

Abstract
Per- and polyfluoroalkyl substances (PFAS) are a class of environmental toxicants, and some, such as perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA), have been associated with hepatic steatosis in rodents and monkeys. It was hypothesized that perfluorosulfonic acids (C4, 6, 8), perfluorocarboxylic acids (C4–14), perfluoro(2-methyl-3-oxahexanoic) acid (HFPO-DA), 1H, 1H, 2H, 2H-perfluorooctanesulfonic acid (6:2 FTS) along with 3 PFOS precursors could induce expression of lipid metabolism genes and lipid deposition in human hepatocytes. Five-donor pooled cryopreserved human hepatocytes were cultured and treated with 0.1% DMSO vehicle or various PFAS (0.25 to 25 μM) in media. After a 48-hr treatment, mRNA transcripts related to lipid transport, metabolism, and synthesis were measured using a Quantigene Plex assay. After 72-hr treatments, hepatocytes were stained with Nile Red dye to quantify intracellular lipids. Overall, PFAS were transcriptionally active at 25 μM. In this model, lipid accumulation was not observed with C8-C12 treatments. Shorter chain PFAS (C4-C5), 6:2 FTS, and PFOS precursor, metFOSA, induced significant liver lipid accumulation, and gene activation at lower concentrations than legacy PFAS. In summary short chain PFAS and other alternative PFAS were more potent gene inducers, and potential health effects of replacement PFAS should be critically evaluated in humans.
Keywords: PFAS, perfluorinated compounds, Human hepatocytes, Lipids
GRAPHICAL ABTRACT
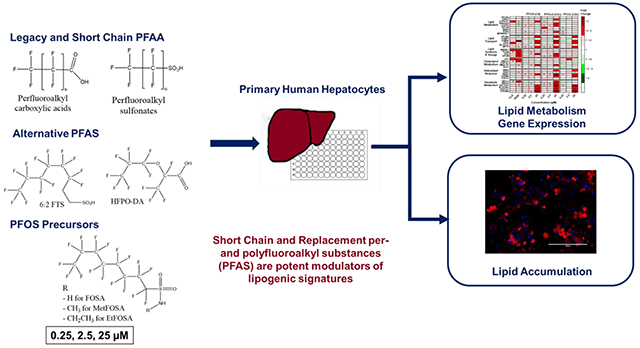
INTRODUCTION
Per- and polyfluoroalkyl substances (PFAS) are synthetic chemicals used in the production of many heat-, water-, and stain-resistant consumer products, and in aqueous film forming foams (AFFF) for fire suppression. PFAS contain strong carbon-fluorine bonds that allow these molecules to resist degradation and bioaccumulate. Two of the most well studied PFAS are eight-carbon (C8) perfluoroalkyl acids (PFAA), perfluorooctanesulfonate (PFOS) and perfluorooctanoic acid (PFOA). PFOS and PFOA have long serum half-lives in humans, with a mean of 5.4 years for PFOS and 3.8 years for PFOA (Olsen et al., 2007). Multiple human studies have found significant associations between PFAS exposure and adverse outcomes such as suppressed immunity, dyslipidemia, and kidney and testicular cancers in areas with extremely high exposures (Barry et al., 2013; Grandjean et al., 2017b, 2017a; Lin et al., 2019; Sunderland et al., 2019; Vieira et al., 2013). In rodents and monkeys, liver is known to be a sensitive organ to PFAS exposure, with PFOS and PFOA exposure causing decreased body weight, increased liver weight, and hepatocellular hypertrophy and lipid vacuolation (Butenhoff et al., 2002; Qazi et al., 2010; Seacat et al., 2003, 2002; Son et al., 2008; Wan et al., 2012). Human studies have found positive associations between PFAS exposure and biomarkers of liver injury, such as alanine aminotransferase (ALT) and cytokeratin 18 (Bassler et al., 2019; Darrow et al., 2016; Gallo et al., 2012; Gleason et al., 2015; Lin et al., 2010). Jin et al., (2020) has also found associations with PFAS exposure and the severity of liver injury in children with non-alcoholic fatty liver disease (NAFLD). PFAS have been shown to cause hepatic steatosis in rodents, however there is limited understanding of the ability of PFASs to induced liver diseases in humans.
In 2006, eight companies voluntarily phased out the use of legacy PFAS in the U.S. Since 1999-2000, serum PFOS and perfluorohexanesulfonate (PFHxS) concentrations in the U.S. population have been declining, whereas the PFOA concentrations remained constant between 2003-2008 and perfluorononanoic acid (PFNA) increased during 1999–2008 (Kato et al., 2011). Although the use of legacy PFAS compounds has decreased, companies have replaced them with short chain PFAA and alternative PFAS structures, such as perfluoro-2-propoxypropanoic acid (HFPO-DA, or GenX), a replacement for PFOA (Sun et al., 2016). Many of these new PFAS have been detected in humans and the environment (Supplemental Table 1). While PFOA and PFOS have been well studied, less is known about the biological activity of new PFAS detected in the environment. Perfluorooctane sulfonamide (FOSA), and like structures (MetFOSA and EtFOSA), are PFOS precursors, and can be metabolized to PFOS, however, like the new PFAS, little is known about their potential toxicity (Gebbink et al., 2016, 2015; Letcher et al., 2014). Short chain PFAS typically have shorter half lives in humans (Supplemental Table 1), and because of this, are generally thought of as safer alternative to legacy PFAS. Rosen et al. (2013) used primary mouse and human hepatocytes to evaluate lipid pathways of many PFAA, however genes that were differentially expressed were inconsistent across compounds, making comparisons of biological activity between compounds difficult. It is unclear whether short chain PFAS have similar gene expression profiles to legacy PFAS, and whether these short chain PFAS have similar potency in human hepatocytes. In general, PFOA, PFOS, and PFHxS are known to activate Peroxisome Proliferator Activated Receptor (PPAR) isoforms, such as PPAR-α and PPAR-γ with PPAR-α activation thought to be more robust in rodents compared to humans (Behr et al., 2020). Additionally, PFOA and PFOS have been shown to upregulate the downstream expression of genes associated with Constitutive Androstane Receptor (NR1I3 or CAR), such as Cytochrome P450 Family 2 Subfamily B Member 6 (CYP2B6), as well as Nuclear Factor E2-Related Factor 2 (NRF2), such as Superoxide Dismutase 1 (SOD1) and NAD(P)H Quinone Dehydrogenase 1 (NQO1) (Abe et al., 2017; Alharthy and Hardej, 2021; Hamilton et al., 2021).
PFOS and PFOA can induce hepatic steatosis in mice and rats. PFOS induces a liver pathology in cynamologus monkey consistent with steatosis. Primary hepatocyte studies have demonstrated increased expression of genes involved in lipogenesis (Bjork et al., 2011; Salter et al., 2021), but have not shown whether the gene expression changes are associated with increased cellular lipid accumulation. In addition, it is important to understand whether PFAS that are still in use (i.e. PFBS, PFHxS) behave similarly to PFAS that have been withdrawn from use (i.e. PFOA and PFOS). We hypothesize that emerging PFAS and PFAS replacements will induce lipogenic gene expression profiles and increase cellular lipid deposition.
Herein, critical compounds in the PFAS family have been screened for modulation of lipogenic gene expression in primary human hepatocytes. In addition to evaluating gene expression, Nile Red staining was included to evaluate intracellular lipid accumulation, since few studies have evaluated the potential for hepatic steatosis in humans. The highest PFAS concentration (25 μM) was selected based previous studies where PFOS and PFOA induced gene expression changes in human hepatocytes (Bjork et al., 2011; Liu et al., 2007). The first reports of human occupational exposure for PFOS, PFOA, and PFHxS have been reported as high 799, 691, and 290 ng/mL in serum, which corresponds to approximately 1.60, 1.67, and 0.73 μM concentrations, respectively (Olsen et al., 2007). Lower concentrations (2.5 and 0.25 μM) were also evaluated to have more environmental relevance, and all concentrations were kept the same across compounds to compare potency. By choosing the same doses for these structurally diverse compounds, this will allow for identification of structural features that produced lipid accumulation and specific gene expression signatures to be compared. Herein we describe that in the hepatocyte culture system used, shorter chain PFAS (C4-C6), 6:2 FTS, and PFOS precursors induced significant liver lipid accumulation, and gene activation at lower concentrations than legacy PFAS.
MATERIALS AND METHODS
Test chemicals.
Legacy PFAS (PFHxS, PFOS, and PFOA), short chain PFAA (PFBA, PFPeA, PFHxA, PFHpA, and PFBS,), long chain PFAA (PFNA, PFDA, PFUnDA, PFDoDA, PFTrDA, and PFTeDA), and alternative PFAS (HFPO-DA and 6:2 FTS), and precursor PFAS (FOSA, MetFOSA, and EtFOSA) as described in Supplemental Table 2 were purchased from AccuStandard Inc. (New Haven, CT, USA) and dissolved in dimethylsulfoxide (DMSO; Sigma Aldrich, St. Louis, MO). Known PPAR activators were purchased from Sigma Aldrich (St. Louis, MO): rosiglitazone (ROSI; PPARγ activation) and clofibrate (CLO; PPARα activation) were prepared in DMSO stocks and diluted in media for treatments of 5 μM and 250 μM respectively based effective responses in previous studies (Moon et al., 2012; Richert et al., 2003). A palmitate and oleate (1:2, P/O) mixture was used as a positive control for lipid accumulation. A 10 mM P/O stock mixture was created using sodium salts of palmitate and oleate from Sigma Aldrich (St. Louis, MO). 3.33 mmol palmitate and 6.66 mmol oleate was dissolved in deionized water in a boiling water bath. Once dissolved, the fatty acids were then complexed to 10% BSA (Bovine Serum Albumin, Fisher Scientific, Catalog# BP1600-100, Fair Lawn, NJ) by gently heating at 37°C. Once cooled, the P/O mixture was filtered through a 0.2 μm syringe filter and stored at −20°C until use. The 10 mM P/O stock was diluted in media to a final concentration of 0.5 mM for hepatocyte treatment.
Cell culture and treatment.
Cryopreserved human hepatocytes (CryostaX 5 donor pool, catalog# HPCH05+, Lot 1510213) and thawing and plating media (kits K8000, K8200, and K8300) were obtained from Sekisui XenoTech (Kansas City, KS). Cells were thawed and plated according to manufacturer protocols. Briefly, cryotube was thawed in a 37°C water bath for ~80 seconds and then place in OptiThaw medium and gently inverted. The cells were centrifuged at 100 x g for 5 min at room temperature and supernatant was discarded. Cells were resuspended in OptiPlate media. Cell viability and yield was assessed using trypan blue exclusion method on a hemocytometer. Hepatocytes were then seeded into collagen I coated 96-well plates at the recommended seeding density (5.6 x 104 cells per well). The plates were then incubated at 37°C in 5% CO2 humidified air for 4 hours. OptiPlate media was replaced with OptiCulture media (supplemented with Pen/Strep mix) and maintained at 37°C in 5% CO2 humidified air for 24 hours. After a 24-hour incubation, cells were treated with either vehicle (0.1% DMSO), PFAS (0.25, 2.5, and 25 μM), or positive controls (5 μM ROSI, 250 μM CLO, and 0.5 mM 1:2 P/O) added to OptiCulture media with a final DMSO concentration of 0.1% in the culture medium. Each plate contained controls and technical replicates of 4 wells per treatment. Every 24 hours, media was removed and replaced by fresh media containing treatments. Cell lysates were collected after 48 hours for evaluation of gene expression. A second set of cells were fixed and stained with Nile Red/DAPI after 72 hours for evaluation of cellular lipid disposition.
QuantiGene Plex Assay.
The QuantiGene plex assay uses Quantigene Branched DNA (bDNA) technology and is highly concordant to qPCR (Canales et al., 2006). Cell lysates were prepared using the QuantiGene Sample Processing Kit (Invitrogen by Thermo Fisher Scientific, Santa Clare, CA) and stored at −80°C until use. Plates were warmed to room temperature and heated to 37°C for 30 min before mRNA was quantified. The quantification of selected mRNAs was performed using the QuantiGene 2.0 Plex Assay kit targeted for 36 individual genes in lipid and xenobiotic metabolism pathways (Invitrogen by Thermo Fisher Scientific, Santa Clare, CA #QCP139-M18041194). The gene targets were selected based on known gene PFAS targets and pathways, such as PPARs, NR1L3, and nuclear receptor subfamily 1, group I, member 2 (NR1I2, or PXR) as describe by Bjork et al. (2011). The assay was conducted according to manufacturer protocols for use with cell lysate. The assay was read on a Bio-Plex® 200 system (Bio-Rad Laboratories, Hercules, CA). The fluorescence intensity (FI; minus background) for each gene was normalized to the housekeeping gene β-actin.
Nile Red Staining.
Cells were fixed in 10% formalin for 10 min at room temperature before staining. Nile Red was reconstituted in DMSO (3.1 mM) and aliquoted to avoid freeze thaw cycles. Nile Red was added to cells at a final concentration of 3.1 μM in phosphate buffered saline (PBS) to image the intracellular lipid stores (Erion et al., 2015). After incubating for 15 min, the solution was removed, and DAPI (4’,6-diamidino-2-phenylindole) was used as a counterstain (300 nM in PBS for 5 min). After washing with PBS, cells were imaged using an EVOS® FL Auto Cell Imaging System (Waltham, MA). Fluorescence was quantified on a SpectroMax M2 plate reader (Molecular Devices, San Jose, CA) with excitation and emission wavelengths of 485/535 nm for Nile Red and 358/461 nm for DAPI. Nile Red fluorescence was normalized to DAPI fluorescence to account for difference in cell number and compared to the DMSO treated cells.
Quantitative structure-activity relationship (QSAR) model development.
To investigate the relationships between PFAS’s structures/properties and lipid accumulation or related gene expression regulation, Quantitative Structure-Activity relationship (QSAR) models were developed (Ciallella et al., 2020). Two types of molecular descriptors (RDKit and Dragon) were combined with three machine learning algorithms including k Nearest Neighbor (kNN), Random Forest (RF) and Support Vector Machine (SVM), for QSAR model developments (Russo et al., 2018). Total 70 physic-chemical property descriptors were calculated using the Dragon software version 6.0. Total 200 RDKit descriptors including compositional and electro-topological state information were calculated using the standard RDKit (www.rdkit.org) package. The fold changes of lipid accumulation and 35 gene expressions under the highest concentration (25 μM) were used to define the activity classifications of tested molecules. For each of the 36 endpoints, the continuous fold change value was converted to binary classifications (0 as inactives or 1 as actives) by defining corresponding thresholds. The thresholds of classifications and the number of actives/inactives were listed in Supplemental Table 3. All the QSAR models were evaluated by five-fold cross validation procedure 1, and the best model of each modeling set with the highest Correct Classification Rate (CCR) are shown in Supplemental Table 4.
Statistical Analysis.
Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Dunnett’s test for multiple comparisons using GraphPad Prism software v8.3.0 (La Jolla, CA). Significance was considered to be p<0.05. All values are means ± SEM; N = 3-4 per group.
RESULTS
PFAS modulate lipid metabolism pathways in human hepatocytes.
Mean fold change and statistics for each individual gene and treatment are included in a Supplemental Excel file. ROSI induced expression of Fatty Acid Binding Protein 4 (FABP4) by 4.3 fold and three of the lipid synthesis and storage pathway genes by ~1.5 fold, which is consistent with PPARγ activation (Lee et al., 2018). CLO induced Cytochrome P450 family 4 subfamily A member 11 (CYP4A11) by 1.5 fold, which is consistent with observed changes in human hepatocytes (Richert et al., 2003). The legacy PFAS (PFOA, PFHxS, and PFOS; Fig. 1) induced at least 7 transcripts out of 35 related to lipid metabolism, lipid transport, and antioxidant response pathways at the highest concentration of 25 μM. The legacy PFAS also highly induced expression of genes related to xenobiotic metabolism, such as Cytochrome P450 2B6 (CYP2B6) and Sulfotransferase 2A1 (SULT2A1) at 25 μM. Unlike the legacy PFAS, the short chain PFAA (e.g. PFBA, PFPeA, PFHxA, PFHpA, and PFBS; Fig. 2) increased the relative transcript abundance for at least 7 of the 35 transcripts measured at all three concentrations tested, with the exception of the PFBA and PFPeA at 25 μM. Short chain PFAA induced the expression of transcripts in all pathways, with the exception of the antioxidant response that only has slight induction (< 1.5-fold). Also, unlike the legacy PFAS, there was a robust induction of transcripts related to lipid synthesis and storage pathways (i.e. Sterol Regulatory Element-Binding Protein 1 (SREBF1); Fatty Acid Synthase (FASN); Stearoyl-CoA Desaturase, (SCD); Glycerol-3-phosphate acyltransferase 1, mitochondrial, GPAM). Of the long chain PFAA (Fig. 3), PFNA only had minimal significant gene expression changes over (< 1.5-fold) and induced CPT1B mRNA over 1.5-fold at 0.25 μM). PFDA (C10) showed a similar profile to legacy PFAS with induction of in 17 out of 35 gene targets over 1.5-fold at 25 μM (Fig. 3). PFUnDA (C11) and PFDoDA (C12), produced similar gene induction profiles to short chain PFAA, however this was observed only at lower concentrations (0.25-2.5 μM). Treatment with C11 at 25 μM had induction of 3 out of 35 gene targets over 1.5-fold, and C12 at 25 μM repressed 14 of 35 gene targets. PFTrDA (C13) and PFTeDA (C14) had much less induction of genes analyzed. C13 induced Phosphoenolpyruvate carboxykinase 2, mitochondrial (PCK2; 25 μM), and GPAM, (25 μM) gene expression, and there was no significant changes in gene expression changes with C14 treatment.
Fig. 1. Legacy PFAA exposure modulates gene expression changes in cryopreserved human hepatocytes.
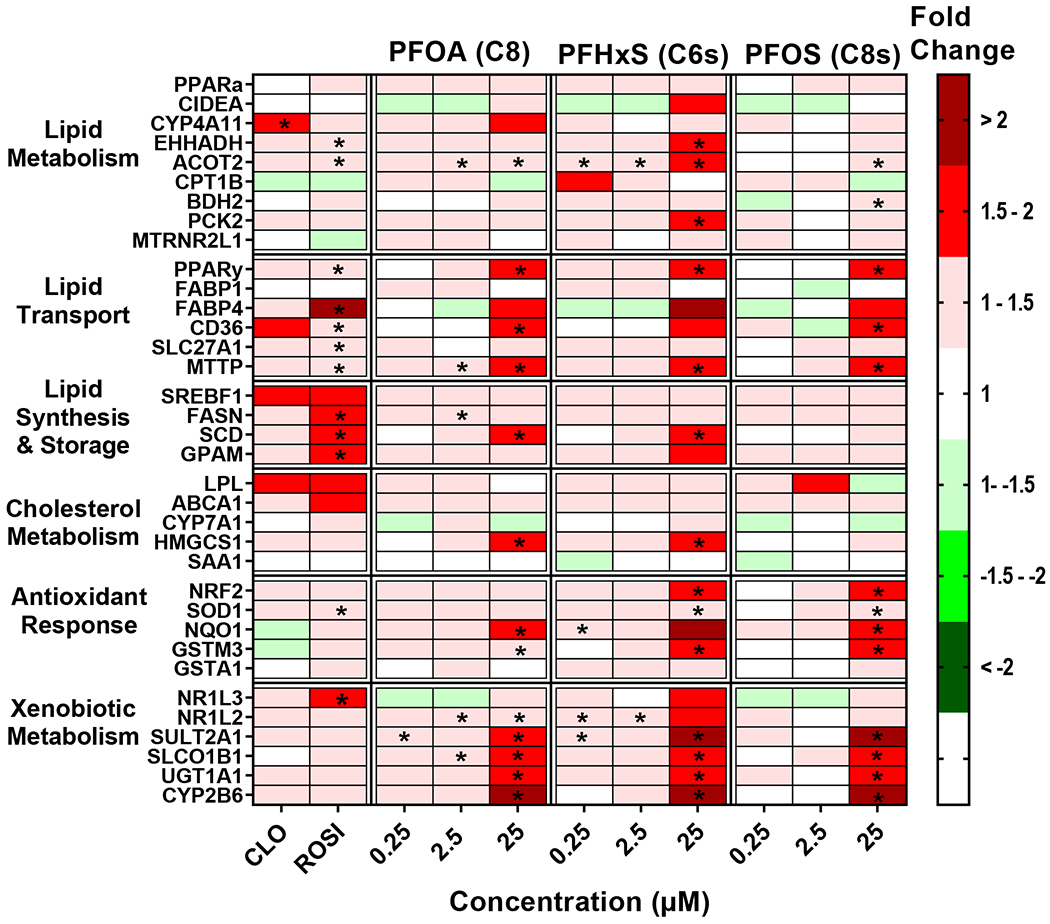
Human hepatocytes were treated with PFOA, PFHxS, and PFOS at concentrations of 0.25-25 μM. Cell lysate was processed, and gene expression was analyzed using a custom QuantiGene bead plex assay and analyzed using BioPlex 200 System according to manufacturer’s protocols. Fluorescence intensity was normalized to β-actin and fold change was calculated compared to the vehicle control. Clofibrate (CLO, 250 μM) and rosiglitazone (ROSI, 5 μM) treatments were used as positive controls. Red indicates gene induction and green indicates gene repression. Fold change was calculated and analyzed using an ANOVA followed by Dunnett’s test compared to the DMSO treated cells. * indicates P < 0.05. All colors represent means; N = 3-4.
Fig. 2. Short chain PFAA exposure modulates gene expression changes in cryopreserved human hepatocytes.
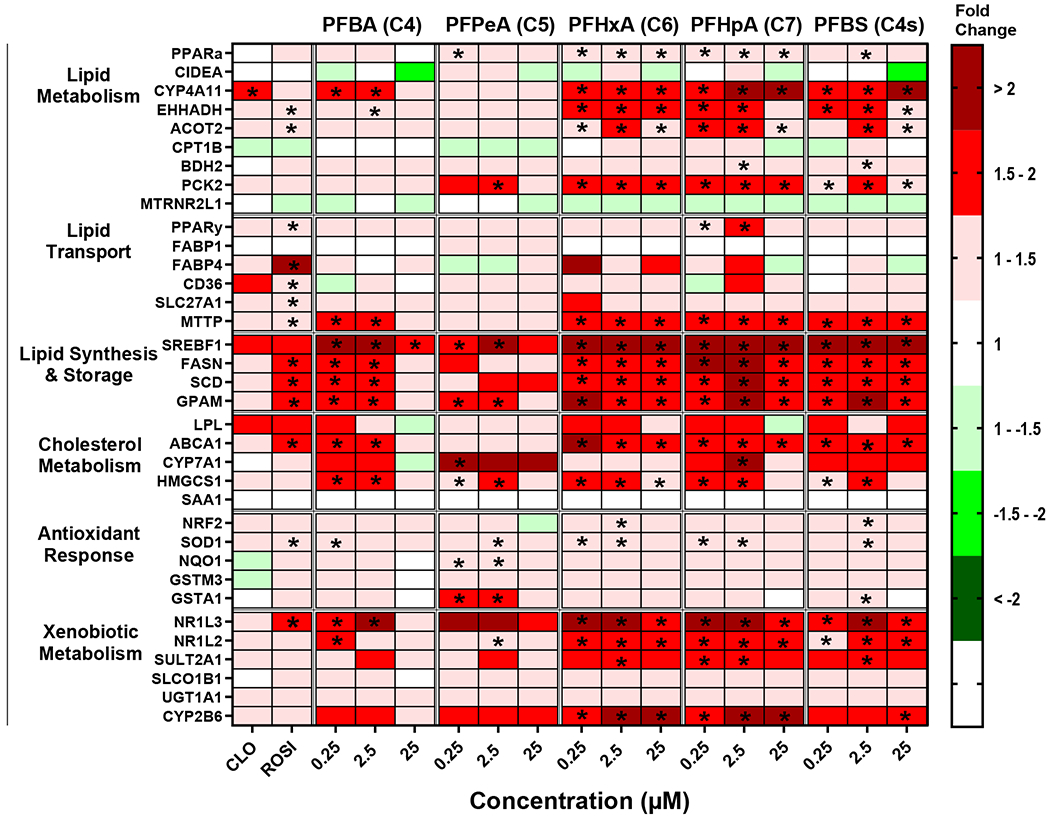
Human hepatocytes were treated with short chain length PFAA (C4-C7, and C4s) at concentrations of 0.25-25 μM. Cell lysate was processed, and gene expression was analyzed using a custom QuantiGene bead plex assay and analyzed using BioPlex 200 System according to manufacturer’s protocols. Fluorescence intensity was normalized to β-actin and fold change was calculated compared to the vehicle control. Clofibrate (CLO, 250 μM) and rosiglitazone (ROSI, 5 μM) treatments were used as positive controls. Red indicates gene induction and green indicates gene repression. Fold change was calculated and analyzed using an ANOVA followed by Dunnett’s test compared to the DMSO treated cells. * indicates P < 0.05. All colors represent means; N = 3-4.
Fig. 3. Long chain carboxylic acid PFAA exposure modulates gene expression changes in cryopreserved human hepatocytes.
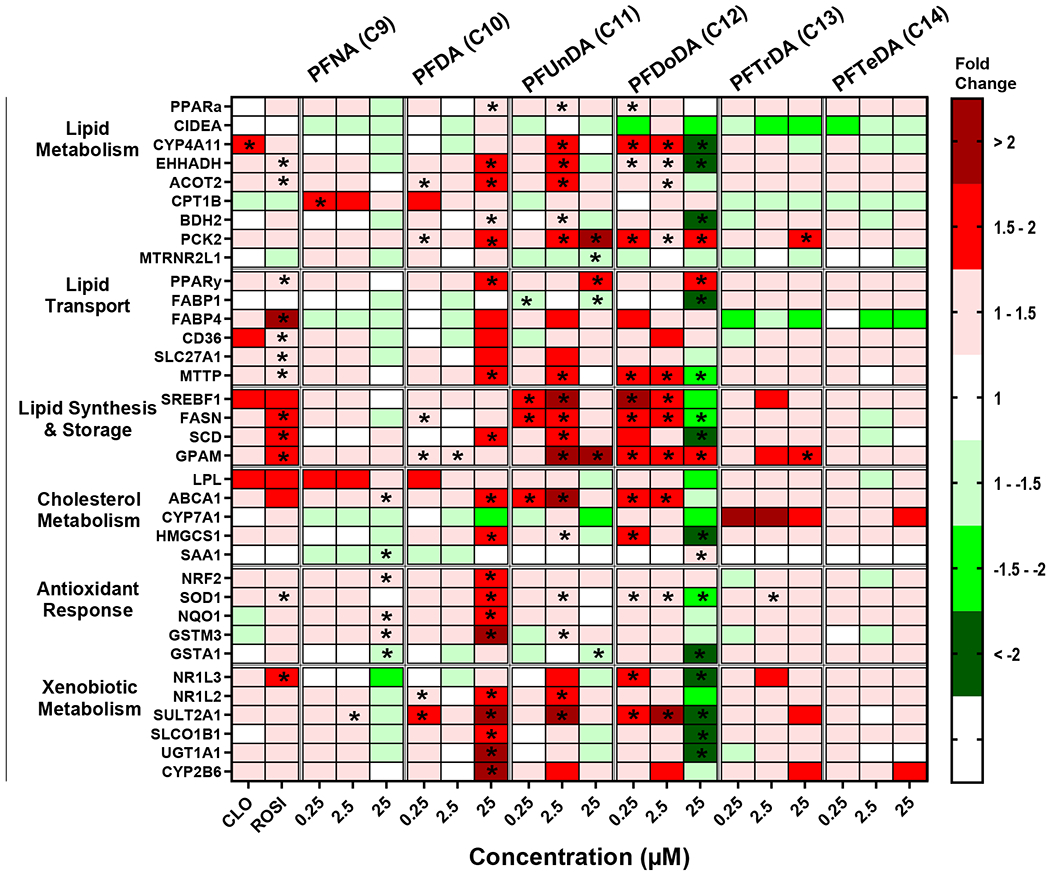
Human hepatocytes were treated with long chain PFAA (C9-C14)at concentrations of 0.25-25 μM. Cell lysate was processed, and gene expression was analyzed using a custom QuantiGene bead plex assay and analyzed using BioPlex 200 System according to manufacturer’s protocols. Fluorescence intensity was normalized to β-actin and fold change was calculated compared to the vehicle control. Clofibrate (CLO, 250 μM) and rosiglitazone (ROSI, 5 μM) treatments were used as positive controls. Red indicates gene induction and green indicates gene repression. Fold change was calculated and analyzed using an ANOVA followed by Dunnett’s test compared to the DMSO treated cells. * indicates P < 0.05. All colors represent means; N = 3-4.
Fig. 4 depicts effects of alternative and precursor PFAS on gene expression in human hepatocytes. HFPO-DA, like the legacy PFAS, only induced genes at 25 μM. HFPO-DA was the only compound to induce cell death activator CIDE-A (CIDEA), and HFPO-DA also induced 6 out of 35 genes in lipid transport, cholesterol, and xenobiotic metabolism pathways. Overall, 6:2 FTS, MetFOSA, and EtFOSA exerted similar gene expression profiles with short chain PFAA. 6:2 FTS, MetFOSA, and EtFOSA upregulated 9, 10, and 8 of 35 transcripts, respectively, at the lowest concentration tested (0.25 μM) and exerted greater than two-fold induction in the lipid synthesis and storage pathway genes. FOSA induced gene expression only at 2.5 μM and increased transcript levels related to lipid synthesis and storage, and cholesterol metabolism pathways.
Fig. 4. Alternative and precursor PFAS exposure modulates gene expression changes in cryopreserved human hepatocytes.
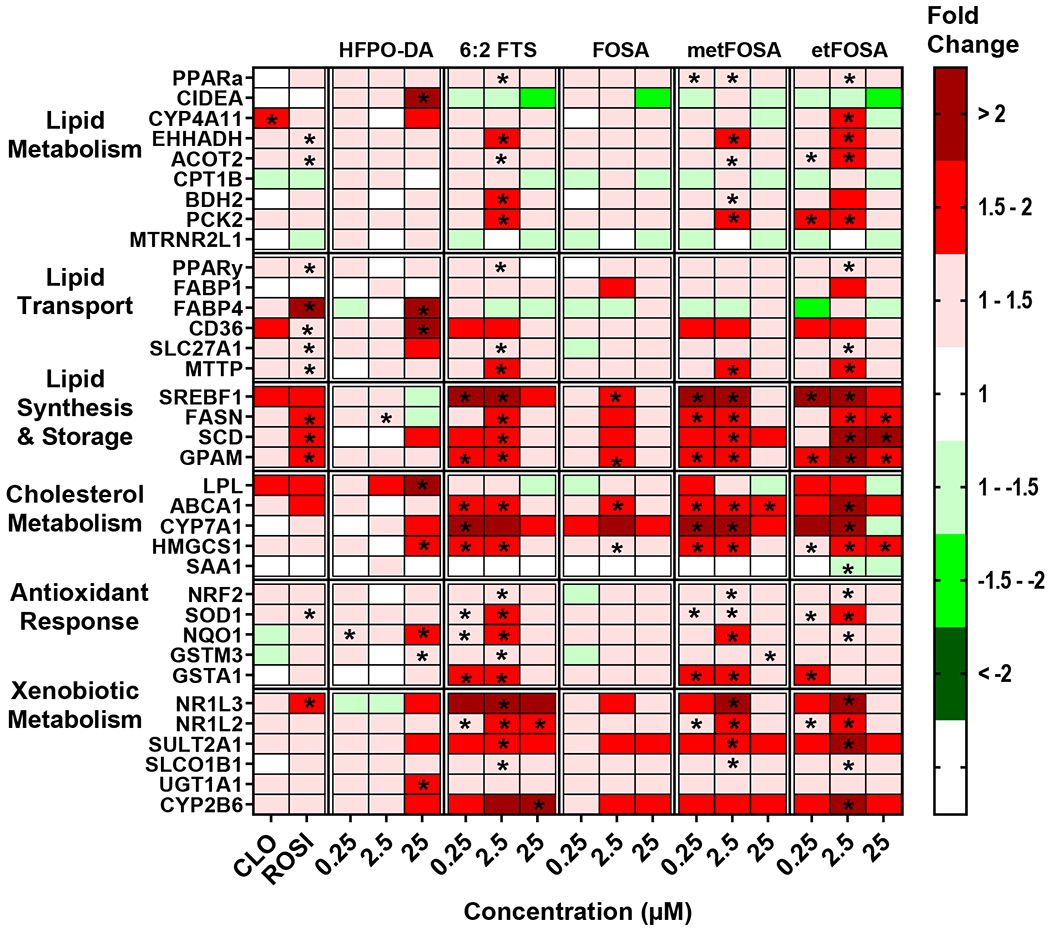
Human hepatocytes were treated with HFPO-DA, 6:2 FTS, FOSA, metFOSA, and etFOSA at concentrations of 0.25-25 μM. Cell lysate was processed, and gene expression was analyzed using a custom QuantiGene bead plex assay and analyzed using BioPlex 200 System according to manufacturer’s protocols. Fluorescence intensity was normalized to β-actin and fold change was calculated compared to the vehicle control. Clofibrate (CLO, 250 μM) and rosiglitazone (ROSI, 5 μM) treatments were used as positive controls. Red indicates gene induction and green indicates gene repression. Fold change was calculated and analyzed using an ANOVA followed by Dunnett’s test compared to the DMSO treated cells. * indicates P < 0.05. All colors represent means; N = 3-4.
Hepatocyte Morphology.
After 72 hours, hepatocytes were stained with Nile Red and DAPI and observed under a fluorescent microscope. Representative images of 25 μM PFAS treatment are shown in Figs. 5–8. The hepatocytes treated with 0.5 mM of palmitate and oleate mixture (1:2, P/O) increased Nile Red staining by approximately 4-fold. The 25 μM treatments of PFNA, PFUnDA and PFDoDA (Fig. 7C, 7E, and 7F) appear to have less cells compared to the DMSO controls. As described in Supplemental Fig. 1, there was no toxicity observed with PFAS treatment, after evaluation of ATP levels.
Fig. 5. Legacy PFAA did not induced liver lipid accumulation.
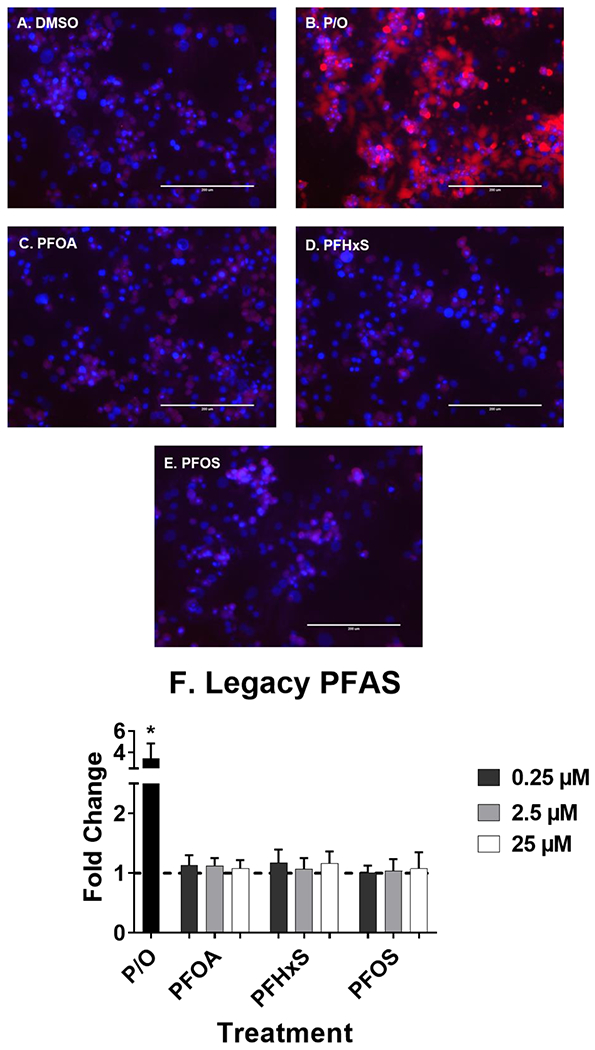
Human hepatocytes were treated with PFOA, PFHxS, and PFOS (0.25-25 μM) for 72 hours to induce liver lipid accumulation. 1:2 Palmitate and Oleate (0.5 mM, P/O) was included as positive control for lipid accumulation. Representative fluorescent images at 25 μM (A-E) were taken using an EVOS® FL Auto Cell Imaging System. Nile Red fluorescence was measured (excitation 485 nm/emission 535 nm) and normalized to DAPI fluorescence (excitation 358 nm/emission 461 nm). Fold change (F) was calculated compared to the DMSO treated cells. Calculations were done using an ANOVA followed by Dunnett’s test and * indicates P < 0.05. All values are means ± SEM; N = 3-4.
Fig. 8. 6:2 FTS and metFOSA induces liver lipid accumulation.
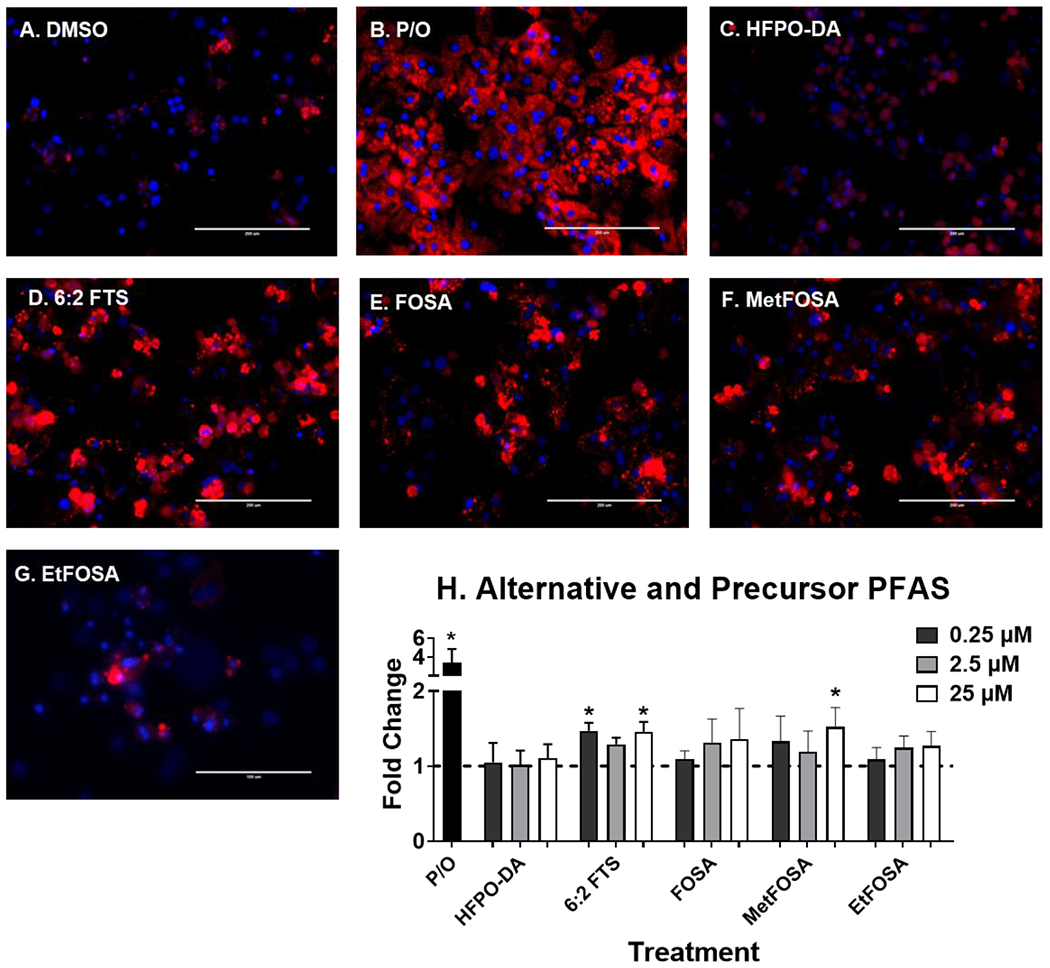
Human hepatocytes were treated with HFPO-DA, 6:2 FTS, FOSA, metFOSA, and etFOSA at 0.25-25 μM for 72 hours to induce liver lipid accumulation. 1:2 Palmitate and Oleate (0.5 mM, P/O) was included as positive control for lipid accumulation. Representative fluorescent images at 25 μM (A-G) were taken using an EVOS® FL Auto Cell Imaging System. Nile Red fluorescence was measured (excitation 485 nm/emission 535 nm) and normalized to DAPI fluorescence (excitation 358 nm/emission 461 nm). Fold change (H) was calculated compared to the DMSO treated cells. Calculations were done using an ANOVA followed by Dunnett’s test and * indicates P < 0.05. All values are means ± SEM; N = 3-4.
Fig. 7. PFTrDA induce liver lipid accumulation.
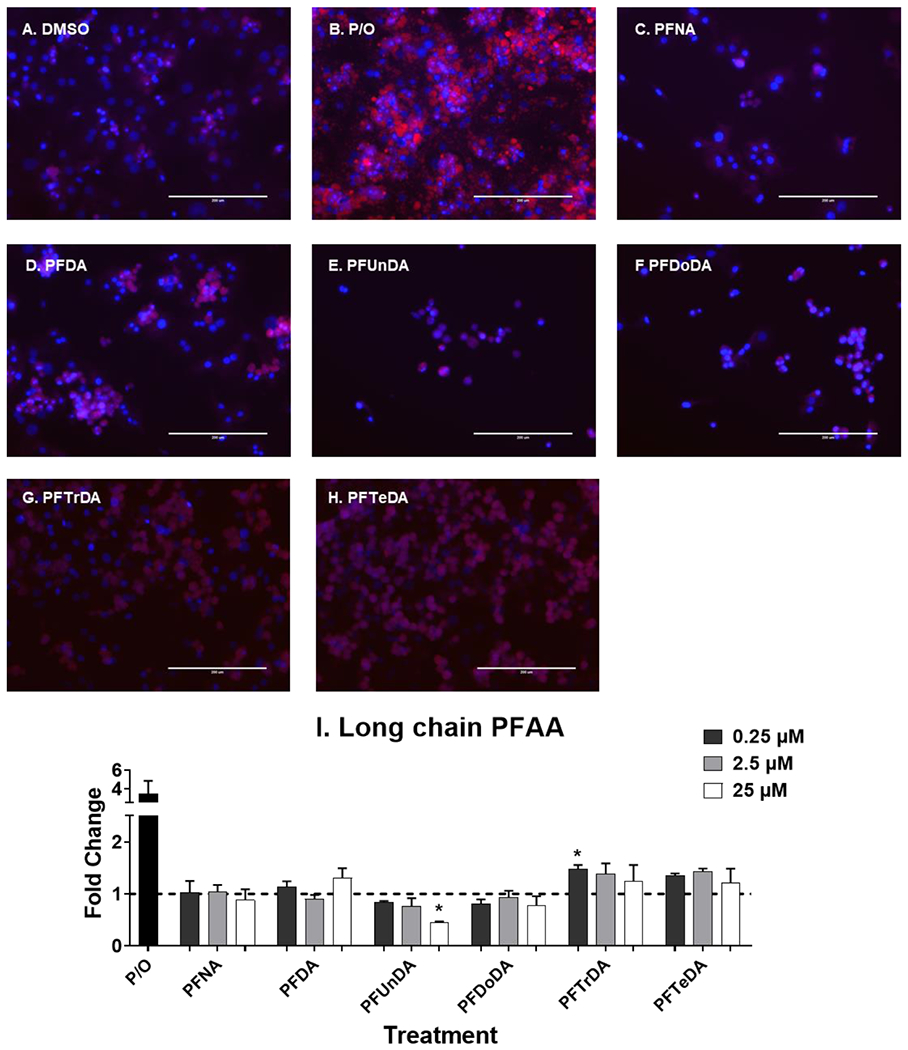
Human hepatocytes were treated with long chain carboxylic acid PFAA (C9-C14) at 0.25-25 μM for 72 hours to induce liver lipid accumulation. 1:2 Palmitate and Oleate (0.5 mM, P/O) was included as positive control for lipid accumulation. Representative fluorescent images at 25 μM (A-H) were taken using an EVOS® FL Auto Cell Imaging System. Nile Red fluorescence was measured (excitation 485 nm/emission 535 nm) and normalized to DAPI fluorescence (excitation 358 nm/emission 461 nm). Fold change (I) was calculated compared to the DMSO treated cells. Calculations were done using an ANOVA followed by Dunnett’s test and * indicates P < 0.05. All values are means ± SEM; N = 3-4.
Short chain PFAA, alternative and precursor PFAS induced lipid accumulation in hepatocytes.
At the concentrations selected, lipid accumulation was not observed for legacy PFAS (Fig. 5). Short chain PFAA (Fig. 6), however, did increase lipid accumulation. Treatment with PFBA (0.25-25 μM), PFPeA (2.5 μM)and PFBS (2.5 and 25 μM) increased cellular lipid staining by ~1.5-fold. Of the long chain PFAA (Fig. 7), PFUnDA at 25 μM decreased lipid accumulation by 2.2-fold. PFTrDA also increased lipid staining at (0.25 μM) by 1.5-fold. Of the alternative PFAS (Fig. 8), 6:2 FTS induced lipid accumulation at 0.25, and 25 μM) by 1.5-fold and MetFOSA induced lipid accumulation at 25 μM by 1.5-fold. HFPO-DA, FOSA, and EtFOSA did not significantly increase lipid staining.
Fig. 6. Short chain PFAA induce liver lipid accumulation.
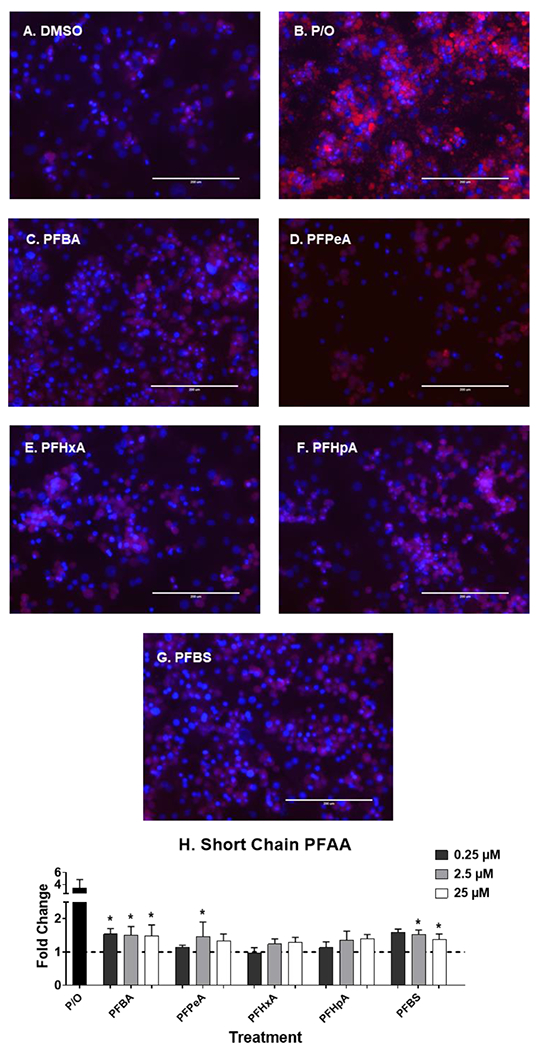
Human hepatocytes were treated with short chain PFAA (C4-C7, and C4s) at 0.25-25 μM for 72 hours to induce liver lipid accumulation. 1:2 Palmitate and Oleate (0.5 mM, P/O) was included as positive control for lipid accumulation. Representative fluorescent images at 25 μM (A-G) were taken using an EVOS® FL Auto Cell Imaging System. Nile Red fluorescence was measured (excitation 485 nm/emission 535 nm) and normalized to DAPI fluorescence (excitation 358 nm/emission 461 nm). Fold change (H) was calculated compared to the DMSO treated cells. Calculations were done using an ANOVA followed by Dunnett’s test and * indicates P < 0.05. All values are means ± SEM; N = 3-4.
Quantitative structure-activity relationship (QSAR) analysis.
To investigate the relationship between PFAS’s structures and lipid accumulation or related gene expression regulation, QSAR models were developed. The best model of each modeling set with the highest Correct Classification Rate (CCR) are shown in Supplemental Table 4. Most QSAR models showed acceptable predictivity (CCR > 0.7) and can be used to predict the lipid accumulation and related gene expression regulations of new PFASs in the future. By performing descriptor analysis of the modeling results (Table 1), it is possible to evaluate how structure features of PFASs affect their activities. For example, molecular weight (MW) was found to have significant contributions to lipid accumulation and key gene expression changes induced by PFAS since MW was ranked as the most important descriptor in most resulted models. The other common physic-chemical properties identified to be important are atomic ionization, polarity, and lipophilicity. Besides, the existence of hydrogen bond donors/acceptors (e.g. oxygens, nitrogens, sulfones, sulfonates, etc) and halogens also played important roles in models of specific endpoints.
Table 1.
Important Features for models of selected endpoints.
| Endpoint | Molecular weight | Atoms or groups | Lipophilicity | Bond | Ionization, polarizability |
|---|---|---|---|---|---|
| Lipid accumulation | Average molecular weight | Percentage of N atoms. Number of N atoms, and sulfones. Percentage of O atoms | Squared Moriguchi octanol-water partition coeff. (logP^2), and Hydrophilic factor | Mean atomic polarizability (scaled on Carbon atom). Topological polar surface area using N,O polar contributions | |
| CYP4A11 | Average molecular weight | Percentage of C atoms, and O atoms. Number of O atoms, S atoms, sulfonates (thio-/dithio-), and heavy atoms | Number of multiple bonds and double bonds | Unsaturation count, and unsaturation index | |
| EHHADH | Average molecular weight ignoring hydrogens | Number of heavy atoms | Wildman-Crippen LogP and surface area contribution | Topological polar surface area | |
| UGT1A1 | Average molecular weight | Percentage of C atoms, and halogen atoms. Number of O atoms and sulfonates (thio-/dithio-) | Moriguchi octanol-water partition coeff. (logP) | Rotatable bond fraction. Number of multiple bonds | Mean first ionization potential (scaled on Carbon atom) and Mean atomic polarizability (scaled on Carbon atom) |
| SULT2A1 | Average molecular weight | Percentage of O atoms. Number of O atoms, sulfonates (thio-/dithio-), S atoms, heavy atoms, and Heteroatoms. | Hydrophilic factor | Number of double bonds, and multiple bonds. | Unsaturation count, and unsaturation index. Mean first ionization potential (scaled on Carbon atom). Topological polar surface area using N,O polar contributions, and using N,O,S,P polar contributions, |
| SREBF1 | Average and exact molecular weights. Average molecular weight ignoring hydrogens | Number of halogens, and alkyl halides | Wildman-Crippen LogP value, and Wildman-Crippen LogP and surface area contribution | Number of rotatable bonds | |
| SCD | Average molecular weight ignoring hydrogens | Number of Heteroatoms and heavy atoms | Wildman-Crippen LogP and surface area contribution | Number of rotatable bonds | |
| MTTP | Average molecular weight | Percentage of C atoms and halogen atoms. Number of O atoms, sulfonates (thio-/dithio-), S atoms, and heavy atoms | Moriguchi octanol-water partition coeff. (logP) | Rotatable bond fraction. Number of multiple bonds, and double bonds | Mean first ionization potential (scaled on Carbon atom) and Mean atomic polarizability (scaled on Carbon atom). Unsaturation count and unsaturation index. Topological polar surface area using N,O,S,P polar contributions and using N,O polar contributions |
DISCUSSION
This work screened critical members of PFAS family to gather information on trends in gene expression and lipid accumulation related to PFAS structure, gene expression, and lipid accumulation in human hepatocytes. Potent induction of gene expression was observed for at least eight of the PFAS tested. Legacy PFAS are known PPARα inducers; Wolf et al. (2012) observed increasing activity of PPARα with increasing chain length of the PFAA up to PFNA and lower activity with longer chain PFAA. These assays showed limited PPARα activation with the legacy PFAS, as there was only slight induction in several PPARα regulated genes, such as CYP4A11, Enoyl-CoA Hydratase and 3-hydroxyacyl CoA Dehydrogenase (EHHADH), and Acyl-CoA Thioesterase 2 (ACOT2). Consistent with Wolf et al. (2012) many of the short chain PFAA exhibited more potent activation in PPARα regulated genes, whereas longer chain PFAA exerted little to no induction. Of the alternative and precursor PFAS, 6:2 FTS, MetFOSA, and EtFOSA, and produced a slight activation in this pathway with inductions of EHHADH and PCK2 over 1.5-fold. Limited PPARα activation is not unexpected in our human model, as rodents have more robust PPARα activation than in humans (Richert et al., 2003). As PFOA in humanized and PPARα-null mouse models has also been described to induced hepatic steatosis (Filgo et al., 2015; Schlezinger et al., 2020), other PPARα-independent pathways has also been investigated.
Rosen et al. (2017) described several PPARα-independent pathways, such as CAR or NR1I3, and PPARγ. Consistent with Rosen et al. (2017), we observed activation of genes regulated by CAR and PPARy. Many of the PFAA increased the transcript levels of Microsomal Triglyceride Transfer Protein (MTTP), Long-chain Fatty Acid Transport Protein 1 (SLC27A1), FABP4 and/or Fatty Acid Translocase (CD36), –all PPARy regulated genes. The alternative and precursor PFAS also followed these trends. PFAA, alternative and precursor PFAS increased CAR, as well as several CAR-regulated genes, such as CYP2B6 and SULT 2A1. The cellular mechanisms that drive PFAS lipid modulation in human liver remain unknown, and activation of multiple of nuclear receptors have been described for several PFAS. Thus, our focus was to identification trends related lipid accumulation and pathways devoted to lipid accumulation. Several studies have evaluated the binding potential for PFAS to bind to nuclear factors, such as the PPARs, (Ishibashi et al., 2019; Li et al., 2019) and PXR (Lai et al., 2020; Zhang et al., 2017), which was evaluated in this study, as well as hepatocyte nuclear factor 4-alpha, HNF4α (Beggs et al., 2016). This knowledge could be used to prioritize PFAS based on activity in these assays and correlate the observed trends in chain length and lipid accumulation to help understand the potential for human health effects.
The 25 μM treatments of PFNA, PFUnDA and PFDoDA (Fig. 7C, 7E, and 7F) appear to have less cells compared to the DMSO controls, which may explain why PFNA, PFUnDA and PFDoDA overall had much less gene activation and/or gene repression at this concentration. As described in Supplemental Fig. 1, there was no toxicity observed with PFAS treatment, however another marker of cell viability may display different results. Apoptosis has been described for PFOS and PFOA in HepG2 cells (Hu and Hu, 2009), and increases in biomarkers of apoptosis have also been associated with serum PFAS levels in humans (Bassler et al., 2019). A limitation of this work is that long chain PFAA were much less soluble in DMSO and required slight heat (37°C) before they were soluble upon visual inspection at the highest concentration. The levels of PFAS and potential metabolites of precursor PFAS in the treatment media were also not evaluated. Further evaluation of longer chain PFAS and cytotoxicity should be conducted. Another limitation of this study is the use of hepatocyte lots that were pooled from the same donors in lieu of using multiple donors. Pooled hepatocytes allowed for less variability in response that would allow us to capture changes in gene expression and screen the responses to the different PFAS. Additional work comparing individual donors, various ethnicities and genotypes is critical to understanding individual responses to PFAS, and potentially sensitive populations.
The PFAS that increased cellular lipid content correlated closely with the PFAS that highly induced lipid synthesis and storage pathways. As described by the pathway map in Supplemental Scheme 1, increasing expression of Sterol Regulatory Element-Binding Proteins (SREBPs), or transcription factors that control lipid synthesis gene expression, can lead to increased amounts of lipid in hepatocytes and hepatic steatosis. We hypothesize that induction of the lipid synthesis pathway is the driving force behind observed increase in hepatic lipid deposition because no additional lipids were added to the culture media, however as the results of mRNA expression and lipid accumulation are not quite consistent with some of the compounds, especially with the long chain PFAA. Currently it is not clear which cellular pathways drive lipid accumulation. As the goal of this work screen critical PFAS structures for known gene activation, a compelling follow up to this body of work would be to use respective inhibitors and determine which pathways are critical for lipid accumulation. Also, there was no observable differences in dose-response with the induction of lipid accumulation, even though contractions span 10-fold differences. The lack of a dose-response may be due to the limited amount of glucose in the media. An interesting study to build upon this observation would be to add additional glucose to the media and measure the dose-response of lipid accumulation. Gene expression in the long chain PFAA displayed an inverted-U shape trend with PFUnDA, and PFDoDA exhibiting more gene activation at concentrations of 0.25 and 2.5 μM, whereas PFNA, PFDA (except for 25 μM), PFTrDA and PFTeDA had much less gene activation. This pattern of activity is likely due to structural differences such chain length and molecular weight. Treatment with PFTrDA and PFTeDA at all three concentrations also inducted lipid accumulation, and unlike the short chain PFAA, PFTrDA and PFTeDA had limited activation of the measured genes. It is possible, especially in these long chain PFAA, that lipid accumulation may be occurring through alternative pathways that were not measured in these assays, such as downregulation of lipid metabolism and transport, and HNF4α.
Surprisingly, the short chain PFAA and some of the alternative and precursor PFAS (6:2 FTS, MetFOSA, and EtFOSA) tested were among the most potent in these assays, with significant gene activation at all three concentrations (0.25-25 μM), whereas many of the legacy PFAS were transcriptionally active only at the 25 μM concentration. Short chain PFAS in general have shorter half lives in humans (Supplemental Table 1), however the results of these assays suggest that these newer compounds should be evaluated carefully. The potency may also be dependent on cellular partitioning, with short chain and replacement PFAS structures potentially being more permeable or have a varying affinity for hepatic transporters. Yang et al. (2009) has shown that short-chain perfluoroalkyl carboxylic acids, such as PFBA, have less affinity for the hepatic transporter, organic anion transporting polypeptide 1a1 (Oatp 1a1) in rats. Another study that evaluated cellular PFAS levels in HepG2 cells, found that short-chain length perfluoroalkyl carboxylic acids had less cellular levels, but PPARα activities were induced at lower cellular concentrations for the short-chain PFAS studies (Rosenmai et al., 2018), which does suggest that short-chain PFAA maybe more potent gene inducers than legacy PFAS. The observed trends differs from several previous studies (Bjork and Wallace, 2009; Buhrke et al., 2013; Rosen et al., 2013). The key difference between these previous studies and the present body of work was whether PFAS treatments were conducted in the presence of serum and serum proteins, such as albumin. In the present study, PFAS treatment was conducted in the presence of serum, which arguably better replicates an in vivo system as PFAS would be taken up by hepatocytes from the blood circulation. Similar to our work, Rosenmai et al. (2018), found that PPARα activities were induced at lower concentrations for the short-chain PFAA compared to the long-chain PFAA in HepG2 cells and include serum with PFAS treatments. To further understand the reason serum may affect trends in hepatocytes, protein binding values to bovine serum albumin from Allendorf et al. (2019) were plotted against key endpoint within the present study. As detailed in Supplemental Fig. 2, there is a significant correlation between the protein binding association constant (Ka) and PFAS molecular weights, suggesting that longer chain PFAS molecule are more liking to bind to serum albumin and reduce the amount available for hepatocyte activity. Although not significant, negative correlation trends were also observed, with lipid accumulation and gene expression.
To investigate the relationships between PFAS’s structures and lipid accumulation or related gene expression regulation, QSAR models were developed, and descriptor analysis of the modeling results (Table 1) fount molecular weight (MW) to have significant contributions to lipid accumulation and key gene expression changes induced by PFAS. For the endpoint of lipid accumulation (Table 1), average molecular weight, percentage of nitrogen, sulfones, and oxygen atoms, hydrophobicity, and polar surface area were the structural features that had significant contributions to lipid accumulation changes induced by PFAS. To the best of our knowledge, this is the first study to evaluate lipid accumulation with PFAS treatment in primary human hepatocytes. Other studies have shown increased lipid accumulation or hepatic steatosis with legacy PFAS in rodents and monkeys (Das et al., 2017; Filgo et al., 2015; Seacat et al., 2002), and this has also be observed with HFPO-DA treatment in mice and fish (Blake et al., 2020; Guillette et al., 2020). In vitro hepatocyte models have only evaluated gene expression changes (Bjork et al., 2011; Rosen et al., 2013), which this study has also evaluated. Although the current training set is too small to determine the actual effects (e.g. the level of effects) of these structure features, the analysis result strongly suggest the importance of structure diversity for PFAS in the current and future studies.
Overall, the goal of this work was to screen critical members of PFAS family to gather information on trends related to PFAS structure in human hepatocytes. We selected cryopreserved primary human hepatocytes as a model because they express uptake transporter proteins, such as OATPs and NTCP, known to mediate PFAS uptake (Han et al., 2012; Nakagawa et al., 2008; Yang et al., 2009). We observed potent induction of gene expression for at least eight PFAS tested, with induction of gene expression occurring at nanomolar concentrations. Our data indicate that many PFAS on the market also behave similarly at the level of cell signaling to PFOS and PFOA, which have been withdrawn. The data also demonstrates that differences in PFAS structure, such as molecular weight, impacts gene expression changes in hepatocytes regarding lipid metabolism and lipid accumulation with PFAS treatment. Overall, our study indicates that replacement PFAS and PFAS still in use can induce cellular changes in gene expression and lipid accumulation in human hepatocytes and suggest that further effects should be examined in humans.
Supplementary Material
Highlights:
19 PFAS were screened for gene expression and lipid accumulation in human hepatocytes
Short chain PFAA, alternative and precursor PFAS were more potent gene activators
Short chain PFAA, alternative and precursor PFAS induced lipid accumulation
Molecular weight and protein binding may impact activity and cellular uptake in hepatocytes
ACKNOWLEDGMENTS
This work was supported by NIH grant P42ES027706. Research was made possible by the use of equipment available through the Rhode Island Institutional Development Award (IDeA) Network of Biomedical Research Excellence from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103430. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would like to thank Vanessa Jabbour and Mackenzie Nigro for their help conducting cell culture, Nile Red staining, and gene expression experiments. This work was presented in part at the Gordon Research Conference and Seminar Cellular and Molecular Mechanisms of Toxicity in Summer 2019 (travel award to EM) and Society of Toxicology 2019 annual meeting (travel awards to EM).
2. ABBREVIATIONS
- PFAS
per- and polyfluoroalkyl substance
- PFAA
perfluoroalkyl acids
- PFBS (C4s)
perfluorobutane sulfonic acid
- PFHxS (C6s)
perfluorohexanesulfonate
- PFOS (C8s)
erfluorooctanesulfonate
- PFBA (C4)
perfluorobutanoic acid
- PFPeA (C5)
perfluoropentanoic acid
- PFHxA (C6)
perfluorohexanoic acid
- PFHpA (C7)
perfluoroheptanoic acid
- PFOA (C8)
perfluorooctanoic acid
- PFNA (C9)
perfluorononanoic acid
- PFDA (C10)
perfluorodecanoic acid
- PFUnDA (C11)
perfluoroundecanoic acid
- PFDoDA (C12)
perfluorododecanoic acid
- PFTrDA (C13)
perfluorotridecanoic acid
- PFTeDA (C14)
perfluorotetradecanoic acid
- HFPO-DA (GenX, PFPrOPrA)
perfluoro(2-methyl-3-oxahexanoic) acid
- FOSA
perfluorooctane sulfonamide
- MetFOSA
N-methyl perfluoro-1-octanesulfonamide
- EtFOSA
N-ethyl perfluoro-1-octanesulfonamide
- 6:2 FTS
1H, 1H, 2H, 2H-perfluorooctane sulfonic acid (6:2)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information.
Supplemental File containing PFAS reference information, cell viability, QSAR analysis, protein binding analysis (PDF)
Supplemental File describing mean fold change and statistics for each gene target and treatment (xlsx)
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT author statement
Author contributions:
Emily Marques: Conceptualization, Formal analysis, Investigation, Data Curation, Writing - Original Draft, Visualization
Marisa Pfohl: Investigation, Writing - Review & Editing
Giuseppe Tarantola: Investigation, Writing - Review & Editing
Lucie Ford: Investigation, Writing - Review & Editing
Ogochukwu Amaeze: Investigation, Writing - Review & Editing
Xuelian Jia: Methodology, Formal analysis, Investigation, Writing - Review & Editing
Jessica Alesio: Methodology, Formal analysis, Writing - Review & Editing
Sangwoo Ryu: Methodology, Formal analysis, Writing - Review & Editing
Hao Zhu: Resources, Writing - Review & Editing, Supervision
Geoffrey D. Bothun: Resources, Writing - Review & Editing, Supervision
Angela L. Slitt: Conceptualization, Resources, Writing - Review & Editing, Supervision, Project administration, Funding acquisition
REFERENCES
- Abe T, Takahashi M, Kano M, Amaike Y, Ishii C, Maeda K, Kudoh Y, Morishita T, Hosaka T, Sasaki T, Kodama S, Matsuzawa A, Kojima H, Yoshinari K, 2017. Activation of nuclear receptor CAR by an environmental pollutant perfluorooctanoic acid. Arch Toxicol 91, 2365–2374. 10.1007/s00204-016-1888-3 [DOI] [PubMed] [Google Scholar]
- Alharthy SA, Hardej D, 2021. The role of transcription factor Nrf2 in the toxicity of perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) in C57BL/6 mouse astrocytes. Environmental Toxicology and Pharmacology 86, 103652. 10.1016/j.etap.2021.103652 [DOI] [PubMed] [Google Scholar]
- Allendorf F, Berger U, Goss K-U, Ulrich N, 2019. Partition coefficients of four perfluoroalkyl acid alternatives between bovine serum albumin (BSA) and water in comparison to ten classical perfluoroalkyl acids. Environ Sci Process Impacts 21, 1852–1863. 10.1039/c9em00290a [DOI] [PubMed] [Google Scholar]
- Barry V, Winquist A, Steenland K, 2013. Perfluorooctanoic acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ. Health Perspect 121, 1313–1318. 10.1289/ehp.1306615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler J, Ducatman A, Elliott M, Wen S, Wahlang B, Barnett J, Cave MC, 2019. Environmental perfluoroalkyl acid exposures are associated with liver disease characterized by apoptosis and altered serum adipocytokines. Environ. Pollut 247, 1055–1063. 10.1016/j.envpol.2019.01.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs KM, McGreal SR, McCarthy A, Gunewardena S, Lampe JN, Lau C, Apte U, 2016. The Role of Hepatocyte Nuclear Factor 4-Alpha in Perfluorooctanoic Acid- and Perfluorooctanesulfonic Acid-Induced Hepatocellular Dysfunction. Toxicol Appl Pharmacol 304, 18–29. 10.1016/j.taap.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr A-C, Plinsch C, Braeuning A, Buhrke T, 2020. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicology in Vitro 62, 104700. 10.1016/j.tiv.2019.104700 [DOI] [PubMed] [Google Scholar]
- Bjork JA, Butenhoff JL, Wallace KB, 2011. Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes. Toxicology 288, 8–17. 10.1016/j.tox.2011.06.012 [DOI] [PubMed] [Google Scholar]
- Bjork JA, Wallace KB, 2009. Structure-Activity Relationships and Human Relevance for Perfluoroalkyl Acid–Induced Transcriptional Activation of Peroxisome Proliferation in Liver Cell Cultures. Toxicological Sciences 111, 89–99. 10.1093/toxsci/kfp093 [DOI] [PubMed] [Google Scholar]
- Blake BE, Cope HA, Hall SM, Keys RD, Mahler BW, McCord J, Scott B, Stapleton HM, Strynar MJ, Elmore SA, Fenton SE, 2020. Evaluation of Maternal, Embryo, and Placental Effects in CD-1 Mice following Gestational Exposure to Perfluorooctanoic Acid (PFOA) or Hexafluoropropylene Oxide Dimer Acid (HFPO-DA or GenX). Environ Health Perspect 128, 27006. 10.1289/EHP6233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrke T, Kibellus A, Lampen A, 2013. In vitro toxicological characterization of perfluorinated carboxylic acids with different carbon chain lengths. Toxicology Letters 218, 97–104. 10.1016/j.toxlet.2013.01.025 [DOI] [PubMed] [Google Scholar]
- Butenhoff J, Costa G, Elcombe C, Farrar D, Hansen K, Iwai H, Jung R, Kennedy G, Lieder P, Olsen G, Thomford P, 2002. Toxicity of Ammonium Perfluorooctanoate in Male Cynomolgus Monkeys after Oral Dosing for 6 Months. Toxicol Sci 69, 244–257. 10.1093/toxsci/69.1.244 [DOI] [PubMed] [Google Scholar]
- Canales RD, Luo Y, Willey JC, Austermiller B, Barbacioru CC, Boysen C, Hunkapiller K, Jensen RV, Knight CR, Lee KY, Ma Y, Maqsodi B, Papallo A, Peters EH, Poulter K, Ruppel PL, Samaha RR, Shi L, Yang W, Zhang L, Goodsaid FM, 2006. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol 24, 1115–1122. 10.1038/nbt1236 [DOI] [PubMed] [Google Scholar]
- Ciallella HL, Russo DP, Aleksunes LM, Grimm FA, Zhu H, 2020. Predictive modeling of estrogen receptor agonism, antagonism, and binding activities using machine- and deep-learning approaches. Laboratory Investigation 1–13. 10.1038/s41374-020-00477-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow LA, Groth AC, Winquist A, Shin H-M, Bartell SM, Steenland K, 2016. Modeled Perfluorooctanoic Acid (PFOA) Exposure and Liver Function in a Mid-Ohio Valley Community. Environ. Health Perspect 124, 1227–1233. 10.1289/ehp.1510391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, Corton JC, Abbott BD, 2017. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 378, 37–52. 10.1016/j.tox.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erion KA, Berdan CA, Burritt NE, Corkey BE, Deeney JT, 2015. Chronic Exposure to Excess Nutrients Left-shifts the Concentration Dependence of Glucose-stimulated Insulin Secretion in Pancreatic β-Cells. J Biol Chem 290, 16191–16201. 10.1074/jbc.M114.620351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filgo AJ, Quist EM, Hoenerhoff MJ, Brix AE, Kissling GE, Fenton SE, 2015. Perfluorooctanoic Acid (PFOA)-induced Liver Lesions in Two Strains of Mice Following Developmental Exposures: PPARα Is Not Required. Toxicol Pathol 43, 558–568. 10.1177/0192623314558463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Leonardi G, Genser B, Lopez-Espinosa M-J, Frisbee SJ, Karlsson L, Ducatman AM, Fletcher T, 2012. Serum perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS) concentrations and liver function biomarkers in a population with elevated PFOA exposure. Environ. Health Perspect 120, 655–660. 10.1289/ehp.1104436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbink WA, Bignert A, Berger U, 2016. Perfluoroalkyl Acids (PFAAs) and Selected Precursors in the Baltic Sea Environment: Do Precursors Play a Role in Food Web Accumulation of PFAAs? Environ. Sci. Technol 50, 6354–6362. 10.1021/acs.est.6b01197 [DOI] [PubMed] [Google Scholar]
- Gebbink WA, Glynn A, Berger U, 2015. Temporal changes (1997–2012) of perfluoroalkyl acids and selected precursors (including isomers) in Swedish human serum. Environmental Pollution 199, 166–173. 10.1016/j.envpol.2015.01.024 [DOI] [PubMed] [Google Scholar]
- Gleason JA, Post GB, Fagliano JA, 2015. Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the US population (NHANES), 2007–2010. Environmental Research 136, 8–14. 10.1016/j.envres.2014.10.004 [DOI] [PubMed] [Google Scholar]
- Grandjean P, Heilmann C, Weihe P, Nielsen F, Mogensen UB, Budtz-Jørgensen E, 2017a. Serum Vaccine Antibody Concentrations in Adolescents Exposed to Perfluorinated Compounds. Environ. Health Perspect 125, 077018. 10.1289/EHP275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean P, Heilmann C, Weihe P, Nielsen F, Mogensen UB, Timmermann A, Budtz-Jørgensen E, 2017b. Estimated exposures to perfluorinated compounds in infancy predict attenuated vaccine antibody concentrations at age 5-years. J Immunotoxicol 14, 188–195. 10.1080/1547691X.2017.1360968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillette TC, McCord J, Guillette M, Polera ME, Rachels KT, Morgeson C, Kotlarz N, Knappe DRU, Reading BJ, Strynar M, Belcher SM, 2020. Elevated levels of per- and polyfluoroalkyl substances in Cape Fear River Striped Bass (Morone saxatilis) are associated with biomarkers of altered immune and liver function. Environment International 136, 105358. 10.1016/j.envint.2019.105358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton MC, Heintz MM, Pfohl M, Marques E, Ford L, Slitt AL, Baldwin WS, 2021. Increased toxicity and retention of perflourooctane sulfonate (PFOS) in humanized CYP2B6-Transgenic mice compared to Cyp2b-null mice is relieved by a high-fat diet (HFD). Food Chem Toxicol 152, 112175. 10.1016/j.fct.2021.112175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Nabb DL, Russell MH, Kennedy GL, Rickard RW, 2012. Renal elimination of perfluorocarboxylates (PFCAs). Chem. Res. Toxicol 25, 35–46. 10.1021/tx200363w [DOI] [PubMed] [Google Scholar]
- Hu X-Z, Hu D-C, 2009. Effects of perfluorooctanoate and perfluorooctane sulfonate exposure on hepatoma Hep G2 cells. Arch Toxicol 83, 851–861. 10.1007/s00204-009-0441-z [DOI] [PubMed] [Google Scholar]
- Ishibashi H, Hirano M, Kim E-Y, Iwata H, 2019. In Vitro and In Silico Evaluations of Binding Affinities of Perfluoroalkyl Substances to Baikal Seal and Human Peroxisome Proliferator-Activated Receptor α. Environ. Sci. Technol 53, 2181–2188. 10.1021/acs.est.8b07273 [DOI] [PubMed] [Google Scholar]
- Jin R, McConnell R, Catherine C, Xu S, Walker DI, Stratakis N, Jones DP, Miller GW, Peng C, Conti DV, Vos MB, Chatzi L, 2020. Perfluoroalkyl Substances and Severity of Nonalcoholic Fatty Liver in Children: An Untargeted Metabolomics Approach. Environ Int 134, 105220. 10.1016/j.envint.2019.105220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Wong L-Y, Jia LT, Kuklenyik Z, Calafat AM, 2011. Trends in exposure to polyfluoroalkyl chemicals in the U.S. Population: 1999-2008. Environ. Sci. Technol 45, 8037–8045. 10.1021/es1043613 [DOI] [PubMed] [Google Scholar]
- Lai TT, Eken Y, Wilson AK, 2020. Binding of Per- and Polyfluoroalkyl Substances to the Human Pregnane X Receptor. Environ. Sci. Technol 54, 15986–15995. 10.1021/acs.est.0c04651 [DOI] [PubMed] [Google Scholar]
- Lee YK, Park JE, Lee M, Hardwick JP, 2018. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Research 2, 209–215. 10.1016/j.livres.2018.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letcher RJ, Chu S, McKinney MA, Tomy GT, Sonne C, Dietz R, 2014. Comparative hepatic in vitro depletion and metabolite formation of major perfluorooctane sulfonate precursors in arctic polar bear, beluga whale, and ringed seal. Chemosphere 112, 225–231. 10.1016/j.chemosphere.2014.04.022 [DOI] [PubMed] [Google Scholar]
- Li C-H, Ren X-M, Cao L-Y, Qin W-P, Guo L-H, 2019. Investigation of binding and activity of perfluoroalkyl substances to the human peroxisome proliferator-activated receptor β/δ. Environ Sci.: Processes Impacts 21, 1908–1914. 10.1039/C9EM00218A [DOI] [PubMed] [Google Scholar]
- Lin C-Y, Lin L-Y, Chiang C-K, Wang W-J, Su Y-N, Hung K-Y, Chen P-C, 2010. Investigation of the associations between low-dose serum perfluorinated chemicals and liver enzymes in US adults. Am. J. Gastroenterol 105, 1354–1363. 10.1038/ajg.2009.707 [DOI] [PubMed] [Google Scholar]
- Lin P-ID, Cardenas A, Hauser R, Gold DR, Kleinman KP, Hivert M-F, Fleisch AF, Calafat AM, Webster TF, Horton ES, Oken E, 2019. Per- and polyfluoroalkyl substances and blood lipid levels in pre-diabetic adults—longitudinal analysis of the diabetes prevention program outcomes study. Environment International 129, 343–353. 10.1016/j.envint.2019.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Yu K, Shi X, Wang J, Lam PKS, Wu RSS, Zhou B, 2007. Induction of oxidative stress and apoptosis by PFOS and PFOA in primary cultured hepatocytes of freshwater tilapia (Oreochromis niloticus). Aquatic Toxicology 82, 135–143. 10.1016/j.aquatox.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Moon JH, Kim HJ, Kim HM, Yang AH, Lee B-W, Kang ES, Lee HC, Cha BS, 2012. Upregulation of hepatic LRP1 by rosiglitazone: a possible novel mechanism of the beneficial effect of thiazolidinediones on atherogenic dyslipidemia. Journal of Molecular Endocrinology 49, 165–174. 10.1530/JME-12-0119 [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Hirata T, Terada T, Jutabha P, Miura D, Harada KH, Inoue K, Anzai N, Endou H, Inui K-I, Kanai Y, Koizumi A, 2008. Roles of organic anion transporters in the renal excretion of perfluorooctanoic acid. Basic Clin. Pharmacol. Toxicol 103, 1–8. 10.1111/j.1742-7843.2007.00155.X [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR, 2007. Half-Life of Serum Elimination of Perfluorooctanesulfonate,Perfluorohexanesulfonate, and Perfluorooctanoate in Retired Fluorochemical Production Workers. Environ Health Perspect 115, 1298–1305. 10.1289/ehp.10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qazi MR, Abedi MR, Nelson BD, DePierre JW, Abedi-Valugerdi M, 2010. Dietary exposure to perfluorooctanoate or perfluorooctane sulfonate induces hypertrophy in centrilobular hepatocytes and alters the hepatic immune status in mice. Int. Immunopharmacol 10, 1420–1427. 10.1016/j.intimp.2010.08.009 [DOI] [PubMed] [Google Scholar]
- Richert L, Lamboley C, Viollon-Abadie C, Grass P, Hartmann N, Laurent S, Heyd B, Mantion G, Chibout S-D, Staedtler F, 2003. Effects of clofibric acid on mRNA expression profiles in primary cultures of rat, mouse and human hepatocytes. Toxicology and Applied Pharmacology 191, 130–146. 10.1016/S0041-008X(03)00231-X [DOI] [PubMed] [Google Scholar]
- Rosen MB, Das KP, Rooney J, Abbott B, Lau C, Corton JC, 2017. PPARα-independent transcriptional targets of perfluoroalkyl acids revealed by transcript profiling. Toxicology 387, 95–107. 10.1016/j.tox.2017.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Das KP, Wood CR, Wolf CJ, Abbott BD, Lau C, 2013. Evaluation of perfluoroalkyl acid activity using primary mouse and human hepatocytes. Toxicology 308, 129–137. 10.1016/j.tox.2013.03.011 [DOI] [PubMed] [Google Scholar]
- Rosenmai AK, Ahrens L, Godec T. le, Lundqvist J, Oskarsson A, 2018. Relationship between peroxisome proliferator-activated receptor alpha activity and cellular concentration of 14 perfluoroalkyl substances in HepG2 cells. Journal of Applied Toxicology 38, 219–226. 10.1002/jat.3515 [DOI] [PubMed] [Google Scholar]
- Russo DP, Zorn KM, Clark AM, Zhu H, Ekins S, 2018. Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol. Pharmaceutics 15, 4361–4370. 10.1021/acs.molpharmaceut.8b00546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter DM, Wei W, Nahar PP, Marques E, Slitt AL, 2021. Perfluorooctanesulfonic Acid (PFOS) Thwarts the Beneficial Effects of Calorie Restriction and Metformin. Toxicol Sci 182, 82–95. 10.1093/toxsci/kfab043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlezinger JJ, Puckett EL, Oliver J, Nielsen G, Heiger-Bernays W, Webster TF, 2020. Perfluorooctanoic acid activates multiple nuclear receptor pathways and skews expression of genes regulating cholesterol homeostasis in liver of humanized PPARα mice fed an American diet. bioRxiv 2020.01.30.926642. 10.1101/2020.01.30.926642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Clemen LA, Eldridge SR, Elcombe CR, Butenhoff JL, 2003. Sub-chronic dietary toxicity of potassium perfluorooctanesulfonate in rats. Toxicology 183, 117–131. 10.1016/S0300-483X(02)00511-5 [DOI] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Olsen GW, Case MT, Butenhoff JL, 2002. Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol. Sci 68, 249–264. [DOI] [PubMed] [Google Scholar]
- Son H-Y, Kim S-H, Shin H-L, Bae HI, Yang J-H, 2008. Perfluorooctanoic acid-induced hepatic toxicity following 21-day oral exposure in mice. Arch Toxicol 82, 239–246. 10.1007/s00204-007-0246-x [DOI] [PubMed] [Google Scholar]
- Sun M, Arevalo E, Strynar M, Lindstrom A, Richardson M, Kearns B, Pickett A, Smith C, Knappe DRU, 2016. Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environ. Sci. Technol. Lett 3, 415–419. 10.1021/acs.estlett.6b00398 [DOI] [Google Scholar]
- Sunderland EM, Hu XC, Dassuncao C, Tokranov AK, Wagner CC, Allen JG, 2019. A Review of the Pathways of Human Exposure to Poly- and Perfluoroalkyl Substances (PFASs) and Present Understanding of Health Effects. J Expo Sci Environ Epidemiol 29, 131–147. 10.1038/s41370-018-0094-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira VM, Hoffman K, Shin H-M, Weinberg JM, Webster TF, Fletcher T, 2013. Perfluorooctanoic acid exposure and cancer outcomes in a contaminated community: a geographic analysis. Environ. Health Perspect 121, 318–323. 10.1289/ehp.1205829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan HT, Zhao YG, Wei X, Hui KY, Giesy JP, Wong CKC, 2012. PFOS-induced hepatic steatosis, the mechanistic actions on β-oxidation and lipid transport. Biochim. Biophys. Acta 1820, 1092–1101. 10.1016/j.bbagen.2012.03.010 [DOI] [PubMed] [Google Scholar]
- Wolf CJ, Schmid JE, Lau C, Abbott BD, 2012. Activation of mouse and human peroxisome proliferator-activated receptor-alpha (PPARα) by perfluoroalkyl acids (PFAAs): Further investigation of C4–C12 compounds. Reproductive Toxicology, A Second Special Issue on Recent Advances in Perfluoroalkyl Acid Research 33, 546–551. 10.1016/j.reprotox.2011.09.009 [DOI] [PubMed] [Google Scholar]
- Yang C-H, Glover KP, Han X, 2009. Organic anion transporting polypeptide (Oatp) 1a1-mediated perfluorooctanoate transport and evidence for a renal reabsorption mechanism of Oatp1a1 in renal elimination of perfluorocarboxylates in rats. Toxicology Letters 190, 163–171. 10.1016/j.toxlet.2009.07.011 [DOI] [PubMed] [Google Scholar]
- Zhang Y-M, Dong X-Y, Fan L-J, Zhang Z-L, Wang Q, Jiang N, Yang X-S, 2017. Poly- and perfluorinated compounds activate human pregnane X receptor. Toxicology 380, 23–29. 10.1016/j.tox.2017.01.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.