

SUMMARY
Infection and vaccination repeatedly expose individuals to antigens that are conserved between influenza virus subtypes. Nevertheless, antibodies recognizing variable influenza epitopes greatly outnumber antibodies reactive against conserved epitopes. Elucidating factors contributing to the paucity of broadly reactive influenza antibodies remains a major obstacle for developing a universal influenza vaccine. Here, we report that inducing broadly reactive influenza antibodies increases autoreactive antibodies in humans and mice and exacerbates disease in four distinct models of autoimmune disease. Importantly, transferring broadly reactive influenza antibodies augments disease in the presence of inflammation or autoimmune susceptibility. Further, broadly reactive influenza antibodies spontaneously arise in mice with defects in B cell tolerance. Together, these data suggest that self-tolerance mechanisms limit the prevalence of broadly reactive influenza antibodies, which can exacerbate disease in the context of additional risk factors.
Graphical Abstract
In brief
Labombarde et al. investigate whether broadly reactive influenza antibodies, which may also bind self-antigens, contribute to autoimmunity. They demonstrate that induction of these antibodies also supports development of autoreactive antibodies in both humans and mice. Transferring broadly reactive monoclonal antibodies exacerbates autoimmunity in the context of inflammation or genetic susceptibility.
INTRODUCTION
Protective antibodies induced by influenza A virus vaccination or infection are primarily directed at epitopes in the variable head region of the viral surface protein, hemagglutinin (HA), and are typically specific for one influenza subtype (Angeletti and Yewdell, 2018a; Krammer, 2019). Conversely, broadly reactive influenza antibodies that bind epitopes conserved between distinct influenza subtypes are considerably less prevalent (Angeletti and Yewdell, 2018a; Corti and Lanzavecchia, 2013; Ellebedy et al., 2014; Krammer, 2019; Sui et al., 2011). Targeting these conserved epitopes is the basis for developing a universal influenza vaccine that provides immunity against distinct influenza strains (Ekiert and Wilson, 2012; Krammer, 2016; Angeletti and Yewdell, 2018a; Corti and Lanzavecchia, 2013; Corti et al., 2011; Ellebedy et al., 2014; Krammer, 2019; Sui et al., 2011; Wrammert et al., 2008). However, it remains unclear why these antibodies are so uncommon, despite repeated exposure to conserved influenza epitopes through vaccination or infection.
Many factors contribute to the paucity of broadly neutralizing influenza antibodies, including steric hindrance and molecular constraints caused by the proximity of some conserved epitopes to the viral membrane (Angeletti and Yewdell, 2018b; Guthmiller et al., 2021; Krammer, 2016; Lee and Wilson, 2015). A prevailing hypothesis was that B cells with high-affinity receptors specific for the variable epitopes outcompete B cells specific for conserved HA epitopes in germinal centers (Angeletti and Yewdell, 2018a). However, we demonstrated that in the absence of germinal center competition, broadly reactive influenza antibodies remained rare (Keating et al., 2020). Additionally, the precursor frequency of B cells specific for conserved and variable HA epitopes are similar, indicating that low precursor frequency does not limit the prevalence of broadly reactive influenza antibodies (Angeletti et al., 2019). Moreover, removal of the HA head from the stalk region supported a robust, high-affinity antibody response to the conserved stalk epitopes, indicating that these epitopes are immunogenic and can elicit an antibody response (Angeletti et al., 2019; Krammer, 2016). Thus, mechanisms that contribute to the scarcity of broadly reactive influenza antibodies remain unknown.
Recent data demonstrate that antibodies targeting conserved HA epitopes are more likely to be polyreactive than antibodies specific for the variable region of HA (Andrews et al., 2015; Bajic et al., 2019; Guthmiller et al., 2020; Khurana et al., 2020). Polyreactivity refers to the ability of an antibody to bind multiple, molecularly distinct antigens and was found to be an inherent property of antibodies binding the conserved HA stalk region (Guthmiller et al., 2020). Broadly reactive influenza antibodies also bound other proteins including insulin, lipopolysaccharide (LPS), and double-stranded DNA (dsDNA), demonstrating a propensity for autoreactivity. These data raise the possibility that self-tolerance mechanisms may limit the antibody response to the conserved regions of influenza virus. In support of this, vaccination or infection with the 2009 pandemic H1N1 strain, which generated more broadly neutralizing antibodies than previous seasonal strains (Andrews et al., 2015; Li et al., 2012; Pica et al., 2012; Thomson et al., 2012; Wrammert et al., 2011), increased the risk of autoimmune disorders, including narcolepsy and Guillain-Barré syndrome (GBS) (Barker and Snape, 2014; Hao et al., 2019; Kwong et al., 2013; Nohynek et al., 2012; Vellozzi et al., 2014; Vogel, 2015). However, direct evidence that enhancing immunity to conserved influenza epitopes promotes autoreactive antibodies is lacking. A critical unanswered question is whether antibodies that bind both influenza and self-antigens increase susceptibility to autoimmune disease. Thus, we utilized multiple mouse models to test whether the induction of broadly reactive influenza antibodies exacerbated autoimmune disease. Our data demonstrate that induction of broadly reactive influenza antibodies enhanced both generation of autoreactive antibodies and susceptibility to autoimmune disease. Transfer of broadly reactive influenza monoclonal antibodies exacerbated autoimmune disease in the context of inflammation or genetic predisposition to autoimmunity. Further, mice with genetic defects in B cell tolerance spontaneously developed broadly reactive influenza antibodies without prior exposure to influenza antigens. Importantly, humans infected with a novel strain of influenza, which induced higher levels of broadly reactive influenza antibodies than prior seasonal strains, also had elevated levels of anti-ganglioside antibodies. Together, our results suggest that tolerance mechanisms limit the generation of broadly reactive influenza antibodies and have important implications for designing safe and effective vaccines against epitopes naturally avoided by the immune system.
RESULTS
Induction of broadly reactive influenza antibodies increases development of autoreactive antibodies
To test whether enhancing broadly reactive influenza antibodies also increased autoreactive antibodies, we utilized a mouse model of vaccination in which C57BL/6 mice were given HKx31, an H3N2 strain of influenza, intraperitoneally (i.p.). The i.p. route is a well-established model of vaccination, as influenza replication is limited, yet the full spectrum of viral proteins is produced (Flynn et al., 1998). We previously demonstrated that mice treated with a low dose of rapamycin, in conjunction with HKx31 (HKx31 + RAP), had more broadly reactive influenza antibodies and better protection against subsequent heterosubtypic infections with H5N1, H7N9, and H1N1 strains than PBS-treated control mice (HKx31 + PBS) (Keating et al., 2013). Importantly, the enhanced protection afforded by rapamycin transferred to naive mice via serum and was due to an altered antibody response to HKx31. Since mice treated with HKx31 + RAP had increased broadly reactive influenza antibodies, we tested whether these mice also had higher levels of autoreactive antibodies than mice that received HKx31 + PBS. Serum was tested on protein arrays for immunoglobulin M (IgM) or IgG reactivity to autoantigens that are commonly targeted in autoimmune diseases (Li et al., 2007). Analyzing IgM reactivity showed that sera from HKx31 + RAP mice, which contained more broadly reactive influenza antibodies, had more autoantigen-binding IgM than sera from HKx31 + PBS mice (Figure 1A). Of the 128 antigens on this array, 47 antigens had significantly higher reactivity to antibodies from HKx31 + RAP mice than from HKx31 + PBS mice. Many of these proteins are commonly targeted in autoimmune arthritis (Figure 1B) and systemic lupus erythematosus (SLE) (Figure 1C). We confirmed reactivity to several of the array autoantigens by ELISA. As controls, we included serum from uninfected, C57BL/6 mice (normal mouse serum [NMS]) and NZBWF1 mice, which have a defect in tolerance and model human SLE (Morel, 2010). We found that HKx31 + RAP-treated mice had increased levels of antibodies specific for collagen (Figure 1D), histone H4 (Figure 1E), histone H1 (Figure 1F), and the Smith (Sm) antigen (Figure 1G) compared with HKx31 + PBS mice. Importantly, mice treated with rapamycin only, in the absence of HKx31, did not have more autoreactive antibodies than mice treated with PBS only (Figures 1D and 1E). Increased levels of IgM antibodies specific for collagen, histones, and GM1 + GD1a gangliosides in HKx31 + RAP mice were sustained for at least 90 days after exposure to HKx31 even though rapamycin was only given for 28 days (Figure 1H). Critically, HKx31 + RAP mice did not have a generalized increase in IgM antibodies that bind non-specifically to many antigens, as reactivity was not increased on a second array using antigens from other pathogens, allergens, and cancer-specific proteins (Figure S1A). Moreover, IgM antibodies reactive to insulin, LPS, dsDNA, anti-nuclear antibodies (ANAs), and keyhole limpet hemocyanin (KLH) by ELISA did not increase in HKx31 + RAP mice relative to HKx31 + PBS mice (Figures 1I–1M). As the extent of reactivity to self-antigens decreased in the presence of albumin in previous studies (Andrews et al., 2015), it is important to note that either FBS or BSA was used as a blocking buffer in the ELISAs presented here.
Figure 1. Induction of broadly reactive influenza antibodies is accompanied by elevated levels of autoreactive antibodies.

(A) C57BL/6 mice were given HKx31 and treated daily with rapamycin or PBS. Serum taken 14 days after infection was analyzed for reactivity to 128 autoantigens via protein arrays. Antigens with a statistically significant increase in reactivity to IgM antibodies in rapamycin-treated mice are depicted. Data are representative of two independent experiments of four to five mice per group per experiment.
(B and C) Signal intensities from the autoantigen array of groups of antigens associated with (B) arthritis and (C) lupus.
(D–G) Sera from mice 14 days after HKx31 exposure was tested by ELISA for reactivity to (D) collagen II (IgM), (E) histone H4 (IgM), (F) histone H1 (IgM), and (G) Smith antigen (IgM).
(H) Sera was taken from mice 90 days after HKx31 and tested for IgM reactivity against collagen, whole histones, GM1 + GD1a, histone H1, and histone H4.
(I–O) Sera from mice 14 days after HKx31 exposure was tested by ELISA for reactivity to (I) insulin (IgM), (J) LPS (IgM), (K) dsDNA (IgM), (L) nuclear antigens (IgM), (M) KLH (IgM), (N) GM1 + GD1a (IgM), and (O) GM1 (IgG).
(P) Sera from mice 14 days after administration of HKx31, MCMV, SARS-CoV-2 RBD/CFA, or OVA/CFA was analyzed by ELISA for GM1/GD1a IgM antibodies.
ELISAs are representative of two to seven independent experiments with at least four mice per group. Data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001 determined by Mann-Whitney test.
We next examined serum for IgG binding to the 128-autoantigen array and found that HKx31 + RAP mice did not have elevated IgG antibodies reactive to the arrayed autoantigens relative to control mice (Figures S1B–S1D). This may be due to a general reduction in IgG antibodies in HKx31 + RAP mice compared with control mice (Keating et al., 2013, 2020) or the lack of relevant antigens on the array.
Since the arrays did not include antigens targeted in GBS, an autoimmune disease associated with the 2009 H1N1 pandemic (Ghaderi et al., 2016; Hao et al., 2019; Kwong et al., 2013; Vellozzi et al., 2014), we assessed these antibodies by ELISA. Antibodies reactive against myelin-associated glycoprotein (MAG) and gangliosides are associated with various neurological pathologies (Cutillo et al., 2020; Lardone et al., 2010). In particular, anti-ganglioside antibodies are present in approximately 50% of individuals with GBS and may correlate with disease severity (Koga et al., 2015). We found that HKx31 + RAP mice had more anti-ganglioside GM1 IgM (Figure 1N) and IgG (Figure 1O) antibodies than HKx31 + PBS mice. Although rapamycin-treated mice typically have reduced IgG antibody levels, anti-GM1 IgG was elevated in HKx31 + RAP mice compared with in HKx31 + PBS mice, indicating that increased levels of broadly reactive influenza antibodies correlate with increases in both IgG and IgM antibodies associated with GBS.
Mice given rapamycin without influenza did not have increased autoreactive antibodies compared with mice given PBS (Figures 1D and 1E), suggesting that generating autoreactive antibodies requires activating an immune response. However, it is not known whether the antigen type impacts the specificity of the antibody response with rapamycin. Therefore, we examined whether immunization with other antigens or infection with a distinct virus with rapamycin increases anti-ganglioside antibodies. Mice were treated daily with rapamycin or PBS and infected with murine cytomegalovirus (MCMV). Additional mice were immunized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein receptor-binding domain (RBD) or ovalbumin (OVA) emulsified in complete Freund’s adjuvant (CFA). Reactivity to GM1 and GD1a was not elevated in rapamycin-treated mice given MCMV, RBD/CFA, or OVA/CFA compared with in control mice (Figure 1P), suggesting that exposure to influenza virus contributes to the specificity of antibodies generated in combination with rapamycin. Interestingly, rapamycin did not increase IgM antibodies specific for MCMV, OVA, or SARS-CoV-2 RBD relative to PBS-treated mice (Figure S2), further demonstrating that the rapamycin-mediated broadening of the antibody response is antigen specific. Together, these data demonstrate that promoting broadly reactive influenza antibodies also induced higher levels of antibodies reactive against self-proteins targeted in autoimmune diseases, including GBS.
Antibodies that bind influenza virus also bind self-proteins
To test whether autoreactive antibodies in the serum of HKx31 + RAP mice also bound influenza, or if these antibodies developed in parallel, we measured reactivity to histones by ELISA before and after serum was adsorbed against influenza proteins. If histone-specific antibodies also bind influenza, incubation with influenza proteins will remove these antibodies. As a negative control, serum was adsorbed against insulin because anti-insulin antibodies did not increase in HKx31 + RAP mice (Figures 1I and S1D). For a positive control, we adsorbed serum against histones, which removed approximately 70% of histone-specific antibodies (Figure 2A). Remarkably, nearly 60% of the histone-specific antibodies were also removed when adsorbed against HKx31, while less than 10% were adsorbed with insulin (Figure 2A), indicating that the antibodies that bind self-proteins also bind influenza.
Figure 2. Antibodies that bind influenza also bind self-proteins.

(A) Serum taken from mice 14 days after HKx31 and daily rapamycin or PBS was analyzed by ELISA for IgM binding to histones after adsorption to HKx31, histones, or insulin.
(B and C) IgG (B) and IgM (C) monoclonal antibodies were generated from mice given HKx31 and rapamycin (red text) or PBS (blue text) and tested by ELISA for reactivity to influenza antigens HKx31 (H3N2), Vn1203 (H5N1), and PR8 (H1N1) and the indicated self-proteins. Optical densities (ODs) for each antibody and antigen are plotted.
(D and E) IgG (D) and IgM (E) monoclonal antibodies were tested by ELISA to rH3 and rH5.
(F and G) Reactivity to HKx31 whole virus was evaluated by western blot with (F) IgG and (G) IgM monoclonal antibodies.
(H and I) Serum taken 14 days after HKx31 was analyzed by ELISA for IgM antibodies specific for (H) PI(4)P or (I) cardiolipin.
(J and K) IgG and IgM monoclonal antibodies were tested for binding to lipids with lipid strips. Lipid strips depicted in Figure S3 were scored for reactivity on a scale of 0–3.
Data are representative of two to four experiments (A, H, I, data are mean ± SEM). *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001 as determined by Mann-Whitney test.
To further examine if autoreactive antibodies also bind influenza, we generated IgM and IgG monoclonal antibodies from HKx31 + PBS and HKx31 + RAP mice by screening hybridomas for binding to influenza, histones, and gangliosides. We performed two screens, and in both experiments, we found more antibodies that bound self-proteins from rapamycin-treated mice than from control mice (Table S1), consistent with our ELISA data. As expected, more IgM than IgG antibodies displayed reactivity to self-proteins (Figures 2B and 2C). Most of the IgG antibodies bound only to HKx31; however, we also identified three IgG antibodies that bound multiple influenza subtypes in addition to albumin and other self-proteins (Figure 2B). We did not isolate any anti-influenza IgM antibodies that exclusively recognized HKx31 (Figure 2C). Most IgM antibodies that bound multiple influenza subtypes also bound self-proteins, with the exception of 1P10J6B11, which primarily bound the three influenza strains. The self-antigen-reactive antibodies typically bound multiple self-antigens, demonstrating that these antibodies may be more “flexible” in their binding requirements, enabling them to bind multiple, molecularly distinct epitopes, consistent with previous data (Guthmiller et al., 2020). However, broadly reactive antibodies did not exhibit similar reactivity to each self-antigen, indicating a degree of specificity. For example, the IgM antibody 24R7A9 had the highest reactivity to histones but had no reactivity to dsDNA or nuclear antigens (Figure 2C). Meanwhile, 24R9H5 bound all three influenza subtypes but showed less reactivity to histones and more reactivity to albumin. More IgM antibodies isolated from HKx31 + RAP mice (Figure 2C, red text) displayed reactivity to self-antigens than IgM antibodies from HKx31 + PBS mice (Figure 2C, blue text). Thus, antibodies induced by influenza bound both influenza and self-antigens, and treatment that enhanced protection against multiple influenza subtypes promoted these cross-reactive antibodies.
To determine which influenza protein the broadly reactive antibodies bound, we tested reactivity to recombinant HA (rHA) by ELISA and whole HKx31 via western blot. Most of the monoclonal antibodies bound to rHA (Figures 2D and 2E). However, four IgM antibodies had significantly reduced reactivity to rHA relative to whole virus, suggesting that they may bind other conserved proteins of influenza viruses (Figure 2E). Additionally, western blot showed that most IgG antibodies bound to the full-length HA (HA0) and the variable head of HA (HA1), as demonstrated by 13P1H7 and 24R3B2 (Figure 2F). One of the broadly reactive IgG antibodies, 13P10C1, only bound to the HA trimer, while the 9R1G9D5 antibody bound to the HA trimer, the HA1, and the HA2 stalk region (Figure 2F). The IgM antibodies preferentially bound the HA1 region, and only 24R10C1 also bound the HA2 region (Figure 2G). Together, these data show that many broadly reactive influenza antibodies also bind self-proteins.
Phospholipid reactivity was recently demonstrated by influenza HA stalk-binding antibodies (Bajic et al., 2019; Guthmiller et al., 2020). In addition, several broadly neutralizing HIV antibodies bind lipids, which may allow these antibodies to access epitopes near the viral membrane (Haynes et al., 2005; Liu et al., 2015; Matyas et al., 2009). Interestingly, many autoreactive antibodies have lipid-binding characteristics that facilitate binding to self-antigens (Kaczmarek et al., 2017; Petrovas et al., 1999; Suurmond and Diamond, 2015). Therefore, we tested whether serum from HKx31 + RAP mice demonstrated lipid reactivity. We observed a significant increase in IgM antibodies reactive against phosphatidylinositol 4-phosphate (PI(4)P) and cardiolipin in sera from HKx31 + RAP mice compared with HKx31 + PBS mice (Figures 2H and 2I). To examine if antibodies that bind influenza also bind lipids, we examined lipid reactivity of the IgM and IgG monoclonal antibodies and found that all broadly reactive IgM and IgG monoclonal antibodies readily bound lipid antigens (Figures 2J, 2K, S3A, and S3B). However, the IgG and IgM monoclonal antibodies that bound influenza with minimal binding to self-proteins did not bind lipids (Figures 2J, 2K, S3A, and S3B). Importantly, the reactivity to various lipids differed among the broadly reactive influenza antibodies, demonstrating that these antibodies do not bind all lipids non-specifically. While these monoclonal antibodies were isolated based on binding to influenza, histones, and gangliosides, all broadly reactive influenza monoclonal antibodies bound a variety of lipids.
Broadly reactive influenza antibodies spontaneously develop in mice with defects in B cell tolerance
The fact that autoreactive antibodies and broadly reactive influenza antibodies increase concurrently, and that broadly reactive influenza monoclonal antibodies also bind self-antigens, suggests that self-tolerance mechanisms may limit the generation and/or maintenance of broadly reactive influenza antibodies. Thus, we tested whether NZBWF1 mice, which have defects in B cell tolerance and are a well-characterized mouse model of SLE (Morel, 2010), generated influenza-specific antibodies without exposure to influenza. Remarkably, 9-week-old NZBWF1 mice had significantly higher levels of IgM antibodies reactive against multiple influenza subtypes than age-matched C57BL/6 mice (Figure 3A). The increase in broadly reactive influenza antibodies in NZBWF1 mice was maintained until at least 28 weeks of age (Figure 3B). Moreover, influenza-specific antibodies that developed spontaneously in NZBWF1 mice reacted to rHA proteins (Figure 3C). NOD mice also spontaneously develop autoimmune disease, but the development of disease in NOD mice depends more on autoreactive T cells than disease in NZBWF1 mice. Accordingly, NOD mice did not spontaneously develop influenza-specific antibodies as observed in NZBWF1 mice (Figure 3C), further suggesting that the defects in B cell tolerance in NZBWF1 mice contribute to the generation of these antibodies.
Figure 3. Defects in B cell tolerance lead to spontaneous generation of broadly reactive influenza antibodies.

(A and B) Serum from uninfected (A) 9-week-old or (B) 28- to 31-week-old NZBWF1 or C57BL/6 mice was analyzed by ELISA for IgM antibodies specific for HKx31, PR8, Vn1203, and histones + GM1.
(C) Serum from uninfected 6- to 11-week-old C57BL/6, NZBWF1, and NOD mice were analyzed by ELISA for IgM antibodies specific for rH1, rH3, and rH5 proteins.
(D) Serum from uninfected NZBWF1 mice was analyzed for IgM binding to histones, HKx31, and Vn1203 after adsorption to HKx31 or histones.
(E) Linear regression of histones/ganglioside IgM versus HKx31, PR8, and Vn1203 IgM in serum from 28- to 31-week-old NZBWF1 mice.
(F) Serum from 18-week-old, uninfected, and HKx31-infected (9 weeks after infection) NZBWF1 and C57BL/6 mice was analyzed in a hemagglutination inhibition assay.
Data are mean ± SEM and are representative of (A–C) two, (D) four, or (F) three independent experiments and analyzed by (A, B, and F) Mann-Whitney test or (C and D) Kruskal-Wallis test with Dunn’s multiple comparison test.
We performed adsorption experiments to test whether influenza-specific antibodies that spontaneously developed in NZBWF1 mice bound to both influenza and self-proteins. We found that most influenza-reactive antibodies in NZBWF1 mice also bound histones (Figure 3D). Notably, adsorption of sera with HKx31 (H3N2) removed antibodies specific for Vn1203, an H5N1 subtype, indicating that influenza-specific antibodies in NZBWF1 mice are broadly reactive against multiple subtypes of influenza viruses (Figure 3D). Further, there was a strong correlation between levels of antibodies reactive against self-antigens (histones and gangliosides) and antibodies specific for three different subtypes of influenza virus, suggesting that defects in self-tolerance that lead to autoreactive antibodies also contribute to influenza-specific antibodies (Figure 3E). Importantly, the influenza-reactive antibodies in uninfected NZBWF1 mice inhibited hemagglutination of red blood cells, a correlate of protection against infection, indicating that these antibodies are functional (Figure 3F). The development of broadly reactive influenza antibodies without exposure to influenza virus in NZBWF1 mice, which have defects in B cell tolerance, further highlights that self-tolerance mechanisms may limit the generation of broadly reactive influenza antibodies.
Enhanced generation of broadly reactive influenza antibodies exacerbates autoimmunity
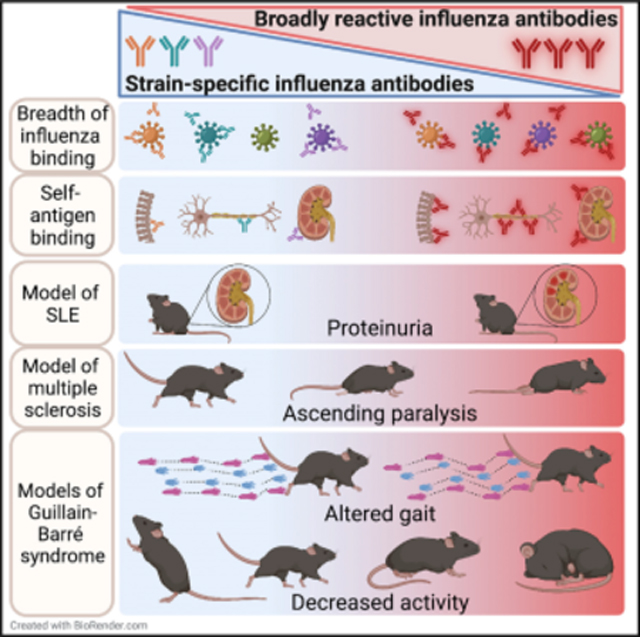
B cells with self-reactive B cell receptors are present at a high frequency in healthy individuals (Tiller et al., 2007). However, various tolerance mechanisms typically prevent autoreactive B cells from causing disease unless other checkpoints fail (Gitlin et al., 2016). While antibodies specific for the conserved portion of the HA stalk are more polyreactive and autoreactive than antibodies to the HA head (Andrews et al., 2015; Bajic et al., 2019; Guthmiller et al., 2020; Khurana et al., 2020), it is unclear whether these antibodies promote autoimmune pathology in the presence of other contributing factors. Thus, we tested whether enhancing generation of broadly reactive antibodies exacerbated autoimmunity in four different mouse models. In experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis, mice received HKx31 + RAP, HKx31 + PBS, rapamycin, or PBS for 14 days and were allowed to rest for 2 weeks prior to EAE induction. Rapamycin alone did not impact disease severity as measured by the development of ascending paralysis (Figure 4A); however, HKx31 + RAP, which protects mice against multiple subtypes of influenza (Keating et al., 2013), significantly exacerbated paralysis compared with HKx31 + PBS (Figure 4B). We next tested susceptibility to experimental autoimmune neuritis (EAN), a model of GBS. Mice were given HKx31 + RAP or HKx31 + PBS for 21 days, allowed to rest for 2 weeks, and then immunized with P0 peptide. Mice were monitored for disease by evaluating activity and stride length as a measure of flaccid paralysis. HKx31 + RAP mice had decreased activity levels and altered gait parameters compared with HKx31 + PBS mice (Figures 4C and 4D), indicating that mice with higher levels of broadly reactive influenza antibodies developed increased paralysis in this model as well.
Figure 4. Mice with increased levels of broadly reactive influenza antibodies are more susceptible to autoimmune disease.

(A and B) C57BL/6 mice were (A) treated daily with rapamycin or PBS or (B) given HKx31and rapamycin or PBS for 14 days, allowed to rest for 2 weeks, immunized with native MBP in CFA, and scored daily for disease.
(C and D) C57BL/6 mice were given HKx31, treated with rapamycin or PBS for 21 days, and immunized with P0 peptide in CFA 2 weeks later. To monitor for disease, activity was measured (C) in an open field test or (D) by measuring stride length 75 days after disease induction.
(E) C57BL/6 mice were given HKx31, treated with rapamycin or PBS for 20 days, allowed to rest for 6 days, and infected with C. jejuni. Control mice were treated with rapamycin or PBS without HKx31 and infected with C. jejuni. Activity was measured in an open field test.
(F–J) Nine-week-old NZBWF1 mice were given HKx31 and rapamycin or PBS for 28 days. Serum was analyzed by ELISA for IgM antibodies specific for (H) HKx31, (G) PR8, (H) Vn1203, or (J) whole histones. Statistical significance between HKx31 + PBS and HK31 + RAP is indicated by an asterisk and between RAP only and HK31 + RAP by a hashtag.
(I) Serum from NZBWF1 mice 10 days after HKx31 was analyzed by ELISA for IgM antibodies specific rH3, rH1, and rH5.
(K and L) Serum from NZBWF1 mice 28–38 days after HKx31 was tested for IgM antibodies specific for (K) dsDNA, (L) GM1 + GD1a, and histones.
(M) The amount of protein in the urine was measured 113 days after HKx31.
All data show mean ± SEM and are representative of at least two experiments and were analyzed by (A–C) two-way ANOVA, (D, I, and K–M) Mann-Whitney test, or (E–H and J) mixed-effects model with Tukey’s multiple corrections.
GBS is associated with Campylobacter jejuni (C. jejuni) infection in both humans and mice (Hao et al., 2019; Malik et al., 2014; St Charles et al., 2017). Therefore, we tested whether increased levels of broadly reactive influenza antibodies exacerbated disease following C. jejuni infection. Mice were given HKx31 + RAP or HKx31 + PBS for 20 days, allowed to rest for 6 days, infected with C. jejuni, and monitored for signs of peripheral nerve demyelination. Similar to the EAN model, HKx31 + RAP mice exhibited decreased activity levels after C. jejuni infection compared with HKx31 + PBS mice (Figure 4E), indicating more severe flaccid paralysis. Importantly, mice given rapamycin without HKx31 did not demonstrate enhanced paralysis (Figure 4E), suggesting that the increase in paralysis requires exposure to influenza virus. These data demonstrate that mice with higher levels of broadly reactive influenza antibodies developed more severe disease in a model caused by a bacterium associated with GBS in humans.
Finally, we examined the potential of broadly reactive influenza antibodies to contribute to disease in the context of a genetic predisposition to autoimmunity. NZBWF1 mice model SLE and spontaneously develop ANAs, proteinuria, hemolytic anemia, and glomerulonephritis beginning at approximately 23 weeks of age (Morel, 2010). This model is distinct, as disease occurs spontaneously in the absence of infection or overt inflammation. Nine-week-old NZBWF1 mice were given HKx31 + RAP, HKx31 + PBS, rapamycin, or PBS for 28 days. We first assessed the immune response to HKx31 and found that NZBWF1 mice treated with rapamycin had significantly higher IgM, but not IgG, antibodies specific for HKx31, PR8 (H1N1) and Vn1203 (H5N1), relative to PBS-treated controls (Figures 4F–4H and S4A–S4C). Critically, the influenza-specific IgM antibodies remained elevated for months after the initial HKx31 exposure (Figures 4F–4H) and bound rHA from multiple influenza subtypes (Figure 4I). Consistent with the increase in broadly reactive influenza antibodies, NZBWF1 mice given HKx31 + RAP were better protected following secondary infection with an H5N1 influenza virus strain than were HKx31 + PBS mice (Figure S4D).
To test whether the increase in broadly reactive influenza antibodies coincided with elevated autoreactive antibodies, we measured anti-histone antibodies. Interestingly, NZBWF1 mice given HKx31 + RAP had increased levels of anti-histone IgM antibodies compared with HKx31 + PBS mice, and these antibodies remained elevated over 100 days (Figure 4J). Antibodies reactive to dsDNA, histones, and GM1/GD1a gangliosides were also increased in HKx31 + RAP NZBWF1 mice compared with in PBS-treated mice (Figures 4K and 4L). Further, HKx31 + RAP NZBWF1 mice had elevated proteinuria compared with PBS controls (Figure 4M), which indicates kidney damage, a hallmark of SLE. Thus, NZBWF1 mice that had higher levels of broadly reactive influenza antibodies also had elevated levels of autoreactive antibodies and greater kidney damage. Together, these data demonstrate that induction of broadly reactive influenza antibodies was associated with greater susceptibility to disease in four distinct models of autoimmunity and suggest that broadly reactive influenza antibodies have the potential to contribute to pathogenesis in the context of underlying tolerance defects or additional assaults on the immune system.
Transfer of broadly reactive monoclonal antibodies increases susceptibility to autoimmune disease
We next tested the impact of broadly reactive influenza antibodies on disease susceptibility by monitoring autoimmunity after the transfer of monoclonal antibodies. To first determine whether broadly reactive influenza monoclonal antibodies bound self-antigens in vivo, we injected cocktails of monoclonal antibodies that were either specific for HKx31 or broadly reactive to multiple influenza subtypes into naive, C57BL/6 mice. We then examined the kidneys for antibody deposits, an indicator of glomerulonephritis. We observed a significant increase in both IgG and IgM kidney deposits in mice that received broadly reactive influenza monoclonal antibodies compared with mice given monoclonal antibodies specific for HKx31 (Figures 5A and 5B), demonstrating that broadly reactive influenza antibodies have a greater potential to bind kidney self-antigens than strain-specific antibodies. To next determine whether these antibody deposits can contribute to pathogenesis, we utilized Balb/c mice, which do not spontaneously develop autoimmunity and are not prone to glomerulonephritis. Balb/c mice were given a cocktail of broadly reactive influenza monoclonal antibodies, a cocktail of monoclonal antibodies that exclusively bind HKx31, or an anti-dsDNA monoclonal antibody (163p.77) that was previously shown to induce glomerulonephritis (Krishnan et al., 2012). Three weeks later, mice received a second injection of antibodies and were monitored weekly for proteinuria. Mice that received broadly reactive influenza antibodies developed proteinuria sooner than mice given HKx31-reactive antibodies, although the incidence was not significantly different between the groups (Figure 5C). As inflammation during the early phase of autoantibody deposition accelerates the development of proteinuria and nephritis in SLE models (Triantafyllopoulou et al., 2010), we examined the pathogenicity of the broadly reactive influenza monoclonal antibodies in the context of inflammation provided by poly I:C. Interestingly, mice given broadly reactive influenza monoclonal antibodies, along with poly I:C, developed significantly more disease than mice that received monoclonal antibodies specific only for HKx31 (Figure 5D). In fact, the extent of proteinuria was similar between mice that received broadly reactive influenza monoclonal antibodies and mice given the anti-dsDNA monoclonal 163p.77 antibody (Figure 5D). Thus, in the absence of genetic susceptibility to autoimmunity, broadly reactive influenza monoclonal antibodies can exacerbate SLE-like disease in the presence of additional inflammation.
Figure 5. Transfer of broadly reactive monoclonal antibodies increases susceptibility to autoimmune disease.

(A) C57BL/6 mice received either broadly reactive or HKx31-specific (single reactive) IgM and IgG monoclonal antibodies. Kidneys were harvested 24 h later and stained for IgM and IgG.
(B) Area of IgM and IgG staining in kidney sections was calculated as a proportion of area encompassing individual glomeruli. Data are mean ± SEM and representative of two independent experiments evaluated by Mann-Whitney test.
(C and D) Six- to eight-week-old Balb/c mice were given two injections of broadly reactive or HKx31-specific IgG monoclonal antibodies with or without poly I:C, which was given three times per week. Proteinuria was measured weekly, and the incidence of mice with 65 mg/dL urine protein or greater is depicted.
(E) NOD mice received two to three injections of broadly reactive or HKx31-specific IgM and IgG monoclonal antibodies. Blood glucose levels were monitored weekly.
Incidence of (C and D) proteinuria and (E) diabetes was compared via log rank (Mantel-Cox) test.
We next tested whether the transfer of broadly reactive influenza monoclonal antibodies increased disease in mice with a genetic predisposition to autoimmune disease. NOD mice spontaneously develop type 1 diabetes due to tolerance defects impacting both the T and B cells (Anderson and Bluestone, 2005). As NOD mice typically become diabetic around 20 weeks of age, we injected 11-week-old mice with cocktails of broadly reactive or single influenza reactive monoclonal antibodies and monitored the mice weekly for diabetes. Remarkably, the incidence of diabetes was significantly enhanced in mice receiving broadly reactive influenza monoclonal antibodies compared with NOD mice given monoclonal antibodies specific for HKx31 (Figure 5E). Overall, these data demonstrate that small amounts of broadly reactive influenza antibodies exacerbate autoimmune disease in the presence of additional inflammation or underlying defects in self-tolerance.
Influenza virus infection in humans correlates with increased autoreactive antibodies
Humans infected or vaccinated with the H1N1 pandemic strain during the 2009–2010 influenza season had more broadly reactive influenza antibodies than individuals infected with prior seasonal influenza virus strains (Andrews et al., 2015; Li et al., 2012; Pica et al., 2012; Thomson et al., 2012; Wrammert et al., 2011). Therefore, we examined whether these individuals had an increase in autoreactive antibodies relative to individuals infected with influenza in the four subsequent influenza seasons. Specifically, we assessed antibodies reactive against gangliosides and MAG because these antibodies are connected with GBS, which was associated with a higher risk in individuals exposed to the 2009 H1N1 pandemic strain (Ghaderi et al., 2016; Kwong et al., 2013; Vellozzi et al., 2014). First, we measured the reactivity of total Ig (IgM + IgG) to these autoantigens and found that MAG-specific antibodies tended to be higher in infected individuals than in uninfected individuals (Figure S5A). To determine the contribution of IgM versus IgG to this reactivity, we then analyzed a subset of individuals for MAG- and ganglioside-specific IgM and IgG separately and saw that IgG antibodies accounted for the majority of this reactivity (Figure S5B). Consequently, we focused our analysis on MAG- and ganglioside-specific IgG antibodies and found that individuals infected with H1N1 in the 2009–2010 influenza season had significantly higher levels of IgG antibodies specific for MAG, GM1, GD1a, GD1b, and GQ1b relative to uninfected individuals (Figure 6A), which is consistent with the increase in GBS cases associated with influenza following the 2009 influenza pandemic. It is intriguing that there were also significantly more anti-MAG and -ganglioside antibodies in individuals infected during the 2012–2013 season with an H3N2 subtype than in uninfected individuals (Figure 6A). Interestingly, the individuals with the highest influenza microneutralization titers exhibited the highest levels of anti-MAG or -ganglioside IgG antibodies (Figure 6B), raising the possibility that the antibodies mediating microneutralization also bind MAG and gangliosides. Together, these data demonstrate that humans infected with a novel strain of influenza, which induced higher levels of broadly reactive influenza antibodies compared to previous years, also exhibited an increase in antibodies reactive to MAG and gangliosides. This finding is consistent with our data from the murine models and is particularly important given that influenza infection during the 2009–2010 season was associated with increased cases of GBS (Ghaderi et al., 2016; Kwong et al., 2013; Vellozzi et al., 2014).
Figure 6. Influenza infection in humans is associated with increased anti-MAG and -ganglioside antibodies.

(A) Plasma from individuals infected with influenza between 2009 and 2014 and uninfected controls was analyzed by ELISA for reactivity to gangliosides and MAG. The normalized ODs were compared via non-parametric, rank-based MANOVA to test for statistical variations of normalized ODs as effects of infection status and year. The overall effect was significant (p = 0.003), and asterisks indicate significance of post hoc Dunnett’s tests comparing infection years with the influenza-negative condition.
(B) Reactivity to each ganglioside was plotted with binned microneutralization log10 titers. Non-parametric, rank-based MANOVA was used to test for statistical significance. Overall effect was significant (p < 0.001), and asterisks indicate significance of post hoc Tukey’s pairwise comparisons.
(C) Monoclonal antibodies isolated from humans were tested for reactivity to MAG, gangliosides, ANA, and phospholipids via ELISA, and normalized ODs are plotted in the heatmap.
(D–F) Normalized ODs of monoclonal antibody reactivity to MAG, ANAs, and phospholipids were plotted based on (D) reactivity to influenza subtypes, (E) location of HA binding, or (F) previously characterized polyreactivity and were tested by multiple Mann-Whitney tests. Mean ± SEM is depicted.
Monoclonal antibodies derived from humans bind both influenza virus proteins and self-proteins
The increased levels of antibodies reactive against MAG and gangliosides in individuals infected with influenza virus could be due to an increase in antibodies that bind to both influenza and MAG or gangliosides. Conversely, the increased levels of these antibodies may be the result of non-specific inflammation that promotes the expansion of anti-MAG and -ganglioside antibodies that do not bind influenza antigens. Therefore, we examined a panel of human influenza-specific monoclonal antibodies for binding to MAG, gangliosides, phospholipids, and nuclear antigens. The monoclonal antibodies analyzed included well-characterized, broadly reactive influenza antibodies and others more recently characterized as binding to only one or multiple subtypes of influenza virus (Andrews et al., 2015; Guthmiller et al., 2020). We identified several influenza-specific antibodies that also bound to MAG, gangliosides, nuclear antigens, or phospholipids (Figure 6C). Interestingly, the monoclonal antibodies that bound self-antigens included both broadly reactive and single HA-reactive antibodies (Figures 6C and 6D). Given that broadly reactive influenza antibodies were more likely to be polyreactive than antibodies specific for a single influenza subtype (Bajic et al., 2019; Guthmiller et al., 2020; Khurana et al., 2020), it was surprising that several single, HA-reactive monoclonal antibodies also bound to self-antigens. It was also unexpected that antibodies specific for the HA stalk did not show more reactivity to self-antigens than the HA head-specific antibodies (Figure 6E). It is important to note that antibodies reactive against one self-protein did not necessarily bind to all self-antigens, indicating that reactivity was not simply due to “sticky” antibodies that bind non-specifically to several antigens. In prior reports demonstrating increased polyreactivity in broadly reactive influenza antibodies relative to single reactive antibodies, polyreactivity was based on binding to dsDNA, insulin, cardiolipin, and LPS. However, it was not clear whether polyreactivity correlated with reactivity to antigens targeted in autoimmune diseases associated with influenza infection such as GBS (Hao et al., 2019). Therefore, we compared reactivity to MAG and gangliosides between influenza-specific monoclonal antibodies previously characterized as polyreactive or non-polyreactive (Andrews et al., 2015). We found that there was not a significant difference in binding to MAG, gangliosides, and phospholipids between polyreactive and non-polyreactive antibodies (Figure 6F). In fact, several of the polyreactive antibodies did not bind these antigens, while a few of the non-polyreactive antibodies showed reactivity to MAG, gangliosides, and phospholipids (Figure 6F). Together, these data demonstrate that, similar to murine models, antibodies isolated from humans can bind to both influenza virus and self-antigens targeted in autoimmunity. Importantly, while polyreactivity may exist among antibodies binding conserved regions of HA, this polyreactivity does not necessarily correlate with autoreactivity to all self-proteins. Further, even antibodies characterized as reactive to a single HA and as non-polyreactive may bind MAG, gangliosides, ANAs, or phospholipids.
DISCUSSION
Understanding factors that regulate the generation and maintenance of broadly reactive influenza antibodies remains a major obstacle for developing a universal influenza vaccine. Our data suggest that self-tolerance mechanisms limit the generation of broadly reactive influenza antibodies, which contributes to the paucity of these antibodies. This conclusion is based on data demonstrating the induction of autoreactive antibodies in conjunction with broadly reactive antibodies, exacerbated autoimmunity, and identification of human and mouse monoclonal antibodies that bound to multiple influenza subtypes and self-proteins. Additionally, mice with defects in B cell tolerance developed broadly reactive influenza antibodies without exposure to influenza virus. These data are consistent with recent reports demonstrating that broadly reactive influenza antibodies are more likely to be polyreactive than antibodies specific for one influenza subtype (Andrews et al., 2015; Bajic et al., 2019; Guthmiller et al., 2020; Khurana et al., 2020). In fact, polyreactivity was shown to be an inherent feature of broadly reactive influenza antibodies (Guthmiller et al., 2020). However, we demonstrate that polyreactive antibodies do not necessarily bind self-antigens associated with autoimmune disease, suggesting that their polyreactive nature alone may not be an indicator of the pathogenic potential of antibodies. Further, our data highlight the importance of investigating reactivity to self-antigens typically targeted in individuals with autoimmunity.
While polyreactive antibodies bind molecularly distinct proteins, we demonstrated that broadly reactive influenza antibodies do not bind non-specifically to all proteins. Broadly reactive influenza antibodies may bind molecular moieties conserved across groups of proteins, such as glycans, large hydrophobic regions, or other post-translational modifications. Similar to the broadly reactive influenza antibodies that we generated, broadly neutralizing HIV antibodies and antibodies associated with many autoimmune disorders demonstrate a high degree of lipid binding (Haynes et al., 2005; Liu et al., 2015; Matyas et al., 2009; Petrovas et al., 1999; Haji-Ghassemi et al., 2017; Suurmond and Diamond, 2015). Conserved regions of HIV-1 and influenza HA are proximal to the viral membrane, and the ability of antibodies to bind lipids may enhance their binding to viral epitopes in this region. However, this property may also promote binding to self-proteins.
As the goal of a universal influenza vaccine is to generate durable, broadly reactive influenza antibodies, a critical question is whether elevated levels of broadly reactive influenza antibodies could exacerbate autoimmune diseases. We demonstrate that the enhanced generation of broadly reactive influenza antibodies also increased autoreactive antibodies and exacerbated disease in four distinct models of autoimmunity. Our data further highlight the physiological significance of previous findings by showing that humans infected with the novel 2009 H1N1 influenza subtype, which induced higher levels of broadly reactive influenza antibodies than infections with prior seasonal influenza subtypes (Andrews et al., 2015; Li et al., 2012; Pica et al., 2012; Thomson et al., 2012; Wrammert et al., 2011), also had increased levels of autoreactive antibodies associated with GBS. This finding is particularly significant as a greater risk of two autoimmune diseases, narcolepsy and GBS, were associated with the pandemic 2009 H1N1 influenza strain relative to strains circulating in prior or subsequent years (Ahmed et al., 2014; Barker and Snape, 2014; Han et al., 2011; Hao et al., 2019; Kwong et al., 2013; Nohynek et al., 2012; Vellozzi et al., 2014; Vogel, 2015).
Importantly, our data demonstrate that the transfer of relatively small amounts of broadly reactive influenza monoclonal antibodies exacerbated autoimmunity in the presence of inflammation or underlying defects in tolerance. Thus, an increase in broadly reactive influenza antibodies alone is not sufficient to induce autoimmune disease. However, in the context of a secondary infection, additional inflammation, or a genetic susceptibility, broadly reactive influenza antibodies can exacerbate autoimmunity. It is important to note that the broadly reactive influenza monoclonal antibodies that enhanced autoimmunity in mice mirrored the binding patterns of broadly reactive influenza monoclonal antibodies isolated from humans.
While broadly reactive influenza antibodies are rare relative to strain-specific antibodies, they tend to occur more frequently after exposure to novel influenza virus strains (Andrews et al., 2015; Ellebedy et al., 2014; Joyce et al., 2016; Li et al., 2012; Pica et al., 2012; Thomson et al., 2012; Wheatley et al., 2015; Whittle et al., 2011; Wrammert et al., 2011). Interestingly, exposure to a novel influenza virus also generated more polyreactive and autoreactive antibodies than encounters with previously circulating viruses (Guthmiller et al., 2020), which could be due to greater activation of naive B cells after exposure to a novel virus. Compared with memory B cells, the activation of naive B cells may promote the generation of antibodies with more flexible binding characteristics. As a result, these antibodies can bind distinct subtypes of influenza; however, they may also bind self-antigens. Indeed, a significant proportion of antibodies generated after exposure to a novel immunogen are autoreactive (Wardemann et al., 2003). However, as the immune response continues, self-tolerance mechanisms operating within clonal selection typically reduce levels of autoreactive antibodies over time (Gitlin et al., 2016; Zhang et al., 2016). Thus, broadly reactive influenza antibodies may not be durable because they are more likely to be polyreactive and therefore subject to self-tolerance mechanisms. In support of this, there is evidence of a significant decline in the levels of broadly reactive influenza antibodies following different vaccine strategies to novel influenza strains (Andrews et al., 2017; Feldman et al., 2019; Fukuyama et al., 2020; Wheatley et al., 2015). Our data reveal that a critical consequence of promoting the maintenance of broadly reactive influenza antibodies is increased susceptibility to autoimmune disease in the event of additional challenges to self-tolerance checkpoints.
Finally, our data highlight how self-tolerance mechanisms contribute to the evolutionary fitness of viruses. Epitopes that are avoided due to self-tolerance will not be subjected to immune pressure, and as a result, these epitopes will be more stable or conserved as viruses evolve. Consequently, the conserved epitopes have a higher probability to induce cross-reactive antibodies against self-proteins. Other viruses likely benefit from self-tolerance mechanisms to avoid immune pressure. For example, while several broadly neutralizing HIV antibodies have been identified, these antibodies are rare and have been shown to be autoreactive (Haynes et al., 2005; Liu et al., 2015). Thus, it is imperative to consider the autoreactive potential of antibodies when designing vaccines aimed at generating durable antibodies to epitopes that are naturally avoided by the immune response and particularly the conserved influenza epitopes that are evaded by immune systems of multiple species as evolutionarily diverged from humans as lampreys (Altman et al., 2015).
Limitations of the study
It is not possible to directly test whether an increase in broadly reactive influenza antibodies that also bind self-proteins precipitate autoimmunity in humans. Nevertheless, accumulating evidence demonstrates that autoreactive antibodies are predicative of certain autoimmune disorders (Scofield, 2004; Xiao et al., 2021). Moreover, there is an association in both humans and animal models of GBS and anti-ganglioside antibodies (Ho et al., 1999; Willison, 2011). Several decades of research utilizing animal models of autoimmunity have revealed fundamental mechanisms that contribute to autoimmune disease in humans (Sakaguchi, 2000). Our data revealing enhanced disease following the transfer of broadly reactive influenza antibodies directly demonstrate the potential of these antibodies to contribute to disease. Although our results do not directly test whether elevated levels of broadly reactive influenza antibodies increase the risk of autoimmunity in humans, the fact that infection with a strain of influenza that generated high levels of broadly reactive influenza antibodies was associated with higher levels of autoreactive antibodies suggests that these antibodies may be a factor that contributes to autoimmunity in humans.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maureen McGargill (maureen.mcgargill@stjude.org).
Materials availability
Hybridomas from which the monoclonal antibodies were generated are available upon request from the lead contact.
Data and code availability
Microarray data have been deposited at NCBI GEO: GSE196366, and are publicly available as of the date of publication. All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original custom code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
The FLU09 cohort was described previously (Oshansky et al., 2014). Briefly, participants met the clinical case definition of influenza virus infection at the time of enrollment or were asymptomatic household contacts of a confirmed, influenza-positive participant. The age of participants in the study ranged from 3.6 weeks to 71 years of age. The study included 79 male and 120 female participants. Written, informed consent was acquired from participants’ parents or guardians and written assent from age-appropriate subjects was acquired at the time of enrollment. Two subjects included in the analyses had immune disorders: one HIV-positive patient, and one patient with rheumatoid arthritis. However, all but one of the significant results we reported remain after excluding these two patients. None of the participants had documented diagnosis of cancer, cardiovascular disease, diabetes, neurological disease, or liver disease. A single control patient (i.e., influenza-negative) had been previously diagnosed with diabetes. One participant had previously been diagnosed with sickle cell anemia. Excluding the single HIV-positive subject, no participants were immunosuppressed. Several individuals included in the study were previously diagnosed with asthma, but the data obtained from all of these individuals generally fell within the range of variation exhibited by non-asthmatic patients. The institutional review boards of St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center/Le Bonheur Children’s Hospital approved the study.
Mice
Female, seven-to eight-week-old C57BL/6J mice, six-week-old NOD/ShiLtJ, six-to eight-week-old Balb/cJ, and nine-week-old NZBWF1/J mice were obtained from The Jackson Laboratory and randomly assigned to experimental groups. All mice were maintained under specific pathogen free conditions at St. Jude Children’s Research Hospital, and all animal studies were approved by the Institutional Animal Care and Use Committee.
Cell lines and primary cultures
Sp2/mIL-6 hybridoma cells (ATCC CRL-2016) (Harris et al., 1992) were used as fusion partner for antibody production as described below. Prior to the fusion, Sp2/mIL-6 hybridoma cells were maintained at 37°C, 5% CO2 in Medium A (Stemcell Technologies) with 10% FCS. MDCK cells (ATCC CCL-34) were used for influenza A microneutralization assays as described below. Prior to the assay cells were maintained at 37°C, 5% CO2 in Eagle’s Minimum Essential Medium, 10% FCS.
Microbe strains
Campylobacter jejuni (C. jejuni, ATCC 700297) cultures were expanded in Mueller Hinton broth from glycerol stocks by anaerobic incubation with agitation (220 rpm, 37°C) for 48 h.
METHOD DETAILS
Virus and infections
The HKx31 and ΔVn1203 viruses were constructed using the eight-plasmid reverse genetics system (Hoffmann et al., 2002) and contained the six internal genes of A/Puerto Rico/8/34 (PR8) and genes encoding the HA and NA surface proteins from either A/Aichi (H3N2) or A/Vietnam/1203/04 (H5N1), respectively. Murine cytomegalovirus (MCMV) was obtained from ATCC (VR-1399). Mice were given 1 × 108 EID50 of HKx31 or 1 × 106 PFU MCMV diluted in PBS by intraperitoneal (i.p.) injection. Beginning one day prior to HKx31 or MCMV, mice received daily i.p. injections of 1.5 μg rapamycin (Rapamune; Wyeth) diluted in PBS, or PBS alone as a control. For secondary challenge infection with ΔVn1203, mice were anesthetized with Avertin (2,2,2-tribromoethanol) and challenged intranasally with 2 × 105 EID50 of ΔVn1203. Mice were monitored daily for weight loss and clinical signs of disease. Mice determined to be moribund were euthanized via CO2 asphyxiation.
Campylobacter jejuni infection
Campylobacter jejuni (C. jejuni, ATCC 700297) cultures were expanded in Mueller Hinton broth from glycerol stocks by anaerobic incubation with agitation (220 rpm, 37°C) for 48 h. Cultures were then spun down, concentrated ten-fold in PBS, and kept on ice protected from oxygen until inoculation. Mice received 0.2 mg of ampicillin via oral gavage for two days prior to C. jejuni infection. The day of infection, mice were fasted from food and water for six hours prior to inoculation with 2 × 107 colony forming units (CFU) of C. jejuni via oral gavage. To confirm infection, stool was collected from mice prior to and three days following infection. Stool samples were homogenized, serially diluted, then track plated onto Campy CVA agar (Fisher Scientific) and incubated at 42°C using a conventional Campy gas pack and pouch system (Hardy Diagnostics) for 48 h.
Autoantigen array and analysis
Autoantigen arrays were performed at Microarray and Immune Phenotyping Core Facility of University of Texas Southwestern Medical Center (UTSW) and carried out as described previously (Li et al., 2007). Briefly, serum samples from HKx31-infected mice, treated with rapamycin or PBS for 14 days, were frozen and sent to UTSW. The sera were pre-treated with DNase I and incubated on autoantigen arrays, and the bound antibodies were detected using a Cy3 labeled anti-mouse IgG (532 nm) or Cy5 labeled anti-mouse IgM (635 nm) to generate Tiff images. Genepix Pro 6.0 software was used to analyze the images and generate data. The normalized fluorescent intensity (NFI) was used to generate the heatmap and to compare the significance of difference between rapamycin and PBS-treated groups using the Mann-Whitney U test with the Benjamini-Hochberg approach to correct for multiple comparisons. Samples detected as outliers by mvoutliers (Gschwandtner, M. & Filzmoser, P. (2009). mvoutlier: Multivariate outlier detection based on robust methods. R package version 1.4. www.statistik.tuwien.ac.at/public/filz/) were excluded. Average linkage and Euclidean distance were used for hierarchical clustering of samples based on the normalized intensity.
Experimental autoimmune encephalitis (EAE)
Mice were given 1 × 108 EID50 HKx31 i.p., and then rapamycin or PBS, daily as described above. Mice received rapamycin or PBS for 14 days, and then were rested for two weeks without additional injections to be sure the rapamycin was not present at the time of EAE induction. EAE was induced by injecting mice subcutaneously (s.c.) with an emulsion of 100 μg of mouse native myelin basic protein (Creative Biomart) with 400 μg of CFA (Chondrex). Mice also received 200 ng of pertussis toxin (List Biologicals) i.p. at the same time as induction, and two days later. The mice were monitored daily for disease, which was measured by the following scoring strategy: 0 - no signs of disease; 1- limp tail; 2 - partial hindlimb paralysis; 3 - complete hindlimb paralysis; 4 - complete hindlimb and partial forelimb paralysis or moribund. Data are representative of two independent experiments of at least four mice per group.
Experimental autoimmune neuritis (EAN)
EAN, a mouse model for Guillain-Barre syndrome (GBS), was carried out as described previously (Gonsalvez et al., 2017). Briefly, mice were given HKx31 i.p. and treated with rapamycin or PBS as described above for 21 days. The mice were rested for two weeks, and disease was induced by injecting mice s.c. with an emulsion of P0 peptide (180–199, SSKRGRQTPVLYAMLDHSRS; synthesized at St. Jude Hartwell center and purified by high-performance liquid chromatography (HPLC)) in CFA. Immunization was carried out twice; at day 0 and day 8 with 120 μg of P0 peptide emulsified in 400 μg of CFA (Chondrex). The mice received 400 ng of pertussis toxin one day prior to the first immunization, and 300 ng of pertussis toxin one and three days after each immunization. Data are representative of four independent experiments with seven to ten mice per group. The mice were monitored for development of EAN utilizing an open field test as described previously (St Charles et al., 2017). Briefly, mice were placed individually in a rat cage that was split into four quadrants and monitored for one minute. Each time the mouse crossed a quadrant or exhibited rearing, they received a score of 1. The cumulative score for one minute was calculated, and the average cumulative score is graphed to indicate activity of the mouse. A lower score is correlated with lower activity level and is indicative of EAN (St Charles et al., 2017). The mouse stride parameters were measured as described previously (Brooks et al., 2012; St Charles et al., 2017). The front and back paws of the mouse were painted with different colors of non-toxic dye. The mice were allowed to walk individually over a strip of Whatmann filter paper placed in a long, narrow chamber with a dark box at the end. The forelimb and hindlimb stride length were determined by measuring the distance between the centers of the forelimb prints and the hindlimb paw prints, respectively, on the Whatmann filter paper. Forelimb and hindlimb width were measured as the distance between the right and left forelimbs and hindlimbs, respectively. The average of two to three measurements is plotted. Measurements were made by personnel blinded to the treatment of the study subjects. An increase in stride length is indicative of ataxia and EAN (Leroy et al., 2009; St Charles et al., 2017).
Protein immunization
Beginning one day prior to immunization, mice received daily i.p. injections of rapamycin or PBS as detailed above. Ovalbumin (OVA) or SARS-CoV-2 receptor binding domain (RBD) protein was mixed one to one with CFA to deliver 50 μg of protein with 100 μg of M. tuberculosis per mouse, i.p. Mice were bled 14 days after immunization, and serum was analyzed by ELISA.
ELISAs
All ELISAs were performed as detailed below using serum dilutions determined empirically by titration analyses or according to the manufacturer’s instructions. Some commercial ELISAs require low dilutions for use in conditions optimized by the manufacturer. In addition, all ELISAs include a positive and negative control sample used at the same dilution as the test samples to demonstrate the range of each ELISA and allow for plate-to-plate comparisons over time.
Anti-histone IgM and IgG ELISAs were based on previously described methods (Hasegawa et al., 1998). Briefly, microtiter plates (Nunc) were coated with 0.15 μg/well of histones H2B, H1, H3 or H4 (New England BioLabs) in PBS. The plates were washed and blocked with 2.5% BSA in PBS for two hours, followed by incubation with mouse serum diluted at 1:100 for 90 min at room temperature. The plates were washed, and anti-mouse IgM (Southern Biotech) or anti-mouse IgG HRP antibodies were added and incubated at room temperature for 30 min. The plates were washed and TMB substrate was added for 15–30 min. The reaction was stopped with 100 μL/well of 1N H2SO4 and the absorbance was measured at 450 nm and 550 nm. Plotted OD is 450 nm–550 nm.
The whole histone, albumin, collagen, ganglioside (GM1+GD1a), LPS, and KLH ELISAs were performed as follows. ELISA plates were coated overnight at 4°C with 0.15 μg per well of whole histones from chicken red blood cells (Immunovision), albumin from mouse sera (Sigma), collagen II (Chondrex), ganglioside GM1 (US Biological) and ganglioside GD1a (Sigma) from bovine brain, LPS from E. coli (Sigma), or KLH native (Sigma). Plates were blocked with 2.5% FBS in PBS for one hour at room temperature. Samples were diluted 1:100 in PBS with 0.1% Tween 20, and were incubated on the plate for one hour at room temperature. Plates were washed again then incubated with anti-mouse IgM-HRP (Southern Biotech) diluted 1:5000 in PBS for 30 min at room temperature. After washing, the TMB reagent was added and incubated for up to 15 min. The reaction was stopped with 100 μL/well of 1N H2SO4 and the absorbance was measured at 450 nm and 550 nm. The OD of 450–550 nm is plotted.
Anti-mouse GM1 IgG (MyBiosource), anti-mouse insulin, ANA, dsDNA, and Sm ELISAs (all from Alpha diagnostics), and BÜHLMANN Gangliocombi™ MAG ELISA for detecting human anti-myelin associated glycoprotein (MAG) and anti-ganglioside antibodies (BÜHLMANN Diagnostics Corp.) were carried out according to manufacturer’s instructions. Samples were diluted 1:2 for anti-mouse GM1 IgG, 1:25 for insulin, 1:100 for dsDNA and ANA ELISA, and 1:25 for Sm ELISAs. For the BÜHLMANN Gangliocombi™ MAG ELISA, the human monoclonal antibodies were obtained from Patrick Wilson (Guthmiller et al., 2020; Andrews et al., 2015; Chen et al., 2018; Corti et al., 2011; Dreyfus et al., 2012; Henry et al., 2019; Henry Dunand et al., 2016; Wrammert et al., 2011) and were normalized at 1 mg/mL prior to testing and the results are expressed as the normalized OD, which is the percent ratio of the sample OD to the OD of the provided calibrator, as recommended by the manufacturer to normalize inter-assay variability.
For HKx31, PR8, and Vn1203 ELISAs, microtiter plates were coated overnight with lysed whole virus diluted in PBS at 20 ng/well (for serum IgM), 10 ng/well (for serum IgG), 80 ng/well (for monoclonal IgM), or 40 ng/well (for monoclonal IgG). Plates were blocked in either 2.5% FBS (for serum samples) or 50% Pierce protein-free blocking buffer in PBS (Thermo Fisher) (for monoclonal antibodies) for one hour, then washed and incubated with diluted samples (1:400 in PBS for serum IgM, 1:800 in PBS for serum IgG, or 5 μg/well in PBS-T for monoclonal antibodies) for one hour. The plates were washed and incubated with goat anti-mouse IgM-HRP (1:5000 in PBS) or IgG-HRP (1:6000 in PBS) (Southern Biotech) for 30 min. The plates were washed and incubated with TMB for up to 15 min, stopped with 1N H2SO4,and absorbances were measured at 450 and 550 nm on a microplate reader, with the final OD plotted as 450–550 nm. ELISAs for recombinant influenza HA proteins were performed as whole influenza ELISAs, with the exceptions that 75 ng of recombinant HA proteins (A/California/04/2009 H1N1, A/Aichi/2/1968 H3N2, and A/Vietnam/1203/2004 H5N1; eEnzyme) were coated per well for serum IgM, 1 μg/mL for monoclonal IgM, and 0.5 μg/mL for IgG. The TMB incubation time for the rHA ELISAs was ten minutes.
The ELISAs for the detection of MCMV (MyBioSource), ovalbumin (OVA) (Sigma), and SARS-CoV-2 Spike receptor binding domain (RBD) were adapted from a previously described protocol (Amanat et al., 2020). Briefly, purified MCMV (75 ng for IgG, 11.25 ng for IgM), OVA (30 ng/well), or SARS-CoV-2 Spike RBD (30 ng/well)) were diluted in PBS and incubated overnight at 4°C on MaxiSorp 384-well plates (Nunc). Plates were then blocked with 3% non-fat milk (AmericanBio) in PBS with 0.1% Tween 20 (PBST) (Fisher) for one hour at room temperature. The serum samples were diluted in 1% non-fat milk in PBST, incubated on the washed plates for one hour at room temperature. Plates were washed and incubated with HRP-conjugated secondary antibodies for 30 min at room temperature. After the final wash, SIGMAFAST OPD (Sigma #P9187) solution was added to each well for eight minutes, at which time the development was stopped by adding 25 μL per well of 3N hydrochloric acid (Fisher). The values of optical density were measured at 490 nm.
Isolation and purification of mouse monoclonal antibodies
Mice were given 1 × 108 EID50 HKx31 i.p. and treated daily with PBS or rapamycin as described above. Mice were boosted with 1 × 108 EID50 HKx31 i.p. on day 26. Two days later, spleens were harvested and 6 × 106 cells from each spleen were fused to an equal number of Sp2/mIL-6 hybridoma cells (ATCC® CRL-2016™) (Harris et al., 1992). The fusion was transferred to 96 or 384 well plates and incubated in HAT-(Sigma, St. Louis, MO) supplemented media (ClonaCell™-HY Medium E, Stemcell Technologies, Vancouver, BC) for ten days. The supernatants from wells containing clones were then screened by a combination ELISA for reactivity to self-proteins (histones, albumin, GM1, and GD1a) and HKx31. Clones that were positive for self-proteins or HKx31 were then single cell sorted, cultured, and rescreened by ELISAs for IgM and IgG reactivity to self-proteins or HKx31. Selected IgG clones were purified using a HiTrap™ Protein G HP (GE Healthcare). IgM clones were adapted to a protein-free media (Gibco) then enriched by size exclusion of 100 kDa or greater. The immunoglobulin concentration of the IgG clones was measured using Qubit or nanodrop, and the immunoglobulin concentration of the IgM clones was quantitated using an immunoglobulin isotyping magnetic bead panel (Milliplex) or by anti-mouse IgM ELISA (RayBiotech). The initial ELISA screens were performed using BSA or FBS as a blocking buffer. After we discovered that some of the monoclonal antibodies bound albumin, all subsequent ELISAs, including all the ELISAs with monoclonal antibodies were performed with a protein and FBS-free blocking buffer (Pierce). The purified monoclonal antibodies were tested by ELISA diluted to 0.225 μg/well in PBS.
Western blots
Purified influenza virus was mixed with reducing sample buffer (Pierce) and heated up to 95°C for five minutes to denature the virus. Total denatured protein (5 μg) was loaded into each lane of an 8–16% Criterion TGX Precast Gel (BioRad) and run according to the manufacturer’s instructions. The proteins were transferred to a PVDF membrane (Millipore) in 1X transfer buffer with constant voltage, 100 V, for one hour at 4°C. Following transfer, the PVDF membrane was cut into strips and blocked with 5% milk in TBST for one hour. Each strip was incubated with either 15 μg IgG monoclonal antibody or 3 μg IgM monoclonal antibody diluted in 3 mL of the blocking buffer for one hour, followed by three washes with TBST. The strips were incubated with anti-mouse IgM (Jackson ImmunoResearch) or IgG (Southern Biotech) secondary antibody conjugated with HRP in 5% milk in TBST for one hour. After three washes with TBST, SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) was added to the strips and images were captured using an Amersham Imager 600 imagers (GE Healthcare Life Sciences).
Detection of lipid reactivity using lipid strips and lipid ELISA
Prior to all incubations, 1 μL of purified HKx31 virus and the recombinant H3 protein of HKx31 (Sino Biological) were spotted on the lipid strips as the binding controls. Either PIP or Membrane lipid strip (Echelon Biosciences) was blocked with 50% Pierce Protein-Free (PBS) Blocking Buffer (ThermoFisher) in PBS for one hour and incubated with 1 μg/mL of each monoclonal antibody diluted in the blocking buffer. After a one-hour incubation, the strips were washed three times with PBST and hybridized with the corresponding secondary antibodies as used in Western blot in the protein-free blocking buffer for one hour. The detection of the binding antibodies was described previously in Western blot.
For the PI(4)P ELISA, MaxiSorp 96-well ELISA plates (Invitrogen #44–2404-21) were coated with PI(4)P (Avanti #840045) in absolute ethanol and dried overnight at room temperature. The plates were blocked with 50% Pierce Protein-Free (PBS) Blocking Buffer in PBS for one hour. Serum samples were diluted in PBST with 2.5% FBS, and 75 μL of diluted sample was incubated on the blocked plates for one hour at room temperature. The plate washing and substrate development were as described in the ELISA protocol for influenza antibody detection above.
Monoclonal antibody transfers to Balb/c mice
Six-to eight-week-old Balb/c mice were given a cocktail of 225 μg of either single (13P1A7, 13P1H7, 24R1C4) or broadly reactive (9R1G9D5, 13P10C1, 24R6C1) mAb IgG i.p., followed three hours later by 200 μg i.p. of Poly I:C (Sigma Aldrich). As a positive control, some mice were injected with 225 μg of 163p.77, an antibody previously shown to bind dsDNA and the kidney basement membrane (Krishnan et al., 2012). Poly I:C injections were repeated three times a week for three weeks. Mice received another 225 μg of the same mAb IgG i.p. as above three weeks after the first injection. Urine was collected weekly by manual expression of the bladder to check for proteinuria via Urastix (Siemens) or Qubit (Invitrogen).
Monoclonal antibody transfers to NOD mice
Seven-to eleven-week-old female NOD mice received two to three injections of monoclonal antibody cocktails containing 200–300 μg of broadly reactive (9R1G9D5, 13P10C1, 24R6C1, 24R7A9, 24R9H5, 1P6L16C2, 9R5H16D9) or HKx31-specific (single reactive) (13P1A7, 13P1H7, 24R1C4, 1P10J6B11) antibodies over the course of two weeks. Blood glucose was monitored weekly with a Contour blood glucose monitoring system (Bayer) by testing a drop of blood from the tail vein. Readings greater than 300 mg/dL were confirmed with a second reading at least 24 h later and scored as diabetic. Animals were euthanized when diabetes developed.
Kidney deposits
C57BL/6 mice were injected i.v. with a cocktail containing 150 μg IgG and 150 μg IgM mAb specific for either HKx31 only (single reactive) (13P1A7, 1P10J6B11) or multiple influenza subtypes (broadly reactive) (9R1G9D5, 13P10C1, 13P3E5, 13P4G3, 9R5H16D9, 24R7A9, 24R9H5). Twenty-four hours after injection, kidneys were harvested and drop fixed in 10% neutral buffered formalin prior to analysis. Tissues were cryosectioned at 10 μm thickness and allowed to air dry prior to antigen unmasking with sodium citrate buffer, pH 6.0 for 20 min at 100°C. Sections were subsequently blocked in buffer comprised of PBS containing 2% bovine serum albumin and 0.5% Tween-20. Tissues were stained overnight in PBS containing 2% BSA and Cy3-conjugated donkey anti-mouse IgM (Jackson ImmunoResearch), and AF647-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch). Slides were washed with PBS and mounted with prolong glass hardset mounting medium containing NucBlue (ThermoFisher). High resolution images were acquired using a Marianis spinning disk confocal microscope (Intelligent Imaging Innovations) comprised of a CSU-W and Super-Resolution via Optical Reassignment (SoRa) module (Yokagowa), equipped with a 40X 1.3NA objective, 405 nm, 561 nm and 647 nm laser lines and Prime 95B CMOS camera (Photometrics). Images were analyzed using Slidebook software (Intelligent Imaging Innovations) where images were segmented in each fluorescent channel, and the area of positive staining pixels was determined separately for IgM and IgG. The square area of IgM and IgG signals were subsequently calculated as a proportion of area encompassing individual glomeruli. Images are representative of experiments with at least four mice per group.
Measurement of urine protein
Urine was collected from the NZBW1 mice by manual expression of the bladder and placed in Eppendorf tubes prior to analysis. Quantitative determination of creatinine concentration was performed by the Veterinary Pathology Core of St. Jude Children’s Research Hospital using ABX Pentra Creatinine 120 CP reagent and Jaffe’s reaction. Quantification of total urine protein was determined by Qubit fluorometer (Invitrogen). Proteinuria was evaluated by Uristix (Siemens).
Adsorption of mouse sera
For adsorption assays, serum from NZBWF1 mice or mice given HKx31 i.p. and treated with rapamycin for 14 days was diluted 1:100 with PBS and incubated for one hour on microtiter plates coated with insulin (Alpha Diagnostics) or 80 ng/well of HKx31, 80 ng/well of Vn1203, 0.6 μg/well of whole histones, which were previously blocked with 2.5% FBS. For the adsorption, 50 μL of diluted serum was added to each well in replicates of four. After one hour, the replicates were pooled and coated on another set of plates for an additional hour. Following the second adsorption, the replicates were pooled, and 75 μL of the adsorbed serum was transferred to plates coated with 20 ng/well of HKx31, 20 ng/well of Vn1203, or 0.15 μg/well of whole histone. The ELISA was carried out as described above.
Hemagglutination inhibition (HI) assay
HI assays were performed based on the protocol from Kaufmann et al. (Kaufmann et al., 2017). Mouse sera (9 μL) was combined with 27 μL of receptor destroying enzyme (RDE) in PCR tubes and incubated overnight at 37°C in a thermocycler followed by a 30-min incubation at 56°C. Sera with RDE (25 μL/well) was then serially diluted in a microtiter plate and incubated with four HA units/well of whole HKx31 virus for 30 min at room temperature, followed by a 30–45-min incubation with 50 μL/well of 0.5% turkey red blood cells.
Microneutralization assay
Microneutralization assay for the human samples were carried out as described previously (Oshansky et al., 2014). Plasma were tested against A/Tennessee/1–560/2009, A/Brisbane/59/2007, A/Brisbane/10/2007, B/Brisbane/60–8/2010, and B/Florida/4/2006. Plasma samples were heat-inactivated at 56°C, serially diluted, and incubated with 100 tissue culture infected dose-50 (TCID50) of the virus for one hour at 37°C. After the incubation, the plasma-virus mixture was transferred to MDCK cells pre-seeded in a 96-well plate. The antibody titer was defined as the reciprocal of the highest dilution that resulted in 50% inhibition of infection compared to control wells. A microneutralization titer of >1:40 is typically considered protective. We compared the highest microneutralization titer of the viruses tested for each sample to reactivity to myelin associated glycoprotein (MAG) and gangliosides via the BÜHLMANN Gangliocombi™ anti-MAG ELISA.
QUANTIFICATION AND STATISTICAL ANALYSIS
The Mann-Whitney tests and the two-way ANOVAs were performed with GraphPad Prism (GraphPad Software, La Jolla, CA). The analysis for the autoantigen arrays is described above. For the BÜHLMANN Gangliocombi™ anti-MAG ELISA, the percent ratio of the OD for each sample to the assay controls was determined according to manufacturer’s instructions. Due to violations of the parametric assumptions underlying these data, we employed non-parametric rank-based MANOVA (Dobler et al., 2019) to detect significant differences in antigen binding ratios as functions of influenza infection by year and binned microneutralization titers. (Microneutralization bins were defined by categorizing a sample’s highest log10 microneutralization titer as <1, 1–3, or >3.) Specifically, we utilized the rankMANOVA (v0.0.6; http://github.com/smn74/rankMANOVA, Sarah Friedrich, Dennis Dobler, Markus Pauly, 2019) R package to test for an overall effect using sample-specific bootstrapping with 10,000 iterations. In the case of a statistically significant overall effect, post hoc Dunnett’s tests were calculated to compare each infection time point to the influenza-negative condition, and post hoc Tukey’s tests were calculated for all potential pairwise comparisons among microneutralization bins. Multiple testing correction for pairwise post hoc tests are unnecessary and not recommended in this context, as explained in the rankMANOVA documentation and in Dobler et al. (Dobler et al., 2019).
Information regarding statistical details for all experiments can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Goat anti-mouse IgM-HRP | Southern Biotech | Cat# 1021-05; RRID: AB_2794240 |
| Goat anti-mouse IgG-HRP | Southern Biotech | Cat# 1036-05; RRID: AB_2794348 |
| Donkey anti-mouse IgM-HRP | Jackson ImmunoResearch | Cat# 715-035-020; RRID: AB_2340768 |
| Cy3-conjugated donkey anti-mouse IgM | Jackson ImmunoResearch | Cat# 715-165-020; RRID: AB_2340811 |
| AF647-conjugated donkey anti-mouse IgG | Jackson ImmunoResearch | Cat# 715-605-151; RRID: AB_2340863 |
| 163p.77 | Tony N Marion; UTHSC; (Krishnan et al., 2012) | N/A |
| 1P10J6B11 | This paper | N/A |
| 1P6L16C2 | This paper | N/A |
| 9R1G9D5 | This paper | N/A |
| 9R3G24E3 | This paper | N/A |
| 9R4D16C1 | This paper | N/A |
| 9R4I3H3 | This paper | N/A |
| 9R5H16D9 | This paper | N/A |
| 13P1A7 | This paper | N/A |
| 13P1H2 | This paper | N/A |
| 13P1H7 | This paper | N/A |
| 13P2A9 | This paper | N/A |
| 13P3E5 | This paper | N/A |
| 13P4G12 | This paper | N/A |
| 13P4G3 | This paper | N/A |
| 13P10C1 | This paper | N/A |
| 24R10C1 | This paper | N/A |
| 24R1C4 | This paper | N/A |
| 24R3B2 | This paper | N/A |
| 24R6C1 | This paper | N/A |
| 24R6D5 | This paper | N/A |
| 24R7A9 | This paper | N/A |
| 24R9H5 | This paper | N/A |
| 047-09 1G05 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 051-09 5A01 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| SFV009 3D04 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 029-09 4A01 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 051-09 4B02 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 051-09 5A02 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 030-09 2B03 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 030-09 3B03 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| SC70 5B03 | Patrick Wilson; University of Chicago; (Wrammert et al., 2011) | N/A |
| SC1009 3B05 | Patrick Wilson; University of Chicago; (Wrammert et al., 2011) | N/A |
| 030-09 1E05 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 045-09 2B06 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| SC70 1F02 | Patrick Wilson; University of Chicago; (Wrammert et al., 2011) | N/A |
| 030-09 2G03 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| SFV005 2G02 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| SC1000 3D04 | Patrick Wilson; University of Chicago; (Wrammert et al., 2011) | N/A |
| 047-09 1F05 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| CR9114 | Patrick Wilson; University of Chicago; (Dreyfus et al., 2012) | N/A |
| F16 | Patrick Wilson; University of Chicago; (Corti et al., 2011) | N/A |
| 051-09 4A03 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 047-09 4E01 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 13001-07 5E01 | Patrick Wilson; University of Chicago; (Henry Dunand et al., 2016) | N/A |
| SFV015 2F04 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 045-09 2B05 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| SFV019 2A02 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 13001-07 5G01 | Patrick Wilson; University of Chicago; (Henry Dunand et al., 2016) | N/A |
| 240 1A04 | Patrick Wilson Patrick Wilson; University of Chicago; Guthmiller et al., 2020 | N/A |
| 217 1A02 | Patrick Wilson Patrick Wilson; University of Chicago; (Guthmiller et al., 2020) | N/A |
| 220 1A04 | Patrick Wilson Patrick Wilson; University of Chicago; (Guthmiller et al., 2020) | N/A |
| 047-09 4G02 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 008-10 5G04 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| DR 2A02 | Patrick Wilson; University of Chicago; (Andrews et al., 2015) | N/A |
| 229 1D05 | Patrick Wilson; University of Chicago; (Chen et al., 2018) | N/A |
| 229 2B04 | Patrick Wilson; University of Chicago; (Chen et al., 2018) | N/A |
| 235 1E06 | Patrick Wilson; University of Chicago; (Chen et al., 2018) | N/A |
| 041 1D04 | Patrick Wilson; University of Chicago; (Henry et al., 2019) | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| A/Brisbane/10/2007 | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
| A/Brisbane/59/2007 | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
| A/Tennessee/1-560/2009 | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
| B/Brisbane/60-8/2010 | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
| B/Florida/4/2006 | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
| Campylobacter jejuni | ATCC | Cat# 700297 |
| HKx31 influenza virus | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
| MCMV (Murine cytomegalovirus) | ATCC | Cat# VR-1399 |
| ΔVn1203 influenza virus | Paul Thomas; St. Jude Children’s Research Hospital | N/A |
|
| ||
| Biological samples | ||
|
| ||
| FLU09 plasma samples | St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center/Le Bonheur Children’s Hospital; (Oshansky et al., 2014) | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Ampicillin | Millipore | Cat# A9518-25G |
| Avertin (2,2,2-tribromoethanol) | Peoples Custom Rx | N/A |
| Complete Freund’s Adjuvant (with 5 mg/mL Mycobacterium tuberculosis) | Chondrex | Cat# 7023 |
| HAT (sodium hypoxanthine, aminopterin and thymidine) media supplement | Sigma | Cat# H0262 |
| Pertussis toxin | List Biologicals | Cat# 180 |
| Pierce protein-free blocking buffer | Thermo Fisher | Cat# 37572 |
| Poly I:C (Polyinosinic–polycytidylic acid) | Sigma Aldrich | Cat# P1530 |
| Rapamune | Wyeth | Cat# NDC 00008-1030-04 |
| Sigmafast OPD (o-Phenylenediamine dihydrochloride) | Sigma | Cat# P9187 |
| SuperSignal West Dura Extended Duration Substrate | Thermo Scientific | Cat# 34076 |
| Peripheral Myelin Protein P0 peptide (180–199, SSKRGRQTPVLYAMLDHSRS) | Hartwell Center; SJCRH | N/A |
| Albumin from mouse sera | Sigma | Cat# A3559-5MG |
| Collagen II (chick type II collagen) | Chondrex | Cat# 2016 |
| Ganglioside GD1a (from bovine brain) | Sigma | Cat# G2392 |
| Ganglioside GM1 | US Biological | Cat# G2006-10 |
| H1 (histone) | New England BioLabs | Cat# M2501S |
| H2B (histone) | New England BioLabs | Cat# M2505S |
| H3 (histone) | New England BioLabs | Cat# M2506S |
| H4 (histone) | New England BioLabs | Cat# M2504S |
| Insulin (human recombinant) | Fitzgerald | Cat# 30-AI51 |
| Insulin (mouse recombinant) | R&D Systems | Cat# 7544-MR |
| LPS (Lipopolysaccharide) from E. coli | Sigma | Cat# L3012 |
| Mouse native myelin basic protein | Creative Biomart | Cat# MBP-6949M |
| Native KLH (Keyhole limpet hemocyanin) | Sigma | Cat# SRP6195-25MG |
| Ovalbumin (OVA) | Sigma | Cat# A5503-10G |
| PI(4)P (Phosphatidylinositol-4-phosphate) | Avanti | Cat# 840045 |
| rHA (A/Aichi/2/1968 H3N2) | eEnzyme | Cat# IA-0043W-005P |
| rHA (A/California/04/2009 H1N1) | eEnzyme | Cat# IA-SW-12P |
| rHA (A/HKx31 H3N2) | Sino Biological | Cat# 40059-V08H |
| rHA (A/Vietnam/1203/2004 H5N1) | eEnzyme | Cat# IA-0051-005P |
| SARS-CoV-2 receptor binding domain (RBD) protein | St. Jude GMP | N/A |
| Whole histones from chicken red blood cells | Immunovision | Cat# HIS-1010 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Anti-mouse ANA ELISA | Alpha Diagnostics | Cat# 5210 |
| Anti-mouse dsDNA ELISA | Alpha Diagnostics | Cat# 5120 |
| Anti-mouse GM1 IgG ELISA | MyBiosource | Cat# MBS7213788 |
| Anti-mouse IgM ELISA | RayBiotech | Cat# ELM-IGM-1 |
| Anti-mouse Insulin ELISA | Alpha Diagnostics | Cat# 3710-MIM |
| Anti-mouse Sm ELISA | Alpha Diagnostics | Cat# 5405 |
| BÜHLMANN Gangliocombi™ MAG ELISA | BÜHLMANN Diagnostics Corp | Cat# EK-GCM-U |
| Immunoglobulin isotyping magnetic bead panel | Milliplex | Cat# MGAMMAG-300K-04 |
| Membrane lipid strip | Echelon Biosciences | Cat# P-6002-2 |
| PIP lipid strip | Echelon Biosciences | Cat# P-6001-2 |
| Uristix | Siemens | Cat# AM-2166 |
|
| ||
| Deposited data | ||
|
| ||
| Microarray data | This paper | GEO: GSE196366 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MDCK cells | ATCC | Cat# CCL-34; RRID: CVCL_0422 |
| Sp2/mIL-6 hybridoma cells | ATCC | Cat# CRL-2016; RRID: CVCL_C561 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Balb/cJ: wildtype | The Jackson Laboratory | Cat# 000651; RRID: IMSR_JAX:000651 |
| Mouse: C57BL/6J: wildtype | The Jackson Laboratory | Cat# 000664; RRID: IMSR_JAX:000664 |
| Mouse: NOD/ShiLtJ: wildtype | The Jackson Laboratory | Cat# 001976; RRID: IMSR_JAX:001976 |
| Mouse: NZBWF1/J: wildtype | The Jackson Laboratory | Cat# 100008; RRID: IMSR_JAX:100008 |
|
| ||
| Software and algorithms | ||
|
| ||
| Genepix Pro 6.0 | Molecular Devices | RRID: SCR_010969 |
| Slidebook | Intelligent Imaging Innovations | RRID: SCR_014300 |
Highlights.
Human and mouse broadly reactive influenza antibodies bind self-antigens
Enhancing broadly reactive antibodies exacerbates disease in models of autoimmunity
Transfer of broadly reactive monoclonal antibodies augments autoimmune disease
Humans infected with novel influenza subtypes have elevated autoreactive antibodies
ACKNOWLEDGMENTS
We thank Jenna Guthmiller and Patrick Wilson for providing the human monoclonal antibodies, the St. Jude Animal Resource Center for animal care, and Ankit Malik for input with EAN and C. jejuni infections. This work was supported by a National Institute of Allergy and Infectious Diseases grant (R01AI114728), NIAID contract to St. Jude Center of Excellence for Influenza Research and Surveillance contract (HHSN272201400006C), and ALSAC to M.A.M.
Footnotes
DECLARATION OF INTERESTS
P.G.T. has consulted for Illumina, 10X, Immunoscape, and Cyotagents. P.G.T. and J.C.C. filed patents related to the treatment of severe respiratory infections (not based on research in this paper).
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110482.
REFERENCES
- Ahmed SS, Schur PH, MacDonald NE, and Steinman L (2014). Narcolepsy, 2009 A(H1N1) pandemic influenza, and pandemic influenza vaccinations: what is known and unknown about the neurological disorder, the role for autoimmunity, and vaccine adjuvants. J. Autoimmun. 50, 1–11. [DOI] [PubMed] [Google Scholar]
- Altman MO, Bennink JR, Yewdell JW, and Herrin BR (2015). Lamprey VLRB response to influenza virus supports universal rules of immunogenicity and antigenicity. Elife 4, e07467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F, Stadlbauer D, Strohmeier S, Nguyen THO, Chromikova V, McMahon M, Jiang K, Arunkumar GA, Jurczyszak D, Polanco J, et al. (2020). A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 26, 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, and Bluestone JA (2005). The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 23, 447–485. [DOI] [PubMed] [Google Scholar]
- Andrews SF, Huang Y, Kaur K, Popova LI, Ho IY, Pauli NT, Henry Dunand CJ, Taylor WM, Lim S, Huang M, et al. (2015). Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 7, 316ra192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SF, Joyce MG, Chambers MJ, Gillespie RA, Kanekiyo M, Leung K, Yang ES, Tsybovsky Y, Wheatley AK, Crank MC, et al. (2017). Preferential induction of cross-group influenza A hemagglutinin stem–specific memory B cells after H7N9 immunization in humans. Sci. Immunol 2, eaan2676. [DOI] [PubMed] [Google Scholar]
- Angeletti D, and Yewdell JW (2018a). Is it possible to develop a “universal” influenza virus vaccine? Outflanking antibody immunodominance on the road to universal influenza vaccination. Cold Spring Harb. Perspect. Biol. 10, a028852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti D, and Yewdell JW (2018b). Understanding and manipulating viral immunity: antibody immunodominance enters center stage. Trends Immunol. 39, 549–561. [DOI] [PubMed] [Google Scholar]
- Angeletti D, Kosik I, Santos JJS, Yewdell WT, Boudreau CM, Mallajosyula VVA, Mankowski MC, Chambers M, Prabhakaran M, Hickman HD, et al. (2019). Outflanking immunodominance to target subdominant broadly neutralizing epitopes. Proc. Natl. Acad. Sci. U S A 116, 13474–13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic G, van der Poel CE, Kuraoka M, Schmidt AG, Carroll MC, Kelsoe G, and Harrison SC (2019). Autoreactivity profiles of influenza hemagglutinin broadly neutralizing antibodies. Sci. Rep. 9, 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker CIS, and Snape MD (2014). Pandemic influenza A H1N1 vaccines and narcolepsy: vaccine safety surveillance in action. Lancet Infect. Dis. 14, 227–238. [DOI] [PubMed] [Google Scholar]
- Brooks SP, Trueman RC, and Dunnett SB (2012). Assessment of motor coordination and balance in mice using the rotarod, elevated bridge, and footprint tests. Curr. Protoc. Mouse Biol. 2, 37–53. [DOI] [PubMed] [Google Scholar]
- Chen Y-Q, Wohlbold TJ, Zheng N-Y, Huang M, Huang Y, Neu KE, Lee J, Wan H, Rojas KT, Kirkpatrick E, et al. (2018). Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D, and Lanzavecchia A (2013). Broadly neutralizing antiviral antibodies. Annu. Rev. Immunol. 31, 705–742. [DOI] [PubMed] [Google Scholar]
- Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, et al. (2011). A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856. [DOI] [PubMed] [Google Scholar]
- Cutillo G, Saariaho A-H, and Meri S (2020). Physiology of gangliosides and the role of antiganglioside antibodies in human diseases. Cell. Mol. Immunol. 17, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobler D, Friedrich S, and Pauly M (2019). Nonparametric MANOVA in meaningful effects. Ann. Inst. Stat. Math. 72, 997–1022. [Google Scholar]
- Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, et al. (2012). Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, and Wilson IA (2012). Broadly neutralizing antibodies against influenza virus and prospects for universal therapies. Curr. Opin. Virol. 2, 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellebedy AH, Krammer F, Li G-M, Miller MS, Chiu C, Wrammert J, Chang CY, Davis CW, McCausland M, Elbein R, et al. (2014). Induction of broadly cross-reactive antibody responses to the influenza HA stem region following H5N1 vaccination in humans. Proc. Natl. Acad. Sci. U S A 111, 13133–13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RA, Fuhr R, Smolenov I, Mick, Ribeiro A, Panther L, Watson M, Senn JJ, Smith M, Almarsson Ӧ, et al. (2019). mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334. [DOI] [PubMed] [Google Scholar]
- Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, and Doherty PC (1998). Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity 8, 683–691. [DOI] [PubMed] [Google Scholar]
- Fukuyama H, Shinnakasu R, and Kurosaki T (2020). Influenza vaccination strategies targeting the hemagglutinin stem region. Immunol. Rev. 296, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaderi S, Gunnes N, Bakken IJ, Magnus P, Trogstad L, and Håberg SE (2016). Risk of Guillain-Barré syndrome after exposure to pandemic influenza A(H1N1)pdm09 vaccination or infection: a Norwegian population-based cohort study. Eur. J. Epidemiol. 31, 67–72. [DOI] [PubMed] [Google Scholar]
- Gitlin AD, von Boehmer L, Gazumyan A, Shulman Z, Oliveira TY, and Nussenzweig MC (2016). Independent roles of switching and hypermutation in the development and persistence of B lymphocyte memory. Immunity 44, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalvez DG, Fletcher JL, Yoo SW, Wood RJ, Murray SS, and Xiao J (2017). A simple approach to induce experimental autoimmune neuritis in C57BL/6 mice for functional and neuropathological assessments. J. Vis. Exp 10.3791/56455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthmiller JJ, Lan LY-L, Fernández-Quintero ML, Han J, Utset HA, Bitar DJ, Hamel NJ, Stovicek O, Li L, Tepora M, et al. (2020). Polyreactive broadly neutralizing B cells are selected to provide defense against pandemic threat influenza viruses. Immunity 53, 1230–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthmiller JJ, Han J, Utset HA, Li L, Lan LY-L, Henry C, Stamper CT, McMahon M, O’Dell G, Fernández-Quintero ML, et al. (2021). Broadly neutralizing antibodies target a hemagglutinin anchor epitope. Nature 602, 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haji-Ghassemi O, Gagnon SML, Müller-Loennies S, and Evans SV (2017). Polyspecificity of anti-lipid A antibodies and its relevance to the development of autoimmunity. Adv. Exp. Med. Biol. 966, 181–202. [DOI] [PubMed] [Google Scholar]
- Han F, Lin L, Warby SC, Faraco J, Li J, Dong SX, An P, Zhao L, Wang LH, Li QY, et al. (2011). Narcolepsy onset is seasonal and increased following the 2009 H1N1 pandemic in China. Ann. Neurol. 70, 410–417. [DOI] [PubMed] [Google Scholar]
- Hao Y, Wang W, Jacobs BC, Qiao B, Chen M, Liu D, Feng X, and Wang Y (2019). Antecedent infections in Guillain-Barré syndrome: a single-center, prospective study. Ann. Clin. Transl. Neurol. 6, 2510–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JF, Hawley RG, Hawley TS, and Crawford-Sharpe GC (1992). Increased frequency of both total and specific monoclonal antibody producing hybridomas using a fusion partner that constitutively expresses recombinant IL-6. J. Immunol. Methods 148, 199–207. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Sato S, Kikuchi K, and Takehara K (1998). Antigen specificity of antihistone antibodies in systemic sclerosis. Ann. Rheum. Dis. 57, 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, et al. (2005). Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308, 1906–1908. [DOI] [PubMed] [Google Scholar]
- Henry C, Zheng N-Y, Huang M, Cabanov A, Rojas KT, Kaur K, Andrews SF, Palm A-KE, Chen Y-Q, Li Y, et al. (2019). Influenza virus vaccination elicits poorly adapted B cell responses in elderly individuals. Cell Host Microbe 25, 357–366.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Dunand CJ, Leon PE, Huang M, Choi A, Chromikova V, Ho IY, Tan GS, Cruz J, Hirsh A, Zheng N-Y, et al. (2016). Both neutralizing and non-neutralizing human H7N9 influenza vaccine-induced monoclonal antibodies confer protection. Cell Host Microbe 19, 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TW, Willison HJ, Nachamkin I, Li CY, Veitch J, Ung H, Wang GR, Liu RC, Cornblath DR, Asbury AK, et al. (1999). Anti-GD1a antibody is associated with axonal but not demyelinating forms of Guillain-Barre syndrome. Ann. Neurol. 45, 168–173. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Krauss S, Perez D, Webby R, and Webster RG (2002). Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 20, 3165–3170. [DOI] [PubMed] [Google Scholar]
- Joyce MG, Wheatley AK, Thomas PV, Chuang G-Y, Soto C, Bailer RT, Druz A, Georgiev IS, Gillespie RA, Kanekiyo M, et al. (2016). Vaccine-induced antibodies that neutralize group 1 and group 2 influenza A viruses. Cell 166, 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek R, Pasciak M, Szymczak-Kulus K, and Czerwinski M (2017). CD1: a singed cat of the three antigen presentation systems. Arch. Immunol. Ther. Exp. (Warsz.) 65, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann L, Syedbasha M, Vogt D, Hollenstein Y, Hartmann J, Linnik JE, and Egli A (2017). An optimized hemagglutination inhibition (HI) assay to quantify influenza-specific antibody titers. J. Vis. Exp 10.3791/2F55833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating R, Hertz T, Wehenkel M, Harris TL, Edwards BA, McClaren JL, Brown SA, Surman S, Wilson ZS, Bradley P, et al. (2013). The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat. Immunol. 14, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating R, Johnson JL, Brice DC, Labombarde JG, Dent AL, and McGargill MA (2020). Broadly reactive influenza antibodies are not limited by germinal center competition with high-affinity antibodies. MBio 11, e01859. 10.1128/mBio.01859-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Hahn M, Klenow L, and Golding H (2020). Autoreactivity of broadly neutralizing influenza human antibodies to human tissues and human proteins. Viruses 12, 1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M, Takahashi M, Yokoyama K, and Kanda T (2015). Ambiguous value of anti-ganglioside IgM autoantibodies in Guillain-Barré syndrome and its variants. J. Neurol. 262, 1954–1960. [DOI] [PubMed] [Google Scholar]
- Krammer F (2016). Novel universal influenza virus vaccine approaches. Curr. Opin. Virol. 17, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F (2019). The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol. 19, 383–397. [DOI] [PubMed] [Google Scholar]
- Krishnan MR, Wang C, and Marion TN (2012). Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. 82, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JC, Vasa PP, Campitelli MA, Hawken S, Wilson K, Rosella LC, Stukel TA, Crowcroft NS, McGeer AJ, Zinman L, et al. (2013). Risk of Guillain-Barré syndrome after seasonal influenza vaccination and influenza health-care encounters: a self-controlled study. Lancet Infect. Dis. 13, 769–776. [DOI] [PubMed] [Google Scholar]
- Lardone RD, Yuki N, Odaka M, Daniotti JL, Irazoqui FJ, and Nores GA (2010). Anti-GM1 IgG antibodies in Guillain-Barré syndrome: fine specificity is associated with disease severity. J. Neurol. Neurosurg. Psychiatry 81, 629–633. [DOI] [PubMed] [Google Scholar]
- Lee PS, and Wilson IA (2015). Structural characterization of viral epitopes recognized by broadly cross-reactive antibodies. Curr. Top. Microbiol. Immunol. 386, 323–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy T, Silva M, D’Hooge R, Aerts J-M, and Berckmans D (2009). Automated gait analysis in the open-field test for laboratory mice. Behav. Res. Methods 41, 148–153. [DOI] [PubMed] [Google Scholar]
- Li G-M,Chiu C,Wrammert J,McCausland M, Andrews SF, Zheng N-Y, Lee J-H, Huang M, Qu X, Edupuganti S, et al. (2012). Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc. Natl. Acad. Sci. U S A 109, 9047–9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q-Z, Zhou J, Wandstrat AE, Carr-Johnson F, Branch V, Karp DR, Mohan C, Wakeland EK, and Olsen NJ (2007). Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin. Exp. Immunol. 147, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Yang G, Wiehe K, Nicely NI, Vandergrift NA, Rountree W, Bonsignori M, Alam SM, Gao J, Haynes BF, et al. (2015). Polyreactivity and autoreactivity among HIV-1 antibodies. J. Virol. 89, 784–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik A, Sharma D, St Charles J, Dybas LA, and Mansfield LS (2014). Contrasting immune responses mediate Campylobacter jejuni-induced colitis and autoimmunity. Mucosal Immunol. 7, 802–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyas GR, Beck Z, Karasavvas N, and Alving CR (2009). Lipid binding properties of 4E10, 2F5, and WR304 monoclonal antibodies that neutralize HIV-1. Biochim. Biophys. Acta 1788, 660–665. [DOI] [PubMed] [Google Scholar]
- Morel L (2010). Genetics of SLE: evidence from mouse models. Nat. Rev. Rheumatol. 6, 348–357. [DOI] [PubMed] [Google Scholar]
- Nohynek H, Jokinen J, Partinen M, Vaarala O, Kirjavainen T, Sundman J, Himanen S-L, Hublin C, Julkunen I, Olsén P, et al. (2012). AS03 adjuvanted AH1N1 vaccine associated with an abrupt increase in the incidence of childhood narcolepsy in Finland. PLoS One 7, e33536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshansky CM, Gartland AJ, Wong S-S, Jeevan T, Wang D, Roddam PL, Caniza MA, Hertz T, Devincenzo JP, Webby RJ, et al. (2014). Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 189, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovas C, Vlachoyiannopoulos PG, Kordossis T, and Moutsopoulos HM (1999). Anti-phospholipid antibodies in HIV infection and SLE with or without anti-phospholipid syndrome: comparisons of phospholipid specificity, avidity and reactivity with beta2-GPI. J. Autoimmun. 13, 347–355. [DOI] [PubMed] [Google Scholar]
- Pica N, Hai R, Krammer F, Wang TT, Maamary J, Eggink D, Tan GS, Krause JC, Moran T, Stein CR, et al. (2012). Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. U S A 109, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S (2000). Animal models of autoimmunity and their relevance to human diseases. Curr. Opin. Immunol. 12, 684–690. [DOI] [PubMed] [Google Scholar]
- Scofield RH (2004). Autoantibodies as predictors of disease. Lancet 363, 1544–1546. [DOI] [PubMed] [Google Scholar]
- St Charles JL, Bell JA, Gadsden BJ, Malik A, Cooke H, Van de Grift LK, Kim HY, Smith EJ, and Mansfield LS (2017). Guillain Barré Syndrome is induced in Non-Obese Diabetic (NOD) mice following Campylobacter jejuni infection and is exacerbated by antibiotics. J. Autoimmun. 77, 11–38. [DOI] [PubMed] [Google Scholar]
- Sui J, Sheehan J, Hwang WC, Bankston LA, Burchett SK, Huang C-Y, Liddington RC, Beigel JH, and Marasco WA (2011). Wide prevalence of heterosubtypic broadly neutralizing human anti-influenza A antibodies. Clin. Infect. Dis. 52, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurmond J, and Diamond B (2015). Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J. Clin. Invest. 125, 2194–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson CA, Wang Y, Jackson LM, Olson M, Wang W, Liavonchanka A, Keleta L, Silva V, Diederich S, Jones RB, et al. (2012). Pandemic H1N1 influenza infection and vaccination in humans induces cross-protective antibodies that target the hemagglutinin stem. Front. Immunol. 3, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, and Wardemann H (2007). Autoreactivity in human IgG+ memory B cells. Immunity 26, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafyllopoulou A, Franzke C-W, Seshan SV, Perino G, Kalliolias GD, Ramanujam M, Rooijen N, Davidson A, and Ivashkiv LB (2010). Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc. Natl. Acad. Sci. U S A 107, 3012–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellozzi C, Iqbal S, and Broder K (2014). Guillain-Barre syndrome, influenza, and influenza vaccination: the epidemiologic evidence. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am 58, 1149–1155. [DOI] [PubMed] [Google Scholar]
- Vogel G (2015). Narcolepsy link to pandemic flu vaccine becomes clearer. Science 349, 17. [DOI] [PubMed] [Google Scholar]
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC (2003). Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377. [DOI] [PubMed] [Google Scholar]
- Wheatley AK, Whittle JRR, Lingwood D, Kanekiyo M, Yassine HM, Ma SS, Narpala SR, Prabhakaran MS, Matus-Nicodemos RA, Bailer RT, et al. (2015). H5N1 vaccine-elicited memory B cells are genetically constrained by the IGHV locus in the recognition of a neutralizing epitope in the hemagglutinin stem. J. Immunol. 195, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle JRR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, et al. (2011). Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. U S A 108, 14216–14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willison HJ (2011). Biomarkers in experimental models of antibody-mediated neuropathies. J. Peripher. Nerv. Syst. 16, 60–62. [DOI] [PubMed] [Google Scholar]
- Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng N-Y, Mays I, Garman L, Helms C, et al. (2008). Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrammert J, Koutsonanos D, Li G-M, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, et al. (2011). Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 208, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao ZX, Miller JS, and Zheng SG (2021). An updated advance of autoantibodies in autoimmune diseases. Autoimmun. Rev. 20, 102743. [DOI] [PubMed] [Google Scholar]
- Zhang R, Verkoczy L, Wiehe K, Alam SM, Nicely NI, Santra S, Bradley T, Pemble CW, Zhang J, Gao F, et al. (2016). Initiation of immune tolerance–controlled HIV gp41 neutralizing B cell lineages. Sci. Transl. Med. 8, 336ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data have been deposited at NCBI GEO: GSE196366, and are publicly available as of the date of publication. All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original custom code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.