

ABSTRACT
The autophagosome has two lipid bilayer membranes. The outer membrane fuses with the lysosome, while the inner membrane is degraded to release autophagic contents for degradation. It remains unclear how the inner vesicle of the autophagosome (called the autophagic vesicle) is disintegrated after autophagosome-lysosome fusion. Here, we identified C. elegans LPLA-2/M05B5.4 as a key enzyme that degrades membranous material in lysosomes. LPLA-2 is homologous to human PLA2G15, a lysosomal phospholipase A2 family protein that catalyzes cleavage of membrane phospholipids. We found that loss of LPLA-2 causes accumulation of large membrane whor ls in enlarged lysosomes and both phenotypes are suppressed by blocking macroautophagy/autophagy. Moreover, autophagic vesicles persisted in enlarged lysosomes in PLA2G15 knockdown cells and lpla-2(lf) mutants, which suggests that the breakdown of the inner autophagosomal membrane in lysosomes is impaired. lpla-2(lf) mutants exhibit severe defects in both embryonic and larval development. Our data suggest that disintegration of the inner autophagosomal membrane by LPLA-2 promotes the release and subsequent degradation of autophagic contents in lysosomes, which is essential for C. elegans development.
Abbreviations: ATG: autophagy related; EPG: ectopic PGL granules; GFP: green fluorescent protein; LGG-1: LC3, GABARAP and GATE-16 family; LPLA-2: lysosomal phospholipase A2; PGL: P granule abnormality protein; PLA2: phospholipase A2; SD: standard deviation; SEPA-1: suppressor of ectopic P granules in autophagy mutant; SQST-1: sequestosome related.
KEYWORDS: Autophagic vesicle, C. elegans, development, inner autophagosomal membrane, lysosomal phospholipase A2, lysosome
Introduction
Macroautophagy (hereafter referred to as autophagy) is a homeostatic process that degrades cytoplasmic contents such as protein aggregates, lipids, damaged organelles and invading pathogens through lysosomes. During autophagy, bulk cytosol or specific substrates are sequestered by a double-membrane structure, termed the phagophore, which expands and closes to form an autophagosome [1–3]. The autophagosome may fuse directly with the lysosome, where cargos are degraded, or it may undergo a maturation process by fusing with early and/or late endosomes to form an amphisome, which then fuses with the lysosome to degrade the autophagic contents [4–7]. Clearance of autophagic substrates and recycling of the resulting catabolites maintain cellular homeostasis and are important for development and immunity [8–10].
Multiple protein complexes including RABs, SNAREs and tethering factors act coordinately to control fusion of autophagosomes with lysosomes [11,12]. STX17-SNAP29-VAMP7 or -VAMP8 and STX7-SNAP29-YKT6 are the two SNARE complexes involved in autophagosome-lysosome fusion, while tethering factors such as HOPS, EPG5 and PLEKHM1 promote fusion by facilitating the formation of SNARE complexes [11,12]. Unlike endosomes and phagosomes, autophagosomes have two lipid bilayer membranes. The outer autophagosomal membrane (OAM) fuses with the lysosomal membrane, while the inner autophagosomal membrane (IAM) is disrupted after autophagosome-lysosome fusion, so that autophagic contents are released and degraded by lysosomal hydrolases [12]. In budding yeast, the putative lipase Atg15/Aut5/Cvt17 is responsible for the lysis of autophagic bodies, Cvt bodies and multivesicular body (MVB) vesicles in the vacuole [13–15]. Atg15 is an integral membrane protein that is localized to the ER and targeted to the vacuole via the MVB pathway [13–15]. It contains a putative lipase active-site motif, which is important for the breakdown of autophagic bodies in the vacuole [13,14]. An in vitro study suggests that Atg15 acts as a phospholipase that prefers phosphatidylserine (PS) as the substrate [16]. Atg15 is unique to yeast, so it is unclear how the single lipid bilayer-enclosed autophagic vesicles are disrupted in lysosomes of higher eukaryotes. The ATG conjugation systems are shown to be important for efficient degradation of the IAM in mammalian cells, probably due to their role in promoting the closure of the autophagosome [17]. However, the lysosomal enzyme(s) that disrupts IAMs remains to be identified in metazoans.
Phospholipase A2 (PLA2) family enzymes are acyl esterases that hydrolyze the sn-2 position of membrane glycerophospholipids to liberate free fatty acids and lysophospholipids [18]. PLA2G15/LPLA2 (phospholipase A2 group XV) is a unique member of the PLA2 family based on its cellular localization to the lysosome and its acidic pH optimum [19,20]. PLA2G15 recognizes phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG) and phosphatidylserine (PS) as substrates and exhibits a broad positional specificity for both sn-1 and sn-2 acyl groups [18,21]. Structural analyses of human PLA2G15 suggest that it utilizes both electrostatic charges and the hydrophobic patch on its membrane-binding domain to bind substrate-containing membranes [18,22]. PLA2G15 is highly expressed in the alveolar macrophages of rats and mice and is implicated in metabolism of pulmonary surfactant phospholipids [19,23,24]. Moreover, studies in pla2g15 knockout mice suggest that PLA2G15 may be involved in the induction of adaptive T-cell immunity to Mycobacterium tuberculosis and the presentation of self-lipid antigens [18,25–27]. However, the role of PLA2G15 in lysosomes is not clearly defined and the mechanism by which PLA2G15 regulates surfactant metabolism or host defense remains unknown.
In this study, we identified LPLA-2, the C. elegans homolog of human PLA2G15, as a key enzyme responsible for degrading membranous material in lysosomes. Our data suggest that LPLA-2 is essential for facilitating the release and degradation of autophagic cargos in lysosomes and for the maintenance of embryonic and larval development. We found that autophagic vesicles persist in enlarged lysosomes in lpla-2 mutants and PLA2G15 knockdown cells, which suggests that lysosomal phospholipase A2 is important for the lysis of inner autophagosomal membranes.
Results
qx348 mutants accumulate abnormal lysosomes
We performed a forward genetic screen in C. elegans for mutants with morphologically abnormal lysosomes. We isolated qx348, which contained abnormally enlarged lysosomes. In wild type, lysosomes labeled by the lysosomal membrane protein LAAT-1::GFP appeared as small puncta and thin tubules, with an average volume of 0.55 μm3 in 4-fold embryos and 0.52 μm3 in L1 larvae (Figures 1A-C and 1G-I) [28]. In qx348, however, lysosomes were significantly bigger than in wild type at embryonic, larval and adult stages, and tubular lysosomal structures were absent (Figure 1D-H). The average volume of lysosomes reached 1.8 μm3 and 1.5 μm3 in qx348 embryos and L1 larvae, and 4.3 μm3 in adult hypodermis, which is about 3 times bigger than in wild type at the same stage (Figure 1I). The lysosomal enlargement and absence of tubular morphology were observed in multiple tissues in qx348 mutants including hypodermis, body wall muscle, intestine and sheath cells (Figure 1F and S1A-F). The lysosomal enlargement in qx348 was seen from the 1.5-fold embryonic stage throughout larval and adult stages (Figure 1D-F and S1G-M). In the adult hypodermis and intestine of qx348, the enlarged LAAT-1-positive structures were visible under DIC microscopy as vesicular compartments with irregular shapes (Figure S1N-Oii). These LAAT-1-positive structures increased with age and became aggregated (Figure S1G-M). In contrast, reporters of the ER (GFP::TRAM-1), Golgi (MANS::GFP), mitochondria (Mito::GFP), early endosomes (YFP::2XFYVE, GFP::RAB-5) and recycling endosomes (GFP::RAB-10) all exhibited a similar pattern in qx348 as in wild type, which indicates that these intracellular organelles are not affected (Figure S2A-L).
Figure 1.
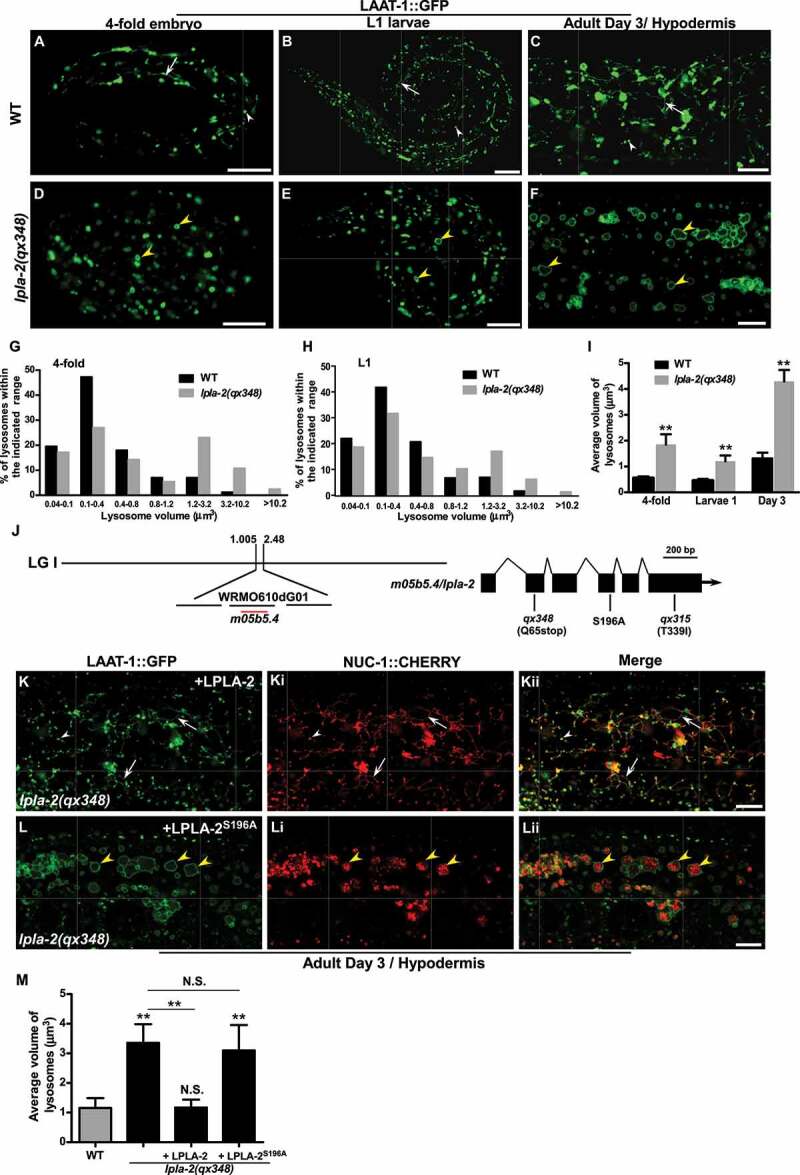
lpla-2 mutants accumulate enlarged lysosomes. (A-F) Confocal fluorescence images of 4-fold embryos (A and D), L1 larvae (B and E) and adult hypodermis (C and F) in wild type (A-C) and lpla-2(qx348) (D-F) expressing LAAT-1::GFP. (G-I) Quantification of lysosome volume in 4-fold embryos, L1 larvae (L1) and adult worms at day 3. (J) Cloning of lpla-2. The left bar indicates the genetic position of lpla-2. The lpla-2 gene structure is shown at the right with filled boxes representing exons and thin lines indicating introns. The mutation sites identified in the two lpla-2 alleles, qx348 and qx315, and the one that affects the serine residue essential for the catalytic activity of the enzyme are indicated. (K-Lii) Confocal fluorescence images of the hypodermis in lpla-2(qx348) adults carrying LAAT-1::GFP and NUC-1::CHERRY, with expression of wild-type LPLA-2 (K-Kii) or LPLA-2S196A (L-Lii). Quantification of the average volume of lysosomes is shown in (M). In (A-F and K-Lii), arrows indicate tubular lysosomal structures. White and yellow arrowheads indicate normal and enlarged globular lysosomes, respectively. In (I and M), at least 10 animals were scored in each strain at each stage, and data are shown as mean ± SD. One-way ANOVA with Tukey’s post hoc test (M) or two-way ANOVA with the Bonferroni post hoc test (I) was performed to compare mutant datasets with wild type or datasets that are linked by lines. **P < 0.001 (I), **P < 0.0001 (M), N.S.: no significance. Scale bars: 10 µm.
qx348 affects LPLA-2, a widely expressed lysosomal phospholipase A2
qx348 was mapped to the center of linkage group I between the map positions 1.005 and 2.48 (Figure 1J). By transformation rescue, we found that expression of m05b5.4, which encodes a previously uncharacterized protein with 417 amino acids, rescued the lysosomal enlargement phenotype in qx348 (Figures 1J–Kii and 1M). The protein encoded by m05b5.4 is homologous to human PLA2G15/LPLA2 (phospholipase A2 group XV) [29] (Figure S2M). We thus named this gene lpla-2 (Figure 1J and S2M). qx348 carried a C-to-T mutation in lpla-2 that resulted in a premature stop codon after Lys 64 (Figure 1J and S2M). The sequence of lpla-2 was also determined in qx315, which is allelic to qx348 and had a similar lysosome enlargement phenotype (Figure 1J, S2N-P and S2R). qx315 contained a C-to-T mutation that caused replacement of Thr 339 by Ile in LPLA-2 (Figure 1J and S2M).
Lysosomal PLA2 family enzymes catalyze cleavage of phospholipids at the sn-2 site in a calcium-independent manner and have an acidic pH optimum [19]. C. elegans LPLA-2 shares 63% sequence similarity and 45% sequence identity with human PLA2G15 (Figure S2M). Expression of LPLA-2, but not LPLA-2S196A, in which the serine residue essential for catalytic activity is disrupted, fully rescued the enlarged lysosome phenotype in qx348, which indicates that LPLA-2 acts as a phospholipase to maintain lysosome morphology (Figures 1J-M,S2M and Table 1) [30]. We generated an LPLA-2::CHERRY reporter driven by the lpla-2 promoter, which fully rescued the enlarged lysosome phenotype in lpla-2(qx348) (Figure S2Q, S2R and Table 1). LPLA-2::CHERRY was widely expressed from embryonic stages throughout larval and adult stages in various tissues, including pharynx, hypodermis, sheath cells, intestine, muscle and tail region (Figure 2A-Hi). LPLA-2::CHERRY labeled both punctate and tubular structures that were positive for the lysosomal membrane proteins LAAT-1 and SCAV-3 (Figure 2I-Jii). This suggests that LPLA-2 localizes to lysosomes.
Table 1.
Primers used for plasmid construction
| Primer | Sequence (5ʹ to 3ʹ) |
|---|---|
| PYL184 | CGGCTAGCCTCAAGTCTTCAATACATCTGGC |
| PYL185 | GGGGTACCAATTAACTGGAAAATGGCTTTTCG |
| PYL396 | GTCACGCTATGGGTAATCCCCTTTC |
| PYL397 | CATAGCGTGACCGACG AGTACCAC |
Figure 2.
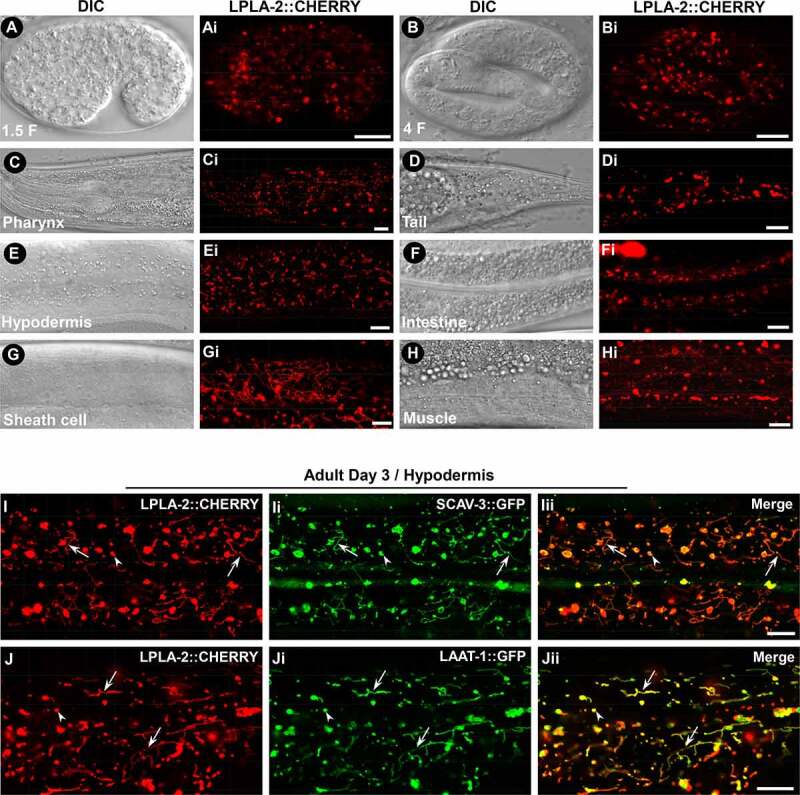
LPLA-2 is widely distributed and localizes to lysosomes. (A-Hi) DIC and confocal fluorescence images of wild type expressing LPLA-2::CHERRY driven by the lpla-2 promoter. (I-Jii) Confocal fluorescence images of the hypodermis in wild type co-expressing LPLA-2::CHERRY and SCAV-3::GFP (I–Iii) or LAAT-1::GFP (J-Jii). LPLA-2 colocalizes with SCAV-3 and LAAT-1 to both vesicular (arrowheads) and tubular (arrows) lysosomes. Scale bars: 10 µm.
Blocking autophagy suppresses lysosome enlargement in lpla-2 mutants
lpla-2 mutants accumulate enlarged lysosomes, which may be caused by defects in cargo degradation. To test this, we blocked autophagy, which delivers cytoplasmic cargos to lysosomes, and examined the lysosome phenotype in lpla-2 mutants. LGG-1 is the C. elegans homolog of Atg8/LC3. It associates with autophagic structures and is essential for autophagosome formation [31]. ATG-13/EPG-1 and EPG-6/WIPI4 regulate autophagy initiation and autophagosome formation, respectively [32,33]. We found that loss of LGG-1, ATG-13 or EPG-6 did not obviously affect the morphology of lysosomes labeled by SCAV-3::GFP, but they greatly suppressed lysosome enlargement in lpla-2 embryos and hypodermis (Figure 3A-I and S3A-I). Lysosome volume, which was greatly increased in lpla-1(qx348), was reduced significantly in lpla-2;lgg-1, lpla-2;atg-13 and lpla-2;epg-6 double mutants (Figure 3I and S3I). Moreover, accumulation of abnormal vesicular structures in the lpla-2 hypodermis was suppressed greatly by mutation in epg-6, lgg-1, atg-18 or epg-5 (Figure S3J-N). In addition, atg-13 or epg-6 mutation led to significantly reduced lysosomal volume in embryonic muscle cells of lpla-2 mutants (Figure S3O-U). In the intestine, loss of atg-13 or epg-6 restored the tubular lysosomal morphology in lpla-2 worms, and the overall lysosomal pattern in lpla-1;epg-6 and lpla-2;atg-13 double mutants resembled that in wild type (Figure S3V-AA). Altogether, these data indicate that blocking autophagy suppresses lysosomal enlargement in lpla-2 mutants. Moreover, we found that elevation of autophagy activity by either starvation treatment or loss of OGT-1, the O-GlcNAc transferase, further increased the lysosomal volume in lpla-2 mutants (Figure 3J-N) [34]. Furthermore, lysosomes purified from lpla-2(qx348) mutants accumulated high levels of LGG-1/LC3, which was suppressed by expression of LPLA-2 (Figure 3O). Taken together, these data suggest that loss of lpla-2 affects degradation of autophagic cargos, which leads to enlargement of lysosomes.
Figure 3.
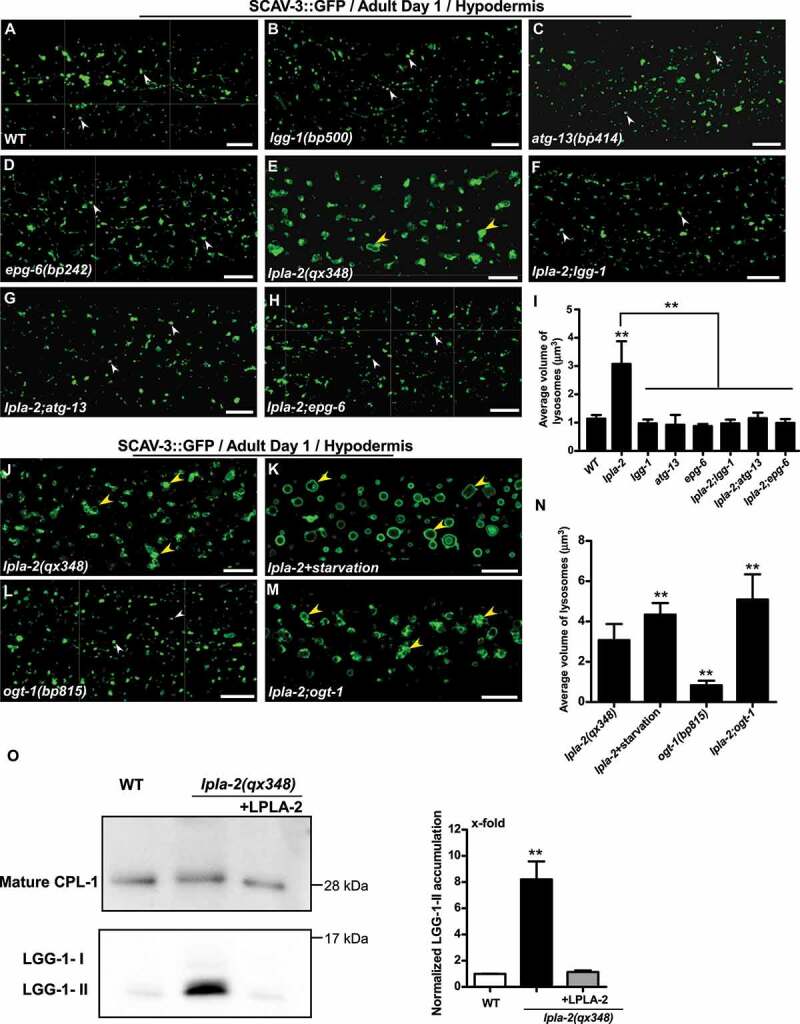
Blocking autophagy suppresses lysosome enlargement in lpla-2 mutants. (A-H and J-M) Confocal fluorescence images of the hypodermis in the indicated strains expressing SCAV-3::GFP. White and yellow arrowheads indicate normal and enlarged vesicular lysosomes, respectively. (I and N) Quantification of lysosome volume. At least 10 animals were scored in each strain. (O) Western blot analysis of LGG-1 accumulation in purified lysosomes from the indicated strains. Fully processed mature CPL-1 (CathePsin L family) was used to normalize the lysosome amount in each strain. LGG-1 accumulation was quantified and normalized to 1-fold in wild type (right panel). At least three independent experiments were performed. In (I, N and O), data are shown as mean ± SD. One-way ANOVA with Tukey’s post hoc test was performed to compare mutant datasets with wild type or datasets that are linked by lines. **P < 0.0001. All other points had P > 0.05. Scale bars: 10 µm.
Loss of LPLA-2 partially impairs autophagy
We next examined whether the autophagy process is affected in lpla-2 mutants. The C. elegans SQSTM1/p62 homolog, SQST-1 (SeQueSTosome-related), associates with various protein aggregates and is removed by autophagy during embryogenesis [31]. SQST-1 is cleared in wild-type embryos but persisted in autophagy-defective epg-6 mutant embryos at both early and late stages (Figure 4A-Di) [31]. In lpla-2(qx348), SQST-1 puncta were detected in 4-fold embryos but not at the 200-cell stage (Figure 4E-Fi). In addition, SQST-1::GFP was absent in wild type but accumulated in lpla-2(qx348) mutants at both larval and adult stages (Figure S4A-Di). LGG-1 associates with autophagic structures and is removed during embryogenesis [31,35]. LGG-1 was observed only at the early stage in wild-type embryos; however, in lpla-2 mutants, it was present in both early and late stages as in epg-6 embryos (Figure 4G-Li). PGL-3, a component of PGL granules, is removed in somatic cells by selective autophagy during embryogenesis [35]. SEPA-1 is the autophagy substrate that mediates aggregation and autophagic degradation of PGL granules in C. elegans embryos [35]. We found that PGL-3 and SEPA-1 were cleared in lpla-2(qx348) embryos as in wild type, but they accumulated in epg-9(bp320) mutant embryos, which are defective in autophagy (Figure S5A-Li) [36]. Altogether, these data suggest that the autophagy process is partially impaired or delayed in lpla-2(lf), causing accumulation of SQST-1 and LGG-1 in late-staged embryos. Consistent with this, we found that lpla-2 mutants accumulated both LGG-1-I and LGG-1-II (the PE-conjugated form of LGG-1) as in epg-6 mutants (Figure 4M). Expression of LPLA-2, but not LPLA-2S196A, the catalytically inactive version of LPLA-2, led to clearance of LGG-1 and SQST-1 puncta in lpla-2(qx348) embryos, and suppressed accumulation of LGG-1-I and LGG-1-II in lpla-2(qx348) worms (Figure 4M and S4E-Li). This indicates that the phospholipase activity of LPLA-2 is important for degradation of autophagic cargos in lysosomes. The survival of C. elegans L1 larvae in the absence of food requires autophagy. We found that lpla-2 larvae had a significantly shortened lifespan compared to wild type in the absence of food, which is consistent with impaired autophagy activity (Figure 4N).
Figure 4.
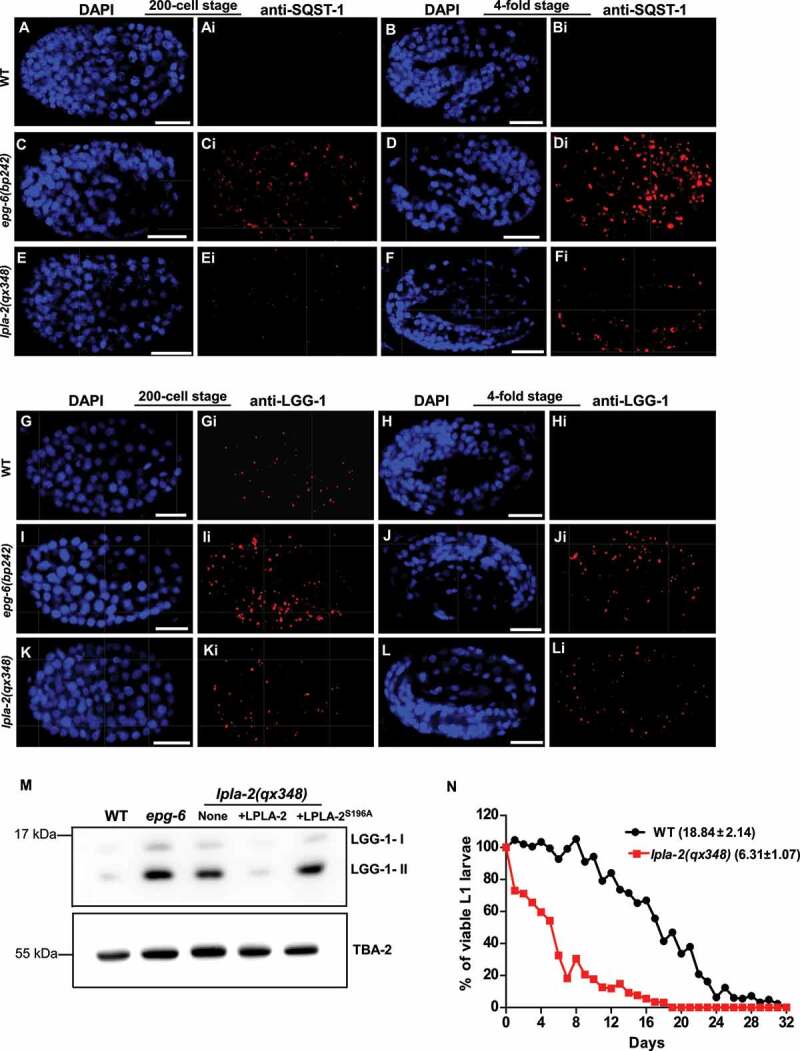
The autophagy process is partially impaired in lpla-2 mutants. (A-Li) Confocal fluorescence images of wild type (WT), epg-6(bp242) and lpla-2(qx348) embryos at the 200-cell and 4-fold stages stained by anti-SQST-1 (A-Fi) or anti-LGG-1 antibodies (G-Li). DAPI staining shows nuclei in each embryo. (M) Western blot analysis of LGG-1-I and LGG-1-II (lipid-conjugated form) in wild type (WT), epg-6(bp242) and lpla-2(qx348) without and with expression of LPLA-2 or LPLA-2S196A. (N) Quantification of the survival of WT and lpla-2(qx348) L1 larvae in the absence of food. At least 200 animals were scored at each time point in each strain. Three independent experiments were performed and the mean lifespan (mean ± SD) is shown in parenthesis. Scale bars: 10 µm.
We next examined whether degradation of endocytic cargo is affected in lpla-2 mutants. CAV-1, a plasma membrane-localized protein, and VIT-2, the yolk lipoprotein, are delivered to lysosomes through the endocytic pathway and are degraded shortly after fertilization and during embryogenesis respectively [37,38]. We found that CAV-1::GFP was absent in both wild type and lpla-2 mutant fertilized embryos, which indicates that CAV-1 degradation is not affected (Figure S6A-Bi). VIT-2::GFP was cleared in late-staged wild-type embryos or L1 larvae, and in the viable lpla-2 embryos and L1 larvae (Figure S6C-J). However, high levels of VIT-2::GFP were observed in lpla-2 embryos that were arrested either before or at the 4-fold stage (Figure S6K-Lii). This suggests that degradation of the yolk lipoprotein VIT-2 may be partially impaired in lpla-2 mutants. Impairing endocytosis by dyn-1 mutation did not reduce lysosomal volume in lpla-2 (Figure S6M-Q).
lpla-2 lysosomes accumulate a large quantity of membranes and contain intact autophagic vesicles
To investigate how loss of lpla-2 affects autophagic cargo degradation in lysosomes, we performed transmission electron microscopy (TEM) analyses. In wild-type hypodermis, lysosomes were observed as spherical, membrane-enclosed vesicles with few membrane layers (Figure 5A). In lpla-2(qx348) mutants, however, lysosomes were significantly enlarged and filled with dense membrane whorls, which indicates accumulation of undigested membranes (Figures 5B and 5F). The soluble lysosomal hydrolases NUC-1 and CPL-1 were diffuse in the lysosomal lumen in wild type and also in cup-5 mutants, which contain big lysosomes due to defective cargo degradation (Figure 5I, 5J and S5M-Nii) [39–41]. In lpla-2 mutants, however, the NUC-1::CHERRY and CPL-1::CHERRY signals were concentrated into discrete foci within lysosomes, consistent with occupation of the lysosomal lumen by large quantities of membranes (Figure 5K, 5L and S5O-Oii). Moreover, GFP::LGG-1, a single-copy reporter of LGG-1 [42], was diffuse in cup-5 lysosomes, but exhibited as discrete foci in lysosomes of lpla-2;cup-5 double mutants (Figure 5G-Hii). Both lysosomal enlargement and accumulation of membranous material in lpla-2 were efficiently suppressed by lgg-1 mutation, which blocks autophagy (Figure 5D-F). The lysosomal size and content in lpla-2;lgg-1 double mutants resembled that in lgg-1 single mutants (Figure 5D-F). These data suggest that loss of lpla-2 affects digestion of autophagic membranous contents in lysosomes. The double-membrane autophagosome fuses with lysosomes to deliver cargo for degradation. In this process, the outer membrane of the autophagosome fuses with the lysosome, while the inner autophagosomal membrane is disintegrated after fusion to release contents for degradation. In addition to large quantities of membranous material, we observed the presence of autophagic vesicles surrounded by a single lipid bilayer membrane in lpla-2 lysosomes (Figure 5C). This suggests that disintegration of the inner autophagosomal membrane is affected in lpla-2 mutants. RNAi inactivation of PLA2G15 in HeLa cells caused lysosome enlargement (Figure 5M-O). The average diameter of lysosomes in PLA2G15 knockdown cells reached 441 nm, which is almost twice as big as in control cells (Figure 5O). Moreover, TEM analyses revealed that 40% of lysosomes in PLA2G15 knockdown cells contained intact autophagic vesicles, which is significantly higher than in control cells (7.6%) (Figures 5N and 5P). Furthermore, we observed accumulation of intraluminal vesicles (ILVs) in lysosomes of PLA2G15 knockdown cells (Figures 5N and 5Q). These data suggest that disruption of autophagic vesicles and ILVs in lysosomes is affected by PLA2G15 knockdown.
Figure 5.
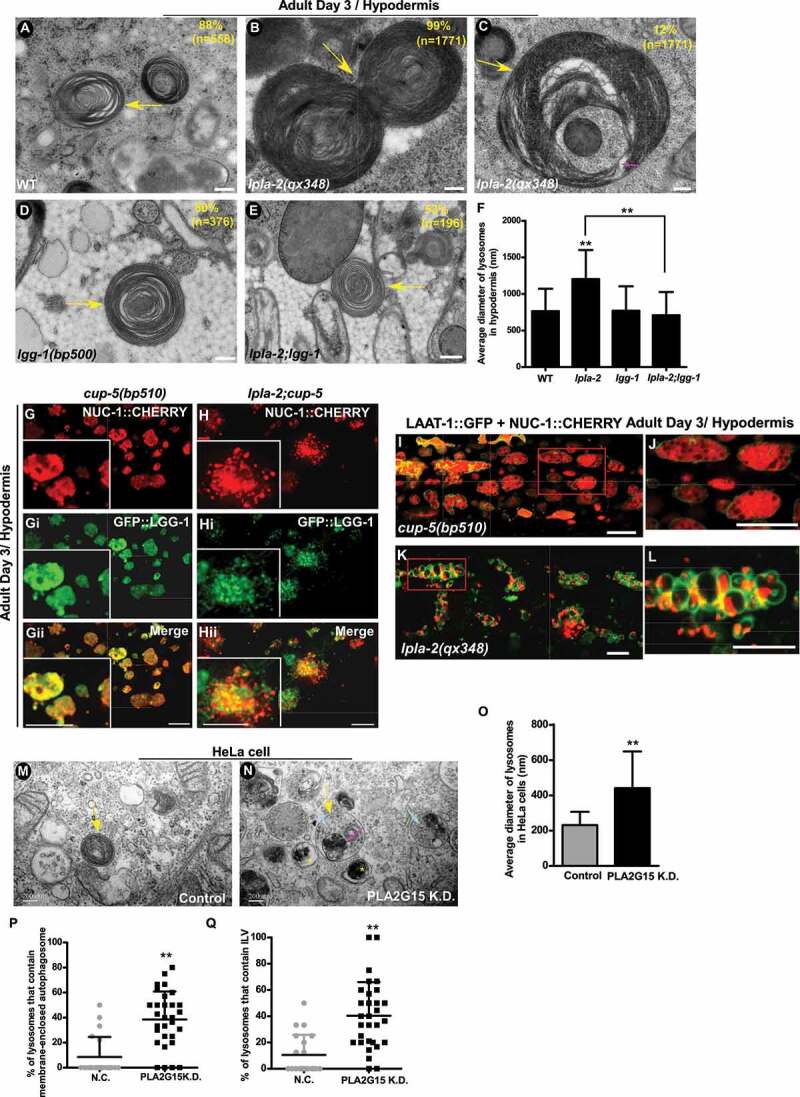
lpla-2 lysosomes accumulate a large quantity of membranous material and contain intact autophagic vesicles. (A-E) Transmission electron micrographs (TEM) of lysosomes in the hypodermis of wild type (A), lpla-2(qx348) (B and C) lgg-1(bp500) (D) and lpla-2;lgg-1 (E). Lysosomes are indicated by yellow arrows. A single lipid bilayer-enclosed autophagic vesicle in a lpla-2 lysosome is indicated by the pink arrow. The percentage of lysosomes with the indicated pattern is quantified and shown at the upper right corner in each panel. (F) Quantification of the diameter of lysosomes in the indicated C. elegans strains. (G-Hii) Confocal fluorescence images of the hypodermis in cup-5(bp510) (G-Gii) and lpla-2(qx348);cup-5 (H-Hii) expressing NUC-1::CHERRY and GFP::LGG-1. (I-L) Confocal fluorescence images of the hypodermis in cup-5(bp510) (I and J) and lpla-2(qx348) (K and L) adults expressing LAAT-1::GFP and NUC-1::CHERRY. Magnified images of the boxed area in (I) and (K) are shown in (J) and (L), respectively. (M and N) TEM of lysosomes in HeLa cells without (M) and with siRNA knockdown of PLA2G15 (N). Lysosomes are indicated by yellow arrows. The intact autophagic vesicle and intraluminal vesicles (ILVs) in lysosomes of PLA2G15 knockdown cells are indicated by pink and blue arrows, respectively. The yellow asterisks indicate double-membrane autophagosomes. The percentage of lysosomes that contain intact autophagic vesicle and ILVs was quantified and shown in (P and Q). (O) Quantification of the diameter of lysosomes in HeLa cells without and with knockdown of PLA2G15. In (F and O-Q), at least 40 lysosomes were scored in each sample. Data are shown as mean ± SD. Student’s two-tailed unpaired t test (O-P) or one-way ANOVA with Tukey’s post hoc test (F) was performed to compare mutant or knockdown datasets with control or wild type, or datasets that are linked by lines. **P < 0.0001. All other points had P > 0.05. Scale bars: 200 nm in (A-E, M and N) and 10 µm in (G-L).
Loss of lpla-2 affects embryogenesis and larval development
lpla-2 mutants exhibited severe defects in embryogenesis. Approximately 71% of lpla-2(qx348) embryos died during embryogenesis, with death occurring at both early and late embryonic stages (Figure 6A-K). Moreover, lpla-2 embryos that hatched grew more slowly than wild-type larvae, and 17% of them were arrested during larval development (Figures 6L and 6M). Both embryonic death and larval growth phenotypes in lpla-2 were fully rescued by expression of LPLA-2 (Figure 6K-M). Autophagy-defective mutants lgg-1, atg-18 and epg-6 exhibited a variable embryonic lethality from 16% to 51% (Figure 6K). Compared with lpla-2 single mutants, the embryonic lethality was not significantly altered in lpla-2;lgg-1 and lpla-2;epg-6 but slightly increased in lpla-2;atg-18 double mutants (Figure 6K). These data further support that LPLA-2 plays an essential role in autophagic cargo degradation. Loss of OGT-1, which increases autophagy activity [34], further increased the lethality of lpla-2 embryos from 71% to 100% (Figure 6K).
Figure 6.
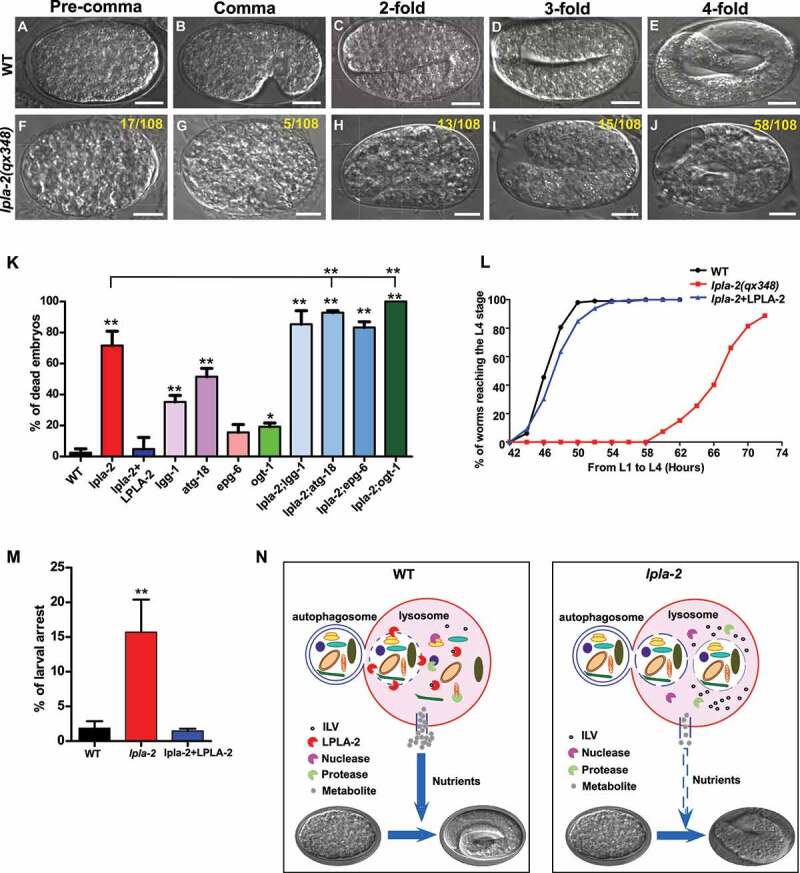
lpla-2 mutants are defective in embryonic and larval development. (A-J) DIC images of embryos in wild type (WT) and lpla-2(qx348) at different embryonic stages. The number of embryos that died at each stage is indicated at the upper right corner of each panel (F-J). Scale bars: 10 µm. (K, L and M) Embryonic lethality (K) and larval development (L and M) were quantified in the indicated strains. At least 100 embryos or 150 larvae were scored in each strain. Three independent experiments were performed, and data are shown as mean ± SD. One-way ANOVA with Tukey’s post hoc test was performed to compare mutant datasets with wild type or datasets that are linked by lines. **P < 0.0001. All other points had P > 0.05. (N) Proposed model of autophagic cargo degradation and catabolite release in wild-type and lpla-2 lysosomes. LPLA-2 promotes disintegration of the autophagic vesicles in lysosomes to release cargo for degradation. The resulting catabolites are recycled from lysosomes to provide energy and building blocks essential for embryogenesis and larval development.
Discussion
Lysosomes degrade macromolecules and recycle catabolites to maintain cellular homeostasis. Here we identified C. elegans LPLA-2, a lysosomal phospholipase A2, as a key enzyme responsible for degrading membranes in lysosomes. Loss of LPLA-2 causes enlarged lysosomes that accumulate large amounts of membranous content, and both phenotypes are suppressed by disruption of autophagy. Elevation of autophagy leads to further enlargement of lysosomes in lpla-2 mutants. This indicates that LPLA-2 is essential for resolving membranous materials delivered by autophagy in lysosomes. Our data suggest that the autophagy process is partially impaired in lpla-2 mutants. This is probably due to defective cargo release and digestion in lpla-2 lysosomes, causing impaired autophagy kinetics.
The outer membrane of the autophagosome fuses with the lysosomal membrane. During this process, autophagic cargos remain enclosed by the inner autophagosomal membrane. Breakdown of the inner membrane is essential for releasing autophagic cargos, which are then degraded by lysosomal hydrolases in the lumen. Our data suggest that LPLA-2 is essential for facilitating the release and degradation of autophagic cargos in lysosomes. We found that autophagic vesicles persist in lysosomes of lpla-2(lf) mutants and PLA2G15 knockdown cells. Moreover, lysosomal accumulation of intraluminal vesicles (ILVs), reminiscent of those in MVBs, is observed in PLA2G15 knockdown cells. These data suggest that lysosomal PLA2 family enzymes play a conserved role in the breakdown of inner autophagosomal membranes in lysosomes, and they may have a general function in lysosomal lysis of vesicular membranes. It is unclear how impaired lysis of autophagic vesicles leads to accumulation of huge membrane whorls as seen in lpla-2 lysosomes. Further studies are needed to determine whether additional lipases are involved and how they coordinate with LPLA-2 to disrupt vesicular membranes in lysosomes.
Development of C. elegans embryos, which are enclosed by impermeable eggshells, relies on degradation of maternally loaded materials, independent of external nutrients. lpla-2 mutants exhibit severe defects in both embryonic and larval development, which indicates that lysosomal degradation of autophagic substrates is essential for C. elegans development. It is conceivable that disintegration of the autophagic vesicles by LPLA-2 promotes release of cargos, which are digested by lysosomal hydrolases to provide energy and essential building blocks for embryogenesis and larval development (Figure 6N, left panel). In lpla-2 mutants, cargo release and subsequent degradation are impaired, causing nutrient deprivation that leads to embryonic lethality (Figure 6N, right panel). Moreover, the buildup of undigested membranes in lpla-2 mutants may further impair lysosome function. Consistent with this, elevation of autophagy by ogt-1 mutation further enhances lpla-2 defects, causing complete embryonic lethality in lpla-2; ogt-1 double mutants. PLA2G15 has been implicated in pulmonary surfactant metabolism and in host response to pulmonary infection and inflammation [18]. pla2g15 knockout mice older than one year exhibit autoimmune phenotypes [19]. Future investigation may address whether defects in release and/or clearance of autophagic substrates contribute to the pulmonary and immune abnormalities in PLA2G15 null mice.
Materials and methods
C. elegans strains
Strains of C. elegans were cultured and maintained using standard protocols [43]. The N2 Bristol strain was used as the wild type strain except for polymorphism mapping in which Hawaiian strain CB4856 was used. The following strains were used in this work: Linkage group I (LG I): lpla-2(qx315), lpla-2(qx348); LG II: lgg-1(bp500), epg-5(tm3425); LG III: cup-5(bp510), epg-1/atg-13(bp414), epg-6(bp242), ogt-1(bp815); LG V: epg-9(bp320), atg-18(gk378). lpla-2(qx348) was used for genetic analysis.
Transgenic animals carrying extrachromosomal arrays (qxEx) were generated using standard microinjection methods, and genome-integrated arrays (qxIs) were obtained by γ-ray irradiation to achieve stable expression from arrays with low copy numbers. The following arrays were used in this study: qxIs354 (Pced-1LAAT-1::GFP), qxIs257 (Pced-1NUC-1::CHERRY), qxIs430 (Pscav-3SCAV-3::GFP), qxIs468 (Pmyo-3LAAT-1::GFP), qxIs520 (Pvha-6LAAT-1::GFP), qxIs439 (Psemo-1GFP::TRAM-1), qxEx3928 (Psemo-1MANS::GFP), qxEx4342 (Psemo-1GFP::RAB-10), yqIs157 (Psemo-1Mito::GFP), qxIs408 (Pced-1GFP::RAB-5), qxEx5512 (Plpla-2LPLA-2::CHERRY), qxIs556 (Plpla-2LPLA-2::CHERRY), qxEx7390 (Plpla-2LPLA-2S196A), yqEx632 (Pcpl-1CPL-1::CHERRY), qxSi13 (Plgg-1GFP::LGG-1), bpIs151 (Psqst-1SQST-1::GFP). We obtained bIs1 (Pvit-2VIT-2::GFP) and pwIs281 (Ppie-1CAV-1::GFP), pwIs116 (Prme-2RME-2::GFP) from Dr. B. Grant (Rutgers University, USA), and opIs334 (Pced-1YFP::2xFYVE) from Dr. K.S. Ravichandran (University of Virginia, USA).
Isolation, mapping and cloning of lpla-2.
LAAT-1::GFP-expressing worms were treated with EMS (Sigma, M0880). Late-staged embryos or early-staged larvae at the F3 generation were examined for accumulation of enlarged LAAT-1::GFP-positive structures. From a screen that covered ~10,000 haploid genomes, 50 mutants were obtained. The recessive mutations qx348 and qx315 failed to complement each other in causing enlargement of LAAT-1::GFP-positive structures, which suggests that they affect the same gene. Both mutant alleles were backcrossed with the N2 strain at least six times before further analyses.
Single nucleotide polymorphism (SNP) mapping placed lpla-2 on linkage group I (LG I), between pkP1057 (+1.005) and pkP1058 (+2.48). 45 fosmid-mixtures that cover this region were injected into lpla-2(qx348) mutants. The enlarged lysosome phenotype in qx348 was reversed in the transgenic animals carrying the target gene. The molecular lesions in lpla-2(qx315) and lpla-2(qx348) were determined by sequencing the corresponding genomic region.
Microscopy and imaging analysis
Differential interference contrast (DIC) images were captured with an Axioimager M2 microscope (Carl Zeiss) equipped with an AxioCam monochrome digital camera (Carl Zeiss). Confocal microscopy images were taken with an inverted confocal microscope (LSM 880; Carl Zeiss, Germany) with 488 (emission filter BP 503–530) and 543 (emission filter BP 560–615) lasers. Images were processed and viewed using LSM Image Browser and ZEN software (Carl Zeiss, Germany).
Quantification of lysosome volume
Fluorescence images of embryos, larvae or adults expressing LAAT-1::GFP or SCAV-3::GFP in 10–15 z series (0.5 μm/section) were captured by spinning-disk microscopy. Serial optical sections were analyzed and the volume of LAAT-1::GFP- or SCAV-3::GFP-positive lysosomes was quantified by Velocity software (PerkinElmer, USA). At least 10 animals were quantified in each strain at each stage.
Examination of embryonic and larval development
To examine embryonic development, ~40 young adult worms (24 h post L4) were placed on an NGM plate (3 g/L NaCl, 2.5 g/L Tryptone [Oxoid, LP0042], 20 g/L Agar [Ausoble, K022], 25 mM K2HPO4,/KH2PO4 [pH 6.0], 1 mM CaCl2, 1 mM MgSO4, 5 mg/L cholesterol [Sigma, C8503]) seeded with Escherichia coli OP50 (provided by Caenorhabditis Genetics Center [University of Minnesota]) for 1.5 h. The worms were removed, and the eggs laid on the plate were followed. After 20 h, newly hatched L1 worms were counted and the percentage of hatched L1 was quantified to determine the level of embryonic lethality. Embryos (200) were followed and quantified in each strain. At least three independent experiments were performed. The average percentage of dead embryos from 3 independent experiments is shown in (Figure 6K).
To analyze larval development, more than 300 L1 worms were collected and cultured at 20°C. After 42-h incubation, newly developed L4 worms were counted and transferred every 2 h until no live larvae were present on the plate. The percentage of larvae that reached the L4 stage was determined at each time point to indicate progression of larval development (Figure 6L). The total percentage of fully developed L4 was quantified to determine the level of larval arrest. At least 3 independent experiments were performed in each strain and the average results from 3 experiments are shown in (Figure 6M).
L1 survival assay
Embryos were collected and placed in M9 buffer (3 g/L KH2PO4, 6 g/L NaH2PO4, 5 g/L NaCl, 1 mM MgSO4) at 20°C to synchronize their growth to the larvae 1 (L1) stage. The L1 worms were transferred to 96-well plates, with about 200 larvae per well. The worms were transferred to NGM plates seeded with fresh Escherichia coli OP50 every 2 d and the surviving larvae were quantified at each time point. At least 3 independent experiments were performed for each strain. The representative survival curve is shown in (Figure 4N) and the mean lifespan from 3 independent experiments is indicated in the parenthesis.
Lysosome purification and examination of LGG-1 accumulation
Lysosomes were purified from adult C. elegans using a Lysosome Isolation Kit (Sigma, LYSISO1) as described previously with minor modifications [28]. Briefly, mix-staged adult worms were collected, and worm pellets were suspended in the extraction buffer supplied in the Lysosome Isolation kit and ruptured by glass beads with a FastPrep24 Instrument (MP Biomedicals, Ohio, USA). The worm lysate was centrifuged at 23,708 g to precipitate the crude lysosomes, which were diluted in 19% Optiprep Density Gradient Medium Solution (Sigma, D1556) and further separated on a sucrose (Sigma, V900116-500 G) density gradient (from bottom to top: 27%, 19%, 16%, 8%). Following centrifugation, enrichment of lysosomes was determined by the amount of processed CPL-1 in different fractions that contained same amount of total proteins (from bottom to top: B1-4). LGG-1 accumulation was examined in the purified lysosome fraction by western blot. At least three independent experiments were performed in each strain.
Immunostaining
Mix-staged embryos were fixed and incubated with anti-LGG-1 (1: 1000; prepared by the antibody center of National Institute of Biological Sciences, Beijing, China), anti-SQST-1 (1: 1000, provided by Hong Zhang lab [Institute of Biophysics, Chinese Academy of Sciences]), anti-PGL-3 (1: 1000, provided by Hong Zhang lab [Institute of Biophysics, Chinese Academy of Sciences]) or anti-SEPA-1 (1: 1000, provided by Hong Zhang lab [Institute of Biophysics, Chinese Academy of Sciences]) antibodies in blocking buffer at 4°C overnight. After washing with PBST (PBS [0.24 g/L KH2PO4, 1.42 g/L Na2HPO4, 8 g/L NaCl, 0.2 g/L KCl] + 0.2% Tween 20 (Amresco, 777)), samples were incubated with Texas red-conjugated and/or FITC-conjugated secondary antibodies (Jackson ImmunoResearch, 112–075-003 and 115–095-003) at a 1:200 dilution for 1 h at room temperature. The samples were then washed and mounted in 15% VECTASHIELD mounting medium with DAPI (VECTOR, H-1200) and visualized using a Zeiss LSM 880 inverted confocal microscope (Carl Zeiss, Germany).
Cell culture and transfection
HeLa cells (1101HUM-PUMC000011, provided by National Infrastructure of Cell line Resources [Chinese Academy of Sciences and Peking Union Medical College]) were cultured in DMEM (Hyclone, SH30022.01B) with 10% fetal bovine serum (FBS; Hyclone, SH30084.03) supplemented with 50 μg/ml penicillin-streptomycin (Gibco, 15,140,122). siRNAs were purchased from GenePharma (PLA2G15 siRNA1: CGAAAGCUACUUCACAAUCTT; PLA2G15 siRNA2: GCGAGAUGAUCGAGGAGAUTT; PLA2G15 siRNA3: ACACUGGAUGGCAAGAAUGTT). Cells were transfected with siRNAs using Lipofectamine RNAi MAX (Life Technologies, 13,778,150) for 72 h according to the manufacturer’s instructions.
Transmission Electron Microscopy (TEM)
Adult C. elegans (day 3 of adulthood) were rapidly frozen using a high-pressure freezer EM ICE (Leica, Germany). Freeze-substitution was carried out in anhydrous acetone containing 1% osmium tetroxide. The samples were kept sequentially at −90°C for 72 h, −60°C for 10 h and −30°C for 10 h and were finally brought to 0°C for 6 h in a freeze-substitution unit EM AFS2 (Leica, Germany). The samples were washed three times (20 min each time) in fresh anhydrous acetone and were gradually infiltrated with Embed-812 resin (Electron Microscopy Sciences, 14,120) in the following steps: 1 (resin):3 (acetone) for 3 h; 1:1 for 5 h; 3:1 overnight, and 100% resin for 4 h. Samples were then kept overnight and embedded at 60°C for 48 h. The fixed samples were cut into 70 nm sections with a microtome EM UC7 (Leica, Germany) and electron stained with uranyl acetate and lead citrate. Sections were observed with a HT7800 (HITACHI, Japan) operating at 80 kV.
Monolayers of HeLa cells were fixed in 0.1 M PBS containing 2.5% glutaraldehyde at 4°C for 2 h. Fixed samples were rinsed with PBS and post-fixed with 1% OsO4 at 4°C for 1 h. The samples were dehydrated in ethanol and embedded in EMbed812. Embedded samples were sectioned using an UC6 ultramicrotome equipped with a 45° diamond knife (Diatome, DU4530) to obtain 70-nm ultrathin sections. The grids were stained at RT with aqueous uranyl acetate and Reynolds lead citrate before imaging. Imaging was performed at 80 kV on a JEM-1400 (JEOL, USA) transmission electron microscope.
Plasmid construction
To construct Plpla-2LPLA-2::CHERRY, CHERRYwas firstly inserted into pPD49.26 vectors (Addgene, 1686; deposited by Andrew Fire lab [Stanford University]) through KpnI and SacI sites, then ligating the lpla-2 promoter and the genomic sequence of lpla-2 amplified using primers PYL184 and PYL185 through the NheI and KpnI sites. To generate Plpla-2LPLA-2S196A::CHERRY, site-directed mutagenesis was performed to introduce the mutation into Plpla-2LPLA-2::CHERRYby primers PYL396/PYL397.
Statistical analysis
The standard deviation (SD) was used as y-axis error bars for bar charts plotted from the mean value of the data. Data derived from different genetic backgrounds were compared by Student’s two-tailed unpaired t test, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test, or two-way ANOVA followed by the Bonferroni post hoc test, as indicated in the figure legends. Data were considered statistically different at P < 0.05. P < 0.05 is indicated with single asterisks and P < 0.0001 with double asterisks (0.001 in two-way ANOVA analysis).
Supplementary Material
Acknowledgments
We thank Drs. B. Grant (Rutgers University, USA) and K.S. Ravichandran (University of Virginia, USA) for reporter strains, Dr. Hong Zhang (Institute of Biophysics, Chinese Academy of Sciences) for antibodies and Dr. Isabel Hanson for editing services. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40OD010440).
Funding Statement
This work was supported by the National Natural Science Foundation of China [91754203, 3163001]; Strategic Priority Research Program of the Chinese Academy of Sciences [XDB19000000]; Ministry of Science and Technology of the People’s Republic of China [2016YFA0500203].
Disclosure of potential conflicts of interest
The authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Carlsson SR, Simonsen A.. Membrane dynamics in autophagosome biogenesis. J Cell Sci. 2015. Jan 15;128(2):193–205. [DOI] [PubMed] [Google Scholar]
- [2].Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012. Jan 10;22(1):R29–34. [DOI] [PubMed] [Google Scholar]
- [3].Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009. Jul;10(7):458–467. [DOI] [PubMed] [Google Scholar]
- [4].Berg TO, Fengsrud M, Stromhaug PE, et al. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998. Aug 21;273(34):21883–21892. [DOI] [PubMed] [Google Scholar]
- [5].Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell. 2016. Dec 1;3(12):588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Longatti A, Tooze SA. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009. Jul;16(7):956–965. [DOI] [PubMed] [Google Scholar]
- [7].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013. Dec;14(12):759–774. [DOI] [PubMed] [Google Scholar]
- [8].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011. Nov 11;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [9].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011. Jan 20;469(7330):323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. [DOI] [PubMed] [Google Scholar]
- [11].Corona AK, Jackson WT. Finding the middle ground for autophagic fusion requirements. Trends Cell Biol. 2018. Nov;28(11):869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020;6(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Epple UD, Suriapranata I, Eskelinen EL, et al. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J Bacteriol. 2001. Oct;183(20):5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Teter SA, Eggerton KP, Scott SV, et al. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J Biol Chem. 2001. Jan 19;276(3):2083–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Epple UD, Eskelinen EL, Thumm M. Intravacuolar membrane lysis in Saccharomyces cerevisiae. Does vacuolar targeting of Cvt17/Aut5p affect its function? J Biol Chem. 2003. Mar 7;278(10):7810–7821. [DOI] [PubMed] [Google Scholar]
- [16].Ramya V, Rajasekharan R. ATG15 encodes a phospholipase and is transcriptionally regulated by YAP1 in Saccharomyces cerevisiae. FEBS Lett. 2016. Sep;590(18):3155–3167. [DOI] [PubMed] [Google Scholar]
- [17].Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016. Nov 25;354(6315):1036–1041. [DOI] [PubMed] [Google Scholar]
- [18].Shayman JA, Tesmer JJG. Lysosomal phospholipase A2. Biochim Biophys Acta Mol Cell Biol Lipids. 2019. Jun;1864(6):932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shayman JA, Kelly R, Kollmeyer J, et al. Group XV phospholipase A(2), a lysosomal phospholipase A(2). Prog Lipid Res. 2011. Jan;50(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abe A, Shayman JA. Purification and characterization of 1-O-acylceramide synthase, a novel phospholipase A2 with transacylase activity. J Biol Chem. 1998. Apr 3;273(14):8467–8474. [DOI] [PubMed] [Google Scholar]
- [21].Abe A, Hiraoka M, Shayman JA. Positional specificity of lysosomal phospholipase A2. J Lipid Res. 2006. Oct;47(10):2268–2279. [DOI] [PubMed] [Google Scholar]
- [22].Glukhova A, Hinkovska-Galcheva V, Kelly R, et al. Structure and function of lysosomal phospholipase A2 and lecithin: cholesterolacyltransferase. Nat Commun. 2015. Mar 2;6(1):6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abe A, Hiraoka M, Wild S, et al. Lysosomal phospholipase A2 is selectively expressed in alveolar macrophages. J Biol Chem. 2004. Oct 8;279(41):42605–42611. [DOI] [PubMed] [Google Scholar]
- [24].Hiraoka M, Abe A, Lu Y, et al. Lysosomal phospholipase A2 and phospholipidosis. Mol Cell Biol. 2006. Aug;26(16):6139–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schneider BE, Behrends J, Hagens K, et al. Lysosomal phospholipase A2: a novel player in host immunity to Mycobacterium tuberculosis. Eur J Immunol. 2014. Aug;44(8):2394–2404. [DOI] [PubMed] [Google Scholar]
- [26].Kolter T, Winau F, Schaible UE, et al. Lipid-binding proteins in membrane digestion, antigen presentation, and antimicrobial defense. J Biol Chem. 2005. Dec 16;280(50):41125–41128. [DOI] [PubMed] [Google Scholar]
- [27].Gilleron M, Lepore M, Layre E, et al. Lysosomal lipases PLRP2 and LPLA2 process mycobacterial multi-acylated lipids and generate T cell stimulatory antigens. Cell Chem Biol. 2016. Sep 22;23(9):1147–1156. [DOI] [PubMed] [Google Scholar]
- [28].Liu B, Du H, Rutkowski R, et al. LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science. 2012. Jul 20;337(6092):351–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hiraoka M, Abe A, Shayman JA. Cloning and characterization of a lysosomal phospholipase A2, 1-O-acylceramide synthase. J Biol Chem. 2002. Mar 22;277(12):10090–10099. [DOI] [PubMed] [Google Scholar]
- [30].Hiraoka M, Abe A, Shayman JA. Structure and function of lysosomal phospholipase A2: identification of the catalytic triad and the role of cysteine residues. J Lipid Res. 2005. Nov;46(11):2441–2447. [DOI] [PubMed] [Google Scholar]
- [31].Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010. Jun 11;141(6):1042–1055. [DOI] [PubMed] [Google Scholar]
- [32].Tian E, Wang F, Han J, et al. epg-1 functions in autophagy-regulated processes and may encode a highly divergent Atg13 homolog in C. elegans. Autophagy. 2009. Jul;5(5):608–615. [DOI] [PubMed] [Google Scholar]
- [33].Lu Q, Yang P, Huang X, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011. Aug 16;21(2):343–357. [DOI] [PubMed] [Google Scholar]
- [34].Guo B, Liang Q, Li L, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014. Dec;16(12):1215–1226. [DOI] [PubMed] [Google Scholar]
- [35].Zhang Y, Yan L, Zhou Z, et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. Elegans Cell. 2009. Jan 23;136(2):308–321. [DOI] [PubMed] [Google Scholar]
- [36].Liang Q, Yang P, Tian E, et al. The C. elegans ATG101 homolog EPG-9 directly interacts with EPG-1/Atg13 and is essential for autophagy. Autophagy. 2012. Oct;8(10):1426–1433. [DOI] [PubMed] [Google Scholar]
- [37].Sato K, Sato M, Audhya A, et al. Dynamic regulation of caveolin-1 trafficking in the germ line and embryo of Caenorhabditis elegans. Mol Biol Cell. 2006. Jul;17(7):3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Grant B, Hirsh D, Kimble J. Receptor-mediated endocytosis in the caenorhabditis elegans oocyte. Mol Biol Cell. 1999. Dec;10(12):4311–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Treusch S, Knuth S, Slaugenhaupt SA, et al. Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc Natl Acad Sci U S A. 2004. Mar 30;101(13):4483–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sun T, Wang X, Lu Q, et al. CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy. 2011. Nov 7;7(11):1308–1315. [DOI] [PubMed] [Google Scholar]
- [41].Miao R, Li M, Zhang Q, et al. An ECM-to-nucleus signaling pathway activates lysosomes for C. elegans larval development. Dev Cell. 2020. Jan 6;52(1):21–37 e5. [DOI] [PubMed] [Google Scholar]
- [42].Liu J, Li M, Li L, et al. Ubiquitination of the PI3-kinase VPS-34 promotes VPS-34 stability and phagosome maturation. J Cell Biol. 2018. Jan 2;217(1):347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974. May;77(1):71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.