from Aedes aegypti mosquito and the recent Indian sample from an outbreak in 2021 (new name: ZB.1.0). A second group obtained in hierBAPS contained 26 sequences from South-Eastern Asia (Cambodia, Indonesia, Philippines, Thailand, and Vietnam), Southern Asia (India and Bangladesh), and Eastern Asia (China and South Korea). This group is clearly under-sampled and potentially harbours many different lineages as indicated in the second level of analysis of hierBAPS that divided this into four subgroups. Due to the small number of samples in this analysis, we restrict the naming proposal to one group (new name: ZB.1.1). A third group was composed of 78 similar sequences from South-Eastern Asia, mostly from the Singaporean outbreak in 2016 (59 sequences from Singapore), 18 from Thailand, and 1 from Cambodia (new name: ZB.1.2). A fourth group included 327 sequences from Oceania (French Polynesian, Samoa, Fiji), South America (Brazil, Colombia, Ecuador, Peru, Suriname, French Guiana, and Venezuela), Caribbean (Barbados, Cuba, Dominican Republic, Haiti, Martinique, Puerto Rico, Saint Barthélemy, Trinidad and Tobago, and Virgin Islands), as well as a small number of sequences from Middle Africa (two from Angola), Western Africa (three from Cape Verde), South-Eastern Asia (one from Singapore and one from Cambodia), Eastern Asia (four from China), Central America (two from Mexico, three from Nicaragua, and four from Panama), Melanesia (one from Fiji), and Northern America (two from the USA). We called this group ZB.2.0 and it includes the basal American sequences and includes the sequences from Oceania. The other group is composed of 231 sequences mostly from Central America (Belize, El Salvador, Guatemala, Honduras, Mexico, and Nicaragua) and 22 from the Caribbean (Cuba and Puerto Rico) and 4 from Northern America (USA) and was named ZB.2.1. Finally, a fifth group included 88 sequences from Caribbean (Cuba, Dominican Republic, Haiti, Jamaica, and Puerto Rico) and Northern American (USA) sequences and was named ZB.2.2.
Figure 3.
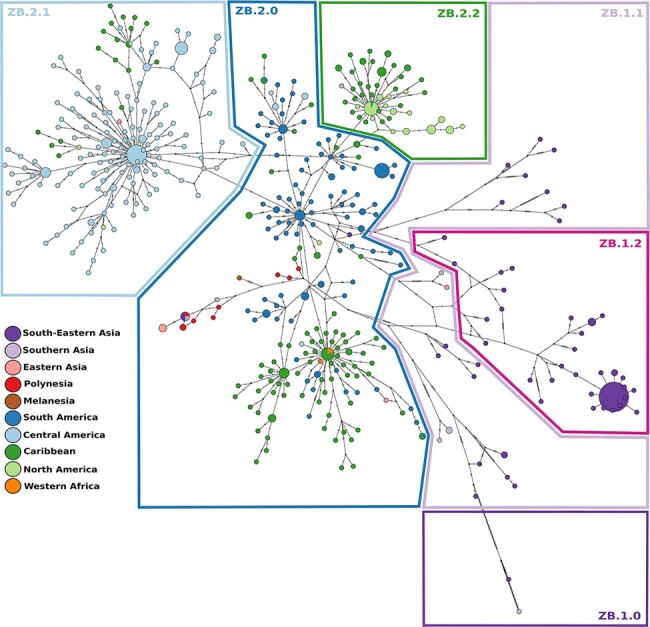
TCS haplotype network, with nodes coloured by geographical region. A total of 610 sequences and 714 segregating sites were used to construct the TCS network. Sizes of the nodes are proportional to the number of sequences in that node. The perpendicular dashes on the branches connecting two nodes represent the number of mutations between those nodes. The drawn polygons represent the proposed classification based on the clusterings from HierBAPS. A version of this network including the node labels is provided in Supplementary Fig. S6.
Figure 4.
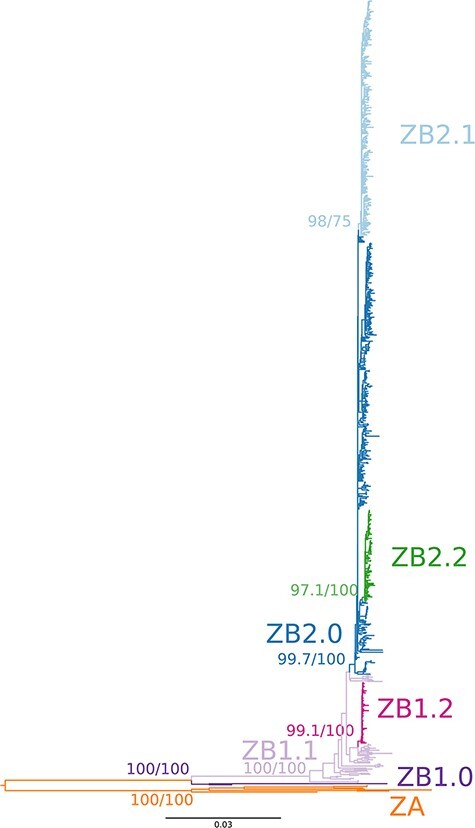
ML tree of the ‘Afro-Asian’ dataset (759 sequences). Colour annotations represent the clusterings and proposed nomenclature. Branch support values for the clusterings were obtained with SH-aLRT test (first value) and ultrafast bootstrap (second value).
Considering the seven groups defined above (ZA, ZB.1.0, ZB.1.1, ZB.1.2, ZB.2.0, ZB.2.1, and ZB.2.2), AMOVA showed a significant between-group genetic differentiation when considering all groups, ZB groups only (ZB.1.0, ZB.1.1, ZB.1.2, ZB.2.0, ZB.2.1, ZB.2.2), ZB.1 groups only (ZB.1.0, ZB.1.1, and ZB.1.2), and ZB.2 groups only (ZB.2.0, ZB.2.1, and ZB.2.2) (AMOVA, P-value <0.001 in all cases; Supplementary Table S7). Mean genetic distances between subgroups from ZA and ZB ranged from 0.117 to 0.127 NT substitutions per site (Table 1). Between ZB subgroups (ZB.1.0, ZB.1.1, ZB.1.2, ZB.2.0, ZB.2.1, and ZB.2.2), it ranged from 0.005 to 0.063, with higher values occurring between the basal Asian sequences (ZB.1.0) and the other groups and with lower values occurring between ZB.2 subgroups. The distribution of pairwise genetic distances within each subgroup was unimodal and with low dispersal for the ZB2 subgroups (ZB2.0, ZB.2.1, and ZB.2.2). For the other subgroups (ZA and ZB1.0, ZB.1.1 and ZB.1.2), it was more dispersed and showed several peaks, which may indicate that further subdivision would be necessary if the sample size was larger (Fig. 5).
Table 1.
Mean (and range) pairwise genetic distances (nucleotide substitutions per site) between and within groups.
| ZA | ZB.1.0 | ZB.1.1 | ZB.1.2 | ZB.2.0 | ZB.2.1 | ZB.2.2 | |
|---|---|---|---|---|---|---|---|
| ZA | 0.056 (0.000–0.080) | 0.117 (0.107–0.127) | 0.127 (0.123–0.135) | 0.126 (0.121–0.134) | 0.127 (0.120–0.137) | 0.127 (0.122–0.135) | 0.126 (0.119–0.133) |
| ZB.1.0 | 0.0469 | 0.062 (0.043–0.079) | 0.061 (0.046–0.078) | 0.062 (0.045–0.083) | 0.062 (0.044–0.080) | 0.063 (0.046–0.079) | |
| ZB.1.1 | 0.017 (0–0.029) | 0.015 (0.009–0.026) | 0.0146 (0.006–0.028) | 0.015 (0.008–0.027) | 0.015 (0.008–0.016) | ||
| ZB.1.2 | 0.003 (0–0.014) | 0.009 (0.006–0.017) | 0.010 (0.007–0.016) | 0.010 (0.008–0.016) | |||
| ZB.2.0 | 0.004 (0–0.011) | 0.005 (0.001–0.010) | 0.005 (0.002–0.010) | ||||
| ZB.2.1 | 0.003 (0–0.006) | 0.005 (0.003–0.009) | |||||
| ZB.2.2 | 0.002 (0–0.004) |
Figure 5.
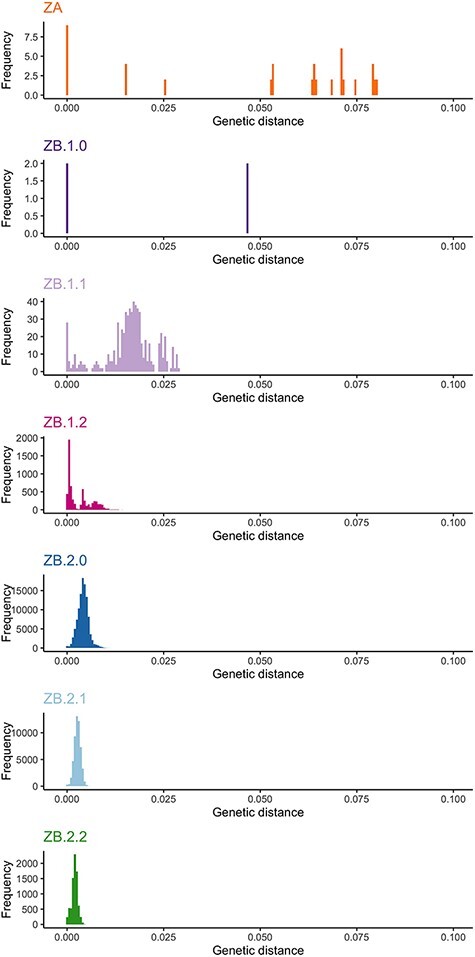
Histograms of the pairwise genetic distances (nucleotide substitutions per sites) for each clustering.
3.6. Phylogeographic reconstruction
Phylogeographic reconstruction was done using the geo-referenced ‘Asian genotype’ dataset (748 sequences). PastML results on the ML rooted tree, as well as the LSD2 time tree, showed generally high probability values for the ancestral state (geographical region) assigned to each node (Fig. 6; Supplementary Fig. S7). A large group of South-East Asian sequences was found to be ancestral to the others, which spread towards Polynesia and from there gave rise to the American lineage. From French Polynesia, the virus likely spread to other islands located in the Pacific and also reached the South American continent, more particularly Brazil. Another route going from Polynesia to Haiti (Caribbean) and then to Brazil (South America) was also suggested but with a smaller expression. From the South American (mainly Brazil) epidemics onwards, multiple exportation and diversification events were inferred to neighbouring countries such as Colombia and Venezuela but also to Nicaragua, Honduras (Central America), and the Dominican Republic and Puerto Rico (Caribbean). ZIKV lineages that caused an outbreak in the Caribbean were passed on to North America. Some other routes were also suggested, between Central American countries, and from Dominican Republic to Haiti. The spread to Cuba was suggested to have been imported from Central America but also from other Caribbean islands. The sequences sampled in Angola (Middle Africa) and Cape Verde (Western Africa) are likely derived from the South American cluster.
Figure 6.
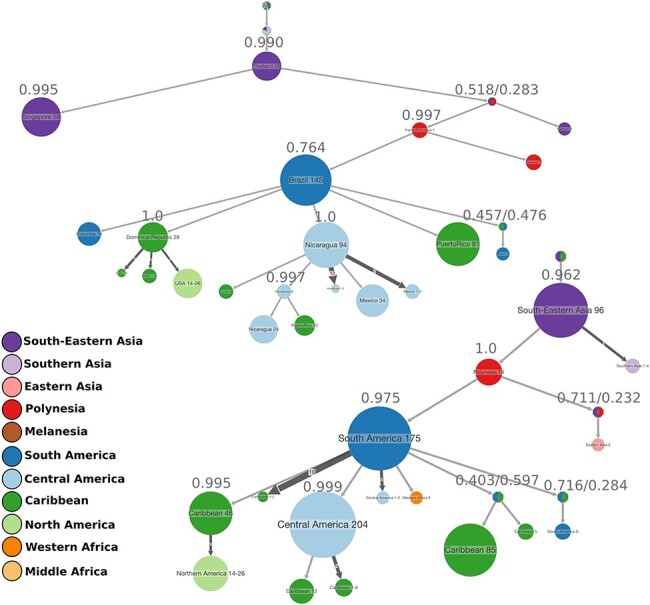
ACR of ZIKV geographical locations, either countries (top) or regions (bottom). The compressed visualizations were obtained in PastML from the rooted ML tree, where each node represents the ancestral state (geographical region), and the size of the node is proportional to the number of tips collapsed into that node. This represents the transmissions happening in the same geographical regions and with the same source within that region. The marginal probability of each node being in the state represented is shown on top of the node. The colours correspond to the geographical regions. The results of ACR for the time tree are found in Supplementary Fig. S7.
3.7. Mutation tracking and selection analyses
In all three datasets analysed for selection (‘Afro-Asian’ dataset—759 sequences, ‘Asian genotype’ dataset—752 sequences, and ‘early Am’ dataset—68 sequences), the FUBAR method detected evidence of negative/purifying selection for many codon sites, uniformly distributed across the genome (Supplementary Fig. S8). Positive selection was detected by MEME for a total of 50 sites (Supplementary Table S8), two of which were also detected by FUBAR. In 36 of these sites, the alternative AA(s) were caused by mutations in at least two positions in the codon. Seven codon sites with signals of positive selection showed high frequencies of the alternative AA in at least one of the genetic groups defined in the previous section K101R (C_101), M2074L (NS3_572), I2445L/M/T (NS4B_176), V2449I/A/F/T (NS4B_180), Y2594H (NS5_74), S3162P (NS5_642), and D3223S/V (NS5_703) (Supplementary Fig. S9). The remaining sites showed very low frequencies of the alternative AA, most of them present in only one to six sequences (Supplementary Table S8).
We explored the mutations that occurred at the major internal nodes of the phylogeny: ‘African genotype’ (ZA) vs. ‘Asian genotype’ (ZB) (node 1); PreAm-ZIKV (Africa + Asia except French Polynesia) vs. Am-ZIKV (French Polynesia + America) (node 2); node leading to Central American group (node 3); and node leading to Caribbean and North American group (node 4) (Fig. 6).
No mutually exclusive mutations (NTs present in all members of one group and absent from the other group) distinguished between sequences from human and sequences from mosquito hosts. Between ‘African’ and ‘Asian’ genotypes, 170 mutually exclusive NT mutations, of which 24 were non-synonymous (Supplementary Table S8). One of these showed evidence of positive selection, K101R (C_101). There were no mutually exclusive NTs between PreAm-ZIKV and Am-ZIKV. In 42 AA positions, the alternative AA occurred in high frequency in at least one of the groups, most of them exclusively in the African sequences (Supplementary Table S8). In 17 of them, the alternative AA was more prevalent in other groups (Supplementary Fig. S9). For example, the alternative AA in position S139N (PR_17) occurs in all Am-ZIKV sequences, while in all other sequences the consensus AA is present (except in one sequence of ZB.1.1 that also has the alternative) (Supplementary Fig. S9). V2634M/T (NS5_114) has AA V in all ZB.2 (except in 22 sequences of ZB.2.0) and the alternative AA M is present in all other groups. The alternative AA in G894A (NS1_100) is present in all ZB.2.1 (except in one sequence) and is absent in all the remaining sequences (except in one sequence from ZB.1.0). The alternative AA in M2074L (NS3_572) occurs in a large proportion of the sequences of ZB.2.1, while in the remaining sequences only the consensus M AA occurs. The alternative AA in R3045C/S (NS5_525) is present in all ZB.2.1 (except in 3 sequences) and in two sequences of ZB.2.0 and is absent in all others (Supplementary Fig. S9).
We also analysed the variation in the proteins that have been targets of vaccine development, E, prM, and NS1 (Pattnaik et al. 2020). The envelope (E) protein in this dataset presented 112 non-synonymous mutations, but all below the 5 per cent frequency threshold. Five of these had mutually exclusive AAs between ZA (‘African genotype’) and ZB (‘Asian genotype’) (Supplementary Table S8). Other two sites in E showed evidence of positive selection with the MEME method, one of them having more than one alternative AA in a few sequences, and another (V784L) showing the alternative AA in five sequences from the Singapore outbreak. The Pr peptide had 35 non-synonymous mutations, but only 1 (S139N—PR_17) had a frequency of the alternative AA higher than 5 per cent as described above. Three of the others had mutually exclusive AA between ZA and ZB. Another AA mutation of Pr had evidence of positive selection (detected by MEME method). Protein M had 22 non-synonymous mutations, all with alternative AA frequency lower than 5 per cent and two of them with mutually exclusive AAs between ZA and ZB. Protein NS1 had 101 non-synonymous AAs, and 4 of them with alternative AA frequency higher than 5 per cent. Three had evidence of positive selection, of which three were detected by the MEME method and one was detected by the FUBAR method.
4. Discussion
Herein, we analyse a curated dataset of ZIKV near-complete genomes, which is representative of the global ZIKV genetic diversity. The detailed genome-wide diversity and differentiation analyses of ZIKV carried out here with a larger number of sequences than in previous studies have allowed identifying the genetic groups and the most relevant mutations to classify and propose a dynamic nomenclature system, as well as to reconstruct the most likely dispersion routes and explore the putative evolutionary processes involved.
4.1. Lineage naming system for ZIKV
Based on an in-depth evolutionary dynamics analysis, we propose here a new nomenclature system for ZIKV that avoids geographic references and can be adapted to future new emerging lineages. The two phylogenetically distinct genotypes, formerly named ‘African’ and ‘Asian’, are here called ZA and ZB genotypes. As with the recent SARS-Cov2 nomenclature, we propose that, within each genotype, lineages are dynamically subcategorized. The African ZA genotype harbours high genetic diversity and thus will likely deserve a detailed subcategorization in the future. The Asian ZB genotype has a basal lineage, here called ZB.1.0, represented by the older and more differentiated Malaysian sequence. Lineage ZB.1.1 is likely under-sampled and may be further subdivided when more sequences become available from Southern and South-eastern Asia. Lineage ZB.1.2 is very localized in the Singaporean outbreak. ZB.2.0 includes the Polynesian sequences and the basal American lineage. ZB.2.1 is a lineage that occurred mainly in Central America and ZB.2.2 in the Caribbean and North America.
4.1.1. Genome-wide diversity and differentiation patterns
The consensus sequences obtained from this dataset have very few differences from the reference genome KJ776791 proposed by Theys et al. (2017) (only one AA change, and 10 NT changes), which confirms this sequence as a good reference for ZIKV for future studies.
Most of the AA positions were highly conserved. The sites with the highest NT and AA diversity were in gene regions C, prM, NS1, NS3, NS4B, and NS5. Previous studies have described non-synonymous mutations that only occurred in certain populations. However, with the larger number of sequences used in this study, we have found some occurring in other previously unexplored populations. For example, the A148P AA change was previously reported to occur in human isolates from both ‘African’ and ‘Asian’ lineages but not in African mosquitoes, suggesting potential relevance in human infectivity (Wang et al. 2016). However, we found it present in mosquito samples from the ‘Asian genotype’, potentially implying a different interpretation concerning infectivity. Few ZIKV genomes from mosquito host and from non-human primates are available, which prevents thorough analyses of the differentiation between viral sequences coming from different host species. The older sequences available in our dataset, from Africa and Malaysia, are from mosquito host species and no viral sequences from humans are available from that time for comparison. In our curated dataset, the recent sample from Guinea (MN025403_Guinea_2018-08) was the only one from the ‘African genotype’ collected in human host and it groups with the older mosquito African sequences. Also, the remaining sequences obtained from mosquito hosts are scattered across the phylogenetic tree and haplotype network and are very similar to the ones sampled in humans, which suggests small between-host viral genetic differentiation. The recent sample from India, also from a human host (OK054351_India_28 July 2021), clustered with an old Malaysia sequence. but with an unexpected high divergence. This finding warrants further investigation to understand the reason for the accumulation of such high divergence.
The ‘African’ and ‘Asian’ genotype lineages, ZA and ZB, were clearly divergent, showing ∼12 per cent pairwise genetic distance. Within ZA (‘African genotype’ lineage), there is likely further differentiation, as already suggested (Faye et al. 2014). However, due to the low number of African sequences, we did not include that differentiation in the current nomenclature proposal. As our nomenclature proposal is dynamic, differentiation patterns coming from future genomes can easily be included to update the classification. Within ZB (‘Asian genotype’ lineage), sequences diverged by up to 6 per cent, on average, with the highest values found between the older Malaysian sequence (ZB.1.0) and the remaining sequences from ZB. Within Pre-Am-ZIKV (ZB.1.0, ZB.1.1, and ZB.1.2 subgroups) divergence was ∼ 6 per cent when involving ZB.1.0, and ∼1.5 per cent between pairs of sequences ZB.1.1—ZB.1.2. Within Am-ZIKV, the mean pairwise divergence between the defined subgroups was much lower ∼ 0.5 per cent, as expected from a very recent diversification. Within each subgroup in the Americas, the divergence was even lower (∼ 0.2–0.3 per cent). Faria et al. (2017) also reported pairwise genetic distance lower within Am-ZIKV strains than within the Pre-Am-ZIKV strains, with values around 0.3 per cent for Am-ZIKV and around 0.5 per cent for Pre-Am-ZIKV (obtained from three sequences from Southeast Asia and one from Micronesia), which is coherent with the older and ancestral nature of Pre-Am-ZIKV.
The genetic diversity values that we obtained (mean entropy at NT level of 0.021) are high when compared to those reported by Collins et al. (2019). However, in this case, the authors calculated within-lineage entropy based on single-NT variants found within the sequencing assemblies of each sequenced strain, producing very low entropy values (<0.004). Contrastingly, here we used consensus sequences available in GenBank and do not have information about the within-host variability, which would also certainly be interesting to look at and identify potential associations with pathogenicity (Rossi 2018).
When establishing the classification and nomenclature proposal here presented, we also took into consideration the diversification events and the most likely dispersion routes of ZIKV. For example, the definition and naming of ZB.2.0 as being the basal American cluster, is supported by the ACR, since it includes the Polynesia sequences that gave origin to this clade. This analysis also supported the naming of the other subgroups within ZB.2 (ZB.2.1 and ZB.2.2) as both derived from the basal ZB.2.0. The dispersion of the pandemic within the ‘Asian genotype’ (ZB) was South-Eastern Asia > Polynesia > South America > the Caribbean and Central America > Northern America. Although the method used in PastML is considered robust to phylogenetic uncertainty and sampling bias (Ishikawa et al. 2019), it still depends on sampling availability and other countries that are not in this dataset may have also contributed to the pandemic. The dispersion patterns indicated by these reconstructions were consistent when using the rooted tree and when using the time tree, but revealed some uncertainties at particular nodes of the tree, in particular involving sequences from Haiti and Cuba. In any of these cases, our reconstruction is consistent with previous studies. Haiti has been previously suggested as a possible ‘steppingstone’ for the arrival in Brazil (Faria et al. 2017), and this ancestral reconstruction does not exclude this hypothesis, at least for some lineages in Brazil. The introduction in Cuba has been previously suggested to have two origins, from Central America and from other islands in the Caribbean (Grubaugh et al. 2019), which was also suggested in this reconstruction.
4.1.2. Selection and recombination
The large number of low-frequency mutations that were detected across epidemic regions (star-like distribution in the haplotype network) and that are distributed homogeneously across the genome point to a demographic expansion scenario. This is consistent with the introduction of a new virus in an immunologically naive population causing an outbreak in the Americas (Metsky et al. 2017). The signal of strong purifying selection, widespread across the genome, which we detected with the FUBAR method, indicates the occurrence of a large number of deleterious mutations being purged. This high degree of negative selection widespread across the genome had already been found for ZIKV (Shrivastava et al. 2018). A high rate of deleterious mutations being eliminated in the long term by strong purifying selection has also been reported in dengue virus by Holmes (2003), who hypothesized that this high rate may be a result of replication of arboviruses occurring in alternate host species. Evidence of strong purifying selection has also been found in another flaviviruses, such as the West Nile virus (Jerzak et al. 2005). The type of transmission cycle, together with the lack of an adaptive immune system in mosquitoes, has been suggested to also explain the small number of sites detected to be under positive selection in ZIKV (Shrivastava et al. 2018), as found here and in other studies on ZIKV (Liang et al. 2017).
The FUBAR method detected two sites under positive selection. This type of method, intended to detect pervasive selection across lineages, may misleadingly attribute negative selection to a site that experienced episodic selection in particular lineages, followed by strong conservation (Murrell et al. 2012; Spielman et al. 2019). The MEME method allowed detecting several more sites under episodic selection. For several of these sites, there were more than one alternative AAs, caused by mutations in at least two positions in the codon. However, most of those sites had very low frequencies of the alternative AAs (in many cases, only one or two sequences harboured those mutations), which suggested that they did not contribute to the viral adaptation during the epidemics. Restricting the analysis to subsets of data, namely the period of the earlier Am-ZIKV expansion and the years just before that expansion, has also allowed detecting one additional site under positive selection, V620L/G.
Three sequences were identified as harbouring potential recombinant regions, two from Singapore and one from Micronesia. These short genomic regions had higher similarity with African sequences. Gong, Xu, and Han (2017) have already detected 19 potential recombination events in African and Singapore ZIKV sequences but did not exclude experimental error. In fact, a clear support for a recombinant event is still lacking. RDP4 signalled these regions as having the breakpoint positions undetermined and indicated that it is possible that this apparent recombination signal could have been caused by an evolutionary process other than recombination. An experimental study has shown that recombination can occur in flaviviruses but at a very low frequency (Taucher, Berger, and Mandl 2010). In dengue virus, it has been occasionally reported (Holmes, Worobey, and Rambaut 1999; Tolou et al. 2001; Craig et al. 2003). In ZIKV, other studies have reported potential recombinant events. For example, Faye et al. (2014) found evidence for 13 recombination events between African strains when sequencing partial E and NS5 regions. Han et al. (2016) reported a recombinant sequence from Brazil having two genomic regions more similar to a Suriname strain, while the remaining genome was more similar to a French Polynesia strain. Shrinet et al. (2016) found evidence of recombination in African isolates and not in the Asian lineage. Shrivastava et al. (2018) found evidence of one recombination event but noticed that both parents and daughter strains were very similar to each other and thus concluded that this was not a real recombination event. Simón et al. (2018) did not find any evidence of recombination in the strains from Latin America. These discrepancies between studies are likely related to the different datasets and methods used. The power to detect recombination is dependent on the recombination rate, level of divergence, age of the event, and the method used (Posada and Crandall 2001). In the case of ZIKV, no striking evidence of recombination has yet been consistently found, but the occurrence of rare recombinant events cannot be excluded. Our results give support to the hypothesis that recombination does not play a significant role in ZIKV evolution (Shrivastava et al. 2018).
4.1.3. Tracking mutations
We have identified the mutations that were segregated into different groups. Seven sites with evidence of positive selection showed a high frequency of the alternative AA in particular lineages/genetic groups (R101K, M2074L, I2445L/M/T, V2449I/F/T, Y2594H, S3162P, D3223S/V), which may point to a role in the adaptive process. In the dataset that we analysed, the substitution S139N (prM) occurred in all members of the Am-ZIKV clade and not in the other clades, except for one sequence in the ZB1.1 clade. This mutation has been reported as being exclusive to the Am-ZIKV clade (e.g. Pettersson et al. 2016). Another interesting mutation is the A982V (188 V in Liu et al. 2017, referring to the position within the NS1 gene). The V AA confers enhanced mosquito infectivity (Liu et al. 2017) and is absent in the early Asian sequences and present in African and recent American strains (Liu, Shi, and Qin 2019). In our study, the MEME method detected this position as being under positive selection, likely due to the occurrence of mutations in the first and second codon positions that lead to three alternative alleles at low frequency (<5 per cent) (Supplementary Table S5). M/T2634V was found in all the sequences from the American outbreak (and was not present in French Polynesia) (as found in Pettersson et al. 2016; Liu, Shi, and Qin 2019), but no evidence of altered pathogenesis in mice was found for this mutation (Zhao et al. 2018).
The level of genetic variability in ZIKV constitutes important information for the development of antibodies or specific vaccines against the virus. The proteins E and prM have low genetic variability, as shown by the very low frequencies of alternative AAs, reinforcing them as good targets for vaccine development (Heinz and Stiasny 2017; Nambala and Su 2018).
Several questions about genome-wide diversity in ZIKV remain to be explored. The untranslated terminal regions 5ʹ- and 3‘-UTR were not analysed here, and changes in these regions may be biologically relevant (Zhu et al. 2016) and deserve attention. Also, epistatic effects (contribution of more than one mutation to the observed phenotype) should be considered (Rossi 2018). The African and Asian continents are strikingly under-sampled, and hidden ZIKV variation remains to be discovered. Also, in the Americas, new mutations continue to be identified in Aedes mosquitoes collected during the 2015/2016 ZIKV epidemics, for example, in the Caribbean island of Barbados (Thannesberger et al. 2021), and the epidemiological and genomic situation of ZIKV should be continuously monitored.
In conclusion, we used a large dataset of near full-genome ZIKV sequences to analyse its genetic diversity, phylogenetics, and phylodynamics and understand its differentiation patterns and guide the development of a dynamic classification system. Our results are consistent and extend the results of previous studies. Based on those, we develop a classification system that avoids geographical references and is flexible to accommodate potential future lineages. It will be a helpful tool for studies that involve ZIKV genomic variation and its association with pathogenicity. Furthermore, it will serve as a starting point to study on-going ZIKV epidemics and outbreaks that lead to the emergence of new variants. The proposed classification will provide guidance for ZIKV surveillance and to implement public health measures to mitigate outbreaks.
Supplementary Material
Acknowledgements
We thank Nuno Faria and Bram Vrancken for sharing data. We thank Stéphane Hue and an anonymous reviewer for comments on our work that allowed us to improve this manuscript.
Contributor Information
Sofia G Seabra, Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical (IHMT), Universidade Nova de Lisboa (UNL), Rua da Junqueira 100, Lisboa 1349-008, Portugal.
Pieter J K Libin, Department of Computer Science, Artificial Intelligence Lab, Vrije Universiteit Brussel, Brussels 1050, Belgium; Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium; Data Science Institute, I-Biostat, Hasselt University, Agoralaan Gebouw D, Diepenbeek 3590, Belgium.
Kristof Theys, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium.
Anna Zhukova, Institut Pasteur, Université Paris Cité, Unité Bioinformatique Evolutive, 25-28 rue du Dr Roux, Paris F-75015, France; Institut Pasteur, Université Paris Cité, Bioinformatics and Biostatistics Hub, 25-28 rue du Dr Roux, Paris F-75015, France.
Barney I Potter, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium.
Hanna Nebenzahl-Guimaraes, Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical (IHMT), Universidade Nova de Lisboa (UNL), Rua da Junqueira 100, Lisboa 1349-008, Portugal.
Alexander E Gorbalenya, Department of Medical Microbiology, Leiden University Medical Center, Postbus 9600, Leiden 2300 RC, The Netherlands; Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow 119899, Russia.
Igor A Sidorov, Department of Medical Microbiology, Leiden University Medical Center, Postbus 9600, Leiden 2300 RC, The Netherlands.
Victor Pimentel, Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical (IHMT), Universidade Nova de Lisboa (UNL), Rua da Junqueira 100, Lisboa 1349-008, Portugal.
Marta Pingarilho, Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical (IHMT), Universidade Nova de Lisboa (UNL), Rua da Junqueira 100, Lisboa 1349-008, Portugal.
Ana T R de Vasconcelos, National Laboratory for Scientific Computing, Av. Getulio Vargas, 333, Quitandinha, Petrópolis, Rio de Janeiro 25651-075, Brazil.
Simon Dellicour, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium; Spatial Epidemiology Lab (SpELL), Université Libre de Bruxelles, CP 264/3, 50 av. F.D. Roosevelt, Bruxelles B-1050, Belgium.
Ricardo Khouri, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium; Instituto Gonçalo Moniz, Fundação Oswaldo Cruz/MS, Salvador, Bahia 40296-710, Brazil.
Olivier Gascuel, Institut Pasteur, Université Paris Cité, Unité Bioinformatique Evolutive, 25-28 rue du Dr Roux, Paris F-75015, France; Institut de Systématique, Evolution, Biodiversité (UMR7205 - CNRS, MNHN, SU, EPHE, UA), Muséum National d’Histoire Naturelle, CP 50, 45 rue Buffon, Paris 75005, France.
Anne-Mieke Vandamme, Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical (IHMT), Universidade Nova de Lisboa (UNL), Rua da Junqueira 100, Lisboa 1349-008, Portugal; Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium.
Guy Baele, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, KU Leuven, Herestraat 49 - box 1030, Leuven 3000, Belgium.
Lize Cuypers, Department of Laboratory Medicine, University Hospitals Leuven, Herestraat 49, Leuven 3000, Belgium.
Ana B Abecasis, Global Health and Tropical Medicine (GHTM), Instituto de Higiene e Medicina Tropical (IHMT), Universidade Nova de Lisboa (UNL), Rua da Junqueira 100, Lisboa 1349-008, Portugal.
Data availability
Sequence alignment files and Python and R scripts are available at https://github.com/seabrasg/zika_diversity.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This research was supported in part by the European Union’s Horizon 2020 research and innovation program ZIKAlliance (Agreement No 734548) and by Fundação para a Ciência e Tecnologia (FCT) through funds GHTM-UID/04413/2020. S.G.S. was funded by FCT, Portugal, through contrato-programa 1567 (CEECINST/00102/2018). K.T. was supported by a Fonds Wetenschappelijk Onderzoek post-doctoral grant. P.L. was supported by a doctoral (1S31916N) and post-doctoral grant (#1242021N) provided by the Fonds Wetenschappelijk Onderzoek and was also supported by funding from the Flemish Government under the ‘Onderzoeksprogramma Artificiele Intelligentie (AI) Vlaanderen’ programme. P.L. also received funding from the research council of the Vrije Universiteit Brussel (OZR-VUB) via grant number OZR3863BOF. S.D. acknowledges funding from the Fonds de la Recherche Scientifique (FNRS, Belgium). S.D. and G.B. acknowledge support from the Research Foundation—Flanders (Fonds voor Wetenschappelijk Onderzoek—Vlaanderen, G098321N). B.P. and G.B. acknowledge support from the Internal Fondsen KU Leuven/Internal Funds KU Leuven (Grant No. C14/18/094). G.B. also acknowledges support from the Research Foundation—Flanders (‘Fonds voor Wetenschappelijk Onderzoek—Vlaanderen,’ G0E1420N).
Conflict of interest:
The authors declare no conflicts of interests.
References
- Alcantara L. C. J. et al. (2009) ‘A Standardized Framework for Accurate, High-throughput Genotyping of Recombinant and Non-recombinant Viral Sequences’, Nucleic Acids Research, 37: 634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry F. et al. (2021) ‘Recent African Strains of Zika Virus Display Higher Transmissibility and Fetal Pathogenicity than Asian Strains’, Nature Communications, 12: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A. et al. (2013) ‘GenBank’, Nucleic Acids Research, 41: 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni M. F., Posada D., and Feldman M. W. (2007) ‘An Exact Nonparametric Method for Inferring Mosaic Structure in Sequence Triplets’, Genetics, 176: 1035–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen T. C., Philippe H., and Bryant D. (2006) ‘A Simple and Robust Statistical Test for Detecting the Presence of Recombination’, Genetics, 172: 2665–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A., and Shen T. J. (2003) ‘Nonparametric Estimation of Shannon’s Index of Diversity When There are Unseen Species in Sample’, Environmental and Ecological Statistics, 10: 429–43. [Google Scholar]
- Cheng L. et al. (2013) ‘Hierarchical and Spatially Explicit Clustering of DNA Sequences with BAPS Software’, Molecular Biology and Evolution, 30: 1224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement M., Posada D., and Crandall K. A. (2000) ‘TCS: A Computer Program to Estimate Gene Genealogies’, Molecular Ecology, 9: 1657–9. [DOI] [PubMed] [Google Scholar]
- Collins N. D. et al. (2019) ‘Inter- and Intra-lineage Genetic Diversity of Wild-type Zika Viruses Reveals Both Common and Distinctive Nucleotide Variants and Clusters of Genomic Diversity’, Emerging Microbes & Infections, 8: 1126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig S. et al. (2003) ‘Diverse Dengue Type 2 Virus Populations Contain Recombinant and Both Parental Viruses in a Single Mosquito Host’, Journal of Virology, 77: 4463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuypers L. et al. (2018) ‘Time to Harmonize Dengue Nomenclature and Classification’, Viruses, 10: 569.doi: 10.3390/v10100569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatorre E., Mir D., and Bello G. (2017) ‘Tracing the Origin of the NS1 A188V Substitution Responsible for Recent Enhancement of ZIKA Virus Asian Genotype Infectivity’, Memorias Do Instituto Oswaldo Cruz, 112: 793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N. R. et al. (2016) ‘Zika Virus in the Americas: Early Epidemiological and Genetic Findings’, Science, 352: 345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2017) ‘Establishment and Cryptic Transmission of Zika Virus in Brazil and the Americas’, Nature, 546: 406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye O. et al. (2014) ‘Molecular Evolution of Zika Virus during Its Emergence in the 20th Century’, PLoS Neglected Tropical Diseases, 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix A. C. et al. (2017) ‘Cross Reactivity of Commercial Anti-dengue Immunoassays in Patients with Acute Zika Virus Infection’, Journal of Medical Virology, 89: 1477–9. [DOI] [PubMed] [Google Scholar]
- Fonseca V. et al. (2019) ‘A Computational Method for the Identification of Dengue, Zika and Chikungunya Virus Species and Genotypes’, PLoS Neglected Tropical Diseases, 13: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs M. J., Armstrong J. S., and Gibbs A. J. (2000) ‘Sister-scanning: A Monte Carlo Procedure for Assessing Signals in Rebombinant Sequences’, Bioinformatics, 16: 573–82. [DOI] [PubMed] [Google Scholar]
- Gong Z., Xu X., and Han G. Z. (2017) ‘The Diversification of Zika Virus: Are There Two Distinct Lineages?’ Genome Biology and Evolution, 9: 2940–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A. E. et al. (2010) ‘Practical Application of Bioinformatics by the Multidisciplinary VIZIER Consortium’, Antiviral Research, 87: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh N. D. et al. (2019) ‘Travel Surveillance and Genomics Uncover a Hidden Zika Outbreak during the Waning Epidemic’, Cell, 178: 1057–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Han J. F. et al. (2016) ‘Homologous Recombination of Zika Viruses in the Americas’, Journal of Infection, 73: 87–8. [DOI] [PubMed] [Google Scholar]
- Heinz F. X., and Stiasny K. (2017) ‘The Antigenic Structure of Zika Virus and Its Relation to Other Flaviviruses: Implications for Infection and Immunoprophylaxis’, Microbiology and Molecular Biology Reviews, 81: 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C. (2003) ‘Patterns of Intra- and Interhost Nonsynonymous Variation Reveal Strong Purifying Selection in Dengue Virus’, Journal of Virology, 77: 11296–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C., Worobey M., and Rambaut A. (1999) ‘Phylogenetic Evidence for Recombination in Dengue Virus’, Molecular Biology and Evolution, 16: 405–9. [DOI] [PubMed] [Google Scholar]
- Huson D. H., and Bryant D. (2006) ‘Application of Phylogenetic Networks in Evolutionary Studies’, Molecular Biology and Evolution, 23: 254–67. [DOI] [PubMed] [Google Scholar]
- Ishikawa S. A. et al. (2019) ‘A Fast Likelihood Method to Reconstruct and Visualize Ancestral Scenarios’, Molecular Biology and Evolution, 36: 2069–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerzak G. et al. (2005) ‘Genetic Variation in West Nile Virus from Naturally Infected Mosquitoes and Birds Suggests Quasispecies Structure and Strong Purifying Selection’, Journal of General Virology, 86: 2175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond S. L., Frost S. D. W., and Muse S. V. (2005) ‘HyPhy: Hypothesis Testing Using Phylogenies’, Bioinformatics, 21: 676–9. [DOI] [PubMed] [Google Scholar]
- Kuno G., and Chang G. J. (2007) ‘Full-length Sequencing and Genomic Characterization of Bagaza, Kedougou, and Zika Viruses’, Archives of Virology, 152: 687–96.doi: 10.1007/s00705-006-0903-z. [DOI] [PubMed] [Google Scholar]
- Lanciotti R. S. et al. (2008) ‘Genetic and Serologic Properties of Zika Virus Associated with an Epidemic, Yap State, Micronesia, 2007’, Emerging Infectious Diseases, 14: 1232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh J. W., and Bryant D. (2015) ‘PopART: Full-feature Software for Haplotype Network Construction’, Methods in Ecology and Evolution / British Ecological Society, 6: 1110–6. [Google Scholar]
- Letunic I., and Bork P. (2021) ‘Interactive Tree of Life (Itol) V5: An Online Tool for Phylogenetic Tree Display and Annotation’, Nucleic Acids Research, 49: W293–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D. et al. (2017) ‘Insights into Intercontinental Spread of Zika Virus’, PLoS One, 12: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libin P. J. K. et al. (2019) ‘VIRULIGN : Fast Codon-correct Alignment and Annotation of Viral Genomes’, Bioinformatics, 35: 1763–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. et al. (2017) ‘Evolutionary Enhancement of Zika Virus Infectivity in Aedes Aegypti Mosquitoes’, Nature Publishing Group, 545: 482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Shi W., and Qin C. (2019) ‘The Evolution of Zika Virus from Asia to the Americas’, Nature Reviews. Microbiology, 17: 131–9. [DOI] [PubMed] [Google Scholar]
- Martin D., and Rybicki E. (2000) ‘RDP: Detection of Recombination Amongst Aligned Sequences’, Bioinformatics, 16: 562–3. [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2021) ‘The Emergence and Ongoing Convergent Evolution of the SARS-CoV-2 N501Y Lineages’, Cell, 184: 5189–200.e7.doi: 10.1016/j.cell.2021.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsky H. C. et al. (2017) ‘Zika Virus Evolution and Spread in the Americas’, Nature, 546: 411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J. et al. (2016) ‘Zika Virus Associated with Microcephaly’, New England Journal of Medicine, 374: 951–8. [DOI] [PubMed] [Google Scholar]
- Murrell B. et al. (2013) ‘FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection’, Molecular Biology and Evolution, 30: 1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2012) ‘Detecting Individual Sites Subject to Episodic Diversifying Selection’, PLoS Genetics, 8: e1002764.doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso D., Ko A. I., and Baud D. (2019) ‘Zika Virus Infection—after the Pandemic’, The New England Journal of Medicine, 381: 1444–57. [DOI] [PubMed] [Google Scholar]
- Nambala P., and Su W. C. (2018) ‘Role of Zika Virus prM Protein in Viral Pathogenicity and Use in Vaccine Development’, Frontiers in Microbiology, 9: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole Á. et al. (2021) ‘Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool’, Virus Evolution, 7: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padidam M., Sawyer S., and Fauquet C. M. (1999) ‘Possible Emergence of New Geminiviruses by Frequent Recombination’, Virology, 265: 218–25. [DOI] [PubMed] [Google Scholar]
- Paraschiv S. et al. (2017) ‘Epidemic Dispersion of HIV and HCV in a Population of Co-infected Romanian Injecting Drug Users’, PLoS One, 12: e0185866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattnaik A., Sahoo B. R., and Pattnaik A.K. (2020) ‘Current status of zika virus vaccines: successes and challenges’, Vaccines, 8: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson J. H. et al. (2016) ‘How Did Zika Virus Emerge in the Pacific Islands and Latin America?’ mBio, 7: e01239–16.doi: 10.1128/mBio.01239-16.Invited. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D., and Crandall K. A. (2001) ‘Evaluation of Methods for Detecting Recombination from DNA Sequences: Computer Simulations’, Proceedings of the National Academy of Sciences, 98: 13757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2020) ‘A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology’, Nature Microbiology, 5: 1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D. et al. (2000) ‘HIV-1 Nomenclature Proposal’, Science, 288: 55–6. [DOI] [PubMed] [Google Scholar]
- Roos R. P. (2016) ‘Zika virus-A Public Health Emergency of International Concern’, JAMA Neurology, 73: 1395–6. [DOI] [PubMed] [Google Scholar]
- Rossi S. L. (2018) ‘Did Zika Virus Mutate To Cause Severe Outbreaks?’ Trends in Microbiology, 26: 877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen M. O. et al. (1995) ‘Identification of Breakpoints in Intergenotypic Recombinants of HIV Type 1 by Bootscanning’, AIDS Research and Human Retroviruses, 11: 1423–5. [DOI] [PubMed] [Google Scholar]
- Shrinet J. et al. (2016) ‘Analysis of the Genetic Divergence in Asian Strains of ZIKA Virus with Reference to 2015–2016 Outbreaks’, Bulletin of the World Health Organization. 10.2471/BLT.16.176065. [Google Scholar]
- Shrivastava S. et al. (2018) ‘Whole Genome Sequencing, Variant Analysis, Phylogenetics, and Deep Sequencing of Zika Virus Strains’, Scientific Reports, 8: 15843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. et al. ICTV Report Consortium . (2017) ‘ICTV Virus Taxonomy Profile: Flaviviridae’, Journal of General Virology, 98: 2–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón D. et al. (2018) ‘An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region’, Viruses, 10: 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin Y. et al. (2017) ‘Differential Virulence between Asian and African Lineages of Zika Virus’, PLoS Neglected Tropical Diseases, 11: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. B. et al. (2014) ‘Expanded Classification of Hepatitis C Virus into 7 Genotypes and 67 Subtypes: Updated Criteria and Genotype Assignment Web Resource’, Hepatology, 59: 318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. R. et al. (2018) ‘African and Asian Zika Virus Isolates Display Phenotypic Differences Both in Vitro and in Vivo’, American Journal of Tropical Medicine and Hygiene, 98: 432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. M. (1992) ‘Analyzing the Mosaic Structure of Genes’, Journal of Molecular Evolution, 34: 126–9. [DOI] [PubMed] [Google Scholar]
- Spielman S. J. et al. (2019) ‘Evolution of Viral Genomes: Interplay between Selection, Recombination, and Other Forces’. In: Anisimova, M. (ed.) Evolutionary Genomics: Statistical and Computational Methods, Methods in Molecular Biology, Vol. 1910. pp. 427–68. New York, NY: Humana.doi: 10.1007/978-1-4939-9074-0_14. [DOI] [PubMed] [Google Scholar]
- Taucher C., Berger A., and Mandl C. W. (2010) ‘A Trans -complementing Recombination Trap Demonstrates A Low Propensity of Flaviviruses for Intermolecular Recombination’, Journal of Virology, 84: 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannesberger J. et al. (2021) ‘Viral Metagenomics Reveals the Presence of Novel Zika Virus Variants in Aedes Mosquitoes from Barbados’, Parasites & Vectors, 14: 343.doi: 10.1186/s13071-021-04840-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theys K. et al. (2017) ‘Zika Genomics Urgently Need Standardized and Curated Reference Sequences’, PLoS Pathogens, 13: e1006528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2018) ‘The Impact of HIV-1 Within-host Evolution on Transmission Dynamics’, Current Opinion in Virology, 28: 92–101. [DOI] [PubMed] [Google Scholar]
- To T.H. et al. (2016) ‘Fast Dating Using Least-Squares Criteria and Algorithms’, Systematic Biology, 65: 82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolou H. J. G. et al. (2001) ‘Evidence for Recombination in Natural Populations of Dengue Virus Type 1 Based on the Analysis of Complete Genome Sequences’, Journal of General Virology, 82: 1283–90. [DOI] [PubMed] [Google Scholar]
- Tonkin-Hill G. et al. (2018) ‘RhierBAPs: An R Implementation of the Population Clustering Algorithm Hierbaps’, Wellcome Open Research, 3: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2019) ‘Fast Hierarchical Bayesian Analysis of Population Structure’, Nucleic Acids Research, 47: 5539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trösemeier J. H. et al. (2016) ‘Genome Sequence of a Candidate World Health Organization Reference Strain of Zika Virus for Nucleic Acid Testing’, Genome Announcements, 4: e00917–16.doi: 10.1128/genomeA.00917-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. et al. (2016) ‘From Mosquitos to Humans: Genetic Evolution of Zika Virus’, Cell Host & Microbe, 19: 561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S. et al. (2018) ‘Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes’, Molecular Biology and Evolution, 35: 773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiller G. F. (1998) ‘Phylogenetic Profiles: A Graphical Method for Detecting Genetic Recombinations in Homologous Sequences’, Molecular Biology and Evolution, 15: 326–35. [DOI] [PubMed] [Google Scholar]
- WHO ⁄OIE ⁄FAO . (2012) ‘Continued Evolution of Highly Pathogenic Avian Influenza A(H5N1): Updated Nomenclature’, Influenza and Other Respiratory Viruses, 6: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder-Smith A., and Osman S. (2020) ‘Public Health Emergencies of International Concern: A Historic Overview’, Journal of Travel Medicine, 27: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L. et al. (2017) ‘A Single Mutation in the prM Protein of Zika Virus Contributes to Fetal Microcephaly’, Science, 106: 933–6. [DOI] [PubMed] [Google Scholar]
- Zhao F. et al. (2018) ‘Negligible Contribution of M2634V Substitution to ZIKV Pathogenesis in AG6 Mice Revealed by a Bacterial Promoter Activity Reduced Infectious Clone’, Scientific Reports, 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z. et al. (2016) ‘Comparative Genomic Analysis of Pre-epidemic and Epidemic Zika Virus Strains for Virological Factors Potentially Associated with the Rapidly Expanding Epidemic’, Emerging Microbes and Infections, 5: e22–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence alignment files and Python and R scripts are available at https://github.com/seabrasg/zika_diversity.