

Abstract
PRMT5 and its substrate adaptor proteins (SAPs), pICln and Riok1, are synthetic lethal dependencies in MTAP-deleted cancer cells. SAPs share a conserved PRMT5 binding motif (PBM) which mediates binding to a surface of PRMT5 distal to the catalytic site. This interaction is required for methylation of several PRMT5 substrates, including histone and spliceosome complexes. We screened for small molecule inhibitors of the PRMT5-PBM interaction and validated a compound series which binds to the PRMT5-PBM interface and directly inhibits binding of SAPs. Mode of action studies revealed formation of a covalent bond between a halogenated pyridazinone group and cysteine 278 of PRMT5. Optimization of the starting hit produced a lead compound, BRD0639, which engages the target in cells, disrupts PRMT5-RIOK1 complexes, and reduces substrate methylation. BRD0639 is a first-in-class PBM-competitive inhibitor that can support studies of PBM-dependent PRMT5 activities and the development of novel PRMT5 inhibitors that selectively target these functions.
Graphical Abstract
Introduction
The protein arginine methyltransferase PRMT5 plays a central role in the maintenance of cellular homeostasis. By catalyzing symmetric dimethylation of arginines in over 100 nuclear and cytoplasmic proteins, PRMT5 participates in the regulation of gene expression, ribosomal biogenesis, mRNA splicing, protein translation, DNA damage response and immune functions1. Not surprisingly, PRMT5 is an essential gene and its complete inactivation is not compatible with survival of most cell lines2.
PRMT5 has also been shown to exert oncogenic functions and its levels are dysregulated in many solid and hematological malignancies, an event that correlates with advanced disease and poor clinical outcomes3. As such, PRMT5 is an attractive drug target in oncology and several potent and selective inhibitors of the catalytic pocket have been developed and are in clinical trials4. However, given the essential role of PRMT5 in normal tissue homeostasis, it is likely that PRMT5 inhibition could have a limited therapeutic window.
A wider therapeutic index might be achieved by selectively targeting PRMT5 in cancers bearing deletions of the methylthioadenosine phosphorylase gene, MTAP, with which PRMT5 has been shown to be synthetic lethal. MTAP deletion is a relatively common cancer genomic aberration that occurs in pancreatic tumors (26%), glioblastoma (53%) and mesotheliomas (67%)5, 6. This loss is due to its proximity to the CDKN2A tumor suppressor locus, with which MTAP is frequently co-deleted. Loss of MTAP has unknown functional consequences for tumorigenesis, but it leads to accumulation of its substrate methylthioadenosine (MTA), which can act as an endogenous competitor of S-adenosylmethionine (SAM), the methyl donor used by all arginine N-methyltransferases. Notably, MTA appears to selectively inhibit PRMT5, but not other PRMT family members, likely due to key structural features within the PRMT5 SAM binding pocket5. The partial inhibition of PRMT5 sensitizes MTAP-deleted cells to further loss of PRMT5 function by siRNA. Thus, pharmacological inhibition of PRMT5 could be a viable strategy with a suitable therapeutic window for clinical use against MTAP-deleted cancers5,7,8. Unfortunately, available inhibitors of the PRMT5 catalytic pocket act with either a SAM cooperative9 or SAM/MTA-competitive mode of action10, 11, 12 and as such do not appear to take advantage of MTAP deletion. MTA-cooperative PRMT5 inhibitors are predicted to better leverage this synthetic lethality. Two such molecules have been recently disclosed with different degrees of selectivity for MTAP-deleted cancer cells in vitro24 and in preclinical studies23. Inhibitors of the upstream enzyme MAT2A, which catalyzes the synthesis of SAM, have also been discovered and optimized to exploit the unbalanced SAM/MTA ratio of MTAP-deleted cancers. Preclinical data support an MTAP-dependent and synergistic anti-tumor activity when these inhibitors are used in combination with taxanes or gemcitabine8. However, SAM is a ubiquitous methyl donor, and a reduction of its levels would alter the activity of multiple methyltransferases, potentially posing a toxicity risk to this approach. The efficacy and safety profile of MAT2A inhibitors are currently being evaluated in clinical trials (NCT03435250).
In addition to PRMT5 itself, large-scale shRNA screens also identified PRMT5 substrate adaptors pICln and RIOK1, as well as its obligate partner WDR77, as synthetic lethalities in MTAP-null cancers2. These results suggested alternative routes for therapeutic modulation of PRMT5 activity, such as direct inhibition of the substrate adaptor proteins or inhibition of the interactions with PRMT5. These mechanisms would be distinct from inhibition of the catalytic site and potentially provide synergy with, or a different therapeutic profile than previously mentioned approaches. We recently elucidated the molecular mechanism mediating the interaction between PRMT5 and its SAPs. Specifically, we identified a conserved 7 residue peptide sequence, GQF(D/E)DA(D/E), present in all three known PRMT5 SAPs (pICln, RIOK1 and COPR5), that mediates substrate adaptor binding to PRMT5, which we termed the PRMT5 Binding Motif (PBM). We solved the structure of a PBM peptide bound to its cognate site on PRMT5, which is distinct and distal from the catalytic domain. We also determined that genetic perturbation of this site results in loss of substrate methylation and a reduction in MTAP-deleted cell growth relative to WT13. Based on these observations, we launched a screening campaign to identify small molecules capable of inhibiting this interaction.
Here we describe the identification and development of the first-in-class PBM competitive covalent compound, BRD0639, a chemical probe with on-target cellular activity that can support the exploration of a new class of PRMT5 inhibitors.
Results
Hit finding activities
To identify inhibitors of the PBM peptide interaction we utilized several hit-finding approaches to screen lead-like or fragment compound libraries (Fig. 1a). As a primary strategy, we utilized a competition fluorescence polarization (FP) assay to measure the ability of compounds to displace the interaction between a fluorophore-labeled RIOK1 PBM peptide and the purified PRMT5:WDR77 hetero-octameric complex. In total, we screened more than 900K small molecules: 850K from the Diversity and Lead-like compound libraries from Charles River Laboratories, 50K from the Chembridge DIVERSet, 14K compounds from the Broad Institute Diversity Oriented Synthesis (DOS) library14 and 1K compounds selected by virtual screening. In parallel, we also carried out an NMR-based fragment screen and an in silico virtual pharmacophore screen (Fig. 1a and SI). These latter approaches did not yield starting points for optimization and were thus deprioritized.
Figure 1. Hit Finding Activities.

(a) flow chart for inhibitors of the PBM:PRMT5 interaction. (b) Chemical structure of 1. (c) Dose-dependent displacement of a fluorescently-labeled RIOK1-derived PBM peptide probe by 1, as measured by FP.
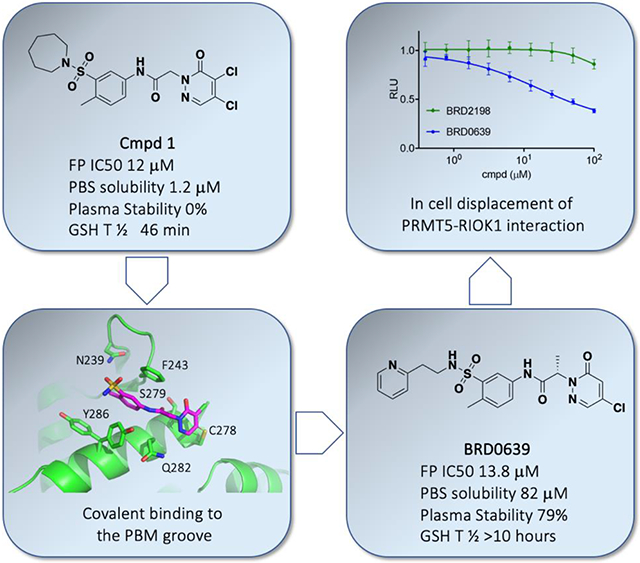
Conversely, initial hits from the FP assay-based screen (with ≥20% displacement) were reassessed in duplicate, and counter-screened against an unrelated protein complex FP assay (MCL1-Noxa) to remove false positives (Fig. 1a and 1c). The IC50 values of confirmed hits were determined using the PRMT5 FP assay across an 8-point dose range. These studies led to the identification of only one cluster, based on a N-Aryl acetamide substituted dichloropyridazinone, as exemplified by compound 1 (Fig. 1b and Table 1) with an FP IC50 of 12 μM and solubility of 1.2 μM in PBS. Compound 1 was validated by SPR, NMR and X-ray crystallography (Fig. 2a).
Table 1.
R1 modifications
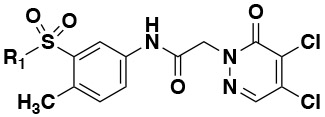
|
||||||
|---|---|---|---|---|---|---|
| Compound | Structure of R1 |
PRMT5 IC50 (μM) FP40min |
PBS Solubility (μM) |
Plasma Stability (%Remaining) | GSH T½ (min) |
|
| Human | Mouse | |||||
| 1 |
|
12.4 | 1.2 | 92 | 0 | 46 |
| 2 |
|
5.4 | 98 | 84 | 0 | ND |
| 3 |
|
12.7 | 0.8 | ND | ND | ND |
| 4 |
|
10.1 | 89 | 72 | 0 | ND |
| 5 |
|
14.1 | 56 | 89 | 0 | 25 |
| 6 |
|
2.2 | 47 | 82 | 14 | 31 |
| 7 |
|
4.6 | 88 | 100 | 21 | ND |
| 8 |
|
2.5 | 93 | 92 | 2 | ND |
| EPZ015666 | >100 | 101 | ND | ND | ND | |
| JNJ-64619178 | >100 | 7 | ND | ND | ND | |
| MTA | >100 | 110 | ND | ND | ND | |
Figure 2. Mechanism of binding.

(a) Compound 1 (magenta) binds distal to the catalytic site (SAM analog, sinefungin, red). A protomer of PRMT5 (light green) and WDR77 (dark green) are shown as cartoons. The 3 other protomer units from the heterooctamer complex are shown in grey (PDB code 6V0P). (b) Crystal structure of compound 1 (magenta) bound to PRMT5 (green). Residues described in the text are labeled. Two alternate conformations of Y286 are shown (PDB code 6V0P). (c) Cryo-EM structure of compound 6 (cyan) bound to PRMT5 (green) (PDB code 7M05).
The PRMT5 catalytic site is distal from the PBM groove and S-Adenosyl Methionine (SAM) binding does not affect the interaction between PRMT5 and the PBM peptide13. Consistent with this observation, we found that SAM competitive (JNJ-64619178) and SAM co-operative (EPZ015666) inhibitors of the PRMT5 catalytic site failed to inhibit this binding (Table 1 and Fig. 2a).
EARLY SAR
Early hit exploration focused on improving solubility and potency of compound 1. Analysis of commercially available analogs highlighted the requirement for the pyridazinone portion and as such we began exploring the SAR of sulfonamide variations (Table 1). The majority of analogs were synthesized in five steps from available reagents (Scheme 1). The azepane could readily be modified and its solubility greatly enhanced by the introduction of a basic nitrogen (compound 2, solubility 98 μM). Smaller rings (compound 4, solubility 89 μM), as well as acyclic sulfonamides (compound 5, solubility 56 μM), were both more amenable to rapid analog synthesis and retained significant potency (FP IC50 10 μM and 14 μM, respectively) opening the path forward for further SAR exploration. While these simple modifications significantly improved compound solubility, we found that pyridine isomers (compounds 6-8) also led to an improvement in potency. Indeed, compound 6 showed a good balance of potency and solubility (Table 1). Modification of the aryl ring (Table 2), either by substitution of the methyl group (compounds 9-11) or by the addition of methyl groups to the unsubstituted carbons (X, Y, or Z, compound 12-14), were tolerated, albeit with modest reduction in either potency, solubility, or both. Pyridine isomer 15 was tolerated, if poorly soluble, whereas other isomers 16, 17 were not, and saturated analog 18 demonstrated no ability to displace the PBM peptide. Modification of the central acetamide (Table 3) afforded significant improvements in potency by the installation of single methyl group alpha to the carbonyl 20, whereas the other enantiomer 21 and the N-methyl analog 19 were decidedly less active. These amide modifications all resulted in increased stability in mouse plasma, presumably by blocking amide hydrolysis by carboxylesterase, which is present in high concentrations in mice15,16.
Scheme 1. (A) Synthesis of 1 and 6 (B) Synthesis of sulfonamides 34 and 36.

Table 2.
Central ring modifications
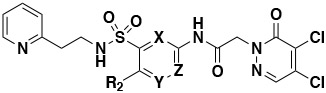
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound Number |
R2 | X | Y | Z | PRMT5 IC50 (μM) FP40min |
PBS Solubility (μM) |
Plasma Stability (%Remaining) |
|
| Human | Mouse | |||||||
| 9 | −H | CH | CH | CH | 19.2 | 19 | ND | ND |
| 10 | −OCH3 | CH | CH | CH | 23.3 | 96 | ND | ND |
| 11 | −CH2CH3 | CH | CH | CH | 4.2 | 12 | ND | ND |
| 12 | −CH3 | CH | CH | C-CH3 | 6.1 | 14 | ND | ND |
| 13 | −CH3 | CH | C-CH3 | CH | 2.5 | 3.7 | ND | ND |
| 14 | −CH3 | C-CH3 | CH | CH | 39.0 | 7.8 | ND | ND |
| 15 | −CH3 | N | CH | CH | 4.5 | 5 | 96 | 75 |
| 16 |
|
>200 | 96 | 70 | 3 | |||
| 17 |
|
200 | 7.6 | ND | ND | |||
| 18 |
|
>200 | 98 | ND | ND | |||
Table 3.
R3 and R4 modification
|
||||||
|---|---|---|---|---|---|---|
| Compound Number |
R3 | R4 | PRMT5 IC50 (μM) FP40min |
PBS Sol (μM) |
Plasma Stability (%Remaining) |
|
| Human | Mouse | |||||
| 19 | −CH3 | −H | >200 | 86 | 89 | 86 |
| 20 | −H | (S)-CH3 | 0.3 | 64 | 92 | 92 |
| 21 | −H | (R)-CH3 | 55.4 | 70 | 95 | 97 |
Mechanism of Binding
To understand the mode of binding and support compound optimization, we generated a high resolution (1.9Å after elliptical truncation of anisotropic data) co-crystal structure of compound 1 with the PRMT5:WDR77 complex (Fig. 2a and 2b, PDB ID: 6V0P, Suppl. Table 1 and Suppl. Fig. 1b). Only one site of new electron density was observed and overlapped with the known PBM binding site, confirming inhibition by direct competition. The refined electron density (Supp. Fig. 1a with 2Fo-Fc) suggested that the 4-position of the pyridazinone ring forms a covalent bond with PRMT5 Cys278 as a result of nucleophilic attack by the Cys278 thiol and subsequent loss of a chlorine, driven by the re-aromatization of the pyridazinone. The crystal structure also points to key non-covalent interactions that are likely to drive initial binding and site specificity. The core aniline of the compound forms a pi-pi-stack with Phe243 and Tyr286, in a manner analogous to that formed with Phe230 of the Riok1 peptide 13 (PDB ID: 6V0N). Several hydrogen bonds are also made between the compound and protein, including (1) between the compound sulfonamide and Asn239, (2) between the compound amide and Ser279, and (3) between Gln282 and the free nitrogen on the pyridazinone.
Despite the high-resolution nature of the crystal structure, no unambiguous density was observed for the azepane functional group which might be due to its location at a crystal contact and this was left unmodelled (Supp. Fig. 1b).
As significant potency improvements were made via substitution of the original azepane group, we sought to understand these changes from a structural perspective. However, none of the improved potency variants examined were amenable to high resolution crystallization likely due to the local crystal contact interface. We therefore solved a Cryo-EM structure of the PRMT5:WDR77 hetero-octameric complex bound to the pyridyl ethyl variant, compound 6 (Fig. 2c, Table 2 and Suppl. Fig. 1c, PDB ID: 7M05). This structure was solved to an overall resolution of 2.4Å, which represents a substantial improvement compared to previously published Cryo-EM structures of PRMT5 resolved at 3.4Å (PDB ID: 6UGH) and 3.7Å (EMD-713717). The structure has excellent side-chain resolution and clear density for compound 6 (Fig. 2c and Suppl. Fig. 1c). The pyridyl ethyl position is well defined in the density and the other functional groups are in similar poses compared to the X-ray structure of compound 1. The pyridyl ethyl side chain is oriented back towards the core of the molecule via rotation of the sulfonamide and flexibility of the ethyl linker. This molecular conformation produces a new 4-ring stack of pi-pi interactions consisting of (from “front to back”) Tyr286-Pyridine-Aryl core-Phe243. Notably, this produces a folded-back structure analogous to the distinct conformation of the PBM peptide13. Compared to the crystal structure of compound 1, the density for Lys241 is better resolved and a hydrogen bond between this sidechain and the compound sulfonamide can now be modeled.
Compound 6 is a covalent binder
We began exploring the putative covalent mode of compound binding by performing a time course FP analysis of compound 6. Here, IC50 values were determined at 10 minutes intervals up to 4 hours. The observed IC50 values decreased more than 10-fold over time, reaching the assay floor of ~100 nM (Fig. 3a). The covalent nature and site specificity of the compound was further verified using a PRMT5 complex with a Cys278 to Ala mutation. In FP competition experiments, this compound series, as exemplified by compound 6 (blue squares), has little or no activity against the C278A mutant (maroon squares, 0.6 vs >100 μM) in contrast to a competitor PBM peptide which is comparable against both WT and C278A proteins (blue and red circles IC50 values 2.0 vs 1.4 μM, respectively) (Fig. 3b).
Figure 3. Compound 6 is a covalent binder.

(a) compound 6 shows time-dependent displacement of a fluorescently-labeled PBM peptide probe as measured by FP (b) FP competition assay with compound 6 (squares) or pICln 13-mer control peptide (circles) displacing the fluorescently-labeled PBM probe from WT PRMT5 (blue) and C278A mutant (green). (c) Deconvoluted mass spectra for WT and C278A PRMT5 +/− compound 6 (d) SPR competition assay, PRMT5:WDR77 complex was pre-incubated with compound 6 or DMSO and then immobilized to the chip surface. Full-length pICln protein was titrated as analyte.
In addition, intact mass measurement of the WT PRMT5:WDR77 complex before and after compound treatment showed an increase for PRMT5 of 460 Da with compound 6 (Fig. 3c) correlating with the expected mass of the compound, less one molecule of HCl that is lost upon covalent attachment of a single adduct to PRMT5. No change in the mass of WDR77 was observed (Supp. Fig. 2). Consistent with the above findings, treatment of PRMT5C278A with compound 6 did not result in a mass shift, indicating that the covalent attachment was taking place at Cys278. For reference, the PRMT5 and WDR77 proteins contain 12 and 13 total cysteine residues, respectively, indicating selective binding.
Finally, we investigated whether pre-incubation with compound 6 was sufficient to block binding, as measured by SPR, between PRMT5 and full-length pICln. To do so, PRMT5 was preincubated with either DMSO or compound 6, and then immobilized to the chip surface without further compound treatment. Titration of pICln to DMSO-treated PRMT5 produced binding with a KD of 32 nM. In contrast, a greatly reduced binding response was observed for compound-treated PRMT5 consistent with linear, non-specific binding (Fig. 3d). Together, these results demonstrate that the compound binds by an irreversible covalent mechanism and reacts with a single cysteine on the PRMT5:WDR77 complex.
Optimization of reactivity
Concerned that the highly reactive nature of these molecules could negatively impact their selectivity, we explored modifications of the warhead with the goal of decoupling potency from reactivity. The electrophilic reactivity of compounds towards the cysteine residue of glutathione (GSH) was used as a surrogate for intrinsic reactivity 18. Not surprisingly, modifications outside of the warhead had negligible effects on reactivity to GSH, but affected FP potency (see acetamide, sulfonamide and phenyl ring substitutions (Tables 1, 2). However, with changes at the pyridazinone warhead we observed a strong correlation between GSH reactivity and FP assay potency (Table 4). Of the various substitutions tested, the most interesting was the monochloro substitution, exemplified by compound 26, which provided an acceptable balance between activity and reactivity (FP IC5040min 72.9 μM; GSH T ½ 693 min). From 26, the addition of a chiral alpha methyl substituent led to 27 (BRD0639), which, as observed before, significantly improved potency while retaining low reactivity (FP IC5040min 13.8 μM; GSH T ½ 916 min).
Table 4.
X and Y Modifications

|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound Number |
R3 | R4 | X | Y | PRMT5 IC50 (μM) |
PBS Sol (μM) |
Plasma Stability (%Remaining) |
GSH T½ (min) |
|
| Human | Mouse | ||||||||
| 22 | −H | (rac)-CH3 | −CI | −CH3 | >200 | 83 | ND | ND | >3469 |
| 23 | −H | −H | −F | −CI | 0.37 | 43 | 18 | 0 | <2 |
| 24 | −H | −H | −F | −CH3 | 72.1 | 110 | ND | ND | 990 |
| 25 | −H | −H | −F | −H | 35.6 | 110 | ND | ND | 35 |
| 26 | −H | −H | −CI | −H | 72.9 | 72 | 100 | 40 | 693 |
| 27 (BRD0639) | −H | (S)-CH3 | −CI | −H | 13.8 | 82 | 87 | 79 | 916 |
| 28 | −H | (R)-CH3 | −CI | −H | 31.7 | 87 | 86 | 79 | 3105 |
| 29 | −CH3 | (S)-CH3 | −CI | −H | >200 | 114 | ND | ND | ND |
| 30 (BRD2198) | −H | −H | −H | −CI | >200 | 90 | ND | ND | 3465 |
To quantify the contribution of various substitutions to both initial reversible binding (KI) and rate of maximum covalent reactivity (kinact), we developed a mass spectrometry-based kinact/KI assay. The kinact/KI ratio is considered the best time-independent measure of covalent compound potency19. In this assay, we incubated purified PRMT5:WDR77 protein with varying concentrations of the inhibitors and quenched over a time course by rapidly reducing the pH with the addition of formic acid. Subsequent LC-MS analysis enabled the quantitation of the loss of unmodified PRMT5 represented as percent occupancy as a function of time across a range of concentrations (Fig. 4a). This allowed the generation of Kobs vs concentration curves (Fig. 4c, Supp. Fig. 3) from which we were able to generate kinact/KI values for a number of inhibitors (Fig. 4c)19.
Figure 4. Kinetic formation of the covalent adducts between 6 and PRMT5.

(a) Dose dependent kinetics of 6-PRMT5 as determined by intact mass spectrometry (b) Kinact/KI analysis of covalent modification by 26 (blue), 25 (red), 20 (green), and 6 (purple) as determined by intact mass spectrometry (c) Kinetic parameters for select compounds.
This assay revealed a high degree of correlation between kinact/KI values and the FP IC5040min (Fig. 4c). For example, the highly potent dichloro compound 20, shows a greater than 30-fold increase in the overall rate of adduct formation, compared to compound 1 (kinact/KI = 14,626 vs 449 (M−1 sec−1), respectively). Here, an improvement in both initial binding (KI) and maximum potential inactivation rate (kinact) contributed significantly to the increased potency of compound 20. A similar correlation is observed between the pair of mono-chloro compounds 27 and 26 (FP IC5040min 13.8 μM vs 72.9 μM and kinact/KI 244 (M−1 sec−1) vs 56 (M−1 sec−1), respectively). However, in this case the improved potency is primarily the result of a higher kinact (0.715 vs 0.123 M−1 sec−1), which compensates for a modest reduction in apparent KI (49 vs 36 μM). Thus, while we do not have a structural understanding of this activity relationship, we hypothesize that the affinity differences observed might arise as a result of the methyl group favoring the formation of an “encounter complex” prior to the covalent bond formation.
Indeed, while we observed a clear correlation between FP values and Kinact/Ki values, reactivity to GSH was not always correlated. For example, a few compounds with similar reactivity to GSH (i.e. dichloro 6 and monofluoro 25: T ½ 31 vs 35 min, respectively) showed more than a 40-fold different rate of covalent modification of PRMT5 (kinact/KI = 2,856 vs 69 (M−1 sec−1)), which otherwise correlates well with the observed 17-fold difference in FP IC5040min (2.2 μM vs 35.6 μM, respectively). This result suggests that the nature of the leaving group, chlorine or fluorine, has significant effects on the overall rate of adduct formation, that is not fully explained by their differences in intrinsic reactivity. The same is true for other modifications in close proximity to the leaving group. Indeed, this kinetic analysis guided us to the monochloro pyridazinone as a low intrinsic reactivity warhead that retains good potency.
Cellular activity of 27: on-target and on-mechanism mode of action
To determine whether 27 could engage PRMT5 in a cellular context, we treated Expi293 cells overexpressing HA-tagged WDR77 and WT PRMT5 for 6 hours and then analyzed anti-HA immunoprecipitates by LC-MS. This approach revealed a dose dependent formation of PRMT5-adducts (Fig. 5a). An EC50 of 3 μM was observed, however only ~40% of total PRMT5 protein was labeled by 27, possibly due to the high rate of protein production in this overexpression system. Similar results were obtained by treating the cells for 24 hours (data not shown).
Figure 5 -. Cell activities of 27.

(a) PRMT5 adduct formation using 27. Expi293 cells were transiently transfected for 48 hrs with an HA-tagged PRMT5:WDR77 complex before treatment with 27. Cultures were harvested at 6 hours, PRMT5 complex isolated by HA-affinity, and analyzed for modification by LC/MS. (b) PRMT5-RIOK1 NanoBiT assay in permeabilized cells. 293T cells stably expressing SmBiT-PRMT5 and LgBiT-RIOK1 proteins. (c) NanoBit assay performed in intact cells after treatment with 27 or 30. (d) Left panel: WB analysis of total symmetric arginine dimethylation levels in MTAP−/− HCT116 cells, in response to 27 or 30. Right panel: WB analysis of total symmetric arginine dimethylation levels in MTAP−/− HCT116 cells overexpressing the PRMT5 ADA mutant which is unable to bind to the PBM peptide, with and without KO of endogenous PRMT5.
To test if compounds were able to disrupt the interaction between PRMT5 and an SAP in cells, we used a PRMT5-RIOK1 NanoBiT complementation assay13. Compounds 1 and 27 were tested alongside related inactive compounds 29 and 30 (BRD2198) in permeabilized cells (Fig. 5b). Both compounds 1 and 27, but neither of the inactive compounds, disrupt the PRMT5-RIOK1 complex with an IC50 of 4 μM and 7.5 μM, respectively. Next, we assessed whether 27 could engage the target in intact, non-permeabilized cells. To this end, we treated PRMT5-RIOK1 NanoBit expressing cells with either 27 or 30 for 40 minutes and then assessed the stability of the PRMT5-RIOK1 complex (Fig. 5c). Consistent with what was observed in permeabilized cells, 27 but not 30 disrupted the complex (IC50 16 μM vs >100 μM). The different IC50 values observed in permeabilized vs intact cells (7.5 μM vs 16 μM) suggests a defect in cellular permeability. Finally, cellular treatment with 27 resulted in inhibition of PRMT5 methyltransferase function, as demonstrated by the reduction in symmetric arginine dimethylation levels by WB (Fig. 5d). We found that methylation of some, but not all substrates, were inhibited by treatment with 27. Notably, albeit modest when compared to the effects of a PRMT5 catalytic inhibitor (Suppl. Fig.4), this phenotype is very consistent and closely recapitulates the one observed following genetic disruption of the PBM-PBM groove interaction using a PRMT5 PBM mutant, which failed to rescue methylation of some, but not all substrates13 (Fig. 5d; compare lanes 2 and 5, arrows). This finding is consistent with the conclusion that 27 induced changes in methylation are on-target and on-mechanism.
Discussion and Conclusions
We report here the discovery and characterization of a first-in-class PRMT5-PBM competitive inhibitor. Our recent characterization of the binding interface between PRMT5 and its SAPs enabled the development of a robust screening system to identify PBM-competitive small molecule inhibitors. Despite considerable efforts in hit identification using three independent screening approaches, we identified only one chemical starting point. As protein-protein interaction sites can be challenging drug targets, it is possible that larger or more diverse libraries, potentially including DNA-encoded libraries, might yield additional scaffolds.
Our initial validated hit was a highly reactive covalent binder. Other liabilities of compound 1 were poor aqueous solubility and high instability in mouse plasma. Our hit-to-lead efforts resolved the solubility and the plasma instability issues and significantly reduced the intrinsic GSH reactivity to levels comparable with or below that for clinically-approved acrylamide warheads20. In addition to a robust FP system for HTS and routine potency measurements, we developed a novel LC-MS assay to quantitatively measure KI and Kinact to understand the effects of compound modifications more fully. This system revealed that key points of compound modification, such as the azepane to ethyl pyridine substitution and S-methyl addition, led to significant improvements in reversible binding without otherwise altering the maximum rate of reactivity. Nonetheless, it is clear that the covalent interaction is a necessary component of compound activity, which proved difficult to remove while retaining potency. However, we were able to tune reactivity to acceptable levels via alteration from the initial dichloro-pyridazinone to a monochloro. Importantly, our lead compound 27 can engage the cellular target and effectively outcompete binding between full-length PRMT5 and RIOK1 proteins with an IC50 of 7.5μM and 16 μM in permeabilized and living cells, respectively. Moreover, we demonstrate a pharmacodynamic response to the most proximal marker of PRMT5 activity, the formation of symmetrically dimethylated arginine. This effect appears to be on-target as closely related compounds with the same warhead, but inactive in FP assays, are also inactive in the cellular context. Consistent with an on-target effect, 27 (BRD0639) reduces SDMA in the same subset of proteins also affected by genetic perturbation of the PBM binding site14.
A PBM-competitive inhibitor may result in a different pharmacodynamic and therapeutic response as compared to that of a catalytic inhibitor. PRMT5 has a key role in regulating multiple essential cellular activities, including transcription, ribosomal biogenesis and mRNA splicing. Genetic disruption of the PBM binding site, however, leads to selective impairment of its splicing function mediated by the SAP pICln14. In contrast, catalytic site inhibitors of PRMT5 are pan-substrate and therefore may be more likely to result in non-tolerated on-target toxicity.
Moving forward, it would be interesting to explore additional strategies for targeting the PRMT5 methylosome. It is notable that the relevance of PRMT5 to MTAP-deleted cancers was observed by shRNA knockdown, suggesting that partial reduction in total PRMT5 protein concentration would also be sufficient to see selective viability effects. As such, PRMT5 degraders could represent an alternative therapeutic approach to this target. It should be noted that the development of heterobifunctional degraders directed against PRMT5 have been reported recently21. However, their potency is limited, possibly since conventional PRMT5 catalytic inhibitors were used as the target bait. Here, we believe that the depth of the catalytic pocket might pose a structural challenge in connecting a catalytic PRMT5 inhibitor, via a linker, to an E3-ligase binder. From this perspective, the discovery of a new surface exposed and druggable site on PRMT5, the development of a fully validated suite of assays and the generation of a tool compound to support screenings for new, non-covalent binders, could offer a unique starting point for the development of potent PRMT5 degraders.
Experimental Section
Recombinant Protein Expression and Purification
Codon optimized DNAs for PRMT5 (uniprot: O14744) and WDR77 (uniprot: Q9BQA1) were cloned into the pFastBac Dual baculovirus vector (Thermo Fisher) under the control of either the p10 or polyhedrin promoter, respectively. The WDR77 protein was engineered with either an N-terminal His6-Strep tag II followed by the HRV 3C protease site (PRMT5WT:WDR77 construct) or a C-terminal ybbR-sortase and His6 tag (PRMT5C278A:WDR77 construct) to aid purification, resulting in the mass difference observed for intact WDR77 in Supplemental Figure 2. The C278A mutation was introduced to PRMT5 using the QuickChange mutagenesis method (Agilent Technologies). PRMT5 was untagged in all constructs. The PRMT5:WDR77 heterocomplex was co-expressed in Sf9 (Spodoptera frugiperda) cells with a BIIC ratio of 1:10,000. Cultures were harvested after 72 hours. Purification began with resuspension of the cell pellet in lysis buffer (25 mM HEPES (pH 7.4), 300 mM NaCl, 1 mM TCEP, 10% glycerol, 0.1% Triton X100, SigmaFast Protease Inhibitor tablets) before breaking with a microfluidizer (Avestin). The lysate was clarified via centrifugation, filtered with a 0.2 um filter, and passed over a Streptactin XT affinity column (IBA GmbH). The column was washed for 20 CV with wash buffer (25 mM HEPES (pH 7.4), 300 mM NaCl, 1 mM TCEP, 10% glycerol) before eluting bound protein with buffer containing 2.5 mM desthiobiotin. The N-terminal tag was cleaved with the addition of HRV 3C protease (AG Scientific) and incubation overnight at 4°C. The PRMT5:WDR77 complex was further purified by passing samples over a Superose 6 (10/300) column (GE LifeSciences) using SEC buffer (50 mM HEPES (pH 7.4), 10% glycerol, 1 mM TCEP, 150 mM NaCl). Aliquots were frozen at −80°C.
Fluorescence polarization
The FP competition assay had final concentrations 200 nM PRMT5:WDR77 protomer, (experimentally determined KD for the interaction13), 10 nM peptide probe, 50 mM HEPES pH 7.4, 100 mM NaCl, 0.5 mM TCEP, and 0.01% v/v Tween 20. For HTS, 1536-well black non-binding surface plates (Corning) were used with a 5 μL assay volume. The assay reagents were added to pre-plated compounds with a final compound concentration of 20 μM. The plates were incubated at room temperature and the final endpoint read at 40 minutes. For SAR data, experiments were performed in triplicate in 384-well black non-binding surface plates (Corning) at a final assay volume of 20 μL. Data were collected using either a Spectramax Paradigm with Rhodamine FP filter set or a Perkin-Elmer Envision with Bodipy TMR FP filter set. The peptide probe was a KU560 fluorophore (KU dyes, catalog KU560-R-6)-labeled peptide derived from the RIOK1 PBM sequence: [acetyl]SRVVPGQFDDADSSD[^KU560][amide]). As a positive control, peptide [acetyl]LMSRVVPGEFDDADSSD[amide] was used at a 20 μM concentration. Fluorescence polarization response was normalized across each plate to 100 (average of DMSO control wells) and 0 (average of unlabeled peptide control).
Nuclear Magnetic Resonance
Experiments were performed on a 600 MHz Bruker Avance III Spectrometer equipped with a 5 mm QCI cryoprobe and a SampleJet for automated sample handling. All experiments were conducted at 280 K. For. For STD NMR (Mayer, 1999; Begley, 2013) final sample conditions involved 200 μM ligand, 2% DMSO, 1 μM protomer, 25 mM HEPES-d, pH=7.4, 150 mM NaCl, 1 mM TCEP-d, followed by competition with 20 μM 13-mer peptide (KD 300 nM). All NMR data was analyzed using Topspin (Bruker). The SPY peptide was synthesized by Thermo-Fisher, sequence: [acetyl]GQF*EDAD[amide], where F* is 3-Fluoro phenylalanine; 19F signal at −113.75 ppm.
Surface Plasmon Resonance
Biacore experiments were performed as previously described13 with the exception that PRMT5 protein was pre-incubated at a concentration of 25 nM with 2 μM compound 6 or DMSO overnight at 4C prior to immobilization. Labeling was confirmed to be complete by LC-MS. Data were fit in Prism using a one site, total and non-specific binding model.
Crystallography
Crystals were produced as previously described13 and then soaked with compound 1 at an approximate concentration of 500 μM for 72 hr before harvesting. Diffraction images were indexed and integrated using XDS and further processed for anisotropy via elliptical truncation using the STARANISO server (Global Phasing). Cambridge, United Kingdom: Global Phasing Ltd.). Refinement was performed in Buster (Global Phasing) and Phenix with manual building/review in Coot. Compound restraints were generated using Glide (Global Phasing).
CryoEM
PRMT5:WDR77 complex at a 5 μM concentration was co-incubated with 25 μM compound 6, and 20 μM JNJ-64619178 overnight at 4C. Covalent modification was confirmed by LC-MS. PRMT5 complex was then purified by size exclusion on a Superose 6 Increase 10/300 GL (GE Healthcare) column in mobile phase buffer 10 mM HEPES pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM TCEP, and 100 nM JNJ-64619178. Protein was concentrated to 10 mg/ml using a Proteus X-spinner with 10 kDa filter (Anatrace) and snap frozen in liquid nitrogen. UltrAuFoil 300 mesh grids were pre-treated by glow discharge for 30 sec. Immediately prior to application, protein samples were thawed and diluted 7-fold in a buffer containing 10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM TCEP and 100 nM JNJ-64619178. Using a FEI Vitrobot Mark IV, 3.5 μl of the protein sample was applied to the grid before blotting and plunge-freezing into liquid ethane. Data were acquired on a Titan Krios microscope with Gatan K2 Quantum detector at a nominal magnification of 130,000x and randomized defocus values of −1.4, −1.7 or −2.1 Å. For each image, 40 movie frames were recorded with a total exposure time of 8 sec and estimated electron dose of 62.4 e−/Å2. A total of 2169 movies were collected from one grid using automatic acquisition. All data processing was performed in the cisTEM software suite22. Frames 4-40 from each movie were motion corrected, summed and then processed by CTF estimation. All images with a CTF fit resolution ≤ 3.1 Å (1836 images) were retained for single particle analysis. A total of 956,646 particles were picked for 2D classification. Due to particle crowding, only 474,404 particles (22 of 50 classes) were selected for 3D refinement. EMD-7137 was used as a starting volume model with an initial high-resolution limit of 20 Å and a defined D2 symmetry. 3D refinement converged at an estimated resolution of 2.39 Å using half-map analysis with a 0.143 FSC cutoff. Phenix Autosharpen was used prior to rigid body docking of the 6V0P crystal structure as a starting model. Manual model building was performed in Coot followed by real space refinement in Phenix.
Stability to GSH
Ten μl of compound at a final concentration of 0.1, 1 and 10 μM, or control working solution, were diluted in 190 μl of 5 mM glutathione (GSH) in PBS (pH 7.4) and incubated at 37°C for 0, 15, 30, 60, 120 and 1440 min, in duplicate. As a negative control, incubations without GSH were carried out at two time-points (0 and 1440 min). Ibrutinib and afatinib were used as positive controls and were tested at a final concentration of 10 μM. At each time-point, the reaction was terminated by adding 600 μl cold acetonitrile containing labetalol as the internal standard. The samples were stored at - 80°C until the last incubation time-point was completed. Sample plates were defrosted and placed on a shaker for 5 min, followed by centrifugation at 4000 rpm for 20 min. 100 μl of each sample, after centrifugation, was further diluted with water before mass spec analysis. Samples with compound concentration at 0.1, 1 and 10 μM were diluted with 100 μl, 300 μl and 600 μl water, respectively. Samples were analyzed by LC-MS/MS (Sciex API 4000). The percent of the parent compound remaining at each time-point was determined based on peak area ratios at the 0 min time-point and half-life was calculated using the first order kinetics equation. In addition, the samples were analyzed for formation of the predicted GSH adduct at each time-point.
Intact mass measurement by LC-MS
Purified PRMT5:WDR77 was thawed on ice and centrifuged to remove potential aggregates from the freeze/thaw process. The protein was then solvent exchanged into the reaction buffer (10 mM HEPES 7.4, 150 mM NaCl, 1 mM TCEP, using a 40 kDa Zeba desalting column (ThermoFisher), and diluted to 10 μM. For single time point experiments, a mixture of 10 μL of protein, 1 μL of compound 6 (0.5 mM in DMSO) or DMSO and 90 μL reaction buffer were incubated at RT for 4 hours. Intact mass measurement of the PRMT5:WDR77 complex with and without compounds was performed using the BioAccord LC-ToF (composed of an ACQUITY I-Class UPLC and RDa detector with ESI source, Waters Corporation). 1 μl of each sample was injected onto a C4 column (ACQUITY UPLC Protein BEH, 300Å, 1.7 μm, 2.1 X 50 mm, Waters Corporation) held at 80 °C. Mobile phases A and B consisted of 0.1% formic acid (MilliporeSigma LiChroPur) in LC-MS grade water or LC-MS grade acetonitrile (JTBaker), respectively, with initial column conditions set to 95% water/5% acetonitrile. Protein was desalted for one minute before elution with a gradient of 5% to 85% mobile phase B in 2.5 min followed by ionization in positive ionization mode with the cone voltage set to 55 V and desolvation temperature of 550 °C. The instrument scan rate was 5 Hz over 50 to 2000 m/z. PRMT5 and WDR77 coeluted at 2.38 minutes and mass spectra were deconvoluted using UNIFI and the MaxEnt1 algorithm.
Covalent modification of PRMT5 and kinetic analysis (kinact/KI) by LC-MS
Purified PRMT5:WDR77 was thawed and treated as described in the previous section. For kinact/ki analyses, PRMT5:WDR77 was diluted to 50 nM and dispensed into 384-well plates (Greiner Bio-One #781280) containing a 12-point dilution series of various compounds to collect 8 time points (0, 2 ,5, 10, 20, 30, 45, and 60 min) using a 384ST-head Agilent Bravo. Time points were quenched with formic acid (LiChropur, MilliporeSigma, final concentration 0.5%). To quantify the unmodified PRMT5 and WDR77 a multiple-reaction monitoring (MRM) method was developed using a Waters ACQUITY UPLC I-Class PLUS chromatography system connected to a Xevo TQ-XS mass spectrometer by focusing on a single charge state for each protein (PRMT5, m/z 855.0 → m/z 854.90, z = 85; WDR77, m/z 870.80 → m/z 870.75, z = 46). ESI source parameters were set as follows: positive ionization mode, capillary voltage 2.5 kV, cone voltage, 50 V; collision energy, 5 V; cone gas flow, 200 L/h; collision gas flow, 0.16 mL/min; source temperature, 150 °C; desolvation temperature, 350 °C; and desolvation gas flow, 900 L/h. Analytes were separated on an Agilent PLRP-S column (5 μm; 2.1 × 50 mm, 1000Å Agilent Technologies) at 60 °C. Sample storage temperature was set to 20 °C. Mobile phases and initial conditions were the same as described above. Proteins were eluted using a 1.5 min gradient of 5% to 98% acetonitrile at 0.4 ml/min. MassLynx and TargetLynx software (Waters, version 4.2) were used for sample acquisition and data quantification, respectively. Analytes were quantified by integration of peak areas for each charge state described above. For analysis, the peak areas were normalized to the 0 min time point. Kinetic analysis of the kinact and KI values were fit in Graphpad Prism 719.
Target engagement in cells
Expi293 cells were cultured in a 1:1 mixture of Expi293:Freestyle media (Thermofisher). At a cell density of ~2.5x106 cells/ml, 250 ml of cells were transfected using 200 μl FectoPRO (Polyplus Transfection) reagent mixed with 100 μg PRMT5 and 100 μg HA-tagged WDR77 plasmids13. One day after transfection, 3 mM valproic acid and 0.4% w/v glucose were added to the culture. Two days after transfection, cells were split into multiple flasks with 30 ml cells each and treated with compound or DMSO at a final 0.2% DMSO concentration. Cell sample was collected after 6 hours, pelleted and washed 3 times with ice cold PBS. Additional aliquots were analyzed for viability and cell number using a Vi-Cell XR Cell Counter (Beckman Coulter) and all samples were determined to have >98% viability based on trypan blue exclusion. Each sample was lysed in buffer 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM TCEP, 1% v/v Tween-20 and 2 mM reduced glutathione. The PRMT5:WDR77 complex was immunoprecipitated by anti-HA agarose resin (Thermofisher) and then eluted by 50 μM 3X-HA peptide (AnaSpec). Each eluate was measured by intact mass LC-MS to determine the percentage of complex with compound adduct.
NanoBiT
HEK293T cells stably expressing PRMT5 tagged with an N-terminal SmBiT peptide and RIOK1 tagged with N-terminal LgBiT peptide were described before13. Cells were cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) supplemented with 10% FBS (Life Technologies) and 1% penicillin-streptomycin (Life Technologies). For the assay, cells were centrifuged at 150 rad/s, media removed, and resuspended in Opti-MEM (Life Technologies). Cells were plated at (10x104 cells/ well) in 96well black wall, clear bottom, tissue culture treated plates (Corning) and treated with the indicated compounds using D300e Digital Printer (Tecan). All wells normalized to highest DMSO concentration (1% v/v). Each experimental condition was tested in triplicate.
NanoBiT in permeabilized cells:
50 uL of lysis buffer was added to each well (10% glycerol, 50 mM Tris-HCL, 150 mM KCl, 2 mM EDTA, 0.1% NP40) and cells were incubated for 5 min at RT. Nano-Glo® Luciferase Assay Substrate (Promega #N1110) was diluted 1:50 in buffer, 100 μL added to each well and pipetted to mix. NanoBiT in intact cells: Cells were treated for 40 min, at which point Nano-Glo® Live Cell Substrate (Promega, # N2011) was diluted 1:20 in buffer, 25 μL mixture was added to each well and the plate tapped gently to mix. Luciferase signal was measured using the EnVision plate reader (Perkin Elmer). Results shown are representative of three independent experiments.
Western Blot
HCT116 MTAP −/− were cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies), supplemented with 10% FBS (Life Technologies) and 1% penicillin-streptomycin (Life Technologies). Cells were plated at 1x106/well in 6-well tissue culture treated plates (Corning), and treated the next day with the indicated compounds to a final concentration of 25 μM. DMSO and non-treated control wells included. 12 hr after treatment, the media was refreshed and cells retreated. 12 hr later, cells were washed 1X with PBS and lysed on ice for 15 min in 50 μL lysis buffer (1mL RIPA, 10% glycerol, 1% protease inhibitor, 1% phosphatase inhibitor). Protein concentration was calculated using Pierce BCA Protein Assay Kit, 40 μg protein per sample was run on a 4-12% Bis-Tris gels (NuPAGE, Life Technologies) using MES buffer. Gels were dry-transferred to nitrocellulose membrane (iBlot system, Life Technologies). Membranes were blocked using Intercept Blocking Buffer (LI-COR) for 1 hr and probed overnight with primary antibodies (Rabbit anti-SDMA, CST13222; mouse anti-vinculin, Sigma, RABBIT anti-PRMT5 (Abcam Ab31751). Blots were washed 3X with 1% TBST buffer and probed with secondary antibodies 680RD goat anti-Mouse and 800CW goat anti-Rabbit (LI-COR) for 1 hr. Membranes were washed 3X with 1% TBST and imaged using LI-COR Odyssey imaging system. Results shown are representative of three independent experiments.
Chemical Synthesis
General experimental conditions for small molecule synthesis
All anhydrous solvents, reagent grade solvents for chromatography and starting materials were purchased from either Sigma Aldrich Chemical Co. or Fisher Scientific. Water was distilled and purified through a Milli-Q water system (Millipore Corp., Bedford, MA). General methods for purification of compounds involved the use of silica cartridges purchased from Grace or Combiflash Purification systems. The reactions were monitored by TLC on precoated Merck 60 F254 silica gel plates and visualized using UV light (254 nm). All compounds were analyzed for purity by reverse phase HPLC [Shimadzu HPLC instrument with a Hamilton reversed phase column (HxSil, C18, 3μm, 2.1 mm × 50 mm (H2)). Eluent A: 5% CH3CN in H2O, eluent B: 90% CH3CN in H2O. A flow rate of 0.2 mL/min was used with photodiode-array detection at 220 and 254 nm] and shown to have ≥ 95% purity. NMR spectra were obtained on a Bruker NanoBay 400 instrument. Chemical shifts are reported in parts per million (ppm, δ) relative to the residual solvent peak in the corresponding spectra (chloroform δ 7.26, methanol δ 3.31, DMSO δ 3.33) and coupling constants (J) are reported in hertz (Hz) (where s = singlet, bs = broad singlet, d = doublet, dd = double doublet, bd = broad doublet, ddd = double doublet of doublet, t = triplet, tt = triple triplet, q = quartet, m = multiplet) and analyzed using ACD NMR or MestReNova data processing. Mass spectra values are reported as m/z. All reactions were conducted under nitrogen unless otherwise noted. Solvents were removed in vacuo on a rotary evaporator.
.Analytical UPLC methods
LRMS (LC-MS) Instrument: Agilent 1200\G1956A; Column: Kinetex EVO C18 30*2.1mm,5um; eluent A: 0.0375% TFA in water (v/v); eluent B: 0.01875% TFA in Acetonitrile (v/v); gradient 0-0.8 min5-95% B, 0.8-1.2 min 95% B, 1.2-1.5 5%B; flow 1.5 mL/min; temperature 50°C; DAD scan: 200-500 nm.
HRMS (LC-MS) was performed on purified compounds, diluted to 0.1 mM in DMSO, injecting 3 μl, reported data is an average of triplicate runs. Instrument: Agilent 1290 UHPLC; Column: Waters Acquity UPLC BEH C18, 1.7 μm, 2.1x50 mm; eluent A: Water with 0.1% formic acid, B: Acetonitrile with 0.1% formic acid; gradient 0-0.1 min 5% B, 0.1-5.0 min 5-95% B, 5.0-5.5 min 95% B, 5.5-5.6 min 5% B, 5.6-6.0 min 5% B; flow 0.5 mL/min; temperature 45°C; DAD scan: 200-500 nm, MS System Agilent 6545 Quadrupole Time of Flight Source Parameters: Gas Temp (°C) 350, Drying Gas (l/min) 10, Nebulizer (psi) 20, Sheath gas Temp (°C) 400, Sheath Gas Flow (l/min) 12, Capillary Voltage (V) 3500, Nozzle Voltage (V) 2000.
Synthesis of 1 (Scheme 1 A).
ethyl 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetate (31).
To a mixture of 4,5-dichloropyridazin-3(2H)-one (CAS 932-22-9, 30 g, 181.84 mmol) and K2CO3 (50.27 g, 363.69 mmol) in DMF (150 mL) was added ethyl 2-bromoacetate (33.40 g, 200.03 mmol, 22.12 mL) at 25 °C, and the mixture was stirred at 50 °C for 2 hours. The reaction mixture was diluted with water (100 ml) and ethyl acetate (300 ml), and the layers separated. The organic layer was washed with saturated aqueous sodium chloride (200 mL), dried over anhydrous sodium sulfate, insoluble materials removed by filtration, and volatiles removed under reduced pressure, the resulting residue was purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to give the title compound as a white solid (38.4 g, 84% yield). LRMS (m/z): Calcd [M+H]+ for C8H10Cl2N2O3 260.0; found 250.9; 1H-NMR (400 MHz, CHLOROFORM-d) δ = 7.82 (s, 1H), 4.90 (s, 2H), 4.28 (q, 2H), 1.30 (t, 3H).
2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetic acid (32):
Ethyl 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetate (31, 15 g, 59.75 mmol) was suspended in aqueous hydrochloric acid (5% w/v, 300 mL) and the mixture was stirred at 105 °C for 90 minutes, cooled to room temperature, and the resulting solid was isolated by filtration to give the title compound (10 g, 75% yield) as a white solid. LRMS (m/z): Calcd [M+H]+ for C6H5Cl2N2O3 223.0; found 223.0; 1H-NMR (400 MHz, CHLOROFORM-d) δ = 7.85 (s, 1H), 4.95 (s, 2H).
Synthesis of 34 ((Scheme 1 B).
1-((2-methyl-5-nitrophenyl)sulfonyl)azepane (33).
To a solution of 2-methyl-5-nitrobenzene-1-sulfonyl chloride (5 g, 21.22 mmol) and azepane (4.32 g, 31.83 mmol, 4.91 mL) in THF (100 mL) at 0 °C was slowly added TEA (6.44 g, 63.66 mmol, 8.86 mL). The mixture was stirred for 12 h at 25°C, volatiles removed under reduced pressure, and the residue partitioned between ethyl acetate and water, the layers were separated and the aqueous phase was extracted twice more with ethyl acetate, the combined organic layers were washed with saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure to provide the title compound as a yellow oil (3.7 g, 58% yield), which was used without further manipulation. LRMS (m/z): Calcd [M+H]+ for C13H19N2O4S 289.1; found 299.0; 1H NMR (400 MHz, CHLOROFORM-d) δ = 8.60 (d, J = 2.4 Hz, 1H), 8.27 (dd, J = 2.4, 8.4 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 3.47 - 3.40 (m, 4H), 2.75 (s, 3H), 1.88 - 1.78 (m, 4H), 1.74 - 1.65 (m, 4H).
3-(azepan-1-ylsulfonyl)-4-methylaniline (34).
A mixture of 1-((2-methyl-5-nitrophenyl)sulfonyl)azepane (33, 3.7 g, 12.40 mmol) and palladium on carbon (100 mg, 10% w/w) in methanol (80 mL) was degassed and purged with hydrogen three times, and the mixture was stirred at 25 °C for 16 hours under hydrogen atmosphere (15 psi). The mixture was filtered through Celite, and volatiles removed under reduced pressure to afford the title compound as a yellow oil (3.4 g, 102% yield). LRMS (m/z): Calcd [M+H]+ for C13H21N2O2S 269.1; found 269.0.
N-(3-(azepan-1-ylsulfonyl)-4-methylphenyl)-2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetamide (1). A mixture of 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetic acid (32, 997.17 mg, 4.47 mmol) and DMF (27.24 mg, 372.61 umol, 28.67 uL) and thionyl chloride (664.95 mg, 5.59 mmol) in THF (20 mL) at 0 °C was heated at 60 °C for 30 minutes. The solution was cooled to 0 °C, treated with 3-(azepan-1-ylsulfonyl)-4-methylaniline (34, 1 g, 3.73 mmol) and TEA (754.10 mg, 7.45 mmol, 1.04 mL) and warmed to 25 °C and stirred for 30 minutes. The reaction mixture was diluted with saturated aqueous ammonium chloride, extracted twice with ethyl acetate, combined organic layers were dried over sodium sulfate, insoluble materials were removed by filtration, volatiles were removed under reduced pressure and the residue was purified by prep-HPLC (column: Kromasil 250*50 mm*10 um; mobile phase: [water (0.225% FA)-ACN]; B%: 37ACN%-67ACN%, 26 min, 85% min) to give the title compound as a white solid (1.4 g, 79% yield). LCMS: Rt=0.805 min, [M+H]+= 418.2; 1H NMR (400 MHz, DMSO) δ 10.60 (s, 1H), 8.29 (s, 1H), 8.06 (d, J = 2.3 Hz, 1H), 7.66 (dd, J = 8.3, 2.3 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 4.98 (s, 2H), 3.27 (t, J = 5.8 Hz, 4H), 2.47 (s, 3H), 1.65 (tt, J = 6.4, 3.8 Hz, 4H), 1.56 (dq, J = 5.8, 4.2, 2.7 Hz, 4H); 13C{1H} NMR (101 MHz, DMSO) δ 164.70, 155.89, 138.32, 136.64, 136.54, 136.11, 133.33, 132.85, 131.26, 122.50, 118.77, 55.79, 47.63, 28.91, 26.35, 19.26. HRMS (ESI/Q-TOF) RT 3.35 min, m/z: [M + H]+ Calcd for C19H22Cl2N4O4S 473.0817; Found 473.0371.
2-methyl-5-nitro-N-[2-(pyridin-2-yl)ethyl]benzene-1-sulfonamide (35). Was made analogously to 33. LRMS (m/z): Calcd [M+H]+ for C14H16N3O4S 322.36; Found 322.0; 1H NMR (400 MHz, Chloroform-d) δ 8.83 (d, J = 2.4 Hz, 1H), 8.48 (ddd, J = 5.0, 1.9, 0.9 Hz, 1H), 8.26 (dd, J = 8.3, 2.5 Hz, 1H), 7.62 (td, J = 7.7, 1.8 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.18 (ddd, J = 7.6, 4.9, 1.1 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.87 (t, J = 5.8 Hz, 1H), 3.42 (q, J = 5.7 Hz, 2H), 3.01 – 2.96 (m, 2H), 2.76 (s, 3H).
5-amino-2-methyl-N-[2-(pyridin-2-yl)ethyl]benzene-1-sulfonamide (36). Made analogously to 34. LRMS (m/z): Calcd [M-H]- for C14H16N3O2S 290.4; Found 290.0; 1H NMR (400 MHz, Chloroform-d) δ 8.47 (dt, J = 4.8, 1.4 Hz, 1H), 7.58 (td, J = 7.7, 1.9 Hz, 1H), 7.33 (d, J = 2.5 Hz, 1H), 7.17 – 7.11 (m, 1H), 7.06 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.72 (dd, J = 8.1, 2.6 Hz, 1H), 6.24 (t, J = 6.0 Hz, 1H), 3.57 (s, 2H), 3.33 (q, J = 6.0 Hz, 2H), 2.92 (t, J = 5.9 Hz, 2H), 2.45 (s, 3H).13C{1H} NMR (101 MHz, CDCl3) δ 159.29, 149.06, 144.64, 138.41, 136.88, 133.47, 126.07, 123.67, 121.89, 118.90, 116.17, 42.13, 36.00, 19.09.
Compounds 2, 4-14 were made analogously to 1. Compound 3 was purchased from Enamine (Catalog ID: Z19024493) and used without further purification.
2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-((4-methyl-1,4-diazepan-1-yl)sulfonyl)phenyl)acetamide (2). LCMS: Rt=0.742 min, [M+H]+= 488.1; 1H NMR (400 MHz, DMSO-d6) δ = 10.66 - 10.53 (m, 1H), 8.33 - 8.25 (m, 1H), 8.12 - 8.01 (m, 1H), 7.77 - 7.62 (m, 1H), 7.49 - 7.29 (m, 1H), 5.12 - 4.91 (m, 2H), 3.34 - 3.30 (m, 2H), 2.60 - 2.52 (m, 6H), 2.49 - 2.44 (m, 3H), 2.29 - 2.20 (m, 3H), 1.83 - 1.75 (m, 2H); HRMS (ESI/Q-TOF) RT: 2.01 min, m/z: [M + H]+ Calcd for C19H24Cl2N5O4S 488.0926; Found 488.0919
2-(4,5-dichloro-6-oxopyridazin-1(6h)-yl)-n-(4-methyl-3-(piperidin-1-ylsulfonyl)phenyl)acetamide (3). HRMS (ESI/Q-TOF) RT: 3.22 min, m/z: [M + H]+ Calcd for C18H21Cl2N4O4S 459.0660; Found 459.0205.
2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-((4-methylpiperazin-1-yl)sulfonyl)phenyl)acetamide (4). LCMS: Rt=0.753 min, [M+H]+= 474.0; 1H NMR (400 MHz, DMSO-d6) δ = 10.66 (s, 1H), 8.30 (s, 1H), 8.11 (d, J = 2.1 Hz, 1H), 7.69 (br d, J = 2.1 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 4.99 (s, 2H), 3.06 (br s, 4H), 2.53 - 2.52 (m, 3H), 2.35 (br s, 4H), 2.16 (s, 3H). HRMS (ESI/Q-TOF) RT: 1.97 min, m/z: [M + H]+ Calcd for C18H22Cl2N5O4S 474.0769; Found 474.0763.
2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(3-(N,N-dimethylsulfamoyl)-4-methylphenyl)acetamide (5). LCMS (ESI): Rt=0.877 min, [M+H]+= 418.9; 1H NMR (400 MHz, DMSO-d6) δ = 10.67 - 10.62 (m, 1H), 8.30 (s, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.70 (dd, J = 2.3, 8.3 Hz, 1H), 7.42 (s, 1H), 4.99 (s, 2H), 2.73 (s, 6H), 2.54 - 2.53 (m, 3H); HRMS (ESI/Q-TOF) RT: 2.72 min, m/z: [M + H]+ Calcd for C15H17Cl2N4O4S 419.0347; Found 419.0345.
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (6). LCMS: Rt=0.732 min, [M+H]+= 496.1; 1H NMR (400 MHz, DMSO) δ 10.58 (s, 1H), 8.40 (ddd, J = 4.8, 1.9, 0.9 Hz, 1H), 8.29 (s, 1H), 8.09 (d, J = 2.3 Hz, 1H), 7.77 (t, J = 5.8 Hz, 1H), 7.69 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (td, J = 7.7, 1.9 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.17 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 7.13 (dt, J = 7.8, 1.1 Hz, 1H), 4.97 (s, 2H), 3.19 – 3.08 (m, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H); 13C{1H} NMR (101 MHz, DMSO) δ 164.61, 158.23, 155.90, 148.97, 138.72, 136.50, 136.45, 136.40, 136.08, 132.99, 132.85, 131.22, 123.20, 122.55, 121.58, 119.29, 55.76, 41.96, 37.28, 19.0;. HRMS (ESI/Q-TOF) RT: 1.95 min, m/z: [M + H]+ Calcd for C20H20Cl2N5O4S 496.0613; Found 496.0606.
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-(N-(2-(pyridin-3-yl)ethyl)sulfamoyl)phenyl)acetamide (7). LCMS: Rt=0.732 min, [M+H]+= 496.1;1H NMR (400 MHz, DMSO-d6) δ = 10.62 (s, 1H), 8.38 (br d, J = 3.6 Hz, 1H), 8.32 (s, 1H), 8.29 (s, 1H), 8.12 (d, J = 1.9 Hz, 1H), 7.80 (br t, J = 5.3 Hz, 1H), 7.66 (dd, J = 1.9, 8.2 Hz, 1H), 7.51 (br d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.24 (dd, J = 4.8, 7.6 Hz, 1H), 4.98 (s, 2H), 3.03 (q, J = 6.5 Hz, 2H), 2.68 (br t, J = 6.9 Hz, 2H), 2.42 (s, 3H); HRMS (ESI/Q-TOF) RT: 1.93 min, m/z: [M + H]+ Calcd for C20H20Cl2N5O4S 496.0613; Found 496.0606.
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-(N-(2-(pyridin-4-yl)ethyl)sulfamoyl)phenyl)acetamide (8). LCMS: Rt=0.752 min, [M+H]+= 496.0; 1H NMR (400 MHz, DMSO-d6) δ = 10.58 (s, 1H), 8.40 (br d, J = 5.0 Hz, 2H), 8.29 (s, 1H), 8.13 (d, J = 2.1 Hz, 1H), 7.79 (t, J = 5.7 Hz, 1H), 7.65 (dd, J = 2.2, 8.2 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.11 (d, J = 5.8 Hz, 2H), 4.98 (s, 2H), 3.08 - 3.03 (m, 2H), 2.70 - 2.66 (m, 2H), 2.41 (s, 3H); HRMS (ESI/Q-TOF) RT: 1.91 min, m/z: [M + H]+ Calcd for C20H20Cl2N5O4S 496.0613; Found 496.0606.
2-(4,5-dichloro-6-oxo-pyridazin-1-yl)-N-[3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (9). 1H NMR (400 MHz, Chloroform-d) δ 8.96 (s, 1H), 8.52 (d, J = 5.1 Hz, 1H), 8.14 (s, 1H), 7.95 (dd, J = 8.3, 1.8 Hz, 1H), 7.92 (s, 1H), 7.79 (d, J = 2.3 Hz, 1H), 7.71 (td, J = 7.7, 1.7 Hz, 1H), 7.54 (d, J = 7.7 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 7.29 (s, 1H), 7.21 (d, J = 7.8 Hz, 1H), 5.06 (s, 2H), 3.41 (t, J = 6.2 Hz, 2H), 3.04 (t, J = 6.3 Hz, 2H);HRMS (ESI/Q-TOF) RT: 1.84 min, m/z: [M + H]+ Calcd for C19H18Cl2N5O4S 482.0456; Found 482.0447.
2-(4,5-dichloro-6-oxopyridazin-1(6h)-yl)-n-(4-methoxy-3-(n-(2-(pyridin-2-yl)ethyl) sulfamoyl)phenyl)acetamide (10). 1H NMR (400 MHz, DMSO-d6) δ = 10.45 (s, 1H), 8.45 (d, J = 3.9 Hz, 1H), 8.29 (s, 1H), 8.00 (d, J = 2.6 Hz, 1H), 7.77 (dd, J = 2.6, 9.0 Hz, 1H), 7.67 (dt, J = 1.8, 7.6 Hz, 1H), 7.31 (t, J = 5.8 Hz, 1H), 7.23 - 7.19 (m, 1H), 7.18 (d, J = 2.1 Hz, 1H), 7.17 - 7.14 (m, 1H), 4.95 (s, 2H), 3.82 (s, 3H), 3.17 - 3.11 (m, 2H), 2.83 (t, J = 7.2 Hz, 2H); HRMS (ESI/Q-TOF) RT: 1.79 min, m/z: [M + H]+ Calcd for C20H20Cl2N5O5S 512.0562; Found 512.0556.
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-ethyl-3-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (11). 1H NMR (400 MHz, DMSO-d6) δ = 10.60 (s, 1H), 8.42 (br d, J = 4.3 Hz, 1H), 8.29 (s, 1H), 8.09 (d, J = 1.8 Hz, 1H), 7.82 (br t, J = 5.6 Hz, 1H), 7.74 (dd, J = 1.8, 8.3 Hz, 1H), 7.65 (dt, J = 1.5, 7.6 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.22 - 7.11 (m, 2H), 4.98 (s, 2H), 3.20 - 3.14 (m, 2H), 2.90 - 2.81 (m, 4H), 1.16 (t, J = 7.5 Hz, 3H); HRMS (ESI/Q-TOF) RT: 2.16 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0760.
2-(4,5-dichloro-6-oxopyridazin-1(6h)-yl)-n-(2,4-dimethyl-5-(n-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (12). 1H NMR (400 MHz, DMSO-d6) δ = 9.82 (s, 1H), 8.40 (br d, J = 4.0 Hz, 1H), 8.29 (s, 1H), 7.90 (s, 1H), 7.69 (br t, J = 5.6 Hz, 1H), 7.63 (dt, J = 1.5, 7.6 Hz, 1H), 7.22 (s, 1H), 7.20 - 7.15 (m, 1H), 7.13 (d, J = 7.8 Hz, 1H), 5.03 (s, 2H), 3.12 (q, J = 6.9 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H), 2.25 (s, 3H); HRMS (ESI/Q-TOF) RT: 1.96 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0762.
2-(4,5-dichloro-6-oxopyridazin-1(6h)-yl)-n-(3,4-dimethyl-5-(n-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (13). 1H NMR (400 MHz, DMSO-d6) δ = 10.49 (s, 1H), 8.43 - 8.36 (m, 1H), 8.29 (s, 1H), 7.97 (d, J = 1.7 Hz, 1H), 7.76 (t, J = 5.7 Hz, 1H), 7.66 - 7.61 (m, 2H), 7.17 (dd, J = 5.1, 6.9 Hz, 1H), 7.11 (d, J = 7.8 Hz, 1H), 4.97 (s, 2H), 3.17 (q, J = 6.9 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H), 2.33 - 2.24 (m, 6H);HRMS (ESI/Q-TOF) RT: 2.08 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0762.
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(2,4-dimethyl-3-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (14). 1H NMR (400 MHz, DMSO-d6) δ = 9.85 (s, 1H), 8.39 (br d, J = 4.0 Hz, 1H), 8.29 (s, 1H), 7.70 (br t, J = 5.6 Hz, 1H), 7.61 (dt, J = 1.8, 7.6 Hz, 1H), 7.36 (br d, J = 8.1 Hz, 1H), 7.22 - 7.16 (m, 1H), 7.16 - 7.12 (m, 1H), 7.09 (br d, J = 7.7 Hz, 1H), 5.01 (s, 2H), 3.20 - 3.12 (m, 2H), 2.81 (br t, J = 7.2 Hz, 2H), 2.56 (s, 3H), 2.42 (s, 3H); HRMS (ESI/Q-TOF) RT: 1.89 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0764.
N-(6-chloro-5-methylpyridin-2-yl)acetamide (37). To a mixture of 6-chloro-5-methylpyridin-2-amine (CAS 442129-37-5, 6 g, 42.08 mmol) and pyridine (4.99 g, 63.12 mmol, 5.09 mL) in DCM (50 mL) was added acetyl chloride (4.29 g, 54.70 mmol, 3.90 mL) and the mixture was stirred at 20 °C for 12 hr. The mixture was diluted with water and extracted twice with ethyl acetate, combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, filtered to remove insoluble materials, and volatiles removed to provide the title compound as a yellow solid (8.1 g, assumed quant.). LRMS (m/z): Calcd [M+H]+ for C8H10ClN2O 185.4; found 185.4; 1H NMR (400 MHz, CHLOROFORM-d) δ = 8.12 - 8.02 (m, 1H), 8.02 - 7.83 (m, 1H), 7.61 - 7.50 (m, 1H), 2.36 - 2.32 (m, 3H), 2.20 - 2.16 (m, 3H).
N-(6-(benzylthio)-5-methylpyridin-2-yl)acetamide (38). To a solution of N-(6-chloro-5-methylpyridin-2-yl)acetamide (37, 10.1 g, 54.71 mmol) in DMF (100 mL) was added potassium carbonate (30.24 g, 218.82 mmol) and phenylmethanethiol (20.38 g, 164.12 mmol, 19.23 mL) at 20°C. The mixture was stirred at 60°C for 12 h, diluted with water and extracted twice with ethyl acetate, the combined organic layers were washed with water, saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue was purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to afford the title compound as a yellow solid (9.33 g, 63% yield).1H NMR (400 MHz, CHLOROFORM-d) δ = 7.88 - 7.77 (m, 1H), 7.77 - 7.63 (m, 1H), 7.42 - 7.37 (m, 2H), 7.36 - 7.29 (m, 3H), 7.28 - 7.23 (m, 1H), 4.49 - 4.35 (m, 2H), 2.29 - 2.21 (m, 3H), 2.21 - 2.18 (m, 3H); 1H NMR (400 MHz, DMSO-d6) δ = 10.33 (s, 1H), 7.73 (br d, J = 8.1 Hz, 1H), 7.49 - 7.43 (m, 3H), 7.33 - 7.28 (m, 2H), 7.26 - 7.21 (m, 1H), 4.50 (s, 2H), 2.13 (s, 3H), 2.11 (s, 3H).
6-acetamido-3-methylpyridine-2-sulfonyl chloride (39). To a solution of N-(6-(benzylthio)-5-methylpyridin-2-yl)acetamide (38, 9.33 g, 34.26 mmol) in DCM (100 mL) was added water (20 mL) and sulfuryl chloride (32.36 g, 239.79 mmol, 23.97 mL) at 0 °C. The reaction mixture slowly warmed to 20°C and stirred for 2 hr. Water was added and the mixture was extracted three times with dichloromethane, combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, and volatiles removed under reduced pressure to afford the title compound as a yellow oil (11.6 g, assumed quant.).
N-[5-methyl-6-[2-(2-pyridyl)ethylsulfamoyl]-2-pyridyl]acetamide (40). To a solution of 6-acetamido-3-methyl-pyridine-2-sulfonyl chloride (39, 6 g, 24.13 mmol) in THF (60 mL) was added triethylamine (7.32 g, 72.38 mmol, 10.07 mL) and 2-(2-pyridyl)ethanamine (4.42 g, 36.19 mmol, 4.33 mL). The mixture was stirred at 25°C for 12 hr, diluted with water, and extracted twice with ethyl acetate. Combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials were removed by filtration, volatiles removed under reduced pressure and the residue purified by column chromatography on silica gel, eluting with a gradient of ethyl acetate in petroleum ether to provide the title compound as a yellow solid (4.7 g, 58% yield). LRMS (m/z): Calcd [M+H]+ for C15H19N4O3S 335.1; found 335.3.
6-amino-3-methyl-N-[2-(2-pyridyl)ethyl]pyridine-2-sulfonamide (41). A mixture of N-[5-methyl-6-[2-(2-pyridyl)ethylsulfamoyl]-2-pyridyl]acetamide (40, 4.7 g, 14.06 mmol, 1 eq) in hydrogen chloride (6M in water, 50 mL) was stirred at 80°C for 12 hr. Volatiles were removed under reduced pressure to provide the title compound as a white solid (4.6 g, HCl salt, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.87 - 8.77 (m, 1H), 8.54 (dt, J = 1.5, 7.9 Hz, 1H), 8.11 (br s, 1H), 8.03 - 7.90 (m, 2H), 7.50 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 8.6 Hz, 1H), 3.61 (q, J = 5.9 Hz, 2H), 3.30 (t, J = 6.5 Hz, 2H), 2.29 (s, 3H).
2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(5-methyl-6-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)pyridin-2-yl)acetamide (15). Prepared in a manner analogous to Compound 6. 1H NMR (400 MHz, DMSO-d6) δ = 11.05 (s, 1H), 8.51 (d, J = 4.2 Hz, 1H), 8.09 (br d, J = 2.2 Hz, 1H), 7.97 (br t, J = 5.7 Hz, 1H), 7.89 (d, J = 8.3 Hz, 1H), 7.74 (dt, J = 1.8, 7.6 Hz, 1H), 7.31 - 7.23 (m, 2H), 5.16 (br s, 2H), 3.62 - 3.50 (m, 2H), 2.99 (t, J = 7.3 Hz, 2H), 2.53 (s, 3H); HRMS (ESI/Q-TOF) RT: 2.08 min, m/z: [M + H]+ Calcd for C19H19Cl2N6O4S 497.0565; Found 497.0558.
3-(benzylthio)-2-methyl-5-nitropyridine (42). A mixture of 3-bromo-2-methyl-5-nitropyridine (2 g, 9.22 mmol) was added phenylmethanethiol (1.49 g, 11.98 mmol, 1.40 mL), N,N-diethyl isopropylamine (2.38 g, 18.43 mmol, 3.21 mL), Tris(dibenzylideneacetone)dipalladium (421.95 mg, 460.79 umol) and 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene (533.24 mg, 921.57 umol) in dioxane (30 mL) was heated at reflux (100°C) for 12 hours. The mixture was cooled to room temperature, diluted with water, extracted three times with ethyl acetate, the combined organic layers were washed with water, saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue purified by column chromatography on silica gel, eluting with a gradient of ethyl acetate in petroleum ether, to afford the title compound as a yellow solid (2.6g, assumed quant.). 1H NMR (400 MHz, CHLOROFORM-d) δ = 9.04 - 8.91 (m, 1H), 8.25 - 8.04 (m, 1H), 7.33 - 7.22 (m, 5H), 4.23 - 4.06 (m, 2H), 2.62 - 2.53 (m, 3H).
2-methyl-5-nitropyridine-3-sulfonyl chloride (43). To 3-(benzylthio)-2-methyl-5-nitropyridine (42, 1.0 g, 3.84 mmol) in DCM (20 mL) was added water (4 mL) and sulfuryl chloride (3.63 g, 26.89 mmol, 2.69 mL) at 0 °C. The reaction mixture slowly warmed to 20°C and stirred for 30 min, volatiles were removed under reduced pressure, the residue was suspended in dichloromethane, dried over sodium sulfate, insoluble materials removed by filtration, and volatiles removed under reduced pressure to afford the title compound as a yellow oil (1.0 g, assumed quant.), which was used without further manipulation.
1-methyl-4-((2-methyl-5-nitropyridin-3-yl)sulfonyl)-1,4-diazepane (44). A mixture of 2-methyl-5-nitropyridine-3-sulfonyl chloride (43, 1.0 g, 4.23 mmol) 1-methyl-1,4-diazepane (723.83 mg, 6.34 mmol, 788.49 uL) and triethylamine (1.28 g, 12.68 mmol, 1.76 mL) in THF (20 mL) was stirred for 12 h at 20°C. Volatiles were removed under reduced pressure and the residue was purified by prep-HPLC (column: Kromasil 250*50mm*10um; mobile phase: [water(0.225%FA)-ACN];B%: 5%-30%,15min) to afford the title compound as a brown solid (430 mg, 32% yield).
6-methyl-5-((4-methyl-1,4-diazepan-1-yl)sulfonyl)pyridin-3-amine (45). A mixture of 1-methyl-4-((2-methyl-5-nitropyridin-3-yl)sulfonyl)-1,4-diazepane (44, 100 mg, 318.11 umol) ammonium chloride (170.16 mg, 3.18 mmol) and Zn dust (83.20 mg, 1.27 mmol) in THF (2 mL) was stirred at 66 °C for 12 hr. After cooling to room temperature, insoluble materials were removed by filtration through Celite, aqueous hydrochloric acid was added to the filtrate, and volatiles removed under reduced pressure to afford the title compound as a yellow solid (75 mg, di-HCl salt, 66% yield).
2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(6-methyl-5-((4-methyl-1,4-diazepan-1-yl)sulfonyl)pyridin-3-yl)acetamide (16). A mixture of 6-methyl-5-((4-methyl-1,4-diazepan-1-yl)sulfonyl)pyridin-3-amine (45, 75 mg, 209.91 umol, 2 HCl salt), 2-(4,5-dichloro-6-oxo-pyridazin-1-yl)acetic acid (32, 56.17 mg, 251.89 umol), 2-chloro-1-methyl-pyridin-1-ium iodide (80.44 mg, 314.86 umol), and N,N-diethyl isopropylamine (135.64 mg, 1.05 mmol, 182.81 uL, 5 eq) in THF (5 mL) was stirred at 20°C for 12 h. Volatiles were removed under reduced pressure and the residue was purified by prep-HPLC (column: Shim-pack C18 150*25*10um;mobile phase: [water(0.225%FA)-ACN];B%: 3%-33%,10min) to afford the title compound as a yellow solid (10 mg, 9% yield). 1H NMR (400 MHz, METHANOL-d4) δ = 8.75 - 8.70 (m, 1H), 8.65 - 8.60 (m, 1H), 8.14 - 8.10 (m, 1H), 5.12 - 5.06 (m, 2H), 3.74 - 3.67 (m, 2H), 3.58 - 3.51 (m, 2H), 3.24 - 3.18 (m, 4H), 2.80 - 2.77 (m, 3H), 2.76 - 2.72 (m, 3H), 2.20 - 2.07 (m, 2H); HRMS (ESI/Q-TOF) RT: 1.74 min, m/z: [M + H]+ Calcd for C18H23Cl2N6O4S 489.0878; Found 489.0872.
2-chloro-5-methyl-pyridine-4-sulfonyl chloride (46). 2-chloro-5-methyl-pyridin-4-amine (500 mg, 3.51 mmol), TFA (5 mL), and hydrochloric acid (12M in water, 2.5 mL) was treated with sodium nitrite (723.40 mg, 10.48 mmol) in water (3.5 mL) and stirred for 1 h at 0°C, insoluble materials were removed by filtration. Sulfur dioxide gas was bubbled through a mixture of copper(I)chloride (34.95 mg, 353.00 umol), copper(II)chloride (234.14 mg, 1.74 mmol) in acetic acid (30 mL) for 30 min at 0°C. The mixtures were combined and stirred at 0°C for 1.5 h, diluted with dichloromethane, washed twice with ice-water, twice with saturated aqueous sodium hydrogen carbonate, and saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, and volatiles removed under reduced pressure to afford the title compound as a yellow oil (530 mg, 67% yield).
2-chloro-N,N,5-trimethyl-pyridine-4-sulfonamide (47). A mixture of 2-chloro-5-methyl-pyridine-4-sulfonyl chloride (46, 530 mg, 2.34 mmol) and N-methylmethanamine (2M in THF, 5 mL) was stirred for 2 h at 15°C. The mixture was diluted with ethyl acetate, washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue was purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petreolum ether to afford the title compound as a white solid (300 mg, 55% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ = 8.41 (s, 1H), 7.69 (s, 1H), 2.90 (s, 6H), 2.58 (s, 3H).
tert-butyl N-[4-(dimethylsulfamoyl)-5-methyl-2-pyridyl]carbamate (48). A mixture of 2-chloro-N,N,5-trimethyl-pyridine-4-sulfonamide (47, 250 mg, 1.07 mmol), tert-butyl carbamate (623.91 mg, 5.33 mmol), cesium carbonate (624.70 mg, 1.92 mmol), 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene (73.96 mg, 127.82 umol) and Tris(dibenzylideneacetone)dipalladium (39.02 mg, 42.61 umol) in dioxane (5 mL) was stirred at 80°C for 4 hr. The mixture was diluted with water, extracted three times with ethyl acetate, combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials were removed by filtration, volatiles removed under reduced pressure and the residue was triturated with petroleum ether:ethyl acetate (1:1) and the title compound isolated by filtration as a yellow solid (260 mg, 77% yield). 1H NMR (400 MHz, DMSO-d6) δ = 10.14 (s, 1H), 8.36 (s, 1H), 8.15 - 8.12 (m, 1H), 2.79 (s, 6H), 2.44 (s, 3H), 1.49 (s, 9H).
2-amino-N,N,5-trimethyl-pyridine-4-sulfonamide (49). Tert-butyl N-[4-(dimethylsulfamoyl)-5-methyl-2-pyridyl]carbamate (48, 240 mg, 760.97 umol) in hydrochloric acid (2M in methanol, 5 mL) was stirred at 25°C for 12 hr. Volatiles were removed under reduced pressure to afford the title compound as a yellow solid (300 mg, HCl salt, assumed quant.). LRMS (m/z): Calcd [M+H]+ for C8H14N3O2S 216.1; found 216.1.
2-(4,5-dichloro-6-oxo-pyridazin-1-yl)-N-[4-(dimethylsulfamoyl)-5-methyl-2-pyridyl]acetamide (17). Prepared analogously to Compound 16, using 49 and purified by prep-HPLC (column: Phenomenex Synergi C18 150*25*10um; mobile phase: [water (0.225%FA)-ACN];B%: 26%-56%,10min) to afford the title compound as a white solid (45 mg, 38% yield). 1H NMR (400 MHz, DMSO-d6) δ = 11.29 (s, 1H), 8.49 (s, 1H), 8.32 (s, 1H), 8.29 (s, 1H), 5.07 (s, 2H), 2.78 (s, 6H), 2.48 (s, 3H); HRMS (ESI/Q-TOF) RT 2.61 min, m/z: [M + H]+ Calcd for C14H16Cl2N5O4S 420.0300; Found 420.0295.
Tert-butyl 3-(2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetamido)piperidine-1-carboxylate (50). A mixture of tert-butyl 3-aminopiperidine-1-carboxylate (179.61 mg, 896.80 umol), 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetic acid (32, 200 mg, 896.80 umol), N,N-diethyl isopropylamine (347.72 mg, 2.69 mmol, 468.62 uL) and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (511.49 mg, 1.35 mmol) in DMF (4 mL) was stirred at 25 °C for 12 hours. Insoluble materials were removed by filtration, volatiles removed under reduced pressure and the residue was purified by prep-HPLC (column: Phenomenex Luna C18 150 *40mm 10 um; mobile phase: [water (0.225%FA)-ACN]; B%: 28%-58%, 8.5min) to afford the title compound as a white solid (220 mg, 61% yield). LRMS (m/z): [M+H]+= 305.1;1H NMR (400 MHz, DMSO-d6) δ = 8.23 (s, 1H), 8.19 (br d, J = 7.3 Hz, 1H), 4.74 (d, J = 6.4 Hz, 2H), 3.58 (br d, J = 2.9 Hz, 2H), 3.32 (s, 1H), 2.90 (br d, J = 2.8 Hz, 2H), 1.80 (br s, 2H), 1.68 (br d, J = 8.3 Hz, 2H), 1.39 (s, 9H).
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(piperidin-3-yl)acetamide (51). To tert-butyl 3-(2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetamido)piperidine-1-carboxylate (50, 120 mg, 296.10 umol) in DCM (1 mL) was added hydrochloric acid (4 M in methanol, 4 mL). The mixture was stirred at 25 °C for 1 hour. Volatiles were removed under reduced pressure to afford the title compound as a white solid (100 mg, HCl salt, 98% yield). LRMS (m/z): Calcd [M+H]+ for C11H15Cl2N4O2 305.1; found 304.8.
2-(4,5-Dichloro-6-oxopyridazin-1(6H)-yl)-N-(1-(N,N-dimethylsulfamoyl)piperidin-3-yl)acetamide (18). A mixture of 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(piperidin-3-yl)acetamide (51, 100 mg, 292.72 umol, HCl salt), N,N-dimethylsulfamoyl chloride (63.05 mg, 439.08 umol, 47.05 uL) and triethylamine (88.86 mg, 878.17 umol, 122.23 uL) in THF (2 mL) was stirred at 25 °C for 2 hours. Volatiles were removed under reduced pressure and the residue was purified by prep-TLC on silica gel, eluting with ethyl acetate to afford the title compound as a white solid (30 mg, 25% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.33 (d, J = 7.3 Hz, 1H), 8.31 (s, 1H), 4.81 (d, J = 4.9 Hz, 2H), 3.83 - 3.66 (m, 1H), 3.56 (dd, J = 3.9, 11.8 Hz, 1H), 3.00 - 2.91 (m, 1H), 2.80 (s, 6H), 2.77 - 2.66 (m, 2H), 1.92 - 1.78 (m, 2H), 1.64 - 1.51 (m, 1H), 1.49 - 1.37 (m, 1H); HRMS (ESI/Q-TOF) RT 2.18 min, m/z: [M + H]+ Calcd for C13H20Cl2N5O4S 412.0613; Found 412.0609.
N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]formamide (52). To a mixture of 5-amino-2-methyl-N-[2-(2-pyridyl)ethyl]benzenesulfonamide (36, 1 g, 3.43 mmol, 1 eq) and formic acid (1.90 g, 41.19 mmol, 1.55 mL) in toluene (10 mL) was stirred at 115 C for 2 hr. The mixture was cooled to room temperature and diluted with water, extracted four times with ethyl acetate, combined organics washed repeatedly with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, and volatiles removed under reduced pressure to afford the title compound as a light-yellow oil (1.23 g, assumed quant.). 1H NMR (400 MHz, DMSO-d6) δ = 10.39 (s, 1H), 8.43 - 8.40 (m, 1H), 8.30 (d, J = 1.8 Hz, 1H), 8.12 (d, J = 2.1 Hz, 1H), 7.79 (br t, J = 5.6 Hz, 1H), 7.71 (dd, J = 2.3, 8.1 Hz, 1H), 7.65 (dt, J = 1.8, 7.7 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.21 - 7.13 (m, 2H), 3.22 - 3.12 (m, 2H), 2.83 (t, J = 7.4 Hz, 2H), 2.44 (s, 3H).
2-methyl-5-(methylamino)-N-[2-(2-pyridyl)ethyl]benzenesulfonamide (53). To a mixture of N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]formamide (52, 1 g, 3.13 mmol) in THF (10 mL) was added borane dimethyl sulfide complex (626.22 uL) drop-wise at 0°C over a period of 15 mins. The mixture was heated to 60°C for 12 hours. The reaction mixture was diluted with water and extracted 4 times with ethyl acetate, combined organic phases were washed three times with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials were removed by filtration, volatiles removed under reduced pressure and the residue purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to afford the title compound as an off-white solid (565 mg, 59% yield). LRMS (m/z): Calcd [M+H]+ for C15H19N3O2S 306.1; found 306.0; 1H NMR (400 MHz, DMSO-d6) δ = 8.45 - 8.41 (m, 1H), 8.45 - 8.41 (m, 1H), 7.66 (dt, J = 1.9, 7.7 Hz, 1H), 7.53 (t, J = 5.8 Hz, 1H), 7.21 - 7.13 (m, 2H), 7.07 - 7.03 (m, 2H), 6.62 (dd, J = 2.6, 8.2 Hz, 1H), 5.90 (q, J = 4.9 Hz, 1H), 3.15 - 3.08 (m, 2H), 2.86 - 2.80 (m, 2H), 2.66 (d, J = 5.0 Hz, 3H), 2.33 (s, 3H).
2-(4,5-dichloro-6-oxo-pyridazin-1-yl)-N-methyl-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (19). Prepared in an analogous manner to 6 using 53. 1H NMR (400 MHz, METHANOL-d4) δ = 8.27 (br d, J = 4.2 Hz, 1H), 7.95 (s, 1H), 7.89 (br s, 1H), 7.62 (br t, J = 7.0 Hz, 1H), 7.52 (br d, J = 7.2 Hz, 1H), 7.42 (br d, J = 7.8 Hz, 1H), 7.21 - 7.10 (m, 2H), 4.64 (s, 2H), 3.31 - 3.27 (m, 2H), 3.25 (br d, J = 1.7 Hz, 3H), 2.86 (br t, J = 7.0 Hz, 2H), 2.51 (s, 3H); HRMS (ESI/Q-TOF) RT 1.98 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0763.
methyl (S)-2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)propanoate (54). To a solution of 4,5-dichloropyridazin-3(2H)-one (5 g, 30.31 mmol) and methyl (2R)-2-hydroxypropanoate (6.31 g, 60.61 mmol, 5.79 mL) in THF (50 mL) was added triphenylphosphine (15.90 g, 60.61 mmol) and stirred at 20°C for 10 min, cooled to 0°C, and diisopropyl azodicarboxylate (12.26 g, 60.61 mmol, 11.79 mL) was added and the reaction mixture was stirred at 20°C for 2h. Volatiles were removed under reduced pressure and the residue was purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to provide the title compound as a white solid (7.6 g, 99% yield). 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.84 (s, 1H), 5.58 (q, J = 7.3 Hz, 1H), 3.76 (s, 3H), 1.70 (d, J = 7.1 Hz, 3H).
(S)-2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)propanoic acid (55). methyl (S)-2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)propanoate (54, 7.6 g, 30.27 mmol) was suspended in aqueous hydrogen chloride (5% w/v, 150 mL) and the mixture was stirred at 105°C for 1.5 h. The reaction was cooled to room temperature and the resulting solid was isolated by filtration and washed with water to afford the title compound as a white solid (5.7 g, 79% yield).
(S)-2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)propanamide (20). A mixture of (2S)-2-(4,5-dichloro-6-oxo-pyridazin-1-yl)propanoic acid (55, 100 mg, 421.87 umol) and 5-amino-2-methyl-N-[2-(2-pyridyl)ethyl]benzenesulfonamide (36, 122.92 mg, 421.87 umol), 2,4,6-Tripropyl-1,3,5,2λ5,4λ5,6λ5-trioxatriphosphinane 2,4,6-trioxide (295.31 mg, 464.06 umol, 275.99 uL, 50% w/w in ethyl acetate) and N,N-diethyl isopropylamine (65.43 mg, 506.24 umol, 88.18 uL) in DMF (1 mL) was stirred at 25°C for 3 hr. Volatiles were removed under reduced pressure and the residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150*25*10um;mobile phase: [water(0.225%FA)-ACN];B%: 13%-43%, 10min) to afford the title compound as a white solid (35 mg, 16% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.40 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 8.32 (s, 1H), 8.07 (d, J = 2.3 Hz, 1H), 7.76 (t, J = 5.8 Hz, 1H), 7.71 (dd, J = 8.3, 2.3 Hz, 1H), 7.64 (td, J = 7.6, 1.9 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.17 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 7.13 (dt, J = 7.8, 1.1 Hz, 1H), 5.44 (q, J = 7.0 Hz, 1H), 3.14 (td, J = 7.2, 5.7 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H), 1.61 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (101 MHz, DMSO) δ 167.93, 158.24, 156.02, 148.97, 138.61, 136.65, 136.40, 136.19, 135.68, 132.87, 132.44, 131.17, 123.21, 122.78, 121.59, 119.52, 59.23, 41.93, 37.28, 19.05, 15.74; SFC (Chiralpac AD-3, 50x4.6 mm I.D., 3uM, Mobile phase: Gradient elution 5%-40% 2-propanol (0.05% diethyl amine) in CO2, Flow rate 3 ml/min, PDA detector, Column Temp 35 C, Back pressure 100Bar) RT: 1.029 min, 96.8% ee; HRMS (ESI/Q-TOF) RT: 2.18 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0759.
(R)-2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)propanamide (21). Was prepared analogously to 20. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.40 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 8.32 (s, 1H), 8.07 (d, J = 2.3 Hz, 1H), 7.76 (t, J = 5.8 Hz, 1H), 7.71 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (td, J = 7.7, 1.9 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.17 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 7.13 (dd, J = 7.8, 1.2 Hz, 1H), 5.44 (q, J = 7.0 Hz, 1H), 3.13 (dd, J = 7.6, 5.9 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H), 1.61 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (101 MHz, DMSO) δ 167.93, 158.24, 156.02, 148.97, 138.61, 136.65, 136.40, 136.19, 135.68, 132.87, 132.44, 131.17, 123.21, 122.77, 121.59, 119.52, 59.23, 41.93, 37.28, 19.05, 15.74; SFC (Chiralpac AD-3, 50x4.6 mm I.D., 3uM, Mobile phase: Gradient elution 5%-40% 2-propanol (0.05% diethylamine) in CO2, Flow rate 3 ml/min, PDA detector, Column Temp 35 C, Back pressure 100Bar) RT: 0.642 min, 97.8% ee; HRMS (ESI/Q-TOF) RT: 2.17 min, m/z: [M + H]+ Calcd for C21H22Cl2N5O4S 510.0769; Found 510.0759.
(rac)- ethyl 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)propanoate (56). To a solution of 4,5-dichloropyridazin-3(2H)-one (CAS 932-22-9, 1 g, 6.06 mmol) and potassium carbonate (1.68 g, 12.12 mmol) in DMF (10 mL) was added ethyl 2-bromopropanoate (1.21 g, 6.67 mmol, 868.36 uL) at 25 °C, and the mixture was stirred at 50 °C for 2 hours. The mixture was diluted with ethyl acetate and water, layers separated, and the organic phase washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether, to afford the title compound as a white oil (1.14 g, 71% yield). LRMS (m/z): Calcd [M+H]+ for C9H10Cl2N2O3 265.0; found 264.9; 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.19 (t, J=7.13 Hz, 4 H) 1.62 (d, J=7.25 Hz, 4 H) 1.97 (s, 1 H) 4.15 (q, J=7.09 Hz, 2 H) 5.47 (q, J=7.21 Hz, 1 H) 7.77 (s, 1 H).
ethyl 2-(4-chloro-5-methyl-6-oxo-pyridazin-1-yl)propanoate (57). To a mixture of ethyl 2-(4,5-dichloro-6-oxo-pyridazin-1-yl)propanoate (56, 14.5 g, 54.70 mmol) and methylboronic acid (3.11 g, 51.96 mmol) in dioxane (150 mL) and H2O (15 mL) was added Cs2CO3 (53.46 g, 164.09 mmol) and [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloride, complex with dichloromethane (2.00 g, 2.73 mmol). The mixture was stirred at 110°C for 2 hr, diluted with aqueous sodium hydrogen carbonate, extracted twice with ethyl acetate. The combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure, and the residue purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to afford the title compound as a white solid (4.12 g, 31% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.10 (s, 1H), 5.40 (q, J = 7.1 Hz, 1H), 4.12 (q, J = 7.0 Hz, 2H), 2.17 (s, 3H), 1.54 (d, J = 7.2 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H).
2-(4-chloro-5-methyl-6-oxopyridazin-1(6H)-yl)propanoic acid (58). Was prepared in an analogous manner to 55.
2-(4-chloro-5-methyl-6-oxo-pyridazin-1-yl)-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]propanamide (22). Prepared in an analogous manner to 6. 1H NMR (400 MHz, DMSO-d6) δ = 10.41 (s, 1H), 8.40 (dd, J = 0.9, 4.8 Hz, 1H), 8.13 - 8.07 (m, 2H), 7.78 - 7.69 (m, 2H), 7.64 (dt, J = 1.8, 7.6 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.20 - 7.11 (m, 2H), 5.39 (q, J = 7.1 Hz, 1H), 3.18 - 3.10 (m, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H), 2.17 (s, 3H), 1.59 (d, J = 7.1 Hz, 3H); HRMS (ESI/Q-TOF) RT: 2.14 min, m/z: [M + H]+ Calcd for C22H25ClN5O4S 490.1315; Found 490.1310.
ethyl 2-(5-chloro-4-fluoro-6-oxopyridazin-1(6H)-yl)acetate (59). A mixture of ethyl 2-(4,5-dichloro-6-oxopyridazin-1(6H)-yl)acetate (31, 1 g, 3.98 mmol), potassium fluoride (1.16 g, 19.92 mmol, 466.53 uL) and silver sulfate (124.19 mg, 398.30 umol, 67.49 uL) in DMF (10 mL) was stirred at 110 °C for 12 hours. The mixture was diluted with water and extracted twice with ethyl acetate, combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to afford the title compound as a white solid (470 mg, 50% yield). LRMS (m/z): Calcd [M+H]+ for C8H8ClFN2O3 235.0; found 235.0; 1H NMR (400 MHz, Chloroform-d) δ 7.85 (s, 1H), 4.89 (s, 2H), 4.23 (q, J = 7.1 Hz, 2H), 1.27 (t, J = 7.2 Hz, 3H); 19F NMR (376 MHz, Chloroform-d) δ -110.33; 13C{1H} NMR (101 MHz, Chloroform-d) δ 166.72, 158.65 (d, J = 6.4 Hz), 158.18 (d, J = 279.6 Hz), 129.70 (d, J = 28.6 Hz), 119.29 (d, J = 9.3 Hz), 62.16, 53.96, 14.17.
2-(5-chloro-4-fluoro-6-oxopyridazin-1(6H)-yl)acetic acid (60). A mixture of ethyl 2-(5-chloro-4-fluoro-6-oxopyridazin-1(6H)-yl)acetate (59, 100 mg, 426.24 umol) in hydrochloric acid (3 M in water, 10 mL) was stirred at 90 °C for 1 hour. The mixture was frozen and volatiles removed by lyophilization to afford the title compound as a white solid (100 mg, assumed quant.), which was used without further manipulation. LRMS (m/z): Calcd [M+H]+ for C6H4ClFN2O3 207.0; found 207.1.
2-(5-chloro-4-fluoro-6-oxo-pyridazin-1-yl)-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (23). A mixture of 5-amino-2-methyl-N-[2-(2-pyridyl)ethyl]benzenesulfonamide (36, 100 mg, 343.21 umol), 2-(5-chloro-4-fluoro-6-oxopyridazin-1(6H)-yl)acetic acid (60, 85.07 mg, 411.85 umol), 2-chloro-1-methyl-pyridin-1-ium iodide (131.53 mg, 514.81 umol) and N,N-diethyl isopropylamine (88.71 mg, 686.42 umol, 119.56 uL) in THF (2 mL) was stirred at 25 °C for 2 hours. Volatiles were removed under reduced pressure and the residue was purified by prep-HPLC (column: Shim-pack C18 150*25*10um; mobile phase: [water (0.225%FA)-ACN]; B%: 5%-38%, 11min) to afford the title compound as a white solid (85 mg, 52% yield). 1H NMR (400 MHz, DMSO-d6) δ = 10.59 (s, 1H), 8.45 - 8.39 (m, 2H), 8.10 (d, J = 2.2 Hz, 1H), 7.78 (t, J = 5.7 Hz, 1H), 7.71 - 7.62 (m, 2H), 7.31 (d, J = 8.4 Hz, 1H), 7.18 (dd, J = 5.3, 7.0 Hz, 1H), 7.14 (d, J = 7.8 Hz, 1H), 5.00 (s, 2H), 3.18 - 3.13 (m, 2H), 2.82 (t, J = 7.3 Hz, 2H), 2.44 (s, 3H); 19F NMR (376 MHz, DMSO-d6) δ -111.08; HRMS (ESI/Q-TOF) RT: 1.75 min, m/z: [M + H]+ Calcd for C20H20ClFN5O4S 480.0908; Found 480.0901.
ethyl 2-(4-fluoro-5-methyl-6-oxopyridazin-1(6H)-yl)acetate (61). A mixture of ethyl 2-(5-chloro-4-fluoro-6-oxopyridazin-1(6H)-yl)acetate (59, 1 g, 4.26 mmol), methylboronic acid (280.66 mg, 4.69 mmol), cesium carbonate (4.17 g, 12.79 mmol) and [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloride, complex with dichloromethane (155.94 mg, 213.12 umol) in dioxane (20 mL) and water (2 mL) was stirred at 110 °C for 6 hours. The mixture was cooled to room temperature, diluted with water, extracted twice with ethyl acetate, combined organic phases were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue was purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to provide the title compound as a pink solid (560 mg, 61% yield). LRMS (m/z): Calcd [M+H]+ for C9H11FN2O3 215.1; found 215.0; 1H NMR (400 MHz, CHLOROFORM-d) δ = 7.79 (s, 1H), 4.89 (s, 2H), 4.29 - 4.23 (m, 2H), 2.15 (d, J = 2.6 Hz, 3H), 1.35 - 1.29 (m, 3H).
2-(4-fluoro-5-methyl-6-oxopyridazin-1(6H)-yl)acetic acid (62). A mixture of ethyl 2-(4-fluoro-5-methyl-6-oxopyridazin-1(6H)-yl)acetate (61, 150 mg, 700.30 umol) in hydrochloric acid (3 M in water, 15 mL) was stirred at 90 °C for 90 minutes. The mixture was frozen and volatiles removed by lyophilized to afford the title compound (120 mg, 92% yield) as a off-white solid. LRMS (m/z): Calcd [M+H]+ for C7H8FN2O3 187.1; found 186.9;
2-(4-fluoro-5-methyl-6-oxopyridazin-1(6h)-yl)-n-(4-methyl-3-(n-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (24). Was prepared analogously to 23. 1H NMR (400 MHz, DMSO-d6) δ = 10.57 (s, 1H), 8.47 - 8.38 (m, 1H), 8.21 (s, 1H), 8.11 (d, J = 2.1 Hz, 1H), 7.78 (t, J = 5.7 Hz, 1H), 7.72 - 7.62 (m, 2H), 7.31 (d, J = 8.4 Hz, 1H), 7.19 (dd, J = 5.3, 7.0 Hz, 1H), 7.14 (d, J = 7.7 Hz, 1H), 4.92 (s, 2H), 3.19 - 3.11 (m, 2H), 2.82 (t, J = 7.3 Hz, 2H), 2.44 (s, 3H), 2.02 (d, J = 2.3 Hz, 3H); HRMS (ESI/Q-TOF) RT: 1.68 min, m/z: [M + H]+ Calcd for C21H23FN5O4S 460.1455; Found 460.1448.
ethyl 2-(4-fluoro-6-oxo-pyridazin-1-yl)acetate (63). A mixture of ethyl 2-(5-chloro-4-fluoro-6-oxopyridazin-1(6H)-yl)acetate (59, 300 mg, 1.21 mmol), palladium on carbon (60 mg, 5% w/w) and triethylamine (366.28 mg, 3.62 mmol, 503.82 uL) in EtOAc (6 mL) was stirred under an atmosphere of hydrogen at 25 °C for 12 hr. Insoluble materials were removed by filtration through Celite, volatiles removed under reduced pressure, and the residue purified by column chromatography on silica gel eluting with a gradient of ethyl acetate in petroleum ether to afford the title compound as a colorless oil (214 mg, 88% yield). LRMS (m/z): Calcd [M+H]+ for C8H9FN2O3 201.1; found 201.0.
2-(4-fluoro-6-oxo-pyridazin-1-yl)-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (25). Was prepared analogously to 23. 1H NMR (400 MHz, DMSO-d6) δ = 10.56 (s, 1H), 8.41 – 8.40 (d, 1H), 8.26 (d, 1H), 8.10 (d, 1H), 7.77 (m, 1H), 7.69 (m, 1H), 7.64 (m, 1H), 7.31 -7.29 (m, 1H), 7.14 – 7.12 (m, 1H), 7.02 – 6.99 (m, 1H), 4.92 (s, 2H), 3.18 - 3.13 (m, 2H), 2.84 – 2.80 (m, 2H), 2.44 (s, 3H); HRMS (ESI/Q-TOF) RT: 1.47 min, m/z: [M + H]+ Calcd for C20H21FN5O4S 446.1298; Found 446.1290.
2-chloro-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (65). A mixture of 5-amino-2-methyl-N-[2-(2-pyridyl)ethyl]benzenesulfonamide (36, 300 mg, 1.03 mmol) and triethylamine (208.38 mg, 2.06 mmol, 286.62 uL) in DCM (5 mL) was treated with 2-chloroacetyl chloride (151.18 mg, 1.34 mmol, 106.46 uL) at 0°C. The mixture was stirred at 20°C for 12h. The mixture was diluted with water and extracted twice with ethyl acetate. Combined organic layers were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials removed by filtration, volatiles removed under reduced pressure and the residue purified by prep-TLC on silica gel, eluting with dichloromethane:methanol 10:1, to give the title compound as a yellow solid (200 mg, 53% yield). LRMS (m/z): Calcd [M+H]+ for C16H18ClN3O3S 368.1; found 368.1; 1H NMR (400 MHz, DMSO-d6) δ = 10.53 (s, 1H), 8.45 - 8.37 (m, 1H), 8.09 (d, J = 2.3 Hz, 1H), 7.82 - 7.72 (m, 2H), 7.65 (dt, J = 1.8, 7.6 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.21 - 7.12 (m, 2H), 4.26 (s, 2H), 3.20 - 3.14 (m, 2H), 2.83 (t, J = 7.4 Hz, 2H), 2.45 (s, 3H).
2-(4-chloro-6-oxopyridazin-1(6H)-yl)-N-(4-methyl-3-(N-(2-(pyridin-2-yl)ethyl)sulfamoyl)phenyl)acetamide (26). A mixture of 4-chloro-1H-pyridazin-6-one (35.51 mg, 272.07 umol), 2-chloro-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (65, 100 mg, 247.33 umol) and potassium carbonate (102.55 mg, 742.00 umol) in DMF (2 mL) was stirred at 50°C for 2 hr. The mixture was diluted with water, extracted twice with ethyl acetate, combined organic phases were washed with saturated aqueous sodium chloride, dried over sodium sulfate, insoluble materials were removed by filtration, volatiles removed under reduced pressure and the residue was purified by prep-HPLC (column: Phenomenex Synergi C18 150*25*10um;mobile phase: [water(0.225%FA)-ACN];B%: 2%-32%,10min) to give the title compound as a yellow solid (45 mg, 36% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.40 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 8.14 (d, J = 2.4 Hz, 1H), 8.09 (d, J = 2.3 Hz, 1H), 7.77 (t, J = 5.8 Hz, 1H), 7.69 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (td, J = 7.7, 1.9 Hz, 1H), 7.35 (d, J = 2.4 Hz, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.17 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 7.13 (dt, J = 7.9, 1.1 Hz, 1H), 4.90 (s, 2H), 3.14 (td, J = 7.2, 5.6 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H); 13C{1H} NMR (101 MHz, DMSO-d6) δ 164.96, 158.53, 158.24, 148.97, 139.12, 138.70, 136.54, 136.51, 136.41, 132.97, 131.10, 127.07, 123.20, 122.47, 121.59, 119.22, 54.62, 41.96, 37.28, 19.06; HRMS (ESI/Q-TOF) RT: 1.68 min, m/z: [M + H]+ Calcd for C20H21ClN5O4S 462.1002; Found 462.0994.
Synthesis of BRD0639 (Scheme 5 B).
Scheme 5. Syntheses of mono-chloropyridazinones (A) Synthesis of 26 (B) Synthesis of 27 and (C) Synthesis of 30.
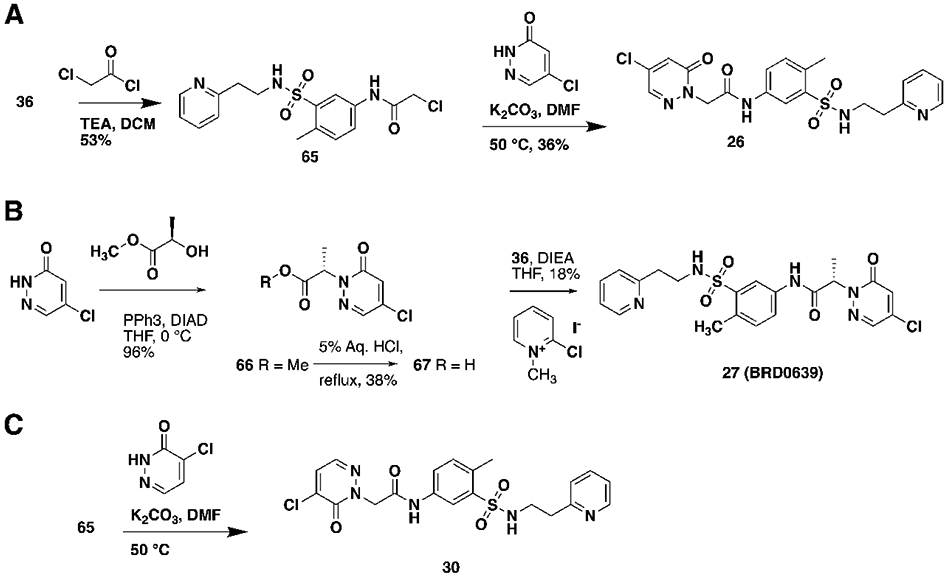
methyl (S)-2-(4-chloro-6-oxopyridazin-1(6H)-yl)propanoate (66).
To a solution of 5-chloro-3(2H)-Pyridazinone (CAS 660425-07-0, 300 mg, 2.30 mmol) and methyl (2R)-2-hydroxypropanoate (478.88 mg, 4.60 mmol, 439.34 uL) in THF (3 mL) was added triphenylphosphine (1.21 g, 4.60 mmol) at room temperature, placed in an ice water bath, and after 10 minutes was added diisopropyl azodicarboxylate (929.46 mg, 4.60 mmol, 893.72 uL) the mixture was removed from the ice water bath, and allowed to warm to 20°C for 2h. Volatiles were removed under reduced pressure and the residue was purified by column chromatography on silica gel, eluting with a gradient of ethyl acetate in petroleum ether to give the title compound as a white solid (480 mg, 96% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.15 (d, J = 2.4 Hz, 1H), 7.33 (d, J = 2.4 Hz, 1H), 5.43 (q, J = 7.2 Hz, 1H), 3.64 (s, 3H), 1.53 (d, J = 7.1 Hz, 3H).
(2S)-2-(4-chloro-6-oxo-pyridazin-1-yl)propanoic acid (67). Methyl (2S)-2-(4-chloro-6-oxo-pyridazin-1-yl)propanoate (66, 480 mg, 2.22 mmol) was suspended in aqueous hydrochloric acid (5% w/v, 4 mL) and heated to 105 °C for 12 hr, cooled to room temperature, and volatiles removed to provide a brown solid, this was suspended in hot toluene, a small amount of materials were removed by filtration, and after cooling to room temperature, the title compound was isolated by filtration as an off white solid (170 mg, 38% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.14 (d, J = 2.3 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 5.34 (q, J = 7.2 Hz, 1H), 1.52 (d, J = 7.2 Hz, 3H).
(2S)-2-(4-chloro-6-oxo-pyridazin-1-yl)-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]propanamide (27)
(BRD0639). To a mixture of (2S)-2-(4-chloro-6-oxo-pyridazin-1-yl)propanoic acid (67, 50.00 mg, 246.80 umol) and 5-amino-2-methyl-N-[2-(2-pyridyl)ethyl]benzenesulfonamide (36, 71.91 mg, 246.80 umol) in THF (1 mL) was added DIEA (127.59 mg, 987.19 umol, 171.95 uL) over 5 minutes, followed by 2-chloro-1-methyl-pyridin-1-ium iodide (94.58 mg, 370.20 umol). The reaction mixture was stirred at 20°C for 1h, volatiles were removed under reduced pressure and the residue was purified by prep-HPLC (column: Phenomenex Gemini NX-C18(75*30mm*3um); mobile phase: [water(10mM NH4HCO3)-ACN];B%: 18%-48%,8min) to give the title compound as a white solid (21 mg, 18% yield). 1H NMR (400 MHz, DMSO) δ 10.42 (s, 1H), 8.43 – 8.38 (m, 1H), 8.17 (d, J = 2.4 Hz, 1H), 8.08 (d, J = 2.3 Hz, 1H), 7.75 (s, 1H), 7.71 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (td, J = 7.7, 1.9 Hz, 1H), 7.32 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.17 (ddd, J = 7.7, 4.9, 1.2 Hz, 1H), 7.13 (d, J = 7.7 Hz, 1H), 5.39 (q, J = 7.1 Hz, 1H), 3.14 (t, J = 7.3 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H), 1.60 (d, J = 7.1 Hz, 3H).;13C{1H} NMR (101 MHz, DMSO) δ 168.39, 158.64, 158.25, 148.97, 138.88, 138.61, 136.75, 136.40, 136.04, 132.85, 131.05, 126.60, 123.21, 122.67, 121.59, 119.43, 57.99, 41.94, 37.29, 19.05, 15.69; SFC (Chiralpac AS-3, 50x4.6 mm I.D., 3uM, Mobile phase: Gradient elution 5%-40% Methanol (0.05% diethylamine) in CO2, Flow rate 3 ml/min, PDA detector, Column Temp 35 C, Back pressure 100Bar) RT 2.037 min, 98.7% ee; HRMS (ESI/Q-TOF) RT: 1.88 min, m/z: [M + H]+ Calcd for C21H23ClN5O4S 476.1159; Found 476.1152.
(2R)-2-(4-chloro-6-oxo-pyridazin-1-yl)-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]propanamide (28). Was prepared analogously to 27. 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.40 (ddd, J = 4.9, 1.9, 0.9 Hz, 1H), 8.17 (d, J = 2.4 Hz, 1H), 8.08 (d, J = 2.3 Hz, 1H), 7.75 (s, 1H), 7.71 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (td, J = 7.6, 1.9 Hz, 1H), 7.32 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.17 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 7.13 (dt, J = 7.8, 1.1 Hz, 1H), 5.39 (q, J = 7.1 Hz, 1H), 3.14 (t, J = 7.3 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H), 1.60 (d, J = 7.1 Hz, 3H); 13C{1H} NMR (101 MHz, DMSO-d6) δ 168.39, 158.64, 158.25, 148.97, 138.88, 138.61, 136.75, 136.40, 136.04, 132.85, 131.05, 126.60, 123.21, 122.67, 121.59, 119.43, 57.99, 41.94, 37.29, 19.05, 15.70; SFC (Chiralpac AS-3, 50x4.6 mm I.D., 3uM, Mobile phase: Gradient elution 5%-40% Methanol (0.05% diethylamine) in CO2, Flow rate 3 ml/min, PDA detector, Column Temp 35 C, Back pressure 100Bar) RT 1.700 min, 95.7% ee; HRMS (ESI/Q-TOF) RT: 1.89 min, m/z: [M + H]+ Calcd for C21H23ClN5O4S 476.1159; Found 476.1152.
(2S)-2-(4-chloro-6-oxo-pyridazin-1-yl)-N-methyl-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]propanamide (29). Was prepared analogously to 20. 1H NMR (400 MHz, DMSO-d6) δ = 8.33 (br d, J = 4.8 Hz, 1H), 7.96 (d, J = 2.0 Hz, 1H), 7.88 (d, J = 1.6 Hz, 1H), 7.68 (dt, J = 1.9, 7.7 Hz, 1H), 7.52 (dd, J = 1.6, 8.0 Hz, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.24 - 7.17 (m, 2H), 6.86 (s, 1H), 5.26 (q, J = 6.8 Hz, 1H), 3.30 (br s, 2H), 3.26 (s, 3H), 2.91 (dt, J = 1.8, 7.2 Hz, 2H), 2.55 (s, 3H), 1.43 (d, J = 7.0 Hz, 3H); SFC (Chiralpac AD-3, 50x4.6 mm I.D., 3uM, Mobile phase: Gradient elution 5%-40% 2-propanol (0.05% diethylamine) in CO2, Flow rate 3 ml/min, PDA detector, Column Temp 35 C, Back pressure 100Bar) RT: 2.072 min, 86.3% ee (minor isomer at 1.895 min); HRMS (ESI/Q-TOF) RT: 1.83 min, m/z: [M + H]+ Calcd for C22H25ClN5O4S 490.1315; Found 490.1308.
2-(5-chloro-6-oxo-pyridazin-1-yl)-N-[4-methyl-3-[2-(2-pyridyl)ethylsulfamoyl]phenyl]acetamide (30). Prepared analogously to 26. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.41 (ddd, J = 4.8, 1.9, 0.9 Hz, 1H), 8.10 (d, J = 2.4 Hz, 1H), 7.95 (d, J = 4.5 Hz, 1H), 7.86 (d, J = 4.5 Hz, 1H), 7.77 (t, J = 5.8 Hz, 1H), 7.69 (dd, J = 8.2, 2.3 Hz, 1H), 7.64 (td, J = 7.6, 1.8 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.17 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 7.13 (dt, J = 7.8, 1.1 Hz, 1H), 4.97 (s, 2H), 3.15 (td, J = 7.3, 5.7 Hz, 2H), 2.81 (t, J = 7.3 Hz, 2H), 2.43 (s, 3H);13C{1H} NMR (101 MHz, DMSO-d6) δ 164.87, 158.24, 156.91, 148.97, 138.71, 136.54, 136.40, 136.07, 135.17, 132.98, 131.11, 130.90, 123.20, 122.47, 121.58, 119.21, 55.85, 41.97, 37.28, 19.07; HRMS (ESI/Q-TOF) RT: 1.52 min, m/z: [M + H]+ Calcd for C20H21ClN5O4S 462.1002; Found 462.0997.
Supplementary Material
Scheme 2. Syntheses of Pyridine analogs (A) Synthesis of 15 (B) Synthesis of 16 (C) Synthesis of 17 and (D) Synthesis of piperidine 18.

Scheme 3. (A) Synthesis of 19 (B) Synthesis of 20 (C) Synthesis of 21.
Scheme 4. (A) Syntheses of Fluoro Pyridazinones (A) synthesis of 23 (B) synthesis of 24 (C) synthesis of 25.
ACKNOWLEDGMENTS
This work was funded as part of an on-going research collaboration between the Broad Institute and an affiliate of Deerfield Management Company, L.P and by the NIH grant 1R01CA233626-01A1 to WRS.
ABBREVIATIONS USED
- CDKN2a
Cyclin Dependent Kinase Inhibitor 2A
- COPR5
Cooperator of PRMT5
- DIEA
Diisopropylethylamine
- FP
fluorescence polarization
- GSH
Glutathione
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- LgBIT
LargeBit
- MAT2a
Methionine Adenosyltransferase 2A
- MCL1
Induced myeloid leukemia cell differentiation protein 1
- MTA
5′-S-Methyl-5′-thioadenosine
- ND
Not determined
- PBM
PRMT5 binding motif
- PBS
phosphate buffered saline
- pICln
Chloride conductance regulatory protein
- PRMT
Protein Arginine Methyltransferases
- PRMT5
Protein Arginine Methyltransferase 5
- RIOK1
RIO kinase 1
- SAM
S-Adenosyl-L-methionine
- SAP
substrate adaptor proteins
- SDMA
symmetrically dimethylated arginine
- SmBiT
SmallBit
- SPR
surface plasmon resonance
- TCEP
Tris(2-carboxyethyl)phosphine
- WDR77
WD repeat-containing protein 77
Footnotes
Supporting Information
The supporting information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00507?goto=supporting-info.
Additional Methods: NMR-based fragment screening; Virtual screening; crystallography data collection and refinement statistics; electron density maps; protein MassSpectra data; kinetic parameters plotted data; 1H and 13C NMR spectra; LCMS chromatograms (PDF)
Molecular formula strings (CSV)
Atomic coordinates of PRMT5:WDR77 bound to compound 1 (PDB code 6V0P), and compound 6 (PDB code 7M05) have been deposited with the Protein Data Bank. Authors will release the atomic coordinates and experimental data upon article publication.
CONFLICTS OF INTEREST: W.R.S. is a Board or SAB member and holds equity in Ideaya Biosciences, Civetta Therapeutics, and Bluebird bio and has consulted for Array, Astex, Epidarex Capital, Ipsen, PearlRiver Therapeutics, Merck Pharmaceuticals, Sanofi, Servier and Syndax Pharmaceuticals and receives research funding from Deerfield Investments, Pfizer Pharmaceuticals, Merck Pharmaceuticals, Ideaya Biosciences and Ridgeline Discovery. W.R.S. is a co-patent holder on EGFR mutation diagnostic patents. P.M. and B.J.M. are employees of and hold equity in Tango Therapeutics. V.K.K. holds equity in Dogma Therapeutics and is consultant for Anji Pharmaceuticals. A.I. consults for Ridgeline Discovery.
REFERENCES
- 1.Musiani D; Bok J; Massignani E; Wu L; Tabaglio T; Ippolito MR; Cuomo A; Ozbek U; Zorgati H; Ghoshdastider U; Robinson RC; Guccione E; Bonaldi T, Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci Signal 2019, 12 (575). [DOI] [PubMed] [Google Scholar]
- 2.McDonald ER 3rd; de Weck A; Schlabach MR; Billy E; Mavrakis KJ; Hoffman GR; Belur D; Castelletti D; Frias E; Gampa K; Golji J; Kao I; Li L; Megel P; Perkins TA; Ramadan N; Ruddy DA; Silver SJ; Sovath S; Stump M; Weber O; Widmer R; Yu J; Yu K; Yue Y; Abramowski D; Ackley E; Barrett R; Berger J; Bernard JL; Billig R; Brachmann SM; Buxton F; Caothien R; Caushi JX; Chung FS; Cortes-Cros M; deBeaumont RS; Delaunay C; Desplat A; Duong W; Dwoske DA; Eldridge RS; Farsidjani A; Feng F; Feng J; Flemming D; Forrester W; Galli GG; Gao Z; Gauter F; Gibaja V; Haas K; Hattenberger M; Hood T; Hurov KE; Jagani Z; Jenal M; Johnson JA; Jones MD; Kapoor A; Korn J; Liu J; Liu Q; Liu S; Liu Y; Loo AT; Macchi KJ; Martin T; McAllister G; Meyer A; Molle S; Pagliarini RA; Phadke T; Repko B; Schouwey T; Shanahan F; Shen Q; Stamm C; Stephan C; Stucke VM; Tiedt R; Varadarajan M; Venkatesan K; Vitari AC; Wallroth M; Weiler J; Zhang J; Mickanin C; Myer VE; Porter JA; Lai A; Bitter H; Lees E; Keen N; Kauffmann A; Stegmeier F; Hofmann F; Schmelzle T; Sellers WR, Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170 (3), 577–592 e10. [DOI] [PubMed] [Google Scholar]
- 3.Xiao W; Chen X; Liu L; Shu Y; Zhang M; Zhong Y, Role of protein arginine methyltransferase 5 in human cancers. Biomed Pharmacother 2019, 114, 108790. [DOI] [PubMed] [Google Scholar]
- 4.Huang A; Garraway LA; Ashworth A; Weber B, Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 2020, 19 (1), 23–38. [DOI] [PubMed] [Google Scholar]
- 5.Mavrakis KJ; McDonald ER 3rd; Schlabach MR; Billy E; Hoffman GR; deWeck A; Ruddy DA; Venkatesan K; Yu J; McAllister G; Stump M; deBeaumont R; Ho S; Yue Y; Liu Y; Yan-Neale Y; Yang G; Lin F; Yin H; Gao H; Kipp DR; Zhao S; McNamara JT; Sprague ER; Zheng B; Lin Y; Cho YS; Gu J; Crawford K; Ciccone D; Vitari AC; Lai A; Capka V; Hurov K; Porter JA; Tallarico J; Mickanin C; Lees E; Pagliarini R; Keen N; Schmelzle T; Hofmann F; Stegmeier F; Sellers WR, Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351 (6278), 1208–1213. [DOI] [PubMed] [Google Scholar]
- 6.Krasinskas AM; Bartlett DL; Cieply K; Dacic S, CDKN2A and MTAP deletions in peritoneal mesotheliomas are correlated with loss of p16 protein expression and poor survival. Mod Pathol 2010, 23 (4), 531–538. [DOI] [PubMed] [Google Scholar]
- 7.Kryukov GV; Wilson FH; Ruth JR; Paulk J; Tsherniak A; Marlow SE; Vazquez F; Weir BA; Fitzgerald ME; Tanaka M; Bielski CM; Scott JM; Dennis C; Cowley GS; Boehm JS; Root DE; Golub TR; Clish CB; Bradner JE; Hahn WC; Garraway LA, MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351 (6278), 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalev P; Hyer ML; Gross S; Konteatis Z; Chen CC; Fletcher M; Lein M; Aguado-Fraile E; Frank V; Barnett A; Mandley E; Goldford J; Chen Y; Sellers K; Hayes S; Lizotte K; Quang P; Tuncay Y; Clasquin M; Peters R; Weier J; Simone E; Murtie J; Liu W; Nagaraja R; Dang L; Sui Z; Biller SA; Travins J; Marks KM; Marjon K, MAT2A inhibition blocks the growth of MTAP-deleted cancer cells by reducing PRMT5-dependent mRNA splicing and inducing DNA damage. Cancer Cell 2021, 39 (2), 209–224. [DOI] [PubMed] [Google Scholar]
- 9.Chan-Penebre E; Kuplast KG; Majer CR; Boriack-Sjodin PA; Wigle TJ; Johnston LD; Rioux N; Munchhof MJ; Jin L; Jacques SL; West KA; Lingaraj T; Stickland K; Ribich SA; Raimondi A; Scott MP; Waters NJ; Pollock RM; Smith JJ; Barbash O; Pappalardi M; Ho TF; Nurse K; Oza KP; Gallagher KT; Kruger R; Moyer MP; Copeland RA; Chesworth R; Duncan KW, A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015, 11 (6), 432–437. [DOI] [PubMed] [Google Scholar]
- 10.Bonday ZQ; Cortez GS; Grogan MJ; Antonysamy S; Weichert K; Bocchinfuso WP; Li F; Kennedy S; Li B; Mader MM; Arrowsmith CH; Brown PJ; Eram MS; Szewczyk MM; Barsyte-Lovejoy D; Vedadi M; Guccione E; Campbell RM, LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med Chem Lett 2018, 9 (7), 612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brehmer D; Wu TF; Mannens G; Beke L; Vinken P; Gaffney D; Sun WM; Pande V; Thuring JW; Millar H; Poggesi I; Somers I; Boeckx A; Parade M; van Heerde E; Nys T; Yanovich C; Herkert B; Verhulst T; Du Jardin M; Meerpoel L; Moy C; Diels G; Viellevoye M; Schepens W; Poncelet A; Linders JT; Lawson EC; Edwards JP; Chetty D; Laquerre S; Lorenzi MV, A novel PRMT5 inhibitor with potent in vitro and in vivo activity in preclinical lung cancer models. Cancer Research 2017, 77. [Google Scholar]
- 12.Wu TF; Millar H; Gaffney D; Beke L; Mannens G; Vinken P; Sommers I; Thuring JW; Sun WM; Moy C; Pande V; Zhou JG; Haddish-Berhane N; Salvati M; Laquerre S; Lorenzi MV; Brehmer D, JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivo activity, demonstrated in several lung cancer models. Cancer Research 2018, 78 (13). [Google Scholar]
- 13.Mulvaney KM; Blomquist C; Acharya N; Li R; O’Keefe M; Ranaghan M; Stokes M; Nelson AJ; Jain SS; Columbus J; Bozal FK; Skepner A; Raymond D; McKinney DC; Freyzon Y; Baidi Y; Porter D; Ianari A; McMillan B; Sellers WR, Molecular basis for substrate recruitment to the PRMT5 methylosome. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerard B; Lee M. D. t.; Dandapani S; Duvall JR; Fitzgerald ME; Kesavan S; Lowe JT; Marie JC; Pandya BA; Suh BC; O'Shea MW; Dombrowski M; Hamann D; Lemercier B; Murillo T; Akella LB; Foley MA; Marcaurelle LA, Synthesis of stereochemically and skeletally diverse fused ring systems from functionalized C-glycosides. J Org Chem 2013, 78 (11), 5160–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukami T; Yokoi T, The emerging role of human esterases. Drug Metab Pharmacokinet 2012, 27 (5), 466–477. [DOI] [PubMed] [Google Scholar]
- 16.Li B; Sedlacek M; Manoharan I; Boopathy R; Duysen EG; Masson P; Lockridge O, Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem Pharmacol 2005, 70 (11), 1673–1684. [DOI] [PubMed] [Google Scholar]
- 17.Timm DE; Bowman V; Madsen R; Rauch C, Cryo-electron microscopy structure of a human PRMT5:MEP50 complex. PLoS One 2018, 13 (3), e0193205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata Y; Chiba M, The role of extrahepatic metabolism in the pharmacokinetics of the targeted covalent inhibitors afatinib, ibrutinib, and neratinib. Drug Metab Dispos 2015, 43 (3), 375–384. [DOI] [PubMed] [Google Scholar]
- 19.Strelow JM, A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov 2017, 22 (1), 3–20. [DOI] [PubMed] [Google Scholar]
- 20.Ward RA; Anderton MJ; Ashton S; Bethel PA; Box M; Butterworth S; Colclough N; Chorley CG; Chuaqui C; Cross DA; Dakin LA; Debreczeni JE; Eberlein C; Finlay MR; Hill GB; Grist M; Klinowska TC; Lane C; Martin S; Orme JP; Smith P; Wang F; Waring MJ, Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J Med Chem 2013, 56 (17), 7025–7048. [DOI] [PubMed] [Google Scholar]
- 21.Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J, Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J Med Chem 2020, 63 (17), 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant T; Rohou A; Grigorieff N, cisTEM, user-friendly software for single-particle image processing. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marx MA, Fragment-Based Discovery Of MRTX9768, A Synthetic Lethal-Based Inhibitor Designed To Bind The PRMT5•MTA Complex And Selectively Target MTAPDEL Tumors. AACR Annual Meeting 2021, abstract LB003. [Google Scholar]
- 24.Malik R; Park PK; Barbieri C; Blat Y; Sheriff S; Weigelt C; Kopcho L; Celiktas M; Ruzanov M; Naglich J; Price J; Harner M; O’Malley K; Den J; Schmitz W; Li G; Ruan Z; Qin L; Duke G; Rodrigo I; Witmer M; Harden D; Arey B; Soars M; Fink B; Gavai A; Vite G; Voliva CF, A novel MTA non-competitive PRMT5 inhibitor AACR Annual Meeting 2021; abstract 1140. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.