

ABSTRACT
Circular RNAs (circRNAs) are a class of non-coding RNA molecules that are associated with not only normal physiological functions but also various diseases, including cardiac diseases such as myocardial infarction (MI). The present study explored the potential role of circRNA_0007059 (circ_0007059) during MI pathogenesis using in vitro studies. Microarray and quantitative PCR analyses demonstrated elevated circ_0007059 expression and downregulated miR-378 and miR-383 expression in H2O2-treated mice cardiomyocytes and infarcted hearts of MI mouse model as compared those in relevant controls. Moreover, circ_0007059 knockdown improved cardiomyocyte viability after H2O2 treatment as revealed by the CCK-8 and colony formation assays. Flow cytometry and caspase activity assays demonstrated that circ_0007059 suppressed H2O2-induced cardiomyocyte apoptosis. Enzyme-linked immunosorbent assays and Western blotting revealed that inflammatory cytokine (interleukin-1β, interleukin-18 and C-C motif chemokine ligand 5) expression was induced by H2O2 treatment and that circ_0007059 repressed H2O2-induced inflammation. Bioinformatics analyses and dual-luciferase reporter assays showed that circ_0000759 acts as a miR-378 and miR-383 sponge. Furthermore, the upregulation or suppression of miR-378 and miR-383 expression in H2O2-treated cardiomyocytes had similar effects on the apoptosis and inflammation of cardiomyocytes as that of circ_0007059 knockdown or overexpression, respectively. Additionally, lentiviral shRNA-circ_0007059 administration to mice with MI considerably reduced the size of infarcted regions and promoted cardiac activity. Collectively, our findings suggest that circ_0007059 expression is upregulated in mice cardiomyocytes in response to oxidative stress and cardiac tissues of MI mouse model, suggesting its involvement in the pathogenesis of MI by targeting miR-378 and miR-383.
KEYWORDS: Myocardial infarction, circ_0007059, miR-378, miR-383, apoptosis, inflammation
Introduction
Myocardial infarction (MI) is a major cause of unexpected deaths worldwide, necessitating the discovery of novel therapeutics. Despite the use of conventional treatments, MI is still associated with high death rates and long-term complications, including heart failure [1]. Acute MI leads to aggressive cardiovascular output after myocardial ischemia, cardiac surgery, and circulatory arrest in coronary heart diseases [2]. Nonetheless, the molecular mechanisms underlying MI-induced injuries are complex and require thorough investigation.
Circular RNAs (circRNAs) are non-coding RNAs that are capable of forming a covalently closed continuous loop that has been universally observed in different organisms [3]. circRNAs are abundantly expressed across eukaryotes [4] and are often conserved, especially between human and mice. Similar to many mRNAs, circRNAs are often expressed in a developmental stage-specific manner [5], suggesting their possible involvement in various biological functions. A recent report has indicated that the Cdr1as circRNA facilitates MI by regulating the expression of miR-7a target genes [6]. Zhou et al. have reported that the autophagy-related circRNA (ACR) represses autophagy and MI by regulating Pink1-mediated phosphorylation of FAM65B [7]. Wang et al. have shown that the mitochondrial fission and apoptosis-related circRNA (MFACR) modulates mitochondrial apoptosis and fission in the heart by directly targeting miR-652-3p, which represses chondriokinesis and cardiomyocyte death by inhibiting the translation of mitochondrial protein 18 [8]. Another study has demonstrated that the hsa_circ_0007059 circRNA represses proliferation and the epithelial–mesenchymal transition of lung cancer cells by suppressing miR-378 expression [9]. However, the role of hsa_circ_0007059 in MI pathogenesis has not yet been reported.
MicroRNAs (miRs) are endogenous single-stranded non-coding RNA molecules (~19–22 nucleotides) that are crucial modulators of pathologic and physiologic processes. Moreover, miRs are important regulators of a variety of cellular processes; they act by post-transcriptionally destabilizing or inhibiting the translation of target mRNAs. Several miRs are expressed in a tissue-specific manner; for example, miR-1, miR-133a, miR-208a/b, and miR-499 are abundantly expressed in myocardium. Increasing evidence indicates that miRNAs are relevant to heart development and certain cardiovascular diseases, including MI [10]. In particular, miR-378 is reported to participate in MI by attenuating ischemia-induced apoptosis through the suppression of caspase-3 expression in cardiomyocytes [11]. Another miRNA, miR-383, is thought to influence proliferation and apoptosis of glioma cells [12]. Interestingly, miR-383 expression suppression ameliorates post focal cerebral ischemia injury [13] and is also thought to play anti-inflammatory roles in response to homocysteine-induced endothelial injury in rat coronary artery through its interaction with IL1R2 [14].
In this study, a MI mouse model and H2O2-induced cardiomyocyte injury cell model have been established to study the role(s) of circ_0007059 in myocardial dysfunction as well as MI-induced apoptosis and inflammation. Our bioinformatics predictions and dual-luciferase reporter assays have identified that circ_0007059 is an important competing endogenous RNA (ceRNA) that sponges miR-378 and miR-383 expression to induce cardiomyocyte inflammation and apoptosis, respectively. Thus, circ_0007059 has the potential to serve as a promising target for the development of MI therapeutics in the future.
Materials and methods
Animals
Healthy adult male BALB/c mice with body weights ranging from 16 to 20 g were bought from Vitalriver Animal Co., Ltd. (Beijing, P. R. China). Mice were housed in standard animal room conditions (humidity, 55 ± 5%; temperature, 23 ± 1°C) and provided free access to water and food for one week prior to initiation of the experiments. All the experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011). This study was approved by Heilongjiang Provincial Hospital, Heilongjiang, P. R. China.
Establishment of the MI model and lentivirus administration
Mice were anesthetized with tribromoethanol (20 mg/kg) through ventilation. Thoracic surgery was conducted through the 4th intercostal space. The main pulmonary artery and aorta ascendens were clamped. The animals were classified into one of the four groups: (1) control group: mice were not subjected to left anterior descending (LAD) ligation or injection; (2/3) lentiviral-shRNA-negative control (NC)/lentiviral-shRNA-circ_0007059 group: mice were injected with lentiviral-shRNA-NC/lentiviral-shRNA-circ_0007059 (400 nmol/kg in a final volume of 50 µL) into the left chest cavity at the cardiac apex. The arteries were occluded for 10s after injection; (4) MI model group: mice were administered 80 µL saline injection and subsequently MI was induced by LAD ligation with 8–0 looping sutures.
Lentiviral transfection
Post phosphorylation and annealing, the circ_0007059 shRNA and scrambled NC shRNA oligonucleotides were cloned into the pLVX-puro vector (Clontech Laboratories, Inc., CA, USA), subsequently referred to as pLVX-si-circ_0007059 and pLVX-siNC, respectively. si-circ_0007059 and siNC lentiviral particles were prepared by transfecting 293 T cells (Invitrogen, Thermo Fisher Scientific Corp., MA, USA) with the pLVX-si-circ_0007059 and pLVX-siNC vectors, respectively, as well as pMD2.G and psPAX2.
Infarct size detection
Heart samples were collected three days after the MI model was established. After washing off the blood, left ventricles were sectioned to obtain 1-mm-thick slices and stained with 1% 2,3,5-triphenyltetrazolium chloride for 30 min at 37°C. The infarcted region displayed no staining, whereas the normal region appeared red. Image analysis was performed using the Image ProPlus 5.0 software, and the results are expressed as the percentage of left ventricular volume per sample.
Determination of cardiac injury
Cytosolic enzyme creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) levels were measured using an auto-biochemical analyzer (Hitachi Ltd., Tokyo, Japan). Serum troponin activity was detected using enzyme-linked immunosorbent assay (ELISA) commercial kits.
Cardiomyocytes cultivation
Ventricular cardiomyocytes were harvested from mice (1–3 d) and cultured [15,16]. In brief, hearts were removed under aseptic conditions, and ventricle specimens were cut into small pieces prior to trypsin digestion (0.25%). The separated cells were resuspended in DMEM containing 10% FBS, centrifuged for 5 min (1,000 rpm), and then resuspended for 2 h. The obtained cells were cultivated in a non-coated flask. Medium containing 0.1 mM bromodeoxyuridine (BrdU) was added to the flask for clearing out non-cardiomyocytes. Cardiomyocytes were cultured in a 5% CO2 at 37°C.
Cell infection and transfection
For lentiviral infection, cardiomyocytes were incubated with polybrene (5 μg/mL) and lentiviral particles. After 6 h, the medium was replaced, and the cells were stored for subsequent experiments. The miR-378 mimic, miR-383 mimic, miR-378 inhibitor, miR-383 inhibitor, NC mimic, and NC inhibitor were synthesized at Genscript Co., Ltd. (Beijing, P. R. China). The vectors were transfected into cardiomyocytes using Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific Corp). After 4 h, the transfected cells were harvested and used for subsequent experiments.
Cell proliferation and colony formation assays (CFA)
The proliferative capacity of indicated cells was assessed using a CCK-8 assay according to the manufacturer’s guidelines. Briefly, indicated cells were seeded into 96-well plates. After incubation, the cells were supplemented with CCK-8 solution (10 μL per), and cultivated for an additional 120 min at 37°C. The optical density at 450 nm was determined using an Infinite M200 (Tecan Group Ltd., Männedorf, Switzerland). For CFA, the indicated cells were cultivated in 6-well plates for 7 d, fixed in 4% formaldehyde for 20 min, and stained with 1% crystal violet solution. Excess staining was washed off gently with either PBS or DPBS. The stained colonies were then imaged using a bright field phase-contrast microscope attached to a CCD camera.
Flow cytometry (FC)
Cardiomyocyte death was assessed using an apoptosis detection kit. Briefly, post transfection, the binding buffer (20 µL) was added to harvested cardiomyocytes and the cells were incubated for 20 min in the dark with Annexin V-FITC (10 µL) and propidium iodide (PI; 5 µL). Cell death was evaluated using FC.
Western blotting (WB)
RIPA buffer supplemented with a protease inhibitor cocktail (Roche Applied Science, Penzberg, Germany) was used to lyse the cells. Protein concentration was measured using a BCA Kit. The digested protein samples were resolved using 10% SDS polyacrylamide gel electrophoresis (Bio-Rad Laboratories, Inc., CA, USA) and then transferred to PVDF membranes (0.45 µm, Merck Millipore, MA, USA). The membranes were then blocked with 5% BSA in TBST or PBST at 25°C for 60 min, incubated with primary antibodies at 4°C overnight, and subsequently incubated with goat anti-rabbit/mouse IgG for 60 min at 25°C. The primary and secondary antibodies used in this experiment included: caspase-3 antibody (1:2000, ab4051, Abcam, Cambridge, UK), cleaved-caspase-3 antibody (1:500, ab2302, Abcam), caspase-9 antibody (1:2000, ab25758, Abcam), cleaved-caspase-9 antibody (1:1000, ab2324, Abcam), interleukin (IL)-1β antibody (1:1000, ab9722, Abcam), IL-18 antibody (1:1000, ab71495, Abcam), C-C motif chemokine ligand 5 (CCL-5) antibody (1:1000, ab9679, Abcam), nuclear factor erythroid 2-related factor 2 (Nrf2) antibody (1:2000, ab137550, Abcam), heme oxygenase 1 (HO-1) antibody (1:2000, ab13248, Abcam,), GAPDH antibody (1:5000, ab8245, Abcam), and HRP-conjugated goat anti-mouse/rabbit antibody (1:5000, ab97023/ab6721, respectively, Abcam). Immunoreactivity was assayed using the Maximum Sensitivity Substrate Kit (Thermo Fisher Scientific Corp) and a C-DiGit Blot Scanner (LI-COR Biosciences, NE, USA).
RNA isolation and quantitative PCR (qPCR)
Total RNA was isolated from cultivated cardiomyocytes using TRIzol (Invitrogen, Thermo Fisher Scientific Corp). RNA was quantified using SYBR Green-based qPCR kit (Roche Diagnostics GmbH, Mannheim, Germany). GADPH was used as an internal control. The thermal cycling program used for qPCR (20 μL) was performed as follows: 10 min at 95°C and 40 cycles of 15 sec at 95°C, 30 sec at 60°C, and 30 sec at 72°C using a Light Cycler 480 instrument (Roche Diagnostics GmbH). The ΔΔCT method was employed to calculate the target mRNA fold change values by normalization to the internal control relative to the calibrator (average of the control samples).
ELISA
The indicated cells were homogenized and maintained on ice. The levels of inflammatory cytokines and chemokines, including IL-1β, IL-18, and CCL-5, were detected using specific ELISA kits (Thermo Fisher Scientific Corp) in accordance with the manufacturer’s instructions.
Reactive oxygen species (ROS) production measurement
ROS generation was measured using a fluorescent probe, DCFH-DA. After incubation with Propionibacterium acnes for the indicated times, the cells were further incubated using 10 μM of DCFH-DA in the dark for 30 min at 37°C. An Olympus BX51 fluorescence microscope (Olympus Corporation, Tokyo, Japan) was used to measure fluorescence intensity at excitation and emission wavelengths of 488 nm and 525 nm, respectively.
Measurement of superoxide dismutase (SOD) activity
The protein concentration in the cell lysates was determined using a standard BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, P. R. China). Activity of anti-oxidative enzyme SOD was measured using the corresponding assay kits from the Beyotime Institute of Biotechnology (Shanghai, P. R. China). The enzyme activities were expressed as units per milligram tissue (U/mg).
Bioinformatics prediction
The prediction algorithm starBase (starbase.sysu.edu.cn) and Circular RNA Interactome (circinteractome.nia.nih.gov/index.html) were used to identify the targets of circ_0007059. The targets predicted were listed based on target prediction efficacy and likelihood of conserved targeting [17].
Dual-luciferase reporter assay (DLRA)
After amplification, the entire circ_0007059-binding region was inserted into the GV126 vector downstream of the luciferase gene. Oriented mutagenesis was conducted based on the predicted circ_0007059-binding sites in miR-378 and miR-383 to exclude the binding sites. Renilla luciferase plasmid pRL-TK vector (TaKaRa, Japan) was used as a control to monitor the transfection efficiencies. miR-378/miR-383 mimics and circ_0007059-wild-type (WT) or mutant (MU) expression vectors were co-transfected with the luciferase reporter vector in cardiomyocytes. After 36 h of incubation, cells were lysed and Firefly luciferase activity was quantified. Renilla luciferase was used for normalization.
Data analysis
The results are presented as the mean ± standard deviation (SD). A two-tailed t-test or one-way ANOVA with Tukey’s post hoc test was performed to investigate statistical differences between two groups or multiple groups, respectively. The differences were considered to be significant at P < 0.05.
Results
Circ_0007059 expression was upregulated in MI mouse model and H2O2-treated cardiomyocytes
To investigate the roles of circRNAs in MI development, we screened for dysregulated circRNA expression in the infarct of MI mice and compared it with that in the normal mice using microarrays. Our results revealed that the expression of several circRNAs, including circ_0007059, was markedly lower in MI mice infarcts than that in the normal mice tissues (Figure 1(a)). Subsequently, we evaluated the circ_0007059 expression in murine cardiac samples three days after the MI model establishment. Total RNA was extracted from the remote and marginal regions of the infarcts to measure the variation in circ_0007059 levels in infarcted heart regions. qPCR analyses revealed that circ_0007059 expression was upregulated in the infarcted murine heart regions relative to that in the normal group, whereas circ_0007059 expression in the border and distant regions was similar in these groups (Figure 1(b)). H2O2 has been reported to cause cardiomyocyte injury in a time- and concentration-dependent manner [15] and was therefore used to establish an in vitro MI model in cardiomyocytes. qPCR analyses revealed that circ_0007059 expression was elevated in H2O2-treated cardiomyocytes as compared to that in non-treated controls (Figure 1(c)). Overall, these findings indicate that the expression of circ_0007059 is augmented in both in vitro and in vivo models of MI in mice.
Figure 1.
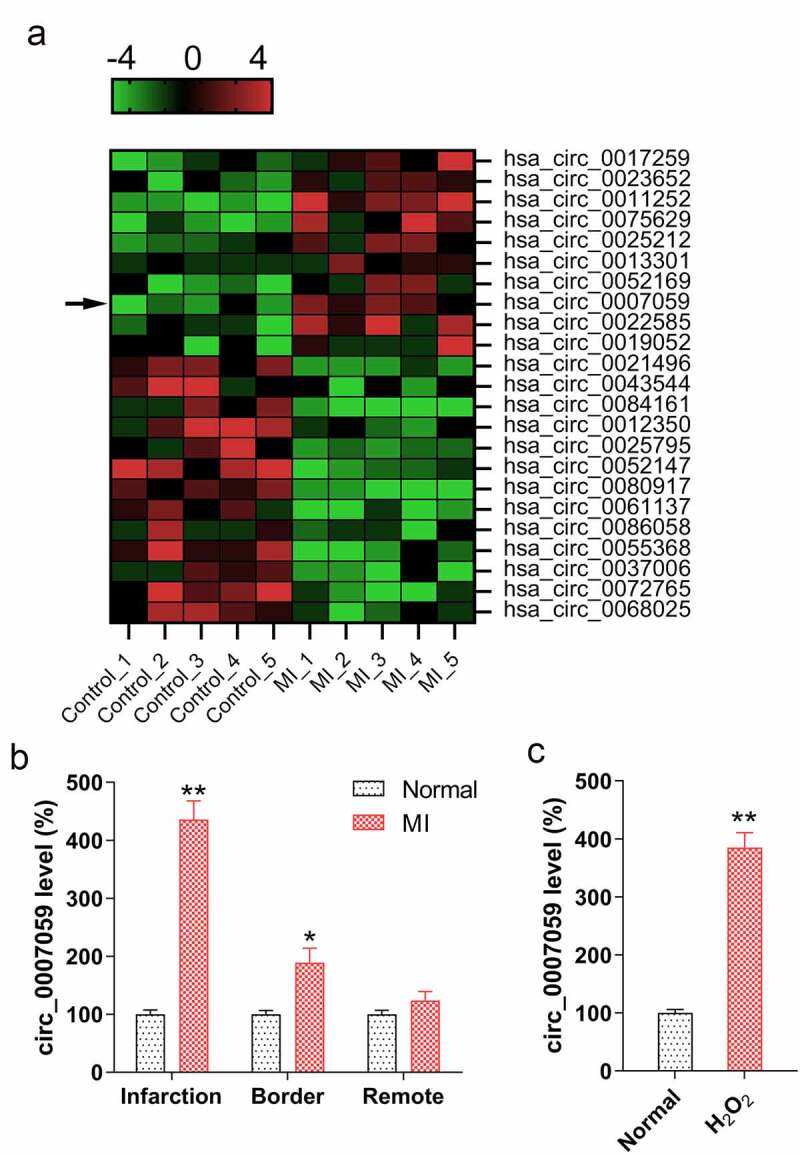
Circ_0007059 expression is upregulated in H2O2-treated cardiomyocytes and MI mice models. (a) Microarray analysis showing genes that are differentially expressed between cardiac tissue from MI mice (n = 5) and normal mice (n = 5). (b) qPCR analysis of circ_0007059 expression in different regions of infarcted hearts of MI mouse models as compared to that in control animals. (c) qPCR analysis for examining circ_0007059 expression in isolated cardiomyocytes treated with 100 μM H2O2. *P < 0.05 and **P < 0.01 versus the control group (CG).
Circ_0007059 knockdown reduces infarcts in MI mice
To explore the effects of circ_0007059 expression on MI mouse heart activity, we evaluated the changes in the cardiac activity and infarct size during MI after circ_0007059 knockdown. The results revealed successful knockdown of circ_0007059 in the cardiac tissue of MI mice (Figure 2(a)). Further measurement of the myocardial infarct area demonstrated that circ_0007059 silencing significantly decreased the infarct size in MI mice as compared to that in the control mice with WT expression of circ_0007059 (Figure 2(b)). The left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF) indicators of cardiac function were measured to determine the effects of circ_0007059 knockdown on cardiac function. The results of echocardiography revealed that LVFS and LVEF were markedly reduced in the MI mice compared to those in normal mice. However, circ_0007059 knockdown elevated LVFS and LVEF in MI mice as compared to that in MI mice with WT expression of circ_0007059 (Figure 2(c,d)). We next investigated the levels of cardiac injury-related enzymes and proteins in the blood (e.g. CK-MB, LDH, and troponin) of MI mice with or without knockdown of circ_00007059. Accordingly, the serum CK-MB activity, LDH activity, and troponin level were examined to evaluate the effect of circ_0007059 downregulation on the cardiac injury-associated enzymes and proteins. The results showed that MI models demonstrated an increase in CK-MB activity, LDH activity, and troponin level in the serum of MI mice, whereas circ_0007059 downregulation led to a significant reduction in all these three indicators (Figure 2(e-g)). These results suggest that circ_0007059 knockdown is an effective strategy in recovering the impaired heart activity induced post MI.
Figure 2.
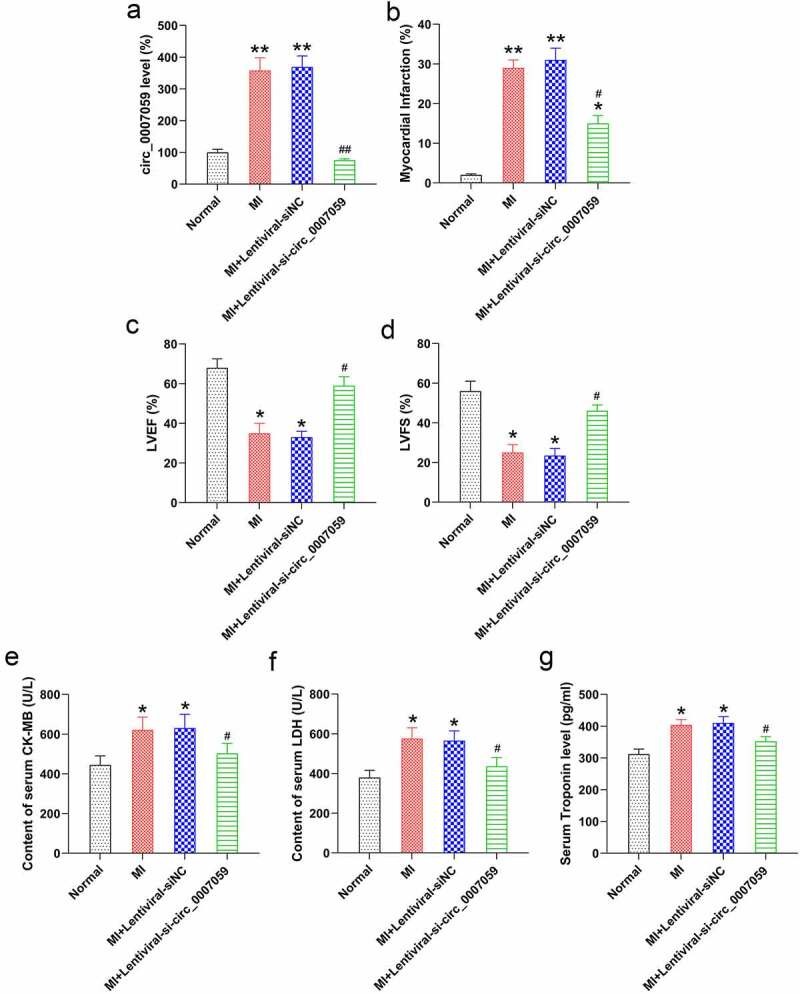
Circ_0007059 knockdown suppresses MI injury in vivo. MI mice were injected with lentiviral vectors bearing shRNA-circ_0007059 or siNC. (a) qPCR analysis of circ_0007059 expression in the heart tissues of MI mouse model. (b) The percentage of the myocardial infarct area for the normal control and MI mice. (c) LVEF in sham-operated and MI mice. (d) LVFS in sham-operated and MI mice. (e, f) Effect of circ_0007059 downregulation on serum CK-MB (CK) and LDH activity after establishing the MI model. (g) Effect of circ_0007059 downregulation on serum troponin content after establishing MI model. *P < 0.05 and **P < 0.01 versus the CG (sham); #P < 0.05 and ##P < 0.01 versus the MI group.
Circ_0007059 knockdown inhibited the viability and promoted the apoptosis and inflammation of H2O2-treated cardiomyocytes
Considering that H2O2 administration inhibits cardiomyocyte survival, we next evaluated whether circ_0007059 knockdown can counteract H2O2-induced cell injury. Consequently, the silencing of circ_0007059 in H2O2-induced cardiomyocytes was verified using qPCR (Figure 3(a)). Subsequently, the CCK-8 assay (Figure 3(b)) and CFA (Figure 3(c)) revealed that the cell viability and proliferation were significantly reduced in cardiomyocytes upon treatment with 100 mM H2O2, and circ_0007059 knockdown significantly restored these defects in viability and growth induced by H2O2.
Figure 3.
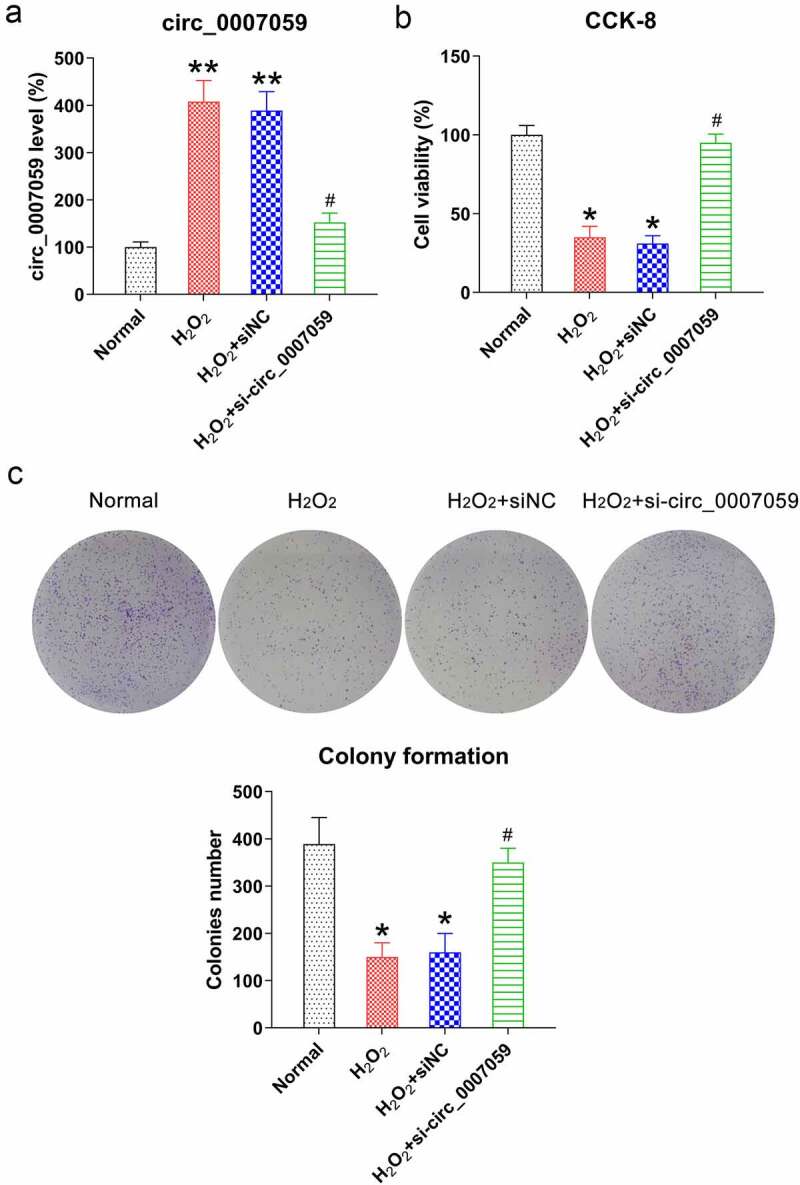
Circ_0007059 knockdown restores cardiomyocyte viability. Cardiomyocytes were infected with lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. (a) qPCR evaluation of the expression of circ_0007059 in cardiomyocytes. (b) CCK-8 assay examining the cell viability, and (c) CFA examining cell growth rate of cardiomyocytes. *P < 0.05 and **P < 0.01 versus the CG; #P < 0.05 versus the H2O2 group.
Since reduced circ_0007059 expression impairs the viability of H2O2-treated cardiomyocytes and MI models, we hypothesized that circ_0007059 silencing mediates the H2O2-induced cell death in cardiomyocytes. FC and Annexin V-FITC/PI staining revealed increased apoptosis of H2O2-exposed cardiomyocytes as compared to that of untreated control cells. However, circ_0007059 knockdown remarkably reduced levels of H2O2-induced cellular apoptosis (Figure 4(a)). WB studies were then conducted to quantify the cleaved-caspase-3 and −9 levels, demonstrating that H2O2 treatment indeed induced the cleavage of caspase-3 and −9, whereas circ_0007059 silencing alleviated the levels of cleaved-caspase-3 and −9 (Figure 4(b)). These results suggest that circ_0007059 silencing protects cardiomyocytes against oxidative stress-induced apoptosis.
Figure 4.
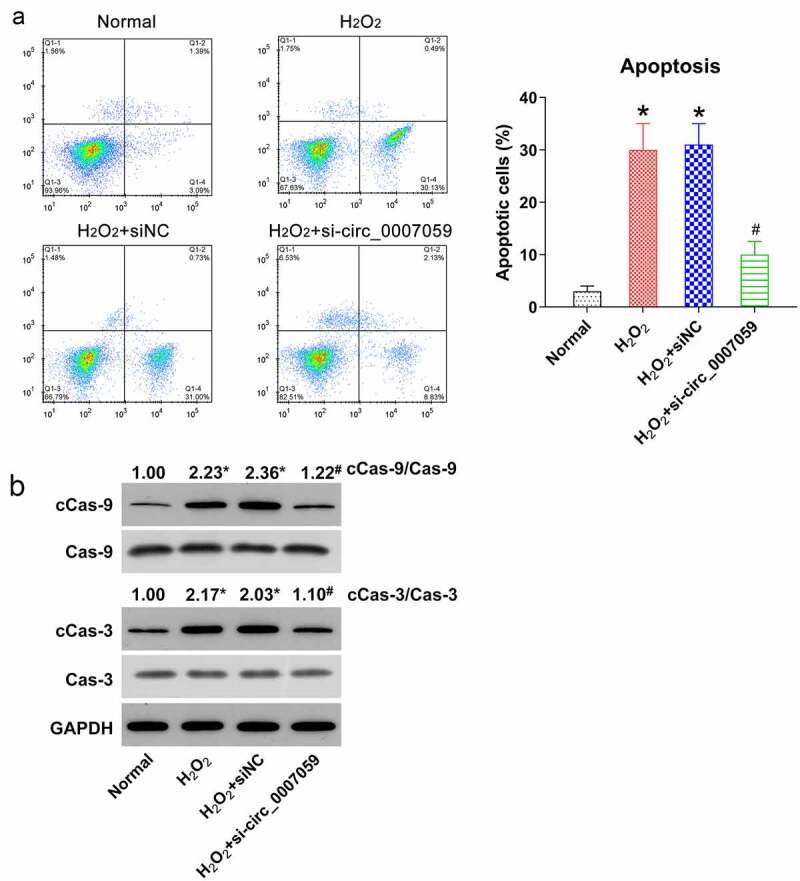
Circ_0007059 knockdown inhibits H2O2-induced apoptosis of cardiomyocytes. Cardiomyocytes were infected with lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. (a) Annexin V-FITC/PI staining and FC were employed to quantify the proportion of apoptotic cardiomyocytes. (b) WB quantification of the levels of caspase-3/9 and cleaved-caspase-3/9. *P < 0.05 versus the CG; #P < 0.05 versus the H2O2 group.
Persistent inflammation can contribute to adverse left ventricular remodeling post MI, making inflammation an important therapeutic target for improving outcomes post MI [18]. Hence, we next examined the potential role of circ_0007059 in regulating H2O2-induced cellular inflammation in cardiomyocytes. Accordingly, WB (Figure 5(a)) and ELISA (Figure 5(b–d)) were employed to examine the expression and release of MI-associated inflammatory cytokines and chemokines such as IL-1β, IL-18, and CCL-5 [18]. Our results revealed that H2O2 treatment induced the expression of IL-1β, IL-18, and CCL5. However, silencing circ_0007059 led to notable reduction in the levels of the investigated cytokines and chemokines as compared to those in the siNC group. Oxidative stress has been reported to be involved in the progression of MI [19]. Hence, we examined ROS production, as well as anti-oxidative SOD enzyme activity and Nrf2/HO-1 activation in cardiomyocytes subjected to H2O2 stimulation and/or circ_0007059 suppression. The results revealed that following H2O2 treatment, the production of ROS was evidently increased (Figure 5(e)), whereas the SOD activity and the Nrf2/HO-1 signal transduction were diminished as compared to those in untreated cells (Figure 5(f,g)). It was also found that circ_0007059 downregulation partially abolishes the effect of H2O2-mediated oxidative reactions (Figure 5(e–g)). Collectively, these data demonstrate that circ_0007059 plays a crucial role in the suppression of H2O2-induced inflammation and oxidative stress in cardiomyocytes.
Figure 5.
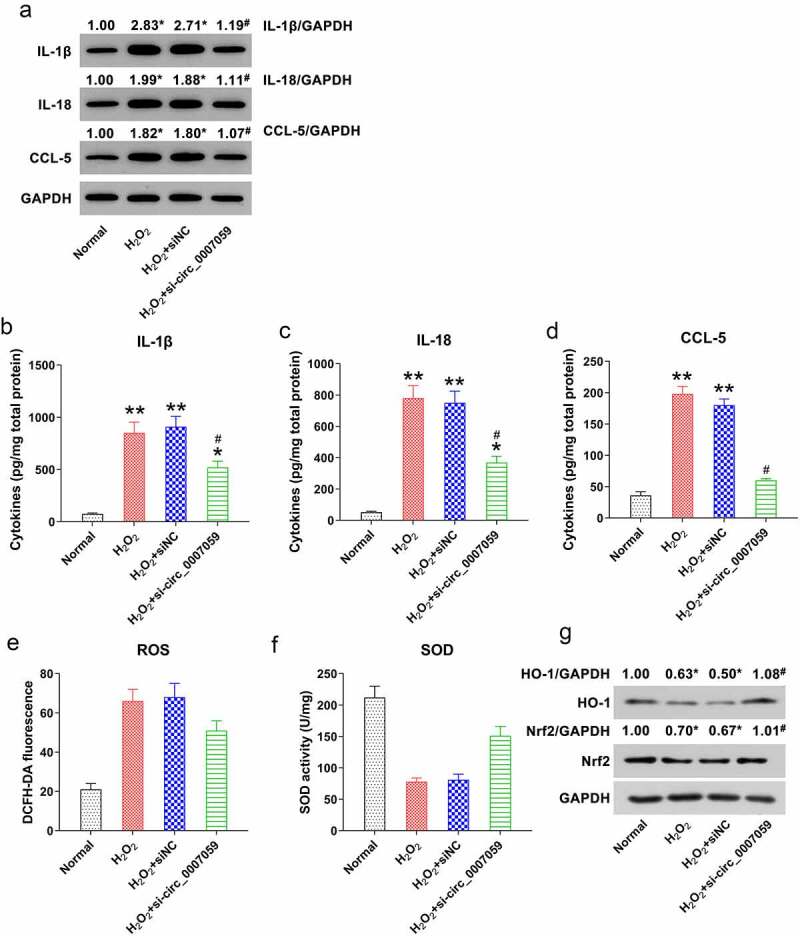
Circ_0007059 knockdown inhibits H2O2-induced cardiomyocyte inflammation. Cardiomyocytes were infected with lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. (a) WB quantification for inflammatory cytokine expression in cardiomyocytes. (B, C, D) ELISA for the quantification of inflammatory cytokines released into the cardiomyocyte medium. (e) DCFH-DA staining was performed to evaluate the intra-cellular ROS production. (f) Activity of anti-oxidative enzyme SOD was determined using SOD assay kits. (g) WB detection of the Nrf2 and HO-1 protein levels in cardiomyocytes. *P < 0.05 and **P < 0.01 versus the CG; #P < 0.05 versus the H2O2 group.
Circ_0007059 acted as a ceRNA that sponges miR-378 and miR-383
Bioinformatics analyses performed to detect the miRNA targets of circ_0007059 established that both miR-378 and miR-383 are putative targets of circ_0007059 (Figure 6(a)). Subsequent DLRAs showed that co-transfection of miR-378/383 mimics suppresses the luciferase activity by 55% and 50%, respectively, when the luciferase gene was fused with the WT circ_0007059-binding region as compared to that with a NC mimic. Moreover, the luciferase activity associated with the circ_0007059 MU binding region displayed no significant differences, suggesting that circ_0007059 directly targets miR-378 and miR-383 (Figure 6(b,c)). Next, we evaluated the expression of miR-378 and miR-383 in the infarcted hearts of MI models. Our qPCR analyses revealed that the levels of both miR-378 and miR-383 were considerably decreased in the infarcted regions of mouse hearts as compared to those of normal mice (Figure 6(d,e)). Furthermore, to verify the effect of circ_0007059 silencing on miR-378 and miR-383 expression in H2O2-treated cardiomyocytes, cells were infected with lentiviral-si-circ_0007059 or -siNC. Subsequently, qPCR studies revealed that the expression of miR-378 and miR-383 was significantly suppressed after H2O2 treatment but elevated after circ_0007059 silencing (Figure 6(f,g)), suggesting that both miR-378 and miR-383 could be downregulated by circ_0007059 expression.
Figure 6.
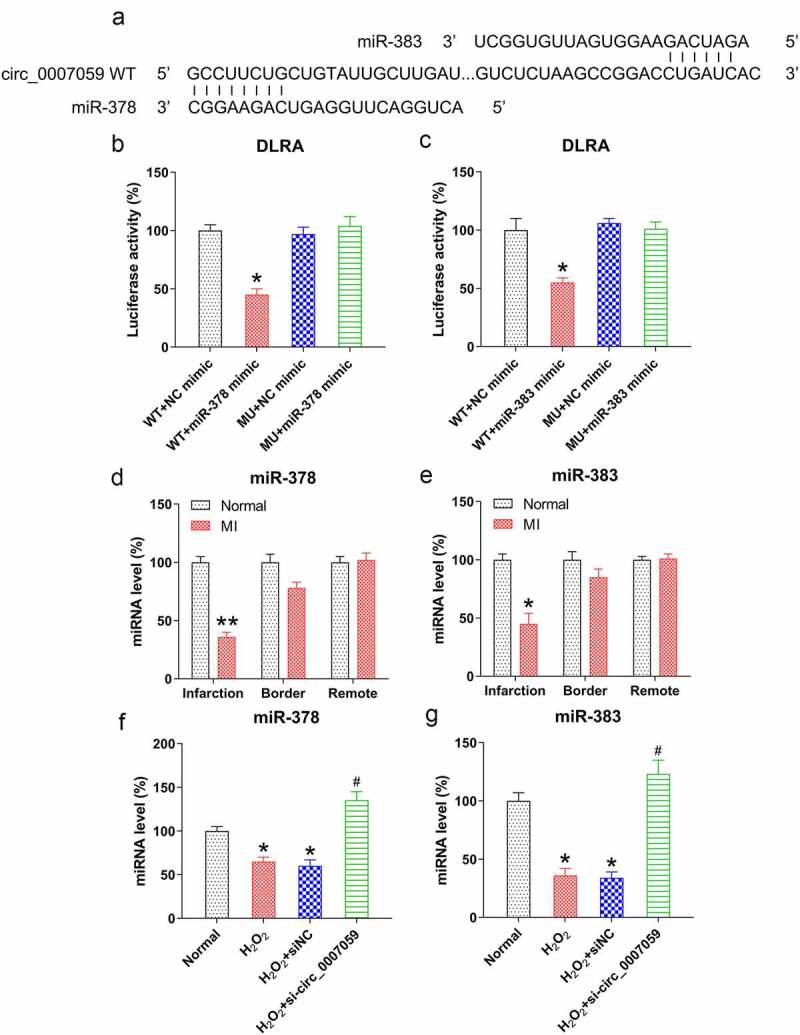
Circ_0007059 is a ceRNA that sponges miR-378 and miR-383. (a) Bioinformatics analyses indicated that circ_0007059 contains a binding site for miR-378 and miR-383. (b, c) DLRA was conducted through co-transfection of a luciferase reporter with WT or MU circ_0007059-binding sites with miR-378 and miR-383 mimics into cardiomyocytes. (d, e) qPCR quantification of miR-378 and miR-383 expression in the infarcted hearts of MI mice. (f, g) Cardiomyocytes were infected with lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. qPCR was used to quantify miR-378 and miR-383 expression. *P < 0.05 versus the CG; #P < 0.05 versus the H2O2 group.
miR-378 and miR-383 regulated circ_0007059-mediated apoptosis and inflammation of cardiomyocytes
Given that miR-378 and miR-383 levels were decreased in H2O2-treated cardiomyocytes but increased upon circ_0007059 knockdown, we aimed to determine whether H2O2-induced circ_0007059-mediated apoptosis and inflammation was attributable to miR-378 and miR-383 and whether the expressions of miR-378 and miR-383 were inhibited after silencing circ_0007059 in H2O2-treated cardiomyocytes. qPCR studies showed the expression of both miR-378 and miR-383 was downregulated by circ_0007059 silencing in H2O2-treated cardiomyocytes. In H2O2-treated cardiomyocytes, transfection of miR-378 and miR-383 mimics also led to increment of (Figure 7(a,b)).
Figure 7.
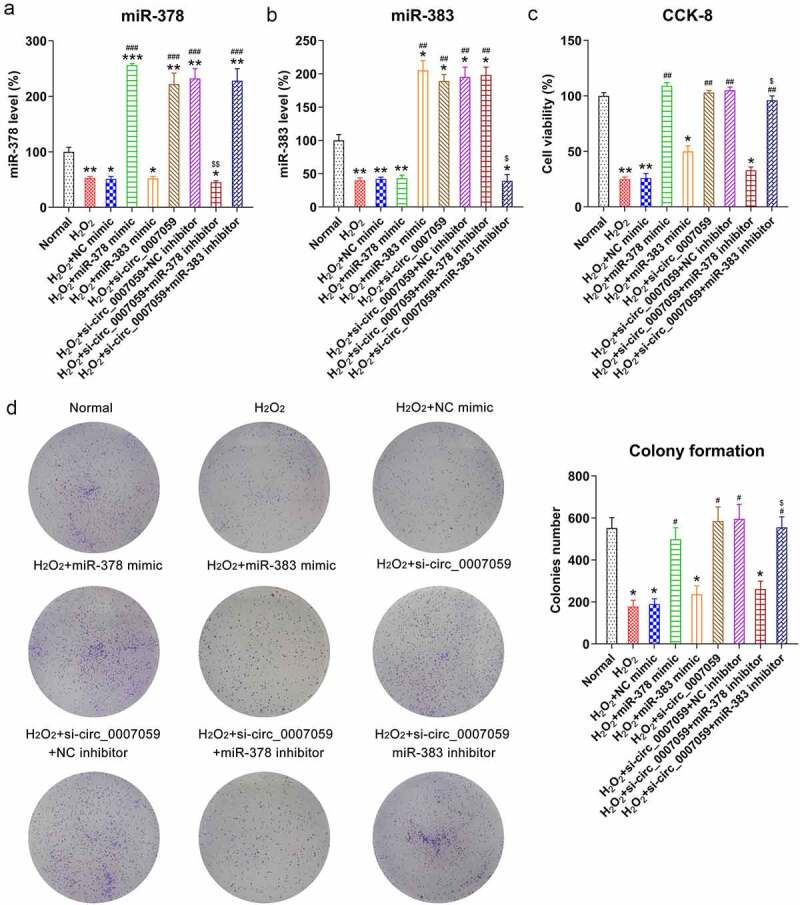
miR-378 influences the viability of H2O2-treated cardiomyocytes. Cardiomyocytes were transfected with miR-378/miR-383 or NC mimic or co-transfected with a miR-378/miR-383 inhibitor and lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. (a, b) qPCR quantification of miR-378 and miR-383 expression in cardiomyocytes. (c) CCK-8 assay examining cell viability. (d) CFA examining cellular growth rate. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the CG; #P < 0.05 and ##P < 0.01 versus the H2O2 group; $P < 0.05 versus the H2O2 + si-circ_0007059 group.
Furthermore, CCK-8 assays and CFAs were conducted to determine the effects of miR-378 and miR-383 on the viability of H2O2-treated cardiomyocytes. Notably, the upregulation of miR-378 led to restored cell viability and growth of H2O2-treated cardiomyocytes, and miR-378 inhibition also counteracted the effects of circ_0007059 knockdown on cell growth and viability as detected by using the MTT assay and CFA. In addition, overexpression or inhibition of miR-383 did not lead to a significant difference in cell viability and growth of H2O2-treated cardiomyocytes (Figure 7(c,d)).
Next, FC was employed to determine the role of these miRNAs in apoptosis. The results revealed that transfection of miR-378 mimics significantly reduced the proportion of cardiomyocytes that were apoptotic, whereas the inhibition of miR-378 restored the levels of apoptosis in circ_0007059-silenced cells (Figure 8(a)). WB studies demonstrated that caspase-3/9 cleavage was significantly decreased by the upregulation of miR-378 expression and elevated after miR-378 inhibition (Figure 8(b)). Interestingly, miR-383 expression did not affect the apoptosis of H2O2-treated cardiomyocytes.
Figure 8.
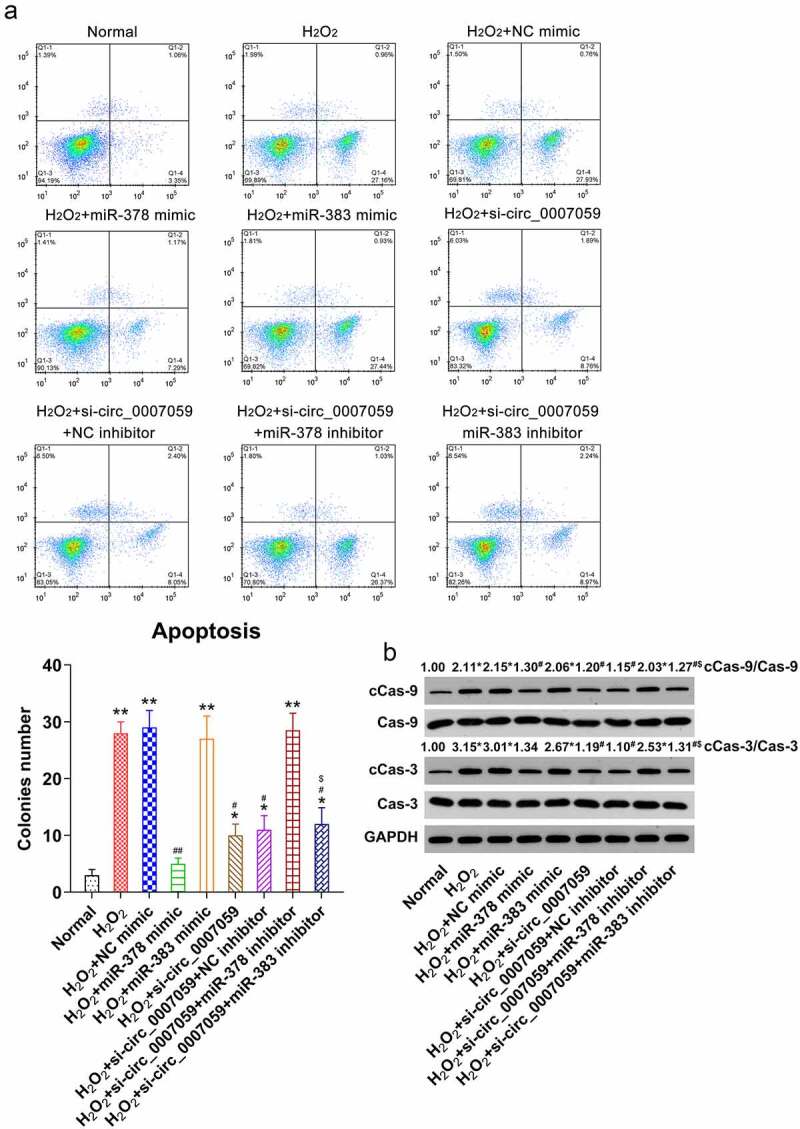
miR-378 influences apoptosis of H2O2-treated cardiomyocytes. Cardiomyocytes were transfected with miR-378/miR-383 or NC mimic or co-transfected with a miR-378/miR-383 inhibitor and lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. (a) Annexin V-FITC/PI staining and FC were used to detect the proportion of apoptotic cardiomyocytes. (b) WB quantification of the levels of caspase-3/-9 and cleaved-caspase-3/-9. *P < 0.05 versus the normal group; #P < 0.05 versus the H2O2 group; *P < 0.05 and **P < 0.01 versus the CG; #P < 0.05 and ##P < 0.01 versus the H2O2 group; $P < 0.05 versus the H2O2 + si-circ_0007059 group.
We also performed WB and ELISA to examine the effect of β-catenin and Keap1 on the production of cytokines in H2O2-treated cardiomyocytes subjected to circ_0007059 knockdown. The results revealed marked reduction in the production of inflammatory cytokines upon transfection of H2O2-treated cardiomyocytes with miR-383 mimics, whereas miR-383 inhibition result in the upregulation of the investigated cytokines and chemokines as compared to that in the respective control groups (Figure 9(a–d)). Thus, our findings reveal that miR-378 does not affect the inflammatory cytokine and chemokine production in H2O2-treated cardiomyocytes, further suggesting that Keap1 plays a pro-inflammatory role in H2O2-treated cardiomyocytes.
Figure 9.
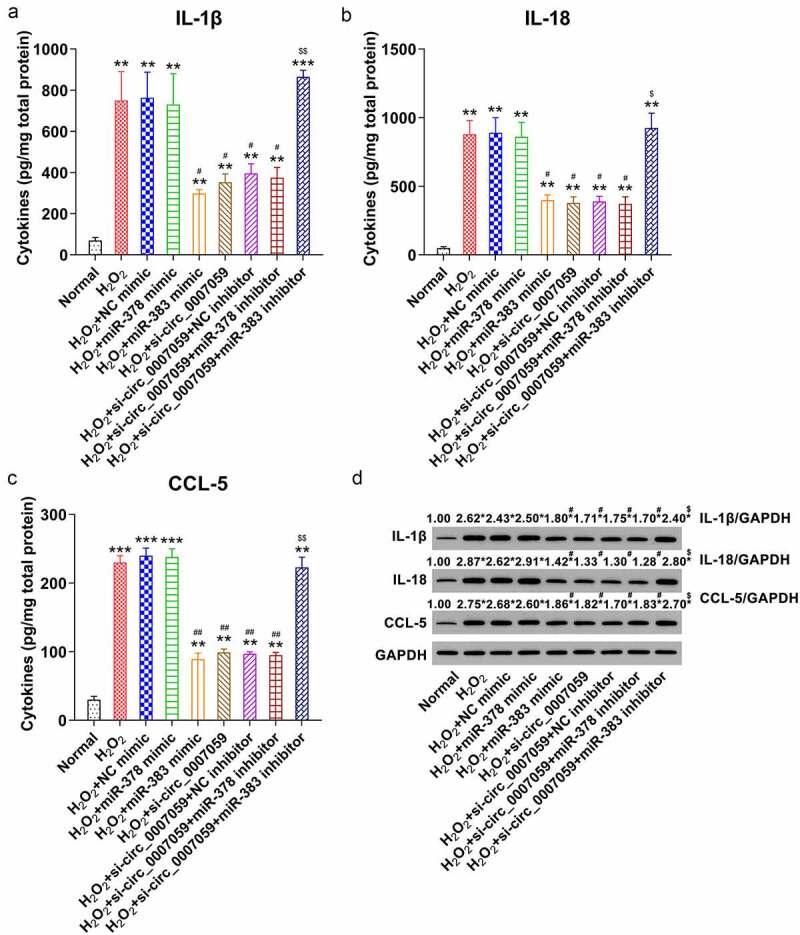
miR-383 influences inflammation in H2O2-treated cardiomyocytes. Cardiomyocytes were transfected with miR-378/miR-383 or NC mimic or co-transfected with a miR-378/miR-383 inhibitor and lentiviral-si-circ_0007059/siNC for 1 d and subsequently exposed to H2O2 (0.1 mM) for 1 d. (a) WB quantification of the inflammatory cytokine expression in cardiomyocytes. (B, C, D) ELISA quantification of inflammatory cytokines released into the cardiomyocyte culture medium. **P < 0.01 and ***P < 0.001 versus the CG; #P < 0.05 and ##P < 0.01 versus the H2O2 group; $P < 0.05 and $$ P < 0.01 versus the H2O2 + si-circ_0007059 group.
Discussion
In this study, we have investigated the role of circ_0007059 in MI injury. Our data shows that circ_0007059 expression is elevated in MI mice and H2O2-treated cardiomyocytes. Furthermore, silencing of circ_0007059 was found to inhibit H2O2-induced apoptosis, inflammation, and oxidative stress in cardiomyocytes. We also found that the suppression of apoptosis and inflammation induced by knockdown of circ_0007059 relied on the expression of miR-378 and miR-383, which were negatively regulated by circ_0007059 (Figure 10). Finally, our results demonstrated that circ_0007059 silencing protected cardiac tissue from MI and improved cardiac function.
Figure 10.
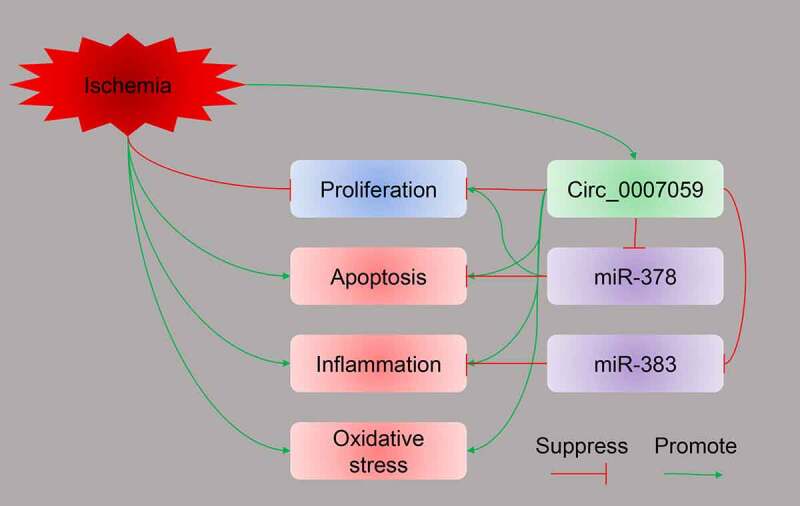
Graphic abstract illustrating the role of circ_0007059 during MI. MI modeling promoted circ_0007059 expression. Silencing of circ_0007059 inhibited H2O2-induced apoptosis, inflammation, and oxidative stress in cardiomyocytes. Furthermore, the suppression of apoptosis and inflammation induced by knockdown of circ_0007059 relied on the expression of miR-378 and miR-383, respectively. Both miR-378 and miR-383 are negatively regulated by circ_0007059.
Apoptosis, inflammation, oxidative stress during MI development are major factors that contribute to declining cardiomyocyte functions [20–22]. In animal MI models, apoptosis has been repeatedly observed in the infarcted heart tissue [23]. Moreover, apoptotic cardiomyocytes in the region bordering the MI area contribute to cardiomyocyte death, cardiac dysfunction, ultimately leading to heart failure and death [24,25]. MI progression is also accompanied by a series of inflammatory reactions that lead to the healing and scar formation during cardiac remodeling [26,27]. Inflammation has been documented to promote myocardial injury. Thus, anti-inflammatory compounds represent an efficient strategy for the therapeutic management of MI [28,29]. For instance, selective COX-2 inhibitors were introduced in the late 1990s in an attempt to develop anti-inflammatory strategies against MI without the risk of gastrointestinal side effects [30,31]. Additionally, MI development increases the generation of ROS from damaged mitochondria, NADPH oxidases, xanthine oxidase, and inflammation [32]. Prolonged oxidative stress, continuous cell death or degeneration, and inflammatory reactions are among the major culprits involved in the clinical complications of acute MI with high risk. ROS causes cell death or tissue degeneration and contributes to inflammation over time, presenting a window of opportunity for novel pharmacological intervention [32]. Therefore, inhibiting cardiomyocyte apoptosis, inflammation, as well as oxidative stress during the early stages of MI is a promising approach to decrease the infarct size and facilitate cardiac repair. Here, it was found that circ_0007059 downregulation reduced apoptosis, inflammation, and oxidative stress in cardiomyocytes upon H2O2 stimulation, providing evidence for the participation of circ_0007059 in the development of MI.
Recent evidence indicates that miRNAs are important mediators of MI pathogenesis. These studies used microarray-based expression profiling in an animal MI model or pathological heart samples from MI patients [33–37]. In cultivated cardiomyocytes experiencing hypoxia-induced apoptosis, miR-378 has been shown to regulate caspase-3 expression [11]. Another study has demonstrated that in homocysteine-treated coronary artery endothelial cells, upregulation of miR-383 expression reduces the levels of pro-inflammatory cytokines, such as IL-1β, IL-6, and IL-18 [14]. Here, we have discovered that circ_0007059 expression is conversely correlated with the expression of miR-378 and miR-383 in cardiomyocytes. Further validation showed that apoptosis and inflammation in H2O2-treated cardiomyocytes were regulated by miR-378 and miR-383, respectively, and that both of these miRNAs were targets of circ_0007059. These data reveal a critical role of circ_0007059 in the induction of apoptosis and inflammation in cardiomyocytes.
In conclusion, our findings suggest that expression of circ_0007059 in cardiomyocytes is promoted by MI injury in vivo and H2O2 exposure in vitro. Circ_0007059 impairs the viability of H2O2-induced cardiomyocytes by mediating apoptosis and inflammation by regulating miR-378 and miR-383 expression, respectively. Therefore, targeting circ_0007059 may serve to be a promising approach to protect the heart during MI pathogenesis.
Funding Statement
This research was supported by a grant entitled, “Effect of atorvastatin on apoptosis of aging myocardium through mitochondrial injury pathway;” No. 2019-140.
Author contributions
Chaorui Xu and Liping An conceived the study and designed the experiments. Zhuowen Jia and Xuefei Cao contributed to data collection. Sha Wang and Jipeng Wang performed the data analysis and interpreted the results. Chaorui Xu drafted the manuscript. Liping An contributed to the critical revision of the article. All authors have read and approved the final draft of the manuscript.
Ethical approval
All experiments were conducted according to the Guide for the Care and Use of Laboratory Animals (see NIH Publication, 8th Edition, 2011). This study was approved by Heilongjiang Provincial Hospital, Heilongjiang, P. R. China.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Fiedler J, Thum T.. MicroRNAs in myocardial infarction. Arterioscler Thromb Vasc Biol. 2013;33(2):201–205. [DOI] [PubMed] [Google Scholar]
- [2].Thind GS, Agrawal PR, Hirsh B, et al. Mechanisms of myocardial ischemia-reperfusion injury and the cytoprotective role of minocycline: scope and limitations. Future Cardiol. 2015;11(1):61–76. [DOI] [PubMed] [Google Scholar]
- [3].Sanger HL, Klotz G, Riesner D, et al. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proceedings of the National Academy of Sciences. 1976;73(11):3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang PL, Bao Y, Yee M-C, et al. Circular RNA is expressed across the eukaryotic tree of life. PloS one. 2014;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(5):453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Geng -H-H, Li R, Su Y-M, et al. The circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of miR-7a on its target genes expression. PloS one. 2016;11(3):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhou L-Y, Zhai M, Huang Y, et al. The circular RNA ACR attenuates myocardial ischemia/reperfusion injury by suppressing autophagy via modulation of the Pink1/FAM65B pathway. Cell Death Differ. 2019;26(7):1299–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang K, Gan T-Y, Li N, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24(6):1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gao S, Yu Y, Liu L, et al. Circular RNA hsa_circ_0007059 restrains proliferation and epithelial-mesenchymal transition in lung cancer cells via inhibiting microRNA-378. Life Sci. 2019;233:116692. [DOI] [PubMed] [Google Scholar]
- [10].Chistiakov DA, Orekhov AN, Bobryshev YV. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J Mol Cell Cardiol. 2016;94:107–121. [DOI] [PubMed] [Google Scholar]
- [11].Fang J, Song X-W, Tian J, et al. Overexpression of microRNA-378 attenuates ischemia-induced apoptosis by inhibiting caspase-3 expression in cardiac myocytes. Apoptosis. 2012;17(4):410–423. [DOI] [PubMed] [Google Scholar]
- [12].Xu D, Ma P, Gao G, et al. MicroRNA-383 expression regulates proliferation, migration, invasion, and apoptosis in human glioma cells. Tumor Biol. 2015;36(10):7743–7753. [DOI] [PubMed] [Google Scholar]
- [13].Pei L, Meng S, Yu W, et al. Inhibition of microRNA-383 ameliorates injury after focal cerebral ischemia via targeting PPARγ. Cell Physiol Biochem. 2016;39(4):1339–1346. [DOI] [PubMed] [Google Scholar]
- [14].Lian Z, Lv FF, Yu J, et al. The anti‐inflammatory effect of microRNA‐383‐3p interacting with IL1R2 against homocysteine‐induced endothelial injury in rat coronary arteries. J Cell Biochem. 2018;119(8):6684–6694. [DOI] [PubMed] [Google Scholar]
- [15].Hang P, Sun C, Guo J, et al. BDNF-mediates Down-regulation of MicroRNA-195 Inhibits Ischemic Cardiac Apoptosis in Rats. Int J Biol Sci. 2016;12(8):979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang W, Tian SS, Hang PZ, et al. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol Ther Nucleic Acids. 2016;5:e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Friedman RC, Farh KK-H, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ong S-B, Hernández-Reséndiz S, Crespo-Avilan GE, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. 2018;186:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Misra MK, Sarwat M, Bhakuni P, et al. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. 2009. Oct;15(10):Ra209–219. [PubMed] [Google Scholar]
- [20].Shah AM, Mann DL. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet. 2011. Aug 20;378(9792):704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu J, Wang H, Li J. Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: a double-edged sword. Clin Med Insights Cardiol. 2016;10:S33164. CMC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bezerra OC, França CM, Rocha JA, et al. Cholinergic stimulation improves oxidative stress and inflammation in experimental myocardial infarction. Sci Rep. 2017;7(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Forteza MJ, Trapero I, Hervas A, et al. Apoptosis and mobilization of lymphocytes to cardiac tissue is associated with myocardial infarction in a reperfused porcine model and infarct size in post-PCI patients. Oxid Med Cell Longev. 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frangogiannis NG. Pathophysiology of Myocardial Infarction. Compr Physiol. 2015. Sep 20;5(4):1841–1875. [DOI] [PubMed] [Google Scholar]
- [25].Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res. 2014. May 1;102(2):240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ji RC. The role of lymphangiogenesis in cardiovascular diseases and heart transplantation. Heart Fail Rev. 2021. Nov;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lodrini AM, Goumans MJ. Cardiomyocytes Cellular Phenotypes After Myocardial Infarction. Front Cardiovasc Med. 2021;8:750510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wen Y-C, Hsiao F-Y, Chan KA, et al. Acute respiratory infection and use of nonsteroidal anti-inflammatory drugs on risk of acute myocardial infarction: a nationwide case-crossover study. J Infect Dis. 2017;215(4):503–509. [DOI] [PubMed] [Google Scholar]
- [29].Huang S, Frangogiannis NG. Anti‐inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol. 2018;175(9):1377–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Libby P, Maroko PR, Bloor CM, et al. Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest. 1973;52(3):599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Boulakh L, Gislason GH. Treatment with non-steroidal anti-inflammatory drugs in patients after myocardial infarction - a systematic review. Expert Opin Pharmacother. 2016. Jul;17(10):1387–1394. [DOI] [PubMed] [Google Scholar]
- [32].Chen QM. Nrf2 for cardiac protection: pharmacological options against oxidative stress. Trends Pharmacol Sci. 2021. Sep;42(9):729–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dong S, Cheng Y, Yang J, et al. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. 2009;284(43):29514–29525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shi B, Guo Y, Wang J, et al. Altered expression of microRNAs in the myocardium of rats with acute myocardial infarction. BMC Cardiovasc Disord. 2010;10(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Boštjančič E, Zidar N, Glavač D. MicroRNA microarray expression profiling in human myocardial infarction. Dis Markers. 2009;27(6):255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu Z, Yang D, Xie P, et al. MiR-106b and MiR-15b modulate apoptosis and angiogenesis in myocardial infarction. Cell Physiol Biochem. 2012;29(5–6):851–862. [DOI] [PubMed] [Google Scholar]
- [37].Huang W, Tian -S-S, Hang P-Z, et al. Combination of microRNA-21 and microRNA-146a attenuates cardiac dysfunction and apoptosis during acute myocardial infarction in mice. Mol Ther Nucleic Acids. 2016;5:e296. [DOI] [PMC free article] [PubMed] [Google Scholar]