

Abstract
Cytokine storm and sterile inflammation are common features of T cell-mediated autoimmune diseases as well as T cell-targeted cancer immunotherapies. While blocking individual cytokines can mitigate some pathology, the upstream mechanisms governing overabundant innate inflammatory cytokine production remain unknown. Here, we have identified a critical signaling node that is engaged by effector memory T cells (TEM) to mobilize a broad pro-inflammatory program in the innate immune system. Cognate interactions between TEM and myeloid cells led to induction of an inflammatory transcriptional profile that was reminiscent, yet entirely independent, of classical pattern recognition receptor (PRR) activation. This PRR-independent ‘de novo’ inflammation was driven by pre-existing TEM engagement of both CD40 and TNFR on myeloid cells. Cytokine toxicity as well as autoimmune pathology could be completely rescued by ablating these pathways genetically or pharmacologically in multiple models of T cell-driven inflammation, indicating that TEM instruction of the innate immune system is a primary driver of associated immunopathology. Thus, we have identified a previously unknown trigger of cytokine storm and autoimmune pathology that is amenable to therapeutic interventions.
One-sentence summary:
Effector memory CD4+ T cells activate myeloid cells to cause innate inflammation bypassing the need for microbial detection
Introduction
Aberrant T cell activation is a driver of pathology in various T cell-mediated autoimmune diseases, including type 1 diabetes, multiple sclerosis, and rheumatoid arthritis (1–4). While self-reactive T cells arising from a breach in tolerance are prevalent in these diseases, inflammatory cytokine production by myeloid cells has been identified as the mediator of substantial pathology (5, 6). Targeting of IL-1β, IL-6, and TNF has been shown to reduce disease severity (6–9), yet poor patient response rates and resulting immunosuppression have been persistent issues with anti-cytokine monotherapies (10, 11). This limitation has presented the need for better mechanistic understanding of myeloid cell-derived cytokine production in the context of T cell-mediated autoimmunity. While previous research has focused on the role of pattern recognition receptors (PRRs) in driving autoimmune disease (12–14), failure of clinical trials targeting PRRs in these diseases (15, 16) demonstrates the non-infectious (sterile) nature of this inflammation and suggests the existence of distinct upstream pathways responsible for production of these damaging innate cytokines.
Following breach in central and peripheral tolerance, auto-reactive T cells become aberrantly activated leading to broad immunopathology. A group of monogenic diseases including IPEX, CHAI, and LATAIE (17) result from disruption of normal tolerance mechanisms (loss of function in FOXP3, CTLA4, or LRBA, respectively) and provide striking clinical examples of such aberrant T cell activation. Complete loss of Foxp3+ regulatory T cells (Tregs) in IPEX patients is an extreme case, and patients exhibit multiple autoimmune manifestations including enteropathy and inflammation of the lung and liver (18, 19). Mouse models of inducible Foxp3+ Treg deletion show similar features of autoimmune pathology (20). While Treg ablation in mice expectedly results in broad T cell activation, widespread activation and tissue infiltration of macrophages and DCs has also been reported (20). Remarkably, no clear correlation has been found between microbial presence and disease severity when Tregs are depleted in adult mice (21).
Innate inflammation associated with activated CD4+ effector memory T cells (TEM) extends to other clinically relevant conditions as well. Cytokine storm is seen following chimeric-antigen receptor (CAR) T cell infusion (22, 23) as well as immune checkpoint blockade therapy (24, 25). In CAR T-cell therapy, the transfer of activated antigen-specific T cells leads to systemic inflammation and elevated levels of IL-6 in the blood (22, 23). While this innate cytokine production is of myeloid origin (26), the driver of such inflammation is unclear. Similar to T cell-mediated autoimmune diseases, severity of cytokine storm in CAR-T cell therapy has not been correlated to microbial recognition by PRRs (27), suggesting existence of a PRR-independent mechanism of myeloid cell activation in this disease. We therefore set out to investigate the mechanism responsible for such damaging innate inflammation associated with presence of activated TEM, but with no links to microbial invasion.
When cognate interactions between pre-formed TEM and myeloid cells (DCs) were induced by a variety of methods, we found that TEM were able to directly instruct de novo innate cytokine production by myeloid cells as part of a broad inflammatory program. Transcriptomic analysis revealed that this TEM-driven activation of myeloid cells overlaps substantially with PRR activation, suggesting that TEM-derived cues essentially replace microbial ligands to drive innate immune activation. Specifically, we found that CD40 signaling is the primary trigger for PRR-independent myeloid cell activation by TEM with contribution from TNFR signaling. Blocking CD40 and TNFR pathways together completely abrogated the cytokine storm induced by activated TEM as well as auto-immune organ pathology associated with the presence of self-reactive T cells. Together, these findings uncover a mechanism of innate immune activation, where TEM instruction, rather than PRR signaling, drives sterile innate inflammatory cytokine storm and auto-immune pathology.
Results
TEM instruct myeloid cells to produce innate cytokines during their cognate interaction
We utilized a previously described in vitro co-culture system to test if interactions between TEM and myeloid cells were sufficient to trigger innate immune activation, in the absence of any PRR ligands (28). Briefly, we co-cultured bone marrow-derived DCs (BMDCs) and polyclonal effector memory TH0 cells that are differentiated in vitro from naïve CD4+ T cells in the presence of IL-2. Cognate interactions were triggered by culturing these cells in the presence of soluble anti-CD3ε antibody (anti-CD3) for 18 hours. Such cognate interaction induced by anti-CD3 led to robust production of not only IL-1β, but also IL-6 and IL-12 (Fig. 1A,B). Other myeloid cell types including bone marrow-derived macrophages (BMDM) and ex vivo CD11c+ splenic DCs also responded to TEM engagement by producing IL-6 (Fig. 1C). Importantly, this induction of inflammatory cytokines, which is typically associated with microbial recognition, was completely independent of TLR signaling, as similar levels of IL-6 and IL-12 were produced by TLR2/4/5xUnc93b13d/3d BMDCs which lack TLR2, TLR4, TLR5 as well as the functional protein, UNC93B1, responsible for TLR3, TLR7, TLR9, TLR11, and TLR13 trafficking (29) (Fig. 1D). We confirmed that BMDCs, and not CD90.2+ TEM, were the sole source of IL-1β, IL-6, and IL-12 in these cultures using intracellular cytokine staining (Fig. 1E, 1F, S1A). Additionally, TH1, TH2 and TH17 TEM cells induced innate cytokine production by BMDCs (Fig. 1G) suggesting that the ability to induce cytokine production by myeloid cells is conserved across all three T cell effector lineages.
Fig. 1: Cognate interactions between BMDCs and TEM drives myeloid production of IL-6 and IL-12.
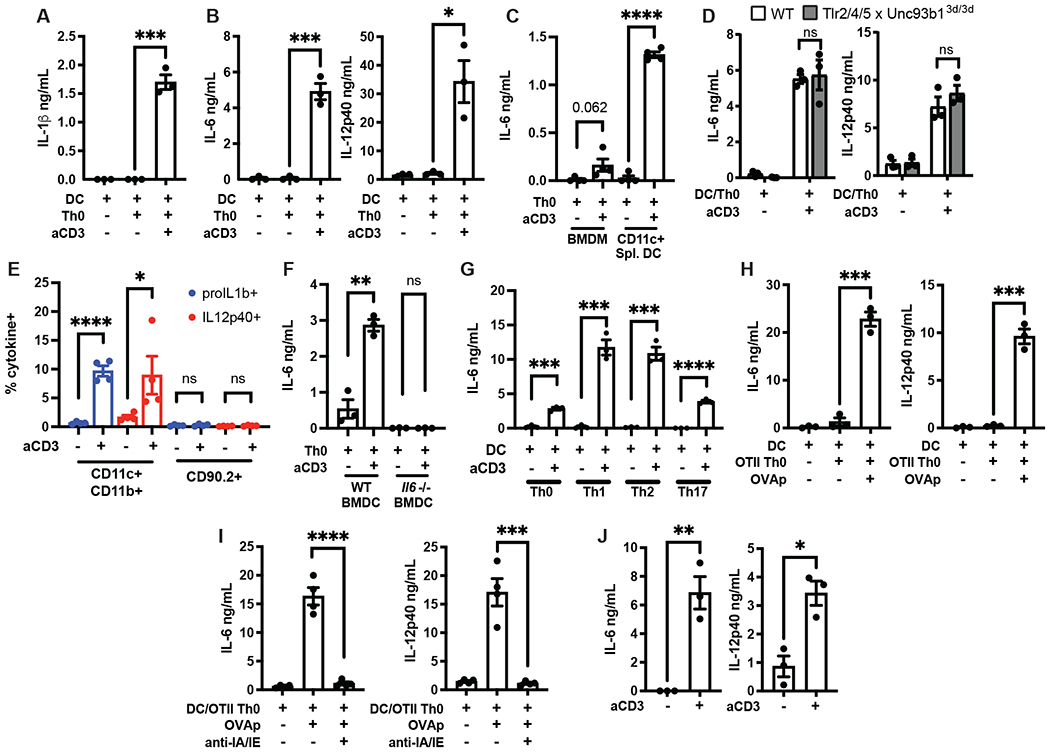
(A) IL-1β, (B) IL-6, and IL-12p40 were measured by ELISA in the supernatants of WT BMDCs cultured with WT TH0 TEM in the presence or absence of anti-CD3 (200ng/mL) for 18hr. (C) IL-6 and IL-12p40 were measured by ELISA in the supernatants of WT BMDMs or CD11c+ splenic DCs cultured with WT TH0 TEM in the presence of anti-CD3 for 18hr. (D) IL-6 and IL-12p40 were measured by ELISA in the supernatants of WT or TLR2/4/5xUnc93b13d/3d BMDCs cultured with WT TH0 TEM in the presence anti-CD3 for 18hr. (E) Quantification of intracellular proIL-1b or IL12p40 production measured by flow cytometry of CD11c+CD11b+ or CD90.2+ cells following culture of WT BMDCs and WT TH0 TEM in the presence of anti-CD3 for 6hr. Representative flow plots shown in Fig. S1A. (F) IL-6 was measured by ELISA in the supernatants of WT or Il6−/− BMDCs cultured with WT Th0 TEM in the presence of anti-CD3 for 6hr. (G) IL-6 was measured by ELISA in the supernatants of WT BMDCs cultured with TH0 or polarized Th1, TH2, or TH17 TEM in the presence of anti-CD3 for 6hr. (H) IL-6 and IL-12p40 were measured by ELISA in the supernatants of WT BMDCs cultured with OT-II TH0 in the presence or absence of 10uM OVA peptide (323-339) (OVAp) and (I) anti-IA/IE antibody (20ug/mL) for 18hr. (J) IL-6 and IL-12p40 levels in the serum of WT mice were quantified by ELISA 6hr following injection with PBS or anti-CD3 (50ug, i.v.). Error bars indicate SEM. (A-D, F-H, J) n=3, (E, I) n= 4, (A-D,F-J) unpaired two-tailed t-test. (E) unpaired one-tailed t-test *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant.
To test if innate inflammation driven by CD3 ligation also occurs during cognate MHC-TCR interactions between T cells and myeloid cells, we utilized an antigen-specific system. We co-cultured OVA-specific OT-II TH0 TEM with WT BMDCs in the presence or absence of OVA peptide 323-339 and found that antigen-specific engagement of TEM with DCs also led to IL-6 and IL-12 production (Fig. 1H). More importantly, secretion of IL-6 and IL-12 was inhibited when blocking antibodies against MHC-II were used, underscoring the requirement for cognate DC-T cell interaction (Fig. 1I). The quantity of cytokine secreted was dictated by the concentration of the OVA peptide (Fig. S1B). This highlights that the avidity of MHC-TCR interaction is a critical determinant for the magnitude of innate inflammation. Overall, these data establish that both polyclonal and antigen-specific interactions between CD4+ TEM and myeloid cells activate the innate immune system to drive production of inflammatory cytokines.
CAR T-cell therapy, where highly activated T cells bearing chimeric receptors with specificity to surface antigens are administered to patients, is known to induce cytokine release syndrome (CRS) marked by elevated levels of circulating IL-6 (22, 23). We therefore asked if highly activated TEM engage myeloid cells and instruct them to drive in vivo production of innate cytokines. To mimic features of CRS induced by CAR-T cell therapy, we injected anti-CD3 intravenously into WT mice to drive widespread T cell activation (30, 31). Indeed, administration of anti-CD3 induced robust levels of circulating IL-6 and IL-12 (Fig. 1J), suggesting that anti-CD3 ligation induced systemic activation of T cells and also drove CRS-like innate inflammation in vivo.
TEM-instructed and PRR-mediated activation of myeloid cells share a substantial transcriptional profile
In addition to cytokine production, BMDCs also upregulated DC maturation markers like CD86 and MHCII upon interaction with TEM (Fig. 2A) pointing to a broader activation of myeloid cells, even in the absence of PRR signaling. To achieve deeper understanding of TEM-induced myeloid cell activation, we performed mRNA sequencing (RNA-seq) of FACS-sorted CD11c+ BMDCs, 3 hours following their interaction with TEM (Fig. S2A). We directly compared the transcriptional profile of such BMDCs to those stimulated with the TLR4 ligand, LPS, for 3 hours. We found that the TEM-induced transcriptional profile of BMDCs (+aCD3) largely overlapped with that of LPS-stimulated BMDCs (+LPS) (Fig. 2B), with noticeable differences in magnitude of gene induction. These two groups shared 704 genes that were significantly downregulated, and 669 genes that were significantly upregulated compared to their respective controls (Fig. 2C). Importantly, TEM also induced a unique set of genes in interacting BMDCs that were not present in LPS-stimulated BMDCs (Fig. S2B, S2C), suggesting involvement of distinct signaling pathways as well as induction of a unique gene program. Together, these data suggest that cognate interaction with TEM leads to activation of a pro-inflammatory gene program in myeloid cells bearing several hallmarks of microbial PRR signaling.
Fig. 2: Broad TEM -induced activation of myeloid cells mimics TLR stimulation.

(A) Expression of CD86 and MHC-II by CD11c+CD11b+ BMDCs measured by flow cytometry following their culture with WT TH0 TEM in the presence of anti-CD3 for 6hr. (B) Heatmap of shared genes up or down regulated found in FACS-sorted CD11c+ BMDCs following their coculture with LPS (+LPS, 100ng/ml) or TH0 TEM (+aCD3, 200ng/ml) compared to unstimulated or coculture with TEM in the absence of anti-CD3 (+TC). (C) Venn diagram of differentially regulated genes in indicated treatment groups. (D) Functional annotation enrichment of genes upregulated in aCD3 verses TC conditions analyzed by EnrichR. (E) GSEA analysis of BMDCs cocultured in the presence or absence of activated TEM. (F) Heatmap of genes representing TNFRSF members. (G) Expression of indicated TNFRSF members on BMDCs following their coculture with activated TH0 TEM. Error bars indicate SEM; (A) representative of 2 independent experiments, (G) n= 3, paired two-tailed t-test. *p<0.05, **p<0.01, ***p<0.001.
We performed pathway analysis on the complete set of genes induced by BMDCs following TEM interaction and found that one of the most enriched pathways was TNF signaling when compared to its respective control (Fig. 2D). GSEA analysis further validated that following their interaction with TEM, CD11c+ BMDCs had significant enrichment of genes associated with TNFRSF signaling and systemic inflammation (Fig. 2E). We looked specifically at TNFRSF expression by BMDCs following TEM interaction and found them to be broadly induced (Fig. 2F). Given the established role of TNFR signaling in NF-kB activation, we hypothesized that TNFRSF ligands, known to be rapidly upregulated by TEM (32), could lead to widespread myeloid cell activation. Tnfrsf1a and Cd40 are prominent TNFR superfamily members associated with inflammation that were notably upregulated by DCs following TEM engagement (Fig 2G). This is consistent with our recent work that demonstrated TEM can engage TNFR on DCs to drive pro-IL-1β synthesis (28). Together, these data led us to hypothesize that TEM engagement of TNFRSF drives myeloid cell activation and innate cytokine production.
CD40 and TNFR signaling pathways are critical for PRR-independent activation of DCs by TEM cells
To test if TNFRSF signaling in DCs drives inflammatory cytokine production during cognate interactions with TEM, we used neutralizing antibodies against several members of the TNF superfamily. While TNFα neutralization (anti-TNFa) only mildly diminished production of IL-6 and IL-12 by BMDCs following their cognate interaction with OT-II Th0 TEM, anti-CD40L treatment significantly reduced the cytokine production (Fig. 3A). However, we found that both TNFα neutralization or CD40L blocking led to significantly abated cytokine production during polyclonal TEM cells interaction with BMDCs in the presence of anti-CD3 (Fig. 3B). More importantly, combined blockade of CD40 and TNFR signaling led to further reduction in innate cytokine production by DCs in vitro, suggesting that CD40 and TNFR signaling are non-redundant in driving this TEM-induced innate inflammation (Fig. 3A, 3B).
Fig. 3: TNFR and CD40 signaling is responsible for TEM induced activation of BMDCs and causes cytokine storm.

(A) IL-6 and IL-12p40 were measured by ELISA in the supernatants of WT BMDCs cultured with OT-II TH0 pretreated with 20ug/mL indicated neutralizing antibodies in the presence or absence of 10uM OVA peptide (323-339) (OVAp)or (B) WT TH0 in the presence of absence of anti-CD3 (200ng/mL) for 18hr. (C) IL-6 was measured by ELISA in the supernatants of WT BMDCs cocultured with total CD4+ T cells (tCD4) isolated from WT mice immunized with OVA for 7 days. Cells were pretreated with indicated neutralizing antibodies and cultured in the presence or absence of endotoxin free OVA (100ug/mL) for 24hr. (D) IL-6 was measured by ELISA in the supernatants of WT BMDCs cocultured with total CD4+ T cells (tCD4) isolated from WT mice immunized with MOG35-55 for 10 days. Cells were pretreated with indicated neutralizing antibodies and cultured in the presence or absence of MOG35-55 (300ug/mL) for 24hr. (E) IL-6 and IL-12p40 levels were quantified by ELISA in the serum of WT mice pretreated with PBS or indicated neutralizing antibodies (200ug, i.p.) for 12 hrs and 6hr following injection of anti-CD3 (50ug, i.p.). (F) Survival rates of WT and TNFaCD40DKO mice where WT mice were pretreated with indicated neutralizing antibodies (200ug, i.p.) for 12 hrs followed by injection of anti-CD3 (200ug, i.p.). (G) Heatmap of genes induced in FACS sorted CD11c+ BMDCs following their coculture with activated TEM and repressed by single or double neutralizing antibody pretreatment. (H) Total number of genes per group in (G). Error bars indicate SEM; (A, B) n = 5, (C-E) n = 3, (F) 3-6 mice per group; (A-E) Ordinary one-way ANOVA with Sidak’s multiple comparisons test. (F) Log-rank Mantel-Cox test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant.
We next tested if antigen-specific polyclonal TEM generated in vivo following immunization could also induce inflammatory cytokine production in a TNFR-CD40 dependent manner. Total CD4+ T cells were isolated from draining lymph nodes of OVA-immunized mice and were cultured with BMDCs in the presence or absence of OVA protein. The polyclonal OVA-specific TEM present in this total CD4+ T cell population induced robust secretion of IL-6 by BMDCs which was largely abrogated when TNF and CD40 signaling was blocked (Fig. 3C). Finally, we investigated if bona fide autoreactive CD4+ T cells are capable of engaging this pathway. We obtained CD4+ T cells from mice immunized with MOG (Myelin Oligodendrocyte Glycoprotein) 35-55 peptide and found that autoreactive antigen-specific CD4+ T cells indeed triggered IL-6 production by MOG peptide-presenting BMDCs in a TNF-CD40L dependent fashion (Fig. 3D). Genetic knockout models were further used to verify the importance of TEM-derived TNF and CD40L in driving innate immune inflammation. TEM deficient in either CD40L or TNFα displayed compromised ability to trigger IL-12 production by BMDCs compared to WT TEM, while CD40L−/− TEM also had compromised ability to trigger IL-6 production (Fig. S3A).
Finally, we asked if blocking CD40-TNFR signaling pathways could rescue innate inflammation following anti-CD3 administration in vivo. WT mice were treated with either anti-TNFα or anti-CD40L antibodies, or combination of the two, for 12 hours prior to anti-CD3 injection. We found that TNF-CD40 signaling blockade attenuated the T cell-induced myeloid cell activation in vivo (Fig. 3E). Administration of high doses of anti-CD3 induces severe CRS, resulting in wasting and death (33–35). Blocking both CD40 and TNFR signaling pathways or using genetic knockouts not only diminished the severity of CRS but also protected mice from succumbing to death resulting from inflammation (Fig. 3F).
To understand the specific effects of CD40 and TNFR signaling on BMDCs during cognate interactions with TEM, we performed RNA-seq on FACS-sorted CD11c+ BMDCs following 3 hours of TEM engagement in the presence of CD40L and/or TNFα blocking antibodies. Hierarchical clustering of genes upregulated following anti-CD3 ligation identified 4 major gene clusters (Fig. 3G, S3B). Cluster I represents genes that are repressed following TNF blockade including Naip1, Fpr1, and Fpr2 (S3C). Genes repressed upon CD40L blockade represent cluster III, and include inflammatory transcripts such as Cxcl1 and Il12b, as well as other TNFSF ligands like Tnfsf9 (41BBL) and Tnfsf15 (TL1A) (Fig. S3C). Cluster II includes genes that are repressed following combination treatment, including numerous inflammatory mediators like Il1b, Il6, Il12a, Cxcl2, and Cxcl3 (Fig. S3C). While clusters I-III were expectedly enriched for genes associated with NF-kB activation (Fig. S3D), we also found significant enrichment for genes associated with extracellular matrix remodeling, suggesting that TNF-CD40 signaling results in vast functional and phenotypic alterations of the myeloid cell. Cluster IV represents genes that are not regulated by either TNF or CD40L blockade (Fig. S3B, S3C). GSEA analysis shows that these genes are enriched for responses to IFNγ (Fig. S3E), suggesting that soluble mediators or additional molecular interactions may be important for complete TEM-induced activation of myeloid cells. Even though TNFR and CD40 signaling seem to additively promote inflammatory gene program, CD40 upregulation itself was independent of TNF signaling (Fig. S3F). Overall, these results show that TNFR-CD40 signaling drive PRR-independent activation of myeloid cells via divergent pathways.
Autoreactive T cells drive inflammatory cytokine production by DCs and macrophages in vivo
During thymic development, central tolerance purges high-affinity autoreactive T cells that could potentially bypass the priming checkpoints (36). However, self-reactive T cells that have escaped negative selection still exist in healthy individuals (37, 38). Foxp3+ regulatory T cells (Tregs) counteract any damaging outcomes of self-reactive T cells, by preventing priming, expansion, and re-activation of self-reactive T cells in the periphery (39). The necessity of this mechanism has been demonstrated by inducible Treg depletion through the use of the Foxp3-DTR mouse model (20, 40–42). Although auto-reactive T cells have been primarily implicated as the direct cause of lung and liver pathology and eventual mortality in this model (20, 41), previous studies have reported alterations in the innate immune compartment (20). More specifically, accumulation of myeloid cells, including DCs, in secondary lymphoid organs, as well as recruitment of inflammatory monocytes and neutrophils to the tissues has been observed in Treg depleted mice. We therefore decided to interrogate if cognate interaction between autoreactive T cells and myeloid cells would result in the features of anti-CD3-induced innate immune activation and inflammation, described above.
To achieve Treg ablation in adult mice, diphtheria toxin (DT) was injected intraperitoneally into Foxp3-DTR mice daily. Following 5 days of treatment and loss of Foxp3+ Tregs, we found robust T cell activation and splenomegaly as reported previously (20) (Fig. S4A, S4B). Within the spleen, we found increased total numbers and relative frequencies of neutrophils, inflammatory monocytes, and macrophages (Fig. 4A–4B, S4C–S4F). We also found increased numbers of DCs, although their proportion of total splenocytes was the same (Fig. 4C, S4F). Our previous results suggest that activated TEM can induce inflammatory cytokine production from myeloid cells, and indeed we were able to detect elevated IL-6 and IL-12 in the serum of Treg-depleted mice (Fig. 4D). To test the sufficiency of expanded autoreactive T cells in inducing innate cytokine production by myeloid cells, we cultured WT BMDCs with CD4+ T cells isolated from Treg-depleted mice, the majority of which displayed an activated profile (Fig. S4A). We found that CD4+ T cells from Treg-depleted mice were able to engage and instruct BMDCs to produce IL-6 (Fig. 4E). Remarkably, this production of IL-6 did not require exogenous addition of antigen or anti-CD3, suggesting that expanded auto-reactive T cells are likely engaging self-antigens presented by MHC complexes on BMDCs. Accordingly, autoreactive T cell-induced IL-6 was compromised by blocking MHC-II interactions (Fig. 4E). Interestingly, the induction of IL-6 seen by self-reactive TEM in vitro was also suppressed when TNFR-CD40 signaling was blocked, or iTregs were added to the co-cultures (Fig. 4F). This is in line with the recent report showing that reinstating Treg function can ameliorate established autoimmune inflammation (43). It is possible that Tregs could directly inhibit upregulation of CD40 on myeloid cells, as previously suggested (44–46), thus preventing its engagement by CD40L expressed on activated self-reactive CD4+ T cells. Importantly, naïve CD4+ T cells (nCD4) isolated from the non Treg-depleted mice did not induce inflammatory cytokine production by BMDCs (Fig. 4F), suggesting that the ability to induce innate inflammation is a distinct property of activated self-reactive TEM. Together, these data show that activated self-reactive T cells instruct interacting myeloid cells, likely through self-peptide recognition, to produce inflammatory cytokines.
Fig. 4: Autoreactive T cells drive inflammatory cytokine production by DCs and macrophages in vivo.

(A) Infiltration of neutrophils (CD11b+Ly6C+Ly6G+), inflammatory monocytes (CD11b+Ly6Chi), (B) macrophages (CD11b+CD11c+), and (C) DCs (CD11c+) into the spleens of Foxp3-DTR mice treated with diphtheria toxin (+DT) or untreated (−DT) for 5 days as quantified by flow cytometry. (D) IL-6 and IL-12 were quantified by ELISA in the serum of mice from (A). (E) IL-6 was measured by ELISA in the supernatants of WT BMDCs cultured with total CD4+ T cells (tCD4) isolated from Foxp3-DTR mice treated with diphtheria toxin (DT) or untreated (−) for 5 days, in the presence or absence of anti-IA/IE antibody (20ug/mL), or (F) indicated neutralizing antibodies (20ug/ml), WT iTregs, or naïve CD4+ (nCD4). Error bars indicate SEM. (A-D) n=4, (E,F) n=4–8. (A-D) unpaired two-tailed t-test, (E, F) Ordinary one-way ANOVA with Sidak’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant.
Autoreactive T cells utilize the CD40-TNFR pathways to activate myeloid cells in vivo and drive auto-immune pathology
Foxp3+ Treg depletion triggers widespread activation of autoreactive T cells resulting in organ-specific damage and systemic inflammation (20). Our data above prompted us to hypothesize that following Treg depletion, the activated and expanded population of self-reactive TEM could engage various antigen-presenting myeloid cell subsets via the TNFR-CD40 pathway to drive innate inflammation and pathology in vivo. In agreement with this hypothesis, we found TNFR expression and upregulation of CD40 in splenic F4/80+ macrophages, Ly6C+ monocytes, cDC1s, and cDC2 but not on non-antigen-presenting myeloid cells such as pDCs or neutrophils (Fig. S5A–S5D). We inhibited TNFR or CD40 signaling in Foxp3+ Treg-depleted mice and found that splenomegaly as well as lymphadenopathy was significantly mitigated in mice treated with anti-CD40L blocking or anti-TNFα neutralizing antibodies (Fig. 5A, 5B, S6A). This was accompanied by reduced neutrophil, monocyte, and CD11c+ DC recruitment to the spleen (Fig. 5C–E, S6B–S6D). Due to vast heterogeneity within myeloid cell populations, we performed a more in-depth analysis of the spleen-infiltrating cell types. We observed TNFR-CD40 dependent infiltration of CD11c+MHCIIhiXCR1+ cDC1s, CD11c+MHCIIhiSIRP1α+ cDC2s, CD11cintMHCIIloPDCA1+ pDCs as well as CD11b+Ly6C+ monocytes, CD11b+F4/80+ macrophages and CD11b+Ly6C+Ly6G+neutrophils into the spleen (Fig. S6E, S6F). In line with our findings on cell infiltration, while individual blockade of TNFR and CD40 signaling only partially attenuated circulating levels of IL-6 and IL-12, combination antibody treatment significantly reduced these inflammatory cytokines in the serum (Fig. 5F). Finally, we confirmed that reduced systemic inflammation was accompanied by diminished cell maturation and cytokine production by various myeloid populations after TNF-CD40L double blockade (Fig 5G, 5H, S6G). These data again point to the combined effects of TNFR and CD40 signaling on driving TEM-induced myeloid cell activation and innate inflammation.
Fig. 5: Blocking TNFR and CD40 can rescue innate inflammation and pathology caused by autoreactive TEM following Treg depletion.

(A, B) Splenomegaly in Foxp3-DTR mice treated with diphtheria toxin (+DT), untreated (−DT), or pretreated with indicated neutralizing antibodies following injections of DT for 5 days. (C) Infiltration of neutrophils (CD11b+Ly6C+Ly6G+), (D) inflammatory monocytes (CD11b+Ly6Chi), and (E) DCs (CD11c+) into the spleens of Foxp3-DTR mice treated with or without DT and indicated neutralizing antibodies for 5 days as quantified by flow cytometry. (F) IL-6 and IL-12p40 were quantified by ELISA in the serum of mice from (A). (G) Expression of CD86 on indicated splenic myeloid populations of Foxp3-DTR mice pretreated with neutralizing antibodies (DT+Ab) and given DT or untreated (NDT) for 3 days. (H) Expression of Il6 and Il12b as quantified by qPCR from indicated splenic myeloid populations isolated from Foxp3-DTR mice pretreated with indicated neutralizing antibodies and given diphtheria toxin (DT) or untreated (−) for 3 days. (I) Representative histology images showing diffuse immune cell infiltration in the liver and lungs in Foxp3-DTR mice treated with DT, compared to those without DT and those treated with neutralizing antibodies for 10 days. Arrows indicate peri-portal and lobular infiltrates in the liver. (J) Scoring quantification of the lungs and livers of Foxp3-DTR mice treated with or without DT and indicated neutralizing antibodies for 10 days. Scale bars: lung (20mm) and liver (50mm). Error bars indicate SEM; (B-F) each dot represents individual mouse, data pooled from 3-4 independent experiments, Ordinary one-way ANOVA with Sidak’s multiple comparisons test. (G, H) data representative of 2 independent experiments with 2 mice each, unpaired one-tailed t-test. (I) data representative of 7-8 mice across 3 experiments. (J) Mann-Whitney test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant.
Tregs actively maintain peripheral tolerance and their depletion is known to result in severe pathology of the skin, lungs, and liver (20). To understand if this TNFR-CD40 mediated instruction of innate cells was directly responsible for pathology in Treg-depleted mice, we monitored disease over 10 days. Indeed, neutralization of TNF and blocking CD40L in Treg-depleted mice significantly dampened lung and liver pathology (Fig. 5I, 5J). Earlier studies have attributed tissue pathology in this model to the overwhelming activation of autoreactive T cells (20). We found that T cell activation was only minimally reduced when TNF and CD40L were blocked (Fig. S6H, S6I) despite dampened pathology, suggesting that activation of self-reactive T cells alone is not sufficient to evoke immunopathology. Together, these data suggest that the autoreactive T cells are not the sole or direct drivers of autoimmune inflammation seen in Treg -depleted mice. Instead, our work reveals that activation of myeloid cells through TNFR-CD40 signaling pathways by autoreactive T cells is the definitive cause of such catastrophic inflammation.
Discussion
The presence of overabundant innate inflammatory cytokines in the context of widespread TEM activation, especially during autoimmunity and T cell-directed immunotherapy, is a major clinical challenge (47). Therapeutic success of anti-cytokine treatment (8, 48, 49) in such conditions shows that innate cytokines are the major driver of pathology in these diseases. However, monotherapies against individual cytokines are not broadly effective (50, 51) and alternate use of general immunosuppressants can render patients prone to infections (10, 11). These limitations underscore the need for mechanistic understanding of T cell-associated innate inflammation and developing upstream interventions to block the broad pro-inflammatory program.
One of the most critical functions of the innate immune system is to respond rapidly to stimuli by producing inflammatory cytokines. While canonical activation of innate immunity stems from pathogen recognition, there are multiple cases by which similar innate cytokine secretion can proceed in the absence of microbial ligands. In auto-inflammatory diseases, for example, gain-of-function mutations in PRRs or their adaptor molecules enable constitutive pathogen-independent activation of signaling pathways (52). Here, we have identified another case of such innate immune activation, whereby TEM can directly instruct interacting myeloid cells to acquire an inflammatory transcriptional program that is independent of, yet mimics, classical PRR activation. We find that both polyclonal as well as antigen-specific in vitro generated TEM have the capacity to engage and activate myeloid cells to drive a pro-inflammatory response. We further show that TNFR and CD40 signaling in myeloid cells together control this PRR-independent myeloid cell activation, producing measurable expression of innate cytokines both in vitro and in vivo. Broad activation of T cells in vivo results in systemic cytokine storm and lethality, features that resemble innate immune activation by PRR ligands (Fig. S7).
The transcriptional profile displayed by myeloid cells upon interaction with TEM largely mirrors that of myeloid cells exposed to microbial ligands. This is rather remarkable and suggests that the innate immune system has the ability to sense existence of immunological memory in the adaptive immune system. In essence, TNF and CD40L presented by TEM appear to serve as surrogates for canonical microbial ligands. The ability of TNFR and CD40 signaling to initiate NF-κB and MAP kinase signaling (53) thus fulfills requirements for inflammatory gene transcription. Our findings further clarify that TNFR and CD40 signaling can trigger de novo inflammation by myeloid cells during TEM interactions, which is distinct from their earlier described role in amplification of cytokine production during naïve T cell priming, which occurs in the presence of PRR ligands (54). While all PRRs trigger measurable activation of myeloid cells, they induce a qualitatively distinct transcriptional profile (55, 56) that is dictated by differential usage of adapters and transcription factors. Analogous to differential outcomes of PRR signaling pathways, we found that TNFR and CD40 also give rise to qualitatively unique myeloid cell activation states, suggesting that members of TNFRSF function as sterile counterparts to PRRs. Interestingly, we have also identified a cluster of genes that are uniquely induced by TEM instruction, but not by PRR activation, signifying a divergent evolution of these two mechanisms of innate activation. This cluster (cluster IV) was also independent of TNF/CD40 signaling, suggesting potential engagement of other TNFRSF members or soluble mediators originating from effector T cells (57).
Innate cytokines are critical for both priming and reactivation of CD4+ T cells (58, 59). The principle of innate control of adaptive immunity dictates that innate cytokine secretion occurs in response to microbial detection by antigen-presenting cells to avoid T cell activation towards self-antigens (Fig. S7) (60). However, certain virulent pathogens have been shown to hijack PRR signaling and broadly suppress cytokine production (61). We postulate that the TNFR-CD40 pathway of myeloid cell activation evolved as a means for antigen-specific TEM to directly instruct the production of innate cytokines needed for their proper function, even when myeloid cells are under the pressure of such immunosuppressive pathogen strategies. This agrees with the observation that innate cytokines such as IL-6 and IL-1β (59, 62), but not PRR activation (63, 64), are required for antigen-specific TEM function. In contrast, we find that naïve T cells are unable to overcome the need for PRR signaling possibly due to lack of constitutive expression or rapid upregulation of TNFRSF ligands as seen in TEM (32, 65, 66). We propose that only TEM are poised to instruct antigen-presenting myeloid cells as they have been previously exposed to PRR dependent signals during priming. In fact, CD4+ TEM were shown to induce protective inflammation in the context of influenza infection without requiring activation of conventional PRRs (63). This ability of TEM ensures that immunological memory is not compromised in the face of virulent pathogens, while still requiring PRR activation for initial generation of pathogen-specific memory.
It is conceivable that the evolution of such mechanism can be inadvertently employed by aberrantly activated T cells in multiple disease settings. Indeed, in the case of auto-reactive TEM arising from depletion of peripheral Tregs, we find that these TEM engage TNFR and CD40 in interacting myeloid cells to induce innate immune activation and inflammatory cytokine production. It is important to note that this innate activation categorically relies on cognate MHC-TCR engagement and is independent of PRR signaling. While auto-reactive T cells can contribute to disease in this model (20), our data provide compelling evidence that substantial pathology is caused by T cell-mediated activation of the innate immune system via TNFR and CD40-induced inflammation. We further show that in a model of CRS as seen following CAR-T cell infusion, presence of widespread activated TEM also leads to TNFR-CD40-dependent innate cytokine storm.
CAR-T cell therapy utilizes both CD4+ and CD8+ T cells (67). Thus, it remains to be explored if CD8+ TEM can also activate interacting myeloid cells to cause innate inflammation and if so, whether similar or distinct mechanisms initiate such PRR-independent innate inflammation.
Antibody therapies targeting CD40 signaling have shown promise in autoimmunity presumably by abating humoral responses as well as T cell priming (54). However, we have now identified CD40L as the initiator of T cell-driven innate inflammation, suggesting that CD40L blockade can also be employed for indications where pre-existing self-reactive memory T cells are drivers of disease such as IPEX syndrome or CAR-T cell therapy. Our findings thus provide a conceptual framework for understanding the evolutionary basis of innate and adaptive immune cross-talk, and more importantly, offer clinically relevant, effective and safer targets for alleviating innate inflammation associated with dysregulated TEM activation.
Materials and Methods
Study Design
The goal of this study was to delineate the mechanisms underlying T cell mediated autoimmune inflammation. To this end, we first used an in vitro system of interaction between T effector memory cells (TEM) and dendritic cells, where in vitro generated TH0 TEM are cocultured with GMCSF derived BMDCs in the presence of anti-CD3 antibody. We found that interactions of BMDCs with TEM can trigger BMDC maturation and cytokine production completely independent of pattern recognition receptor (PRR) signaling. We performed RNA sequencing of BMDCs to characterize and compare their T cell induced transcriptional profile with that of classical PRR signaling. Using both in vitro and in vivo settings, we found that CD40L and TNFα expressed by TEM are critical to activate interacting myeloid cells. This study also includes RNA seq of BMDCs following their interaction with T cells where either CD40 or TNFα or when both are blocked using neutralizing antibodies. We used two in vivo models to comprehensively test the impact of T cell driven innate inflammation. In the first model we activated TEM cells in vivo by injecting anti-CD3 antibody. In the second model, we depleted Treg cells to cause activation and expansion of auto-reactive T cells that is known to lead to T cell driven auto-immunity. Both models cause systemic activation of TEM in vivo that led to overabundant production of innate inflammatory cytokines with dramatic autoimmune pathology in organs such as lung and liver which could be rescued by blocking of TNFα and CD40L. All methods and the statistical approaches are explained in detail in the methods section as well as the figure legends.
Mice
C57BL/6 (Jax:00064) mice were purchased from Jackson Laboratories. Mice with Foxp3-DTR mutation have been described (20). TLR2/4/5xUnc93b13d/3d (29) mice were a gift from Greg Barton. TNFa−/− (Jax:005540) x CD40−/− (Jax:002928) mice were bred in house. Il6−/− (Jax:002650) were a gift from David Hildeman. All mice were bred and maintained in SPF conditions at Cincinnati Children’s Hospital Medical Center in accordance with protocols approved by IACUC. Age- and sex- matched mice between 6 and 12 weeks were used for all experiments. Both males and females were used for all experiments. For Treg depletion experiments, Foxp3-DTR mice were given diphtheria toxin (DT) 25ug/kg on day 0 and 10ug/kg subsequently (diluted in PBS) intraperitoneally every day for 5-10 days. For blockade experiments, mice were treated with 500ug of neutralizing antibodies on day −1 and every 3 days subsequently. For anti-CD3 experiments, WT mice were given intraperitoneal anti-CD3ε antibody (145-2C11, BioXcell), 50ug for serum ELISAs or 200ug for survival. For blockade experiments, mice were pretreated with 500ug of neutralizing antibodies (anti-mouse TNFα (XT3.11); anti-mouse CD154 (MR-1)) intraperitoneally for 12 hours prior to anti-CD3 or DT injections.
Generation of BMDCs
Following RBC lysis, bone marrow cells were plated at .75x10^6/ml in BMDC media (5% FCS containing complete RPMI + 20ng/ml rGMCSF (Biolegend)). Media was replaced on day 2, 4, and 6 then cells were harvest on day 7 by gently flushing each well.
Isolation and differentiation of CD4 T cells
Spleen and mesenteric LNs were harvested and made intro single cell suspensions. Following RBC lysis, naïve CD4 T cells were isolated according to the Mojosort kit protocol (Biolegend). Cell culture plates were coated with anti-CD3ε (5ug/ml, Biolegend, 145-2C11) and anti-CD28 (5ug/ml, Tonbo, 37.51) for 3-4 hours at 37C. Naïve CD4 T cells were plated in coated wells at .5x10^6/ml for 5 days in appropriate Th polarizing conditions (TH0: IL-2 (50U/ml); TH1: IL-2 (50U/mL), IL-12 (10ng/mL), anti-IL-4 (10ug/mL, 11B11); TH2: IL-2 (50U/mL), IL-4 (4ng/mL), anti-IFNg (10ug/mL, XMG1.2); TH17: IL-6 (20ng/mL), TGFb (5ng/mL), anti-IL-4 (10ug/mL, 11B11), anti-IFNg (10ug/mL, XMG1.2), IL-23 (20ng/mL), IL-1b (20ng/mL)). Following differentiation, cells were replated at 1x10^6/ml in the presence of IL-2 (10U/ml) and rested for 2 additional days to allow differentiation into TEM. For generation of iTregs, naïve CD4 T cells were plated in anti-CD3ε (3ug/ml, 145-2C11) coated wells in the presence of soluble anti-CD28 (1ug/ml, 37.51), IL-2 (50U/ml), and TGF-b (5ng/ml, Biolegend) for 3 days.
In vitro cocultures
BMDCs and TH0 cells were harvested and plated at a ratio of 1:4 (DC:T cells) in 10% FCS containing complete RPMI. For blocking experiments, cells were preincubated with anti-TNFα (20ug/ml, Biolegend, MP6-XT22) or anti-CD40L (20ug/ml, Biolegend, MR1) for 30 minutes prior to addition of anti-CD3ε (200ng/ul, Biolegend, 145-2C11), OVA peptide (10uM, Invivogen), or EndoFit OVA protein (100ug/mL, Invivogen). For iTreg cocultures, iTregs were added at 1:2 ratio of Th0. Cells and supernatant were harvested 18 hours later. For generation of polyclonal OVA-specific TEM, WT mice were immunized in the footpad with 5ug LPS and 50ug OVA (Sigma) emulsified in IFA for 7 days. Total CD4 T cells were isolated from draining LNs and cocultured with BMDCs as described previously. For generation of polyclonal MOG35-55-specific TEM, WT mice were immunized subcutaneously with 200uL of MOG35-55 emulsion (Hooke Labs) for 10 days. Total CD4 T cells were isolated from the spleen and cocultured with BMDCs as described previously.
ELISA and serum ELISA
Capture antibodies for IL-6 (Biolegend, MP5-20F3) and IL-12p40 (Invitrogen, G23-8) were diluted and used to coat 96-well flat bottom plates overnight at 4C. Plates were blocked with PBS containing 1% BSA for 2 hours at room temperature. Samples were diluted in blocking buffer and loaded in duplicate then incubated overnight at 4C. Detection antibodies for IL-6 (Biolegend, MP5-32C11) and IL-12p40 (Invitrogen, C17.8) were diluted and used according to standard procedure. Protein concentration was quantified using OPD colorimetric assay.
Blood was collected from mice via heart puncture and left to clot at room temperature for 1 hour. Serum was isolated following centrifugation at 2000g for 10 minutes at 4C. Serum was loaded in duplicate, and ELISA was performed as described above.
Flow cytometry and cell sorting
Following RBC lysis, cells were washed with PBS containing 2mM EDTA and 1% BSA. For surface marker staining, cells were blocked with FC Shield (anti-mouse CD16/32, Tonbo) for 10 minutes then incubated with antibodies of interest for 30 minutes. Surface antibodies include: anti-mouse CD11c (N418), anti-mouse CD11b (M1/70), anti-mouse CD90.2 (30-H12), anti-mouse CD86 (GL1), anti-mouse CD40 (3/23), anti-mouse IA/IE (M5/114.15.2), anti-mouse Ly6C (HK1.4), anti-mouse Ly6G (1A8), anti-mouse Sirpa (P84), anti-mouse XCR1 (ZET), anti-mouse F4/80 (BM8), anti-mouse PDCA1 (927). For intracellular cytokine staining, cells were cultured in the indicated conditions for 6 hours, with Brefeldin A added for the last 4 hours. Cells were immediately harvested on ice, stained with fixable Zombie Yellow (Biolegend), then fixed with Foxp3 Transcription Factor Staining Set (Invitrogen) and stained with anti-mouse proIL-1b (eBioscience, NJTEN3) or anti-mouse IL-12/23 p40 (Biolegend, C15.6) according to manufacturer protocol. Samples were analyzed using Novocyte 2001 (ACEA Biosciences). Cells were gated on singlets and dead cells were excluded. Data were analyzed using FlowJo software. For cell sorting, samples were stained, washed, incubated with DAPI, and sorted by Sony MA900 (maintained by the Research Flow Cytometry Core in the Division of Rheumatology at CCHMC) directly into 10% FCS containing media.
Quantitative PCR
Cells were pelleted, washed with PBS, lysed using Trizol, and stored at −80C. Total RNA was isolated with chloroform and isopropanol, then cleaned with RNeasy mini kit (Qiagen). cDNA synthesis was performed with M-MLV reverse transcriptase (Invitrogen) in the presence of RNase inhibitor (Promega). qPCR was performed using SYBR Green 2x Mastermix (applied Biosystems) and measured using the QuantStudio 7 Flex Real-Time PCR system (Thermo Fisher). Data was normalized to Hprt. The primers used in this study are as follows: mouse Il6 forward: 5’-CCGGAGAGGAGACTTCACAG-3’; mouse Il6 reverse: 5’-GGAAATTGGGGTAGGAAGGA-3’; mouse Il12b forward: 5’-GGTGTAACCAGAAAGGTGCG-3’; mouse Il12b reverse: 5’-TTGGGGGACTCTTCCATCCT-3’; mouse Hprt forward: 5’-CAGTCCCAGCGTCGTGATTA-3’; mouse Hprt reverse: 5’- TGGCCTCCCATCTCCTTCAT-3’.
RNA sequencing
CD11c+ cells were magnetically sorted from CD45.1 GMCSF-derived BMDCs using Miltenyi Automacs. Sorted CD11c+ cells were then cultured in each indicated condition (alone, LPS (100ng/ml), CD45.2 TH0 cells, or TH0 and anti-CD3 (200ng/ml)) for 3 hours. Live CD45.1+CD90.2-CD11c+ were then FACS sorted (Fig. S1B). Sorted cells were washed with cold PBS and immediately lysed in Trizol. cDNA libraries were constructed from purified RNA (either by the lab of Dr. Edward K. Wakeland, UTSW, or BGI) and sequenced, pair-end, using Hi-seq Illumina platform. Adapter sequences and low-quality reads were trimmed using Trim Galore (68), then aligned to the mm10 genome using STAR (69). Duplicate reads were removed using MarkDuplicates. FeatureCounts (70) was used to produce a counts matrix from genes with mapped reads. Differential gene expression between groups was evaluated using DESeq2 (71), along with Enrichr (72) and GSEA (73) for further gene set enrichment analyses. Heatmaps of genes/gene set enrichments were generated in R using ggplot2.
Isolation and histopathological scoring of liver
Liver was obtained from each mouse after perfusion with PBS and fixed with 4% paraformaldehyde overnight and paraffin embedded. Sections of 10-μm thickness were subsequently stained with hematoxylin and eosin for light microscopy and scored in a blinded fashion. Histopathologic changes of necro inflammatory activity were scored by five grades of 0 to 4 as previously reported (74). Grade 0, no inflammation, or necrosis (0%); Grade 1, portal inflammation (1% – 25%) and/or lobular inflammation without necrosis; Grade 2, mild periportal inflammation (26% – 50%) and/or lobular focal hepatocellular necrosis; Grade 3, moderate periportal inflammation (51% – 75%) and/or lobular more extensive necrosis; Grade 4, severe periportal inflammation (>75%) and/or necrosis includes bridging necrosis.
Isolation and histopathological scoring of lung
Animals were sacrificed, perfused with PBS, and lungs inflated at 15 cmH2O with 4% paraformaldehyde and removed for paraffin embedding. Slices at 10-μm thickness were subsequently stained with hematoxylin and eosin. Lung injury scores were quantified by an investigator blinded to the treatment groups using criteria published by the American Thoracic Society (75). Lung injury was assessed on a scale of 0–2 for each of the following criteria: (i) neutrophils in the alveolar space, (ii) neutrophils in the interstitial space, (iii) number of hyaline membranes, (iv) amount of proteinaceous debris, and (v) extent of alveolar septal thickening. The final injury score was derived from the following calculation: score = (20 × i + 14 × ii + 7 × iii + 7 × iv + 2 × v)/ (number of fields × 100).
Statistics
Tests to determine significance are shown in figure legends, with corresponding p values. Parameters for statistical testing are shown in the raw data file.
Supplementary Material
Acknowledgments:
We thank all the members of the Pasare lab for their insight, helpful discussions, and critical reading of this manuscript. Special thanks to Lisa Waggoner for maintaining the mouse colony and the proper functioning of the lab.
Funding:
This work was supported by grants from the National Institutes of Health (AI123176, and AI155426) to C.P. and by the National Science Foundation Graduate Research Fellowship under Grant No. 2017220107 to M.M.M.
Footnotes
Competing interests: Chandrashekhar Pasare is inventor on patent application PCT/US20200/60090 submitted by Cincinnati Children’s Hospital Medical Center that covers methods for treating diseases by blocking TNF and CD40
Data material availability: Data generated in this study have been deposited in the Gene Expression Omnibus (GSE184607 and GSE184608). All other data needed to support the conclusions of the paper are present in the paper or the Supplementary Materials.
References
- 1.Bluestone JA, Bour-Jordan H, Cheng M, Anderson M, T cells in the control of organ-specific autoimmunity. J Clin Invest 125, 2250–2260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabrera SM, Henschel AM, Hessner MJ, Innate inflammation in type 1 diabetes. Transl Res 167, 214–227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu F, Shi M, Zheng C, Shen D, Zhu J, Zheng X, Cui L, The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol 318, 1–7 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Buckley CD, Ospelt C, Gay S, Midwood KS, Location, location, location: how the tissue microenvironment affects inflammation in RA. Nat Rev Rheumatol, (2021). [DOI] [PubMed] [Google Scholar]
- 5.Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M, et al. , Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol 18, 1797–1801 (1988). [DOI] [PubMed] [Google Scholar]
- 6.Lopalco G, Cantarini L, Vitale A, Iannone F, Anelli MG, Andreozzi L, Lapadula G, Galeazzi M, Rigante D, Interleukin-1 as a common denominator from autoinflammatory to autoimmune disorders: premises, perils, and perspectives. Mediators Inflamm 2015, 194864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haraoui B, Differentiating the efficacy of the tumor necrosis factor inhibitors. Semin Arthritis Rheum 34, 7–11 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Poddubnyy D, Rudwaleit M, Efficacy and safety of adalimumab treatment in patients with rheumatoid arthritis, ankylosing spondylitis and psoriatic arthritis. Expert Opin Drug Saf 10, 655–673 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R, Investigators O, Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 371, 987–997 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Fallahi-Sichani M, Flynn JL, Linderman JJ, Kirschner DE, Differential risk of tuberculosis reactivation among anti-TNF therapies is due to drug binding kinetics and permeability. J Immunol 188, 3169–3178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawar A, Desai RJ, Solomon DH, Santiago Ortiz AJ, Gale S, Bao M, Sarsour K, Schneeweiss S, Kim SC, Risk of serious infections in tocilizumab versus other biologic drugs in patients with rheumatoid arthritis: a multidatabase cohort study. Ann Rheum Dis 78, 456–464 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Waldner H, The role of innate immune responses in autoimmune disease development. Autoimmun Rev 8, 400–404 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Allam R, Anders HJ, The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol 20, 538–544 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Needell JC, Zipris D, Targeting Innate Immunity for Type 1 Diabetes Prevention. Curr Diab Rep 17, 113 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Anwar MA, Shah M, Kim J, Choi S, Recent clinical trends in Toll-like receptor targeting therapeutics. Med Res Rev 39, 1053–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monnet E, Choy EH, McInnes I, Kobakhidze T, de Graaf K, Jacqmin P, Lapeyre G, de Min C, Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis 79, 316–323 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG, Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol 142, 1679–1695 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME, X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27, 18–20 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD, The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27, 20–21 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Kim JM, Rasmussen JP, Rudensky AY, Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8, 191–197 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Chinen T, Volchkov PY, Chervonsky AV, Rudensky AY, A critical role for regulatory T cell-mediated control of inflammation in the absence of commensal microbiota. J Exp Med 207, 2323–2330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brudno JN, Kochenderfer JN, Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frey N, Porter D, Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant 25, e123–e127 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Oda H, Ishihara M, Miyahara Y, Nakamura J, Kozuka Y, Iwasa M, Tsunoda A, Yamashita Y, Saito K, Mizuno T, Shiku H, Katayama N, First Case of Cytokine Release Syndrome after Nivolumab for Gastric Cancer. Case Rep Oncol 12, 147–156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao L, Yang Y, Li W, Li T, Gao Q, Nivolumab-induced cytokine-release syndrome in relapsed/refractory Hodgkin’s lymphoma: a case report and literature review. Immunotherapy 10, 913–917 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M, CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med 24, 731–738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sentman ML, Murad JM, Cook WJ, Wu MR, Reder J, Baumeister SH, Dranoff G, Fanger MW, Sentman CL, Mechanisms of Acute Toxicity in NKG2D Chimeric Antigen Receptor T Cell-Treated Mice. J Immunol 197, 4674–4685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR, Philip NH, Oberst A, Chervonsky AV, Katz JD, Pasare C, T cells instruct myeloid cells to produce inflammasome-independent IL-1beta and cause autoimmunity. Nat Immunol 21, 65–74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sivick KE, Arpaia N, Reiner GL, Lee BL, Russell BR, Barton GM, Toll-like receptor-deficient mice reveal how innate immune signaling influences Salmonella virulence strategies. Cell Host Microbe 15, 203–213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferran C, Sheehan K, Dy M, Schreiber R, Merite S, Landais P, Noel LH, Grau G, Bluestone J, Bach JF, et al. , Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: further evidence for transient in vivo T cell activation. Eur J Immunol 20, 509–515 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Bugelski PJ, Achuthanandam R, Capocasale RJ, Treacy G, Bouman-Thio E, Monoclonal antibody-induced cytokine-release syndrome. Expert Rev Clin Immunol 5, 499–521 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Casamayor-Palleja M, Khan M, MacLennan IC, A subset of CD4+ memory T cells contains preformed CD40 ligand that is rapidly but transiently expressed on their surface after activation through the T cell receptor complex. J Exp Med 181, 1293–1301 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthys P, Dillen C, Proost P, Heremans H, Van Damme J, Billiau A, Modification of the anti-CD3-induced cytokine release syndrome by anti-interferon-gamma or anti-interleukin-6 antibody treatment: protective effects and biphasic changes in blood cytokine levels. Eur J Immunol 23, 2209–2216 (1993). [DOI] [PubMed] [Google Scholar]
- 34.Sevmis S, Emiroglu R, Karakayali F, Yagmurdur MC, Dalgic A, Moray G, Haberal M, OKT3 treatment for steroid-resistant acute rejection in kidney transplantation. Transplant Proc 37, 3016–3018 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Staedtke V, Bai RY, Kim K, Darvas M, Davila ML, Riggins GJ, Rothman PB, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S, Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 564, 273–277 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kappler JW, Roehm N, Marrack P, T cell tolerance by clonal elimination in the thymus. Cell 49, 273–280 (1987). [DOI] [PubMed] [Google Scholar]
- 37.Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW, Autoreactive T cells in healthy individuals. J Immunol 172, 5967–5972 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Cebula A, Kuczma M, Szurek E, Pietrzak M, Savage N, Elhefnawy WR, Rempala G, Kraj P, Ignatowicz L, Dormant pathogenic CD4(+) T cells are prevalent in the peripheral repertoire of healthy mice. Nat Commun 10, 4882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shevach EM, Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, Rudensky A, Sparwasser T, Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol 183, 7631–7634 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Boehm F, Martin M, Kesselring R, Schiechl G, Geissler EK, Schlitt HJ, Fichtner-Feigl S, Deletion of Foxp3+ regulatory T cells in genetically targeted mice supports development of intestinal inflammation. BMC Gastroenterol 12, 97 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D, How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity 31, 654–664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu W, Wang ZM, Feng Y, Schizas M, Hoyos BE, van der Veeken J, Verter JG, Bou-Puerto R, Rudensky AY, Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunity. Nat Immunol 22, 1163–1174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, Flores M, Li N, Schweighoffer E, Greenberg S, Tybulewicz V, Vignali D, Clynes R, Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol 180, 5916–5926 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S, Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A 105, 10113–10118 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML, Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med 203, 505–511 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saferding V, Bluml S, Innate immunity as the trigger of systemic autoimmune diseases. J Autoimmun 110, 102382 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, Alecock E, Lee J, Kremer J, IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis 67, 1516–1523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X, Fang L, Guo TB, Mei H, Zhang JZ, Drug targets in the cytokine universe for autoimmune disease. Trends Immunol 34, 120–128 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Browne SK, Anticytokine autoantibody-associated immunodeficiency. Annu Rev Immunol 32, 635–657 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Seymour HE, Worsley A, Smith JM, Thomas SH, Anti-TNF agents for rheumatoid arthritis. Br J Clin Pharmacol 51, 201–208 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manthiram K, Zhou Q, Aksentijevich I, Kastner DL, The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 18, 832–842 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Hayden MS, Ghosh S, Regulation of NF-kappaB by TNF family cytokines. Semin Immunol 26, 253–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grewal IS, Flavell RA, The role of CD40 ligand in costimulation and T-cell activation. Immunol Rev 153, 85–106 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Hu W, Jain A, Gao Y, Dozmorov IM, Mandraju R, Wakeland EK, Pasare C, Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc Natl Acad Sci U S A 112, 13994–13999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takeda K, Kaisho T, Akira S, Toll-like receptors. Annu Rev Immunol 21, 335–376 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Soudja SM, Chandrabos C, Yakob E, Veenstra M, Palliser D, Lauvau G, Memory-T-cell-derived interferon-gamma instructs potent innate cell activation for protective immunity. Immunity 40, 974–988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwasaki A, Medzhitov R, Control of adaptive immunity by the innate immune system. Nat Immunol 16, 343–353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain A, Song R, Wakeland EK, Pasare C, T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Commun 9, 3185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janeway CA Jr., Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54 Pt 1, 1–13 (1989). [DOI] [PubMed] [Google Scholar]
- 61.Sweet CR, Conlon J, Golenbock DT, Goguen J, Silverman N, YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell Microbiol 9, 2700–2715 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Longhi MP, Wright K, Lauder SN, Nowell MA, Jones GW, Godkin AJ, Jones SA, Gallimore AM, Interleukin-6 is crucial for recall of influenza-specific memory CD4 T cells. PLoS Pathog 4, e1000006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strutt TM, McKinstry KK, Dibble JP, Winchell C, Kuang Y, Curtis JD, Huston G, Dutton RW, Swain SL, Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med 16, 558–564, 551p following 564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pasare C, Medzhitov R, Toll-dependent control mechanisms of CD4 T cell activation. Immunity 21, 733–741 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Muller J, Baeyens A, Dustin ML, Tumor Necrosis Factor Receptor Superfamily in T Cell Priming and Effector Function. Adv Immunol 140, 21–57 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Koguchi Y, Thauland TJ, Slifka MK, Parker DC, Preformed CD40 ligand exists in secretory lysosomes in effector and memory CD4+ T cells and is quickly expressed on the cell surface in an antigen-specific manner. Blood 110, 2520–2527 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krueger F, in Babraham Bioinformatics. (2019). [Google Scholar]
- 69.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang CC, Cheng PY, Peng YJ, Wu ES, Wei HP, Yen MH, Naltrexone protects against lipopolysaccharide/D-galactosamine-induced hepatitis in mice. J Pharmacol Sci 108, 239–247 (2008). [DOI] [PubMed] [Google Scholar]
- 75.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM, Acute G Lung Injury in Animals Study, An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44, 725–738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.