

Abstract
Objective:
To characterize the effect of efavirenz on bupropion hydroxylation as a marker of cytochrome P450 (CYP) 2B6 activity in healthy subjects.
Methods:
Thirteen subjects received a single oral dose of bupropion SR 150 mg before and after 2 weeks of efavirenz administration for comparison of bupropion and hydroxybupropion pharmacokinetics. Efavirenz plasma concentrations were also assessed. Subjects were genotyped for CYP2B6 (G516T, C1459T, and A785G), CYP3A4 (A-392G), CYP3A5 (A6986G), and multidrug resistance protein 1 (C3435T).
Results:
The area under the concentration vs. time curve ratio of hydroxybupropion:bupropion increased 2.3-fold after efavirenz administration (P = 0.0001). Bupropion area under the concentration vs. time curve and Cmax decreased by 55% and 34%, respectively (P < 0.002). None of the CYP2B6 or CYP3A genotypes evaluated were associated with a difference in bupropion or efavirenz clearance. The 2 individuals homozygous for multidrug resistance protein 1 3435-T/T had 2.5- and 1.8-fold greater bupropion and efavirenz clearance, respectively, relative to C/C and C/T individuals (P < 0.05).
Conclusions:
Our results confirm that efavirenz induces CYP2B6 enzyme activity in vivo, as demonstrated by an increase in bupropion hydroxylation after 2 weeks of efavirenz administration.
Keywords: efavirenz, bupropion, metabolism, CYP2B6, drug interaction, induction
INTRODUCTION
Originally thought to have a limited role in drug metabolism, cytochrome P450 (CYP) 2B6 has gained greater attention in recent years, having been found to catalyze the metabolism of several commonly used medications. Drugs metabolized partially or completely by CYP2B6 include bupropion, cyclophosphamide, methadone, meperidine, sertraline, and nevirapine.1–5 Efavirenz, a commonly used HIV-1 nonnucleoside reverse transcriptase inhibitor, is also primarily metabolized by CYP2B6.6 A well-characterized inducer of CYP3A4 metabolism, the effect of efavirenz on CYP2B6 activity has not been clearly defined. In vitro studies utilizing liver microsomes or purified CYP2B6 demonstrated inhibition of CYP2B6-mediated bupropion hydroxylation by efavirenz.7,8 However, a subsequent study conducted with human hepatocytes showed induction of CYP2B6 gene expression by efavirenz via activation of human constitutive androstane receptor.9 The effect of efavirenz on CYP2B6 activity has not been evaluated in vivo.
Bupropion is metabolized to 3 active metabolites, including threohydrobupropion, erythrohydrobupropion, and hydroxybupropion.1 Threohydrobupropion and erythrohydrobupropion are formed via nonmicrosomal pathways and are approximately 20% as potent as bupropion.10 In contrast, hydroxybupropion is formed via CYP2B6 and possesses approximately one-half the pharmacologic activity of bupropion.10 Although less potent than bupropion, area under the concentration vs. time curve (AUC) of hydroxybupropion is approximately 17-fold higher than AUC of bupropion at steady state.10 Therefore, hydroxybupropion is likely responsible for much of the pharmacologic activity observed with bupropion administration. Hydroxylation of bupropion is a validated and commonly used probe reaction for CYP2B6 activity in vivo.11
Like other enzymes of the CYP2 family, CYP2B6 is highly polymorphic.12 Genetic variation in CYP2B6 contributes to the large interpatient variability observed in efavirenz pharmacokinetics (PK).13 The most prevalent of the mutations, A785G, G516T, and C1459T, are present in 32.6%, 28.6%, and 14% of whites, respectively.14 Across studies, efavirenz exposure is consistently greater in individuals homozygous for 516-T/T or in those subjects carrying the CYP2B6*6 allele, which is comprised of A785G in combination with G516T.15–18 Other CYP genotypes which may be associated with differences in efavirenz exposure include CYP3A4*1B and CYP3A5*3. In addition, genetic polymorphisms in the multidrug resistance protein 1 (MDR1) gene, which expresses the efflux protein P-glycoprotein, have been implicated in affecting efavirenz disposition.15,19
The primary purpose of this study was to characterize the influence of efavirenz on CYP2B6 activity in healthy volunteers by measuring bupropion and hydroxybupropion plasma concentrations after single-dose bupropion administration before and after 2 weeks of efavirenz administration. A secondary aim of the study was to assess the potential influence of CYP2B6, CYP3A4/5, and MDR1 genetic polymorphisms on efavirenz and bupropion PK parameters.
METHODS
Subjects
Healthy male and female volunteers between the ages of 18 and 55 years were eligible for study participation. Evaluation of potential subjects included a medical history, physical examination, and laboratory analyses (serum electrolytes, complete blood count, and liver function tests) to rule out any medical conditions that could increase subject risk or affect study results. Subjects with a history of or current psychiatric illness or symptoms, including depression or eating disorder, were excluded from participation, as were subjects with a history of alcohol or drug abuse or tobacco use within 4 weeks of study participation. Participants were also required to have a negative HIV enzyme-linked immunosorbent assay test. Females of childbearing potential were required to practice abstinence or use 2 nonhormonal methods of contraception throughout the study; they also received a serum pregnancy test within 1 day of starting efavirenz administration.
Subjects were prohibited from taking any medications, including nonprescription drugs, herbal supplements, and oral contraceptives, within 14 days of study participation. Additional exclusion criteria included a history of intolerance to any of the study medications and persistent diarrhea. Acetaminophen and ibuprofen were allowed as needed throughout the study to treat study-related side effects; however, subjects refrained from taking either of these medications on PK sampling days. Volunteers were prohibited from ingesting grapefruit or grapefruit juice during the entire study period.
Informed consent was obtained from all study participants, and clinical research was conducted in accordance with guidelines for human experimentation as specified by the US Department of Health and Human Services. The study was approved by the National Institute of Allergy and Infectious Diseases Investigational Review Board.
Study Design
This was an open-label, 2 phase, sequential study to evaluate the effect of 2 weeks of efavirenz administration on bupropion hydroxylation in healthy volunteers. Subjects who met inclusion criteria were genotyped for the following alleles at the time of enrollment: CYP2B6 (G516T, C1459T, and A785G), MDR1 (C3435T), CYP3A4 (A-392G), and CYP3A5 (A6986G).
Treatment and Blood Sampling
On the first day of study participation, subjects were administered a single oral dose of bupropion SR 150 mg (1 × 150 mg Wellbutrin SR tablet; GlaxoSmithKline, Research Triangle Park, NC) in the fasted state (phase 1). Blood samples were collected for the determination of bupropion and hydroxybupropion plasma concentrations at the following time points: 0 (predose), 1, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 24 (±2), and 48 (±2) hours postdose. After collection, all samples were immediately centrifuged, separated, and frozen at −80°C until chromatographic analysis.
Within 6 days after the completion of bupropion PK sampling, subjects began taking efavirenz 600 mg (1 × 600 mg Sustiva tablet; Bristol-Myers Squibb, Princeton, NJ) once daily at bedtime. In accordance with clinical recommendations for HIV-infected patients, subjects were instructed to take their daily efavirenz dose without food. Thirteen to 15 days after initiating efavirenz, subjects returned to the clinic for repeat PK sampling (phase 2). Subjects were instructed to take their previous evening’s efavirenz dose before midnight and to refrain from eating or drinking anything other than water after midnight. Upon arriving at the clinic, subjects were administered single doses of efavirenz 600 mg and bupropion SR 150 mg together on an empty stomach. One set of blood samples was collected serially over 48 hours for determination of bupropion and hydroxybupropion plasma concentrations, as in phase 1. A second set of samples was collected at the following time points for quantitation of efavirenz plasma concentrations: 0 (predose), 1, 2, 4, 6, 8, 12, 24 (±2), and 48 (±2) hours after drug administration. Adherence to efavirenz was assessed by pill count, patient interview, and efavirenz plasma concentrations.
Analytical Methods
Bupropion, hydroxybupropion, and diethylpropion (internal standard) were isolated from plasma via liquid–liquid extraction with hexane/isoamyl alcohol (96:4, vol/vol). Briefly, a 100-μL plasma sample was mixed with 20 μL diethylpropion solution (1.0 μg/mL) followed by 30 μL of 1.0 M NH4OH and then vortexed for 5 seconds. Next, 3.0 mL hexane/isoamyl alcohol (96:4, vol/vol) was introduced, and the sample was vortexed for 30–35 seconds. The sample was immediately centrifuged at 3200 rpm for 10 minutes at 4°C, and the organic layer was transferred to a clean 13 × 100-mm test tube. Subsequently, 250 μL of 0.10 M HCl in methanol was added to each sample and vortexed for 30–35 seconds. The samples were then centrifuged again at 3200 rpm for 5 minutes at 4°C, and the top organic layer was discarded. The remaining extract was then evaporated to dryness, followed by reconstitution with 70 μL of mobile phase, transferred into a vial, and placed in the autosampler tray at 4°C.
Bupropion, hydroxybupropion, and diethylpropion were separated using a newly developed high-performance liquid chromatography (HPLC) method and detected by tandem mass spectrometry (MS) using multiple reaction monitoring (MRM). The HPLC-MS-MS analysis was performed using an Acquity Ultra-Performance Liquid Chromatography liquid handling system and a Quattro Premier XE triple quadrupole mass spectrometer (Waters Corp, Milford, MA) controlled by MassLynx 4.1 MS manager software. The separation was performed on an Acquity HSS T3, 2.1 × 50 mm, 1.7-μm analytical column preceded by an Acquity column in-line filter kit using a mobile phase consisting of 30:70 (vol/vol) acetonitrile:3 mM ammonium formate, adjusted to pH 3.50 with formic acid at a flow rate of 0.200 mL/min. The mass spectrometer was used in positive ion electrospray mode with a source temperature of 125°C, desolvation temperature of 350°C, desolvation gas flow of 600 L/h, cone gas flow of 10 L/h, capillary voltage of 0.50 kV, cone voltages at 22–27 V, and collision energy at 12.0 for MRM experiments. Nitrogen was used as the nebulizer, auxiliary, and desolvation gas, whereas argon was used as the collision gas. The resolution was set at 0.60 amu width at half-height in both Q1 and Q3, and the analytes were detected using MRM with a 500-ms dwell time. The MRM transitions were optimized by direct infusion of bupropion, hydroxybupropion, and diethylpropion at a concentration of 100 ng/mL and a syringe pump flow rate of 10 μL/min. The optimal transitions were 240 m/z → 185 m/z for bupropion, 256 m/z → 238 m/z for hydroxybupropion, and 206 m/z → 105 m/z for diethylpropion.
Calibration curves for bupropion and hydroxybupropion were linear from 1.0 to 250 ng/mL (R2 > 0.998) and 5.0 to 800 ng/mL (R2 > 0.997), respectively. Percent errors, as a measure of accuracy, were <15%, and the inter- and intra-assay coefficients of variation for bupropion and hydroxybupropion were 2.31%–8.48% and 6.53%–12.03%, respectively, at 3 different concentrations. The limits of quantitation for bupropion and hydroxybupropion were 1.0 and 5.0 ng/mL, respectively; the limits of detection were 0.20 and 1.0 ng/mL, respectively. During the validation, short-term stability of the drug in plasma and repeated freeze–thaw stability of plasma were evaluated. The overall recovery of all 3 analytes was >85%.
Efavirenz concentrations were determined using a newly developed HPLC method. The bioanalytical assay was performed using a liquid–liquid extraction. The HPLC system consisted of a Waters 2795 Alliance HT separations module and a 2996 photodiode array detector set at λ = 270 nm (Waters Corp). The HPLC system and the assay parameters were controlled using the Empower (Version 5.0) chromatography manager software. The analytical column was an XTerra RP18, 5 μm, 3.9 × 150-mm reverse-phase analytical column and was preceded by an XTerra RP18, 5 μm, 3.9 × 20-mm guard column. Efavirenz and carbamazepine (internal standard) were isolated from human plasma via liquid–liquid extraction. Briefly, a 300-μL plasma sample was mixed with 20 μL of carbamazepine (20.0 μg/mL) followed by 50 μL of 1.0 M NH4OH and then vortexed for 5 seconds. Next, 4.0 mL of [80:19:1] ethyl acetate/hexane/isoamyl alcohol was introduced, and the sample was vortexed for 30–35 seconds. The sample was subsequently centrifuged at 3200 rpm for 10 minutes at 4°C, and the organic layer was transferred to a clean 13 × 100-mm test tube and evaporated to dryness. The sample was reconstituted with 120 μL of 50% methanol and transferred into an HPLC vial. Subsequently, 50 μL was injected into the HPLC system and eluted isocratically at 1.0 mL/min (T = 20°C) for 7 minutes using a mobile phase consisting of (40:60, vol/vol) acetonitrile and 3.0 mM pyrrolidine buffer at pH 10.2.
Calibration curves were linear from 0.050 to 15.0 μg/mL (R2 > 0.997). Percent errors, as a measure of accuracy, were <15%, and the inter- and intra-assay coefficients of variation were 2.08%–5.72% and 2.11%–9.85%, respectively, at 3 different concentrations. The limit of quantitation was 0.050 μg/mL, and the limit of detection was 0.025 μg/mL. During the validation, short-term stability of the drug in plasma and repeated freeze–thaw stability were evaluated. The overall recovery of efavirenz and carbamazepine was >90%.
Genotypic Analysis
After collection of venous blood samples from all subjects, DNA was isolated from peripheral leukocytes with the Qiamp system (Qiagen Inc, Valencia, CA). CYP2B6 genetic polymorphisms (A785G, C1459T, and G516T) were identified using polymerase chain reaction–restriction fragment length polymorphism as previously described.14 Polymerase chain reaction–restriction fragment length polymorphism analysis was also used to identify MDR1 (C3435T), CYP3A4*1B (A-392G), and CYP3A5*3 (A6986G) polymorphisms as previously described.20–22
PK Analysis
Plasma concentrations of bupropion, hydroxybupropion, and efavirenz were analyzed by noncompartmental methods using WinNonlin pharmacokinetic software, version 5.1 (Pharsight Corporation, Mountain View, CA). Maximum plasma concentration (Cmax) and time to reach Cmax (Tmax) were obtained by direct inspection of the plasma concentration–time profiles. Trough (minimum) plasma concentrations (Cmin) for efavirenz were also obtained by direct observation of the plasma concentration–time profiles. The elimination rate constant (λZ) was determined by calculating the absolute value of the slope of the log-linear regression using at least 3 points on the plasma concentration–time plot. The AUC to the last quantifiable concentration (AUC0–t) was determined by the log-linear trapezoidal rule and extrapolated to infinity (AUC0–∞) by dividing the last measured concentration by λZ and adding this value to AUClast. Apparent oral clearance (Cl/F) was estimated as dose/AUC0–∞.
Statistical Analysis
A difference in the ratio of hydroxybupropion:bupropion AUC of at least 35% was considered to be clinically relevant for the purpose of establishing sample size. Assuming a mean hydroxybupropion:bupropion ratio of 18, an SD of 0.4 × 18,10 an α value of 0.05, and a pre- and posttherapy correlation coefficient of 1/2, a sample size of 13 was determined necessary to detect a difference in the ratio of hydroxybupropion:bupropion with 80% power.
Results are reported as geometric means with SDs and geometric mean ratios ± 90% confidence intervals. The PK parameter values for bupropion and hydroxybupropion at baseline (phase 1) and after 2 weeks of efavirenz administration (phase 2) were compared using a 2-tailed, paired t test, except for Tmax, which was analyzed using the Wilcoxon signed rank test. Differences in PK parameter values of bupropion and efavirenz were compared between genetic groups using a 2-tailed, unpaired t test. The P values <0.05 were considered statistically significant for all analyses. SYSTAT Software (version 11, Richmond, CA) was used for sample size calculation and inferential statistics; Microsoft Excel 2003 (Microsoft Corp, Redmond, WA) was used to generate descriptive statistical data.
RESULTS
Subjects
A total of 13 subjects (10 males) were enrolled in the study and completed participation. The median age of subjects was 39 years (range 21–54 years) and the mean (SD) weight was 86 kg (±17 kg). Races were distributed as follows: 69.2% white, 15.4% African American, and 15.4% others. There were no missed doses of efavirenz or bupropion reported. Adherence to efavirenz was further confirmed by pill counts and by determination of plasma concentrations.
Burpropion and Hydroxybupropion PK
The AUC ratio of hydroxybupropion:bupropion increased 2.3-fold from phase 1 to phase 2 (P = 0.0001) (Table 1). Similarly, the Cmax ratio of hydroxybupropion: bupropion increased 2.2-fold (P = 0.00004). Bupropion AUC0–∞ and Cmax decreased by 55% and 34%, respectively (P < 0.002). Hydroxybupropion AUC0–∞ was unchanged, whereas Cmax increased by 50% (P = 0.008). Mean bupropion and hydroxybupropion plasma concentration vs. time curves before and after efavirenz administration are shown in Figures 1 and 2. All the subjects had a decrease in bupropion AUC0–∞ after 2 weeks of efavirenz, as demonstrated in Figure 3A. The effect on hydroxybupropion AUC0–∞ was largely heterogeneous, with 6 subjects experiencing an increase and 7 subjects a decrease after efavirenz administration (Δ AUC0–∞ range −13,031 to 13,143 ng h/mL).
TABLE 1.
Bupropion and Hydroxybupropion PK Parameter Values Before and After 2 Weeks of EFV Administration*
| PK Parameter | Phase 1 Bupropion | Phase 2 Bupropion + EFV | GMR (90% CI) | P |
|---|---|---|---|---|
| Bupropion | ||||
| AUC0−∞ (ng h/mL) | 673 (289) | 304 (152) | 0.45 (0.52 to 0.38) | 0.00002 |
| Cmax (ng/mL) | 81.7 (26.0) | 54.3 (24.2) | 0.66 (0.53 to 0.79) | 0.001 |
| Tmax (h)† | 2.5 (1–3) | 2.5 (2–3.5) | — | 0.28 |
| T1/2 (h) | 8.62 (4.31) | 4.69 (6.22) | 0.54 (0.33 to 0.75) | 0.045 |
| Cl/F (L/h) | 223 (134) | 493 (262) | 2.2 (1.8 to 2.6) | 0.00001 |
| Hydroxybupropion | ||||
| AUC0−∞ (ng h/mL) | 16,000 (5745) | 16,356 (5720) | 1.0 (0.81 to 1.2) | 0.83 |
| Cmax (ng/mL) | 423 (199) | 626 (253) | 1.5 (1.2 to 1.8) | 0.008 |
| Tmax (h)† | 6 (3.5–12) | 4 (2–8) | — | 0.014 |
| T1/2 (h) | 23.8 (9.03) | 15.8 (6.43) | 0.66 (0.58 to 0.74) | 0.0002 |
| Cl/F (L/h) | 9.38 (4.99) | 9.17 (6.20) | 0.98 (0.77 to 1.18) | 0.98 |
| Hydroxybupropion:bupropion | ||||
| AUC0−∞ ratio | 23.8 (12.6) | 53.8 (33.3) | 2.3 (1.8 to 2.7) | 0.0001 |
| Cmax ratio | 5.17 (2.30) | 11.5 (5.74) | 2.2 (1.7 to 2.7) | 0.00004 |
CI, confidence interval; Cl/F, apparent oral clearance; EFV, efavirenz; GMR, geometric mean ratio.
Values are geometric means (SD).
Median (range).
FIGURE 1.
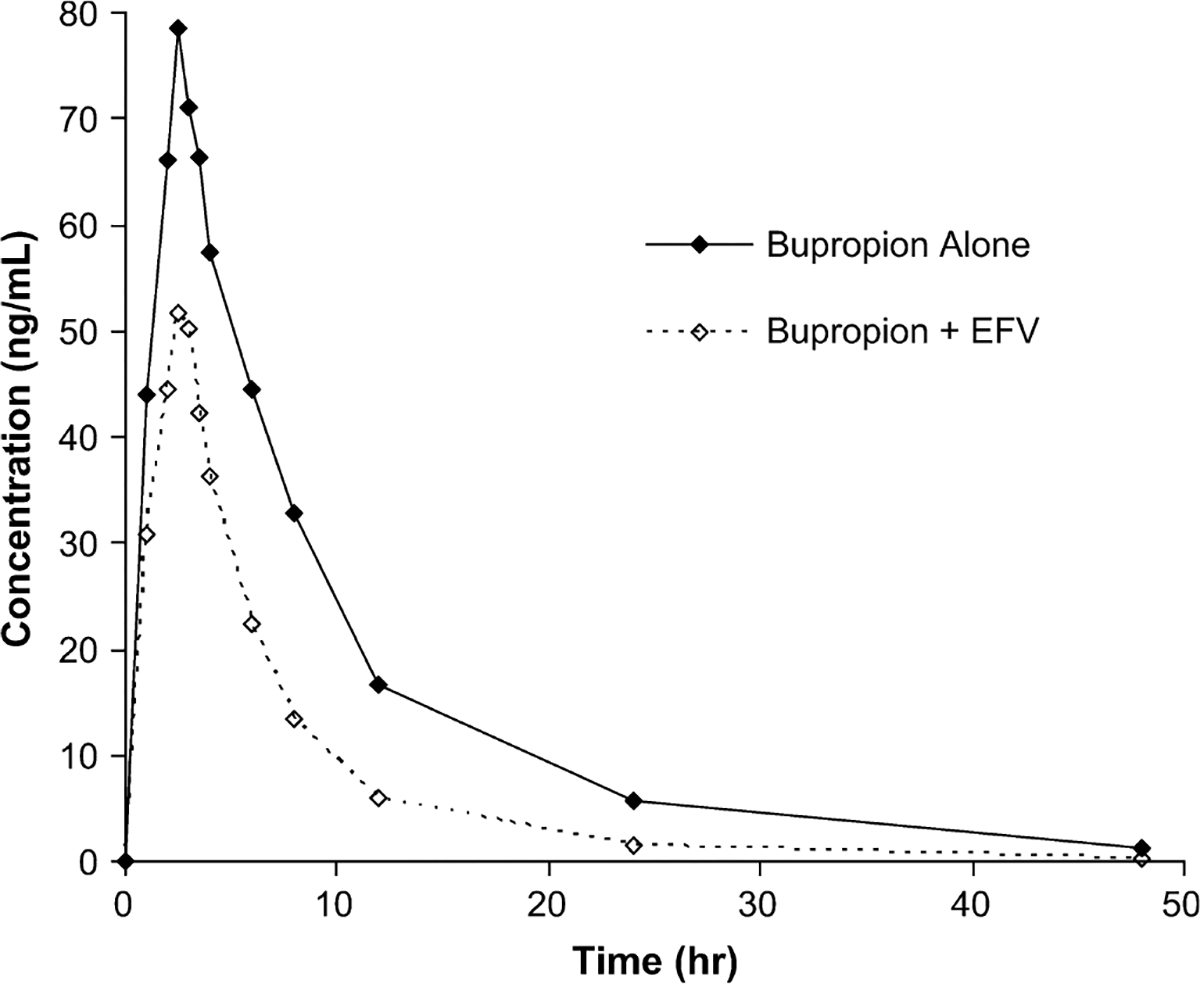
Mean bupropion plasma concentrations vs. time before and after EFV administration. EFV, efavirenz.
FIGURE 2.
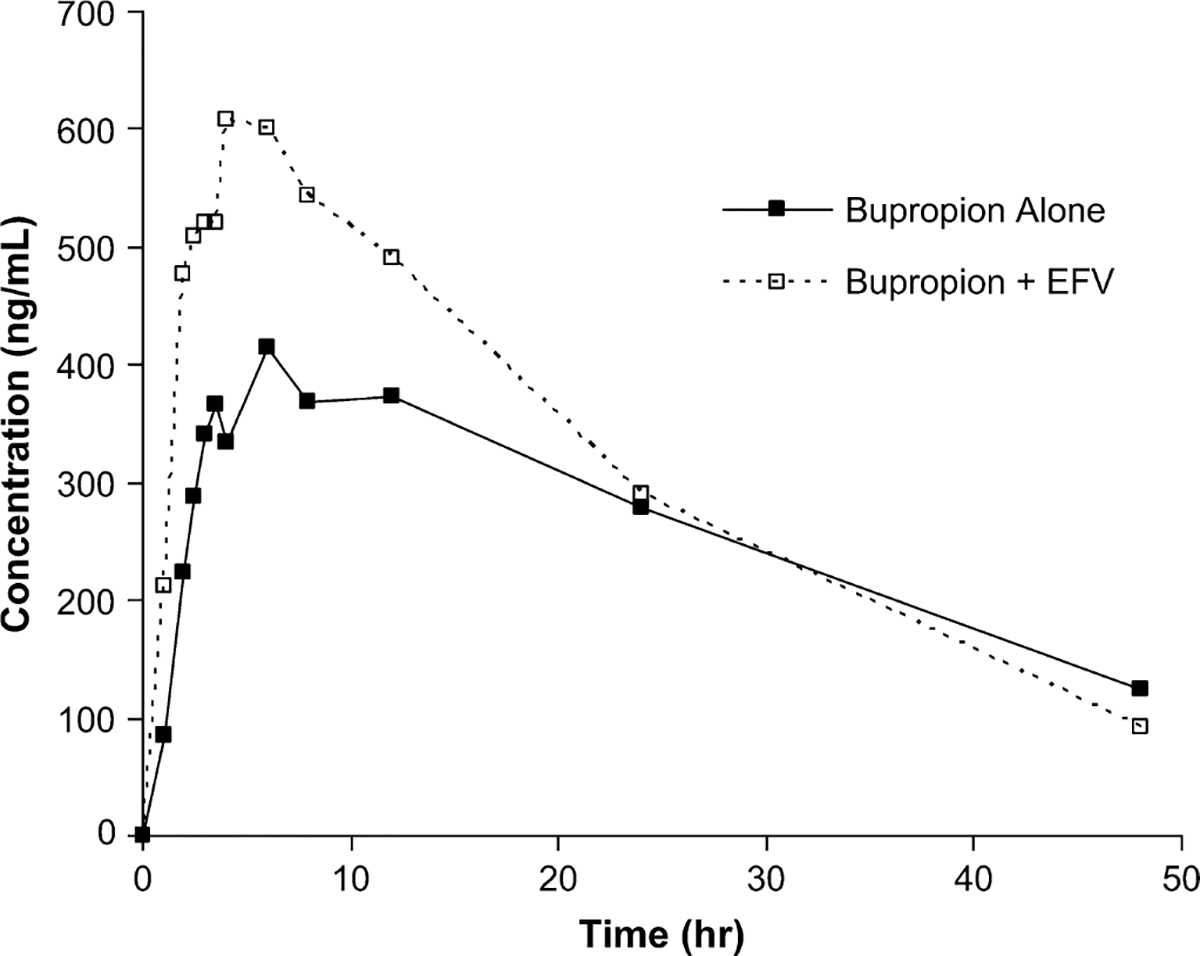
Mean hydroxybupropion plasma concentrations vs. time before and after EFV administration. EFV, efavirenz.
FIGURE 3.
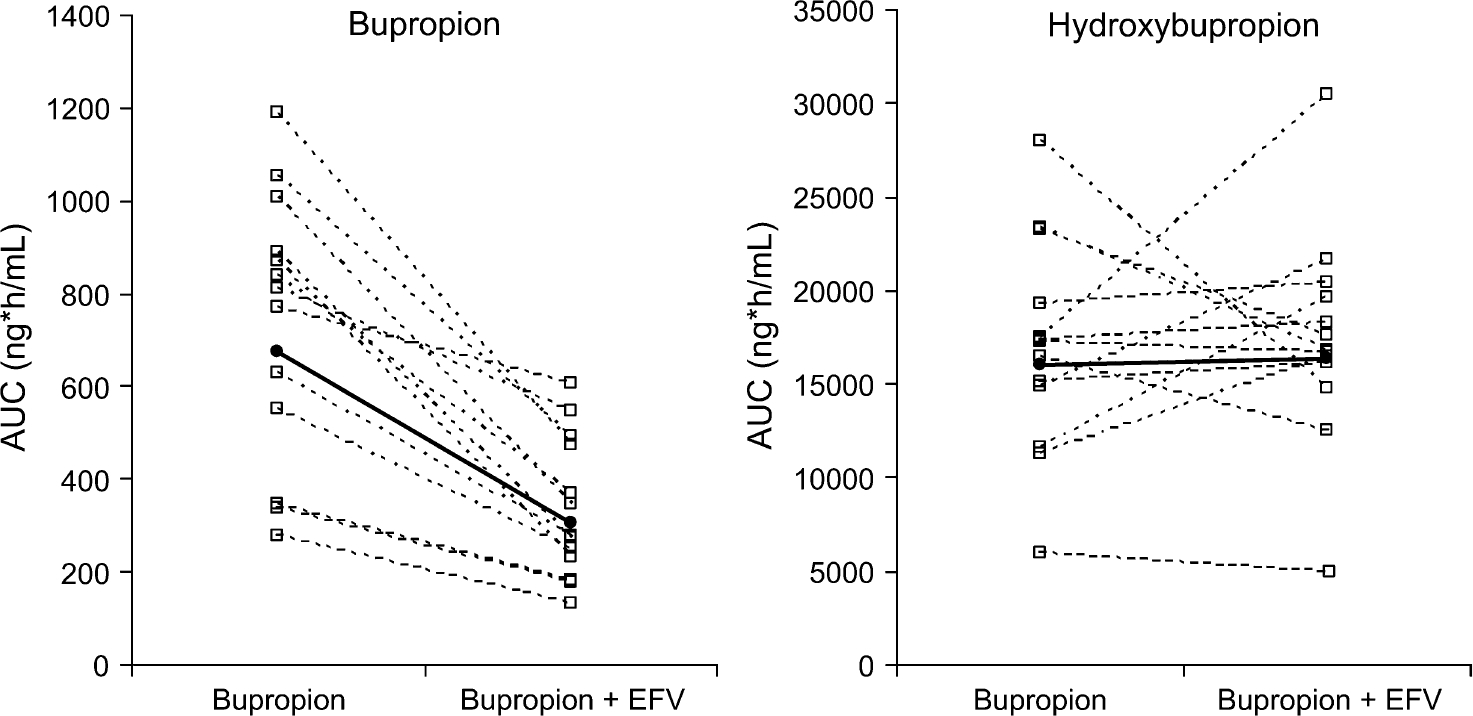
Individual (—□—) and mean (—●—) AUC0–∞ values for bupropion and hydroxybupropion before and after EFV administration. EFV, efavirenz.
Efavirenz PK
The efavirenz exposure observed in this study (AUC, Cmax) is similar to what has been reported previously in HIV patients (Table 2).23,24 The relatively large interpatient variability in efavirenz PK parameters is also consistent with previous observations.25,26
TABLE 2.
EFV PK Parameter Values at Steady State (n = 12)*
| PK Parameter | Geometric Mean | % CV |
|---|---|---|
| AUC0−48 (μg h/mL) | 74.6 | 43 |
| Cmax (μg/mL) | 4.60 | 34 |
| Tmax (h)† | 2.00 | 0.00, 4.00 |
| T1/2 (h) | 30.0 | 46 |
| Cl/F (L/h) | 9.47 | 42 |
Cl/F, apparent oral clearance; CV, coefficient of variation; EFV, efavirenz.
One subject was not included in the EFV PK analysis due to early discontinuation of efavirenz.
Median (range).
Safety
Bupropion was well tolerated with no reported adverse events. Adverse effects attributed to efavirenz were mostly grade 1 central nervous system toxicities, such as vivid dreams, decreased concentration, headache, and dizziness (n = 6). Grade 1 or 2 rash was reported in 3 subjects (all females). The one subject with grade 2 rash discontinued efavirenz early, after 12 days of administration, and was treated with fexofenadine and topical hydrocortisone. However, she was not withdrawn from the study. Because the efavirenz was discontinued only 1 day early, phase 2 of bupropion PK sampling was carried out as planned the following day. There were no abnormal laboratory findings reported for any of the subjects.
Pharmacogenetics
The distribution of genetic variation among subjects is displayed in Table 3. In general, the subjects were largely similar with respect to the genetic variants evaluated. There were no differences in bupropion clearance or exposure observed among the variants in CYP2B6, CYP3A4, or CYP3A5, either before or after concomitant efavirenz. Likewise, there were no differences in efavirenz clearance or exposure among CYP2B6, CYP3A4, or CYP3A5 variants. The 2 subjects homozygous for MDR1 3435-T/T had a 2.5-fold greater bupropion clearance (P = 0.047) and a 1.8-fold greater efavirenz clearance (P = 0.0009) relative to 3435-C/C and C/T individuals. In addition, the AUC ratio of hydroxybupropion:bupropion was ~50% lower in these 2 subjects (P = 0.0004). The magnitude of the difference in their bupropion PK was unchanged after efavirenz administration.
TABLE 3.
Genetic Polymorphisms in Healthy Subjects (N = 13)
| Genotype | n (%) | ||
|---|---|---|---|
| CYP2B6 | |||
| G516T | A785G | C1459T | |
| GG | AA | CT | 7 (54) |
| GT | AG | CT | 6 (46) |
| CYP3A4*1B (A-392G) | |||
| AA | 11 (85) | ||
| AG | 2 (15) | ||
| CYP3A5*3 (A6986G) | |||
| GG | 8 (61.5) | ||
| AG | 4 (31) | ||
| AA | 1 (7.5) | ||
| MDR1 (C3435T) | |||
| CC | 5 (39) | ||
| CT | 6 (46) | ||
| TT | 2 (15) | ||
DISCUSSION
The effect of efavirenz administration on CYP2B6 metabolism in vivo has not previously been reported. Studies have demonstrated the potential for efavirenz to modulate CYP2B6 based on in vitro findings of either metabolic inhibition or induction, depending on the study methodology employed.7–9 Efavirenz is already well characterized as a potent inducer of CYP3A4 metabolism. As CYP3A4 and CYP2B6 are coinduced via transcriptional activation by many of the same compounds, including rifampin, phenobarbital, phenytoin, and clotrimazole,27 it might be reasonably hypothesized that efavirenz induces both CYP3A4 and CYP2B6. Findings by Faucette et al28 support this supposition. The investigators reported that efavirenz, carbamazepine, and nevirapine preferentially activated human constitutive androstane receptor relative to human pregnane X receptor in vitro. Further, the 3 compounds induced CYP2B6 mRNA expression in primary human hepatocytes to a greater magnitude than that of CYP3A4. Nevirapine and carbamazepine have already been established as clinically relevant inducers of CYP2B6 metabolism.29,30
The conversion of bupropion to hydroxybupropion is a validated probe reaction for assessing CYP2B6 activity in vivo. The AUC ratio of hydroxybupropion:bupropion observed at baseline in our study (23.8) is consistent with data from previous studies (range 12.5–41).31–34 After 2 weeks of efavirenz therapy, the AUC ratio increased significantly from 23.8 to 53.8. This finding confirms that efavirenz is an inducer of CYP2B6 metabolism in humans when administered at a therapeutic dose (600 mg daily).
The efficacy of bupropion in smoking cessation is directly related to plasma concentrations of bupropion and its metabolites.35,36 Thus, a 55% decrease in bupropion exposure may have clinical consequences for individuals using bupropion for smoking cessation while on concomitant efavirenz. The pharmacologic activity of hydroxybupropion is about half as potent as bupropion.37 Because hydroxybupropion AUC was unchanged, it is uncertain to what degree the efficacy of bupropion may be impacted by efavirenz. Other CYP2B6 substrates whose plasma concentrations, and thus therapeutic efficacy, may be altered by efavirenz include cyclophosphamide, methadone, ifosfamide, selegiline, meperidine, sertraline, and propofol. Further study is necessary to determine whether interactions between these agents and efavirenz are clinically significant.
A similarly designed study by Loboz et al38 evaluated the effect of 7 days of rifampin therapy on bupropion hydroxylation in healthy white and Chinese subjects. The investigators reported a 1.8-fold increase in the hydroxybupropion:bupropion AUC ratio after rifampin administration—slightly lower than the 2.3-fold increase observed in our study. A cross-study comparison would suggest that efavirenz may be a more potent inducer of CYP2B6 than rifampin. Consistent with this observation, Faucette et al28 reported that efavirenz exhibited slightly greater induction of CYP2B6 relative to rifampin in cultured human hepatocytes.
In both our study and that by Loboz et al,38 hydroxybupropion Cmax increased after administration of bupropion with a CYP2B6 inducer—a 50% increase with efavirenz and a 40% increase with rifampin administration. Hydroxybupropion AUC0–∞ decreased approximately 40% after administration of rifampin but was unchanged in our study. Loboz et al38 theorize that rifampin may also increase the elimination of hydroxybupropion, though the route of hydroxybupropion elimination has not been clearly delineated. Our study findings suggest that efavirenz may also induce hydroxybupropion elimination, though not to the extent of rifampin. Despite a 100% increase in bupropion clearance, hydroxybupropion AUC was unchanged from phase 1 to phase 2; one would have expected an increase in hydroxybupropion AUC. Although hydroxybupropion clearance was not increased by efavirenz, the half-life did decrease significantly from ~24 to 16 hours. The putative mechanism may be induced glucuronidation of hydroxybupropion.39 It is necessary to determine the precise route of hydroxybupropion elimination to better define the role of bupropion hydroxylation as a phenotypic marker for CYP2B6 induction in future studies.
Our study included an exploratory analysis of the relationship between CYP2B6, CYP3A4, CYP3A5, and MDR1 genotypes and bupropion and efavirenz PK. As 9 of the 13 enrolled subjects were whites, it is not surprising that subjects were largely homologous with respect to the genetic variants evaluated. Our findings did not reveal a difference in efavirenz or bupropion PK between individuals heterozygous for CYP2B6 516-G/T and 785-A/G, as compared with G/G and A/A homozygotes. None of the subjects were homozygous for 516-T/T, 785-G/G, or 1459-T/T. Had we enrolled a larger, more diverse subject population, it would have been more likely that a difference in bupropion or efavirenz PK may have been detected between CYP2B6 genotypes. There were also no differences in bupropion or efavirenz PK observed for either CYP3A4*1B or CYP3A5*3. However, a previous, larger study demonstrated a weak association between both genetic variants and plasma efavirenz exposure.15
An MDR1 variant, 3435-TT, is associated with lower P-glycoprotein expression relative to C/C and C/T. Though only 2 subjects were homozygous for T/T, both bupropion and efavirenz clearance were significantly greater in these subjects vs. C/C and C/T individuals. Bupropion and efavirenz AUC and Cmax values were also significantly lower in the T/T homozygotes. The role of P-glycoprotein in efavirenz distribution and elimination remains controversial. However, consistent with our findings, previous studies have demonstrated a significant increase in efavirenz clearance and/or a decrease in exposure in 3435-T/T relative to C/T and C/C genotypes.19,40
In conclusion, results from the current study indicate that efavirenz markedly induces CYP2B6 activity in human subjects. The magnitude of this induction is predictive of a potentially clinically relevant interaction between efavirenz and bupropion and possibly between efavirenz and other CYP2B6 substrates. Further studies are necessary to examine this hypothesis in greater detail. Until these data become available, clinicians should use caution when administering a CYP2B6 substrate in combination with efavirenz because the coadministered drug may undergo enhanced metabolism and reduced therapeutic efficacy.
Acknowledgments
Supported by the Society of Infectious Diseases Pharmacists/Pfizer Award for Research in Infectious Disease Pharmacotherapy; awarded December 2005.
Footnotes
Preliminary data were presented at the following meeting: Efavirenz induces CYP450 2B6 activity as measured by bupropion hydroxylation in healthy subjects. Poster presented at 5th Conference on Retroviruses and Opportunistic Infections, February 5, 2008, Boston, MA.
REFERENCES
- 1.Hesse LM, Venkatakrishnan K, Court MH, et al. CYP2B6 mediates the in vitro hydroxylation of bupropion: potential drug interactions with other antidepressants. Drug Metab Dispos. 2000;28:1176–1183. [PubMed] [Google Scholar]
- 2.Roy P, Yu LJ, Crespi CL, et al. Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based on cDNA-expressed activities and liver microsomal P-450 profiles. Drug Metab Dispos. 1999; 27:655–666. [PubMed] [Google Scholar]
- 3.Kharasch ED, Hoffer C, Whittington D, et al. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76: 250–269. [DOI] [PubMed] [Google Scholar]
- 4.Obach RS, Cox LM, Tremaine LM. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: an in vitro study. Drug Metab Dispos. 2005;33: 262–270. [DOI] [PubMed] [Google Scholar]
- 5.Ramirez J, Innocenti F, Schuetz EG, et al. CYP2B6, CYP3A4, and CYP2C19 are responsible for the in vitro N-demethylation of meperidine in human liver microsomes. Drug Metab Dispos. 2004;32:930–936. [PubMed] [Google Scholar]
- 6.Ward BA, Gorski JC, Jones DR, et al. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther. 2003;306:287–300. [DOI] [PubMed] [Google Scholar]
- 7.Hesse LM, von Moltke LL, Shader RI, et al. Ritonavir, efavirenz, and nelfinavir inhibit CYP2B6 activity in vitro: potential drug interactions with bupropion. Drug Metab Dispos. 2001;29:100–102. [PubMed] [Google Scholar]
- 8.Bumpus NN, Kent UM, Hollenberg PF. Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J Pharmacol Exp Ther. 2006;318:345–351. [DOI] [PubMed] [Google Scholar]
- 9.Faucette SR, Zhang TC, Moore R, et al. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zyban® (bupropion hydrochloride) Sustained-Release Tablets, Prescribing Information [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2007. [Google Scholar]
- 11.Faucette SR, Hawke RL, Lecluyse EL, et al. Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab Dispos. 2000;28:1222–1230. [PubMed] [Google Scholar]
- 12.Zanger UM, Klein K, Saussele T, et al. Polymorphic CYP2B6: molecular mechanisms and emerging clinical significance. Pharmacogenomics. 2007;8:743–759. [DOI] [PubMed] [Google Scholar]
- 13.Desta Z, Saussele T, Ward B, et al. Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics. 2007;8: 547–558. [DOI] [PubMed] [Google Scholar]
- 14.Lang T, Klein K, Fischer J, et al. Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics. 2001;11:399–415. [DOI] [PubMed] [Google Scholar]
- 15.Haas DW, Ribaudo HJ, Kim RB, et al. Pharmacogenetics of efavirenz and central nervous system side effects: an Adult AIDS Clinical Trials Group study. AIDS. 2004;18:2391–2400. [PubMed] [Google Scholar]
- 16.Tsuchiya K, Gatanaga H, Tachikawa N, et al. Homozygous CYP2B6*6 (Q172H and K262R) correlates with high plasma efavirenz concentrations in HIV-1 patients treated with standard efavirenz-containing regimens. Biochem Biophys Res Commun. 2004;319:1322–1326. [DOI] [PubMed] [Google Scholar]
- 17.Rotger M, Colombo S, Furrer H, et al. Influence of CYP2B6 polymorphism on plasma and intracellular concentrations and toxicity of efavirenz and nevirapine in HIV-infected patients. Pharmacogenet Genomics. 2005;15:1–5. [DOI] [PubMed] [Google Scholar]
- 18.Wang D, Sonnerborg A, Rane A, et al. Identification of a novel specific CYP2B6 allele in Africans causing impaired metabolism of the HIV drug efavirenz. Pharmacogenet Genomics. 2006;16:191–198. [DOI] [PubMed] [Google Scholar]
- 19.Fellay J, Marzolini C, Meaden ER, et al. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet. 2002;359:30–36. [DOI] [PubMed] [Google Scholar]
- 20.Eiselt R, Domanski TL, Zibat A, et al. Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics. 2001;11:447–458. [DOI] [PubMed] [Google Scholar]
- 21.Tsuchiya N, Satoh S, Tada H, et al. Influence of CYP3A5 and MDR1 (ABCB1) polymorphisms on the pharmacokinetics of tacrolimus in renal transplant recipients. Transplantation. 2004;78:1182–1187. [DOI] [PubMed] [Google Scholar]
- 22.Kurata Y, Ieiri I, Kimura M, et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin Pharmacol Ther. 2002;72:209–219. [DOI] [PubMed] [Google Scholar]
- 23.Sustiva® (efavirenz) Capsules and Tablets, Prescribing Information [package insert] Princeton, NJ: Bristol-Myers Squibb Company; 2007. [Google Scholar]
- 24.López-Cortés LF, Ruiz-Valderas R, Marín-Niebla A, et al. Therapeutic drug monitoring of efavirenz: trough levels cannot be estimated on the basis of earlier plasma determinations. J Acquir Immune Defic Syndr. 2005;39:551–556. [PubMed] [Google Scholar]
- 25.Barrett JS, Joshi AS, Chai M, et al. Population pharmacokinetic metaanalysis with efavirenz. Int J Clin Pharmacol Ther. 2002;40:507–519. [DOI] [PubMed] [Google Scholar]
- 26.Pfister M, Labbe L, Hammer SM, et al. Population pharmacokinetics and pharmacodynamics of efavirenz, nelfinavir and indinavir: Adult AIDS Clinical Trial Group Study 398. Antimicrob Agents Chemother. 2003;47: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faucette SR, Wang H, Hamilton GA, et al. Regulation of CYP2B6 in primary human hepatocytes by prototypical inducers. Drug Metab Dispos. 2004;32:348–358. [DOI] [PubMed] [Google Scholar]
- 28.Faucette SR, Zhang TC, Moore R, et al. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viramune® (nevirapine) Tablets, Prescribing Information [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc; 2007. [Google Scholar]
- 30.Tegretol® (carbamazepine USP), Prescribing Information [package insert]. East Hanover, NJ; Novartis Pharmaceuticals Corp; 2007. [Google Scholar]
- 31.Turpeinen M, Tolonen A, Uusitalo J, et al. Effect of clopidogrel and ticlopidine on cytochrome P450 2B6 activity as measured by bupropion hydroxylation. Clin Pharmacol Ther. 2005;77:553–559. [DOI] [PubMed] [Google Scholar]
- 32.Palovaara S, Pelkonen O, Jouko U, et al. Inhibition of cytochrome P450 2B6 activity by hormone replacement therapy and oral contraceptive as measured by bupropion hydroxylation. Clin Pharmacol Ther. 2005;74: 326–333. [DOI] [PubMed] [Google Scholar]
- 33.Hogeland GW, Swindells S, McNabb JC, et al. Lopinavir/ritonavir reduces bupropion plasma concentrations in healthy subjects. Clin Pharmacol Ther. 2007;81:69–75. [DOI] [PubMed] [Google Scholar]
- 34.Turpeinen M, Koivuviita N, Tolonen A, et al. Effect of renal impairment on the pharmacokinetics of bupropion and its metabolites. Br J Clin Pharmacol. 2007;64:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston JA, Fiedler-Kelly J, Glover ED, et al. Relationship between drug exposure and the efficacy and safety of bupropion sustained release for smoking cessation. Nicotine Tob Res. 2001;3:131–140. [DOI] [PubMed] [Google Scholar]
- 36.Johnston AJ, Ascher J, Leadbetter R, et al. Pharmacokinetic optimisation of sustained-release bupropion for smoking cessation. Drugs. 2002;62: 11–24. [DOI] [PubMed] [Google Scholar]
- 37.Jefferson JW, Pradko JF, Muir KT. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin Ther. 2005;27:1685–1695. [DOI] [PubMed] [Google Scholar]
- 38.Loboz KK, Gross AS, Williams KM, et al. Cytochrome P450 2B6 activity as measured by bupropion hydroxylation: effect of induction by rifampin and ethnicity. Clin Pharmacol Ther. 2006;80:75–84. [DOI] [PubMed] [Google Scholar]
- 39.Haynes K, Faucette SR, Lindley CM, et al. Evaluation of bupropion hydroxylation as a probe of cytochrome P450 2B6 activity in cultured human hepatocytes. Paper presented at: American College of Clinical Pharmacy; October 20–23, 2002; Savannah, GA. Abstract 165. [Google Scholar]
- 40.Csajka C, Marzolini C, Fattinger K, et al. Population pharmacokinetics and effects of efavirenz in patients with human immunodeficiency virus infection. Clin Pharmacol Ther. 2003;73:20–30. [DOI] [PubMed] [Google Scholar]