

ABSTRACT
Very little is known about the mechanisms that restrict neurotropic herpesviruses such as herpes simplex virus-1 (HSV-1) from infecting the central nervous system (CNS) and causing widespread death of neurons. Likewise, HSV-1 is thought to play a role in chronic neurodegeneration, yet a direct association has remained elusive. To address these issues, we recently showed that the selective macroautophagy/autophagy receptor OPTN (optineurin) specifically targets HSV-1 proteins VP16 and gB for degradation to prevent viral spread in the brain. OPTN deficiency alters host cytokine expression and tissue-specific immune signaling, and enhances necroptotic death of infected neurons. HSV-1-infected optn knockout mice show higher susceptibility to lethal CNS infection and the surviving animals demonstrate cognitive deficiency. Our research suggests that OPTN-mediated autophagy provides an intrinsic immune barrier against neurotropic viruses and protects the CNS from neurodegenerative stress.
KEYWORDS: Herpesvirus, HSV-1, necroptosis, neurodegeneration, neuroprotection, optineurin, selective autophagy
HSV-1 causes lifelong infection in a large section of the human population. Over 60% of adults harbor this neurotropic virus for their entire lives. Mucosal epithelial cells form the initial replication site from where the virus finds its way to peripheral nerves and eventually becomes latent in the trigeminal ganglion (TG). In the TG, it periodically reactivates causing recurrent clinical, such as orofacial and corneal ulcers, or subclinical manifestations. Human, and more directly animal studies, have shown that the virus can also infect the CNS causing herpes simplex encephalitis (HSE). The latter is extremely rare but potentially life threatening for untreated individuals. For most others, asymptomatic reactivation is relatively common and perceived to be a contributing factor in chronic neuronal damage similar to that reported with neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), Alzheimer disease (AD), and glaucoma.
Genetic screens have implicated OPTN in many neurodegenerative disorders including ALS, AD and glaucoma. The OPTN protein is expressed by a variety of cell types including neurons. OPTN selectively participates in protein degradation, which requires its ubiquitin-binding domain for binding to polyubiquinated proteins for recycling via autophagy. Despite its high expression in neurons, well-established role in autophagy, and the fact that HSV-1 encodes a virulence factor, ICP34.5/γ134.5, to inhibit autophagy in neurons, the significance of OPTN during a neurotropic viral infection is poorly studied. Our recent study [1] shows that OPTN restricts HSV-1 infection in vitro and in vivo, regulates tissue-specific immunity, and protects against necroptotic death of infected neurons (Figure 1).
Figure 1.
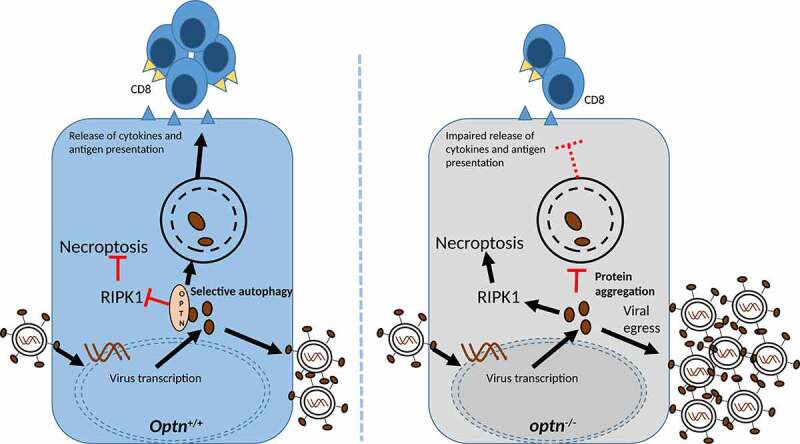
During HSV-1 infection, OPTN centrally regulates necroptosis, immune signaling and degradation of HSV-1-specific proteins. In the absence of OPTN, the intrinsic regulation fails to restrict proliferation of HSV-1.
We found that in cultured cells, which include the natural target epithelial and neuronal cell types, the optn gene knockout (optn KO) or downregulation results in a higher rate of cell-to-cell spread of HSV-1 virions. This increased rate of spread is observed for multiple strains of HSV-1 and a neurotropic animal herpesvirus, pseudorabies virus. Upon further investigation, we noticed that the levels of HSV-1 transactivator protein, VP16, and an essential glycoprotein, gB, are particularly higher in optn KO cells. This effect is specific because the level of another essential viral protein, ICP0, does not alter significantly. Similarly, in vivo both gB and VP16 but not ICP0 are found accumulating in the brainstem and TG of HSV-1 infected optn KO mice. The control mice infected similarly with HSV-1 do not accumulate viral proteins. We therefore proposed that the effect is due to selective autophagy especially because the use of an autophagy inhibitor, bafilomycin A1 (BafA1), reverses the loss of VP16 and gB in wild-type cells. Super-resolution microscopy of cells infected with a recombinant HSV-1 strain reveals OPTN-stained aggregates containing VP16 and, similarly, immunostaining shows VP16-bound OPTN in lysosomal vesicles. Additional results show that autophagic degradation of HSV-1 proteins via OPTN is regulated by TBK1 and the pathogenic effect of a HSV-1 ICP34.5/γ134.5 deletion mutant remains subdued in optn KO cells.
As a hallmark feature, the study found striking differences in severity of infection in optn KO vs. wild-type control animals. Whereas the HSV-1 titer in the TG is undeterred for control or the test mice, severe infection of the brainstem and brain as well as necroptotic death of neurons occurs in optn KO mice only. This is in agreement with the known OPTN function to regulate necroptosis by targeting RIPK1 for autophagic degradation. As a result, treatment of the HSV-1-infected optn KO mice or primary mouse neuronal cells with a RIPK1 inhibitor, necrostatin-1s/Nec-1s, rescues HSV-1-induced death. The severity of infection in terms of viral load and neuronal necroptosis in optn KO mice suggest a neuroprotective role of OPTN. Implicating a possible defect in autophagic recycling causing neuronal damage, the study found OPTN-stained cytosolic aggregates in the brainstem of an ALS patient and an HSE patient but not in a healthy patient. In support of an accelerated neurodegeneration in HSV-1-infected optn KO mice, we found a key behavior-related issue that is specific to the KO animals. After 30 days, HSV-1-infected optn KO mice fail a novel object recognition test, a well-established test for memory-related loss of cognition, which measures their ability to explore new objects. In contrast, the non-infected optn KO animals or infected controls do not show any measureable loss of cognitive ability.
Finally, to address whether an altered immune system also contributes to exacerbated neuronal damage, we found evidence of tissue-specific differences in the cytokine pool and an impaired recruitment of CD4+ and CD8+ T lymphocytes to the CNS of optn KO mice. This observation raises a combined possibility that OPTN regulates tissue-specific immune determinants via selective degradation of viral proteins, antigen presentation, and, correspondingly, a well-tailored cytokine response. The impaired immune response in OPTN-deficient conditions also hints toward its likely contribution in activation of pattern recognition receptors to mount an effective anti-HSV-1 response through type I interferons. In the future, we plan to study molecular details of OPTN-mediated selective autophagy; particularly, in elucidating its role in immune regulation and herpesvirus-dependent neurodegeneration. We also hope to identify naturally occurring OPTN mutations in humans, which might confer higher susceptibility to HSE- and HSV-dependent neurodegeneration.
Funding Statement
This research was supported by the National Institutes of Health and National Eye Institute grants; [K08-EY021520-02, RO1 EY029426, P30 EY001792 and RO1 EY024710] as well as the Butner Pioneer Award, and Duke Health Scholars. This content is solely the responsibility of the authors and doesnot necessarily represent the official views of the NIH.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Ames J, Yadavalli T, Suryawanshi R, et al. OPTN is a host intrinsic restriction factor against neuroinvasive HSV-1 infection. Nat Commun. 2021;12:5401. [DOI] [PMC free article] [PubMed] [Google Scholar]