

Abstract
This work discusses the possibility of designing multilayer oil-in-water emulsions to introduce the maximum possible amount of an antioxidant at the droplet interfaces for the optimal protection of a linseed oil core against oxidation, using a systematic three-step colloidal procedure. An antioxidant (here Tannic Acid – TA) is chosen and its interactions with a primary emulsifier (here Bovine Serum Albumin – BSA) and several polysaccharides are first examined in solution using turbidity measurements. As a second step, LbL deposition on solid surfaces is used to determine which of the polysaccharides to combine with BSA and tannic acid in a multilayer system to ensure maximum presence of tannic acid in the films. From UV-vis and polarization modulation infrared reflection–absorption (PM-IRRAS) spectroscopic measurements it is suggested that the best components to use in a multilayer emulsion droplet, together with BSA and TA, are chitosan and pectin. BSA, chitosan and pectin are subsequently used for the formation of three-layer linseed oil emulsions, and tannic acid is introduced into any of the three layers as an antioxidant. The effect of the exact placement of tannic acid on the oxidative stabilization of linseed oil is assessed by monitoring the fluorescence of Nile red, dissolved in the oil droplets, under the attack of radicals generated in the aqueous phase of the emulsion. From the results it appears that the three-stage procedure presented here can serve to identify successful combinations of interfacial components of multilayer emulsions. It is also concluded that the exact interfacial placement of the antioxidant plays an important role in the oxidative stabilization of the valuable oil core.
Three-step method optimizes multilayer emulsion for maximum tannic acid (TA) amount at surfaces. (1) TA–emulsifier bulk interactions assessed. (2) LbL films built for optimal TA presence. (3) Emulsions built as per LbL design and TA action evaluated.
1. Introduction
Modern food technology depends on intelligent formulation systems for the medium to long-term storage of sensitive food ingredients, the protection of ingredients from oxidation, the incorporation of important nutraceuticals into functional foods, and the guided release of the encapsulated active material. Several storage and delivery media have been exploited in the food industry, including various emulsion types, protein and polymer nanoparticles, hydrogels, and lipid aggregates.1–3 Innovative emulsion types, such as multiple emulsions, nanoemulsions, and multilayer emulsions (MEs) have appeared.2,4–8 MEs are a relatively recent idea in food encapsulation.2,5,7,9–11 They are based on the layer-by-layer deposition of polyelectrolytes, first applied to solid surfaces.12–14 Multilayers of alternatively charged polyelectrolytes can be stable on curved particle surfaces,15 leading to smart nanocapsules.16 It was a natural step to apply them to emulsion droplet surfaces, preventing emulsion coalescence and creaming through electrostatic and steric stabilization. Polyelectrolyte nanocapsules attracted immediately the attention of food technologists.1–3,5–7,9,11,17,18 MEs are attractive to researchers because their interfacial composition is extremely versatile, which allows to stabilize the emulsions under broad ranges of pH, salinity and temperature, not easily attainable with conventional emulsions.9,19–21 They are however tedious to prepare, cannot contain large volumes of the oil phase due to instabilities, and are expensive to produce in large volumes, and as such can be technologically useful only for valuable core materials.9
In the present work the focus is on MEs of oils rich in the omega-3 fatty acids docosahexanoic acid (DHA) and eicosapentaenoic acid (EPA). These compounds have attracted much attention since their beneficial effects on patients with cardiovascular diseases were found.22 Despite recent scepticism about their positive effects,23 a huge market of dietary supplements has been built around omega-3 fatty acids, especially around fish-oil and linseed oil rich in EPA and DHA. Unfortunately, these unsaturated fatty acids are easily oxidized. To circumvent this problem food technology has turned to encapsulation methods, some of which are based on MEs. It has been found that the nature of the wall material plays an important role in the oxidative stabilization of fish oil.24–27 In general however, additional antioxidants must be incorporated in ME formulations as one cannot rely solely on the antioxidant protection of the interfacial layers for long-term stability. In addition, in certain methods used for the long-term storage of omega-3-rich oil phases, such as spray-drying,28–31 one should rely on antioxidants located preferentially on the droplet walls, as one cannot rely on the protective action of molecules dissolved in the continuous phase. In recent years, researchers are trying to assess if the localization of antioxidants at the emulsion interfaces is advantageous.32,33 Several groups have examined the effect of localization on antioxidant action, and have often found surface-active antioxidants advantageous.34–36 The idea of localizing antioxidants at the walls of the emulsion droplets by taking advantage of their complexes with appropriate wall materials has only recently begun to be exploited in the literature.32,33,37–39 In principle, this would best be achieved by covalently binding the antioxidant to the biopolymer; a few such attempts have indeed been made.21,36,38,40 In the present work, however, it is attempted to enhance the interfacial placement of antioxidant molecules in MEs by intermolecular forces alone, using a systematic sequential approach, based on the principles of colloid and interface science, which is described in Fig. 1:
Fig. 1. Use of colloid science principles for the systematic design of MEs with maximum placement of antioxidants at the droplet shells.

In this three-step colloidal approach, one first chooses an antioxidant that is not soluble in the oil phase and is preferably surface-active. First, it is verified that the chosen antioxidant forms strong “physical” complexes in solution with the various biopolymers that can be components of the interfacial multilayers of the ME droplets. This is achieved by measuring antioxidant–biopolymer interactions in aqueous solutions, and carrying out various measurements, and in particular measuring turbidity to probe potential larger-scale aggregation phenomena, which must be carefully assessed before using materials for emulsion-wall formation. Second, a layer-by-layer methodology on solid surfaces is used to prove that the antioxidant–biopolymer complexes will form high-quality layer-by-layer structures with the other selected biopolymers on flat solid surfaces. The successful formation of multilayer structures is monitored using UV-vis and PM-IRRAS (polarization modulation infrared reflection–adsorption) spectroscopy. The use of PM-IRRAS for interfacial component quantification is an innovation, since it is not typically applied to layer-by-layer films of biopolymers. It is a powerful method, which is mostly applied to lipid and biopolymer monolayers at water surfaces,41 and very infrequently used for polymer or lipid layers on electrodes.42–44 The LbL work serves to choose the best layer materials by limiting the large number of potential choices to those systems that demonstrate the best synergy at a solid surface. Finally, MEs with the chosen components in combination with the antioxidant in various potential positions are built. The antioxidant action of the ME layers is examined by monitoring the fluorescence decay of Nile red, solubilized in the oil droplets (ORAC method,36). Other possible ways to probe the antioxidant action are not used in this work, because the focus is really on the oxidation barrier at the surface of the droplets, where it is hoped that much of the antioxidant will be located.
The success of this three-step approach strongly depends on the selection of a suitable antioxidant. Recent innovative work45–47 suggests that tannic acid, an efficient natural antioxidant, may be used as a wall component in conjunction with proteins and cationic polyelectrolytes, and leads to excellent emulsion stabilization. Tannic acid is a large, flexible molecule, which is both water-soluble and interfacially active.48,49 Tannic acid is the ideal antioxidant to validate the colloidal approach presented in Fig. 1 for many reasons: (a) it is a molecule that can be obtained in pure form and in large quantities from natural sources. (b) It is known to form strong, even insoluble complexes with a variety of proteins,50 including those used as primary emulsifiers in MEs.51–56 It also interacts quite strongly with many polysaccharides that are used at ME interfaces, such as chitosan,57 pectin,58 alginate,59 and carboxymethylcellulose.60 In many of the quoted works, tannic acid has been used as a cross-linker of proteins and polysaccharides for the formation of stable nanoparticles. Given the information in the literature, it is not necessary to prove the interaction of TA with proteins and polysaccharides in solution, which is an important part of the necessary first step in Fig. 1! (c) Tannic acid is known to form robust multilayers with a large variety of polymers and proteins on solid substrates using the layer-by-layer method adopted in the present investigation.61–69 (d) Good antioxidant activity has been reported for multilayer films67,69 and for emulsions54,70–72 containing tannic acid. (e) The ME work mentioned before proves that tannic acid can be a valuable component of MEs simultaneously providing interfacial stabilization and antioxidant protection.45–47
A central goal in the present work is to probe if and how the location of the antioxidant (here of tannic acid) at the droplet interface of a ME affects the stability of the encapsulated oil towards oxidation. To examine this issue, linseed oil as the omega-3-rich oil is used to produce MEs, using BSA as the primary emulsifier and various polysaccharides as the layer materials. Tannic acid is used in conjunction with BSA, or with polysaccharides in any of the layers. The colloidal approach of Fig. 1 suggests the optimal polysaccharides for ME formulation (from a set of four different molecules), which turn out to be chitosan and pectin, a combination that has been used before successfully for the formation of MEs.73,74
To summarize, the first goal of this work is to prove the general principle that formulations based on strong antioxidant–biopolymer complexation interactions in the bulk, which give high quality LbL films on surfaces with optimal antioxidant loading, are also highly successful in stabilizing the oil component of MEs towards oxidation. The second major goal is to prove that the localization of tannic acid at the ME droplet surfaces at well-defined molar ratios to the ME wall components is important and may fine-tune the antioxidant properties of the interfaces. In the end, an efficient way of generating ME systems with excellent antioxidant action from rationally predetermined sets of suitable wall components will be demonstrated.
2. Materials and methods
2.1. Materials
Monobasic and dibasic potassium phosphate and tannic acid (TA) were procured from Sigma-Aldrich. 2,2′-Azobis-2-methylpropanimidamide dihydrochloride (AAPH) was obtained from Sigma-Aldrich. Glacial acetic acid, sodium acetate and chloroform were purchased from Merck. Nile red was obtained from ACROS and used without purification. Bovine serum albumin Fraction V (BSA 66.5 kDa) was obtained from Roche Diagnostics. Sodium alginate (SA) was from Sigma-Aldrich (W201502) and is reported (personal communication) to have a molecular weight distribution from 1.2 to 40 kDa. Chitosan (CH) (low molecular weight, estimated at 120 kDa with a distribution of 50 kDa and medium molecular weight, estimated at 250 kDa) was purchased from Sigma Aldrich. Chondroitin sulfate sodium salt (CS) (CAS-9007-28-7) was obtained from Xi'an Huisun Bio-Tech Co. Ltd., China. It was analyzed by GPC and has an average molecular weight of 150 kDa. Pectin (PE) was from ACROS (lot A0388115); it has an esterification degree of about 70% and an approximate molecular weight of 150 kDa (personal communication). Linseed oil (KTC – 100% Pure Cold-Pressed Linseed/Flaxseed Oil) was purchased from a local supermarket. All experiments were performed at pH = 4 and pH = 7 using 0.01 M acetate and phosphate buffer solutions. When necessary, the pH was adjusted to the desired value by the addition of HCl or NaOH. All chemicals were used without further purification.
2.2. Methods
2.2.1. Turbidity
Solutions of TA and proteins or polysaccharides were prepared in acetate (pH = 4) and phosphate (pH = 7) buffer solutions at concentrations in the range 7.5 × 10−6 M to 6.0 × 10−5 M for BSA, 6.25 × 10−6 M for chitosan, 4.0 × 10−6 M for pectin, 3.85 × 10−5 M for sodium alginate and 5.71 × 10−6 M for chondroitin sulfate. Chitosan solutions were prepared only at pH = 4 in acetate buffer. Various amounts of the stock solution of tannic acid (1.8 × 10−3 M) and buffer solution as needed were respectively added into the protein solution (0.5 mL) to a total volume of 1.5 mL, in order to vary the final molar ratio of tannic acid to protein or polysaccharide. After 5 min shaking at room temperature, the absorbance of the mixture was recorded (UV-1900 Shimadzu spectrophotometer) at 600 nm.
2.2.2. Layer-by-layer formation and study using UV-vis and PM-IRRAS spectroscopy
2.2.2.1. Multilayer preparation
Regarding the deposition process, the substrate was sequentially immersed in a solution of polysaccharide (1 g L−1) and in a solution made from stock solutions of tannic acid (1 g L−1) and BSA (1 g L−1), such that tannic acid is in 15-fold molar excess over BSA (n(TA)/n(BSA) = 15). Different molar ratios of TA to BSA from 7.5 to 30 have also been used in some experiments. The immersion speed was 100 mm s−1 and the plate removal speed was 25 mm s−1. The duration of each immersion was 15 minutes, followed by 15 minutes of air-drying at room temperature. Throughout the immersion the solutions were stirred continuously, as in the absence of stirring significant precipitation was observed from BSA/TA solutions with molar ratios of TA to BSA larger than 20. The immersion cycle was repeated to obtain up to 4 bilayers. The progress of protein/polysaccharide multilayer assembly on solid surfaces was monitored using UV-vis and PM-IRRAS spectroscopies. Following a typical deposition process, a multilayer film was deposited on a quartz plate (5 × 3 cm) for the UV-vis studies or on a gold-coated stainless steel plate for the PM-IRRAS studies, using a dip coating device (Nima Technologies). For the UV-vis studies, the quartz plates were first cleaned with aqueous nitric acid (50%) for 30 minutes, then rinsed with ultra-pure water, and then air-dried. For the PM-IRRAS studies, the plates were cleaned with ultra-pure water and ethanol.
2.2.2.2. Multilayer characterization
UV-vis spectra were obtained for each layer, following each immersion (UV-1900 spectrophotometer, Shimadzu). TA has an absorbance maximum at 305 nm, while BSA absorbs more strongly in the range 270–290 nm. The maximum absorbance thus depends on both TA and BSA, while the intermediate polysaccharide layers contribute very little around 300 nm. PM-IRRAS spectra were recorded using a KSV-NIMA instrument with a ZnSe photoelastic modulator (PEM) and a MCT detector. The rinsed and dried plates with the deposited films were mounted on a home-made sample holder. The wavelength setting of the PEM was fixed at 2500 cm−1. The sample was scanned for 300 s with 8 cm−1 resolution and the resulting absorption spectra were recorded between 1400 and 2000 cm−1. The incident beam angle used in all experiments was 83°, where the signal is maximized for the geometry used. Manipulations of the resulting spectra, including baseline correction and subtraction and spectral deconvolution, were performed using the Origin.Pro software.
Powder FTIR spectra of BSA, TA and chitosan were obtained with a JASCO 460 Plus FTIR spectrophotometer. The spectra were recorded using the KBr pellet technique, the sensitivity was 1 cm−1 and the scanning range was 1000–2000 cm−1.
2.2.3. ME formation and characterization
2.2.3.1. Method for stripping linseed oil
The aim of the stripping process was to remove endogenous tocopherols and other potential antioxidants present in commercial linseed oil.75 The stripping method used is based on the affinity chromatography principle and is solvent-free. 600 g of aluminum oxide were previously activated at 200 °C for at least 3 h, and then cooled down in a dessicator (high temperatures may promote oil oxidation during the stripping process). Then 700 g of the commercial linseed oil were stripped by passing the sample through the activated aluminum oxide (200 g) placed in a Büchner funnel. Vacuum was applied, with an initial pressure of 75 mbar, reduced progressively to 30 mbar. This process was repeated twice and the aluminum oxide was changed for each new passage.76
2.2.3.2. Preparation of multilayer emulsions
A primary emulsion of linseed oil was obtained by dispersing 10 mL linseed oil in 90 mL of the aqueous phase (BSA 4 g L−1 by itself or in the presence of tannic acid so that n(TA)/n(BSA) = 9, in a 0.01 M acetate buffer solution at pH = 4), and mixing with a simple laboratory mixer for 2 minutes at room temperature. The mixture was subsequently passed through a high-pressure homogenizer (EmulsiFlex-C5) three times at a pressure of 1000–1200 bar.
To prepare the secondary emulsions 30 mL of a polysaccharide solution (chitosan 0.5 g L−1 by itself or in the presence of tannic acid so that n(TA)/n(CH) = 130 in a 0.01 M acetate buffer at pH = 4) and 30 mL of the same acetate buffer solution were added to 30 mL of the primary emulsions and passed through the high-pressure homogenizer three times, at a pressure of 1000–1200 bar. Tannic acid was added in the secondary emulsion only if it was not already incorporated in the primary emulsion.
To form tertiary emulsions, 15 mL of the polysaccharide solution (pectin 0.25 g L−1 by itself or in the presence of tannic acid so that n(TA)/n(PE) = 90) with 45 mL of acetate buffer solution, were added to 30 mL of the secondary emulsions and were homogenized as described above. Tannic acid was added in the tertiary emulsion only if it was not already incorporated in the primary or secondary emulsions.
For all these three-layer emulsions, the final composition was BSA (0.44 g L−1), tannic acid (0.1 g L−1), chitosan (0.16 g L−1) and pectin (0.16 g L−1), based on total solution volume. The equality of total component concentrations is necessary for a direct comparison of the antioxidant activity of tannic acid in these emulsions. In some cases, pectin was used as the second layer of the MEs and chitosan as the third layer. The protocol was modified to ensure that the final ternary MEs contain the same quantities of BSA, TA and polysaccharides as those quoted above.
Oil-soluble, peroxyl-radical sensitive dye Nile red was added to the linseed oil before emulsification (250 μL of a 5 g L−1 chloroform solution of the dye to 10 mL linseed oil) followed by the multilayer shell assembly, as described.
2.2.3.3. Emulsion characterization
The stability of the multilayer emulsions towards creaming or flocculation was visually assessed over a period of 1–2 weeks, within which all relevant measurements of antioxidant action were performed. Following the high-pressure homogenization stage, the emulsions were very stable in most cases, with the exception of emulsions with pectin as the second layer (see Results section). Following the deposition of each new layer the size distribution of the emulsion droplets and their zeta potential were measured using a Zetasizer Nano ZS system (Malvern Instruments) at 25 °C. Prior to these measurements, all emulsions were diluted 2000 times with their respective buffer solution (at pH = 4 or pH = 7). Disposable polystyrene cuvettes were used, and each measurement was repeated three times.
2.2.3.4. Assessment of antioxidant action of the MEs
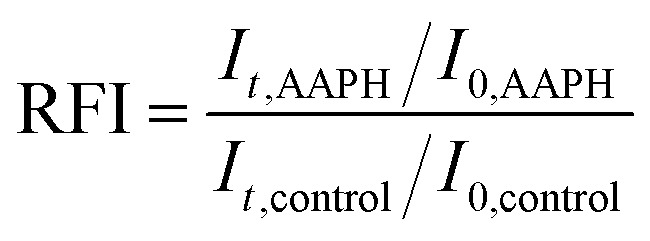
AAPH was selected as a peroxyl radical generator in the aqueous phase of emulsions. An AAPH solution was prepared by dissolving 0.1 g AAPH in 10 mL ultra-pure water. 2.75 mL of 2000-fold diluted emulsion samples were mixed with 0.25 mL of the AAPH solution. Immediately after addition of AAPH, the sample was placed in the Fluorescence Spectroscopy Instrument (Jasco FP-8300). Changes in the fluorescence intensity of Nile red were measured at a regular time interval of 10 seconds for a total of 12 hours. The excitation and emission wavelengths for the fluorescence measurements were 535 nm and 582 nm, respectively. The control sample, which consisted of 2.75 mL of the emulsion with 0.25 mL ultra-pure water, was measured at the same time. The relative fluorescence intensity (RFI) was calculated using the following equation:
 |
1 |
where It,AAPH is the fluorescence intensity of emulsion after ‘t’ min of exposure to AAPH, I0,AAPH is the fluorescence intensity of emulsion immediately after addition of AAPH, and It,control and I0,control are the corresponding quantities for the control solution. Duplicate or triplicate experiments were run for some emulsion wall combinations to check reproducibility.
3. Results and discussion
3.1. Turbidity
As mentioned before, TA interacts strongly with several proteins that are often used as primary emulsifiers in MEs, e.g. BSA, whey protein isolate (WPI), and sodium caseinate among others.51–56 It was decided to use BSA as a model emulsifier, because it has been used successfully in many emulsion formulations, in particular together with tannic acid.45–47 The interaction of tannic acid with polysaccharides or the disruption of protein–tannin interactions by polysaccharides is very significant, and has been documented in the literature in various contexts,57–60,77–80 although complete physicochemical evaluation of the interactions in solution is scarce in the case of polysaccharides. We have opted to use four different polysaccharide molecules in this work, which are chitosan (a polycation, being a linear copolymer of d-glucosamine and N-acetyl-d-glucosamine), chondroitin sulfate (a polyanion, being a sulfated glucosaminoglycan), pectin (a heteropolysaccharide with carboxylic groups, but source-dependent composition), and sodium alginate (a copolymer of β-d-mannuronic acid and α-l-guluronic acid).
Turbidity measurements were carried out keeping the protein or polysaccharide concentration fixed and changing tannic acid concentration. These measurements establish the concentration ranges above which large scale aggregation between TA and the biopolymers take place. It is an important exercise, because TA often serves as a crosslinking agent for proteins and polysaccharides. In Fig. 2 the turbidity at 600 nm is plotted as a function of the molar ratio of tannic acid to BSA for different values of the latter's concentration at pH = 4. The y-axis is in fact the quantity Δτ600 = τ600([TA],[BSA]) − τ600(0,[BSA]) for any BSA concentration (τ stands for turbidity). This quantity will be equal to zero, unless the presence of tannic acid induces a measurable increase in the turbidity of a biopolymer solution.
Fig. 2. Turbidity of tannic acid/BSA solutions at pH = 4 as a function of the molar ratio, for different fixed concentrations of BSA. Inset: critical molar ratio values for the onset of increased turbidity as a function of BSA concentration.

As expected, turbidity depends on both the concentration ratios of the two components and the absolute values of the concentrations. For each BSA concentration there is a critical tannic acid concentration, at which turbidity due to larger-scale aggregation begins to appear. This is plotted as a function of BSA concentration in the inset, and appears to be anticorrelated with the BSA concentration. Fig. S1† in the ESI section shows that the interactions between BSA and tannic acid are much weaker at neutral pH. Critical molar ratios at pH = 4 are close to 10 while at pH = 7 they are in the range of 100. This is probably related to charge repulsion between BSA (pI = 4.7 (ref. 81)) and TA (pKa = 6, although a range of acidity has been quoted for TA82) at neutral pH.
In Fig. 3 one sees that the situation is different for the four polysaccharides examined in this work. The data shown are for solutions of polysaccharides of 1 g L−1 at pH = 4. The abscissa is the ratio of moles of tannic acid per moles of monomer of the polysaccharide. The reason for this choice is that for these natural biopolymers the molecular weight distribution is quite wide and uncertain.
Fig. 3. Turbidity (600 nm) of TA/polysaccharide solutions vs. the molar ratio of tannic acid to polysaccharide monomer. 1 g L−1 polysaccharide solutions at pH = 4.

The turbidity increases almost linearly even at very low tannic acid concentrations and no critical threshold is visible as in the BSA case. The strongest interactions of tannic acid are with chitosan and pectin. Chitosan is nearly insoluble at pH = 7, but the rest of the polysaccharides exhibit similar turbidities at pH = 7. At pH = 4 chondroitin sulfate is still strongly negatively charged, hence its considerably weaker interaction with tannic acid.
3.2. Layer-by-layer work, UV-vis and PM-IRRAS
The formation of layers of emulsifiers and polysaccharides on solid surfaces is by no means equivalent to the situation encountered at the droplet interfaces of MEs, but the investigation of such LbL systems is enlightening. As stated, notable goals of this part of the investigation are: (a) to examine the stability of a layer when a subsequent layer of a different material is deposited. (b) To find out the best possible combinations of components and concentration ranges that ensure maximum deposition of materials (especially the antioxidant) at the surface.
The protocol behind the surface deposition experiments was kept as simple as possible, given the huge number of possible combinations of the layers. The pH was equal to 4 in all deposition solutions, since TA–biopolymer interactions are stronger at this pH. Only chitosan was used as the first layer to take advantage of its known good adhesion to negative glass surfaces.83,84 Tannic acid was always deposited together with BSA, although tannic acid is by itself an excellent component of LbL films.61–66 It was examined if the deposition is linear or not, and if subsequent layers have a negative effect on previous layers. The UV-vis absorbance at 280–310 nm was used to assess the deposited amount of BSA/TA, since both these compounds absorb at this wavelength range while the four polysaccharides used do not. In principle, one could try to distinguish between BSA and TA by more carefully monitoring the absorbance and deconvoluting the spectra above 300 nm, but it turned out that PM-IRRAS measurements reported below offer an excellent alternative.
3.2.1. UV-vis investigation
Fig. S2† shows the absorbance of alternating layers of chitosan and mixtures of BSA and tannic acid (1 g L−1 BSA, with TA being in 15 fold molar excess over BSA n(TA)/n(BSA) = 15). The deposition of several layers proceeds successfully. However, some deposited BSA/TA material is lost when the next chitosan layer is deposited, and the absorbance exhibits a zig-zag behaviour (Fig. S3†), similar to that observed in other LbL systems.85–88 This behaviour is related to the reported effect of polysaccharides on protein–tannic acid interactions.77–80 It may be attributed to the changing charge density at the surface as subsequent layers are deposited, and can be avoided by using very high ionic strength in the deposition solutions, something not wanted in the present experiments in view of the subsequent emulsion application. A related reason for this behaviour may be the changing water content in the films as the film is built. Very similar results are obtained for a more “structured” deposition, where a layer of chitosan is deposited first and then alternating layers of pectin (from 1 g L−1 solutions) and BSA/TA (from 1 g L−1 BSA solutions and n(TA)/n(BSA) = 15 as before). UV-vis spectra of such films are shown in Fig. 4.
Fig. 4. UV-vis absorbance from LbL-deposited films at pH = 4 with chitosan as the first layer and a sequence of alternating layers of pectin and BSA/TA.

Similar experiments were run for different molar ratios of tannic acid to BSA in the BSA/TA layers (ranging from 7.5 to 30), as well as for films in which the chitosan layers are completely replaced by pectin, chondroitin sulfate, or sodium alginate. The results are similar to those in Fig. S2 and 4,† so they are not presented. Fig. 5 summarizes the deposition results for alternating layers of the four different polysaccharides used in this work with BSA/TA mixtures, when all polymers were at 1 g L−1 in their respective solutions and TA was present together with BSA with tannic acid always being in 15 fold molar excess over BSA (n(TA)/n(BSA) = 15). The graph shows that more BSA/TA is deposited when chitosan or pectin are used as intermediate layer materials. It also shows that the deposition is not entirely linear, although for such a small number of layers it is impossible to tell if it is eventually becomes exponential as in other systems.86,88,89 The results of Fig. 5 indicate that chitosan and pectin should be preferable for emulsion work over chondroitin sulfate, and sodium alginate. The amount of TA actually deposited on the interfacial layers is an important quantity that should be optimized for emulsion work. Polarization-modulation infrared reflection–absorption spectroscopy (PM-IRRAS) has been used to determine this issue.
Fig. 5. Deposited amount of BSA/TA for 1–4 bilayers of LbL films of alternating BSA/TA and various polysaccharides.

3.2.2. PM-IRRAS examination of the LbL-films
As far as we know, PM-IRRAS is seldom used to characterize LbL films of polymers on solid surfaces,42–44 for which ATR-IR spectroscopy is more frequently used.90,91 Since the focus here is on the antioxidant, the challenge of using PM-IRRAS, or any IR spectroscopy, in these systems is to identify peaks that belong to tannic acid and are distinct from the peaks of proteins and polysaccharides. This is nontrivial, because all these organic compounds have similar IR-active groups. In Fig. 6 one sees the powder FTIR spectra of BSA, chitosan and TA, and superimposed a scaled PM-IRRAS spectrum from a film with one layer of chitosan and one of BSA/TA (at a molar ratio of TA to BSA equal to 15). There is a significant spectral overlap of several of the peaks, although tannic acid has a considerably different spectrum. Both BSA and chitosan have characteristic amide peaks at 1650 and 1500–1550 cm−1, while the carboxylic ester peaks of tannic acid are located at 1720 and 1625 cm−1. The black filled circles under the PM-IRRAS spectrum identify BSA peaks, the grey filled stem from chitosan while the empty circles are TA peaks. For the analysis of BSA–TA/polysaccharide multilayer films different peaks for potential TA quantitation were evaluated, but most of them turned out to be too weak for accurate work. We ended up by focusing on the region from 1400 to 1800 cm−1 and using the carbonyl peak above 1700 cm−1, which was obtained by spectral deconvolution using Origin.Pro, as demonstrated in Fig. S4.† The method cannot be applied in a straightforward way to films containing polysaccharides with significant absorption in the carbonyl region, such as pectin or sodium alginate. As will be seen however, it is still possible to evaluate films with such components, subtracting the absorbance of the pectin from the total to calculate the absorbance of pure TA in the layers.
Fig. 6. FTIR spectra of powders of BSA, chitosan and tannic acid compared to a scaled PM-IRRAS spectrum of a bilayer of chitosan and BSA/TA (molar ratio n(TA)/n(BSA) = 15) deposited on a reflective surface.

Fig. S5,† obtained for a system with alternating layers of pectin and BSA/TA, shows that the PM-IRRAS signal is much higher for a gold-coated substrate than for an uncoated stainless-steel substrate. For some systems the laser beam of the IRRAS instrument was focused on two different positions of the sample to ensure that the deposition is homogeneous. Such results from a film with alternating layers of chitosan and BSA/TA 1 : 15 are shown in Fig. S6.† Small differences in the readings from the two spots are obvious, but the overall reproducibility in this and other cases examined is satisfactory.
The analysis of adsorbed layers for the TA peak above 1700 cm−1 provides a good measure of the relative amount of tannic acid incorporated in the layers. This is shown in Fig. 7, where the height of this deconvoluted peak is plotted as a function of layer number for an even number of layers. A polysaccharide is used as the sole component of the odd layers while BSA/TA mixtures with a molar ratio n(TA)/n(BSA) = 15 : 1 is used for the even layers. As mentioned before, even for a polysaccharide like pectin, which adsorbs strongly in the carbonyl region, it is possible to evaluate the absorbance due to tannic acid alone, by subtracting the signal introduced by the odd (pectin) layers. The assumption is that each new layer does not lead to a loss of pectin previously deposited. This is not necessarily correct, so for pectin the results in Fig. 7 are only indicative.
Fig. 7. PM-IRRAS signal from layered films where the odd layers are polysaccharides (adsorbed from 1 g L−1 solutions) and the even layers are BSA/TA mixtures (adsorbed from 1 g L−1 solutions of BSA containing TA at a molar ratio TA/BSA = 15 : 1).

The results in Fig. 7 show that chitosan is a better interlayer material than chondroitin sulfate, and is comparable to pectin. With chitosan and pectin as interlayers, it is possible to transfer and keep larger TA amounts at the surface. The deposited amount of TA increases with its concentration (or molar ratio to BSA) in solution. However, TA/BSA ratios higher than 20 give cloudy solutions and the deposition fails in the absence of stirring due to precipitation of the BSA–TA coacervates. For this reason, but also to compare emulsions with different layers (see below), the actual TA/BSA molar ratio used in the next part is closer to 10.
3.2.2.1. Work on multilayer emulsions
Following the work in the bulk aqueous phase and the LbL deposition, it is now time to examine how multilayer emulsions behave, when tannic acid is used as an antioxidant. The comparison is made by inserting Nile red in the oil droplets of multilayer emulsions and examining its oxidation rate in the presence and absence of the radical generator molecule AAPH (2,2′-azobis-2-methylpropanimidamide dihydrochloride). Since the focus of this work is on the oxidation barrier for radicals at the droplet surfaces, it was not deemed necessary to assess the antioxidant action in other ways (e.g., via the measurement of oxidation products, see ref. 27, 39, 45 and 92).
In contrast to the layer-by-layer experiments on solid surfaces, the multilayer emulsions are first stabilized with a layer of BSA, which is an established emulsifier. Chitosan or pectin are not ideal emulsifiers by themselves, although some recent reports examine this issue in more detail as a function of their degree of deacetylation (for chitosan) or methyl esterification (for pectin).93,94 After a first emulsifying layer of BSA, second and third layers of chitosan and pectin or vice versa were added as discussed in the Experimental section† to prepare a three-layer ME. Table 1 contains the results of emulsion characterization in such MEs, including systems in which TA has been introduced in the formulation of any of the three layers. Both dynamic light scattering and zeta potential measurements show that the emulsions with BSA as a first layer, chitosan as a second layer and pectin as a third layer are stable and produce droplets with similar sizes and electrical properties. The expected variation of the zeta potential is observed and essentially proves the multilayer nature of the droplet interfaces, i.e. the BSA layer has a positive potential at pH = 4, chitosan makes the potential more positive and pectin less positive. Addition of tannic acid with the polysaccharide of the second or the third layer does not lead to considerable variations, but it leads to a much larger size when present in the first layer, proving a deleterious effect on the emulsifying properties of BSA. If pectin is used as the second layer and chitosan as the third, it is found that the emulsions are less stable, as implied by the larger droplet sizes. This may also be due to weaker electrostatic stabilization, since the zeta potential value in the BSA–PE–CH system is much lower than that in the BSA–CH–PE system.
Droplet sizes and zeta potentials for the three-layer MEs examined.
| Layer composition | Layer number | D (sD)/nm | ζ (sζ)/mV |
|---|---|---|---|
| BSA–CH–PE | 1 | 267 (127) | 33.8 (4.6) |
| 2 | 217 (97) | 40.2 (5.5) | |
| 3 | 294 (121) | 24.9 (4.1) | |
| BSA/TA–CH–PE | 1 | 664 (98) | 29.1 (3.5) |
| 2 | 353 (240) | 42.3 (4.3) | |
| 3 | 354 (153) | 30.9 (3.8) | |
| BSA–CH/TA–PE | 1 | 267 (127) | 33.8 (4.6) |
| 2 | 250 (125) | 42.9 (6.5) | |
| 3 | 279 (120) | 29.9 (4.8) | |
| BSA–CH–PE/TA | 1 | 267 (127) | 33.8 (4.6) |
| 2 | 217 (97) | 40.2 (5.5) | |
| 3 | 337 (151) | 21.1 (3.7) | |
| BSA–PE–CH | 1 | 267 (127) | 33.8 (4.6) |
| 2 | 914 (189) | 10.4 (3.1) | |
| 3 | 982 (204) | 31.1 (3.3) |
The presence of tannic acid at the droplet interfaces is verified indirectly by these results, especially by the destabilization of the first layer.
Fig. S7† shows how the oxidation behavior is assessed in the present work. It is the result of two parallel experiments, in which the normalized fluorescence intensity of Nile red is monitored in the presence and in the absence of the radical generator AAPH. The normalized (with respect to time zero) intensities of the two experiments are divided at all times to obtain the relative fluorescence intensity (RFI) of eqn (1). In Fig. 8 the effect of placing chitosan as a second layer and pectin as a third layer or vice versa on the stabilization of the droplets against oxidation is examined. Results of MEs with one layer, two layers or three layers are juxtaposed. It is clear that BSA by itself does not provide any protection to the oil core, while any second layer improves the oxidative stability considerably, a fact that is known from other emulsion systems.95,96 Surprisingly, while pectin is satisfactory as a single second layer, it deteriorates when chitosan is added as a third layer. This could be due to a chitosan-pectin coacervate formation in the bulk.97 The sequence BSA–CHI–PE appears to provide maximum protection to the droplets, even in the absence of a specific antioxidant in the system. This is the sequence that will be subsequently used as the basis to probe the effect of positioning of the tannic acid antioxidant in the emulsions.
Fig. 8. Relative fluorescence intensity of Nile red in various multilayer emulsions created using BSA as the primary emulsifier and chitosan and pectin as layer materials in the absence of a specific antioxidant.

The relative antioxidant action of TA in BSA–CHI–PE multilayer emulsions as a function of tannic acid position in the layers is finally examined. Since tannic acid interacts quite strongly with all three components, it is expected to be present to a large extent in the layers at pH = 4. The percentage of tannic acid that exists in the layers has not been measured in this work, since this is usually done in indirect and rather uncertain ways involving emulsion break-up. For its presence at the interfaces only indirect indications exist, from DLS and zeta potential measurements and from the knowledge that tannic acid interacts quite strongly with BSA, chitosan and pectin. In Fig. 9 it is demonstrated that indeed it matters how the antioxidant is administered to the system. Surprisingly, preferential placement of the antioxidant at the first layer actually decreases the oxidative stability of the system. This may stem from a modification of the emulsifying ability of BSA, since tannins are known to affect BSA conformation in solution,51,53 which might have an effect on the compactness of the BSA layer at the droplet interfaces. Using isothermal titration calorimetry, fluorescence, circular dichroism and spectroscopy, it has been verified that the interaction between tannic acid and BSA is strong and extends to molar ratios of tannic acid to BSA as large as 30–40 (in preparation for submission – but see also98). A definite indication for such a structural effect of tannic acid is seen in the DLS results of Table 1, where much larger droplets are observed when tannic acid is introduced together with BSA at the first layer. The rest of the results are in agreement with the picture generated so far from the bulk and solid surface measurements. Introduction of the TA in the second (chitosan) layer definitely provides the strongest antioxidant action, while incorporation in the third layer (with pectin) is also advantageous. RFI values larger than unity observed in the BSA–CHI/TA–PE case should be viewed with caution, but they definitely highlight the excellent oxidative stabilization observed for this system.
Fig. 9. Relative fluorescence intensities of Nile red in various three-layer (BSA–CHI–PE) emulsions where tannic acid has been added as antioxidant.

4. Conclusions
From the results presented in this work, it appears that the colloidal approach outlined in the beginning of this paper holds considerable promise for the systematic design of multilayer emulsions with the goal to improve the stability of the encapsulated oils towards oxidation, despite the fact that the present investigation has been limited to a small set of possible systems. Tannic acid has been used throughout as a model antioxidant. First, it was verified using turbidity measurements that tannic acid interacts quite strongly with BSA, which is the primary emulsifier used exclusively in the present work. From these measurements it was possible to identify the optimal molar ratios of TA/BSA to use in emulsion work (in the range of 10, as for larger ratios one observes considerable aggregation in solution). It was also established that chitosan and pectin are the polysaccharides that interact more strongly with TA, compared to sodium alginate and chondroitin sulfate, hence they are preferable in multilayer emulsions where it is attempted to incorporate tannic acid at the droplet interfaces. At the second stage, in layer-by-layer deposition experiments on solid surfaces it was verified that chitosan and pectin are the optimal layer components for a multilayer configuration that can maximize the interfacial concentration of tannic acid in such systems. Finally, three-layer O/W emulsions with BSA as the primary emulsifier, and chitosan and pectin as layer materials were constructed and tested for their ability to stabilize the sensitive linseed oil towards oxidation. The results demonstrate that the precise location of TA is also important. Surprisingly, its introduction in the first emulsifying layer (with BSA) leads to stability deterioration, presumably because TA affects BSA conformation and emulsifier action. The introduction of TA in the second layer together with chitosan provides excellent stabilization against attack by radicals generated in the aqueous phase.
Further generalization of this work should involve a precise quantification of the interfacial concentration of the antioxidant for these systems, for correlation with the antioxidant action in the layers. The strongest validation of the three-stage design concept presented here should involve a different antioxidant, a different primary emulsifier, and different combinations of layer materials. In particular, working with natural antioxidants (e.g. anthocyanins or betalains), for which the precise bulk interactions with proteins and polysaccharides have not been extensively documented, would prove the importance of the first stage (study of bulk interactions), which plays a relatively minor role in the present work. It would also provide a stronger proof of the validity of the colloidal approach for the design of multilayer emulsions presented in this paper.
EL was responsible for funding acquisition, conceptualization, methodological design, supervision, writing, reviewing and editing the manuscript. SA contributed to the methodological design, and carried out the experimental investigation, data curation and formal analysis of the results.
Conflicts of interest
The authors declare that they do not have a conflict of interest of any type regarding this work.
Supplementary Material
Acknowledgments
EL would like to thank the University of Cyprus for an internal research grant, which allowed the purchase of the high-pressure homogenizer used in this work. SA would like to thank IKYK (the foundation for graduate scholarships of Cyprus), which supported her for the last three years of her PhD thesis. The authors are thankful to Prof. Ioannis Sarigiannis of the University of Nicosia in Cyprus for allowing them to use the Malvern zeta-sizer for zeta-potential measurements.
Electronic supplementary information (ESI) available: Graphs S1–S7. These show turbidity comparisons at two different pH values for the BSA – TA system, LbL deposition information for the chitosan – BSA/TA films, the deconvolution procedure of PM-IRRAS spectra, PM-IRRAS supporting measurements, and an illustration of how the relative fluorescence intensity (RFI) in emulsions in the absence and presence of AAPH is calculated. See DOI: 10.1039/d1ra03512f
References
- Sagalowicz L. Leser M. E. Cur. Op. Colloid Int. Sci. 2010;15:61–72. doi: 10.1016/j.cocis.2009.12.003. [DOI] [Google Scholar]
- McClements D. J. Adv. Colloid Int. Sci. 2015;219:27–53. doi: 10.1016/j.cis.2015.02.002. [DOI] [PubMed] [Google Scholar]
- McClements D. J. Adv. Colloid Int. Sci. 2018;253:1–22. doi: 10.1016/j.cis.2018.02.002. [DOI] [PubMed] [Google Scholar]
- McClements D. J. Rao J. Crit. Rev. Food Sci. Nutr. 2011;51:285–330. doi: 10.1080/10408398.2011.559558. [DOI] [PubMed] [Google Scholar]
- McClements D. J. Cur. Op. Colloid Int. Sci. 2012;17:235–245. doi: 10.1016/j.cocis.2012.06.002. [DOI] [Google Scholar]
- McClements D. J., Food Emulsions: Principles, Practices, and Techniques, CRC Press, Boca Raton, 3rd edn, 2016 [Google Scholar]
- McClements D. J. Jafari S. M. Adv. Colloid Int. Sci. 2018;251:55–79. doi: 10.1016/j.cis.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Berton-Carabin C. Schroën K. Cur. Op. Food Sci. 2019;27:74–81. doi: 10.1016/j.cofs.2019.06.006. [DOI] [Google Scholar]
- Guzey D. McClements D. J. Adv. Colloid Int. Sci. 2006;128–130:227–248. doi: 10.1016/j.cis.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Grigoriev D. O. Miller R. Cur. Op. Colloid Int. Sci. 2009;14:48–59. doi: 10.1016/j.cocis.2008.03.003. [DOI] [Google Scholar]
- Shchukina E. M. Shchukin D. G. Cur. Op. Colloid Int. Sci. 2012;17:281–289. doi: 10.1016/j.cocis.2012.06.003. [DOI] [Google Scholar]
- Decher G. Hong J. D. Schmitt J. Thin Solid Films. 1991;210/211:831–835. doi: 10.1016/0040-6090(92)90417-A. [DOI] [Google Scholar]
- Lvov Y. Ariga K. Ichinose I. Kunitake T. J. Am. Chem. Soc. 1995;117:6117–6123. doi: 10.1021/ja00127a026. [DOI] [Google Scholar]
- Decher G. Science. 1997;277:1232–1237. doi: 10.1126/science.277.5330.1232. [DOI] [Google Scholar]
- Sukhorukov G. B. Lichtenfeld H. Knippel E. Budde A. Möhwald H. Colloids Surf. A. 1998;137:253–266. doi: 10.1016/S0927-7757(98)00213-1. [DOI] [Google Scholar]
- Peyratout C. S. Dähne L. Ang. Chem. Int. Ed. 2004;43:3762–3783. doi: 10.1002/anie.200300568. [DOI] [PubMed] [Google Scholar]
- Jiménez-Colmenero F. Food Res. Int. 2013;52:64–74. doi: 10.1016/j.foodres.2013.02.040. [DOI] [Google Scholar]
- Cuomo F. Ceglie A. Piludu M. Miguel M. G. Lindman B. Lopez F. Langmuir. 2014;30:7993–7999. doi: 10.1021/la501978u. [DOI] [PubMed] [Google Scholar]
- Schmelz T. Lesmes U. Weiss J. McClements D. J. Food Hydrocoll. 2011;25:1181–1189. doi: 10.1016/j.foodhyd.2010.11.005. [DOI] [Google Scholar]
- Fioramonti S. A. Arzeni C. Pilosof A. M. R. Rubiolo A. C. Santiago L. G. J. Food Eng. 2015;156:31–38. doi: 10.1016/j.jfoodeng.2015.01.013. [DOI] [Google Scholar]
- Liu F. Wang D. Sun C. McClements D. J. Gao Y. Food Chem. 2016;205:129–139. doi: 10.1016/j.foodchem.2016.02.155. [DOI] [PubMed] [Google Scholar]
- Dyerberg J. Bang H. O. Lancet. 1979;314:433–435. doi: 10.1016/S0140-6736(79)91490-9. [DOI] [PubMed] [Google Scholar]
- Fodor J. G. Helis E. Yazdekhasti N. Vohnout B. Can. J. Cardiol. 2014;30:864–868. doi: 10.1016/j.cjca.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Hu M. McClements D. J. Decker E. A. J. Agric. Food Chem. 2003;51:1435–1439. doi: 10.1021/jf0203794. [DOI] [PubMed] [Google Scholar]
- Katsuda M. S. McClements D. J. Miglioranza L. H. S. Decker E. A. J. Agric. Food Chem. 2008;56:5926–5931. doi: 10.1021/jf800574s. [DOI] [PubMed] [Google Scholar]
- Charoen R. Jangchud A. Jangchud K. Harnsilawat T. Decker E. A. McClements D. J. Food Chem. 2012;131:1340–1346. doi: 10.1016/j.foodchem.2011.09.128. [DOI] [Google Scholar]
- Berton-Carabin C. C. Ropers M. H. Genot C. Comp. Rev. Food Sci. Food Saf. 2014;13:945–977. doi: 10.1111/1541-4337.12097. [DOI] [Google Scholar]
- Hogan S. A. O'Riordan E. D. O'Sullivan M. J. Microencapsul. 2003;20:675–688. doi: 10.3109/02652040309178355. [DOI] [PubMed] [Google Scholar]
- Klinkesorn U. Sophanodora P. Chinachoti P. McClements D. J. Decker E. A. J. Agric. Food Chem. 2005;53:8365–8371. doi: 10.1021/jf050761r. [DOI] [PubMed] [Google Scholar]
- Shaw L. A. McClements D. J. Decker E. A. J. Agric. Food Chem. 2007;55:3112–3119. doi: 10.1021/jf063068s. [DOI] [PubMed] [Google Scholar]
- Encina C. Vergara C. Gimenez B. Oyarzún-Ampuero F. Robert P. Trends Food Sci. Technol. 2016;56:46–60. doi: 10.1016/j.tifs.2016.07.014. [DOI] [Google Scholar]
- Laguerre M. Bayrasy C. Panya A. Weiss J. McClements D. J. Lecomte J. Decker E. A. Villeneuve P. Crit. Rev. Food Sci. Nutr. 2015;55:183–201. doi: 10.1080/10408398.2011.650335. [DOI] [PubMed] [Google Scholar]
- McClements D. J. Decker E. J. Agric. Food Chem. 2018;66:20–35. doi: 10.1021/acs.jafc.7b05066. [DOI] [PubMed] [Google Scholar]
- Viljanen K. Kylli P. Hubbermann E. M. Schwarz K. Heinonen M. J. Agric. Food Chem. 2005;53:2022–2027. doi: 10.1021/jf047975d. [DOI] [PubMed] [Google Scholar]
- Gonzalez M. J. Medina I. Maldonado O. S. Lucas R. Morales J. C. Food Chem. 2015;183:190–196. doi: 10.1016/j.foodchem.2015.03.035. [DOI] [PubMed] [Google Scholar]
- Pan Y. Nitin N. Colloids Surf. B. 2015;135:472–480. doi: 10.1016/j.colsurfb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Wan Z. L. Wang J. M. Wang L. Y. Yang X. Q. Yuan Y. J. Agric. Food Chem. 2013;61:4433–4440. doi: 10.1021/jf4003945. [DOI] [PubMed] [Google Scholar]
- Wan Z. L. Wang J. M. Wang L. Y. Yuan Y. Yang X. Q. Food Chem. 2014;161:324–331. doi: 10.1016/j.foodchem.2014.04.028. [DOI] [PubMed] [Google Scholar]
- Von Staszewski M. Pizones Ruiz-Henestrosa V. M. Pilosof A. M. R. Food Hydrocoll. 2014;35:505–511. doi: 10.1016/j.foodhyd.2013.07.008. [DOI] [Google Scholar]
- Zhao Y. Wang X. Li D. Tang H. Yu D. Wang L. Jiang L. Proc. Biochem. 2020;89:89–97. doi: 10.1016/j.procbio.2019.10.021. [DOI] [Google Scholar]
- Mendelsohn R. Mao G. Flach C. R. Biochim. Biophys. Acta. 2010;1798:788–800. doi: 10.1016/j.bbamem.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonazzola C. Calvo E. J. Nart F. C. Langmuir. 2003;19:5279–5286. doi: 10.1021/la026528u. [DOI] [Google Scholar]
- Boddohi S. Almodóvar J. Zhang H. Johnson P. A. Kipper M. J. Colloids Surf. B. 2010;77:60–68. doi: 10.1016/j.colsurfb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Tagliazucchi M. Méndez De Leo L. P. Cadranel A. Baraldo L. M. Völker E. Bonazzola C. Calvo E. J. Zamlynny V. J. Electroanal. Chem. 2010;649:110–118. doi: 10.1016/j.jelechem.2010.02.013. [DOI] [Google Scholar]
- Lomova M. V. Sukhorukov G. B. Antipina M. N. ACS Appl. Mater. Int. 2010;2:3669–3676. doi: 10.1021/am100818j. [DOI] [PubMed] [Google Scholar]
- Lau H. H. Murney R. Yakovlev N. L. Novoselova M. V. Lim S. H. Roy N. Singh H. Sukhorukov G. B. Haigh B. Kiryukhin M. V. J. Colloid Int. Sci. 2017;505:332–340. doi: 10.1016/j.jcis.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Sadovoy A. V. Lomova M. V. Antipina M. N. Braun N. A. Sukhorukov G. B. Kiryukhin M. V. ACS Appl. Mater. Int. 2013;5:8948–8954. doi: 10.1021/am401871u. [DOI] [PubMed] [Google Scholar]
- Chibowski E. Espinosa-Jiménez M. Ontiveros-Ortega A. Giménez-Martin E. Langmuir. 1998;14:5237–5244. doi: 10.1021/la971107b. [DOI] [Google Scholar]
- Lin D. Xing B. Environ. Sci. Technol. 2008;42:5917–5923. doi: 10.1021/es800329c. [DOI] [PubMed] [Google Scholar]
- Van Buren J. P. Robinson W. B. J. Agric. Food Chem. 1969;17:773–777. doi: 10.1021/jf60164a003. [DOI] [Google Scholar]
- Ishtikhar M. Ahmad E. Siddiqui Z. Ahmad S. Khan M. V. Zaman M. Nusrat M. S. Chandel T. I. Ajmal M. R. Khan R. H. Int. J. Biol. Macromol. 2018;107:2450–2464. doi: 10.1016/j.ijbiomac.2017.10.136. [DOI] [PubMed] [Google Scholar]
- Thongkaew C. Gibis M. Hinrichs J. Weiss J. Food Hydrocoll. 2014;41:103–112. doi: 10.1016/j.foodhyd.2014.02.006. [DOI] [Google Scholar]
- Xie L. Wehling R. L. Ciftci O. Zhang Y. Food Res. Int. 2017;102:195–202. doi: 10.1016/j.foodres.2017.10.007. [DOI] [PubMed] [Google Scholar]
- Yang F. Yang J. Qiu S. Xu W. Wang Y. Food Chem. 2021;346:128762. doi: 10.1016/j.foodchem.2020.128762. [DOI] [PubMed] [Google Scholar]
- Zhan F. Li J. Wang Y. Shi M. Li B. Sheng F. J. Agric. Food Chem. 2018;66:6832–6839. doi: 10.1021/acs.jafc.8b00503. [DOI] [PubMed] [Google Scholar]
- Zhan F. Yang J. Li J. Wang Y. Li B. Food Hydrocoll. 2018;75:81–87. doi: 10.1016/j.foodhyd.2017.09.010. [DOI] [Google Scholar]
- An X. Kang Y. Li G. Chem. Phys. 2019;520:100–107. doi: 10.1016/j.chemphys.2018.12.009. [DOI] [Google Scholar]
- Chauhan S. S. Shetty A. B. Hatami E. Chowdhury P. Yallapu M. M. Pharmaceutics. 2020;12:285. doi: 10.3390/pharmaceutics12030285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulateefeh S. R. Taha M. O. J. Microencapsul. 2015;32:96–105. doi: 10.3109/02652048.2014.985343. [DOI] [PubMed] [Google Scholar]
- Chen Y. Li Z. Yi X. Kuang H. Ding B. Sun W. Luo Y. LWT--Food Sci. Technol. 2020;118:108778. doi: 10.1016/j.lwt.2019.108778. [DOI] [Google Scholar]
- Ball V. Colloid Interface Sci. Commun. 2014;3:1–4. doi: 10.1016/j.colcom.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Chen C. Chen G. Wan P. Chen D. Zhu T. Hu B. Sun Y. Zeng X. J. Agric. Food Chem. 2018;66:11141–11150. doi: 10.1021/acs.jafc.8b04381. [DOI] [PubMed] [Google Scholar]
- Erel-Unal I. Sukhishvili S. A. Macromolecules. 2008;41:3962–3970. doi: 10.1021/ma800186q. [DOI] [Google Scholar]
- Lau H. H. Yakovlev N. L. Ooi C. P. Kiryukhin M. V. Colloid Polym. Sci. 2019;297:417–422. doi: 10.1007/s00396-018-4426-4. [DOI] [Google Scholar]
- Ringwald C. Ball V. RSC Adv. 2016;6:4730–4738. doi: 10.1039/C5RA24337H. [DOI] [Google Scholar]
- Shutava T. Prouty M. Kommireddy D. Lvov Y. Macromolecules. 2005;38:2850–2858. doi: 10.1021/ma047629x. [DOI] [Google Scholar]
- Yang S. Wang Y. Wu X. Sheng S. Wang T. Zan X. ACS Biomater. Sci. Eng. 2019;5:3582–3594. doi: 10.1021/acsbiomaterials.9b00717. [DOI] [PubMed] [Google Scholar]
- Zhang T. Fang L. Lin N. Wang J. Wang Y. Wu T. Song P. Green Chem. 2019;21:5405–5413. doi: 10.1039/C9GC02454A. [DOI] [Google Scholar]
- Shutava T. G. Prouty M. D. Agabekov V. E. Lvov Y. M. Chem. Lett. 2006;35:1144–1145. doi: 10.1246/cl.2006.1144. [DOI] [Google Scholar]
- Gulcin I. Huyut Z. Elmastaş M. Aboul-Enein H. Y. Arab. J. Chem. 2010;3:43–53. doi: 10.1016/j.arabjc.2009.12.008. [DOI] [Google Scholar]
- Wang Y. Yang F. Yang J. Bai Y. Li B. Carbohydr. Pol. 2021;254:117292. doi: 10.1016/j.carbpol.2020.117292. [DOI] [PubMed] [Google Scholar]
- Zhou B. Gao S. Li X. Liang H. Li S. Int. J. Food Sci. Technol. 2020;55:1924–1934. doi: 10.1111/ijfs.14419. [DOI] [Google Scholar]
- Chun J.-Y. Choi M.-J. Min S.-G. Weiss J. Food Hydrocoll. 2013;30:249–257. doi: 10.1016/j.foodhyd.2012.05.024. [DOI] [Google Scholar]
- Ogawa S. Decker E. A. McClements D. J. J. Agric. Food Chem. 2004;52:3595–3600. doi: 10.1021/jf034436k. [DOI] [PubMed] [Google Scholar]
- Bozan B. Temelli F. Biores. Technol. 2008;99:6354–6359. doi: 10.1016/j.biortech.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Sánchez Hernández M. R. Cuvelier E. M. Turchiuli C. Food Res. Int. 2016;88:32–41. doi: 10.1016/j.foodres.2016.04.035. [DOI] [PubMed] [Google Scholar]
- McManus J. P. Davis K. G. Beart J. E. Gaffney S. H. Lilley T. H. Haslam E. J. Chem. Soc. Perkin Trans. II. 1985:1429–1438. doi: 10.1039/P29850001429. [DOI] [Google Scholar]
- de Freitas V. Carvalho E. Mateus N. Food Chem. 2003;81:503–509. doi: 10.1016/S0308-8146(02)00479-X. [DOI] [Google Scholar]
- Soares S. Mateus N. de Freitas V. J. Agric. Food Chem. 2012;60:3966–3972. doi: 10.1021/jf3002747. [DOI] [PubMed] [Google Scholar]
- Lou W. Bezusov A. Li B. Dubova H. Food Sci. Technol. 2019;13:63–69. [Google Scholar]
- Salis A. Boström M. Medda L. Cugia F. Barse B. Parsons D. F. Ninham B. W. Monduzzi M. Langmuir. 2011;27:11597–11604. doi: 10.1021/la2024605. [DOI] [PubMed] [Google Scholar]
- Ghigo G. Berto S. Minella M. Vione D. Alladio E. Nurchi V. M. Lachowicz J. Daniele P. G. New J. Chem. 2018;42:7703–7712. doi: 10.1039/C7NJ04903J. [DOI] [Google Scholar]
- Yi H. Wu L.-Q. Bentley W. E. Ghodssi R. Rubloff G. W. Culver J. N. Payne G. F. Biomacromol. 2005;6:2881–2894. doi: 10.1021/bm050410l. [DOI] [PubMed] [Google Scholar]
- Antunes J. C. Leite Pereira C. Molinos M. Ferreira-da-Silva F. Dessì M. Gloria A. Ambrosio L. Gonçalves R. M. Barbosa M. A. Biomacromol. 2011;12:4183–4195. doi: 10.1021/bm2008235. [DOI] [PubMed] [Google Scholar]
- Ariga K. Lvov Y. Kunitake T. J. Am. Chem. Soc. 1997;119:2224–2231. doi: 10.1021/ja963442c. [DOI] [Google Scholar]
- Benbow N. L. Webber J. L. Pawliszak P. Sebben D. A. Karpiniec S. Stringer D. Tobin M. J. Vongsvivut J. Krasowska M. Beattie D. A. J. Colloid Int. Sci. 2019;553:720–733. doi: 10.1016/j.jcis.2019.06.036. [DOI] [PubMed] [Google Scholar]
- Kolarik L. Furlong D. N. Joy H. Struijk C. Rowe R. Langmuir. 1999;15:8265–8275. doi: 10.1021/la990413h. [DOI] [Google Scholar]
- Serizawa T. Kawanishi N. Akashi M. Macromolecules. 2003;36:1967–1974. doi: 10.1021/ma020734r. [DOI] [Google Scholar]
- Picart C. Mutterer J. Richert L. Luo Y. Prestwich G. D. Schaaf P. C Voegel J.- Lavalle P. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12531–12535. doi: 10.1073/pnas.202486099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Nkwantabisah S. Gammana M. Tripp C. P. Langmuir. 2014;30:11696–11703. doi: 10.1021/la5031558. [DOI] [PubMed] [Google Scholar]
- Torger B. Müller M. Spectrochim. Acta A. 2013;104:546–553. doi: 10.1016/j.saa.2012.11.080. [DOI] [PubMed] [Google Scholar]
- Nuchi C. D. Hernandez P. McClements D. J. Decker E. A. J. Agric. Food Chem. 2002;50:5445–5449. doi: 10.1021/jf020095j. [DOI] [PubMed] [Google Scholar]
- Klinkesorn U. Food Rev. Int. 2013;29:371–393. doi: 10.1080/87559129.2013.818013. [DOI] [Google Scholar]
- Verkempinck S. H. E. Kyomugasho C. Salvia-Trujillo L. Denis S. Bourgeois M. Van Loey A. M. Hendrickx M. E. Grauwet T. Food Hydrocoll. 2018;85:144–157. doi: 10.1016/j.foodhyd.2018.07.014. [DOI] [Google Scholar]
- Huang J. Wang Q. Li T. Xia N. Xia Q. J. Sci. Food Agric. 2018;98:3513–3523. doi: 10.1002/jsfa.8870. [DOI] [PubMed] [Google Scholar]
- Ogawa S. Decker E. A. McClements D. J. J. Agric. Food Chem. 2003;51:5522–5527. doi: 10.1021/jf026103d. [DOI] [PubMed] [Google Scholar]
- Nordby M. H. Kjøniksen A.-L. Nyström B. Roots J. Biomacromol. 2003;4:337–343. doi: 10.1021/bm020107+. [DOI] [PubMed] [Google Scholar]
- Frazier R. A. Papadopoulou A. Mueller-Harvey I. Kissoon D. Green R. J. J. Agric. Food Chem. 2003;51:5189–5195. doi: 10.1021/jf021179v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.