

ABSTRACT
TBK1 (TANK-binding kinase 1) is an essential receptor protein required for the innate immune response, but the mechanisms underlying TBK1 stability, especially those regulated via autophagy, remain poorly understood. Here, we demonstrate that USP19 (ubiquitin specific peptidase 19) interacts with and promotes TBK1 lysosomal degradation via chaperone-mediated autophagy (CMA). We observed that TBK1 had a canonical CMA motif, knocking down key proteins involved in CMA (HSPA8/HSC70 or LAMP2A) or inhibiting CMA-prevented USP19-mediated TBK1 degradation. Furthermore, USP19 deficiency in macrophages caused an elevation of TBK1 and the activation of the type-I interferon signaling pathway after vesicular stomatitis virus (VSV) infection. Consistently, macrophage-specific usp19 knockout in mice resulted in attenuated VSV replication and resistance to VSV infection in vivo. Altogether, our results suggest that USP19 is a key regulator of TBK1 and uncovers a previously uncharacterized role for USP19 in CMA-mediated TBK1 degradation and infectious diseases.
KEYWORDS: Antiviral immunity, autophagic degradation, hspa8/hsc70, lamp2a, type i interferon
Introduction
Autophagy is a conserved cellular catabolic process that involves the phagocytosis of cytoplasmic proteins or organelles [1]. These cellular components fuse with lysosomes to form autolysosomes, where the encapsulated content is degraded [2,3]. Autophagy plays a crucial role in cellular physiology, permitting adaptation to metabolic stresses, such as starvation, hypoxia, and infection [4]. For this reason, autophagy is implicated in numerous diseases, including cancer, neurodegeneration, aging, and infectious diseases [5]. There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Each of these types differs in terms of the mode of cargo delivery to the lysosome [6,7]. CMA is a selective form of autophagy that targets proteins for lysosomal degradation. To achieve this, a KFERQ motif (Lys-Phe-Glu-Arg-Gln) present on the target proteins is recognized by the intracellular chaperone protein HSPA8/HSC70 (heat shock protein 8), and the complexes are then transported into the lysosome via LAMP2A (lysosomal-associated membrane protein 2A) [8]. The properties of the residues that constitute the motif, rather than the specific amino acids, determine whether HSPA8 can bind to this motif or not [8–10]. The motif is always flanked by a glutamine on one of the ends and contains one or two of the positively charged residues K and R; one or two of the hydrophobic residues F, L, I, or V; and one of the negatively charged E or D residues [8,10]. After translocation, the substrate proteins are rapidly degraded into single amino acids by various lysosomal hydrolases.
CMA is a dynamic process that is tightly regulated by numerous factors and signaling pathways, including NFAT and calcium signaling [11], RARA/RARα signaling [12], the TORC2-AKT1-PHLPP1 axis [13], and endoplasmic reticulum (ER) stress [14]. Furthermore, LAMP2A and other chaperone proteins involved in CMA are regulated at the transcriptional and translational levels [10]. The ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway (ALP) are the two main intracellular degradation systems. These systems are not mutually exclusive; for example, in cancer cells, degradation of the pro-survival protein MCL1 by UPS can be induced by inhibiting HSPA8 and LAMP2A expression [15]. Ubiquitin ligases can also associate with molecular chaperones, such as the ubiquitin ligase STUB1/CHIP, which binds to HSPA8, HSPA/HSP70, and HSP90, and tags associated substrates with the degradation marker ubiquitin [16]. Thus, ubiquitin ligases have potential roles in CMA; however, the function of deubiquitination enzymes in CMA is largely unknown.
TBK1 (TANK-binding kinase 1) is a serine/threonine kinase involved in innate immune signaling pathways, including antiviral responses and host defenses against cytosolic bacterial infections. During viral infection, various viral structural components, including viral DNA, dsRNA, ssRNA, and surface glycoproteins, are recognized as pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs) [17–19]. PRRs recruit different receptor proteins, including TICAM1/TRIF (toll-like receptor adaptor molecule 1), MAVS (mitochondrial antiviral signaling protein), or STING1 (stimulator of interferon response cGAMP interactor 1) to activate TBK1. Activated TBK1 then phosphorylates IRF3 (interferon regulatory factor 3) and IRF7 (interferon regulatory factor 7) and triggers their dimerization and nuclear translocation; this effect results in the promotion of type I interferon gene expression [20]. TBK1 kinase activity is regulated by various mechanisms, including phosphorylation and ubiquitination. For example, the E3 ubiquitin ligase DTX4 promotes Lys48-linked TBK1 polyubiquitination at Lys670, leading to TBK1 degradation [21]. USP38 inhibits type I interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome [22]. TBK1 exhibits E3 ubiquitin ligase activity and can become self-ubiquitinated both in vitro and in vivo [23]. To date, however, the role of the autophagy-lysosome pathway in regulating TBK1 stability has not been explored.
Given the important role of TBK1 in innate immunity, an in-depth analysis of its regulation is warranted. Here, we aimed to identify potential TBK1 regulators in vitro by co-transfecting HEK293T cells with plasmids encoding TBK1 and 43 deubiquitinating enzymes (DUBs), and revealed that USP19 could promote TBK1 degradation. USP19 is a DUB that has an important function in the innate immune response and in muscle cell differentiation [24–26]. Amino acid sequence alignment analysis revealed that TBK1 has a canonical CMA motif (KFDKQ). Using confocal microscopy analysis, co-immunoprecipitation (co-IP), and inhibitor experiments, we found that USP19 interacts with and promotes TBK1 degradation via CMA. TBK1 is essential for IFNB/IFN-β (interferon beta 1, fibroblast) production and innate immunity. Immunoblotting, enzyme-linked immunosorbent assay (ELISA), and flow cytometry analysis demonstrated that USP19 deficiency in macrophages enhanced TBK1 stability, IRF3 activation, IFNB production, and the antiviral innate immune response to viruses both in vitro and in vivo. These findings suggest a critical role for USP19 in CMA-mediated TBK1 degradation and infectious diseases.
Results
USP19 promotes TBK1 lysosomal degradation
TBK1 is crucial for the innate immune response to viral and bacterial infections. To identify the deubiquitinating enzymes that might regulate TBK1 stability, we co-transfected HEK293T cells with TBK1 and DUB expression plasmids and analyzed the deubiquitinating enzyme(s) via immunoblotting to determine TBK1 degradation. After testing 43 DUBs, we identified that USP19 could promote TBK1 degradation (Figure S1A). To confirm this result, we expressed TBK1 and USP19 in HEK293T cells and obtained a similar result (Figure S1B). Furthermore, immunoblotting showed that TBK1 protein levels were reduced in a USP19 dose-dependent manner (Figure 1A). Moreover, overexpression of USP19 in 293 T cells decreased the TBK1 half-life compared to that in the control group (Figure 1B, 1C). These results suggest that USP19 promoted TBK1 degradation.
Figure 1.
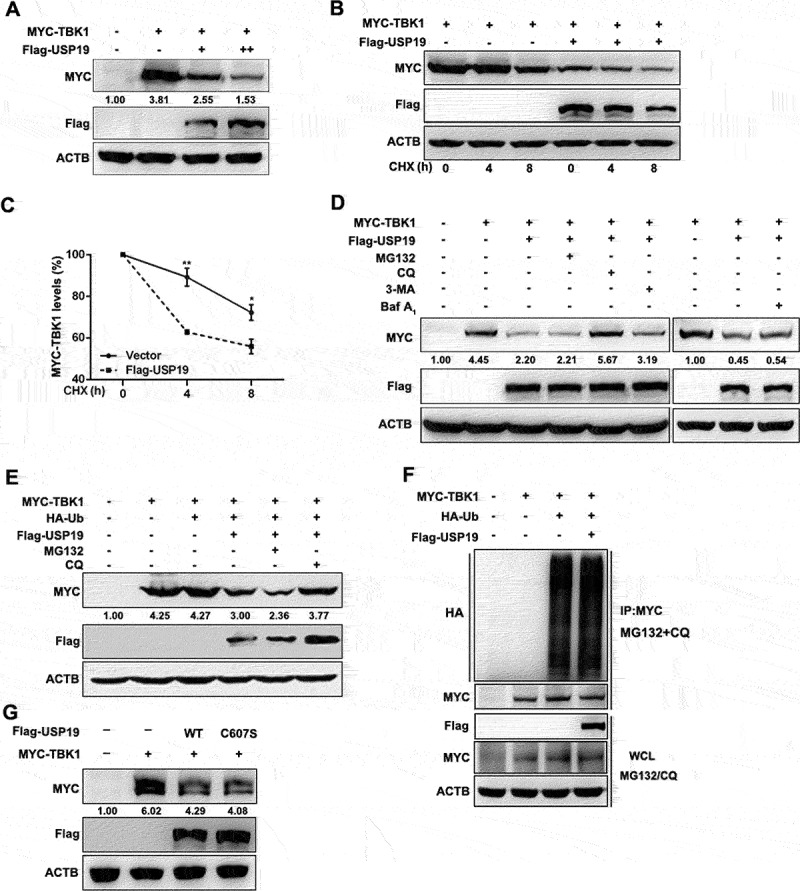
USP19 promotes TBK1 degradation in a lysosome-dependent manner. (A) HEK293T cells were transfected with a MYC-TBK1 plasmid and increasing amounts of a Flag-USP19 expression plasmid and the extracted proteins were detected via immunoblotting. (B and C) HEK293T cells were transfected with MYC-TBK1 and Flag-USP19, then treated with CHX (100 μg/mL) for the indicated times after 24 h transfection. MYC-TBK1 protein levels were analyzed via immunoblotting (B). Myc-TBK1 expression was normalized to ACTB (C). (D and E) HEK293T cells were transfected with the indicated plasmids without (D) or with (E) a HA-Ub plasmid for 24 h, and then treated with MG132 (20 μM), CQ (50 μM), 3-MA (5 mM) or Baf-A1 (20 nM) for 6 h. The proteins were then detected via immunoblotting. (F) HEK293T cells were transfected with MYC-TBK1, HA-Ub, and Flag-USP19 plasmids for 24 h, then the cells were treated with MG132 (20 μM) and CQ (50 μM) for 6 h before harvest. The ubiquitination experiment was performed and the results were analyzed via immunoblotting. (G) HEK293T cells were transfected with MYC-TBK1, Flag-USP19, or Flag-USP19C607S plasmids, and the extracted proteins were detected via immunoblotting. The data are representative of three independent experiments. Error bars show the means ± SD. *p < 0.05, **p < 0.01 using the student’s t test.
Protein degradation predominantly occurs via two pathways: a proteasome-dependent pathway and a lysosome-dependent pathway. To investigate USP19-mediated TBK1 degradation, we co-transfected USP19 and TBK1 into HEK293T cells and then treated the cells with a proteasome inhibitor (MG132), a lysosomal inhibitor (chloroquine [CQ]), a phosphatidylinositol 3-kinase inhibitor (3-methyladenine [3-MA]), or an autophagosome-lysosome fusion inhibitor, bafilomycin A1 (Baf-A1). Only CQ treatment suppressed TBK1 degradation (Figure 1D), suggesting that USP19 promoted TBK1 degradation in a lysosome-dependent manner.
As USP19 is a deubiquitinating enzyme, we next investigated whether TBK1 protein ubiquitination status is affected by USP19. HEK293T cells were transfected with ubiquitin, USP19, and TBK1 expression plasmids, and then treated with MG132 or CQ, and we found that USP19 regulated TBK1 degradation independently of ubiquitin (Figure 1E). Furthermore, USP19 overexpression did not affect TBK1 polyubiquitination (Figure 1F). To confirm these results, we next examined whether USP19 deubiquitinating enzymatic activity affected TBK1 protein levels. We constructed an enzymatically inactive USP19 mutant plasmid containing a C607S substitution (Flag-USP19C607S) [24]. Immunoblot analysis demonstrated that USP19 promoted TBK1 degradation independently of its deubiquitinating enzymatic activity (Figure 1G). Taken together, these results reveal that USP19 promotes TBK1 degradation in a lysosome-dependent manner but is independent of USP19 deubiquitinating enzymatic activity.
TBK1 interacts with the USP19 ubiquitin-specific peptidase (USP) domain via its kinase domain (KD)
To investigate whether USP19 could interact with TBK1, we expressed the Flag-USP19 and MYC-TBK1 plasmids in 293 T cells and performed a co-immunoprecipitation assay. We observed that USP19 was associated with TBK1 (Figure 2A). As TBK1 is an essential receptor involved in the antiviral immune response, we also detected an endogenous interaction between USP19 and TBK1 in HeLa and THP1 cells with or without VSV infection (Figure 2B, C). To further confirm these results, we performed in vitro binding assays, and the results showed that USP19 could interact with TBK1 in vitro (Figure 2D). Our results also demonstrated that USP19C607S (enzymatically inactive USP19 mutant) could interact with TBK1 (Figure 2E). Moreover, we constructed three truncated mutants of TBK1 to explore which domain(s) mediate this interaction with USP19: KD (aa 1–299), KD + ubiquitin-like domain (ULD) (aa 1–382), ULD + coiled-coil (CC) domain (aa 299–729) (Figure 2F, top panel). Co-immunoprecipitation experiments with these truncated mutants revealed that the TBK1 ULD + CC domain truncation mutant was unable to interact with USP19, whereas the TBK1 WT, KD, and KD + ULD could still interact with USP19 (Figure 2F). These data support the hypothesis that TBK1 interacts with USP19 via its kinase domain.
Figure 2.
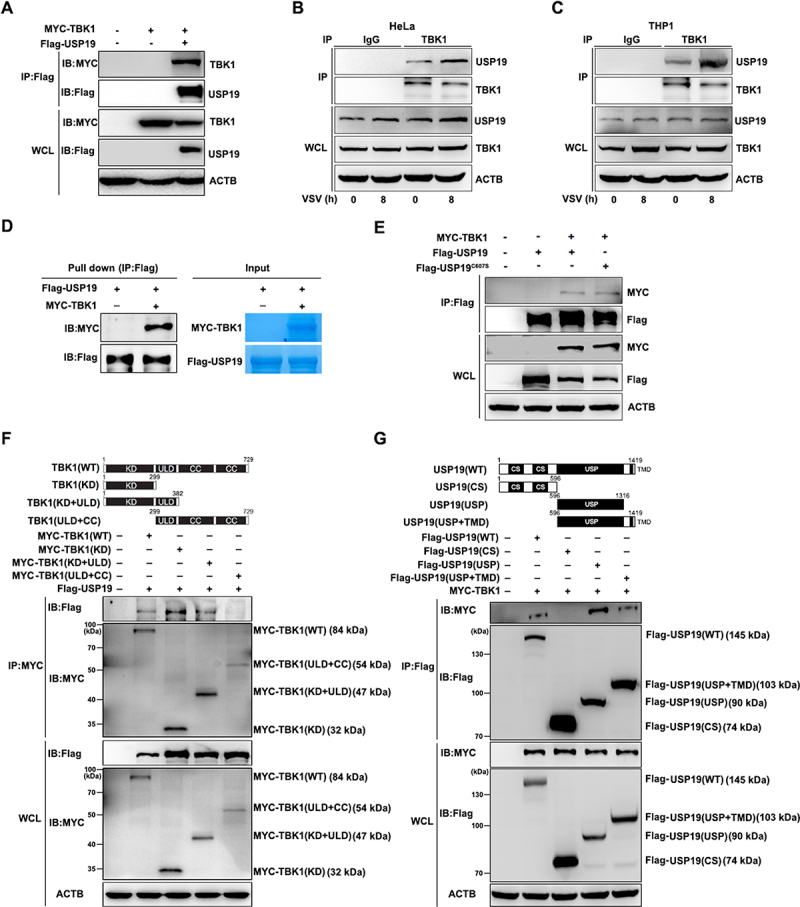
USP19 interacts with TBK1. (A) HEK293T cells were transfected with MYC-TBK1 and Flag-USP19 plasmids, and then subjected to immunoprecipitation with a Flag antibody before immunoblot analysis. (B and C) HeLa (B) or THP1 cells (C) were infected with VSV for the indicated times, and then subjected to immunoprecipitation with a TBK1 antibody before immunoblot analysis of endogenous TBK1and USP19 protein levels. (D) HEK293T cells were transfected with MYC-TBK1 or Flag-USP19 plasmids, using immunoprecipitation to purify the proteins using Flag or MYC beads. The purified proteins, including Flag-usp19 and MYC-TBK1, were mixed in a Co-IP buffer to perform an in vitro pull-down assay and the results were analyzed via Coomassie Brilliant Blue staining and an immunoblot assay. (E) HEK293T cells were transfected with MYC-TBK1, Flag-USP19, or Flag-USP19C607S plasmids, and then subjected to immunoprecipitation with a Flag antibody before immunoblot analysis. (F) Schematic of TBK1 and the derivatives used (top). HEK293T cells were transfected with MYC-TBK1 (WT), MYC-TBK1 (KD), MYC-TBK1 (KD+ULD), MYC-TBK1 (ULD+CC), and Flag-USP19 plasmids, and then analyzed via immunoprecipitation and immunoblotting with the indicated antibodies (bottom). (G) Schematic of USP19 and the derivatives used (top). HEK293T cells were transfected with Flag-USP19 (WT), Flag-USP19 (CS), Flag-USP19 (USP), Flag-USP19 (USP+TMD), and MYC-TBK1, and then analyzed via immunoprecipitation and immunoblotting with the indicated antibodies (bottom). The data are representative of three independent experiments.
Next, we constructed three truncated USP19 mutants to explore which USP19 domain(s) interact with TBK1: tandem CHORD-SGT1 (CS) domain (aa 1–596), USP domain (aa 596–1316), and USP domain + transmembrane domain (TMD) (aa 596–1419). Co-immunoprecipitation experiments showed that USP19 interacted with TBK1 via its USP domain (Figure 2G). In summary, USP19 and TBK1 interact via their USP and kinase domains, respectively.
USP19 promotes TBK1 degradation through chaperone-mediated autophagy
Thus far, our results indicate that USP19 promotes TBK1 degradation in a lysosome-dependent manner. Next, we investigated the mechanisms underlying the negative regulation of USP19 on TBK1. A previous study showed that USP19 promoted autophagy [25], and our data confirmed that overexpression of USP19, but not USP19C607S (enzymatically inactive USP19 mutant) increased LC3B cleavage in HeLa cells (Figure 3A). However, we also found that only the lysosomal inhibitor CQ, but not the autophagosome formation inhibitor 3-MA or autophagosome-lysosome fusion inhibitor Baf-A1, could suppress TBK1 degradation, indicating that USP19 promotes TBK1 degradation independent of macroautophagy. In addition, upon comparing the amino acid sequences of TBK1 between different species, we found that TBK1 exhibited a canonical CMA motif (KFDKQ) (Figure 3B), suggesting that USP19 might promote TBK1 degradation in a CMA-dependent manner. LAMP2A and HSPA8 are central proteins involved in CMA: HSPA8 recognizes and interacts with the CMA motif of target proteins while LAMP2A located on the lysosome membrane facilitates the transport of target proteins into lysosomes for degradation [9]. Immunofluorescence staining revealed that USP19 co-localized with TBK1 and LAMP2A (Figure 3C), and co-immunoprecipitation experiments indicated that USP19 interacted with TBK1, LAMP2A, and HSPA8 (Figure 3D). In addition, we also demonstrated the presence of TBK1 and USP19 proteins in the lysosomes via a lysosome isolation assay (Figure 3E).
Figure 3.
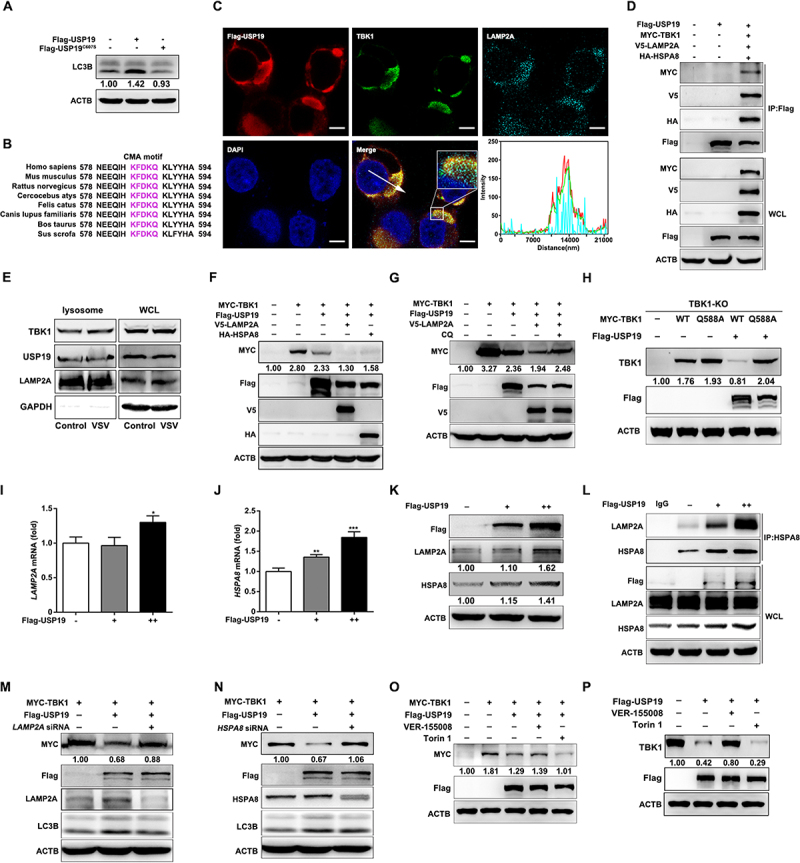
USP19 promotes TBK1 degradation through the CMA-dependent autophagy pathway. (A) HeLa cells were transfected with Flag-USP19 or Flag-USP19C607S plasmid and the LC3B levels were analyzed via immunoblotting. (B) Alignment of TBK1 orthologs. The pink shading indicates the conserved CMA motif. (C) HEK293T cells were transfected with MYC-TBK1 and Flag-USP19 plasmids for 24 h, labeled with the indicated antibodies, and analyzed via confocal microscopy. (D) HEK293T cells were transfected with MYC-TBK1, Flag-USP19, V5-LAMP2A, and HA-HSPA8 plasmids for 24 h, and then the lysates were immunoprecipitated with an anti-Flag antibody and analyzed via immunoblotting. (E) HeLa cells were infected with or without VSV, then the lysosomes were isolated and enriched before the proteins were analyzed via immunoblotting. (F) HeLa cells were transfected with MYC-TBK1, Flag-USP19, V5-LAMP2A, and HA-HSPA8 plasmids before analysis via immunoblotting with the indicated antibodies. (G) HeLa cells were transfected with MYC-TBK1, Flag-USP19, and V5-LAMP2A plasmids, and then treated with CQ (50 μM) before the proteins were analyzed via immunoblotting. (H) Tbk1-knockout Raw 264.7 cells were transfected with Flag-USP19, MYC-TBK1, or MYC-TBK1Q588A plasmids, and the indicated proteins were analyzed via immunoblotting. (I and J) HeLa cells were transfected with increasing amounts of Flag-USP19 plasmids for 24 h before qPCR analysis of LAMP2A (I) and HSPA8 (J) mRNA expression. (K) HeLa cells were transfected with increasing amounts of Flag-USP19 plasmids for 24 h before analysis of LAMP2A and HSPA8 protein levels via immunoblotting. (L) HeLa cells were transfected with increasing amounts of Flag-USP19 plasmids for 24 h, subjected to immunoprecipitation with an anti-HSPA8 antibody, and the results were analyzed via immunoblotting. (M and N) HeLa cells were transfected with gene-specific siRNA to knock down LAMP2A (M) or HSPA8 (N) mRNA expression, and were then transfected with MYC-TBK1 and Flag-USP19 plasmids. MYC-TBK1 protein levels were analyzed via immunoblotting. (O and P) HEK293T cells were transfected with a Flag-USP19 plasmid, with (O) or without (P) MYC-TBK1 plasmids, and then treated with VER-155,008 (5 μM) or Torin 1 (250 nM). MYC-TBK1 or endogenous TBK1 protein levels were analyzed via immunoblotting. Scale bars: 5 μm. The data are representative of three independent experiments. Error bars show the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
In terms of their effect on TBK1 stability, LAMP2A or HSPA8 overexpression enhanced USP19-mediated TBK1 degradation (Figure 3F) while CQ treatment rescued it (Figure 3G). As the CMA-motif is important to CMA-dependent protein degradation, we next constructed a TBK1 CMA-motif mutant plasmid containing a Q588A substitution (MYC-TBK1Q588A) and transfected a MYC-TBK1 or a MYC-TBK1Q588A plasmid into tbk1-knockout RAW264.7 cells with or without the Flag-USP19 plasmid. The results showed that USP19 did not promote TBK1 CMA-motif mutant protein degradation (Figure 3H). Interestingly, USP19 overexpression increased LAMP2A and HSPA8 expression both at the mRNA and protein levels (Figure 3(I-K)) and promoted the interaction between HSPA8 and LAMP2A (Figure 3L). This finding suggests that USP19 might have multiple biological functions, including the regulation of LAMP2A and HSPA8 mRNA expression. Importantly, the siRNA (small interfering RNA)-mediated knockdown of LAMP2A or HSPA8 expression prevented USP19-mediated TBK1 degradation (Figure 3(M, N)). VER-155,008 is an inhibitor of HSPA8 that can inhibit CMA, and Torin 1 is a potent mTOR inhibitor that can induce CMA [13,27]. When we treated HeLa cells with VER155,008, we observed that USP19-regulated TBK1 degradation was greatly reduced; conversely, Torin 1 treatment significantly promoted USP19-regulated TBK1 degradation (Figure 3(O, P)). Collectively, these results indicate that USP19 promotes TBK1 degradation via CMA.
USP19 negatively regulates IFNB production and the cellular antiviral response by promoting TBK1 degradation
TBK1 is an essential serine/threonine kinase involved in virus-triggered type I IFN signaling [28]. Therefore, we tested whether USP19 regulated the TBK1-dependent antiviral response. We designed a USP19-specific siRNA and found that it efficiently inhibited USP19 expression in THP-1 and HeLa cells (Figure 4A and Figure S2A). As anticipated, qPCR showed that USP19 knockdown enhanced VSV-induced IFNB1 expression in THP-1 or HeLa cells, but the CMA inhibitor (VER-155,008) did not further promote IFNB1 expression (Figure 4B and Figure S2B). Similar results were obtained via ELISA (Figure 4C and Figure S2C). USP19 knockdown in THP-1 or HeLa cells also markedly promoted VSV-induced CCL5 (C-C chemokine ligand 5) and CXCL10 (C-X-C motif chemokine ligand 10) mRNA expression, while the mRNA levels of CCL5 and CXCL10 were not significantly increased in the VER-155,008-pretreated USP19-knockdown THP-1 or HeLa cells compared with those in the DMSO-pretreated USP19-knockdown cells (Figure 4D and Figure S2D), suggesting that USP19 negatively regulates the cellular antiviral immune response by promoting the CMA-mediated degradation of TBK1. Virus infection can stimulate macrophages to produce type I IFNs, and then trigger the activation of the Janus kinase (JAK) – signal transducer and activator of transcription (STAT) pathway and the expression of interferon-stimulated genes (ISGs), such as ISG15, IFIT2/ISG54 (interferon-induced protein with tetratricopeptide repeats 2), IFIT1/ISG56 (interferon-induced protein with tetratricopeptide repeats 1), and MX1 (MX dynamin-like GTPase 1) [29,30]. Consequently, we found that the expression of these ISGs increased in USP19-knockdown cells, and there was no significant difference between USP19-knockdown cells pretreated with VER-155,008 and DMSO (Figure 4E and Figure S2E). To observe VSV replication in cells, we infected THP-1 and HeLa cells with GFP-tagged VSV. Confocal microscopy analysis revealed that the GFP fluorescence intensity markedly decreased in USP19 knockdown cells compared to control cells (Figure 4F and Figure S2F). Flow cytometry analysis confirmed that the percentage of GFP+ cells was significantly reduced in USP19 knockdown cells compared with control cells (Figure 4G and Figure S2G). These data also showed that USP19-knockdown cells with or without VER-155,008 pretreatment had no significant effect on the antiviral response against VSV infection. To further explore the related mechanism, we tested endogenous TBK1 level and the results showed that USP19 knockdown obviously inhibited endogenous TBK1 degradation compared to the control group (Figure 4H and Figure S2H). These results demonstrate that USP19 knockdown enhances IFNB production and the cellular antiviral response by inhibiting TBK1 degradation.
Figure 4.
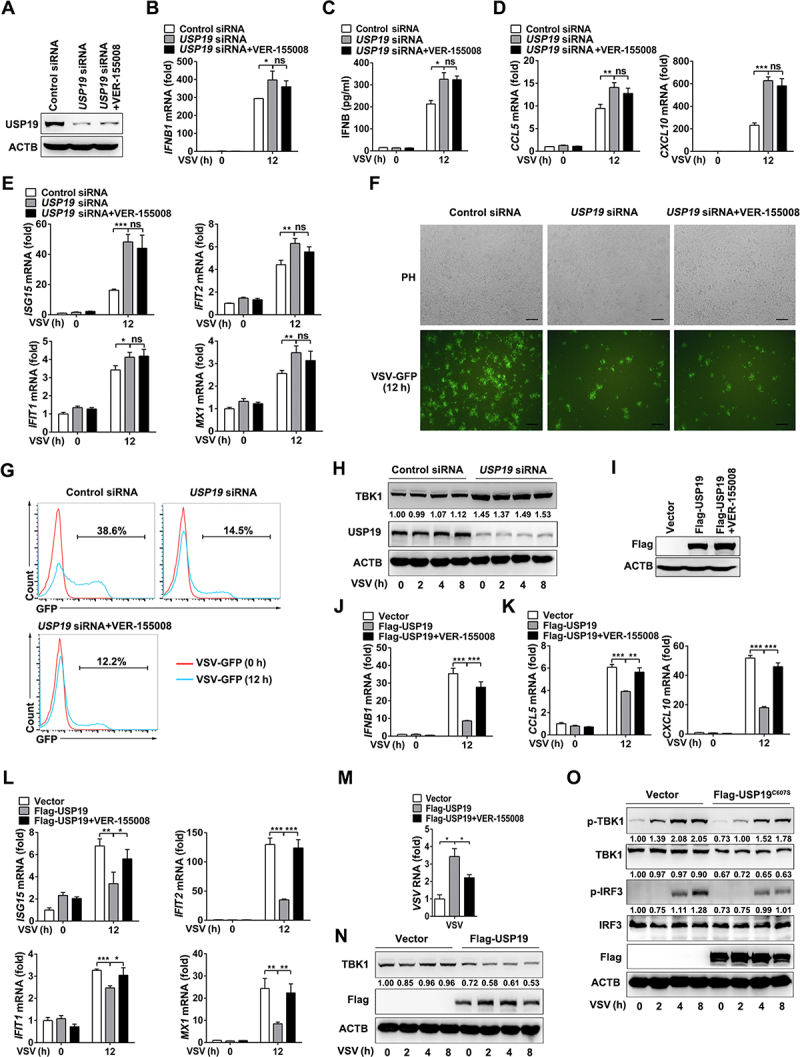
USP19 negatively regulates VSV-induced IFNB production and the antiviral response in THP1 cells. (A-G) THP1 cells were transfected with control or USP19 siRNA for 48 h, with or without pretreatment with VER-155,008 (5 μM) for 30 min, then USP19 protein levels were analyzed via immunoblotting (A). qPCR analysis of IFNB1 expression for the indicated times after VSV infection (MOI = 1) (B). ELISA analysis of IFNB production in the cell culture supernatants 12 h after VSV infection (C). qPCR analysis of CCL5 and CXCL10 expression 12 h after VSV infection (MOI = 1) (D). qPCR analysis of ISG15, IFIT2, IFIT1, and MX1 expression 12 h after VSV infection (MOI = 1) (E). THP-1 cells were infected with VSV-GFP (MOI = 1) for 12 h and the cells were imaged under a confocal microscope (F). The percentage of GFP+ cells was determined via flow cytometry (G). (H) THP1 cells were transfected with control or USP19 siRNA, and then infected with VSV. Immunoblotting was performed to analyze TBK1 protein levels. (I-M) THP1 cells were transfected with Flag-USP19 plasmids or an empty vector for 24 h, with or without pretreatment with VER-155,008 (5 μM) for 30 min. USP19 expression was analyzed via immunoblotting using a Flag antibody (I). qPCR analysis of IFNB1 (J), CCL5, CXCL10 (K), ISG15, IFIT2, IFIT1, and MX1 (L) expression and VSV replication (M) for the indicated times after VSV infection (MOI = 1). (N) THP1 cells were transfected with Flag-USP19 plasmids or an empty vector, and then infected with VSV. Immunoblotting was performed to analyze TBK1 protein levels. (O) THP1 cells were transfected with Flag-USP19C607S plasmids or an empty vector, and then infected with VSV. Immunoblotting was performed to analyze indicated proteins levels. The data are representative of three independent experiments. Scale bars: 100 μm. Error bars show the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
We also transfected a Flag-USP19 plasmid into THP1 and HeLa cells (Figure 4I and Figure S2I). Flag-tagged USP19 overexpression resulted in reduced IFNB1 (Figure 4J and Figure S2J), CCL5, CXCL10 (Figure 4K and Figure S2K), and ISGs (Figure 4L and Figure S2L) mRNA expression, and treatment with the inhibitor VER-155,008 rescued the downregulation of these genes after VSV infection. Furthermore, VSV replication was enhanced in VSV-infected THP1 or HeLa cells overexpressing Flag-USP19, and VER-155,008 treatment significantly promoted the antiviral response in USP19-overexpressing cells (Figure 4M and Figure S2M). Similarly, USP19 overexpression promoted endogenous TBK1 degradation (Figure 4N and S2N). Furthermore, we verified that USP19 negatively regulated the VSV-induced phosphorylation of TBK1 and IRF3, independent of USP19 deubiquitinating enzymatic activity (Figure 4O and Figure S2O). These results indicate that USP19 overexpression suppresses IFNB production and the cellular antiviral response by promoting TBK1 degradation.
To show that USP19 targets TBK1 to negatively regulate the antiviral response, we performed an IFNB1 and interferon-stimulated response element (ISRE) promoter reporter assay. Overexpressing USP19 inhibited TBK1 and the upstream regulators DDX58/RIG-I and MAVS overexpression induced IFNB1 and ISRE promoter activity (Figure 5(A-F)). Results of the co-immunoprecipitation assay revealed that there was no interaction between USP19 and DDX58, MAVS, IRF3, IFIH1/MDA5, CGAS, or STING1 (Figure 5G). These data suggest that USP19 negatively regulates the TBK1-dependent cellular antiviral response by promoting TBK1 degradation.
Figure 5.
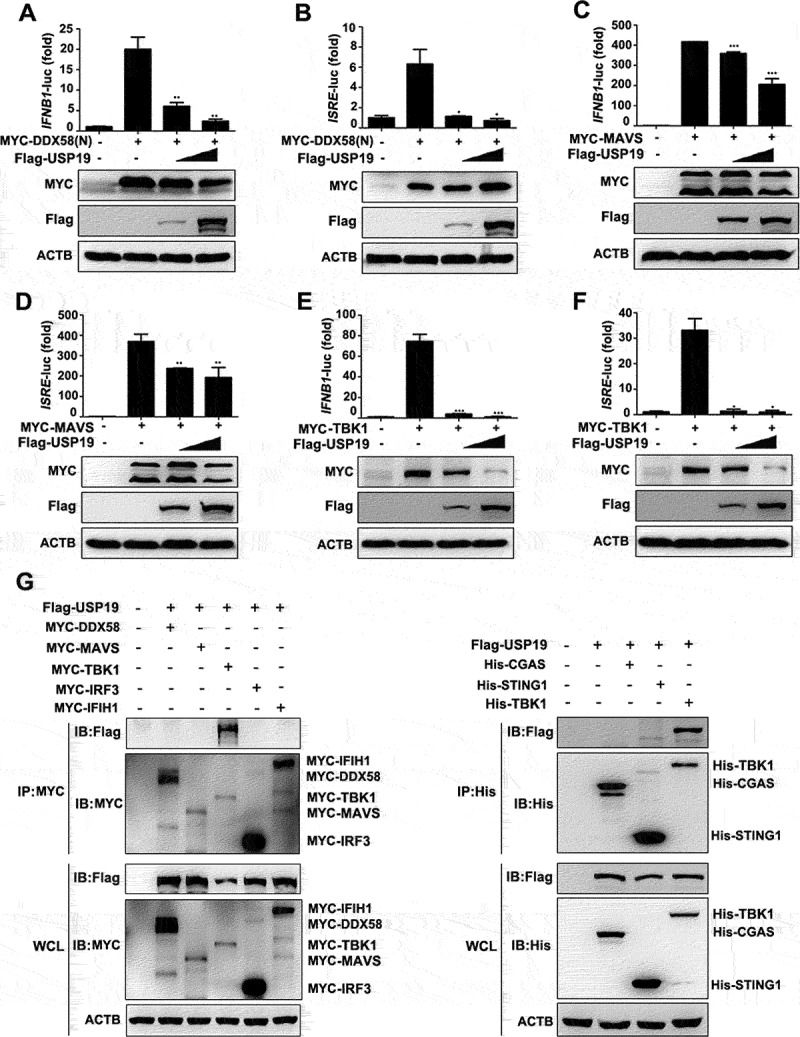
USP19 negatively regulates antiviral signaling by targeting TBK1. (A-F) HEK293T cells were transfected with different concentrations of USP19 plasmids and DDX58 (A and B), MAVS (C and D) or TBK1 (E and F) plasmids. Dual-luciferase reporter assays were performed 24 h after transfection. Exogenous USP19 protein levels were analyzed via immunoblotting. (G) HEK293T cells were transfected with MYC-DDX58, MYC-MAVS, MYC-TBK1, MYC-IRF3, MYC-IFIH1, His-CGAS, His-STING1, His-TBK1, and Flag-USP19 expression plasmids for 24 h before the lysates were subjected to immunoprecipitation with an anti-MYC or His antibody, and the results were analyzed via immunoblotting. The data are representative of three independent experiments. Error bars show the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
Knockdown of USP19 in human PBMCs promotes VSV-induced IFNB production and cellular antiviral response
Thus far, we have shown that USP19 promotes TBK1 degradation via CMA and regulates antiviral innate immunity. To explore the potential clinical value of these findings, we next investigated the effect of USP19 on cellular antiviral responses in primary human peripheral blood mononuclear cells (PBMCs). USP19-siRNA treatment inhibited USP19 expression in the PBMCs (Figure 6(A, B)). Upon infection with VSV, USP19-knockdown PBMCs and VER-155,008-pretreated USP19-knockdown PBMCs expressed higher levels of IFNB1 mRNA and subsequently produced more IFNB protein than VSV-infected control cells (Figure 6(C, D)). Viral replication was inhibited in USP19-knockdown PBMCs infected with VSV. Similarly, the VER-155,008-pretreated USP19-knockdown PBMCs did not exhibit a stronger antiviral response than the USP19-knockdown PBMCs (Figure 6E). Next, we investigated the effects of USP19 on VSV-induced signaling pathway activation. Expression of the TBK1 protein was substantially elevated in USP19-knockdown PBMCs (Figure 6F), suggesting that USP19 negatively regulates the antiviral response by targeting TBK1. Finally, as interferon downstream signaling molecules, the phosphorylation of STAT1 and STAT2 significantly increased in USP19-knockdown PBMCs infected with VSV (Figure 6G). Taken together, these data suggest that USP19 negatively regulates IFNB production and the cellular antiviral response.
Figure 6.
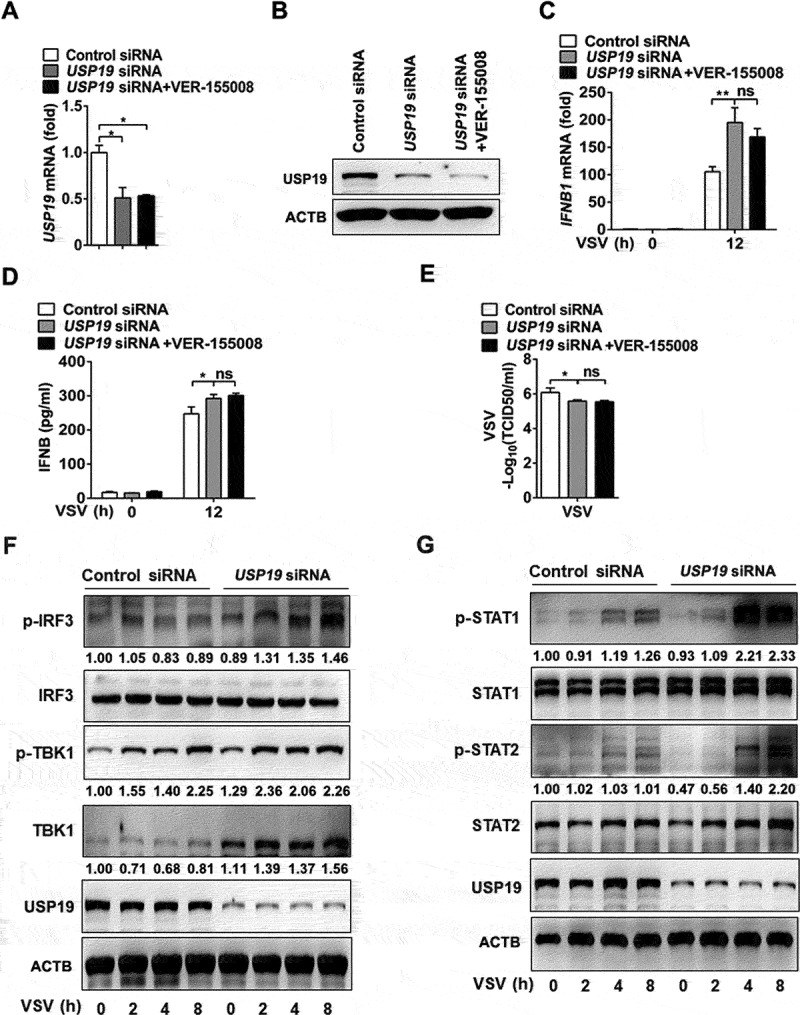
Knockdown of USP19 promotes cellular antiviral response in human PBMCs. (A-E) PBMCs were transfected with control or USP19 siRNAs for 48 h, with or without pretreatment with VER-155,008 (5 μM) for 30 min. USP19 expression was analyzed via qPCR (A) and immunoblotting (B). qPCR analysis of IFNB1 expression 12 h after VSV infection (MOI = 1) (C). ELISA analysis of IFNB production in PBMC supernatants 12 h after VSV infection (MOI = 1) (D). qPCR analysis of VSV replication 12 h after VSV infection (MOI = 1) (E). (F and G) PBMCs were transfected with control or USP19 siRNAs for 48 h, VSV infection (MOI = 1) was performed at the indicated times, and the indicated proteins were analyzed via immunoblotting. The data are representative of three independent experiments. Error bars show the means ± SD. *p < 0.05, **p < 0.01 using Student’s t test.
USP19 deficiency enhanced type I interferon production and antiviral response in mouse peritoneal macrophages by suppressing CMA-mediated TBK1 degradation
To further investigate the role of USP19 in antiviral signaling in vitro and in vivo, we generated Usp19fl/+ mice via CRISPR-Cas9-mediated genome editing (Figure S3A). Southern blot analysis indicated that the targeting vector was successfully recombined with the wild-type (WT) allele (Figure S3B). Genotype identification showed that transgenic mice were successfully constructed (Figure S3C). We crossed Usp19fl/+ mice with Lyz2-Cre mice to obtain Usp19fl/fl and usp19fl/fl Lyz2-Cre mice, and found that the usp19fl/fl Lyz2-Cre mice exhibited USP19 ablation in their macrophages (Figure S3D, S3E). qPCR analysis suggested that usp19 knockout significantly promoted VSV or transfected poly(I:C) (recognized by DDX58)-induced Ifna4 and Ifnb1 expression in mouse peritoneal macrophages, while VER-155,008 did not significantly promote Ifna4 and Ifnb1 expression in the usp19-knockout macrophages (Figure 7(A-D)). In the peritoneal macrophages from the Usp19fl/fl and usp19fl/fl Lyz2-Cre mice, we found that USP19 negatively regulates the herpes simplex virus (HSV)-induced Ifnb1, Ifna4, and Isgs (e.g., Isg15, Ifit2, Ifit1, and Mx1) mRNA expression (Figure S3F-S3H). In addition, our results demonstrated that Ifnb1 mRNA was expressed at higher levels in usp19-knockout macrophages stimulated with toll-like receptor stimuli such as poly(I:C) (recognized by TLR3) and LPS (recognized by TLR4) (Figure S3I and S3J). These results verified that USP19 negatively regulates Ifnb1 expression by targeting TBK1.
Figure 7.
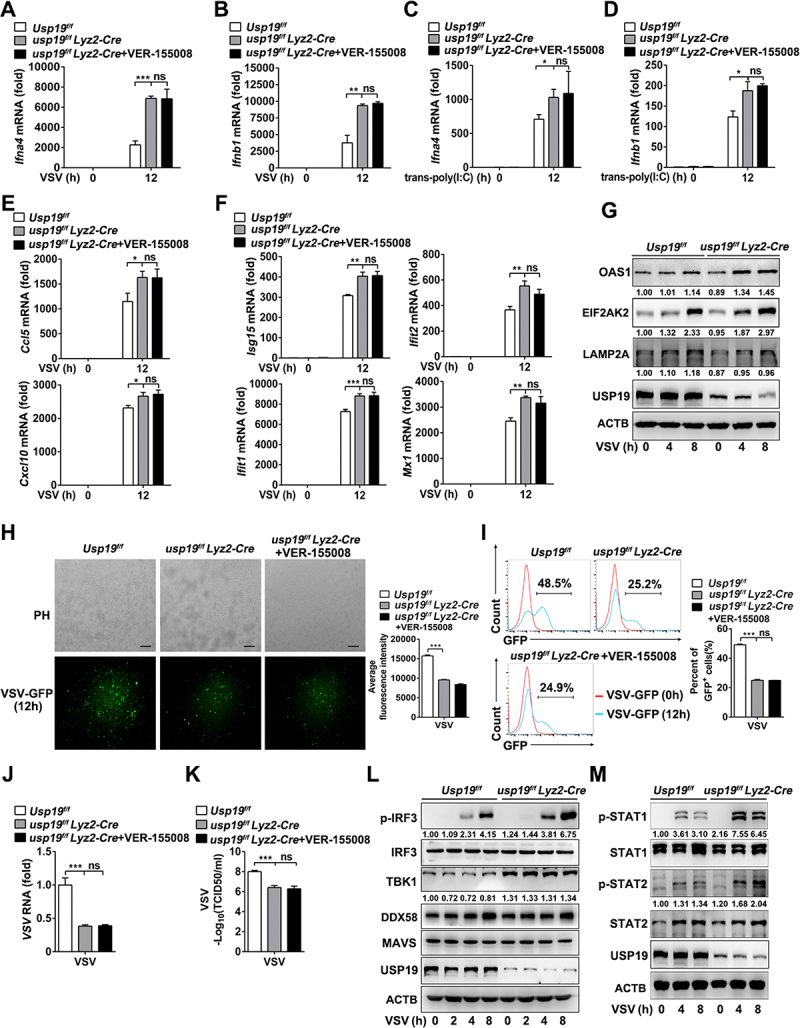
USP19 deficiency in macrophages enhanced VSV-induced type I interferon production and antiviral response by suppressing CMA-mediated TBK1 degradation. (A and B) qPCR analysis of Ifna4 (A) and Ifnb1 (B) expression in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV (MOI = 1) for the indicated times with or without VER-155,008 (5 μM) pretreatment. (C and D) qPCR analysis of Ifna4 (C) and Ifnb1 (D) expression in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages transfected with poly(I:C) (1 μg/mL) for the indicated hours with or without pretreatment with VER-155,008 (5 μM). (E and F) qPCR analysis of Ccl5, Cxcl10 (E), Isg15, Ifit2, Ifit1, and Mx1 expression (F) in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV (MOI = 1) for 12 h with or without pretreatment with VER-155,008 (5 μM). (G) Immunoblot analysis of OSA1, EIF2AK2, and LAMP2A expression in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV (MOI = 1) for the indicated durations. (H and I) Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages were infected with VSV-GFP for 12 h with or without VER-155,008 (5 μM) pretreatment, and images were captured under a fluorescence microscope (H). The percentage of GFP+ cells was determined via flow cytometry (I). (J) qPCR analysis of VSV replication in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV (MOI = 1) with or without VER-155,008 (5 μM) pretreatment. (K) Determination of VSV titers in supernatants via a TCID50 assay of Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV with or without VER-155,008 (5 μM) treatment. (L) Immunoblot analysis of the indicated proteins in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV (MOI = 1) for the indicated durations. (M) Immunoblot analysis of STAT1 and STAT2 phosphorylation in Usp19flox/flox and usp19flox/flox Lyz2-Cre peritoneal macrophages infected with VSV (MOI = 1) for the indicated hours. The data are representative of three independent experiments. Scale bars: 100 μm. Error bars show the means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test.
Similarly, knockout of usp19 in VSV-infected macrophages expressed higher levels of Ccl5 and Cxcl10 mRNA (Figure 7E), and Isg mRNA, such as Isg15, Ifit2, Ifit1, and Mx1 (Figure 7F). The data also showed that VER-155,008 treatment had no additional effects on the expression of these genes in usp19-knockout macrophages with VSV infection (Figure 7 (E-F)). In addition,usp19-knockout mouse peritoneal macrophages expressed higher levels of type I interferon downstream of OAS1 (2ʹ-5′-oligoadenylate synthetase) and EIF2AK2/PKR (eukaryotic translation initiation factor 2-alpha kinase 2) (Figure 7G). Finally, a loss of USP19 in macrophages resulted in a decrease in LAMP2A protein expression (Figure 7G), suggesting that usp19 knockout weakens CMA in macrophages.
We next monitored VSV replication via fluorescence microscopy and flow cytometry analysis to detect GFP-positive cells after VSV-GFP infection. The results indicated that usp19 deficiency reduced the GFP intensity and the percentage of GFP-positive cells compared to Usp19 WT cells (Figure 7 (H, I)). qPCR analysis of VSV RNA levels also suggested that usp19 knockout significantly suppressed VSV replication (Figure 7J). The TCID50 assay indicated that VSV titers in supernatants of usp19 knockout peritoneal macrophages were significantly decreased (Figure 7K). Similarly, we also found that the antiviral response against VSV infection was not significantly different between usp19-knockout macrophages with and without VER-155,008 pretreatment. These results suggest that USP19 regulates the antiviral response.
We further investigated the effects of USP19 on VSV-induced signaling pathway activation. IRF3 phosphorylation and the expression of the TBK1 protein were substantially elevated after VSV infection in usp19-knockout mouse peritoneal macrophages (Figure 7L), while other proteins involved in VSV-induced signaling pathways, such as DDX58 and MAVS, were unaffected (Figure 7L), suggesting that USP19 targets TBK1 and promotes its degradation to negatively regulate antiviral responses. Finally, STAT1 and STAT2 phosphorylation significantly increased after VSV infection in usp19-knockout mouse peritoneal macrophages (Figure 7M). These results demonstrate that USP19 deficiency enhances type I interferon production and promotes VSV-induced antiviral signaling activation in mouse peritoneal macrophages.
USP19 deficiency augments the host response against VSV infection
To investigate the role of USP19 in the antiviral innate response in vivo, we challenged Usp19fl/fl mice, usp19fl/fl Lyz2-Cre mice, and VER-155,008-pretreated usp19fl/fl Lyz2-Cre mice with VSV via intraperitoneal injection. qPCR analysis showed that usp19fl/fl Lyz2-Cre mice expressed higher levels of Ifna4 and Ifnb1 in various organs (including the liver, lung, and spleen) than Usp19fl/fl mice after VSV infection, and VER-155,008 treatment did not further promote Ifna4 and Ifnb1 expression in usp19fl/fl Lyz2-Cre mice after VSV infection (Figure 8(A, B)). VSV titers and replication in these organs from usp19fl/fl Lyz2-Cre mice or VER-155,008-pretreated usp19fl/fl Lyz2-Cre mice were significantly lower than those in Usp19fl/fl mice (Figure 8C). Results from an ELISA assay also indicated that IFNB production in the sera of usp19fl/fl Lyz2-Cre mice or VER-155,008 pretreated usp19fl/fl Lyz2-Cre mice was significantly increased compared to that in the sera of Usp19fl/fl mice (Figure 8D). We also observed less inflammatory cell infiltration and slight tissue injury in the lungs of usp19fl/fl Lyz2-Cre mice after infection with VSV compared with Usp19fl/fl mice, while that in the usp19fl/fl Lyz2-Cre mice showed no significant difference compared with the VER-155,008 pretreated usp19fl/fl Lyz2-Cre mice (Figure 8E). More importantly, usp19fl/fl Lyz2-Cre mice showed a high level of resistance to VSV infection in terms of overall survival compared to Usp19fl/fl mice (Figure 8F). Together, these results suggest that USP19 suppresses IFNB production and inhibits the antiviral innate response in the context of VSV infection in vivo.
Figure 8.
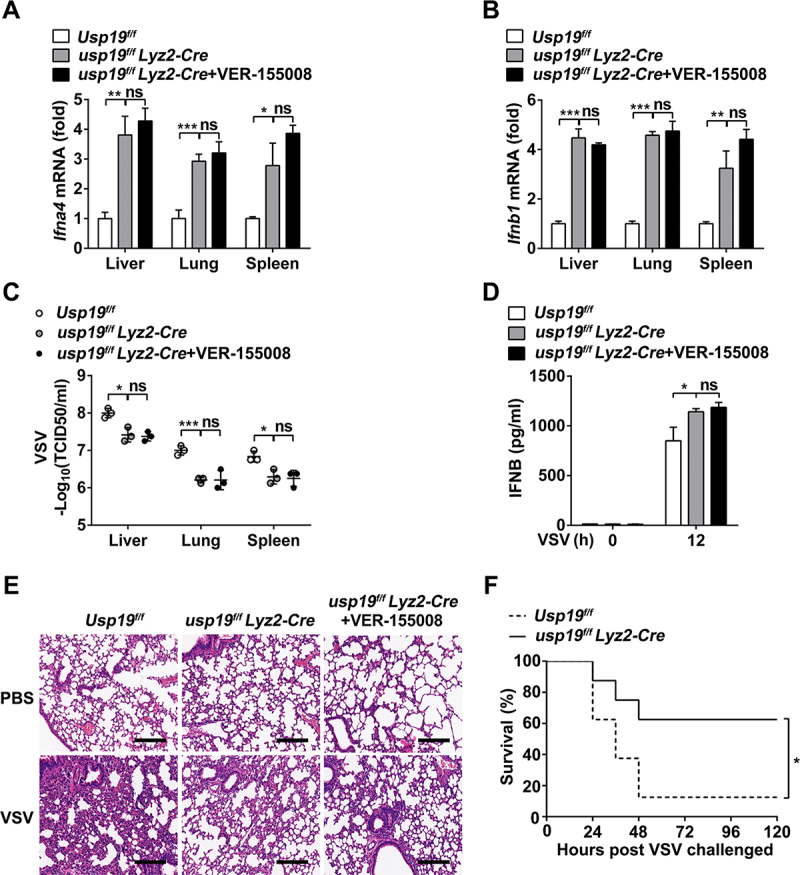
USP19 macrophage-deficient mice show resistance to VSV infection. (A and B) qPCR analysis of Ifna4 (A) and Ifnb1 (B) mRNA expression in organs from Usp19flox/flox and usp19flox/flox Lyz2-Cre mice 12 h after VSV administration (1 × 108 PFU/g) via intraperitoneal injection (n = 3 per group) with or without VER-155,008 (40 mg/kg) treatment. (C) Determination of VSV loads in organs through the TCID50 assay using Usp19flox/flox and usp19flox/flox Lyz2-Cre mice (n = 3 per group). (D) ELISA analysis of IFNB production in sera from Usp19flox/flox and usp19flox/flox Lyz2-Cre mice (n = 3 per group). Error bars show means ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 using Student’s t test. (E) Pathology of Usp19flox/flox and usp19flox/flox Lyz2-Cre mice in response to VSV infection with or without pretreatment with VER-155,008 (40 mg/kg). Scale bar: 200 μm. (F) Survival of Usp19flox/flox and usp19flox/flox Lyz2-Cre mice administered with VSV (1 × 108 PFU/g) via intraperitoneal injection (n = 8–10 per group). Statistical significance was calculated using the Log-rank (Mantel-Cox) test. The data are representative of three independent experiments.
Discussion
TBK1 is an essential adapter protein for the innate immune defense against viruses [31,32]. Phosphorylated TBK1 promotes the phosphorylation and nuclear translocation of IRF3, resulting in type I IFN production [33]. The availability and activity of TBK1 are strictly controlled by several post-translational modifications, such as phosphorylation and ubiquitination, to exert sufficient protective immunity and avoid an excessive harmful immune response [28,34]. Autophagy has also been reported to play important roles in regulating type I IFN production and the innate immune response [35]. Prior to this study, however, the role of CMA in regulating TBK1 is largely unknown.
By performing an unbiased screen comprising co-transfection and immunoblotting, we identified that USP19 could promote TBK1 degradation. USP19 is a deubiquitinating enzyme involved in many biological processes [24–26]. By inhibiting cellular proteasomal activity, lysosomal activity, autophagosome formation, or autophagosome-lysosome fusion, only the lysosomal inhibitor CQ was able to repress USP19-mediated TBK1 degradation. Interestingly, USP19, as a deubiquitinating enzyme, did not affect the ubiquitination of TBK1. Moreover, USP19C607S (enzymatically inactive USP19) still interacted with TBK1 and promoted TBK1 degradation. Based on these experimental results, we deduced that USP19 promotes TBK1 degradation in a lysosome-dependent manner, independent of its deubiquitinase activity. Our results showed that USP19 promoted the degradation of total TBK1 protein, independent of VSV infection. Furthermore, we analyzed the densitometry-based quantification of the total and phosphorylated forms of TBK1 (p-TBK1) then calculated the ratio of phosphorylated protein to total protein. The results showed that even though the total TBK1 protein was significantly upregulated in the USP19-knockdown cells, the ratio of p-TBK1 to TBK1 was not increased in the USP19-knockdown cells compared to the control cells. These data suggest that the phosphorylated form of TBK1 might not be directly degraded by USP19, and that the changes in phosphorylated TBK1 were mainly due to changes in the total protein. Overexpressing USP19 elevated the cleavage of LC3B, which is consistent with a previous study that showed USP19 modulates autophagy [25]. By comparing the amino acid sequences of TBK1 among different species, we found that TBK1 had a conserved CMA-motif (KFDKQ). This finding led us to hypothesize that USP19 promotes TBK1 degradation via CMA. CMA is a strictly selective protein degradation process that is dependent on HSPA8 and LAMP2A [9,36]. Through co-immunoprecipitation and immunofluorescence staining, we found that USP19 could interact with TBK1, HSPA8, and LAMP2A and promote the expression of HSPA8 and LAMP2A. More importantly, knockdown of HSPA8 or LAMP2A in HeLa cells restored TBK1 protein level in USP19-overexpressiong cells. These results were the first to identify that USP19-regulated CMA promotes TBK1 protein degradation. This finding has physiological significance in terms of regulating the TBK1-mediated innate immune response (Figure 9).
Figure 9.
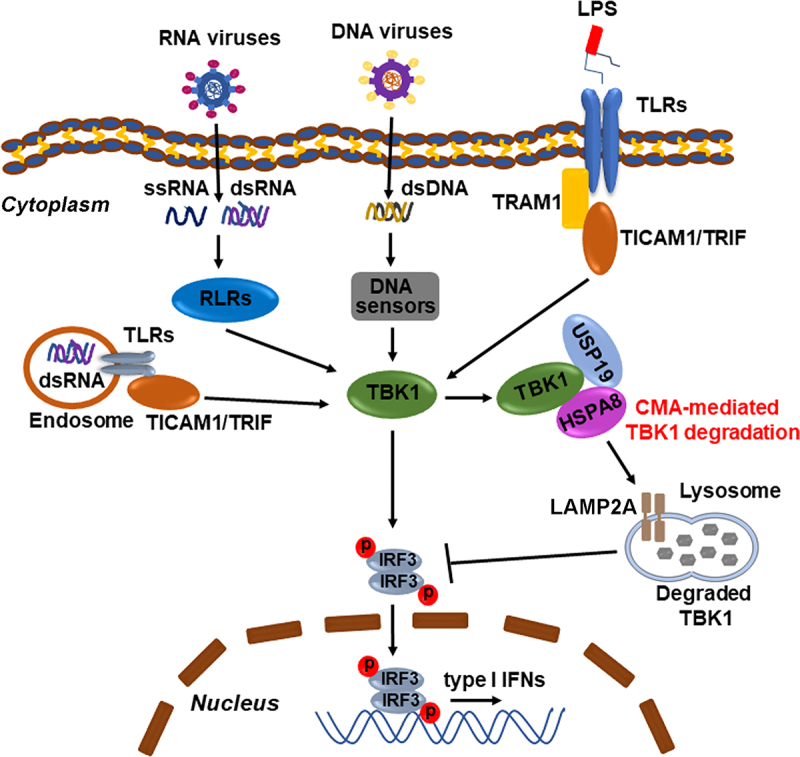
Schematic representation of the role of USP19 in the regulation of TBK1 degradation via CMA. Pattern recognition receptors (PRRs, e.g., RLRs, TLRs, and CGAS) can recognize pathogen-associated molecular patterns (PAMPs, e.g., ssRNA, dsRNA, dsDNA, and LPS) and recruit different receptor proteins (e.g., MAVS, STING1, and TICAM1/TRIF) to activate TBK1. USP19 interacts with the TBK1-HSPA8 complex and then promotes TBK1 degradation via CMA, negatively regulating type I interferon production.
TBK1 belongs to the IKK kinase family and is involved in innate immune signaling pathways [37,38]. Upon viral infection, viral DNA or RNA is recognized by PRRs and recruits different receptor proteins to activate TBK1, which then activates IRF3 and IRF7, and promotes the production of type I IFN [39,40]. We found that USP19-knockdown or deficiency increased type I IFN expression during VSV infection in various cell types, including HeLa cells, THP-1 cells, PBMCs, and mouse peritoneal macrophages. This finding is consistent with a previous study showing that USP19 negatively regulates virus-triggered type I IFN induction. Mechanistically, that study verified that USP19 deubiquitinated BECN1/beclin 1 and impaired the association between DDX58 and MAVS [25]. Meanwhile, other research has shown that USP19 regulates TICAM1 deubiquitination and impairs TLR3/4 signaling, but has no effect on virus-triggered type I IFN induction in RAW264.7 cells [24]. It is possible that USP19 has distinct functions in RAW264.7 cells compared with that in primary macrophages and in other cell lines. Moreover, our results showed that a USP19 deficiency in macrophages enhanced TBK1 protein levels. Data from our experimental results and previous studies support the idea that USP19 has multiple functions in the cellular immune response as a result of interacting with different target proteins. Using our usp19 conditional knockout mice, we found that the loss of USP19 in macrophages did not affect survival or growth; however, these mice were more resistant to VSV infection, as evidenced by high type I IFN production in the serum, a low VSV load in the major organs, and a high survival rate after VSV infection. Moreover, we also found that there was no significant difference in type I IFN production and antiviral response between VSV-infected usp19-knockout macrophages with and without CMA inhibitor VER-155,008 pretreatment. Surprisingly, we found that VER-155,008 treatment alone inhibited IFNB1 expression in HeLa or THP1 cells upon VSV infection but has slightly upregulated VSV-induced Ifnb1 expression in usp19-knockout mouse peritoneal macrophages (data not shown). However, VER-155,008 treatment in USP19-knockdown HeLa cells, THP1 cells, or usp19-knockout macrophages showed similar effects on the regulation of IFNB1 expression as induced by VSV infection. These results suggest that VER-155,008 treatment may have different functions in different cells, not just in the regulation of CMA by inhibiting HSPA8 and HSPA. Based on these data, our results revealed that USP19 negatively regulated the cellular antiviral immune response by promoting the CMA-mediated degradation of TBK1, but not directly depending on CMA, to regulate the antiviral immune response. In addition, usp19-knockout macrophages also produced more type I IFN when the cells were stimulated with HSV, poly(I:C), or LPS, all of which depend on TBK1 activation.
In summary, our results identified a novel function of USP19 in promoting TBK1 degradation via CMA and suggest that USP19 facilitates CMA by upregulating LAMP2A and HSPA8 expression, but the detailed mechanism still needs further investigation. This finding demonstrates that USP19 plays a decisive role in innate immune responses against viruses and provides a potential therapeutic target for some infectious diseases.
Materials and methods
Mice
Usp19fl/+ mice were generated by the Nanjing Biomedical Research Institute of Nanjing University via CRISPR-Cas9-mediated gene editing. In brief, guide RNAs (gRNAs, 5′-TCAGCCTTAAAGATTAGGAGTGG-3′ and 5′-GGCCTCGAGCCCATCGTTTG-3′) were obtained through in vitro transcription and purification. The gRNAs and Cas9 protein were mixed and injected into one-cell stage fertilized eggs together with the targeting vector containing two loxP sites flanking exons 2 and 26 of the Usp19 gene. The injected fertilized eggs were then transplanted into pseudo-pregnant mice. The targeted genomes of the F0 mice were amplified via PCR and sequenced. The chimeras were crossed with wild-type C57BL/6 mice to obtain F1 Usp19fl/+ mice.
Lyz2-Cre mice were purchased from the Nanjing Biomedical Research Institute of Nanjing University. Usp19fl/+ mice were crossed with Lyz2-Cre mice to obtain usp19fl/fl Lyz2-Cre mice. Usp19fl/fl littermates and usp19fl/fl Lyz2-Cre littermates (8 weeks) were used throughout the study. Murine genotypes were determined via PCR analysis of tail DNA, using the following primers to amplify the wild-type (WT) allele (300 bp) and the floxed allele (346 bp): Usp19 forward: 5′-CTATCACCGTGTCTTAAAACTGGAG-3′, Usp19 reverse: 5′-GCTTCCTGCCGGCTCCTATTT-3′; Lyz2-Cre forward: 5′-GAACGCACTGATTTCGACCA-3′, Lyz2-Cre reverse: 5′-GCTAACCAGCGTTTTCGTTC-3′.
Mice were maintained and bred under pathogen-free conditions. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of Shenzhen University, Shenzhen, China.
Cell culture
Mouse peritoneal macrophages were extracted and cultured as described previously [41]. THP-1 cells were cultured in RPMI1640 (Keyi Biotechnology, C0003) with 10% fetal bovine serum, while HEK293T and HeLa cells were cultured in DMEM (Keyi Biotechnology, C0006) containing 10% fetal bovine serum. Enriched populations of human peripheral blood mononuclear cells (PBMCs) were obtained from the blood of healthy donors as described previously [42], cultured in RPMI1640 with 10% fetal bovine serum, and differentiated for 5 days into macrophages using human macrophage colony-stimulating factor (100 ng/mL; Preprotech Group, AF-300-25). THP-1 (TIB-202), HEK293T (CRL-3216), and HeLa (CRM-CCL-2) cells were obtained from ATCC. The tbk1-knockout RAW264.7 cell line was a gift from Dr. Zhou Yu (Center of Systems Medicine, Chinese Academy of Medical Sciences, Suzhou, China) [43]. All cell cultures were maintained at 37°C in 5% CO2 and 95% humidity in the appropriate media.
Plasmids and reagents
Flag-tagged full-length and deletion mutants of USP19, MYC-tagged TBK1 (MYC-TBK1), deletion mutants of TBK1-encoding plasmids, V5-tagged LAMP2A (V5-LAMP2A), and HA-tagged HSPA8 (HA-HSPA8) plasmids were constructed using standard molecular biology techniques. Flag/HA-tagged DUB plasmids were purchased from Addgene (catalog numbers and depositors are shown in Table S1). Antibodies specific to p-IRF3 (4947), IRF3 (4302), p-TBK1 (5483), TBK1 (3504), p-STAT1 (9167), STAT1 (14,994), p-STAT2 (88,410), STAT2 (72,604), DDX58 (3743), and MAVS (4983) were purchased from Cell Signaling Technology. Anti-USP19 (ab93159) was purchased from Abcam. Anti-Flag (M185-3) antibody was purchased from MBL Life Sciences. Anti-HA (51,064), LAMP2A (27,823), HSPA8 (10,654), OAS1 (14,955-1-AP), EIF2AK2 (18,244-1-AP), MYC (10,828-1-AP), Flag 20,543-1-AP, and ACTB/β-actin (60,008) were purchased from the Proteintech Group. Anti-MYC (A00704) was purchased from Genscript Biotechnology. Poly(I:C) (tlrl-pic) and LPS (tlrl-b5lps) were purchased from InvivoGen. Chloroquine (C6628) was purchased from Sigma-Aldrich. MG132 (S2619) and bafilomycin A1 (S1413) were purchased from Selleck Chemicals. VER-155008 (HY-10,941) and Torin 1 (HY-13,003) were purchased from MedChemExpress.
Co-immunoprecipitation and immunoblot analyses
Immunoblot analysis was performed as described previously [44]. For co-immunoprecipitation, cells were lysed in NP-40 lysis buffer (Beyotime Biotechnology, P0013F) containing a protease inhibitor cocktail (B14001; Biomake). The cell lysates were centrifuged at 12,000 × g for 5 min, incubated overnight at 4°C, and immunoprecipitated using IgG or the appropriate antibodies. For the ubiquitination assay, cells were lysed in immunoprecipitation buffer (with 1% SDS) containing a protease inhibitor mixture and boiled for 5 min at 95°C, and the supernatant was collected and diluted 1:10 in immunoprecipitation buffer, followed by incubation overnight at 4°C and immunoprecipitation using IgG (Beyotime Biotechnology, A7028) or the appropriate antibodies. The precipitates were washed seven times with lysis buffer containing 500 mM NaCl and analyzed through an immunoblot assay.
In vitro binding assays
HEK293T cells were plated into 10-cm dishes (2 × 106 cells per dish) and transfected with Flag-USP19 or MYC-TBK1 for 24–36 h, and then lysed in a NP-40 lysis buffer (Beyotime Biotechnology, P0013 F). The cell lysates were centrifuged at 12,000 × g for 5 min, incubated overnight at 4°C, and immunoprecipitated with Flag (Sigma-Aldrich, M8823-1ML) or MYC beads (Biomake, B26302). The beads were washed five times with a lysis buffer containing 500 mM NaCl. We added an elution buffer (0.1 M glycine, pH 3) to the beads for 5 min and then added a neutralization buffer (0.1 M glycine, pH 8). The mixtures were then centrifuged at 1,000 × g for 5 min, 5 μL of the supernatant was collected and subjected to gel electrophoresis, and Coomassie Brilliant Blue staining was performed to confirm the purified proteins. Next, purified Flag-USP19 and MYC-TBK1 proteins were mixed in the NP-40 lysis buffer (Beyotime Biotechnology, P0013 F) for co-immunoprecipitation and analyzed via an immunoblot assay.
Dual-luciferase reporter assays
HEK293T cells (2 × 104 cells per well of a 96-well plate) were co-transfected with luciferase reporter plasmids (IFNB1 or ISRE luciferase reporter), the Renilla luciferase construct pRL-TK (Promega Corporation, E2241; for normalizing the transfection efficiency), and the indicated expression plasmids. The cells were harvested after 24 h. Luciferase assays were performed using a dual-specific luciferase assay kit (Promega Corporation, E1960) according to the manufacturer’s protocol. All experiments were performed in triplicate, and the fold changes were calculated relative to the baseline control in each experiment.
Lysosome enrichment assays
A lysosome enrichment assay was performed using a lysosome enrichment kit for tissues and cultured cells (Thermo Fisher Scientific, 89,839), according to the manufacturer’s protocol.
Quantitative PCR (qPCR) and ELISA
Total RNA was extracted from cells using RNAiso Plus (Takara Biotechnology, 9109), and the first-strand cDNA was reverse-transcribed using SMART MMLV Reverse Transcriptase (Takara Biotechnology, 639,524) according to the manufacturer’s instructions. Gene expression was examined using Hieff qPCR SYBR Green Master Mix (without ROX) (YEASEN Biotechnology, 11201ES03), and gene expression was normalized to Actb using the ΔΔCt method. The gene-specific primers were as follows: HsUSP19 forward, 5′-TAAATCCAAGGCACGATCTGAGG-3′ and reverse, 5′-GCTTTGGGGTTACATGCTCCA-3′; HsIFNB1 forward, 5′- CATTACCTGAAGGCCAAGGA-3′ and reverse, 5′-CAATTGTCCAGTCCCAGAGG-3′; HsLAMP2A forward, 5′-ATTTGGTTAATGGCTCCGTTT-3′ and reverse, 5′-CACATTGAAAGGCTGAACCC-3′; HsHSPA8 forward, 5′-CCTCATCAAGCGTAATACCAC-3′ and reverse, 5′-TCATAAACCTGAATAAGCACACC-3′; MmUsp19 forward, 5′-TGTCAAGAACGACTCGTATGAGA-3′ and reverse, 5′- CTGGAAGATCAGTGTGAAGTCC-3′; MmIfnb1 forward, 5′-ATGAGTGGTGGTTGCAGGC-3′ and reverse, 5′- TGACCTTTCAAATGCAGTAGATTCA-3′; MmIfna4 forward, 5′-ACCCACAGCCCAGAGAGTGACC-3′ and reverse, 5′-AGGCCCTCTTGTTCCCGAGGT-3′; HsACTB forward, 5′-TGGAGAAAATCTGGCACCACACC-3′ and reverse, 5′- GATGGGCACAGTGTGGGTGACCC-3′; MmActb forward, 5′- AGTGTGACGTTGACATCCGT-3′ and reverse, 5′- GCAGCTCAGTAACAGTCCGC −3′.
ELISA kits (BioLegend, 439,407) were used to detect the IFNB concentration in the culture supernatants and sera, according to the manufacturer’s protocols.
Plasmid transfection and RNA interference assay
The plasmids was transfected into cells using jetPRIME (PolyPlus Transferion, 114–15), according to the manufacturer’s protocol. Cells were grown for up to 60%–80% confluence in the 12-well plate plates. DNA was diluted into jetPRIME buffer and mixed via vortexing. The appropriate amount of jetPRIME reagent was then added to the diluted DNA, vortexed for 10 s, and spun down briefly. The cells were the incubated for 10 min at RT, and the transfection mix was added dropwise onto the cells per well with serum-containing medium and distributed evenly. The plates were gently rocked back and forth and from side to side, and were analyzed after 24 h or later. For THP-1 cells, PMA (phorbol 12-myristate 13-acetate, 100 ng/mL) was used to promote the differentiation of THP-1 monocytes into macrophages before transfection. The siRNA oligos were synthesized and transfected into cells using jetPRIME (PolyPlus Transferion, 114–15), according to the manufacturer’s protocol. After 48 h of transfection, the cells were harvested or stimulated with VSV (or VSV-GFP) and analyzed via qPCR, immunoblotting, or flow cytometry (detection VSV-GFP). The siRNA sequences were as follows: Control siRNA: 5′-UUCUCCGAACGUGUCACGUUU-3′; USP19 siRNA: 5′-GAUCAAUGACUUGGUGGAGUU-3′; LAMP2A siRNA: 5′-GCUCGUUCUGGUCUGCCUAUU-3′; HSPA8 siRNA: 5′-UAAUUCUAAGUACAUUGAGACCAGC-3′.
Stimulants and virus infection
Stimulants were used at the following concentrations: LPS, 100 ng/mL; poly(I:C), 10 μg/mL; or transfected with poly(I:C), 1 μg/mL. Cells were infected with VSV (multiplicity of infection [MOI] = 1), GFP-labeled VSV (MOI = 1), or HSV (MOI = 5) for the indicated times. The cell lysates were analyzed via qPCR or immunoblotting, the supernatants were analyzed via ELISA or the TCID50 assay, and the cells were harvested for flow cytometry (for the detection of VSV-GFP). The TCID50 assay was performed as described previously [45]. For in vivo studies, age-and sex-matched littermates were intraperitoneally injected with VSV (1 × 108 PFU/g) with or without pretreatment with VER-155008 (40 mg/kg). The sera of the mice were collected for ELISA detection 12 h after VSV infection. Mice were euthanized 12 h after viral infection to obtain the liver, lung, and spleen tissues for qPCR analysis. Lungs from control or virus-infected mice were dissected, fixed and stained with hematoxylin-eosin using standard procedures, and examined via light microscopy for histological changes. For survival experiments, Usp19fl/fl and usp19fl/fl Lyz2-Cre mice were intraperitoneally injected with VSV and then monitored for survival after viral infection.
Statistical analyses
Statistical significance between two groups was determined using two-tailed Student’s t-test. For the mouse survival study, Kaplan-Meier survival curves were generated and analyzed for statistical significance. The data are presented as the mean ± SD from three independent experiments using GraphPad Prism. Statistical significance was set at p < 0.05; significance levels are presented in the figures.
Acknowledgments
We thank Jessica Kate Tamanini (Scientific Editor, Shenzhen University School of Medicine) for editing manuscript. We thank Dr. Zhou Yu (Center of Systems Medicine, Chinese Academy of Medical Sciences, Suzhou, China) for sharing the tbk1-knockout RAW264.7 cell.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China [No. U1801283, 31870908, 31670914], a grant from the Zhejiang Provincial Natural Science Foundation of China [No. LZ17H100001], a grant from the Shenzhen Science and Technology Innovation Commission [JCYJ20180507182253653], the Guangdong Provincial Science and Technology Program [No. 2019B030301009] and the SZU Top Ranking Project [No. 86000000210] awarded to Dr. Weilin Chen.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Yu L, Chen Y, Tooze SA.. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ktistakis NT, Tooze SA.. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26:624–635. [DOI] [PubMed] [Google Scholar]
- [3].Shibutani ST, Saitoh T, Nowag H, et al. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. 2015;16:1014–1024. [DOI] [PubMed] [Google Scholar]
- [4].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [5].Levine B, Kroemer G. Biological Functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang H, Baehrecke EH. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015;25:376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. [DOI] [PubMed] [Google Scholar]
- [10].Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Valdor R, Mocholi E, Botbol Y, et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. 2014;15:1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Anguiano J, Garner TP, Mahalingam M, et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. 2013;9:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Arias E, Koga H, Diaz A, et al. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59:270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li W, Nie T, Xu H, et al. Chaperone-mediated autophagy: advances from bench to bedside. Neurobiol Dis. 2019;122:41–48. [DOI] [PubMed] [Google Scholar]
- [15].Suzuki J, Nakajima W, Suzuki H, et al. Chaperone-mediated autophagy promotes lung cancer cell survival through selective stabilization of the pro-survival protein, MCL1. Biochem Biophys Res Commun. 2017;482:1334–1340. [DOI] [PubMed] [Google Scholar]
- [16].Demand J, Alberti S, Patterson C, et al. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr Biol. 2001;11:1569–1577. [DOI] [PubMed] [Google Scholar]
- [17].Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. [DOI] [PubMed] [Google Scholar]
- [18].Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. [DOI] [PubMed] [Google Scholar]
- [19].Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. [DOI] [PubMed] [Google Scholar]
- [20].Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lin M, Zhao Z, Yang Z, et al. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination through NLRP4 Signalosome. Mol Cell. 2016;64:267–281. [DOI] [PubMed] [Google Scholar]
- [23].Li D, Yang W, Ren J, et al. The E3 ubiquitin ligase TBK1 mediates the degradation of multiple picornavirus VP3 proteins by phosphorylation and ubiquitination. J Virol. 2019;93:e01438-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu X, Lei C, Xia T, et al. Regulation of TRIF-mediated innate immune response by K27-linked polyubiquitination and deubiquitination. Nat Commun. 2019;10:4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jin S, Tian S, Chen Y, et al. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 2016;35:866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wiles B, Miao M, Coyne E, et al. USP19 deubiquitinating enzyme inhibits muscle cell differentiation by suppressing unfolded-protein response signaling. Mol Biol Cell. 2015;26:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang X, Ni L, Wan S, et al. Febrile temperature critically controls the differentiation and pathogenicity of T helper 17 cells. Immunity. 2020;52:328–341. [DOI] [PubMed] [Google Scholar]
- [28].Deng M, Tam JW, Wang L, et al. TRAF3IP3 negatively regulates cytosolic RNA induced anti-viral signaling by promoting TBK1 K48 ubiquitination. Nat Commun. 2020;11:2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Porritt RA, Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015;36:150–160. [DOI] [PubMed] [Google Scholar]
- [30].Jia M, Qin D, Zhao C, et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat Immunol. 2020;21:727–735. [DOI] [PubMed] [Google Scholar]
- [31].Zhou R, Zhang Q, Xu P. TBK1, a central kinase in innate immune sensing of nucleic acids and beyond. Acta Biochim Biophys Sin (Shanghai). 2020;52:757–767. [DOI] [PubMed] [Google Scholar]
- [32].Zhao C, Zhao W. TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin Ther Targets. 2019;23:437–446. [DOI] [PubMed] [Google Scholar]
- [33].Hiscott J. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007;18:483–490. [DOI] [PubMed] [Google Scholar]
- [34].Liu S, Cai X, Wu J, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. [DOI] [PubMed] [Google Scholar]
- [35].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018;16:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Catarino S, Pereira P, Girao H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017;61:663–674. [DOI] [PubMed] [Google Scholar]
- [37].Zhao P, Wong KI, Sun X, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172:731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lafont E, Draber P, Rieser E, et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol. 2018;20:1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang C, Shang G, Gui X, et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019;567:394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Weil R, Laplantine E, Genin P. Regulation of TBK1 activity by Optineurin contributes to cell cycle-dependent expression of the interferon pathway. Cytokine Growth Factor Rev. 2016;29:23–33. [DOI] [PubMed] [Google Scholar]
- [41].Zhao X, Zhu H, Yu J, et al. c-Cbl-mediated ubiquitination of IRF3 negatively regulates IFN-beta production and cellular antiviral response. Cell Signal. 2016;28:1683–1693. [DOI] [PubMed] [Google Scholar]
- [42].Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–238. [DOI] [PubMed] [Google Scholar]
- [43].Li X, Yang M, Yu Z, et al. The tyrosine kinase Src promotes phosphorylation of the kinase TBK1 to facilitate type I interferon production after viral infection. Sci Signal. 2017;10:eaae0435. [DOI] [PubMed] [Google Scholar]
- [44].Zhao X, Pu D, Zhao Z, et al. Teuvincenone F suppresses LPS-induced inflammation and NLRP3 inflammasome activation by attenuating NEMO ubiquitination. Front Pharmacol. 2017;8:565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen W, Han C, Xie B, et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell. 2013;152:467–478. [DOI] [PubMed] [Google Scholar]